

Abstract
Artificial intelligence (AI) approaches in cancer analysis typically utilize a ‘one-size-fits-all’ methodology characterizing average patient responses. This manner neglects the diverse conditions in the pancancer and cancer subtypes of individual patients, resulting in suboptimal outcomes in diagnosis and treatment. To overcome this limitation, we shift from a blanket application of statistics to a focus on the explicit recognition of patient-specific abnormalities. Our objective is to use multiomics data to empower clinicians with personalized molecular descriptions that allow for customized diagnosis and interventions. Here, we propose a highly trustworthy multiomics learning (HTML) framework that employs multiomics self-adaptive dynamic learning to process each sample with data-dependent architectures and computational flows, ensuring personalized and trustworthy patient-centering of cancer diagnosis and prognosis. Extensive testing on a 33-type pancancer dataset and 12 cancer subtype datasets underscored the superior performance of HTML compared with static-architecture-based methods. Our findings also highlighting the potential of HTML in elucidating complex biological pathogenesis and paving the way for improved patient-specific care in cancer treatment.
Keywords: multiomics Integration, dynamic learning, cancer diagnosis and prognosis, biomarker identification
INTRODUCTION
Unlocking the full potential of artificial intelligence (AI) in cancer analysis requires a departure from the conventional ‘one-size-fits-all’ approach. The concept of generalizing patient responses fails to acknowledge the complex and diverse conditions of individuals during diagnosis and treatment. This antiquated viewpoint inevitably leads to ineffective treatments, where success stories are overshadowed by disappointing outcomes. To truly revolutionize cancer analysis through AI, we must recognize and embrace the individuality of each patient.
Gone are the days when we could rely on generalized patient responses as a basis for treatment decisions. This outdated approach often leads to suboptimal outcomes, where some patients experience remarkable success while others face disappointing results. The future lies in AI-enabled personalized medicine [1], offering a solution to the uncertainties that plague cancer diagnosis and treatment planning. By shifting our focus from mere statistics and the law of averages, we can now delve into the intricate molecular-level multiomics data of each patient [2–4], pinpointing abnormalities with precision and tailoring treatment strategies accordingly [5].
This powerful tool is poised to significantly enhance the capabilities of clinicians, positioning them at the cutting edge of proactive and therapeutic interventions [6]. By harnessing the power of AI, healthcare professionals can predict the likelihood of disease onset, ensuring that intervention happens at the earliest possible stage. Beyond just detection, AI aids in tailoring treatments to individual patients, heralding the age of personalized medicine. This ensures that treatments are optimized for each patient’s unique genetic makeup and health profile, leading to more effective and fewer side effects. The integration of AI into medical practice brings with it a renewed sense of hope. As we delve deeper into the vast complexities of diseases like cancer, the promise of AI-driven solutions hints at a future where these formidable challenges can be comprehensively understood and addressed [7].
Our study proposes a highly trustworthy multiomics learning (HTML) framework (Supplementary Figure S1 available online at http://bib.oxfordjournals.org/), a multiomics dynamic learning framework that is meticulously designed to address the critical features and requirements essential for cancer analysis [8–11]. HTML offers efficiency, interpretability, and reliability in integrating multiomics data by processing each sample with data-dependent architectures and computational flows, opening new avenues for research and clinical practice. We introduced sample-adaptive feature selection and modality selection dynamic learning modules to allocate different weights to features and modalities according the input information. Inspired by biological knowledge, we devised DNA methylation-guided attention and triple contrastive learning modules within HTML, enabling us to replicate hierarchical relationships and align representations of biological components. These approaches not only enhance our understanding of various genes and data modalities but also facilitate personalized diagnosis and treatment through a sample-adaptive process. Compared with other methods, HTML stands out with its superior performance, remarkable interpretability and biologically-guided mechanisms, excelling in capturing intricate biological relationships and identifying biomarkers effectively.
To demonstrate the power of HTML, we construct a comprehensive dataset encompassing a 33-type pancancer dataset and 12 cancer subtype datasets. The results are astounding. HTML surpasses current state-of-the-art approaches, achieving a remarkable increase of 3.51% in pancancer classification accuracy and an impressive 6.8% increase in cancer subtype classification accuracy compared to previous state-of-the-art methods. Moreover, HTML proficiently classifies patients into high-risk and low-risk groups, accurately determining disease risk levels within individual cancer subtypes. Through feature dynamic weights, we identify 849 potential biomarkers across all cancer subtype datasets, many of which have already been highlighted in published articles for their significance in cancer research.
In the spirit of promoting collaboration and facilitating further scientific advancement, we have made the complete HTML package available online. This package includes tutorials, demo cases, training data, pretrained models and test results from our study, all accessible to the broader scientific community. The HTML package can be found at https://github.com/YuxingLu613/HTML.
RESULTS
A highly trustworthy multiomics learning model
In this paper, we present HTML, a highly trustworthy multiomics learning model explicitly designed for multiomics classification tasks (as depicted in Figure 1b). The proposed framework comprises five primary modules:
Figure 1.

Framework of HTML. (a) Illustration and application of our work. (b) The overall pipeline of the HTML model. HTML is an end-to-end framework that integrates multiomics data to perform cancer diagnosis and prognosis. (c) Dynamic learning on both features and modalities. The same feature may play diverse roles across samples, and it is crucial to allocate a samplewise adaptive weight to each feature rather than relying on a fixed weight vector. Similarly, varying weights should be assigned to modalities to indicate their distinct contributions to the final results. (d) DNA methylation-guided attention mechanism. DNA methylation can affect the expression levels of mRNA and miRNA. This guided attention mechanism reflects the interrelationships among different modalities and outputs a coordinated representation that integrates the biological information among modalities. (e) Triple contrastive learning module. Multiomics learning tasks are typically conducted in an unsupervised learning setting, where the data in each modality must be subject to the overall label of the sample. In this module, modality embeddings from a given sample are considered positive pairs, while those from different samples are considered negative pairs. This approach effectively aligns the representations of different modalities, facilitating the integration of multiple data sources. (f) Dirichlet uncertainty prediction. If the probability distribution of a classification result is evenly distributed, it may indicate that the model has a limited capacity to differentiate between various categories. Conversely, when the probability distribution of a specific label is significantly high, it suggests that the model has a higher degree of confidence in its classification result and a more precise prediction. The contribution of each module in HTML was comprehensively addressed through the ablation study in Supplementary Table 4.
An inner-omics feature dynamic module that self-adaptively selects informative features (Figure 1c),
An interomics DNA methylation guided attention module that appropriately integrates complementary information among diverse modalities (Figure 1d),
A triple contrastive learning module that aligns the features of diverse modalities (Figure 1e),
A modality dynamic module that quantifies the relative contribution of each modality toward the final classification results (Figure 1f),
An uncertainty module for prediction and integration employs robust statistical tools (Dirichlet distribution and Dempster–Shafer theory) to provide reliable predictions and enhance the explainability of the model (Figure 1f).
When all five modules of the proposed HTML architecture are integrated with each other, the resulting framework offers both high performance and significant explainability, making it an ideal multiomics integration model for clinical applications such as cancer diagnosis and survival analysis.
HTML is an end-to-end model that can effectively improve the performance of multiomics classification tasks, enabling personalized diagnosis and treatment and facilitating the discovery of novel biomarkers. Our extensive research has led us to firmly believe that HTML constitutes the foremost approach to multiomics integration in biomedical data classification, characterized by high performance, trustworthiness and sensitivity to individual variations.
Datasets and experimental settings
Dataset
We constructed a large-scale pancancer classification dataset of 33 different cancer types and 12 cancer subtype classification datasets from TCGA to showcase the effectiveness of HTML (Methods). Three types of omics data (DNA methylation (DNA meth), mRNA expression (mRNA), and micro-RNA expression (miRNA) except for the pancancer dataset) were used to enable multiomics integration and classification, leading to the elaboration of complementary information pertaining to cancers.
In our experiments, only the samples that matched omics data were included in our study (Supplementary Tables 1–3 available online at http://bib.oxfordjournals.org/). In light of the potential impact of noise and redundant features on the overall quality, we conducted preprocessing and feature preselection on each type of omics data independently. Supplementary Tables 2 and 3 (available online at http://bib.oxfordjournals.org/) display the precise feature quantity before and after preprocessing.
Experimental settings
We conducted a comparative analysis of the classification accuracy of HTML in relation to preexisting multiomics integration algorithms (Table 1 and Supplementary Table 4 available online at http://bib.oxfordjournals.org/) and performed a comprehensive set of ablation studies (Supplementary Table 5 available online at http://bib.oxfordjournals.org/) to demonstrate the crucial roles played by distinct components in HTML. The hyperparameter selection comparison for each dataset is shown in Supplementary Table 6 available online at http://bib.oxfordjournals.org/. To provide a more valid estimate of HTML’s performance, we executed a 5-fold cross-validation and subsequently reported the mean and standard deviation (SD) of the results on the validation fold.
Table 1.
Performance of HTML under different cancer subtypes. To evaluate the effectiveness of HTML, we conducted experiments on 12 different datasets from TCGA and compared it with ten prevalent multiomics classification methods. Our results show that HTML outperforms all other methods in terms of accuracy, F1-macro, and F1-weighted scores on all datasets and achieves the best AUROC and AUPRC scores on most datasets. To ensure the reliability of our results, we applied 5-fold validation and calculated the mean value and standard deviation of each metric for each method.
| Traditional ML methods | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NB | LR | SVM | RF | XGBoost | ||||||||||||||||||||||
| Dataset | Accuracy | F1-macro | F1-weighted | AUC score | PRC score | Accuracy | F1-macro | F1-weighted | AUC score | PRC score | Accuracy | F1-macro | F1-weighted | AUC score | PRC score | Accuracy | F1-macro | F1-weighted | AUC score | PRC score | Accuracy | F1-macro | F1-weighted | AUC score | PRC score | |
| COADREAD | 65.36 (3.27) | 61.62 (2.96) | 67.96 (2.69) | 68.26 (0.86) | 74.44 (0.69) | 86.21 (4.40) | 74.49 (6.45) | 81.76 (4.79) | 77.80 (7.41) | 83.75 (4.98) | 76.80 (3.35) | 45.88 (4.06) | 67.53 (5.18) | 51.19 (1.65) | 73.12 (5.13) | 80.11 (2.98) | 64.39 (5.84) | 76.94 (4.06) | 62.74 (4.51) | 74.75 (7.19) | 82.13 (4.38) | 73.45 (6.19) | 83.20 (5.00) | 76.11 (6.21) | 83.15 (4.54) | |
| ESCA | 89.61 (5.88) | 89.40 (5.78) | 89.62 (5.87) | 90.09 (5.56) | 92.50 (3.88) | 93.99 (3.54) | 93.79 (3.58) | 94.02 (3.54) | 94.33 (3.72) | 95.46 (2.64) | 93.99 (3.54) | 93.82 (3.55) | 94.03 (3.53) | 94.52 (3.52) | 95.49 (2.61) | 95.08 (3.01) | 94.95 (2.99) | 95.10 (3.00) | 95.54 (2.92) | 96.37 (2.11) | 92.91 (5.60) | 92.72 (5.57) | 92.93 (5.59) | 93.37 (5.41) | 94.81 (3.82) | |
| GBMLGG | 51.87 (3.80) | 51.15 (2.94) | 53.05 (3.42) | 64.17 (2.02) | 60.76 (2.00) | 52.84 (7.71) | 46.70 (6.50) | 50.51 (8.62) | 62.51 (4.50) | 55.83 (5.15) | 61.24 (7.14) | 47.72 (5.60) | 53.49 (8.76) | 67.11 (3.08) | 63.18 (6.87) | 57.73 (5.66) | 49.35 (4.43) | 53.94 (6.86) | 65.52 (2.82) | 58.52 (2.27) | 56.75 (4.52) | 50.32 (3.12) | 54.57 (4.68) | 65.03 (2.26) | 58.69 (1.87) | |
| SARC | 64.97 (2.15) | 63.76 (3.27) | 65.98 (2.37) | 74.29 (2.56) | 71.18 (3.84) | 82.90 (2.75) | 81.43 (2.36) | 82.62 (2.85) | 85.83 (2.24) | 85.46 (1.57) | 77.04 (3.53) | 73.56 (5.01) | 76.22 (4.26) | 80.59 (3.88) | 80.73 (3.59) | 78.21 (2.18) | 75.46 (2.94) | 77.87 (2.18) | 81.71 (1.64) | 81.20 (2.85) | 75.85 (5.20) | 72.85 (5.87) | 75.45 (5.29) | 79.71 (3.79) | 77.93 (5.47) | |
| STAD | 53.37 (0.68) | 53.02 (0.50) | 53.14 (0.89) | 54.33 (1.61) | 66.06 (1.80) | 62.53 (4.66) | 61.78 (4.49) | 62.47 (4.71) | 62.07 (4.48) | 71.47 (3.27) | 56.86 (5.78) | 54.90 (6.26) | 55.67 (6.69) | 56.30 (5.46) | 67.48 (4.35) | 60.11 (3.88) | 59.47 (4.04) | 59.90 (4.52) | 60.33 (3.70) | 70.32 (2.57) | 63.60 (7.15) | 62.91 (7.69) | 63.41 (7.51) | 63.66 (7.53) | 72.78 (5.61) | |
| STES | 90.17 (3.15) | 90.00 (3.17) | 90.14 (3.15) | 89.88 (3.16) | 92.54 (2.36) | 94.55 (3.30) | 94.49 (3.32) | 94.55 (3.30) | 94.56 (3.40) | 95.88 (2.53) | 95.11 (3.53) | 95.04 (3.60) | 95.11 (3.53) | 95.16 (3.60) | 96.30 (2.69) | 93.45 (1.43) | 93.38 (1.51) | 93.46 (1.43) | 93.57 (1.60) | 95.10 (1.17) | 91.83 (4.64) | 91.74 (4.67) | 91.84 (4.64) | 91.84 (4.67) | 93.86 (3.55) | |
| THCA | 77.20 (6.76) | 70.16 (5.32) | 78.62 (6.01) | 74.39 (6.25) | 77.05 (4.16) | 81.40 (5.13) | 67.55 (7.10) | 80.06 (5.55) | 66.00 (6.69) | 73.37 (5.88) | 81.60 (4.51) | 64.69 (6.86) | 79.17 (4.86) | 62.70 (5.68) | 72.83 (7.41) | 83.80 (2.59) | 71.02 (7.17) | 82.58 (2.82) | 69.46 (7.34) | 76.46 (4.95) | 82.20 (4.76) | 69.40 (6.75) | 81.20 (4.54) | 67.93 (6.78) | 74.75 (5.35) | |
| UCEC | 71.40 (4.24) | 47.62 (6.00) | 72.50 (4.04) | 67.59 (4.20) | 54.44 (5.35) | 83.49 (5.03) | 53.40 (4.61) | 82.61 (5.57) | 71.66 (4.56) | 59.61 (9.93) | 85.12 (4.30) | 54.39 (4.13) | 82.86 (5.22) | 71.41 (4.61) | 73.75 (3.45) | 83.95 (4.53) | 52.94 (4.10) | 81.15 (5.35) | 68.69 (4.51) | 72.77 (3.09) | 84.65 (3.80) | 54.50 (2.79) | 82.63 (4.92) | 71.47 (4.12) | 67.42 (8.91) | |
| BRCA | 74.97 (3.44) | 74.16 (4.71) | 76.42 (2.89) | 87.33 (3.09) | 79.09 (3.93) | 78.63 (2.87) | 74.72 (4.96) | 78.86 (3.26) | 83.57 (2.87) | 77.66 (4.44) | 82.06 (3.80) | 78.77 (5.77) | 82.32 (3.75) | 86.57 (3.40) | 82.20 (5.08) | 74.06 (3.22) | 66.75 (3.87) | 73.43 (3.37) | 78.78 (3.25) | 70.24 (3.35) | 74.97 (3.06) | 68.23 (2.49) | 74.67 (3.19) | 80.37 (1.93) | 71.40 (2.35) | |
| LGG | 63.35 (3.54) | 61.24 (4.32) | 61.20 (4.01) | 63.89 (4.59) | 74.93 (4.01) | 66.79 (4.47) | 66.61 (4.45) | 66.76 (4.46) | 66.71 (4.59) | 75.04 (3.50) | 68.50 (5.27) | 68.00 (5.37) | 68.07 (5.24) | 68.74 (5.90) | 77.24 (5.04) | 66.59 (4.39) | 66.32 (4.39) | 66.45 (4.38) | 66.58 (4.54) | 75.09 (3.51) | 65.83 (8.01) | 65.63 (8.01) | 65.78 (7.99) | 65.77 (8.18) | 74.38 (6.25) | |
| KIPAN | 85.15 (2.10) | 82.90 (3.15) | 85.35 (1.94) | 89.39 (2.20) | 86.41 (2.60) | 95.19 (1.37) | 94.02 (1.45) | 95.21 (1.37) | 96.26 (1.32) | 94.95 (1.31) | 93.78 (2.14) | 92.24 (2.46) | 93.80 (2.09) | 94.66 (1.44) | 93.34 (2.05) | 91.66 (2.55) | 90.46 (2.88) | 91.67 (2.53) | 93.18 (1.79) | 91.98 (2.49) | 93.78 (1.95) | 92.88 (2.17) | 93.80 (1.92) | 95.29 (1.61) | 94.09 (1.88) | |
| ROSMAP | 66.37 (5.48) | 66.10 (5.62) | 66.12 (5.61) | 66.92 (5.80) | 75.56 (4.45) | 67.80 (4.26) | 67.47 (4.66) | 67.55 (4.57) | 68.19 (4.60) | 76.44 (3.33) | 67.52 (4.00) | 67.22 (4.22) | 67.28 (4.26) | 67.94 (4.17) | 76.27 (3.00) | 66.94 (5.45) | 66.53 (6.03) | 66.58 (5.85) | 67.49 (6.44) | 76.02 (4.98) | 68.68 (5.01) | 68.46 (5.02) | 68.62 (5.14) | 68.72 (5.21) | 76.56 (3.80) | |
| Deep Neural Network | ||||||||||||||||||||||||||
| MOGONET (2021) | MMDynamic (2022) | EMOGI (2021) | Subtype-DCC (2023) | TMC (2022) | ||||||||||||||||||||||
| Dataset | Accuracy | F1-macro | F1-weighted | AUC score | PRC score | Accuracy | F1-macro | F1-weighted | AUC score | PRC score | Accuracy | F1-macro | F1-weighted | AUC score | PRC score | Accuracy | F1-macro | F1-weighted | AUC score | PRC score | Accuracy | F1-macro | F1-weighted | AUC score | PRC score | Uncertainty |
| COADREAD | 79.79 (6.39) | 60.53 (9.36) | 74.61 (9.26) | 73.21 (11.09) | 70.77 (7.36) | 76.48 (6.03) | 43.27 (1.96) | 66.42 (8.14) | 73.94 (5.62) | 69.74 (3.87) | 79.20 (6.83) | 58.73 (14.22) | 73.94 (10.58) | 73.24 (6.19) | 68.89 (4.78) | 82.21 (4.28) | 64.18 (14.17) | 78.05 (7.69) | 77.73 (7.63) | 74.51 (7.14) | 77.38 (6.49) | 49.90 (7.16) | 69.48 (9.23) | 77.80 (2.71) | 73.11 (2.43) | 65.08 (1.92) |
| ESCA | 94.35 (1.35) | 94.33 (1.36) | 94.35 (1.35) | 94.74 (1.68) | 94.46 (2.27) | 96.68 (4.08) | 96.67 (4.10) | 96.69 (4.06) | 98.75 (1.83) | 96.27 (2.01) | 96.70 (2.70) | 96.67 (2.72) | 96.70 (2.69) | 98.51 (2.08) | 98.10 (2.91) | 96.17 (2.77) | 96.11 (2.80) | 96.15 (2.78) | 98.73 (1.10) | 98.77 (1.10) | 97.36 (2.17) | 97.35 (2.18) | 97.36 (2.17) | 97.87 (1.61) | 96.68 (2.01) | 65.04 (3.95) |
| GBMLGG | 54.60 (5.76) | 42.35 (5.59) | 47.41 (7.42) | 72.51 (2.07) | 58.09 (2.51) | 57.14 (4.05) | 46.19 (5.85) | 50.95 (5.20) | 70.69 (5.22) | 54.50 (5.12) | 60.26 (4.84) | 45.81 (2.71) | 51.84 (5.67) | 71.80 (2.86) | 57.26 (4.41) | 58.11 (7.44) | 44.80 (4.64) | 50.47 (7.63) | 69.48 (4.78) | 52.90 (5.24) | 54.76 (9.92) | 39.89 (9.30) | 45.20 (11.01) | 69.05 (9.86) | 53.46 (9.97) | 75.53 (4.18) |
| SARC | 83.70 (6.50) | 82.70 (5.81) | 83.61 (6.62) | 91.85 (2.92) | 85.83 (2.33) | 78.27 (8.19) | 74.93 (13.74) | 75.70 (12.01) | 91.59 (3.59) | 86.34 (6.41) | 78.21 (7.73) | 70.24 (11.74) | 75.39 (9.32) | 91.90 (4.12) | 84.19 (8.14) | 79.00 (7.67) | 75.14 (10.48) | 76.74 (9.27) | 91.66 (3.60) | 86.26 (5.59) | 81.31 (2.72) | 80.66 (3.26) | 80.67 (3.46) | 92.72 (1.36) | 88.42 (2.62) | 64.24 (0.81) |
| STAD | 63.08 (2.84) | 56.51 (7.82) | 58.19 (5.12) | 63.71 (6.17) | 63.68 (5.52) | 64.42 (3.17) | 59.81 (9.49) | 61.47 (6.80) | 64.26 (3.26) | 63.24 (3.46) | 62.28 (4.67) | 55.84 (8.08) | 57.75 (6.77) | 64.09 (4.16) | 63.25 (3.15) | 64.70 (5.50) | 60.93 (7.06) | 61.70 (7.28) | 64.12 (5.70) | 64.59 (5.41) | 60.38 (2.42) | 49.03 (7.92) | 51.84 (6.51) | 61.73 (4.03) | 61.38 (3.42) | 52.77 (5.39) |
| STES | 89.10 (14.08) | 84.21 (23.33) | 86.18 (19.70) | 82.42 (29.99) | 84.65 (23.82) | 94.02 (4.32) | 93.78 (4.51) | 94.03 (4.32) | 97.63 (2.27) | 97.48 (2.40) | 89.11 (9.49) | 88.72 (9.69) | 88.79 (10.06) | 98.28 (2.27) | 97.73 (3.39) | 98.33 (2.22) | 98.28 (2.29) | 98.33 (2.24) | 99.55 (0.63) | 99.52 (0.70) | 88.60 (6.66) | 88.21 (6.73) | 88.51 (6.66) | 97.34 (2.22) | 96.98 (2.64) | 46.96 (2.55) |
| THCA | 83.20 (6.05) | 62.09 (13.36) | 78.30 (9.02) | 84.53 (4.11) | 77.05 (3.95) | 80.40 (4.22) | 50.28 (7.29) | 72.92 (6.70) | 83.50 (3.43) | 75.94 (2.14) | 86.00 (2.28) | 75.03 (6.10) | 85.07 (2.57) | 85.73 (2.78) | 78.88 (4.68) | 86.20 (1.17) | 75.38 (3.97) | 85.23 (1.70) | 85.33 (2.47) | 79.58 (2.11) | 83.20 (3.43) | 62.21 (7.06) | 78.97 (5.01) | 84.51 (4.73) | 78.22 (5.15) | 36.05 (1.63) |
| MOGONET (2021) | MMDynamic (2022) | EMOGI (2021) | Subtype-DCC (2023) | TMC (2022) | ||||||||||||||||||||||
| UCEC | 86.51 (5.38) | 54.82 (7.78) | 83.21 (7.06) | 83.80 (5.08) | 65.53 (1.79) | 81.16 (4.44) | 47.25 (7.34) | 76.51 (6.59) | 84.79 (3.30) | 63.43 (3.54) | 84.88 (4.65) | 52.45 (8.22) | 81.40 (5.86) | 85.47 (4.69) | 67.20 (5.71) | 84.19 (5.48) | 55.18 (6.29) | 81.44 (6.90) | 86.91 (0.95) | 64.44 (1.92) | 79.53 (6.18) | 45.70 (9.72) | 74.40 (10.25) | 85.78 (3.13) | 62.97 (3.34) | 43.71 (1.07) |
| BRCA | 64.69 (7.26) | 38.57 (11.43) | 55.57 (9.94) | 89.37 (2.16) | 71.48 (6.75) | 57.60 (6.77) | 26.82 (4.07) | 44.82 (7.05) | 85.56 (3.43) | 61.41 (3.89) | 63.54 (2.81) | 34.24 (5.35) | 51.86 (5.09) | 88.59 (1.51) | 68.41 (4.92) | 67.43 (4.47) | 42.52 (6.32) | 59.44 (7.70) | 90.80 (1.63) | 76.15 (4.14) | 57.94 (6.45) | 25.01 (8.41) | 43.65 (7.56) | 88.59 (2.68) | 67.22 (7.93) | 48.18 (0.67) |
| LGG | 75.38 (2.00) | 74.56 (2.27) | 74.98 (2.06) | 76.19 (3.13) | 76.23 (2.28) | 85.87 (4.30) | 74.67 (14.65) | 84.29 (5.73) | 95.33 (3.86) | 87.08 (11.03) | 69.84 (3.38) | 67.42 (3.28) | 67.89 (3.87) | 75.90 (3.61) | 76.78 (3.76) | 71.57 (5.53) | 69.51 (8.57) | 69.94 (8.29) | 78.36 (2.26) | 78.07 (1.43) | 66.05 (5.42) | 65.02 (5.64) | 65.14 (5.68) | 70.44 (5.42) | 70.64 (5.54) | 48.38 (6.84) |
| KIPAN | 94.20 (2.63) | 87.00 (12.61) | 93.56 (3.71) | 98.92 (0.43) | 97.01 (1.84) | 67.37 (5.29) | 63.96 (8.37) | 64.88 (7.72) | 75.24 (3.56) | 74.89 (3.64) | 93.06 (3.23) | 90.92 (3.78) | 93.01 (3.32) | 98.75 (0.51) | 95.86 (2.71) | 94.48 (3.31) | 92.91 (3.58) | 94.50 (3.28) | 98.97 (0.31) | 95.69 (3.39) | 89.80 (5.52) | 82.96 (12.35) | 88.79 (6.52) | 98.38 (0.90) | 94.50 (3.38) | 43.62 (0.68) |
| ROSMAP | 71.52 (2.29) | 70.29 (2.08) | 70.69 (2.15) | 75.36 (3.09) | 76.88 (2.83) | 56.99 (5.32) | 46.52 (11.58) | 47.82 (10.57) | 66.69 (6.48) | 67.15 (6.12) | 66.99 (7.05) | 66.13 (7.08) | 66.20 (7.23) | 73.27 (6.40) | 73.91 (5.91) | 62.43 (9.86) | 55.71 (17.19) | 55.63 (17.13) | 76.63 (3.24) | 77.69 (2.94) | 58.11 (3.16) | 49.27 (7.13) | 50.74 (6.33) | 65.17 (12.62) | 67.40 (11.03) | 48.85 (4.84) |
| HTML (Ours) | ||||||||||||||||||||||||||
| Dataset | Accuracy | F1-macro | F1-weighted | AUC score | PRC score | Uncertainty | ||||||||||||||||||||
| COADREAD | 86.43 (6.30) | 76.87 (10.33) | 84.79 (7.97) | 89.20 (5.19) | 86.00 (9.92) | 58.76 (1.13) | ||||||||||||||||||||
| ESCA | 98.81 (2.07) | 98.79 (2.10) | 98.80 (2.08) | 98.93 (1.69) | 97.27 (2.70) | 53.88 (5.06) | ||||||||||||||||||||
| GBMLGG | 65.16 (3.84) | 55.93 (4.83) | 59.91 (6.26) | 70.95 (3.69) | 69.34 (2.09) | 71.69 (0.40) | ||||||||||||||||||||
| SARC | 88.35 (2.65) | 87.95 (1.77) | 88.34 (2.70) | 94.76 (1.72) | 88.70 (3.77) | 48.49 (1.33) | ||||||||||||||||||||
| STAD | 68.48 (4.13) | 67.18 (4.73) | 67.84 (4.64) | 68.73 (3.04) | 64.00 (2.02) | 47.93 (1.14) | ||||||||||||||||||||
| STES | 98.90 (1.34) | 98.86 (1.40) | 98.91 (1.33) | 99.55 (0.76) | 97.85 (1.75) | 53.75 (13.66) | ||||||||||||||||||||
| THCA | 85.40 (1.85) | 72.98 (2.89) | 84.00 (1.66) | 85.12 (3.27) | 77.49 (3.12) | 34.60 (0.52) | ||||||||||||||||||||
| UCEC | 87.91 (3.57) | 57.77 (3.18) | 85.61 (3.81) | 82.88 (2.50) | 65.32 (6.13) | 20.62 (0.44) | ||||||||||||||||||||
| BRCA | 88.21 (3.57) | 88.73 (3.16) | 85.86 (4.27) | 92.26 (1.47) | 86.33 (3.55) | 44.34 (0.33) | ||||||||||||||||||||
| LGG | 77.10 (2.14) | 76.94 (2.26) | 77.10 (2.07) | 79.92 (1.48) | 79.50 (1.65) | 36.90 (0.93) | ||||||||||||||||||||
| KIPAN | 96.71 (1.46) | 95.76 (1.99) | 96.22 (1.44) | 99.16 (0.43) | 96.84 (3.02) | 37.86 (0.65) | ||||||||||||||||||||
| ROSMAP | 84.91 (2.99) | 84.97 (2.97) | 89.88 (3.00) | 87.48 (3.22) | 88.59 (2.96) | 61.78 (1.84) | ||||||||||||||||||||
We employed a battery of metrics for the evaluation of the selected methods: accuracy, average F1 score weighted by support (F1-weighted), macroaveraged F1 score (F1-macro), average area under the receiver operating characteristic curve (AUROC) and average area under the precision-recall curve (AUPRC) for each classification task. It is worth mentioning that we have incorporated an uncertainty metric in HTML to convey the model’s level of confidence in the classification results. In all of our experiments, we conducted the research on a single Tesla V100 GPU.
HTML outperforms existing methods in pancancer and cancer subtype diagnosis
To evaluate HTML’s performance in cancer-subtype classification, we compared the classification performance of HTML with the following ten existing classification algorithms (five are traditional machine learning models and five are deep neural networks):
Naive Bayes (NB) [12].
Linear regression (LR) [13].
Support vector machine classifier (SVM) [14].
Random forest classifier (RF) [15].
Gradient tree boosting-based classifier (XGBoost) [16].
Multi-Omics Graph cOnvolutional NETworks (MOGONET) [8].
Multimodal Dynamics (MMDynamics) [17].
Explainable Multiomics Graph Integration (EMOGI) [9].
Subtype-DCC [18].
Trusted Multi-View Classification (TMC) [19].
All the above methods were trained with the same preprocessed data. All the evaluation metrics of the cancer subtype classification tasks are shown in Table 1, and all the evaluation metrics of the pancancer classification tasks are shown in Supplementary Table 4 available online at http://bib.oxfordjournals.org/.
As shown in Figure 1a and Supplementary Figure 2 available online at http://bib.oxfordjournals.org/, HTML exhibits outstanding performance in the 33-class pancancer classification task, achieving an overall accuracy of 93.34 and an F1-weighted score of 92.22. Notably, we observed that the majority of misclassifications occurred within the same cancer meta-type, such as colon adenocarcinoma (COAD) and rectal adenocarcinoma (READ), both of which are malignancies that manifest in the large intestine. Furthermore, a subset of ESCA samples is erroneously classified as HNSC, implying a potential correlation between these cancer subtypes.
With regard to cancer subtype classification, it is evident that HTML outperformed other methods for most cancer subtype classification datasets. However, some exceptional cases emerged. For instance, the AUROC of MOGONET exceeded that of HTML on the GBMLGG dataset by 1.56%, but HTML still performed better than MOGONET regarding all other metrics. Moreover, although HTML exhibited incompetence with SVM and MOGONET in the STAD and KIPAN datasets, it surpassed all other models across the remaining metrics. Notably, the data imbalance in the UECE dataset, where the largest subtype contains ~15 times more samples than the smallest subtype, may inherently introduce instability into the AUROC and AUPRC results, which may explain the relatively weak results of HTML in the UECE dataset. Overall, HTML demonstrated a strong classification performance across the 12 cancer-subtype classification tasks while surpassing other state-of-the-art methods in all accuracy, F1-macro, and F1-weighted metrics.
Regarding individual datasets, our HTML model achieved high cancer subtype classification results in some relatively straightforward binary classification tasks, such as ESCA, STES and KIPAN. However, the model faced challenges in tougher classification tasks such as GBMLGG and STAD. In the GBMLGG dataset, the model may have experienced difficulty distinguishing and classifying the three subtypes of astrocytoma (ASTRO), oligodendroglioma (ODG) and oligoastrocytoma (OA) due to their common origin from glial cells of the brain and spinal cord. Similarly, in the STAD dataset, there existed ambiguous relationships between the labels adenocarcinoma (ADC) and intestinal adenocarcinoma (IAC), wherein IAC identifies a distinct ADC subtype found in the mucous membrane of the intestines forming a subset of ADCs. This complex label relationship may have contributed to the model’s difficulties in accurately classifying these subtypes.
HTML predicts accurate cancer subtype prognosis
HTML is an adaptable framework that can be proficiently utilized for cancer prognosis tasks. To elucidate this point, we chose four datasets, namely, GBMLGG, UCEC, COADREAD and SARC, to predict patient cancer development across diverse cancer subtypes. The Kaplan–Meier curves for risk prediction in these patients are presented in Figure 2b.
Figure 2.

HTML is a prominent model in multiomics learning. (a) We visualized the classification confusion matrix of HTML on the pancancer dataset, and the results demonstrate that HTML exhibits exceptional performance in pancancer classification. (b) We performed survival prediction tasks on the GBMLGG, UCEC, COADREAD and SARC datasets to identify the high-risk and low-risk groups. The predicted risk groups are divided by the cancer subtypes and visualized by Kaplan–Meier curves. The dashed line represents the high-risk group, and the solid line represents the low-risk group. The P-value is annotated in the graph, where * means P-value  , ** means P-value
, ** means P-value  , and ‘ns’ indicates not significant. (c) We illustrate the distribution of feature dynamic weights under different conditions. We randomly selected the 66th sample’s feature dynamic weight in different modalities from the ESCA dataset (upper 3 charts) and found that their distribution was significantly different. The same phenomena were also found in the same feature dynamic weights across samples (lower 3 charts). (d) We extracted the modality dynamic weights’ distribution in all datasets and determined that the modality dynamic weights are all related to the data distribution in the specific sample. (e) We tested all possible multiomics input combinations and examined their corresponding classification performance.
, and ‘ns’ indicates not significant. (c) We illustrate the distribution of feature dynamic weights under different conditions. We randomly selected the 66th sample’s feature dynamic weight in different modalities from the ESCA dataset (upper 3 charts) and found that their distribution was significantly different. The same phenomena were also found in the same feature dynamic weights across samples (lower 3 charts). (d) We extracted the modality dynamic weights’ distribution in all datasets and determined that the modality dynamic weights are all related to the data distribution in the specific sample. (e) We tested all possible multiomics input combinations and examined their corresponding classification performance.
Our analysis enabled us to effectively differentiate between high-risk and low-risk cohorts for each cancer subtype across all datasets. The low-risk group is represented by a solid line, while the high-risk group is denoted by a dotted line. For example, in the GBMLGG dataset, patients categorized as low-risk for oligodendroglioma (ODG), astrocytoma (AST) and oligoastrocytoma (OAC) demonstrated significantly superior cancer progression outcomes relative to their high-risk counterparts, and this difference was most pronounced in OAC subtypes, where the P value between low-risk and high-risk groups was <0.01. Similarly, in the UCEC dataset, patients with serous endometrial adenocarcinoma (SEAC) and mixed serous and endometrioid (MSEAC) were effectively stratified based on cancer progression risk. However, the prognostic performance for endometrioid endometrial adenocarcinoma (EEA) patients is not statistically significant, which is possibly due to a dearth of available samples. Moreover, the results from the COADREAD and SARC datasets further reinforce HTML’s prognostic capabilities. Taken together, these findings provide compelling evidence of HTML’s ability to accurately predict cancer progression in patients.
HTML enables personalized diagnosis and treatment
Intuitively, (i) the variation of impact for different features within a given sample on the model’s results should be distinct. (ii) The variation in attention given to a particular feature across different samples should be distinct. (iii) The variation in attention given to each feature during different training stages for a given sample should be distinct. Most existing encoder models learn a single set of weights for all inputs, which constrains their ability to fully explore the inherent characteristics of the features and provides understandable interpretations of the original data. To overcome this limitation, we introduced the feature dynamic concept, which adaptively learns feature dynamic weights based on the feature’s characteristics and distribution. In contrast to data preprocessing, which aims to convert raw data into an analysis-ready format by eliminating any anomalies or inconsistencies present in the dataset, feature engineering is devised to allot varying weights to different features across individual samples, enabling the model to focus intently on the most informative features of the dataset.
Based on the analysis of the multiomics data, HTML can be used not only for the diagnostic and prognostic prediction of pancancer or cancer subtypes but also for self-adaptive assignment of cancer-related genes in specific samples through dynamic learning, thereby achieving dynamic monitoring of cancer progression and providing real-time guidance for individual precision treatment. In the randomly selected 66th sample (Figure 2c), HTML assigned different dynamic weights to the genes associated with cancer subtypes. Among the genes in this sample, DNA methylation of B-cell lymphoma-2-like protein 1 (BCL2L1) and hepatocyte nuclear factor 1 homeobox A (HNF1A) were given the highest feature dynamic weights. As a member of the antiapoptotic Bcl-2 family, BCL2L1 is hypomethylated and highly expressed in chemotherapy drug (e.g. cisplatin and paclitaxel) -resistant tumor cells in vitro and has been proven to be associated with drug resistance and recurrence of solid tumor cancer [20–23]. In contrast, aberrant hypermethylation of HNF1A downregulates the expression of UDP-glucuronosyltransferase 1A1 (UGT1A1), which remodels drug metabolism and transport pathways, locally inactivating anticancer drugs by glucuronidation in colon cancer cells [24]. In terms of mRNA expression, microtubule affinity regulating kinase 4 (MARK4) and Laminin Subunit Gamma 2 (LAMC2), with the highest dynamic weights in sample 66, may become potential high-efficiency targets for cancer treatment. MARK4 can promote the proliferation and migration of cancer cells by inhibiting Hippo signaling, the targeted inhibition of which is a strategy to treat cancers [25]. However, its absence restricts the tumorigenicity of cancer cells [26]. Upregulated expression of LAMC2, an indicator of progression in esophageal squamous cell carcinoma (ESCC), is caused by lncRNA Cancer Susceptibility 9 (CASC9) interacting with CREb-binding proteins and is common in patients [27]. LAMC2 and CASC9 have been shown to be important biomarkers for metastasis therapy and the prognosis of ESCC, confirming the potential for HTML to play a role in cancer diagnosis and treatment [28]. HTML can also dynamically evaluate miRNA expression based on multiomics data analysis. The sponging of miR-1304 has been studied in many types of cancer, and this phenomenon that is regulated by multiple circRNAs leads to cancer proliferation and invasion [29–31]. However, miR-1307 is involved in tumor development by influencing target genes, such as Forkhead box O3a (FOXO3A), SET And MYND Domain Containing 4 (SMYD4) and Disabled homolog 2-interacting protein (DAB2IP), to further promote cancer invasion [32–34]. These genes, which were given the highest dynamic weight, may be potential targets for precision cancer therapy. We also extracted feature dynamic weights from three different genes in each sample shown in Figure 2c, with variations being exhibited across different samples. Depending on the sensitivity of HTML to individual differences, cancer diagnosis and treatment will likely be more personalized and effective in the future.
Similar to feature dynamic learning, the quality and noise level of data across modalities and samples vary. Therefore, there is a need for modulated prerequisites in the contribution of each modality toward the ultimate outcome along with the model’s confidence toward each modality that aligns with data quality. As a solution, we devised a modality dynamic learning module that imparts a confidence rating for each modality. As illustrated in Figure 2d, the modality dynamic weights for different modalities across various datasets are visualized with individual data points representing each sample. Our model reveals that miRNA plays a primarily subordinate role in the classification of most datasets, with the exception of ROSMAP, which aligns with the well-established association between various miRNAs and Alzheimer’s disease. For example, Cogswell et al. [35] reported that the expression level of hsa-miR-423 underwent significant changes in the hippocampus and medial frontal gyrus of early- and late-stage AD patients compared with control samples. Some studies also identified an overlap in the expression patterns of specific miRNAs (hsa-miR-155, hsa-miR-126a, hsa-miR-23a, hsa-miR-34a, hsa-miR-9, hsa-miR-27a and hsa-miR-146a) in the retina of a rat model of age-related macular degeneration (AMD) (αβ intravitreal injection) and in the serum of AMD patients. These miRNAs are also recognized as potentially useful biomarkers of AD pathology [36–38].
It is worth noting that our analysis of unimodality data classification (Supplementary Table 7 available online at http://bib.oxfordjournals.org/ and Figure 2e) revealed that higher classification accuracy was achieved for mRNA expression than for DNA methylation and miRNA. This could be attributed to the fact that among these three modalities, mRNA expression is most relevant to the phenotype of the corresponding cancer. However, in our analysis of modality dynamics, we observed that weights for DNA methylation were comparatively higher than those for mRNA expression in the SARC, COADREAD and BRCA datasets and took the leading positions in all other datasets. This finding provides valuable insight into the critical upstream role that DNA methylation may play in the biological processes and pathogenesis of these cancers. It is important to note that this could also be attributed to HTML’s alignment process in the methylation-guided attention mechanism.
HTML provides aligned representations and interpretable predictions
The various omics data, analogs to different biological processes, should adhere to the central dogma of molecular biology, whereby mRNA and miRNA expression is regulated by the DNA methylation process. Consequently, it is crucial to harmonize the representation of mRNA and miRNA expression with that of DNA methylation. In accordance with this tenet, we developed a guided attention module (Figure 1d). To exemplify the interconnectivity among genes, we randomly selected an interomics attention map (first 20 genes in each modality) from the SARC dataset of a patient afflicted with leiomyosarcoma cancer (LMS). As illustrated in Figure 1a, diverse DNA methylation spots may exhibit varying attention weights toward mRNAs and miRNAs, thus mirroring the intricate interrelationships among these genes. Moreover, we employed the concept of skip connections [39] to facilitate enhanced generalization capacity and superior performance after the guided attention module (Figure 1b).
Multiomics learning incorporates a weakly supervised learning assumption, where each modality of multiomics data pertaining to a particular sample, such as DNA methylation, mRNA expression, and miRNA expression, represents a distinct biological perspective. An inherent correlation exists among the different modalities, and they can be mapped to an overall label separately. To increase the accuracy of the overall task, representation vectors for each modality of a given sample should be as close as possible. However, current contrastive learning methods [40, 41] (Figure 3c) consider little about different elements belonging to the same cluster. They tend to choose negative samples randomly from the entire dataset, which can drive away the representation of different elements of the same cluster. To address this challenge, we proposed a triple contrastive learning (TCL) method 1e. We further compared the cosine similarities among representations of different modalities with and without contrastive learning in Figure 3d. We selected the first 50 samples from the STES dataset for display. The value of each point in Figure 3d is the result of the average cosine similarity among the three modalities’ representation vectors in the sample. It can be seen that in most cases (except for one sample), the vector representations of the three modalities became more concentrated and their similarity to each other was greatly increased after being trained with TCL, where the most evident one changed from the similarity of 0.0216 to 0.3424.
Figure 3.

Representation alignment across different modalities. (a) We extracted the attention weight matrices (among the first 20 genes) from the methylation-guided attention module in the SARC dataset, which provides insights into the relationships and interactions between the genes. The upper matrix represents attention between DNA methylation and mRNA, and the lower matrix represents attention between DNA methylation and miRNA. The depth of colour in the matrix indicates the strength of these relationships. These insights can be useful in understanding the mechanisms behind gene regulation and identifying potential targets for therapeutic interventions. (b) We devised the triple contrastive loss (TCL) function to enhance the alignment of vector representations among different modalities within the same sample. In comparison to other contrastive learning loss functions, such as InfoNCE loss and the InfoNCE loss between triples, our TCL approach is more effective in aligning the representation of a given sample and increasing the distance between different samples. (c) We visualized the average cosine similarity among different modalities’ representations, and we discovered that our TCL method leads to a more aligned representation of different modalities within the same sample.
The Softmax operator is widely employed in determining the classification evidential probability [42]. This involves normalizing the neural network output using  and subsequently selecting the class with the maximum confidence probability. Nonetheless, the conventional approach to Softmax operation places inadequate focus on the overall probability distribution of classification results, with emphasis solely on the class with the highest probability [43]. Notably, the class probabilities other than the highest one can actually be informative with respect to classification outcome. Thus, considering the entire probability distribution of classification results is crucial for gaining comprehensive insight into the problem under investigation, we introduced the reliability of the classification results.
and subsequently selecting the class with the maximum confidence probability. Nonetheless, the conventional approach to Softmax operation places inadequate focus on the overall probability distribution of classification results, with emphasis solely on the class with the highest probability [43]. Notably, the class probabilities other than the highest one can actually be informative with respect to classification outcome. Thus, considering the entire probability distribution of classification results is crucial for gaining comprehensive insight into the problem under investigation, we introduced the reliability of the classification results.
Additionally, other late fusion strategies simply concatenate the classification results for each modality or apply average pooling. HTML applies the Dempster–Shafer Theory of Evidence, which follows a trustworthy multimodal integration (TMI) rule for multimodal fusion and uncertainty calculation. The goal of TMI can be summarized in three points: (i) A modality’s classification result should be referenced to its confidence score; (ii) When the uncertainty of all modalities is high, the final prediction must be of low certainty, and vice versa; (iii) The final prediction and uncertainty score should be considered together and dynamically change according to each modality’s predicted outcome.
To validate the efficacy of the uncertainty metrics, we visualized the changes in the performance of HTML using the in−/out-of-distribution (ID/OOD) samples. Here, we set the original samples as ID and the samples with added Gaussian noise as OOD. Specifically, we added Gaussian noise with varying standard deviations (i.e.  ) to the test samples. We conducted 5-fold cross-validation on all ID and OOD samples for each dataset in the 12 datasets and calculated the mean value of classification accuracy, AUROC, and uncertainty score of HTML for each dataset under each distribution setting as well as their 95% confidence intervals, as presented in Figure 4. The information in Figure 4 effectively confirms the usefulness and validity of the uncertainty metrics. In all datasets, in-distribution data gain the highest classification performance and lowest uncertainty score. As the data distribution deviates to a greater extent, the classification performance of the model gradually deteriorates, while the uncertainty scores of the model’s classification results continue to increase. We can infer that in situations where there is a significant amount of data noise, the model’s ability to accurately and consistently predict and classify may be compromised. As such, the uncertainty metric can serve as a reliable compass in evaluating the credibility of the data distribution.
) to the test samples. We conducted 5-fold cross-validation on all ID and OOD samples for each dataset in the 12 datasets and calculated the mean value of classification accuracy, AUROC, and uncertainty score of HTML for each dataset under each distribution setting as well as their 95% confidence intervals, as presented in Figure 4. The information in Figure 4 effectively confirms the usefulness and validity of the uncertainty metrics. In all datasets, in-distribution data gain the highest classification performance and lowest uncertainty score. As the data distribution deviates to a greater extent, the classification performance of the model gradually deteriorates, while the uncertainty scores of the model’s classification results continue to increase. We can infer that in situations where there is a significant amount of data noise, the model’s ability to accurately and consistently predict and classify may be compromised. As such, the uncertainty metric can serve as a reliable compass in evaluating the credibility of the data distribution.
Figure 4.

Uncertainty analysis and biomarker identification. (a) Dirichlet uncertainty analysis of multiomics data with different levels of noise. To validate the effectiveness of the uncertainty metrics, we conducted an experiment where we added Gaussian noise with varying degrees to each dataset and observed the classification performance of HTML. The results are plotted on a graph, where three lines represent accuracy, AUROC (AUC), and uncertainty respectively. To ensure the reliability of our results, we calculated the 95% confidence interval of the results based on 5-fold cross-validation and represented it with shaded areas. The results showed that as the noise level in the data increased, the classification performance of the model continued to decline, accompanied by a continuous increase in the model’s uncertainty. This trend was observed in all 12 datasets. (b) We investigated the expression difference of STC1 between normal and COAD as well as that between normal and READ. Expression alteration is absent in normal and COAD samples but exists in normal and READ samples (P value = 0.0019). (c) We investigated the expression differences in STRN4 between normal and COAD tissues as well as between normal and READ tissues. Expression alteration is absent in normal and READ samples but exists in normal and COAD samples (P value = 0.0363).
HTML identifies important biomarkers
Our research on subtype classification has led to the identification of a variety of DNA methylation, mRNA, and miRNA biomarkers that warrant further investigation for their potential role in the diagnosis and treatment of distinct cancer subtypes. A selection of noteworthy biomarkers is presented in Supplementary Tables 8–19 available online at http://bib.oxfordjournals.org/ and Table 2. To showcase the value of our findings, we chose certain biomarkers from the COADREAD dataset (comprising COAD and READ cancer subtypes) as examples.
Table 2.
Important biomarkers identified by HTML. We listed the important biomarkers found by HTML for further investigation of each cancer subtype in 10 datasets. These biomarkers will serve as potential targets for future research and may lead to novel diagnostic or therapeutic approaches for cancer treatment
| COADREAD | COAD | meth | LRMP, TCF4, OXTR, FLJ39609, UGT2B10 |
| mRNA | TNKS1BP1, BTBD6, TMEM129, STRN4, DGKQ | ||
| miRNA | hsa-mir-411, hsa-mir-101-2, hsa-mir-1185-1, hsa-mir-135a-1, hsa-mir-134 | ||
| READ | meth | DDIT4L, ZNF461, FSTL5, NRAP, GPR150 | |
| mRNA | ZFP36L2, SLC25A39, SLC37A1, PLAUR, SPON1 | ||
| miRNA | hsa-mir-765, hsa-mir-541, hsa-mir-1295, hsa-mir-514-1, hsa-mir-181c | ||
| ESCA | ESCC | meth | SPRR1B, GIPC2, KRT73, ETNK1, GZMH |
| mRNA | CDCA4, SENP5, C10orf58, USP5, FXR1 | ||
| miRNA | hsa-mir-1294, hsa-let-7a-3, hsa-mir-200c, hsa-mir-10b, hsa-mir-184 | ||
| EAC | meth | ME3, RDH5, BCL2L15, CLCA3P, IL18 | |
| mRNA | GPR87, PFN2, STON2, PL-5283, MLF1 | ||
| miRNA | hsa-mir-24-1, hsa-mir-339, hsa-mir-151, hsa-mir-205, hsa-mir-3605 | ||
| GBMLGG | AST | meth | PHLDA2, ARHGEF5L, IMPACT, LOC113230 |
| mRNA | LPPR1, BAP1, ANAPC5, ZNF33B, VAPA | ||
| miRNA | hsa-mir-380, hsa-mir-548o, hsa-mir-3199-1, hsa-mir-204, hsa-mir-1301 | ||
| ODG | meth | PTENP1, SLMO1 | |
| mRNA | SLC24A3, HMP19, GNAI1, SF1, RNASEH2C | ||
| miRNA | hsa-mir-3909, hsa-mir-383, hsa-mir-3662, hsa-mir-3200, hsa-mir-2116 | ||
| OAC | meth | MLH3, CYP51A1, GPR19, HSPA1A | |
| mRNA | PTCD3, ECE2, LETMD1, FXR2, PRPF6 | ||
| miRNA | hsa-mir-338, hsa-mir-22 | ||
| SARC | LMS | meth | TNFSF4, NPB, COL12A1, SNX22, SLCO2A1 |
| mRNA | PLP2, LSM7, U2AF1, DLGAP4, ADRBK1 | ||
| miRNA | hsa-mir-1228, hsa-mir-196a-1, hsa-mir-744, hsa-mir-551b, hsa-mir-664 | ||
| DDLPS | meth | GPR19, TRIM58, SNORD17, LOC391322 | |
| mRNA | MCTS1, NPLOC4, MED16, RNF167, FAM125A | ||
| miRNA | hsa-mir-195, hsa-mir-215, hsa-mir-1292, hsa-mir-548 k, hsa-mir-409 | ||
| others | meth | FAM115A, CYR61, FLJ13224, SEPT10, MTAP | |
| mRNA | WHSC1, C1orf151, PRPF19, HMGN2, THOC5 | ||
| miRNA | hsa-mir-668, hsa-mir-363, hsa-mir-720, hsa-mir-590, hsa-mir-133a-2 | ||
| STAD | `ADC | meth | RPL39L, HIST1H3J, KIT, LOC80154, NSUN7 |
| mRNA | C1orf144, RHOA, TOMM20, ARPP19, IER5L | ||
| miRNA | hsa-mir-328, hsa-let-7f-2, hsa-mir-374b, hsa-mir-149, hsa-mir-3130-1 | ||
| IAC | meth | NCRNA00152, MDS2, CCDC15, B3GALT4, NTN3 | |
| mRNA | RACGAP1, CTBP2, YIPF3, RRM2B, C9orf152 | ||
| miRNA | hsa-mir-489, hsa-mir-551a, hsa-mir-378, hsa-mir-582, hsa-mir-34a | ||
| STES | ESCC | meth | MIR192, ETNK1, LOC80054, KRT71, FZD5 |
| mRNA | MARK4, GLI3, KRI1, TMEM189, ABCF3 | ||
| miRNA | hsa-mir-1292, hsa-mir-30c-2, hsa-mir-128-1, hsa-mir-103-1 | ||
| EAC | meth | LCE2D, POSTN, GFM1, FAM153A, CFTR | |
| mRNA | DGKA, A4GALT, ALS2CR4, DFNA5, DRAP1 | ||
| miRNA | hsa-mir-192, hsa-mir-3942, hsa-mir-125b-1 | ||
| THCA | Usual type | meth | DNAH1, SLC23A1, TUBB6, KLHDC5, OR6Q1 |
| mRNA | APOC1, FAM171B, CCDC149, MPPE1 | ||
| miRNA | hsa-mir-526b, hsa-mir-3117, hsa-mir-433, hsa-mir-16-2, hsa-mir-132 | ||
| Unusual type | meth | TAGLN, TRIP4, PTGIS, NCRNA00152, LTF | |
| mRNA | WSB2, TMEM80, KIAA1191, RPN1 | ||
| miRNA | hsa-mir-449b, hsa-mir-186, hsa-mir-148a, hsa-mir-3926-1, hsa-mir-190b | ||
| UCEC | EEA | meth | DMC1, MIR7–1, PRAMEF21, MIR138–1, TREX1 |
| mRNA | ZNF512 | ||
| miRNA | hsa-mir-485, hsa-mir-1266, hsa-mir-16-1, hsa-mir-3926-1, hsa-mir-15b | ||
| SEA | meth | UBLCP1, KRTAP5–6, TEC | |
| mRNA | |||
| miRNA | hsa-mir-3191, hsa-mir-3944, hsa-mir-346, hsa-mir-616, hsa-mir-138-1 | ||
| MSEAC | meth | SLC18A1, KLHDC7A, FAM167A, ARID1A, LOC400931 | |
| mRNA | RPS18, EPCAM, PAX8, DHX36, HDAC5 | ||
| miRNA | hsa-mir-1910, hsa-mir-3913-1, hsa-mir-27b, hsa-mir-143, hsa-mir-431 | ||
| BRCA | Normal-like | meth | CXCL1, COL11A2, RAET1L |
| mRNA | ALDH1A3, ZNF80, SLC30A10 | ||
| miRNA | hsa-mir-3676, hsa-mir-489, hsa-mir-320b-2 | ||
| Basal-like | meth | MAPK4, ABCC11, TRIML2, LEMD1, GJB3 | |
| mRNA | MIR190B, VLDLR | ||
| miRNA | hsa-mir-3194 | ||
| HER2-enriched | meth | PI3, C16orf57, MIA, MGC3771 | |
| mRNA | TMEM22, ZSWIM2 | ||
| miRNA | hsa-mir-26a-1, hsa-mir-144, hsa-mir-19b-1, hsa-mir-199a-1 | ||
| Luminal B | meth | RGS20 | |
| mRNA | C15orf62, HAS2AS | ||
| miRNA | hsa-mir-1266 | ||
| KIPAN | KICH | meth | FOXL1, GJC2, C5orf4, COMT, NRG4 |
| mRNA | CDH1, CTDSPL, CCL2, FOXI2, ASTN2 | ||
| miRNA | hsa-mir-1291, hsa-mir-660, hsa-mir-548 s, hsa-mir-363, hsa-mir-3934 | ||
| KIRC | meth | LGALS9B | |
| mRNA | KLHL3, RPL28, CLU, INSR, THY1 | ||
| miRNA | hsa-mir-342, hsa-mir-146b, hsa-mir-514-2, hsa-mir-495, hsa-mir-320d-2 | ||
| KIRP | meth | IL18BP, RPS6 | |
| mRNA | ANTXR1, GPI,FKBP10, COBLL1, TACC1 | ||
| miRNA | hsa-mir-153-2, hsa-mir-1229, hsa-mir-30c-1, hsa-mir-3944, hsa-mir-148a | ||
| LGG | Grade 2 | meth | TPPP3, ITPRIPL1, PSG1 |
| mRNA | TMEM30A, RTN3, HNRNPH1, STMN3, PDLIM5 | ||
| miRNA | hsa-mir-218-1, hsa-mir-3609, hsa-mir-3614, hsa-mir-378c, hsa-mir-27a | ||
| Grade 3 | meth | UGT2B28, DNASE1L3, ELSPBP1 | |
| mRNA | OSGIN2, CASC4, ATPAF1, STIP1, LOC643763 | ||
| miRNA | hsa-mir-448, hsa-mir-3200, hsa-mir-370, hsa-mir-627, hsa-mir-200b |
Within the COAD biomarkers, LRMP has been identified as hypermethylated and downregulated in a subpopulation of highly mobile cells, known as MG cells, derived from a COAD cell line. These cells are distinguished by their migratory capacity and epithelial-mesenchymal transition attributes, which are indicative of the metastatic potential and malignancy of COAD. Tanaka et al. [44] conducted a study that demonstrated the significant impairment of MG cell migration with the demethylation and upregulation of LRMP and other genes, suggesting a correlation between LRMP methylation and COAD. This relationship is further supported by the findings of various other studies, including the identification of COAD marker genes through single-cell transcriptomic analysis [45] and gene expression analysis of COAD and normal samples via RT-PCR assay [46]. In addition, genes identified in mRNA expression features, such as TNKS1BP1 [47], which encodes a protein involved in telomere replication, and STRN4 [48], which encodes a scaffolding protein with multiple functions, have also been reported to play a role in COAD tumorigenesis. To further investigate this, we analysed the expression differences of STRN4 between normal and COAD samples using data from Snipstad et al. [49] and compared them to those between normal and READ samples using data from Zuurbier et al. [50], as shown in Supplementary Table 20 available online at http://bib.oxfordjournals.org/ and Figure 4b. We discovered a consistent result, with a significant expression alteration present in normal and COAD samples (P value = 0.03565) but absent in normal and READ samples. In terms of miRNA expression features, hsa-mir-101-2 was identified as one of the prognostic miRNA signatures for COAD in a study by Lv et al. [51]. The targets of this signature are predicted to enrich biological process terms, such as focal adhesion and transcription disorders in cancer.
In terms of biomarkers for READ, STC1 is a member of the secretory glycoprotein family and is involved in processes such as apoptosis and inflammation. Its association with READ has been reported [52], and its secretion by tumor stromal cells may contribute to READ metastasis by mediating PDGF receptor signaling [53]. We analysed the same dataset mentioned earlier to examine the differential expression of STC1 in normal and COAD samples or normal and READ samples, and the results are shown in Supplementary Table 21 available online at http://bib.oxfordjournals.org/ and Figure 4c. We found a significant difference in STC1 expression between normal and READ with a P value close to 0.001, which suggests that STC1 could be a biomarker for READ. Among the genes identified in the mRNA expression features, SPON1 is a coding gene that produces a product predicted to be secreted to the extracellular matrix and is involved in cell adhesion, a process that could be dysregulated in tumorigenesis. Supiot et al. [54] examined its expression before and after preoperative radiotherapy in READ patients and found a significant upregulation. Similarly, upregulation of hsa-mir-765 was found to occur in READ patients in response to neoadjuvant chemoradiotherapy [55]. These findings suggest that SPON1 and hsa-mir-765 could be potential biomarkers for READ. Moreover, TargetScan predicted possible target genes of hsa-mir-765, including PDX1 [56], KLK4 [57] and LHPP [58], all of which have been reported to correlate with READ.
DISCUSSION
In this study, we proposed a highly trustworthy multiomics learning framework (HTML), for personalized pancancer and cancer subtype diagnosis and prognosis. Through training on pancancer and cancer subtype datasets, HTML has the potential to seamlessly integrate multiple modalities of data, thereby enabling personalized medical diagnosis. This innovative technology is poised to overcome the existing barriers between pancancer diagnosis and cancer subtype determination, thereby achieving a comprehensive diagnosis pipeline. This pipeline is capable of processing one or more pieces of omics information from patients. It initially predicts the presence of cancer and the specific type of cancer and subsequently identifies the specific subtype of the patient’s cancer, which has the potential to integrate the entire diagnostic process into one model.
Moreover, conventional models are unable to conduct sample adaptive analysis based on the unique data distribution characteristics of each sample and lack the ability to validate and verify the reliability of their own classification results. To address this challenge, we developed the HTML model, which leverages dynamic learning at both the feature and modality levels and Dirichlet uncertainty learning. This innovative model efficaciously integrates information across diverse modalities of data, enabling it to make cutting-edge classification predictions with remarkable accuracy, enabling the precise identification and diagnosis of cancer-causing genes for individuals, thereby making personalized treatment possible.
HTML’s feature dynamic weight mechanism assigns a unique weight to each feature. This innovative approach enables us to identify potential biomarkers that can differentiate between various types of cancers and cancer subtypes, thereby laying the groundwork for further exploration of the biological mechanisms that underpin cancer development.
Despite utilizing only DNA methylation, mRNA expression and miRNA expression data for our multiomics classification tasks, our HTML framework boasts impressive extensibility and can be customized to accommodate various types of data (such as SNVs, copy number or even medical images and texts) by adjusting the model parameters accordingly. In this way, HTML represents a highly adaptable supervised multiomics classification framework with exceptional interpretability and extensibility.
METHODS
Feature dynamic learning
The multiomics input can be formulated as  . Here,
. Here,  represent
represent  distinct modalities, with each modality containing
distinct modalities, with each modality containing 
 -dimensional vectors. The quantity
-dimensional vectors. The quantity  denotes the number of samples, and the feature volume
denotes the number of samples, and the feature volume  varies across different modalities. The symbol
varies across different modalities. The symbol  represents the set of real numbers.
represents the set of real numbers.
Suppose we have a single omic data input  , where each sample
, where each sample  is represented by a
is represented by a  -dimensional vector
-dimensional vector  . Our objective is to learn a feature dynamic module that generates a dynamic weight vector
. Our objective is to learn a feature dynamic module that generates a dynamic weight vector 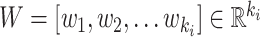 that performs self-adaptive feature selection. We can achieve this by defining a transformation with the input feature to obtain the dynamic feature embedding
that performs self-adaptive feature selection. We can achieve this by defining a transformation with the input feature to obtain the dynamic feature embedding  as follows:
as follows:
 |
where  is generated through an MLP network and has the same dimension as the input
is generated through an MLP network and has the same dimension as the input  ,
,  represents the sigmoid function, and the sigmoid and hyperbolic tangent functions here aim to introduce nonlinear parameters in the learning process.
represents the sigmoid function, and the sigmoid and hyperbolic tangent functions here aim to introduce nonlinear parameters in the learning process.
The concept of feature dynamics can effectively address the issue of overfitting, in which the model tends to fit the noise in the input data rather than the underlying patterns [59]. Through the adaptive selection of features based on their significance to the prediction task, the model can improve its performance when encountering new samples and can achieve a more meaningful interpretation of the input features.
Methylation-guided attention
Through the feature selection module, we obtained a better interomics feature representation  for each sample. Considering the uniqueness of multiomics data and their biological meaning, DNA methylation levels largely affect the expression levels of mRNA and miRNA. Therefore, we proposed a DNA methylation-guided attention mechanism to model the interomics relationships among different omics data.
for each sample. Considering the uniqueness of multiomics data and their biological meaning, DNA methylation levels largely affect the expression levels of mRNA and miRNA. Therefore, we proposed a DNA methylation-guided attention mechanism to model the interomics relationships among different omics data.
In HTML, the original DNA methylation input features serve as the query vectors  , while the mRNA or miRNA input features
, while the mRNA or miRNA input features  act as key and value vectors. Both
act as key and value vectors. Both  and
and  have been projected into the same vector space. The guided attention context vector
have been projected into the same vector space. The guided attention context vector  is then calculated as:
is then calculated as:
 |
where  represents the attention weight between
represents the attention weight between  and
and  and
and  represents the feature numbers of both vectors. The mRNA context vector
represents the feature numbers of both vectors. The mRNA context vector  is computed by the sum of all attention values on DNA methylation features as follows:
is computed by the sum of all attention values on DNA methylation features as follows:
 |
where the alignment  function indicates how well the elements of the DNA features align with the mRNA features at the position and the weights
function indicates how well the elements of the DNA features align with the mRNA features at the position and the weights  are computed by applying a Softmax operation to the previously computed alignment scores as follows:
are computed by applying a Softmax operation to the previously computed alignment scores as follows:
 |
where  is the scaling factor and
is the scaling factor and  is the multiplicative attention weight matrix.
is the multiplicative attention weight matrix.
After the feature dynamic module and methylation-guided attention, we obtained both the inner-omics feature representation  and interomics feature representation
and interomics feature representation  . We then took the average of vectors
. We then took the average of vectors  and
and  (
( for DNA methylation) to obtain the final representation vector
for DNA methylation) to obtain the final representation vector  for each omics.
for each omics.
Triple contrastive learning
To address the interactions among multiomics data and improve the classification task accuracy, we proposed a triple contrastive loss (TCL) function for learning comparisons among multiple modalities. This loss function brings together the distances among the representations  and
and  of all modalities (
of all modalities ( in our experiment) of a given sample while pulling away the representations
in our experiment) of a given sample while pulling away the representations 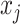 of other samples:
of other samples:
 |
where  is the number of samples,
is the number of samples,  is a temperature parameter,
is a temperature parameter,  is the representation of the
is the representation of the  -th DNA methylation feature,
-th DNA methylation feature,  is the representation of the
is the representation of the  -th mRNA expression feature,
-th mRNA expression feature,  is the representation of the
is the representation of the  -th miRNA expression feature, and
-th miRNA expression feature, and  is the number of negative samples. This process optimizes the representation vectors of each modality and improves the accuracy of the overall task.
is the number of negative samples. This process optimizes the representation vectors of each modality and improves the accuracy of the overall task.
Modality dynamic learning
As with feature dynamic learning, the contribution of individual features varies in each modality toward the final classification outcome. This holds true for disparate modalities as well. To effectively account for each modality’s contribution to the final classification, HTML introduces a modality dynamic learning module to compute the model’s confidence (MCC) for different modalities. Take the DNA methylation feature  as an example:
as an example:
 |
where  is the weight parameter for DNA methylation features of the modality dynamic learning module, and each MCC function returns a confidence score for each modality feature of each sample.
is the weight parameter for DNA methylation features of the modality dynamic learning module, and each MCC function returns a confidence score for each modality feature of each sample.
Dirichlet uncertainty learning
The Dirichlet distribution is a prevalent multivariate probability model used for random variable synthesis [60]. It is expressed in terms of  concentration parameters, denoted
concentration parameters, denoted 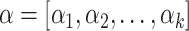 . The probability density function of the Dirichlet distribution is given by:
. The probability density function of the Dirichlet distribution is given by:
 |
where  represents the beta distribution parameterized by
represents the beta distribution parameterized by  , and
, and  denotes the
denotes the  dimensional unit simplex, defined as:
dimensional unit simplex, defined as:
 |
It is noteworthy that each element in  corresponds to a different category concentration, and
corresponds to a different category concentration, and  is
is  -dimensional. From the Dirichlet distribution equation definition, we inferred that each unit of the vector representing the category concentration can parameterize a corresponding Dirichlet distribution. By associating the
-dimensional. From the Dirichlet distribution equation definition, we inferred that each unit of the vector representing the category concentration can parameterize a corresponding Dirichlet distribution. By associating the  vector with the input class probabilities, one obtains a full perspective of the overall distribution of class probability beyond simply relying on the highest classification probability.
vector with the input class probabilities, one obtains a full perspective of the overall distribution of class probability beyond simply relying on the highest classification probability.
We introduce the Dempster–Shafer Theory of Evidence (DST) [61], which utilizes belief mass to allocate subjective probabilities in describing an opinion’s credibility in order to assign an uncertainty score for each modality [19]. A belief mass indicates how reliable the predicted label is, while the overall uncertainty denotes how much the total probability of the class should be questioned. The belief mass and overall uncertainty can be quantified through:
 |
where  represents the number of classification types, and
represents the number of classification types, and  indicates supporting evidence for the belief mass. In the case of multitype classification, the evidence
indicates supporting evidence for the belief mass. In the case of multitype classification, the evidence  is supported by the output from the neural network. The belief mass function and the overall uncertainty are related as follows:
is supported by the output from the neural network. The belief mass function and the overall uncertainty are related as follows:
 |
This supports the notion that the belief mass and overall uncertainty interact complementarily. In our experiments, the Dirichlet distribution allowed modelling of secondary probability and uncertainty beyond relying solely on the highest probability as the selected class [43]. Utilizing the probabilistic output  as evidence from the neural network, we defined the Dirichlet distribution parameters
as evidence from the neural network, we defined the Dirichlet distribution parameters  to ensure that the value of
to ensure that the value of  satisfies the mathematical requisite that
satisfies the mathematical requisite that  in the Dirichlet distribution must be nonnegative.
in the Dirichlet distribution must be nonnegative.
Dempster–Shafer multiomics integration
Once the determination of the belief mass, overall uncertainty and evidence is complete, a quantitative measure of the reliability of opinions can be obtained. Nevertheless, in multiple modality tasks, integrating the belief mass and overall uncertainty from various sources proves to be a challenging task. The integration rule should consider the uncertainty value instead of simply reconciling the belief mass values from different sources. Additionally, multimodal integration must account for conflicting opinions across diverse modalities. Given these requirements, we proposed the following fusion rule. The integrated uncertainty value is calculated as follows:
 |
The variable  represents a measure proposed to assess the conflicting results of different opinions. In cases where opinions are at odds with each other, the sum of the belief mass product is calculated. As the frequency of conflicting scenarios increases, the value of
represents a measure proposed to assess the conflicting results of different opinions. In cases where opinions are at odds with each other, the sum of the belief mass product is calculated. As the frequency of conflicting scenarios increases, the value of  rises, resulting in an increase in the integrated uncertainty value, leading to a less persuasive integrated result. Concerning the fused belief mass, we set the integration rule as follows:
rises, resulting in an increase in the integrated uncertainty value, leading to a less persuasive integrated result. Concerning the fused belief mass, we set the integration rule as follows:
 |
The cross-relation between the belief mass and the overall uncertainty distributes different weights for distinct modalities linked to their respective uncertainties. For instance, when the uncertainty value of modality 1 is higher than that of modality 2, the integrated  may rely more heavily on the input of
may rely more heavily on the input of  thanks to the introduction of
thanks to the introduction of  , and vice versa. The DST integration rule can be easily extended to multiomics scenarios where the number of omics exceeds 2. If we use M to denote both the belief mass and the overall uncertainty:
, and vice versa. The DST integration rule can be easily extended to multiomics scenarios where the number of omics exceeds 2. If we use M to denote both the belief mass and the overall uncertainty:  , we can summarize the integration process between two modalities
, we can summarize the integration process between two modalities  and
and  as:
as:
 |
where the operator  represents the integration process. This integration rule is suitable for commutativity and associativity. The fusion result is independent of the order of fusion and can effectively be applied to more than two modalities as follows:
represents the integration process. This integration rule is suitable for commutativity and associativity. The fusion result is independent of the order of fusion and can effectively be applied to more than two modalities as follows:
 |
The loss function for DST integration methods comprises the cross-entropy loss in each modality:
 |
where  is the number of modalities, and each
is the number of modalities, and each  represents the predicted probability. The loss function for the final classification outcome contains a cross-entropy loss and a KL divergence loss
represents the predicted probability. The loss function for the final classification outcome contains a cross-entropy loss and a KL divergence loss
 |
where
 |
where  is the gamma function and
is the gamma function and  is the Dirichlet distribution after replacing the
is the Dirichlet distribution after replacing the  corresponding to the ground truth label with 1, thus avoiding penalizing the Dirichlet parameter of the ground truth class to 1, and
corresponding to the ground truth label with 1, thus avoiding penalizing the Dirichlet parameter of the ground truth class to 1, and  is the unit vector of length
is the unit vector of length  .
.
We formulated our objective function for HTML by combining the triple contrastive loss, individual cross-entropy loss, and integrated loss. To further enhance the classification results, we incorporated uncertainty metrics and an  regularization term to mitigate overfitting. This comprehensive objective function aims to reduce uncertainty and improve the accuracy of the classification results:
regularization term to mitigate overfitting. This comprehensive objective function aims to reduce uncertainty and improve the accuracy of the classification results:
 |
Key Points
We present a novel and highly personalized approach to cancer diagnosis and prognosis
Our method outperforms static-architecture-based methods in various clinical tasks.
Our method showcases the effectiveness of individualized treatment with a strong emphasis on explainability
Our method incorporates uncertainty metrics to provide reliable indications of the model’s output
Our method contributes to advancing biological research by identifying potential biomarkers
Supplementary Material
Author Biographies
Yuxing Lu is currently a PhD student at the Department of Big Data and Biomedical AI, College of Future Technology, Peking University, Beijing, China. His research primarily focuses on multi-omics data integration and metabolomic knowledge graphs.
Rui Peng is currently a PhD student in the Academy for Advanced Interdisciplinary Studies, Peking University. His research interests focus on multi-omics integration and computational biology.
Lingkai Dong is a masters student in chemical and biomolecular engineering at The Hong Kong University of Science and Technology. His research interests include drug delivery and cancer targeting therapy.
Kun Xia is a master student in School of Software and Microelectronics, Peking University.
Renjie Wu is a bachelor student in School of Life Science, Peking University.
Shuai Xu is currently a PhD student at the Institute of Molecular Medicine, College of Future Technology, Peking University, Beijing, China. His research primarily focuses on the comprehensive analysis of multi-omics data obtained from single-cell sequencing.
Jinzhuo Wang is a research assistant professor in the Department of Big Data and Biomedical AI, College of Future Technology, Peking University. His research interest is multimodal biomedical AI.
Contributor Information
Yuxing Lu, Department of Big Data and Biomedical AI, College of Future Technology, Peking University, Beijing, China.
Rui Peng, Department of Big Data and Biomedical AI, College of Future Technology, Peking University, Beijing, China.
Lingkai Dong, Department of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Hong Kong SAR, China.
Kun Xia, Department of Big Data and Biomedical AI, College of Future Technology, Peking University, Beijing, China.
Renjie Wu, School of Life Sciences, Peking University, Beijing, China.
Shuai Xu, Institute of Molecular Medicine, College of Future Technology, Peking University, Beijing, China.
Jinzhuo Wang, Department of Big Data and Biomedical AI, College of Future Technology, Peking University, Beijing, China.
AUTHOR CONTRIBUTIONS
J.W., Y.L., and R.P. collected and analysed the data. J.W. and Y.L. conceived and designed the project and HTML. J.W. supervised the project. J.W. and Y. L. wrote the manuscript with the assistance of L.D. and R.W., S.X., L.D., and R.W. helped revise the parts of personalized treatment and biomarker discovery.
FUNDING
This work was supported by the Young Elite Scientist Sponsorship Program by BAST (No. BYESS2023026) and the National Natural Science Foundation of China (No. 6220071694).
DATA AVAILABILITY STATEMENT
The BRCA and ROSMAP datasets were obtained from the GitHub repository of MOGONET (https://github.com/txWang/MOGONET). The omics data of the other datasets (COADREAD, ESCA, GBMLGG, SARC, STAD, STES, THCA, UCEC, LGG, KIPAN), as well as the survival time information, were obtained from The Cancer Genome Atlas (TCGA) Program through Broad GDAC Firehose (https://gdac.broadinstitute.org/). The PAM50 breast cancer subtypes of TCGA BRCA patients were obtained through the TCGAbiolinks R package (v2.12.6, http://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html). We have made our preprocessed data for the 12 cancer subtype dataset available through Google Drive (https://drive.google.com/drive/folders/1_tJ2ekhTmWp7ZcRVjUVGx0cqGMRKEhNo?usp=share_link).
CODE AVAILABILITY
The source codes, trained models, demos and tutorials for this work can be found at GitHub (https://github.com/YuxingLu613/HTML).
References
- 1. Lipkova J, Chen RJ, Chen B, et al. Artificial intelligence for multimodal data integration in oncology. Cancer Cell 2022;40:1095–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu MY, Chen TY, Williamson DFK, et al. AI-based pathology predicts origins for cancers of unknown primary. Nature 2021;594:106–10. [DOI] [PubMed] [Google Scholar]
- 3. Lipkova J, Chen TY, Lu MY, et al. Deep learning-enabled assessment of cardiac allograft rejection from endomyocardial biopsies. Nat Med 2022;28:575–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Connor AA, Gallinger S. Pancreatic cancer evolution and heterogeneity: integrating omics and clinical data. Nat Rev Cancer 2022;22:131–42. [DOI] [PubMed] [Google Scholar]
- 5. Berest I, Arnold C, Reyes-Palomares A, et al. Quantification of differential transcription factor activity and multiomics-based classification into activators and repressors: diffTF. Cell Rep 2019;29:3147–59.e12. [DOI] [PubMed] [Google Scholar]
- 6. Lazar AJ, Demicco EG. Human and machine: better at pathology together? Cancer Cell 2022;40:806–8. [DOI] [PubMed] [Google Scholar]
- 7. Ge Z, Leighton JS, Wang Y, et al. Integrated genomic analysis of the ubiquitin pathway across cancer types. Cell Rep 2018;23:213–26.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang T, Shao W, Huang Z, et al. MOGONET integrates multi-omics data using graph convolutional networks allowing patient classification and biomarker identification. Nat Commun 2021;12:3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schulte-Sasse R, Budach S, Hnisz D, Marsico A. Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms. Nat Mach Intell 2021;3:513–26. [Google Scholar]
- 10. Crawford J, Greene CS. Incorporating biological structure into machine learning models in biomedicine. Curr Opin Biotechnol 2020;63:126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaushik AC, Mehmood A, Dai X, Wei D-Q. Pan-cancer analysis and drug formulation for GPR139 and GPR142. Front Pharmacol 2021;11:521245. 10.3389/fphar.2020.521245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murphy KP. Naive bayes classifiers. Univ B C 2006;18:1–8. [Google Scholar]
- 13. Buckley J, James I. Linear regression with censored data. Biometrika 1979;66:429–36. [Google Scholar]
- 14. Suykens JAK, Vandewalle J. Least squares support vector machine classifiers. Neural Process Lett 1999;9:293–300. [Google Scholar]
- 15. Breiman L. Random forests. Mach Learn 2001;45:5–32. [Google Scholar]
- 16. Chen T, Guestrin C. XGBoost: a scalable tree boosting system. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2016. pp.785–94. Association for Computing Machinery, San Francisco, California, USA. [Google Scholar]
- 17. Han Z, Yang F, Huang J, et al. Multimodal dynamics: dynamical fusion for trustworthy multimodal classification. In: 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), 2022. pp.20675–85. IEEE, New Orleans, LA, USA. [Google Scholar]
- 18. Zhao J, Zhao B, Song X, et al. Subtype-DCC: decoupled contrastive clustering method for cancer subtype identification based on multi-omics data. Brief Bioinform 2023;24:bbad025. [DOI] [PubMed] [Google Scholar]
- 19. Han Z, Zhang C, Fu H, Zhou JT. Trusted multi-view classification with dynamic evidential fusion. IEEE Trans Pattern Anal Mach Intell 2022;45:2551–66. [DOI] [PubMed] [Google Scholar]
- 20. Yu Wei, et al. Global analysis of DNA methylation by Methyl-Capture sequencing reveals epigenetic control of cisplatin resistance in ovarian cancer cell. PloS one 2011;6.12:e29450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang X, Feng Y, Gao Y, Hu J. Long noncoding RNA LINC00634 functions as an oncogene in esophageal squamous cell carcinoma through the miR-342-3p/Bcl2L1 axis. Technol Cancer Res Treat 2020;19:153303382092850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shoemaker AR, Oleksijew A, Bauch J, et al. A small-molecule inhibitor of Bcl-XL potentiates the activity of cytotoxic drugs in vitro and in vivo. Cancer Res 2006;66:8731–9. [DOI] [PubMed] [Google Scholar]
- 23. Williams J, Lucas PC, Griffith KA, et al. Expression of Bcl-xL in ovarian carcinoma is associated with chemoresistance and recurrent disease. Gynecol Oncol 2005;96:287–95. [DOI] [PubMed] [Google Scholar]
- 24. Bélanger AS, Tojcic J, Harvey M, Guillemette C. Regulation of UGT1A1 and HNF1 transcription factor gene expression by DNA methylation in colon cancer cells. BMC Mol Biol 2010;11:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adnan M, Anwar S, DasGupta D, et al. Targeting inhibition of microtubule affinity regulating kinase 4 by Harmaline: strategy to combat Alzheimer's disease. Int J Biol Macromol 2023;224:188–95. [DOI] [PubMed] [Google Scholar]
- 26. Arash EH, Shiban A, Song S, et al. MARK4 inhibits hippo signaling to promote proliferation and migration of breast cancer cells. EMBO Rep 2017;18:420–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yulianto A. Pengaruh Literasi Keuangan Syariah Terhadap Keputusan Penggunaan Produk Atau Layanan Lembaga Keuangan Syariah. Indonesia: Universitas Islam Indonesia, 2018. [Google Scholar]
- 28. Shou JZ, Hu N, Takikita M, et al. Overexpression of CDC25B and LAMC2 mRNA and protein in esophageal squamous cell carcinomas and premalignant lesions in subjects from a high-risk population in China. Cancer Epidemiol Biomarkers Prev 2008;17:1424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pan Y, Xu T, Liu Y, et al. Upregulated circular RNA circ_0025033 promotes papillary thyroid cancer cell proliferation and invasion via sponging miR-1231 and miR-1304. Biochem Biophys Res Commun 2019;510:334–8. [DOI] [PubMed] [Google Scholar]
- 30. Liu G, Shi H, Deng L, et al. Circular RNA circ-FOXM1 facilitates cell progression as ceRNA to target PPDPF and MACC1 by sponging miR-1304-5p in non-small cell lung cancer. Biochem Biophys Res Commun 2019;513:207–12. [DOI] [PubMed] [Google Scholar]
- 31. Du W, Li D, Guo X, et al. Circ-PRMT5 promotes gastric cancer progression by sponging miR-145 and miR-1304 to upregulate MYC. Artif Cells Nanomed Biotechnol 2019;47:4120–30. [DOI] [PubMed] [Google Scholar]
- 32. Qiu X, Dou Y. miR-1307 promotes the proliferation of prostate cancer by targeting FOXO3A. Biomed Pharmacother 2017;88:430–5. [DOI] [PubMed] [Google Scholar]
- 33. Han S, Zou H, Lee JW, et al. miR-1307-3p stimulates breast cancer development and progression by targeting SMYD4. J Cancer 2019;10:441–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen S, Wang L, Yao B, Liu Q, Guo C. miR-1307-3p promotes tumor growth and metastasis of hepatocellular carcinoma by repressing DAB2 interacting protein. Biomed Pharmacother 2019;117:109055. [DOI] [PubMed] [Google Scholar]
- 35. Cogswell JP, Ward J, Taylor IA, et al. Identification of miRNA changes in Alzheimer's disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis 2008;14:27–41. [DOI] [PubMed] [Google Scholar]
- 36. Romano GL, Platania CB, Forte S, et al. MicroRNA target prediction in glaucoma. Prog Brain Res 2015;220:217–40. [DOI] [PubMed] [Google Scholar]
- 37. Romano GL, Platania CBM, Drago F, et al. Retinal and circulating miRNAs in age-related macular degeneration: an in vivo animal and human study. Front Pharmacol 2017;8:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burgaletto C, Platania CBM, Di Benedetto G, et al. Targeting the miRNA-155/TNFSF10 network restrains inflammatory response in the retina in a mouse model of Alzheimer's disease. Cell Death Dis 2021;12:905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He K, Zhang X, Ren S, et al. Deep residual learning for image recognition. In: 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), 2016, pp.770–8. IEEE, Las Vegas, NV, USA. [Google Scholar]
- 40. He K, Fan H, Wu Y, Xie S, Girshick R. Momentum Contrast for Unsupervised Visual Representation Learning. In 2020 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR). Seattle, WA, USA, 2020, pp. 9726–35.
- 41. Chen T, Kornblith S, Norouzi M, et al. A simple framework for contrastive learning of visual representations. In: Proceedings of the 37th International Conference on Machine Learning, 2020, pp. 1597–607.
- 42. Moon J, Kim J, Shin Y, Hwang S. Confidence-aware learning for deep neural networks. In international conference on machine learning. PMLR, 2020, 7034–44.
- 43. Sensoy M, Kaplan L, Kandemir M. Evidential deep learning to quantify classification uncertainty. Adv Neural Inf Process Syst 2018;31:01768. [Google Scholar]
- 44. Tanaka H, Kuwano Y, Nishikawa T, et al. ZNF350 promoter methylation accelerates colon cancer cell migration. Oncotarget 2018;9:36750–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sun R, Yang Y, Lü W, et al. Single-cell transcriptomic analysis of normal and pathological tissues from the same patient uncovers colon cancer progression. Cell Biosci 2023;13:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Spisák S, Kalmár A, Galamb O, et al. Genome-wide screening of genes regulated by DNA methylation in colon cancer development. PloS One 2012;7:e46215. 10.1371/journal.pone.0046215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yin H, Hardikar S, Lindstroem S, et al. Telomere maintenance variants and survival after colorectal cancer: smoking- and sex-specific associations. Cancer Epidemiol Biomarkers Prev 2020;29:1817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Y, Gu X, Jiang F, et al. Altered expression of striatin-4 is associated with poor prognosis in bladder transitional cell carcinoma. Oncol Lett 2021;21:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Snipstad K, Fenton CG, Kjaeve J, et al. New specific molecular targets for radio-chemotherapy of rectal cancer. Mol Oncol 2010;4:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zuurbier L, Rahman A, Cordes M, et al. Apelin: a putative novel predictive biomarker for bevacizumab response in colorectal cancer. Oncotarget 2017;8:42949–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lv Y, Duanmu J, Fu X, et al. Identifying a new microRNA signature as a prognostic biomarker in colon cancer. PloS One 2020;15:e0228575. 10.1371/journal.pone.0228575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hou J, Cheng J, Dai Z, et al. Molecular and clinical significance of stanniocalcin-1 expression in breast cancer through promotion of homologous recombination-mediated DNA damage repair. Front Cell Dev Biol 2021;9:731086. https://doi:10.3389/fcell.2021.731086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Peña C, Céspedes MV, Lindh MB, et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res 2013;73:1287–97. [DOI] [PubMed] [Google Scholar]
- 54. Supiot S, Gouraud W, Campion L, et al. Early dynamic transcriptomic changes during preoperative radiotherapy in patients with rectal cancer: a feasibility study. World J Gastroenterol 2013;19:3249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Azizian A, Gruber J, Ghadimi BM, Gaedcke J. MicroRNA in rectal cancer. World J Gastrointest Oncol 2016;8:416–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee Y, Dho SH, Lee J, et al. Hypermethylation of PDX1, EN2, and MSX1 predicts the prognosis of colorectal cancer. Exp Mol Med 2022;54:156–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gratio V, Beaufort N, Seiz L, et al. Kallikrein-related peptidase 4: a new activator of the aberrantly expressed protease-activated receptor 1 in colon cancer cells. Am J Pathol 2010;176:1452–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang X, Cheng H, Zhao J, et al. Long noncoding RNA DLGAP1-AS2 promotes tumorigenesis and metastasis by regulating the Trim21/ELOA/LHPP axis in colorectal cancer. Mol Cancer 2022;21:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Han Y, Huang G, Song S, et al. Dynamic neural networks: a survey. IEEE Trans Pattern Anal Mach Intell 2021;44:7436–56. [DOI] [PubMed] [Google Scholar]
- 60. Spiegelhalter DJ, Lauritzen SL. Sequential updating of conditional probabilities on directed graphical structures. Networks 1990;20:579–605. [Google Scholar]
- 61. Dempster AP. A generalization of bayesian inference. J R Stat Soc B Methodol 1968;30:205–32. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The BRCA and ROSMAP datasets were obtained from the GitHub repository of MOGONET (https://github.com/txWang/MOGONET). The omics data of the other datasets (COADREAD, ESCA, GBMLGG, SARC, STAD, STES, THCA, UCEC, LGG, KIPAN), as well as the survival time information, were obtained from The Cancer Genome Atlas (TCGA) Program through Broad GDAC Firehose (https://gdac.broadinstitute.org/). The PAM50 breast cancer subtypes of TCGA BRCA patients were obtained through the TCGAbiolinks R package (v2.12.6, http://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html). We have made our preprocessed data for the 12 cancer subtype dataset available through Google Drive (https://drive.google.com/drive/folders/1_tJ2ekhTmWp7ZcRVjUVGx0cqGMRKEhNo?usp=share_link).