

Summary
Tumor cell phenotypes and anti-tumor immune responses are shaped by local metabolite availability, but intratumoral metabolite heterogeneity (IMH) and its phenotypic consequences remain poorly understood. To study IMH, we profiled 187 tumor/normal regions from 31 clear cell renal cell carcinoma (ccRCC) patients. A common pattern of IMH transcended all patients, characterized by correlated fluctuations in the abundance of metabolites and processes associated with ferroptosis. Analysis of intratumoral metabolite-RNA covariation revealed that the immune composition of the microenvironment, especially the abundance of myeloid cells, drove intratumoral metabolite variation. Motivated by the strength of RNA-metabolite covariation and the clinical significance of RNA biomarkers in ccRCC, we inferred metabolomic profiles from RNA sequencing data of ccRCC patients enrolled in 6 clinical trials, ultimately identifying metabolite biomarkers associated with response to anti-angiogenic agents. Local metabolic phenotypes therefore emerge in tandem with the immune microenvironment, influence ongoing tumor evolution, and associate with therapeutic sensitivity.
Graphical Abstract
Introduction
Changes to the metabolism of clear cell renal cell carcinoma (ccRCC) tumors are evolutionarily early events in tumorigenesis that occur in the background of widespread intratumoral genetic diversification and extensive immune infiltration1–4. Such metabolic changes, including the upregulation of glucose uptake, increases in antioxidant biosynthesis, and accumulation of lipid droplets, underlie ccRCC malignancy, metastatic competency, and are potentially determinants of its response to metabolically-targeted therapies1,5,6. However, technical challenges associated with the collection of metabolomic data from primary tumors (especially the need for significant amounts of fresh-frozen tissue) have precluded both a large-scale analysis of the association of metabolite levels with genomic/microenvironmental tumor features, and an assessment of the clinical utility of metabolomic measurements for prognostication and therapeutic stratification of patients.
The metabolism of ccRCC has overwhelmingly been studied using single region metabolomic profiling1,6–8. However, two emerging hallmarks of ccRCC biology, intratumoral heterogeneity and abundant infiltration by non-malignant immune cells2,3, fundamentally confound our understanding of the culprit tumor/non-tumor cell populations driving metabolic phenotypes of interest. Recent work suggests that metabolic heterogeneity (e.g. in PHGDH9 and MCT110) can mechanistically influence the competency of tumor cells to metastasize, supporting a rational therapeutic strategy that limits heterogeneity or targets heterogeneous pathways supporting aggressive behavior. However, the aforementioned studies do not address the full extent of tumor-cell-intrinsic metabolic heterogeneity in human cancers. Furthermore, these phenomena imply that the hallmark features of ccRCC metabolism discovered through measurements of cellularly heterogeneous bulk tumors, including aerobic glycolysis and upregulation of the antioxidant response, are themselves heterogeneous and vary significantly across the spatial extent of the tumor. This raises the possibility that some ccRCC metabolic phenotypes arise either directly from non-tumor cells, or indirectly via metabolic reprogramming of tumor cells in specific immune microenvironments. However, existing studies of intratumoral metabolic heterogeneity in ccRCC have been limited to small numbers (typically, two) of tumor regions11,12 and have largely lacked the orthogonal measurements of the genome, transcriptome, and microenvironment necessary to test this hypothesis. Thus, neither the intratumoral heterogeneity of metabolic phenotypes, nor their cellular source (i.e. from tumors or immune cells), are well understood.
Here, to characterize metabolic intratumor heterogeneity and its drivers, we assembled a cohort of 123 tumor and 64 adjacent normal regions from 31 clear cell renal cell carcinoma patients with associated metabolomic, genomic, and transcriptomic profiling. Through analysis of this spatial, multimodal dataset, we discovered that elevated intratumoral metabolomic heterogeneity (IMH) was associated with specific transcriptomic and immunologic features, including lower cytotoxic cell infiltration, but comparatively few distinguishing genomic features. Joint analysis of the entire dataset revealed that all patients evolved a similar pattern of IMH dominated by metabolic program resembling compensation to ferroptotic susceptibility, with tumor regions primarily stratified by their abundance of cysteine, glutathione, and polyunsaturated fatty acids. Studying in detail the molecular features associated with intratumoral metabolite variation across tumor regions, we found that gene expression profiles associated with immune infiltration, and direct measurements of immune cells, significantly correlated with the intratumoral variation of numerous metabolites, indicating that the immune microenvironment and metabolism coevolve in ccRCC to produce spatially-restricted niches with defined nutrient abundance and immune profiles. Motivated by the strong covariation between the metabolome and transcriptome and the clinical significance of the immune microenvironment in ccRCC, we applied a machine learning algorithm (MIRTH) to impute metabolite levels from RNA sequencing data, ultimately identifying metabolites associated with response to systemic therapy in ccRCC.
Results
Multiregional metabolomic, transcriptomic, and genomic profiling of ccRCC
To interrogate metabolomic heterogeneity in RCC, we assembled a mixed cohort of fresh frozen high-quality tumor/adjacent normal specimens covering both (1) patients with only a single region profiled (“single-region”) and (2) patients with multiple tumor and normal regions profiled (“multiregion”). The total cohort included samples from 156 patients, with 33 patients undergoing multiregion profiling. Our cohort included tumor regions which were untreated (n=144), treated with Nivolumab (anti-PD-1) only (n=106), Cabozantinib or Crizotinib (tyrosine kinases inhibitors) (n=8), and with combination therapy (n=10). In tumors from treated patients, therapies were administered prior to surgical resection. Metabolomic profiling by mass spectrometry was performed in two batches, with one batch (“M4”) containing 68 patients (18 multiregion) and the other (“M5”) containing 88 patients (15 multiregion). A total of 268 tumor regions and 78 adjacent normal regions were collected. Nivolumab-exposed tumors were profiled 8 weeks after therapy, while combination-exposed tumors were profiled an average of 7.5 months after therapy.
Principal component analysis (PCA) of the metabolomics data showed clear separation between tumor and normal samples (Supplemental Figure 1a). Using a false detection rate-corrected Wilcoxon test we identified 404 and 507 metabolites that display differential abundance between tumor and normal tissue samples in M4 and M5 respectively (FDR-adjusted p-value < 0.05) which largely overlapped with previously published data (M1)1 (Supplemental Figure 1b). Interestingly, we observed little separation of tumors on principal components analysis based on their exposure to immunotherapy treatment (Supplemental Figure 1c), and while a small fraction of metabolites (33/602, 6%) demonstrated differential abundance in treated (across all treatment groups) versus untreated tumors (FDR-adjusted p-value < 0.05), these metabolites were not enriched in any one metabolic pathway. These data are consistent with prior knowledge on the relatively limited effect of single-agent nivolumab on ccRCC gene expression13; indicating that exposure to immunotherapy produces (over relatively short clinical time scales) comparatively small metabolomic effects.
To examine the extent of metabolomic heterogeneity across regions and patients, we completed unsupervised hierarchical clustering of metabolomic data across tumor regions (Supplemental Figure 2a). Consistent with a prior report14, the majority of tumor regions clustered by patient-of- origin in both M4 and M5 cohorts. However, a small number of regions (derived from numerous patients) clustered separately and were distinct on PCA analysis (Supplemental Figure 2b) and upon inspection were found to have exceptionally low ion counts relative to other tumor samples in the cohort (Wilcoxon p-value 5×10−10) (Supplemental Figure 2c). Based on prior literature15, we hypothesized that these outlier regions with low ion counts may correspond to bona fide depletion of metabolites resulting from tissue necrosis. Differential abundance analysis identified 323 metabolites that distinguish outlier and non-outlier tumor samples (FDR-adjusted p-value < 0.05 and absolute log2 fold-change > 0.5) (Supplemental Figure 2d). Interestingly, the metabolomics data found that one of the most upregulated metabolic pathways was sphingomyelins, a lipid species which can be degraded to release pro-apoptotic ceramide molecules16 (Supplemental Figure 2e, Supplemental Figure 2f). An expert pathologist reviewed 88 samples, five of which were identified as outlier computationally (Supplemental Figure 2g). Review by an expert pathologist (which we consider the gold-standard for necrosis detection) found 5/5 computationally identified outliers were necrotic, while 76/83 non-outliers were found to have no evidence of necrosis (Fisher’s exact test p-value = 2×10−5). Necrosis, therefore, is a dominant mode of metabolic heterogeneity in ccRCC whose metabolic characteristics transcend individual patients. For all further analysis, we focused exclusively on multiregion clear cell tumor and normal regions, excluding necrotic regions (n = 19 regions), non-clear patients (n = 44 regions), and single-sample patients (n = 98 regions). This left a total of 31 ccRCC tumors with associated multiregion profiling (123 tumor regions, 64 normal regions; 17 patients in M4 and 14 patients in M5) (Figure 1a,b). Details on metabolomic, transcriptomic, and genomic data processing are provided in the Methods.
Figure 1. Clinical and Metabolic Features of the Multi-Omics Cohort.
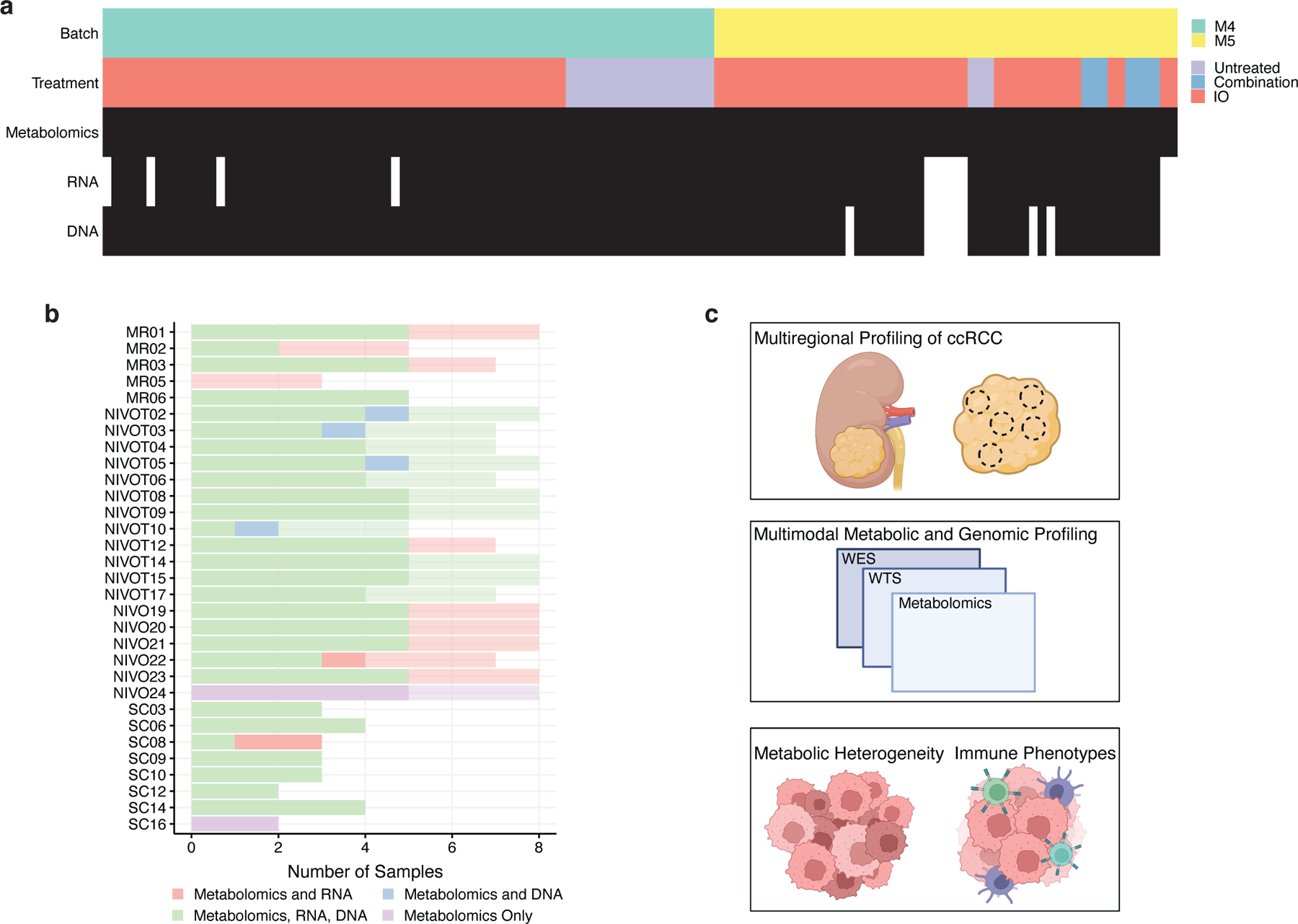
a) Overview of 123 tumor regions collected from 31 patients. b) Sample size of 31 multiregion patients. Normal samples are less opaque. c) Schematic of study workflow showing the collection of multiregion ccRCC samples, followed by multi-omic profiling for metabolic heterogeneity and immune phenotype analyses.
The landscape of intratumoral metabolic heterogeneity (IMH) in ccRCC
To quantify metabolomic heterogeneity both within- and between-patients, we calculated an “h-score” for each metabolite corresponding to the the expected log2 fold-change for a given metabolite i (“h-score”) between two randomly chosen regions (Supplemental Figure 3a). Summarizing h-scores across all regions of a patient, we found that IMH varied widely by patient: in the most heterogeneous patient (NIVO10), a randomly chosen metabolite could be expected to vary 3-fold between regions, compared to 1.3-fold in the least heterogeneous patient (NIVO4). Interpatient metabolomic heterogeneity was greater than intratumoral metabolomic heterogeneity (Figure 2a). Both intrapatient and interpatient heterogeneity was comparable in tumors and adjacent normal kidney. A small number of metabolites (6%, 54/602) demonstrated differences in their extent of intrapatient metabolomic heterogeneity in tumor compared to normal kidney, but these metabolites were not concentrated in any one pathway (Figure 2a). IMH was also not significantly associated with tumor stage (Kruskal-Wallis p-value = 0.45) or grade (Kruskal-Wallis p-value = 0.31).
Figure 2. Tumors With Elevated IMH Are Molecularly Distinct.
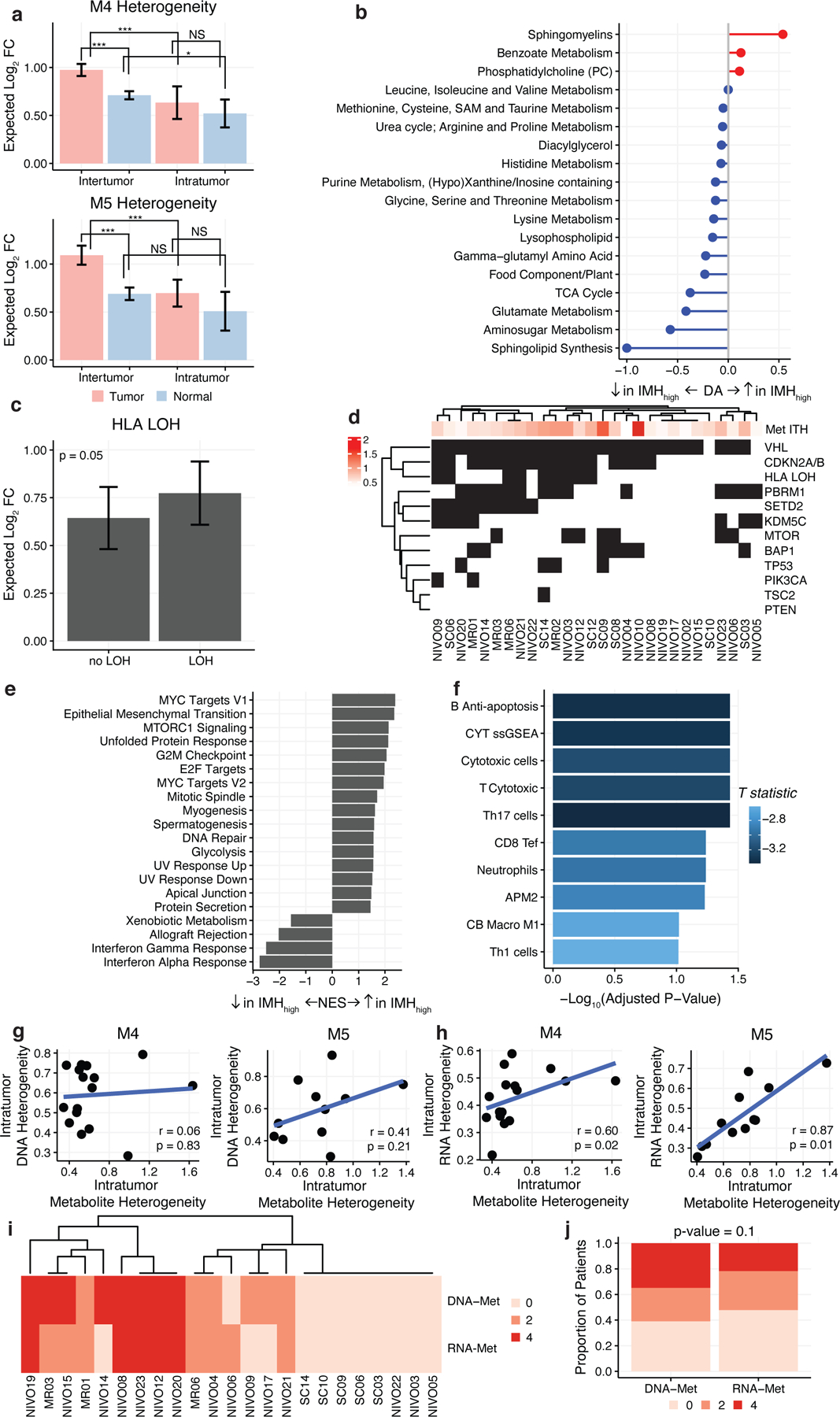
a) Barplots comparing levels of interpatient and intrapatient heterogeneity in tumor and normal tissue (* = p-value < 0.01, *** = p-value < 0.0001, NS = not significant). Heterogeneity values represent log2-fold abundance difference between two randomly chosen regions. b) Pathway-based analysis of metabolic changes in metabolically heterogeneous tumors. Only pathways with 3 or more metabolites are shown. Equation to calculate DA score can be found in Methods. c) IMH is weakly elevated in tumors with HLA LOH. d) Driver mutations in multiregional cohort are not significantly associated with IMH. e) GSEA results of normalized enrichment scores of HALLMARK pathways of genes that are significantly associated with IMH calculated using linear regression. All pathways shown are significant (FDR-adjusted p-value < 0.05). f) Barplot of -log10 FDR-adjusted p-values comparing immune signature expression based on IMH levels. Numerous immune signatures are depleted in IMH-elevated tumors. Blue color indicates that the fold-change between IMH-high and IMH-low tumors is negative. (g-h)IMH is not significantly correlated to intratumoral genetic diversity (g) but significantly correlated to intratumoral transcriptomic diversity (h) across both batches of metabolomic data (M4 and M5). i) Heatmap of Robinson-Foulds distances between DNA, RNA, and metabolite subclonal phylogenies by patient. j) Subclonal phylogenies reconstructed from metabolomic and transcriptomic data show a higher degree of similarity than those reconstructed from metabolomic and genomic data.
We hypothesized that elevated IMH may define a class of ccRCC tumors with distinct genomic, transcriptomic, and microenvironmental features. To test this hypothesis, we performed linear regression on IMH scores of each patient and compared their clinical, metabolomic, transcriptomic, and genomic profiles. There were no significant differences in IMH based on clinical stage (p-value = 0.55) or grade (p-value = 0.31). Comparing the incidence of key driver mutations in ccRCC (VHL, PBRM1, SETD2, BAP1, KDM5C, MTOR, TP53, PIK3CA, TSC2, PTEN, HLA-LOH, and CDKN2A/B loss), patients with loss-of-heterozygosity of HLA had higher IMH (median = 0.77) than patients without HLA LOH (median = 0.64), but this result did not survive multiple hypothesis correction (uncorrected p-value = 0.05) (Figure 2c, 2d). Thus, elevated IMH is not strongly associated with specific genetic alterations in ccRCC tumors.
In contrast, patients demonstrated stark metabolomic differences based on IMH: 137/602 (23%) metabolites significantly correlated with IMH (FDR-adjusted p-value < 0.05), with exceptionally strong enrichment of sphingomyelins (Figure 2b). After controlling for age and sex, we found that significant metabolic correlation with ITH remained (Pearson correlation coefficient comparing FDR-adjusted p-values in the two models = 0.95, p-value < 2.2×10−16). In complement to this, the transcription of 619/19570 (3%) of genes were significantly associated with patient-level IMH scores (FDR-adjusted p-value < 0.05). Performing GSEA, immune pathways (specifically interferon response) were negatively enriched with IMH while pathways associated with cell growth and proliferation were positively enriched (Figure 2e). Comparing the expression of RNA-based immune signatures with IMH, we found statistically significant negative association in cytotoxic cells (FDR-adjusted p-value = 0.04) and Th17 cells (FDR-adjusted p-value = 0.04) (Figure 2f). In total, these data argue that elevated IMH defines a molecularly distinct group of ccRCC tumors with distinct metabolic and microenvironmental features but limited differences in genotype.
Intratumoral metabolomic evolution mirrors transcriptomic evolution
We considered the possibility that IMH may reflect the overall intratumoral molecular diversity of the whole tumor. To test this, we calculated a measure of intratumoral heterogeneity for matched DNA and RNA sequencing data for 28 patients with sufficient data (number of profiled regions >= 2). Interestingly, the magnitude of patient-specific transcriptomic heterogeneity, but not patient-specific genomic heterogeneity, was correlated to patient-specific metabolomic heterogeneity (transcriptomic heterogeneity: Spearman correlation M4 p-value = 0.02, M5 p-value = 0.01, genomic heterogeneity: Spearman correlation M4 p-value = 0.83, M5 p-value = 0.21) (Figure 2g,h). Importantly, there was no significant correlation between transcriptomic and genomic heterogeneity (Spearman correlation M4 p-value = 0.55, M5 p-value = 0.18).
We next investigated the relationship between intratumoral patterns of metabolomic, transcriptomic, and genomic heterogeneity, we reconstructed data-modality-specific evolutionary phylogenies for each patient with at least 3 tumor regions profiled (n=23 total). By examining the similarity of phylogenies reconstructed from different data modalities (using the Robinson-Foulds metric, see Methods), we observed that in cases where the 3 phylogenetic reconstructions diverged, DNA- and metabolite-based evolutionary phylogenies consistently showed less similarity than RNA- and metabolite-based phylogenies, although this result did not reach statistical significance (paired t-test p-value = 0.1) (Figure 2j). Thus, we observe associations in both (1) the magnitude of intratumoral transcriptomic and metabolomic heterogeneity at the level of the whole patient (Figure 2g) and (2) the similarity of transcriptomic/metabolomic profiles between spatially circumscribed tumor regions (Figure 2i), but fail to observe analogous associations when comparing genomic and metabolomic profiles. The data above thus support a model whereby gene expression and metabolite availability can fluctuate and adapt in response to regional changes in a manner potentially independent of genomic diversification, and implies that tumor subclones are unlikely to exhibit broad, whole-metabolome differences in phenotypes.
A heterogeneity program driven by cysteine accumulation, polyunsaturated fatty acids, and ROS tolerance dominates ccRCC tumors
We sought to understand the detailed metabolites and metabolic pathways which ultimately drove IMH across the tumor regions of each patient. To do so, we calculated IMH of each metabolite in tumor regions (see Methods), focusing special attention on metabolites in glycolysis, the TCA cycle, and other central metabolic pathways that have previously been implicated in the genesis and metastatic progression of ccRCC. Within individual pathways of interest, we observed considerable variation in IMH, with specific metabolites (e.g. glucose-6-phosphate) demonstrating exceptionally high heterogeneity relative to other metabolites in the pathway (Figure 3a). Cysteine was an especially remarkable outlier: IMH of cysteine was >1.5-fold higher than the IMH of any other proteinogenic amino acid (Figure 3a/g).
Figure 3. A Ferroptotic-Compensation Program Underlies IMH in ccRCC.
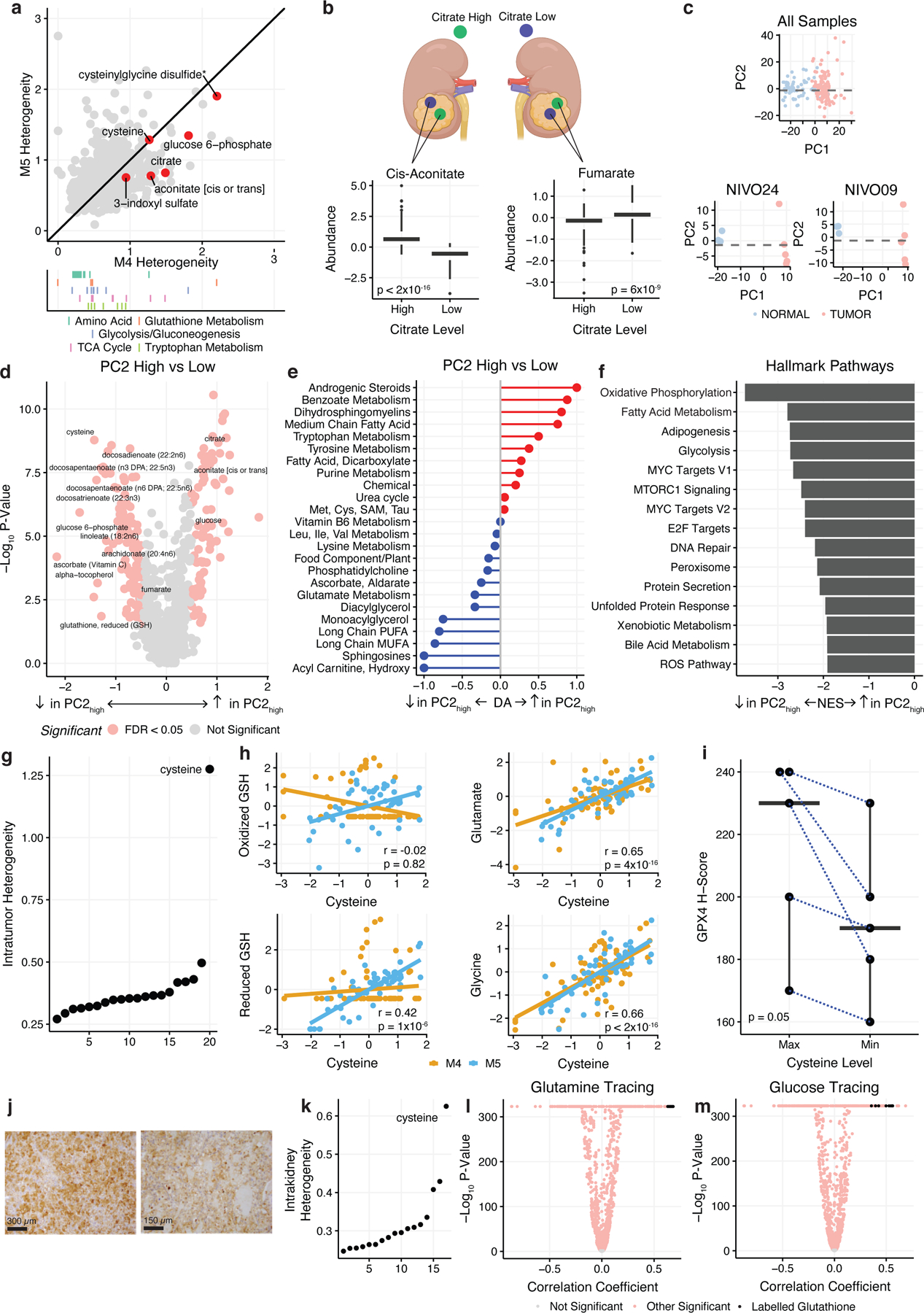
a) Top: Scatterplot of intratumor heterogeneity of individual metabolites in M4 vs M5. Bottom: Rug plot highlighting metabolites in important metabolic pathways. b) Top: Schematic demonstrating citrate high and citrate low regions within a single tumor. Bottom: Boxplots comparing cis-aconitate and fumarate levels in citrate high vs citrate low regions. c) Top: PCA analysis of all multiregion samples colored by tumor and normal. Bottom: PCA analysis of samples from single patients, NIVO09 and NIVO24 colored by tumor and normal. PC1 separates tumor and normal samples while PC2 separates tumor regions within individual patients. d) Volcano plot of metabolites differentially expressed in PC2low vs. PC2high tumor regions colored by significance. Metabolites in red are significant (FDR-adjusted p-value < 0.05 and absolute log2 fold-change > 0.5). e) Pathway-based analysis of metabolic changes in PC2high vs PC2low regions. f) GSEA results comparing PC2low to PC2high regions. All pathways shown are statistically significant (FDR-adjusted p-value < 0.05). g) Cysteine is exceptional in the magnitude of IMH relative to other proteinogenic amino acids. h) Scatterplots of scaled cysteine expression vs oxidized glutathione, reduced glutathione, glutamate, and glycine. Yellow represents samples from the M4 batch, blue represents samples from M5 batch. i) Boxplot comparing GPX4 H-scores in maximum cysteine expression regions vs minimum cysteine expression regions. Maximum and minimum samples selected are paired from the same patient. GPX4 protein expression is elevated in cysteine-high regions. j) GPX4 staining of a cysteine maximum (top) and cysteine minimum (bottom) region from a single patient (MR06). Bar represents 300 µm (top) and 150 µm (bottom). k) Intrakidney heterogeneity of amino acids from spatial mouse kidneys. l) Volcano plots from correlating cysteine abundance with labeling patterns from experiments using uniformly-labeled glutamine. Black dots indicate labelled glutathione isotopologues (i.e. excluding glutathione m+0). k) Intrakidney heterogeneity of amino acids from spatial mouse kidneys. m) Volcano plots from correlating cysteine abundance with labeling patterns from experiments using uniformly-labeled glucose. Black dots indicate labelled glutathione isotopologues (i.e. excluding glutathione m+0).
To understand the consequences of elevated heterogeneity in cysteine and other metabolites, we examined correlated intratumoral fluctuations in metabolite levels. Interestingly, after controlling for baseline metabolite abundance in a given patient, we observed highly similar patterns of intratumoral metabolite covariation across patients. For example, across all patients, citrate-high regions in tumors demonstrated consistently higher levels of cis-aconitate (p-value < 2×10−16) and lower levels of fumarate (p-value = 6×10−9) than citrate-low regions (Figure 3b). This raised the novel possibility that ccRCC patients evolve a common, universal pattern of IMH, manifesting in correlated fluctuations of metabolites around their baseline level in a patient. To test this hypothesis, we transformed our metabolomic data to capture the fluctuation of each metabolite relative to its mean abundance in a given patient and completed principal components analysis (PCA, see Methods). This analysis revealed that the first principal component (“PC1”, explaining 26% of the variance in the data) separated tumor and normal regions (Figure 3c) and KEGG pathway-based analysis showed an enrichment of fatty acids and a decrease in amino acid levels in PC1high regions, consistent with prior metabolomic features of ccRCC tumors (Supplemental Figure 3b).
In contrast to PC1, the second principal component (“PC2”, explaining 12% of the variance) separated tumor regions, but not normal regions, from the majority of patients into two distinct classes (Figure 3c). PC2low tumor regions demonstrated characteristics associated with elevated uptake and phosphorylation of free glucose (high levels of glucose-6-phosphate (FDR-adjusted p-value = 1×10−5) and lactate (FDR-adjusted p-value = .003) and high levels of the TCA cycle intermediates fumarate (FDR-adjusted p-value = 0.001) and malate (FDR-adjusted p-value = 5×10−4). Additionally, PC2low regions also demonstrated higher anti-oxidant levels, including high reduced glutathione (FDR-adjusted p-value = 0.03), alpha-tocopherol (FDR-adjusted p-value = 0.006), and ascorbate (FDR-adjusted p-value = 3×10−4). In contrast, PC2high regions had comparatively higher levels of free glucose (FDR-adjusted p-value = 5×10−6), citrate (FDR-adjusted p-value = 2×10−7), and cis-aconitate (FDR-adjusted p-value = 1×10−6), suggesting that these regions are more glucose-replete and possibly undergo less aerobic glycolysis (Figure 3d). At a pathway level, PC2low tumor regions showed elevation in multiple fatty acid species including long-chain polyunsaturated fatty acids (PUFAs, DA score = −0.8) and hydroxy acylcarnitines (DA score = −1) (Figure 3e), and PC2low regions demonstrated a generic tendency to accumulate lipids (Supplemental Figure 4b). Differential expression of multiregion RNA-sequencing data (controlling for patient-of-origin) identified reactive oxygen species (FDR-adjusted p-value = 6×10−4) and oxidative phosphorylation (FDR-adjusted p-value = 6×10−10) as elevated in PC2low tumor regions (Figure 3f) but no difference in either EIF2AK2 expression or an RNA signature capturing the magnitude of the integrated stress response (ISR p-value = 0.59, EIF2AK2 p-value = 0.28). Investigating defined MitoPathways from MitoCarta 3.0, we found 57/61 (93%) of pathways were significantly depleted in PC2low tumor regions, providing further evidence of increased cellular oxidation in these regions. Importantly, we found no significant differences between tumor purity (p-value = 0.8) (Supplemental Figure 4a), and no significant differences in cellular proliferation as quantified by either RNA signature of proliferation (p-value = 0.31, Supplemental Figure 4c) or Ki67 staining (p-value = 0.19, Supplemental FIgure 4d). We similarly observed no differences in angiogenesis between regions as evaluated using either an angiogenic RNA signature (p-value = 0.29) or CD31 immunofluorescence staining (p-value = 0.11) (Supplemental Figure 4e,f). These data demonstrate that PC2low regions are characterized by elevated rates of glycolysis, high abundance of PUFAs and antioxidants, increased OXPHOS expression, and signatures of elevated ROS production, and that the features of PC2low regions cannot be explained by changes in tumor cellularity, proliferation, or vascularization.
Among the metabolites with the highest contribution to PC2 were reduced glutathione and its rate-limiting precursor cysteine (Supplemental Figure 3c), as well as numerous polyunsaturated fatty acids (PUFAs). These metabolites are each associated with the molecular process of ferroptosis, an iron-mediated form of cell death triggered by peroxidation of PUFAs and counteracted by glutathione17–20. Based on the data in Figures 3b–3f, we hypothesized that cysteine levels may be elevated in a compensatory manner to produce glutathione and prevent ferroptosis in tumor regions with high PUFA levels and OXPHOS expression. Consistent with this, high tumor cysteine levels were correlated with increased levels of reduced glutathione (rho = 0.42, p-value = 1×10−6), as well as increased levels of the glutathione precursors glutamate (rho = 0.65, p-value = 4×10−16) and glycine (rho = 0.66, p-value < 2×10−16) (Figure 3h), but no similar effects were observed for the related disulfide cystine (Supplemental Figure 4g–i).
To directly evaluate the functional consequences of cysteine accumulation in tumor regions, we undertook two complementary analyses. First, to understand the functional effects of cysteine on metabolic flux, we leveraged recent spatial metabolomic isotope tracing data from mouse kidneys21. We recapitulated that, in analogy to Figure 3a, cysteine displays exceptional spatial heterogeneity across the mouse kidney relative to all other proteinogenic amino acids (Figure 3k). Then, by correlating cysteine abundance with labeling patterns from experiments using uniformly-labeled glucose and glutamine, we investigated how flux patterns change in cysteine-rich versus cysteine-depleted regions (Figure 3l, m). Interestingly, we found that regions with elevated cysteine demonstrated decreased glucose- and glutamine-derived labeling of cysteine (suggesting reduced cysteine biosynthesis and increased de novo uptake of cysteine via cystine), and increased glucose-derived and glutamine-derived labeling of glutathione. Together, these data suggest that cysteine-high regions are associated with increased de novo uptake of cysteine (e.g. via SLC7A11/cystine), as well as increased flux from extracellular glucose/glutamine to intracellular glutathione biosynthesis. Second, we performed immunohistochemical staining of GPX4, a critical regulator of ferroptotic resistance17. For 5 patients, we identified tumor regions demonstrating the highest and lowest abundance of cysteine within a given patient, and immunohistochemically stained these regions for GPX4. Consistent with our model above, cysteine-high tumor regions demonstrated elevated GPX4 intensity (paired Wilcoxon p-value 0.05) (Figure 3i, j). Together, these data establish that regions with elevated cysteine levels have distinct metabolic flux patterns and are associated with the upregulation of GPX4. Moreover, these data suggest that PC2low regions are defined by an increased susceptibility to ferroptotic cell death (characterized by higher PUFA levels and OXPHOS activity) and concomitant compensation against ferroptosis in the form of upregulated antioxidant biosynthesis.
Immune cell populations in the ccRCC TME remodel bulk tumor metabolism
When comparing expression of various immune signatures in PC2high regions to PC2low regions, we found significant enrichment of T cell and macrophage cell populations in PC2high regions (Supplemental Figure 4j). Based on this evidence and observations by others that immune and tumor cells compete for nutrients and signaling factors in the ccRCC microenvironment22–27, we therefore hypothesized that the metabolome and immune microenvironment might coevolve to produce niches with defined patterns of metabolite abundance and immune cell composition. To systematically test this hypothesis, we studied the interaction between the immune microenvironment and metabolic phenotypes using a previously validated RNA signature of generic immune infiltration (Immune Score) in each sample28. To control for our repeat measurements of different regions from the same patient, we used a mixed effects model with patient as a random effect. Significance was determined by parametric bootstrapping (100,000 iterations) (Figure 4a and Methods).
Figure 4. Immune Infiltration in the ccRCC Tumor.
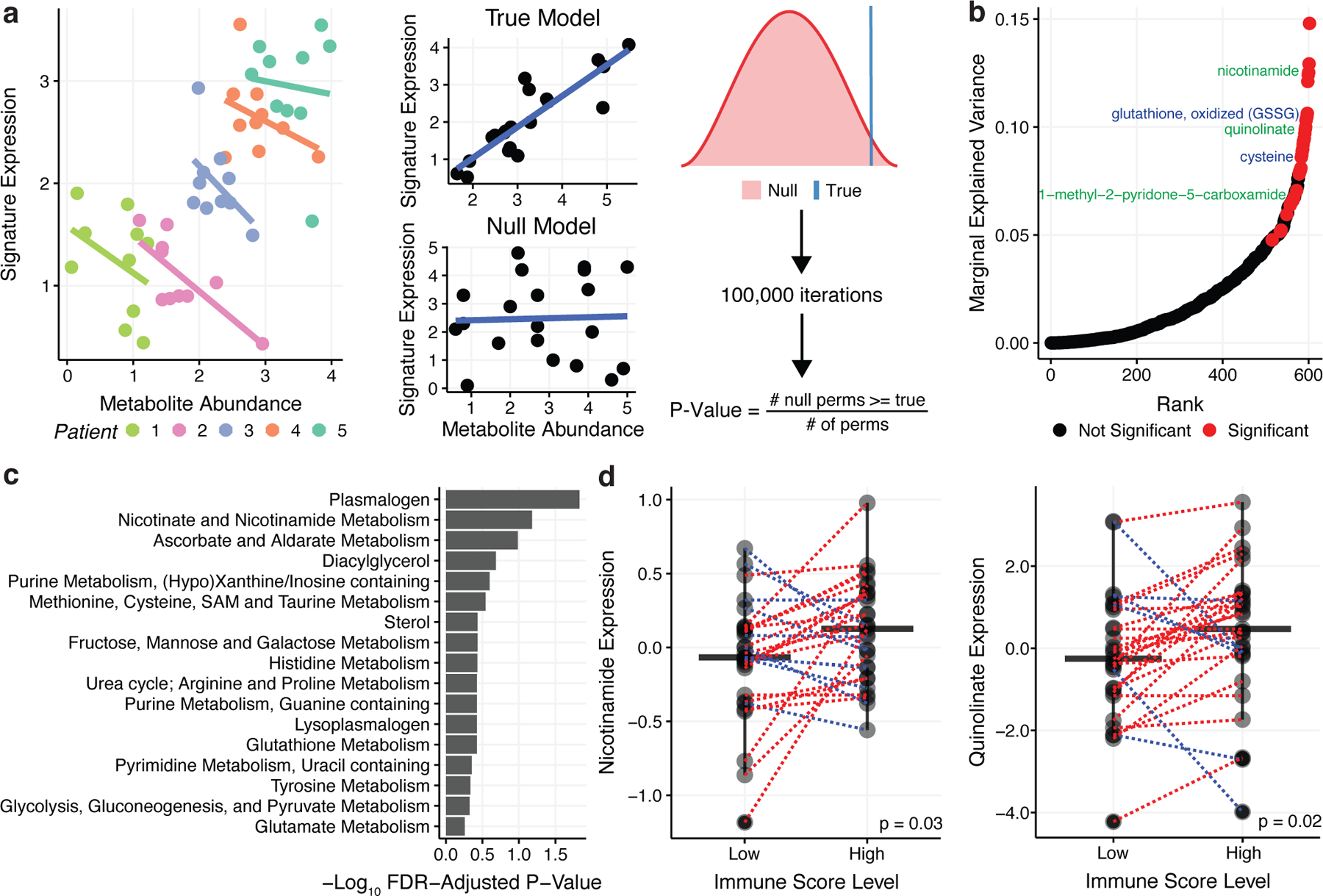
a) Left: Scatterplot illustrating Simpson’s paradox, where non-independence in data due to a patient-level random effect, confounds the co-expression pattern between two variables. Right: In parametric bootstrapping the true model is compared to the null model where the p-value is the fraction of times the null model a test statistic from the null model exceeds the true test statistic. 100,000 bootstraps were performed. b) Marginal explained variance in a mixed-effects linear model of metabolite levels predicting Immune Score with patient added as a random effect. Each dot represents a single metabolite. Significant metabolites in the NAD+ metabolism pathway are labeled in green while significant metabolites in the glutathione pathway are labeled in blue. c) Fisher’s exact test was performed on pathways with 3+ metabolites to determine pathways with a significantly higher proportion of their metabolites associated with Immune Score based on the mixed effects model. The barplot shows FDR-adjusted p-values from Fisher’s exact test. d) Nicotinamide and quinolinate levels are elevated in tumor regions with high immune infiltration relative to paired tumor regions with low immune infiltration.
In total, 33/602 (5%) metabolites were significantly associated with Immune Score across patients (FDR-adjusted p-value < 0.05) (Figure 4b). These immune-associated metabolites included both glutathione and cysteine, which were differentially expressed when comparing PC2high vs PC2low regions (Figure 3c), as well as several metabolites associated with NAD+ metabolism (nicotinamide, quinolinate, and 1-methyl-2-pyridone-5-carboxamide). Pathway analysis revealed that plasmalogens and NAD+ metabolism pathways were enriched for Immune Score coregulation (Figure 4c). Confirming that mixed effects modeling capture intrapatient relationships between metabolites and Immune Score, the correlative relationship between metabolites and Immune Score was preserved intratumorally: taking nicotinamide and quinolinate as examples, regions with the highest levels of Immune Score expression also demonstrated higher levels of quinolinate (Wilcoxon paired p-value = 0.02) and nicotinamide (Wilcoxon paired p-value = 0.03). (Figure 4d). Importantly, the association between metabolite levels and immune infiltration (via Immune Score) was corroborated in an a recently published multimodal, single-region study by of ccRCC29(p = 1×10−5, rho = 0.53, Supplemental Figure 4k). Together, these analyses suggest that the metabolome can be altered by increased immune infiltration and specific metabolites, including but not limited to NAD+ metabolism, are strongly correlated to the extent of immune cell abundance.
Metabolic features of prognostic immune signatures in ccRCC
The Immune Score analysis above represents a crude estimation of the total immune infiltration of a tumor, summing together diverse myeloid and lymphoid cell populations with both pro- and anti-tumorigenic properties. Importantly, emerging data now indicates that RNA signatures capturing certain aspects of the ccRCC microenvironment, including angiogenic gene expression and myeloid/T-effector abundance, are associated with response to therapy in ccRCC30–32. Given the association between the TME and the metabolome (Figure 4b) and generically between transcript levels and metabolite levels (Figure 2), we hypothesized that these signatures may themselves also reflect distinct metabolic phenotypes. Three different signatures, qualitatively reflecting the abundance of cytotoxic (“JAVELIN”)32 cells, myeloid (“Myeloid”)30 cells, and the activity of angiogenesis (‘Angiogenesis”)31 in the tumor, have been identified through retrospective analysis of response data in several clinical trials of immunotherapy and/or anti-VEGF therapy in ccRCC. We confirmed that the expression levels of these signatures were significantly correlated to immunofluorescence staining (Angiogenesis Signature/CD31, rho = 0.63, p-value = 3×10−4, Figure 5a; Myeloid Signature/CD68, rho = 0.58, p-value = 0.001, Figure 5b). Representative images of immunofluorescence staining are in Figure 5c.
Figure 5. Metabolic Features of Prognostic Immune Signatures in ccRCC.
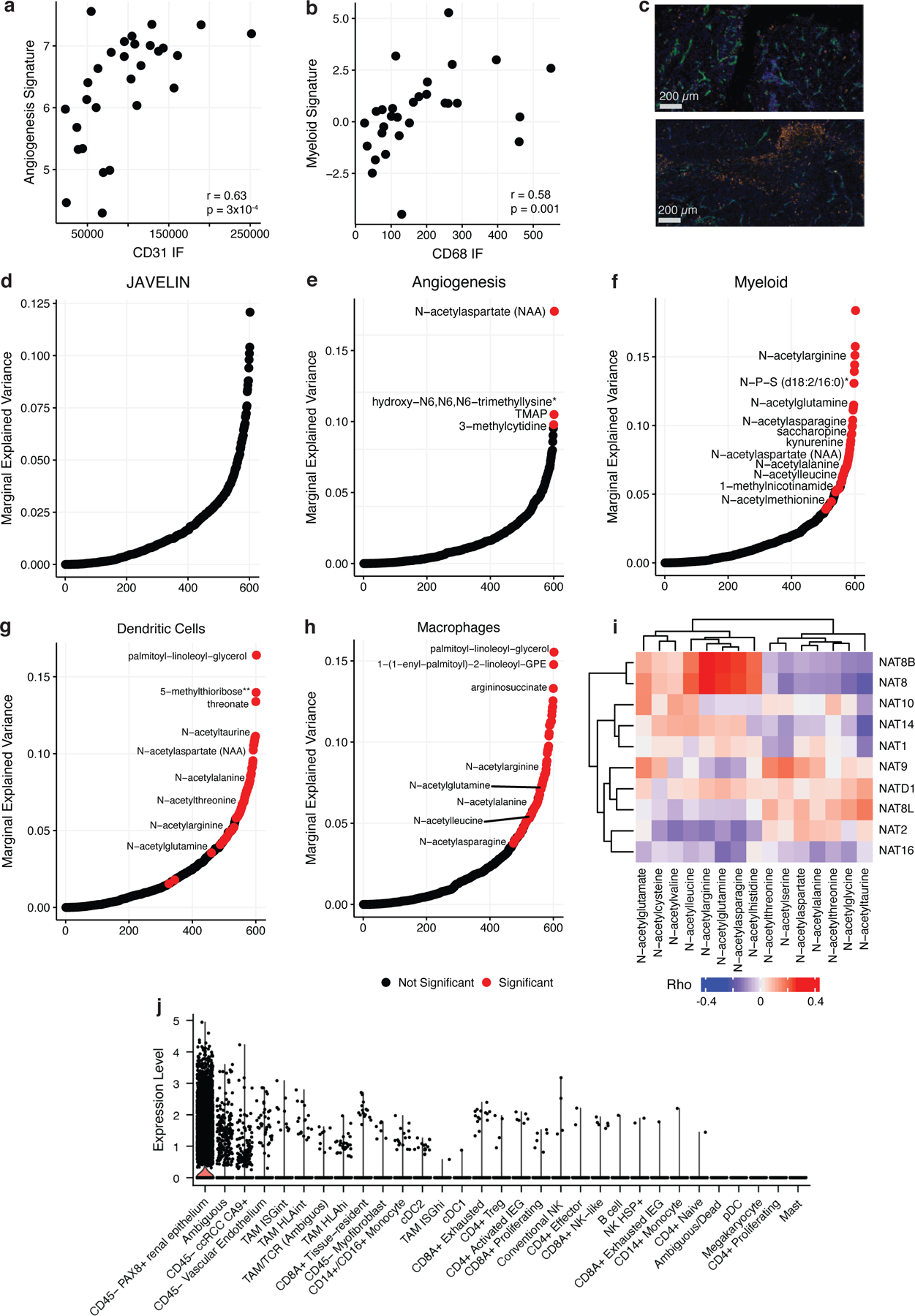
a) Left: Scatterplot comparing CD31 levels from IF staining to expression levels of the McDermott Angiogenesis signature. b) Left: Scatterplot comparing CD68 levels from IF staining to expression levels of the McDermott Myeloid signature. c) Top: Representative IF image of a tumor region (MR01 RB) with high CD31 expression (green) and low CD68 expression (orange). Bottom: Representative IF image of a tumor region (MR01 RE) with low CD31 expression and high CD68 expression. Bar represents 200 µm. Marginal explained variance in a mixed-effects linear model of metabolite levels predicting JAVELIN (d), McDermott Angiogenesis (e), McDermott Myeloid (f), dendritic cell (g), and macrophage (h) signature expression with patient added as a random effect. Each dot represents a single metabolite. Red dots indicate statistically significant associations. i) Heatmap Spearman correlation coefficients between NAT genes and n-acetylated amino acids. j) Boxplots of NAT8 expression levels across cell types in a single-cell ccRCC dataset.
We again used mixed effects models to identify metabolites associated with the angiogenesis, myeloid, and JAVELIN scores. There was significant variability in the association between metabolite levels and prognostic RNA signatures. In total, we identified no metabolites associated with the JAVELIN signature (Figure 5d) and 4 metabolites significantly associated with the angiogenesis signature (Figure 5e). In contrast, 47/602 (8% of the measured metabolome) metabolites associated with the myeloid signature (Figure 5f). The myeloid signature has been associated with inferior outcomes in patients treated with systemic therapy and with recurrence in high-risk localized disease33,34. Metabolites associated with myeloid infiltration concentrated into certain pathways, including most prominently n-acetylated amino acids (NAAs, including n-acetylarginine, n-acetylasparagine, n-acetylglutamine, n-acetylleucine, n-acetylalanine, n-acetylaspartate, and n-acetylmethionine). Most notably, the immunomodulatory metabolite kynurenine35,36, which was the target of recent negative clinical trials of inhibitors of its upstream enzyme IDO1 (which is significantly positively associated with kynurenine, p-value < 2.2×10−16), was significantly negatively associated with the myeloid signature (FDR-adjusted p-value = 5×10−3). These findings indicate that the presence of myeloid cells in the immune microenvironment is strongly associated with a unique pattern of metabolite abundance.
Next, we more granularly investigated the specific myeloid cell populations exhibiting strong correlation with NAA abundance using RNA-based signatures for mast cells, dendritic cells, neutrophils, and macrophages. This analysis revealed that dendritic cells and macrophages were significantly associated with 71/602 (12%) and 80/602 (13%) of metabolites, including several NAAs, respectively (Figure 5g,h). To identify the putative cellular source of NAAs, we first investigated the association between NAAs and the expression of N-acetyltransferase genes acting on them, which identified two genes NAT8 and NAT8B, exhibiting strong and broad correlation to numerous NAAs (Figure 5i). To understand the microenvironmental cellular source driving NAA levels, we used single cell data to evaluate the cell-type-specific expression of NAT8/NAT8B across cell types. While NAT8B was not abundantly expressed, we found that NAT8 expression was highest in tumor and kidney epithelial cells, potentially implying that epithelial, rather than myeloid, cells drove NAA abundance (Figure 5j). We further reasoned that, if myeloid cells did indeed drive the variation in NAA abundance, then the correlation between myeloid cell infiltration and NAA abundance would be preserved in adjacent-normal kidney tissue. However, when tested, no significant association between myeloid infiltration and NAA abundance was found in normal kidney. From these analyses, we concluded that the association between myeloid and NAAs most likely arose from tumor-cell-intrinsic modulation of expression of N-acetyltransferase genes in the presence of myeloid cells in the TME.
Machine learning predicts metabolic profiles of response to anti-VEGF drug sunitinib
Based on the association between metabolite levels and the prognostically significant myeloid signature, we hypothesized that certain metabolomic profiles may directly associate with patients’ response to anti-angiogenic or immune checkpoint blockade therapy. However, the identification of metabolic biomarkers of response to therapy has historically been limited due to the scarcity of metabolomics data itself, which relies on significant quantities of fresh-frozen tumor tissue. Premised on the strong association between transcript and metabolite levels in Figure 4a, we hypothesized that the abundance of a subset of metabolites could be imputed from transcriptomics data (in a manner similar to RNA signatures and immune deconvolution). To evaluate this possibility, we applied a previously published machine learning algorithm (Metabolite Imputation via Rank-Transformation and Harmonization, “MIRTH”) developed by our group37. In this context, MIRTH imputes unmeasured metabolites in a target dataset by learning the relationship between RNA and metabolite levels in a reference dataset where both RNA and metabolites are measured (Supplemental Figure 8a and Methods).
We first sought to benchmark if MIRTH could impute biologically reasonable metabolic phenotypes. To do so, we treated three batches of in-house ccRCC samples with matched RNA and metabolomics data (including both M4 and M5 datasets, and an additional M1 dataset related to a prior publication) as reference datasets, and the TCGA ccRCC cohort (for which only RNA data was available) as the target dataset. We then applied MIRTH to impute 606 tumor and adjacent normal samples from TCGA. Imputed metabolite levels in the TCGA preserved ground-truth metabolic differences between both tumor/normal samples (Spearman’s rho = 0.85, p-value= 1.6×10−31) and high-stage/low-stage samples (Spearman’s rho = 0.39, p-value = 3.0×10−5) (Figure 6a). To further ascertain the accuracy of MIRTH in imputing metabolite levels from RNA data, we benchmarked it on a large dataset of over 900 cancer cell lines jointly profiled by RNA sequencing and metabolomic sequencing in the CCLE. By artificially partitioning the CCLE data into a reference dataset (50% of all cell lines, matched RNA/metabolite data) and a target dataset (50% of all cell lines, RNA data only), we found that MIRTH accurately imputed 96% of metabolites (FDR-corrected Spearman p-value < 0.05 and Spearman’s rho > 0.3) (Figure 6b). Immunomodulatory metabolites, such as 1-methylnicotinamide, were imputed with exceptionally high accuracy (Figure 6c).Together, these data establish the value of MIRTH as a tool to impute otherwise unmeasured metabolite levels in a diverse set of diseases beyond ccRCC data (Figure 6b). Altogether, these data establish that MIRTH successfully imputes metabolite levels from RNA sequencing data across diverse cancer types in solid tumors and cell lines, preserves metabolic relationships between subsets of tumor/normal samples, and learns signatures of progression to aggressive disease.
Figure 6. Metabolic profiles of response to sunitinib predicted by MIRTH.
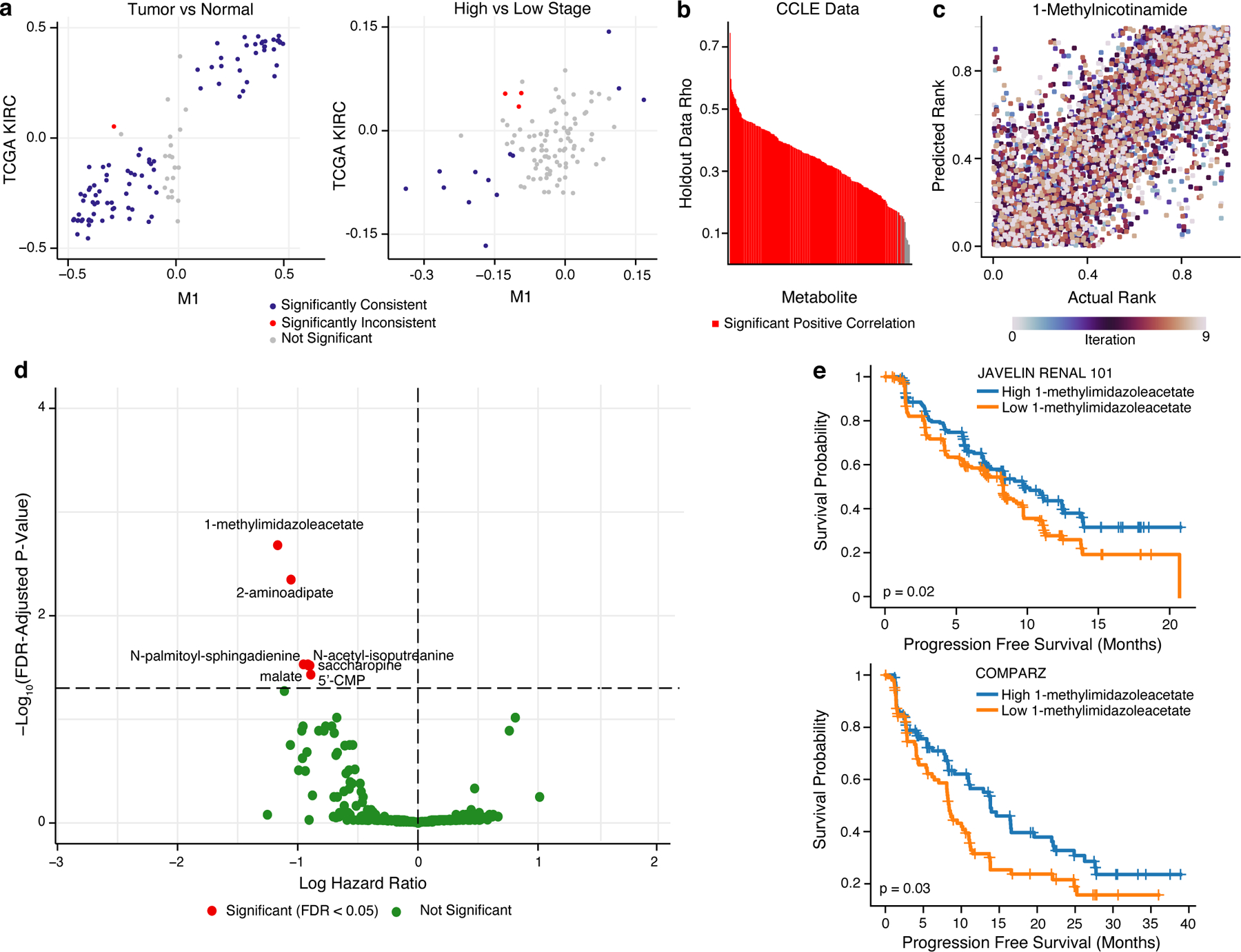
a) Scatterplot comparing the differential abundance of imputed metabolites in TCGA KIRC to true metabolites in M1 between high and low stage tumors (left) or between tumor and normal samples (right). Metabolites in blue are consistently and significantly differentially enriched (FDR-adjusted p-value < 0.05 and rank difference > 0 or < 0 in both datasets, Wilcoxon rank sum test). b) Barplot of median Spearman’s correlation values for each simulated-missing metabolite in CCLE data across 10 MIRTH iterations. Metabolites whose predicted ranks are significantly correlated with ground-truth ranks in >90% of iterations are labeled red. Metabolites with significant negative correlations are discarded. c) Scatterplot of actual vs predicted ranks for 1-methylnicotinamide in CCLE data. Dots are coloured by iteration. d) Volcano plot of 262 reproducibly well-predicted metabolites associated with PFS in sunitinib arms of ccRCC clinical trials, colored by significance (Cox’s proportional-hazards test, adjusted by age and sex). Metabolites in red are significant (FDR-adjusted p-value < 0.05). X-axis is in natural log space. e) Kaplan–Meier plot showing sunitinib-treated ccRCC patients with a high level of 1-methylimidazole acetate (based on median level of 1-methylimidazole acetate) had improved PFS than patients with low level of 1-methylimidazole acetate in JAVELIN RENAL 101 (top) and COMPARZ (bottom) trial.
We then applied MIRTH to impute metabolite levels from RNA sequencing data in 7 publicly available advanced ccRCC clinical trials where patients were treated with either immunotherapy (in combination or alone) or sunitinib (a tyrosine kinase inhibitor, typically used in the control arm of these trials). For 262 metabolites with accurate imputation on cross-validation, we verified that MIRTH imputation preserved the correlative relationship between metabolites and Immune Score signature relative to data from M4/M5 (Supplemental Figure 5d). Next, we evaluated the association between each of the 262 well-predicted metabolites and progression free survival (PFS) by multivariate Cox proportional-hazards models (evaluating different treatment arms separately.) The number of metabolites associated with response varied across each trial and arm, motivating us to identify metabolites with consistent patterns across the trials. Meta-analysis results across 7 clinical trials identified 7 metabolites significantly associated with improved PFS in the sunitinib arms (FDR-adjusted p-value < 0.05) (Figure 6d) and no metabolites significantly associated with PFS in immunotherapy treatment arms (Supplemental Figure 5e). Among the metabolites associated with sunitinib were saccharopine and N-palmitoyl-sphingadienine (d18:2/16:0)*4, which were also significantly associated with the myeloid signature (Figure 5f). The strongest association with sunitinib response was observed in patients with high 1-methylimidazoleacetate (Figure 6e). 1-methylimidazoleacetate is a catabolite of the inflammation-mediating metabolite histamine, suggesting that local inflammation in the tumor may in part mediate response to sunitinib38. Thus, by training a model to impute metabolite levels from transcriptomic data, MIRTH enables the identification of otherwise cryptic metabolite biomarkers in publicly available trial data.
Discussion
Intratumoral genotypic and phenotypic diversification of ccRCC tumors2,39 influences future tumor evolution and likely shapes the development of metastasis and response to therapy40–47. Motivated by the numerous studies implicating remodeled metabolism as a hallmark of ccRCC tumorigenesis and progression, we investigated how intratumoral metabolic heterogeneity (IMH) manifests and studied the underlying genetic, transcriptomic, and microenvironmental factors driving metabolic diversification. Using multiregional sampling of the metabolome, genome, and transcriptome, we found highly heterogeneous tumors have elevated HLA loss of heterozygosity and identified a metabolic program which compensates for ferroptotic susceptibility which drives a substantial fraction of total IMH. Transcriptomic factors, reflecting in part the cellular composition of the TME, influenced metabolomic variation within tumors more than genetic factors. In turn, a large fraction of the metabolome was correlated to the extent of both generic immune infiltration in the tumor, and more specifically to the abundance of myeloid cells in the TME. Seizing on the association between the transcriptome and metabolome, we applied a machine learning algorithm (MIRTH) to impute metabolite levels from RNA-sequencing data and identified a set of metabolites that are reproducibly associated with response to anti-VEGF therapy in ccRCC patients. Importantly, MIRTH naturally generalizes to other cancer types (Figure 6b) and diseases with paired metabolomics and transcriptomics data, and can therefore be used to democratize metabolomic data analysis in contexts where the availability of fresh-frozen primary tissue for metabolomics is limited. MIRTH is thus a means for the discovery of new metabolomic biomarkers and/or targets for therapeutic intervention.
Recent functional data suggests that stochastic heterogeneity in the metabolic phenotypes of tumor cells can endow a subset of cells with the capacity to metastasize9,10. Complementing these data, our group and others have shown that the abundance of glutathione and its related metabolites is associated with metastatic disease and the risk of recurrence in ccRCC1. Here, we discovered that all ccRCC tumors evolve along a common axis of IMH that produces spatially-delimited tumor regions defined by elevation of metabolites associated with glutathione and its related precursors (e.g. cysteine). These data motivate a new model whereby metabolic diversification of ccRCC ultimately produces tumor subclones with elevated glutathione levels that can ultimately seed distant metastases. Importantly, identifying the selective pressures placed on metabolism in metastases will likely require multimodal data and integrative analysis to reconcile apparently conflicting data: while a comparison of transcriptional phenotypes of metastatic and primary tissues from the ImMotion151 Phase III clinical trial reveals no difference in the expression of OXPHOS genes between primary and metastatic tissues (mean difference = 69.9, p-value = 0.055), more direct measurements of metabolic flux suggests ccRCC metastases increase their respiratory capacity relative to primary tumors.
Among the most significant, but also comparatively provocative, limitations of this and other metabolomic studies of primary tumors is that measurements of the tumor metabolome are nearly always taken in bulk and therefore represent a complex mixture of metabolite signals from the constituent tumor and non-tumor (immune, stromal) cells in the microenvironment. Our data here suggests that the cellular composition of the immune microenvironment is associated with specific metabolomic features, and complements newly emerging data on the partitioning of nutrient consumption across cellular compartments23. The coevolution of the tumor metabolome and immune microenvironment likely arises from a combination of several phenomena: (1) cell-type-specific accumulation/depletion of specific metabolites (such as those previously described for 1-methylnicotinamide in ovarian cancer24), (2) competition among different cell populations for nutrients in the circulation48, and (3) systemic features of the metabolism of the patient, including but not limited to factors such as obesity and metabolic syndrome49,50. There is ample evidence of metabolite-level effects on immune response, including the downregulation of glycolysis by CTLA-4 in T-cells25, the inhibition of T-cell proliferation by expression of 2,3-dioxygenase in tumor cells48, and the suppression of T-cell function by tumor-derived lactate secretion48. In support of these findings, large-scale analysis of public clinical trial data here (Figure 6) suggests that a subset of metabolites may be associated with response to approved therapies in ccRCC. These discoveries may provide a means for stratifying patients according to their likelihood to respond to metabolically targeted therapies (e.g. IDO1/epacadostat51). Our findings, in combination with these prior discoveries, suggest that therapeutic strategies which rationally target the metabolome and immune microenvironment in combination may improve response to therapy in ccRCC.
Limitations
Several limitations must be considered when interpreting the conclusions of our study. Due to the multiregional nature of our study, the sample size in terms of patients is relatively small, and statistical power is further diluted by the separation of metabolomic profiling into two batches. Such sample size limitations may soon be overcome as spatial metabolomic technologies capable of sampling intratumoral heterogeneity at significantly higher resolution become more widely adopted. Although we validated several RNA signatures of immune cell infiltration with matched immunofluorescence data, caution must nevertheless be taken in interpreting the covariation of RNA-based signatures of immune infiltration and metabolite levels. More robust signatures, learned from newly abundant single cell sequencing data, may potentially overcome this challenge. Finally, the causal source of covariation between the metabolome and the immune microenvironment remains unclear. Further mechanistic experiments are required to evaluate whether certain metabolic milieu are caused by, or may engender the development of, immune microenvironments with defined cellular composition.
STAR Methods
Resource Availability
Lead Contact
For further information and requests for resources, please contact the lead contact, Ed Reznik (reznike@mskcc.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Metabolomics data generated and used in this study are published at 10.5281/zenodo.7986891. RNA and DNA sequencing data and clinical information was retrieved from Golkaram et al95. All data used to generate display items in this manuscript are available in Data S1. All original code has been deposited at https://github.com/reznik-lab/rcc_coevolution and is publicly available as of the date of publication. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Tumor Sampling
Informed consent was acquired and with Memorial Sloan Kettering Cancer Center institutional review board approval, partial or radical nephrectomies were performed at Memorial Sloan Kettering Cancer Center (New York) and stored at the MSK Translational Kidney Research Program (TKCRP). Samples were flash frozen and stored at −80 degrees Celsius until molecular characterization. Clinical metadata was recorded for all tumor samples. Samples were thawed and extracted as described in detail below, which removed proteins, dislodged small molecules bound to the protein or physically trapped in the protein matrix, and recovered a wide range of chemically diverse metabolites. Samples were then frozen, dried under vacuum and prepared for mass spectrometry. Sex, gender, age, and other clinical data about patients in this study can be found in Golkaram et al95.
Method Details
Sample Preparation and Metabolomic Profiling
The sample extract was split in two and reconstituted in acidic and basic LC-compatible solvents where the acidic extracts were gradient eluted with water and methanol containing 0.1% formic acid, while the basic extracts, which also used water and methanol, contained 6.5 mM ammonium bicarbonate. One aliquot was analyzed using acidic positive ion optimized conditions while the second aliquot used basic negative ion optimized conditions. Two independent injections into separate dedicated columns were performed. MS and data-dependent MS/MS scans using dynamic exclusion were used alternatively to perform MS analysis. The LC/MS portion of the platform was based on a Waters ACQUITY UPLC and a Thermo-Finnigan LTQ-FT mass spectrometer, which had a linear ion-trap (LIT) front end and a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer backend. Ions with counts greater than 2 million provided accurate mass measurements and average mass error was less than 5 ppm. Ions with fewer than 2 million counts required more effort to characterize. Fragmentation spectra (MS/MS) were typically generated in a data-dependent manner but targeted MS/MS was employed as necessary, as in the case with lower level signals.
Raw mass spectrometry files were loaded into a relational database, which was then examined and appropriate QC limits were imposed. Peaks were identified using Metabolon’s proprietary peak integration software and component parts were stored in a separate data structure. An in-house library of standards from Metabolon was used to compare metabolites. Data on each of these standards were based on retention index, mass-to-charge ratio, and MS/MS spectra. For each compound in the metabolomic data, parameters were compared to analogous parameters in the standard library. As described in Evans et al.52, compounds were identified based on three criteria: retention index within 75 RI units of the proposed identification, mass within 0.4 m/s, and MS/MS forward and reverse match scores.
Data Normalization
Block normalization was performed when metabolomics data was measured across several days. Each compound was corrected in run-day blocks by registering the medians to equal one and normalizing each data point accordingly. Minimal measured level of a metabolite across all samples was imputed when metabolite levels were below the detection limit. Probabilistic quotient normalization was then performed, accounting for an overall estimation on the most probable dilution factor53. Data was then log2-transformed.
Necrosis Pathology Review
Hematoxylin and eosin (H&E) stained slides were reviewed for morphologic features including coagulative-type necrosis (Supplemental Figure 2g). Extent of tumor necrosis was subjectively quantified by a pathologist (SG) for all cases that were reviewed.
Whole Transcriptome Sequencing
Thank you for your comment. Extracted RNA samples were quantified with Qubit RNA HS assay Kit, and 100 ng of RNA was used for each library preparation input with Illumina TruSeq Stranded Total RNA with Ribo-zero. Each resulting library was quantified individually with Qubit dsDNA HS Kit and normalized to 4 nM prior to sequencing. RNA sequencing libraries were prepared in batches of 24, with each individual RNA sample uniquely indexed using Illumina TruSeq RNA UD Indexes. Final libraries were pooled for sequencing in up to 36-plex on NovaSeq 6000 S2 flow cell, or up to 72-plex on NovaSeq 6000 S4 flow cell. All RNA samples in the cohort were selected for library preparation and sequencing. Each sample was assigned a unique identifier during sample accessioning to prevent sample selection bias. A paired-end 76 base pair sequencing recipe was used to produce ~200 million total paired reads per sample. Batch effects were not observed (Supplemental Figure 6a).
RNA-seq Data Processing Pipeline
STAR 2-pass alignment54 was used to align RNA-sequencing reads against human genome assembly hg19. Numerous QC metrics, such as general sequencing statistics, gene feature, and body coverage, were calculated through RSeQC55 based on the alignment result. The R package GenomicAlignments56 over aligned reads with UCSC KnownGene57 in hg19 as the base gene model was used to compute raw RNA-seq gene level count values. Union counting mode was used and mapped paired reads after alignment quality filtering were used. Finally, the R package DESeq258 was utilized to estimate the sample size factor (using the estimateSizeFactors function), and to generate the sample size normalized read count matrix (using the counts function). DESeq2’s fpkm function was then used to generate the FPKM normalized values which were derived based on the sample size normalized counts along with the gene model used in raw count quantification.
Whole Exome Sequencing
40ng input of DNA per sample was used to generate libraries for whole exome sequencing with TruSight Oncology DNA Library Prep Kit, and TruSight Oncology index PCR products were used for enrichment. Target exome enrichment was performed using the IDT xGen Universal Blockers and IDT xGen Exome Research High Sensitivity assay (Biotium) was used for library quantification of the post-enriched libraries. Libraries were normalized post-enrichment using bead-based normalization and pooled. Illumina NovaSeq™ 6000 S4 flow cell using the XP workflow for individual lane loading (12-plex per lane) was used to sequence samples using 101 bp paired-end reaches. Each sample yielded an average of 500 million reads and a median target coverage depth of 360X. No differences in TMB were observed between batches (Supplemental Figure 6b).
WES Data Processing Pipeline
Alignment of raw sequencing data to the hg19 genome build was performed using Burrows-Wheeler Aligner (BWA) version 0.7.1759. Genome Analysis Toolkit (GATK) version 3.860 was used following raw reads alignment guidelines from DePristo et al.61 to realign indels, recalibrate base-quality scores and remove duplicate reads. Small variant calling was performed using VarScan 262, Strelka v2.9.1063, Platypus 0.8.164, Mutect2 - part of GATK 4.1.4.161 and SomaticSniper version 1.0.5.0 (SNVs only)65. Variants called by a combination of 2 out of 5 callers are reported, following recommendations from the Cancer Genome Atlas Research Network66. The following criteria were used to filter variants:
Tcov > 10 and Taf >= 0.4, Ncov > 7 and Naf <= 0.01, Tac > 4.
Common SNPs were identified by snp142.vcf and eliminated.
Rare variants identified in dbSNP are kept if Naf = 0.
Low confidence variants had Tcov < 20 or Tac < 4.
Variants must be called by more than a combination of 2 or more callers.
Common variants were identified by gnomAD v2.1.1 and excluded.
Variants were annotated using Variant Effect Predictor (VEP)67. As per Amemiya et al.68, INDELS in blacklisted and/or low mappability regions (such as repeat regions) were excluded. FACETS v.6.169 was used to perform allele-specific copy number analysis while allele-specific HLA loss of heterozygosity was determined using LOHHLA as described by McGranahan et al.70.
Bulk RNA-seq deconvolution analysis
Immune features and signature gene lists of immune cell types were obtained according to Bindea et al.71 and Senbabaoglu et al.3. ssGSEA72 and CiberSort73 methods were used for bulk RNA-seq deconvolution analysis. ssGSEA takes FPKM values as its input and calculates enrichment scores for given gene lists compared to all other genes in the sample’s transcriptome. Cibersort also uses FPKM expression values as its input but has its own published LM22 gene expression matrix of immune cell types of interest, which it uses as a reference to compute the infiltration level of each immune cell type. Based the work of Rooney et al.74, immune cytolytic score (‘CYT’) was calculated using the geometric mean of TPM (transcripts per million) of granzyme A (GZMA) and perforin (PRF1), two cytolytic effector genes. Immune Score, Stromal Score, and ESTIMATE Score were calculated through the estimate R package28. Along with the Angiogenesis score published by Masiero et al.31, the role of stromal and immune compartments in the tumor microenvironment were dissected.
Bulk Gene Set Enrichment Analysis
DESeq2 was used to test for differential expression using negative binomial generalized linear models. T-statistics derived from differential expression analysis using DESeq2 were fed into the fgsea package (which expects ranked statistics) in order to run gene set enrichment analysis.
Ranked gene set enrichment analysis (GSEA) was run on all genes using the t-statistic calculated from the DESeq2 package. 50 gene sets based on Hallmark75 were used. GSEA analysis was performed using the fgsea function from the fgsea R package (v1.18)76.
Metabolomics and RNA ITH scores
Gene- and patient-wise intra-patient heterogeneity scores were calculated using multiregion data. Data was first median-centered to remove any metabolite/gene-level bias. For each metabolite/gene, the difference between each pair of samples from the same tumor were calculated. The median difference between the paired-differences was taken, yielding a metabolite/gene-specific, patient-specific measure of heterogeneity. This was repeated for all metabolites/genes, across all tumors, generating a matrix of metabolite/gene by patient values. Metabolite/gene intratumor heterogeneity values are summarized as the median value per metabolite/gene across all tumors in the cohort. Patient intratumor heterogeneity values are summarized as the median value per tumor across all metabolites/genes. Patient intratumor heterogeneity values represent the expected value of the absolute log2-fold change for a randomly chosen metabolite/gene within a given tumor. This measure of heterogeneity is not biased by the number of multiregion samples per tumor (Spearman correlation p-value = 0.23 in metabolomics, Spearman correlation p-value = 0.15 in transcriptomics) (Supplemental Figure 6c). Inter-patient heterogeneity scores were calculated by taking the difference between each pair of samples from all other patients.
DNA ITH scores
For each pair of regions, the percentage of unique mutations between regions from a patient and across patients was calculated. Genetic ITH scores for each patient were defined as the median proportion of private somatic mutations (SNVs and INDELs) not shared between regions from a single patient, equivalent to 1-Jaccard Similarity Index. Here, a score of 0/1 corresponded to complete overlap/complete lack of overlap in mutational profiles. Similar to RNA/metabolomics ITH scores, this measure of heterogeneity is not biased by the number of multiregion samples per tumor (Spearman correlation p-value = 0.72) (Supplemental Figure 10d).
Differential abundance score
The differential abundance (DA) score evaluates whether a metabolic pathway is differentially regulated between two groups. The score is calculated by applying a Wilcoxon rank sum test to all metabolites in a pathway. P-values were then corrected using the Benjamini-Hochberg method (FDR-adjusted p-value < 0.05). The DA score for each pathway is calculated as: (#significantly enriched metabolites - #significantly depleted metabolites)/ #total metabolites. Only pathways with 3 or more significantly altered metabolites were scored.
Robinson-Foulds Distance
Robinson-Foulds distances in Figure 2i, j were calculated using the phangorn R package (v2.8.1)77. To calculate the Robinson-Foulds distance between two trees (T_1 and T_2) with n tips:
where i(T_1, T_2) represents the number of internal edges and v_s(T_1, T_2) represents the number of shared internal splits. The reported Robinson-Foulds distance is calculated by dividing d(T_1, T_2) by the maximal possible distance i(T_1) + i(T_2) to get a normalized distance.
GPX4 Immunohistochemistry
Immunohistochemistry for glutathione peroxidase 4 (GPX4) was performed in 5 µm FFPE tissue sections using an automated staining system (Leica Bond RX) with 3,3′ diaminobenzidine detection (Figure 3h, i). Antigen retrieval was conducted for 30 minutes using Leica Bond epitope retrieval solution 1 (ER1; citrate, pH 6; cat. #AR9961), followed by an incubation with a rabbit monoclonal GPX4 antibody (EPNCIR144, Abcam, #ab125066, dilution 1:1200) for 30 minutes, and a subsequent incubation of the secondary antibody (Leica Bond Polymer Refine Detection cat. #DS9800). GPX4 staining was quantified as H-Scores [H= intensity (0–3) x percentage of positive cells (1–100)] by a pathologist (YBC) who was blinded to the metabolite levels of the samples.
Linear Mixed Modelling of Immune Phenotypes
A linear regression was performed where metabolite levels were used to predict immune signature expression. To account for repeated measures, a linear mixed model was used, with a random effect for each patient. Marginal r2 was calculated to consider only the variance of the fixed effects78. Functions from performance79 and lme480 packages were used to conduct the modelling. To assess significance, parametric bootstrapping was performed with 100,000 iterations using the pbkrtest package81.
The following equations were used to fit the linear mixed models. Actual model: Metabolite ~ ImmuneSignature + (1 | Patient)
Null model: Metabolite ~ (1 | Patient)
Immunofluorescence for CD31 and CD68
Immunofluorescence detections of CD31 and CD68 (Figure 5a–c) was performed by the Molecular Cytology Core Facility at Memorial Sloan Kettering Cancer Center using Discovery Ultra processor (Ventana Medical Systems.Roche-AZ). Heat and CC1 (Cell Conditioning 1, Ventana cat #950–500) retrieval were performed for 32 minutes. The tissue sections were then blocked for 30 minutes in Background Blocking reagent (Innovex, catalog#: NB306). Next, a mouse monoclonal anti-CD31 primary antibody (Ventana-Roche, cat#760–4378) was incubated for 5 hours, followed by a 16-minute secondary antibody incubation of biotinylated anti-mouse secondary (Vector Labs, MOM Kit BMK-2202) in 5.75 ug/mL Blocker D, Streptavidin-HRP, and TSA Alexa488 (Life Tech, cat#B40932) prepared according to manufacturer instructions in 1:150. 0.02 ug/mL of a mouse monoclonal IgG1 anti-CD68 antibody (DAKO, cat#M0814) was used and incubated for five hours, followed by a 16-minute secondary incubation using a biotinylated goat anti-mouse secondary antibody (Vector Labs, MOM Kit BMK-2202) in 5.75 ug/mL. Blocker D, Streptavidin-HRP, and CF 543 (Biotium, cat#92172) were prepared according to manufacturer instructions in 1:500. To counterstain, 5ug/mL of DAPI [dihydrochloride(2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride] (Sigma D9542) was used for 5 minutes at room temperature. The tissue was then mounted with anti-fade mounting medium Mowiol [Mowial 4–88 (CALBIOCHEM code: 475904)] and coverslipped.
To scan slides, a Pannoramic P250 Flash scanner (3DHistech, Hungary) with 20x/0.8NA objective lens was used. CaseViewer software (3DHistech, Hungary) was used to visualize digitized images. One representative region of interest (ROIs) was selected for CD31 and CD68 and then exported as .tif files. Regions were analyzed using ImageJ/Fiji software (v2.1.0; NIH USA)82.
Imputation of metabolites by RNA with MIRTH
MIRTH (Transcriptomics-Metabolite Imputation via Rank-Transformation and Harmonization) is a method to impute missing metabolites by jointly modeling metabolite covariation across heterogeneously covered metabolomics datasets37. Intuitively, MIRTH operates by learning the covariation between metabolite and transcript levels in a reference dataset where both are measured and then transferring that knowledge to other datasets where metabolites are unmeasured.
Before applying MIRTH, the metabolomics data in each dataset are preprocessed by total ion count (TIC) normalization and transcripts levels are transformed into units of TPM (Transcripts Per Million). Metabolomics and transcriptomics are then rank-transformed, resulting in values between 0 and 1 for each feature across all samples in a batch. Preprocessed metabolomics and transcriptomics data are then aggregated into a single aggregate multiple-dataset matrix X, which then undergoes nonnegative matrix factorization (NMF). As described in detail in 37, MIRTH implements a modified NMF which omits missing data from the element-wise object function. Optimization produces two embedding matrices W and H which are full rank and whose product yields a new matrix with no missing data. Ten-fold cross validation is used to determine the optimal number of embedding dimensions.
Three ccRCC datasets with paired metabolomics and transcriptomics data: M1 (N=32), M4 (N=144), and M5 (N=76) were used to assess MIRTH’s performance (Supplemental Figure 5a). For each dataset, we repeated the following procedure. We masked metabolites in the “target” dataset treating them as effectively missing. We then trained a MIRTH model (where the input data contained both metabolites and RNA from non-target datasets and RNA data only from the target dataset), and then calculated imputed metabolite levels from the target dataset. We repeated such experiments 10 times with different seeds to remove random bias. Finally, we compared for each (sample,metabolite) pair the average of imputed metabolite values by MIRTH with the true metabolite values in the testing set by calculating Spearman’s rank correlation coefficient. Metabolites that satisfied criteria for statistical significance (FDR-adjusted p-value < 0.05, average rho > 0.3) were defined as well-predicted in that dataset (Supplemental Figure 5b). For instance, 1-methylimidazole acetate and 2-aminoadipate were reproducibly well-predicted with an average rho of 0.67 and 0.51 across 3 datasets respectively (Supplemental Figure 5c). Metabolites that were well-predicted in at least 2 datasets are defined as reproducibly well-predicted. This yielded 262 reproducibly well-predicted metabolites.
Survival analysis
We collected RNA sequencing data and patient-level clinical data from 7 published trials of immunotherapeutic vs systemic agents in advanced ccRCC (IMmotion151 (N=823), NCT0242082183; JAVELIN Renal 101 (N=726), NCT0268400684; CheckMate 214 (N=167), NCT0223174985; COMPARZ (N=412), NCT0072094186; CheckMate 9ER (N=16), NCT0314117787; CheckMate 010 (N=45), NCT0135443188; CheckMate 025 (N=248), NCT0166878489).
Considering the different drug effect of treatments in clinical trials, we performed statistical analysis on immuno-therapy arms and sunitinib arms separately (Immuno-therapy drugs: Avelumab + Axitinib, Atezolizumab + Bevacizumab, Nivolumab, Ipilimumab + Nivolumab). For the 262 reproducibly well-predicted metabolites, we tested the association between levels of individual metabolites and progression free survival (PFS) in a multivariate Cox proportional-hazards model (adjusted by age and sex; PFS ~ metabolite level + age + gender). The Python package lifelines90 was used for survival regression. Regression results across multiple clinical trials were then aggregated by a meta-analysis method, restricted maximum likelihood (REML) random effects model in R package metafor91. All p-values were Benjamini-Hochberg corrected92.
Quantification and Statistical Analysis
Statistical analysis
All statistical tests were performed in R. Tests comparing distributions were performed using wilcox.test or t.test. All statistical analyses were two-sided, unless otherwise specified, and p-values were Benjamini-Hochberg corrected92. The survcomp package93 was used to perform the meta-analysis to combine results from the two metabolomic batches using Fisher’s method94.
Supplementary Material
Data S1: Source Data Tables, related to all main and supplementary figures
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal GPX4 | Abcam | #ab125066 |
| Mouse monoclonal CD31 | Vetana-Roche | #760–4378 |
| Mouse monoclonal IgG1 CD68 | DAKO | #M0814 |
| Deposited data | ||
| RNA-sequencing data | Golkaram et. al. | PMID: 36536472 |
| DNA-sequencing data | Golkaram et. al. | PMID: 36536472 |
| Metabolomics data | This paper | |
| Software and algorithms | ||
| DESeq2 | Love et al. | PMID: 25516281 |
| ssGSEA | Barbie et al. | PMID: 19847166 |
| CiberSort | Newman et al. | PMID: 25822800 |
| ESTIMATE | Yoshihara et al. | PMID: 24113773 |
| fgsea | Korotkevich et al. | 10.18129/B9.bioc.fgsea |
| phangorn | Schliep et al. | PMID: 21169378 |
| performance | Lüdecke et al. | DOI: 10.21105/joss.03139 |
| lme4 | Bates et al. | DOI: 10.18637//jss.v067.i01 |
| pbkrtest | Halekoh et al. | DOI: 10.18627//jss.v059.i09 |
| lifelines | Davidson-Pilon | DOI: 10.21105/joss.01317 |
| survcomp | Schröder et al. | PMID: 21903630 |
Highlights.
Multimodal measurements of the ccRCC in 187 tumor and adjacent-normal regions
ccRCC demonstrates a pattern of ferroptosis-associated intratumoral heterogeneity
Local metabolomic phenotypes associate with specific patterns of immune infiltration
Discovery of metabolite biomarkers of therapy response via RNA-metabolite covariation
Acknowledgements
We thank Sohrab Shah, Kayvan Keshari, and Jan Krumsiek for their thoughtful feedback. Graphics were created with www.BioRender.com. ER was supported by the Geoffrey Beene Cancer Research Center Grant Award, Department of Defense Kidney Cancer Research Program (no. W81XWH-18-1-0318), Cycle For Survival Equinox Innovation Award, Kidney Cancer Association Young Investigator Award, Brown Performance Group Innovation in Cancer Informatics Fund, and NIH R37 CA276200. AAH was supported by NIH R01 CA258886, DOD KCRP grant W81XWH-21-1-0941, the Mazumdar-Shaw Kidney Cancer Fund, and the Weiss Family Fund. This work was supported by NIH/NCI Cancer Center Support Grant (P30 CA008748).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
R.R.K is a consultant and/or on the Advisory Board for Eisai and reports receiving institutional research funding from Pfizer, Takeda, Novartis, Xencor, and Allogene Therapeutics. Y.C. is a consult for Inspirna, Inc. M.H.V. is a consultant and/or on the Advisory Board of Eisai, Exelixis, Merck, Calithera, Aveo, Genentech, Oncorena, Affimed, MICU Rx Aravive, onQuality, Astra Zeneca, and Mertelsmann Foundation. M.H.V has received research funding from Pfizer, BMS, and Genentech. C.H.L has received research funding from AstraZeneca, BMS, Calithera, Eisai, Eli Lilly, Exelixis, and Merck. C.H.L is a consultant for Aadi, Aveo, BMS, Exelixis, Eisai, Merck, Pfizer, EMD Serono, and Cardinal Health. C.H.L has received honoraria from AiCME, IDEOlogy Health, Intellisphere, Medscape, MJH, and Research to Practice. M.G. was an employee and shareholder of Illumina Inc. during the preparation of this study. M.G. is currently an employee and shareholder of Roche. R.J.M. is a consultant and/or on the Advisory Board of AstraZeneca, Aveo, Calithera Biosciences, Eisai, Exelixis, Genentech/Roche, Incyte, Pfizer, and Merck. R.J.M. has received research funding from Aveo, Bristol Myers Squibb, Eisai, Exelixis, Genentech/Roche, Novartis, Pfizer, and Merck. A.A.H. is on the Advisory Board for Merck. E.R. is a consultant for Xontogeny, LLC.
Additional Resources
Normalized metabolomics data matrices and sample information can be found at 10.5281/zenodo.7986891.
Using multimodal, multiregional profiling, Tang et al. show that intratumoral metabolomic heterogeneity in clear cell renal cell carcinoma (ccRCC) is driven by the oxidative stress response and coevolves with specific patterns of immune infiltration.
References
Bibliography
- 1.Hakimi AA, Reznik E, Lee C-H, Creighton CJ, Brannon AR, Luna A, Aksoy BA, Liu EM, Shen R, Lee W, et al. (2016). An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell 29, 104–116. 10.1016/j.ccell.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, et al. (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892. 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Şenbabaoğlu Y, Gejman RS, Winer AG, Liu M, Van Allen EM, de Velasco G, Miao D, Ostrovnaya I, Drill E, Luna A, et al. (2016). Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol 17, 231. 10.1186/s13059-016-1092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reznik E, Luna A, Aksoy BA, Liu EM, La K, Ostrovnaya I, Creighton CJ, Hakimi AA, and Sander C (2018). A Landscape of Metabolic Variation across Tumor Types. Cell Syst 6, 301–313.e3. 10.1016/j.cels.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network (2013). Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49. 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li B, Qiu B, Lee DSM, Walton ZE, Ochocki JD, Mathew LK, Mancuso A, Gade TPF, Keith B, Nissim I, et al. (2014). Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 513, 251–255. 10.1038/nature13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jing L, Guigonis J-M, Borchiellini D, Durand M, Pourcher T, and Ambrosetti D (2019). LC-MS based metabolomic profiling for renal cell carcinoma histologic subtypes. Sci. Rep 9, 15635. 10.1038/s41598-019-52059-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reigle J, Secic D, Biesiada J, Wetzel C, Shamsaei B, Chu J, Zang Y, Zhang X, Talbot NJ, Bischoff ME, et al. (2021). Tobacco smoking induces metabolic reprogramming of renal cell carcinoma. J. Clin. Invest. 131. 10.1172/JCI140522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossi M, Altea-Manzano P, Demicco M, Doglioni G, Bornes L, Fukano M, Vandekeere A, Cuadros AM, Fernández-García J, Riera-Domingo C, et al. (2022). PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature 605, 747–753. 10.1038/s41586-022-04758-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, Murphy MM, Gu Z, Gu W, Martin M, et al. (2020). Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120. 10.1038/s41586-019-1847-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okegawa T, Morimoto M, Nishizawa S, Kitazawa S, Honda K, Araki H, Tamura T, Ando A, Satomi Y, Nutahara K, et al. (2017). Intratumor Heterogeneity in Primary Kidney Cancer Revealed by Metabolic Profiling of Multiple Spatially Separated Samples within Tumors. EBioMedicine 19, 31–38. 10.1016/j.ebiom.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Udayakumar D, Cai L, Hu Z, Kapur P, Kho E-Y, Pavía-Jiménez A, Fulkerson M, de Leon AD, Yuan Q, et al. (2017). Addressing metabolic heterogeneity in clear cell renal cell carcinoma with quantitative Dixon MRI. JCI Insight [DOI] [PMC free article] [PubMed]
- 13.Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bossé D, Wankowicz SM, Cullen D, et al. (2018). Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806. 10.1126/science.aan5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biswas D, Birkbak NJ, Rosenthal R, Hiley CT, Lim EL, Papp K, Boeing S, Krzystanek M, Djureinovic D, La Fleur L, et al. (2019). A clonal expression biomarker associates with lung cancer mortality. Nat. Med 25, 1540–1548. 10.1038/s41591-019-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green DR, Galluzzi L, and Kroemer G (2014). Cell biology. Metabolic control of cell death. Science 345, 1250256. 10.1126/science.1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogretmen B (2018). Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 18, 33–50. 10.1038/nrc.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang X, Stockwell BR, and Conrad M (2021). Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol 22, 266–282. 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L, Hobeika CS, Khabibullin D, Yu D, Filippakis H, Alchoueiry M, Tang Y, Lam HC, Tsvetkov P, Georgiou G, et al. (2022). Hypersensitivity to ferroptosis in chromophobe RCC is mediated by a glutathione metabolic dependency and cystine import via solute carrier family 7 member 11. Proc Natl Acad Sci USA 119, e2122840119. 10.1073/pnas.2122840119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng Y-Y, Deasy R, Kost-Alimova M, Dančík V, et al. (2019). A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun 10, 1617. 10.1038/s41467-019-09277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Xing X, Zeng X, Jackson SR, TeSlaa T, Al-Dalahmah O, Samarah LZ, Goodwin K, Yang L, McReynolds MR, et al. (2022). Spatially resolved isotope tracing reveals tissue metabolic activity. Nat. Methods 19, 223–230. 10.1038/s41592-021-01378-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lau AN, and Vander Heiden MG (2020). Metabolism in the tumor microenvironment. Annu. Rev. Cancer Biol 4. 10.1146/annurev-cancerbio-030419-033333. [DOI] [Google Scholar]
- 23.Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, Sugiura A, Cohen AS, Ali A, Do BT, et al. (2021). Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288. 10.1038/s41586-021-03442-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kilgour MK, MacPherson S, Zacharias LG, Ellis AE, Sheldon RD, Liu EY, Keyes S, Pauly B, Carleton G, Allard B, et al. (2021). 1-Methylnicotinamide is an immune regulatory metabolite in human ovarian cancer. Sci. Adv 7. 10.1126/sciadv.abe1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zappasodi R, Serganova I, Cohen IJ, Maeda M, Shindo M, Senbabaoglu Y, Watson MJ, Leftin A, Maniyar R, Verma S, et al. (2021). CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature 591, 652–658. 10.1038/s41586-021-03326-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moffett JR, Arun P, Puthillathu N, Vengilote R, Ives JA, Badawy AA-B, and Namboodiri AM (2020). Quinolinate as a marker for kynurenine metabolite formation and the unresolved question of NAD+ synthesis during inflammation and infection. Front. Immunol 11, 31. 10.3389/fimmu.2020.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A, et al. (2018). Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab 27, 977–987.e4. 10.1016/j.cmet.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun 4, 2612. 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Lih T-SM, Dhanasekaran SM, Mannan R, Chen L, Cieslik M, Wu Y, Lu RJ-H, Clark DJ, Kołodziejczak I, et al. (2023). Histopathologic and proteogenomic heterogeneity reveals features of clear cell renal cell carcinoma aggressiveness. Cancer Cell 41, 139–163.e17. 10.1016/j.ccell.2022.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, Fong L, Joseph RW, Pal SK, Reeves JA, et al. (2018). Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 24, 749–757. 10.1038/s41591-018-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masiero M, Simões FC, Han HD, Snell C, Peterkin T, Bridges E, Mangala LS, Wu SY-Y, Pradeep S, Li D, et al. (2013). A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 24, 229–241. 10.1016/j.ccr.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motzer RJ, Robbins PB, Powles T, Albiges L, Haanen JB, Larkin J, Mu XJ, Ching KA, Uemura M, Pal SK, et al. (2020). Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med 26, 1733–1741. 10.1038/s41591-020-1044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hakimi AA, Voss MH, Kuo F, Sanchez A, Liu M, Nixon BG, Vuong L, Ostrovnaya I, Chen Y-B, Reuter V, et al. (2019). Transcriptomic Profiling of the Tumor Microenvironment Reveals Distinct Subgroups of Clear Cell Renal Cell Cancer: Data from a Randomized Phase III Trial. Cancer Discov 9, 510–525. 10.1158/2159-8290.CD-18-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rappold PM, Vuong L, Leibold J, Chakiryan NH, Curry M, Kuo F, Sabio E, Jiang H, Nixon BG, Liu M, et al. (2022). A Targetable Myeloid Inflammatory State Governs Disease Recurrence in Clear-Cell Renal Cell Carcinoma. Cancer Discov 12, 2308–2329. 10.1158/2159-8290.CD-21-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Capece L, Lewis-Ballester A, Marti MA, Estrin DA, and Yeh S-R (2011). Molecular basis for the substrate stereoselectivity in tryptophan dioxygenase. Biochemistry 50, 10910–10918. 10.1021/bi201439m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.King NJC, and Thomas SR (2007). Molecules in focus: indoleamine 2,3-dioxygenase. Int. J. Biochem. Cell Biol 39, 2167–2172. 10.1016/j.biocel.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 37.Freeman BA, Jaro S, Park T, Keene S, Tansey W, and Reznik E (2022). MIRTH: Metabolite Imputation via Rank-Transformation and Harmonization. Genome Biol 23, 184. 10.1186/s13059-022-02738-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Branco ACCC, Yoshikawa FSY, Pietrobon AJ, and Sato MN (2018). Role of histamine in modulating the immune response and inflammation. Mediators Inflamm 2018, 9524075. 10.1155/2018/9524075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swanton C (2012). Intratumor heterogeneity: evolution through space and time. Cancer Res 72, 4875–4882. 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. (2011). Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94. 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerlinger M, and Swanton C (2010). How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br. J. Cancer 103, 1139–1143. 10.1038/sj.bjc.6605912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee AJX, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IPM, Howell M, et al. (2011). Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 71, 1858–1870. 10.1158/0008-5472.CAN-10-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, and Sawyers CL (2002). Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117–125. 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 44.Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, Laï J-L, Philippe N, Facon T, Fenaux P, and Preudhomme C (2002). Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood 100, 1014–1018. 10.1182/blood.v100.3.1014. [DOI] [PubMed] [Google Scholar]
- 45.Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, and Downing JR (2008). Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 322, 1377–1380. 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campbell PJ, Pleasance ED, Stephens PJ, Dicks E, Rance R, Goodhead I, Follows GA, Green AR, Futreal PA, and Stratton MR (2008). Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci USA 105, 13081–13086. 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin M-L, et al. (2010). The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–1113. 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJW, et al. (2015). Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241. 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berbudi A, Rahmadika N, Tjahjadi AI, and Ruslami R (2020). Type 2 diabetes and its impact on the immune system. Curr. Diabetes Rev 16, 442–449. 10.2174/1573399815666191024085838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andersen CJ, Murphy KE, and Fernandez ML (2016). Impact of obesity and metabolic syndrome on immunity. Adv. Nutr 7, 66–75. 10.3945/an.115.010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitchell TC, Hamid O, Smith DC, Bauer TM, Wasser JS, Olszanski AJ, Luke JJ, Balmanoukian AS, Schmidt EV, Zhao Y, et al. (2018). Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol 36, 3223–3230. 10.1200/JCO.2018.78.9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Evans AM, DeHaven CD, Barrett T, Mitchell M, and Milgram E (2009). Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem 81, 6656–6667. 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- 53.Dieterle F, Ross A, Schlotterbeck G, and Senn H (2006). Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem 78, 4281–4290. 10.1021/ac051632c. [DOI] [PubMed] [Google Scholar]
- 54.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 56.Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118. 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, and Kent WJ (2004). The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32, D493–6. 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498. 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576. 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, Chen X, Kim Y, Beyter D, Krusche P, et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 64.Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SRF , WGS500 Consortium, Wilkie AOM, McVean G, and Lunter G (2014). Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet 46, 912–918. 10.1038/ng.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, and Ding L (2012). SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317. 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018). Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst 6, 271–281.e7. 10.1016/j.cels.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, and Cunningham F (2016). The ensembl variant effect predictor. Genome Biol 17, 122. 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep 9, 9354. 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shen R, and Seshan VE (2016). FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 44, e131. 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J, et al. (2017). Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 171, 1259–1271.e11. 10.1016/j.cell.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 72.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48– 61. 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2016). Fast gene set enrichment analysis. BioRxiv 10.1101/060012. [DOI]
- 77.Schliep KP (2011). phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593. 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakagawa S, and Schielzeth H (2013). A general and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol. Evol 4, 133–142. 10.1111/j.2041-210x.2012.00261.x. [DOI] [Google Scholar]
- 79.Lüdecke D, Ben-Shachar M, Patil I, Waggoner P, and Makowski D (2021). performance: An R Package for Assessment, Comparison and Testing of Statistical Models. JOSS 6, 3139. 10.21105/joss.03139. [DOI] [Google Scholar]
- 80.Bates D, Mächler M, Bolker B, and Walker S (2015). Fitting linear mixed-effects models using lme4. J. Stat. Softw 67, 1–48. 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- 81.Halekoh U, and Højsgaard S (2014). A Kenward-Roger Approximation and Parametric Bootstrap Methods for Tests in Linear Mixed Models - The R Packagepbkrtest. J. Stat. Softw 59. 10.18637/jss.v059.i09. [DOI] [Google Scholar]
- 82.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rini BI, Powles T, Atkins MB, Escudier B, McDermott DF, Suarez C, Bracarda S, Stadler WM, Donskov F, Lee JL, et al. (2019). Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet 393, 2404–2415. 10.1016/S0140-6736(19)30723-8. [DOI] [PubMed] [Google Scholar]
- 84.Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, Venugopal B, Kollmannsberger C, Negrier S, Uemura M, et al. (2019). Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med 380, 1103–1115. 10.1056/NEJMoa1816047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthélémy P, Porta C, George S, et al. (2018). Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med 378, 1277–1290. 10.1056/NEJMoa1712126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, Nathan P, Staehler M, de Souza P, Merchan JR, et al. (2013). Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med 369, 722–731. 10.1056/NEJMoa1303989. [DOI] [PubMed] [Google Scholar]
- 87.Choueiri TK, Powles T, Burotto M, Escudier B, Bourlon MT, Zurawski B, Oyervides Juárez VM, Hsieh JJ, Basso U, Shah AY, et al. (2021). Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med 384, 829–841. 10.1056/NEJMoa2026982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, Vaishampayan UN, Drabkin HA, George S, Logan TF, et al. (2015). Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J. Clin. Oncol 33, 1430–1437. 10.1200/JCO.2014.59.0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. (2015). Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med 373, 1803–1813. 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davidson-Pilon C (2019). lifelines: survival analysis in Python. Journal of Open Source Software 4, 1317. [Google Scholar]
- 91.Viechtbauer W (2010). Conducting Meta-Analyses in R with the metafor Package. J. Stat. Softw 36. 10.18637/jss.v036.i03. [DOI] [Google Scholar]
- 92.Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B (Methodological) 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 93.Schröder MS, Culhane AC, Quackenbush J, and Haibe-Kains B (2011). survcomp: an R/Bioconductor package for performance assessment and comparison of survival models. Bioinformatics 27, 3206–3208. 10.1093/bioinformatics/btr511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mosteller F, and Fisher RA (1948). Questions and Answers. The American Statistician 2, 30. 10.2307/2681650. [DOI] [Google Scholar]
- 95.Golkaram M, Kuo F, Gupta S, Carlo MI, Salmans ML, Vijayaraghavan R, Tang C, Makarov V, Rappold P, Blum KA, et al. (2022). Spatiotemporal evolution of the clear cell renal cell carcinoma microenvironment links intra-tumoral heterogeneity to immune escape. Genome Med 14, 143. 10.1186/s13073-022-01146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Source Data Tables, related to all main and supplementary figures
Data Availability Statement
Metabolomics data generated and used in this study are published at 10.5281/zenodo.7986891. RNA and DNA sequencing data and clinical information was retrieved from Golkaram et al95. All data used to generate display items in this manuscript are available in Data S1. All original code has been deposited at https://github.com/reznik-lab/rcc_coevolution and is publicly available as of the date of publication. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.