

Abstract
Gene expression differences between distinct cell types are orchestrated by specific sets of transcription factors and epigenetic regulators acting upon the genome. In plants, the mechanisms underlying tissue-specific gene activity remain largely unexplored. Although transcriptional and epigenetic profiling of individual organs, tissues, and more recently, of single cells can easily detect the molecular signatures of different biological samples, how these unique cell identities are established at the mechanistic level is only beginning to be decoded. Computational methods, including machine learning, used in combination with experimental approaches enable the identification and validation of candidate cis-regulatory elements driving cell-specific expression. Synthetic biology shows great promise not only as a means of testing candidate DNA motifs, but also for establishing the general rules of nature driving promoter architecture and for the rational design of genetic circuits in research and agriculture to confer tissue-specific expression to genes or molecular pathways of interest.
Introduction
In plants and other eukaryotes, gene activity is regulated at multiple levels, with transcriptional control being the primary and most studied mode of gene expression regulation [1]. Whether a given gene is transcribed in a particular cell type, developmental stage, or growth condition is dictated by: (1) the presence and chromatin accessibility of cis-regulatory elements (CREs) in the gene’s promoter and distal regulatory regions, (2) the availability of corresponding trans-acting transcription factors (TFs) that recognize these CREs, and (3) the physical interactions between these TFs, the basal transcriptional machinery (RNA polymerase II, the Mediator complex, and general TFs), and epigenetic regulators (such as chromatin remodelers or histone modifiers) [1]. Epigenetic factors, some positive and some negative, directly or indirectly alter chromatin structure and make CREs either accessible or inaccessible for binding by TFs and the basal transcriptional apparatus, ultimately regulating the frequency of transcription initiation and elongation [2]. Noteworthy, many of the TFs and epigenetic regulators are themselves expressed in a developmental stage-, tissue-, or condition-dependent manner, thus orchestrating what target genes are, versus are not, expressed in a given cell [3].
In the past 20 years, genome-wide transcriptome and epigenome profiling have become increasingly technically accessible, leading to ample quantitative data having been collected by the scientific community on a variety of species, tissues, and growth conditions. Nonetheless, in plants and other eukaryotes, little is known about how tissue-specific expression is established. Hypothetically, to generate a narrow pattern of gene expression that is restricted to only some tissues, developmental stages, or growth conditions by a combination of positive and negative TFs (Figure 1a), two general mechanistic scenarios are possible. A limited set of narrowly expressed transcriptional activators and positive chromatin remodelers can work together to turn the gene “on” in only some cell types, conditions, or growth stages (Figure 1b). Alternatively, a combination of broadly expressed transcriptional activators and remodelers can turn the gene “on” ubiquitously/constitutively, but another set of more narrowly expressed transcriptional repressors and epigenetic regulators then turn the gene “off” in most tissues/stages/conditions, leaving the gene of interest “on” only in some spatiotemporal domains that lack these negative regulators (Figure 1c). Presumably, in native promoters, a combination of CREs bound by positive and negative, broadly expressed and spatio-temporally restricted regulators is at play in the regulation of a majority of genes in plants and other multicellular organisms (Figure 1d). Hence, for many promoters, the identification of individual CREs conferring tissue-specific expression may not be trivial or at all possible when using classical transgene promoter bashing [4] or the latest CRISPR version of this top-down strategy with the targeted elimination of candidate DNA elements directly in the genome [5]. Demonstrating that a CRE is overrepresented in genes expressed in a particular tissue, stage, or condition (by using computational methods) and is required for that pattern of expression (by showing that mutating the element abolishes the expression enrichment) does not necessarily mean that the CRE is capable of and sufficient for conferring the tissue- or stage-specific expression. The latter needs to be experimentally demonstrated and this is where synthetic biology methods can help.
Figure 1.
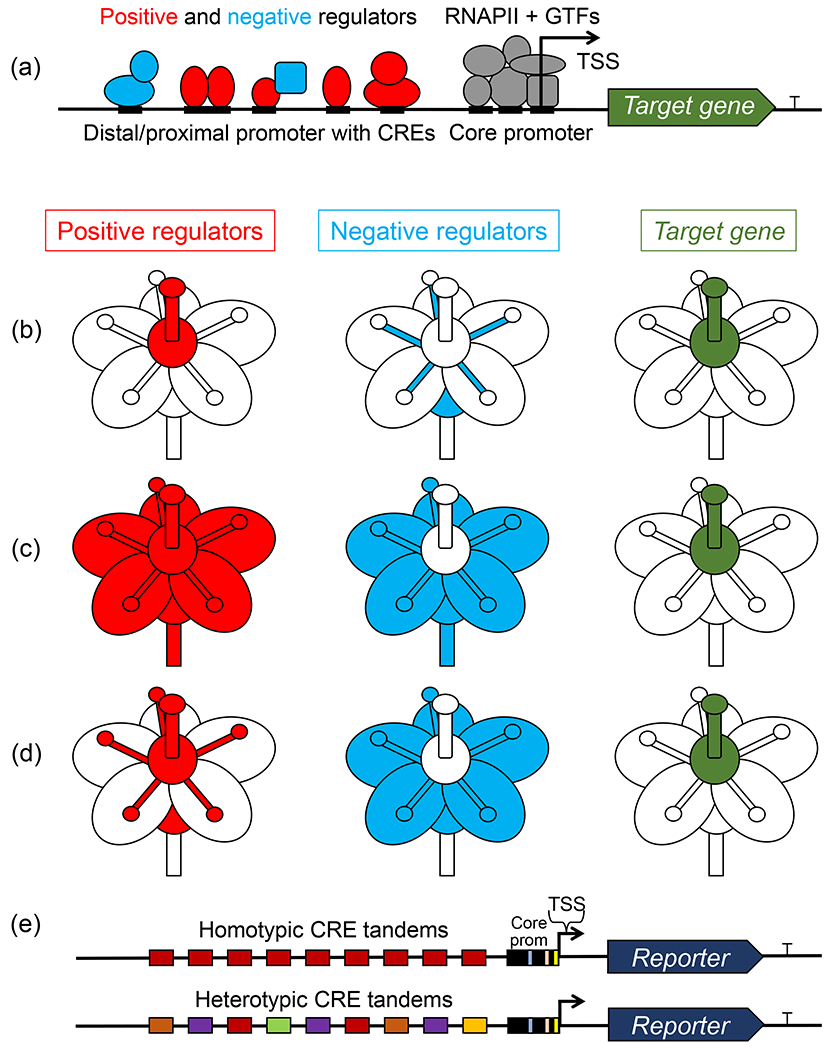
Transcriptional regulation underlying tissue specificity of gene expression. (a) Native promoters harbor multiple cis-regulatory elements (CREs) that bind a combination of positive (red) and negative (cyan) regulators (transcription factors, co-factors, and epigenetic effectors) that, respectively, assist and interfere with the RNA polymerase II (RNAPII) and general TF (GTF) (gray) recruitment to the core promoter. Arrow marks the transcription start site (TSS). (b, c, d) Different expression patterns of positive (red) and negative (cyan) regulators can result in restricted domains of target gene expression (green) if negative regulators negate the effects of positive regulators. (e) Typical synthetic reporter constructs harboring tandems of identical or divergent CREs placed upstream of a well-characterized core promoter, such as (−46)35S, driving a reporter gene, such as GFP, Luciferase, or GUS.
In a bottom-up approach, synthetic distal and proximal promoters or enhancers are constructed from tandems of individual CREs and placed upstream of a well-characterized natural or synthetic core promoter driving a gene of interest, usually a fluorescent or luminescent reporter, or a histochemical marker, thus allowing for convenient visualization of promoter activity patterns (Figure 1e). This approach makes the assumption that the CRE of interest can recruit a tissue- or stage-specific transcriptional activator that interacts with positive epigenetic regulators and general TFs bound at the core promoter to help bring RNA polymerase II and trigger transcription (Figure 1a). Indeed, such an experimental strategy has proven to be successful at conferring hormone-, stress- and pathogen-inducible behavior to synthetic promoters made out of homo- and heteromeric repeat tandems of the TF binding site [6]. In contrast, stacking candidate CREs enriched in specific cell types does not always produce narrowly expressed synthetic tissue-specific promoters [7], with gene activity detected in several cell types (e.g., most green tissues, or most of the root) and leaking to non-target organs. Perhaps, this is not surprising since many CREs are recognized by several sequence-related TFs that are members of a gene family with overlapping domains of expression and similar DNA binding preferences [8]. Furthermore, even the best promoters or tissue-specific cis-elements identified computationally and/or experimentally do not always translate well to other plant species. For example, the ATATT CRE from the Agrobacterium rhizogenes rolD gene initially described as a root-specific DNA motif in plants [9] and employed in the generation of synthetic root-expressed promoters in tobacco [10] was later shown to function as a green tissue element in rice [11]. In addition, since most classical studies that explore the cell specificity of a CRE typically test a limited number of tissues and growth conditions in a single model species, the specificity and orthologous function of the sequence cannot be reliably inferred beyond pointing to possible expression enrichment relative to other tested tissues in one or a handful of plant species.
In this review, we describe how the tissue specificity of gene expression and the role of epigenetic regulators have been approached historically, what state-of-the-art experimental technologies and computational methods are available currently, and the first strides synthetic biology has made to move this field of plant molecular genetics forward.
Molecular genetics approaches
Tissue-specific gene expression differences between plant organs have been traditionally studied by classical molecular techniques such as northern blots, RT-PCRs, microarrays or RNA-seq. These methods were typically performed on whole organs and thus lacked the resolution needed to distinguish between the different cell types that make up an organ or a tissue. To hone in on specific cell types of interest, laser capture microdissection (LCM) was developed, where a tissue fragment of interest is physically excised from a larger frozen or fixed tissue section using a laser [12], and then RNA is extracted and one of the aforementioned transcriptomic methods is applied (Figure 2a). Although mRNA expression analysis on LCM samples provides more refined tissue-specific gene expression information than whole-tissue samples [12,13], the relatively limited spatial resolution, high labor, technological demands, and sample size limitations prompted the development of alternative approaches [14]. Thus, for example, to collect cell-type-specific information, organs could be protoplasted and the cells sorted by fluorescence activated cell sorting (FACS, Figure 2b) based on the expression of tissue-specific fluorescent marker genes [15–17]. This approach led to the generation of tissue-specific gene expression maps and the association of gene expression patterns with cell fate [18].
Figure 2.
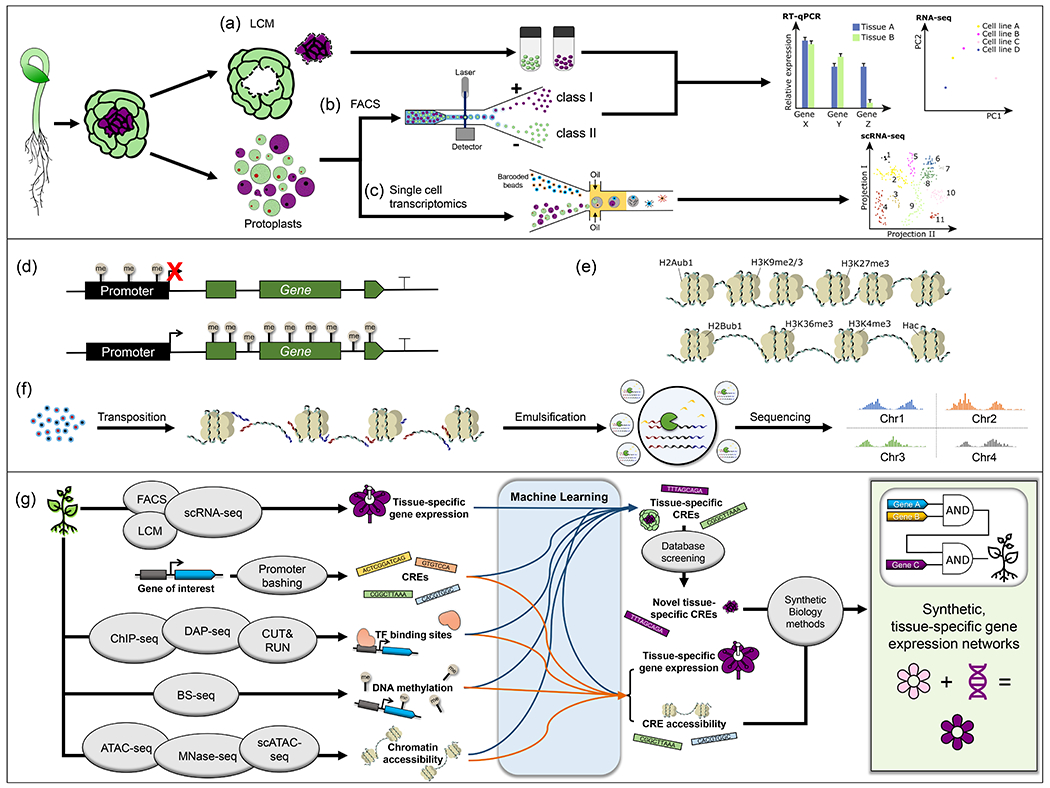
Technical approaches conferring spatial resolution to cell/tissue-specific transcriptome profiling. (a) In LCM, a target cell or group of cells is isolated from a tissue section using a UV laser beam. (b) FACS enables single cell (protoplast) or cell lineage isolation, typically based on the expression of a fluorescent marker, but depends upon the availability of fluorescent cell lines or a cell-type specific fluorescent signal. (c) Single cell transcriptome profiling encapsulates single cells or nuclei into uniquely barcoded nanodroplets that are subjected to next-generation sequencing. (a-c) Gene expression profiling is achieved via one of several techniques. RT-qPCR, RNA-seq and scRNA-seq are currently some of the most relevant transcriptomic methods. RT-qPCR measures the expression of a limited number of genes of interest. Expression values represent an average of a whole sample, potentially masking variability between cell subpopulations or individual cell types. RNA-seq provides a more detailed view of the tissue-specific transcriptome as it collects expression information for all genes in the genome in parallel. The number and quality of the data points are limited by the same constraints as RT-qPCR, with cell dissection/sorting providing only bulk tissue-level resolution. scRNA-seq enables the identification of tissue-specific gene expression profiles at a whole-genome level, but with single-cell resolution, allowing for clustering of groups of cells based on their common expression characteristics. (d) CG cytosine methylation in the promoter region is associated with transcriptionally repressed genes, as are CHG and CHH methylation (not shown). CG cytosine methylation in the gene body is associated with transcriptionally active genes. (e) H2Aub1, H3K9me2/3, and H3K27me3 histone modifications are associated with transcriptionally repressed genes, while H2Bub1, H3K44me3, and H3K36me3, and histone acetylation modifications are associated with transcriptionally active genes. (f) scATAC-seq identifies the genomic locations of accessible chromatin, which correlates with transcriptional activity, with single-cell resolution. Open chromatin is readily accessed by a transposase that cuts and inserts adapters into the genomic DNA, creating adapter-flanked fragments. Single cells or nuclei are then encapsulated in individual droplets where the genomic fragments of each cell are amplified and barcoded prior to sequencing. (g) ML integrates the data from one or many methods of analyzing tissue-specific expression to make predictions about tissue-specific expression, regulation by CREs, or CRE accessibility, and to identify novel CREs. These data can then be used to leverage synthetic biology approaches for testing ML models, furthering the discovery of the regulatory framework of tissue-specific expression, and engineering novel regulatory circuits.
The tissue-specific transcriptomes obtained from the aforementioned studies were, indeed, average transcriptomes from pools of cells. However, no two cells are identical. Therefore, a new technology with the ability to map the transcriptomes of individual single cells was pursued. In 2009, Tang and collaborators developed single cell mRNA sequencing, scRNA-seq (Figure 2c) [19]. This new method uncovered the complexity of individual cell transcriptomes within the same tissue showing, for example, that different transcript isoforms could be expressed in the same cell. The throughput of these initial scRNA-seq experiments was, however, relatively low. Further improvements in cell isolation and sorting by assigning unique DNA barcodes to each cell, along with the encapsulation of a single cell in a nanoliter droplet where genome-wide mRNA sequencing takes place, gave rise to a high-throughput version of scRNA-seq we know today [20,21]. The advent of scRNA-seq and subsequent modifications of this technology [22] allowed the unraveling of the average transcriptome of a highly complex tissue into clusters of single-cell gene expression profiles, which led to the identification of new cell subtypes and the association of transcriptional programs with developmental stages and cell fates in species such as Arabidopsis [23,24], maize [25,26], and rice [27–29].
Cell-specific gene expression patterns and, therefore, cell identities and their differentiation trajectories unveiled by scRNA-seq are programmed by both DNA sequence and chromatin modifications (see below). These DNA-based programs consist of combinations of CREs capable of recruiting suites of TFs and epigenetic regulators, both positive and negative, that cumulatively dictate the dynamics of an individual gene’s activity (Figure 1a,d, [30]). Thus, identifying CREs and deciphering the syntax rules that govern their function are central to understanding gene regulation. Experimentally, the identification of CREs targeted by TFs or epigenetic effectors of interest in vivo is usually carried out by chromatin immunoprecipitation (ChIP)-seq [31,32,33] or DNA affinity purification (DAP)-seq [34]). Furthermore, enzyme- and immuno-tethering methods, such as DNA adenine methyltransferase identification (DamID; [35,36]), chromatin immunocleavage (ChIC), chromatin endogenous cleavage (ChEC; [37])), cleavage under targets and release using nuclease (CUT&RUN; [38]), and cleavage under targets and tagmentation (CUT&Tag; [39]), have been developed to map TF binding sites at a whole-genome level [40]. On the other hand, classical studies aiming to determine the role of putative CREs in the regulation of a specific gene of interest have typically leveraged promoter bashing, i.e., the systematic analysis of a series of truncated promoters driving a reporter gene. By monitoring the effect of the presence/absence of candidate CREs on reporter activity, the function of CREs as enhancers (increased activity) or silencers (decreased activity) of gene expression can be deduced [4]. More recently, CRISPR/Cas9-mediated deletions of CREs in the native genomic context in vivo have been carried out, resulting in the generation of a series of promoter alleles in genes of interest via genome editing [41]. This new approach can mimic an accelerated process of domestication in crops such as tomato [5,42,43], maize, barley, rice [44] or the orphan Solanaceae crop, groundcherry [45]. Despite these practical advances, the complexity of the interactions between different CREs, TFs and epigenetic environments makes the dissection of complex promoters a formidable challenge, and the prediction of the effects of specific allelic variants difficult. Nonetheless, better understanding of these CRE networks would be helpful for the generation of more targeted promoter edits. Overlaying cistrome and epicistrome maps with cell-specific distribution patterns of DNA methylation, histone modifications, and chromatin accessibility (see below), the availability of CREs for TF and epigenetic regulator binding can start to be inferred. This information is fundamental to our understanding of how transcriptional programs are associated with cell identity and fate and, therefore, how CREs could be leveraged by synthetic biology to reprogram crops of interest.
Epigenetic approaches
Cell and tissue identity is determined not only by the genes that are expressed, but also by the genes that are transcriptionally inactive. The transcriptional status of much of the genome is established and maintained via epigenetic mechanisms including maintenance and de novo DNA methylation (Figure 2d), RNA-directed DNA methylation (RdDM), changes in chromatin composition, and histone modifications (Figure 2e) [46]. The three-dimensional (3-D) organization of chromatin also plays a part in determining the accessibility of genes by placing genes in proximity to their regulatory elements, and patterns of 3-D topology can be reflective of the transcriptional status. Recent methodological advances, such as Hi-C, make this a growing area of research [47]. The ability to engineer epigenetic changes and control chromatin structure and 3-D organization in plants has the potential to answer interesting scientific questions and to provide new tools and strategies for crop improvement [48].
Methylation of the fifth position of the cytosine ring (5mC) is a common epigenetic mark and can occur in several sequence contexts: CG, CHG, or CHH (where H refers to A, T or C) [46]. CG methylation in the gene body is generally associated with active expression, while CG methylation of the promoter and transcription start site (TSS), as well as CGH, and CHH methylation, are all generally associated with transcriptionally inactive DNA (Figure 2d) [46]. DNA can also be methylated at adenine (6mA), although the role of 6mA in the regulation of gene expression in plants is still largely unclear [49]. Experimentally, methylated DNA sites are typically detected with the help of methylation-sensitive restriction enzymes, anti-5mC or anti-6mA antibodies, bisulfite conversion of unmethylated cytosines, or nanopore sequencing [50,51]. Bisulfite sequencing (BS-seq or MethylC-seq) has been initially applied at the single-cell level in mammalian systems [52]. Li et al. [53] developed a single-cell approach to measure methylation in plants called bisulfite-converted randomly integrated fragments sequencing (BRIF-seq). BRIF-seq was used to measure methylation in the microspores from four maize tetrads, and while the microspores within a tetrad were highly similar, the methylation patterns between tetrads showed a level of heterogeneity that suggests tetrads undergo differential methylation reprogramming [53].
The methylation pattern of DNA has been shown to help define the transcriptional profile and, therefore, the identity of a cell. Kawakatsu et al. [54] used FACS to isolate six different cell types from the Arabidopsis root meristem. These different cell types were then subjected to BS-seq and RNA-seq to compare the methylation and gene expression patterns, revealing cell-type specific methylation patterns [54]. Recently, the CLASSY (CLSY) family of putative chromatin remodeling factors was found to regulate tissue-specific methylation in Arabidopsis [55]. Data from CLSY reporters, MethylC-seq, and small-RNA seq in Arabidopsis flower buds, ovules, mature leaves, and young rosettes revealed that different tissues display unique patterns of DNA methylation, small RNAs, and CLSY expression. Using combinations of CLSY and RdDM mutants, it was found that specific CLSY expression profiles define tissue-specific epigenomes, primarily via the RdDM pathway [55], providing a mechanism whereby the expression of specific chromatin remodelers or combinations of proteins establishes tissue identity through epigenetic marks.
The mechanisms of DNA methylation establishment and maintenance have been manipulated to achieve targeted epigenetic modifications in plants. Targeted demethylation of 5mC in Arabidopsis has been demonstrated using zinc finger and CRISPR technologies [56]. Likewise, heritable gene-specific CHH [57] and CG [58] methylation has also been achieved in Arabidopsis using CRISPR. For example, using dCas9 fused to a bacteria-derived methyltransferase led to de novo methylation of both CG and CHH sites in the FLOWERING WAGENINGEN (FWA) promoter and was able to rescue the associated late-flowering mutant phenotype [58]. In combination with cell- or tissue-specific Cas9 proteins or gRNAs, this kind of engineering would allow for the epigenetic modification of a gene of interest with cellular or tissue-type resolution [59–61].
DNA methylation is not the only epigenetic parameter that controls gene expression. While 5mC methylation can directly prevent some TFs from accessing their respective CREs, it also recruits chromatin remodeling complexes that bring about changes in chromatin structure and, ultimately, compactness that affects the physical accessibility of DNA to the transcriptional machinery. Chromatin accessibility can be experimentally assessed at a whole-genome level using high-throughput methods such as the assay for transposase accessible chromatin (ATAC)-seq (Figure 2f), micrococcal nuclease (MNase)-seq, DNase-seq, and formaldehyde-assisted isolation of regulatory elements (FAIRE)-seq [62]. ATAC-seq [63] can also be employed at the single-cell (sc) level, revealing cell-specific chromatin accessibility differences. ATAC-seq experiments using single nuclei from Arabidopsis roots found that the chromatin accessibility patterns of cell type-specific marker genes mirror transcription levels, suggesting that cell identity is regulated at the level of chromatin accessibility [24]. ATAC-seq of single nuclei has also been experimentally demonstrated in soybean (Glycine max) [64]. Dorrity et al. [65] coupled scATAC-seq of Arabidopsis roots with published scRNA-seq data to uncover endodermal cell types that could not be resolved by scRNA-seq alone. This combined dataset was also used to identify TFs and CREs that may define cell-type specific expression [65]. Marand et al. [66] employed scATAC-seq in maize to map CREs on genomic scale. By grouping single cells based on chromatin accessibility, these researchers were then able to use chromatin accessibility as a surrogate for, or in coordination with, RNA expression data from known cell-type or tissue-specific markers. This study revealed that maize domestication has relied on the selection of agronomically favorable CRE alleles [66].
The structure of chromatin is cumulatively controlled via multiple post-translational modifications of histone proteins. Some of these are associated with repressed (e.g., H3K9me2/3, H3K27me3, H2Aub1) and some with transcriptionally active (e.g., H3K4me3, H3K36me3, H2Bub1, and histone acetylation) chromatin (Figure 2e) [67]. Commercial antibodies are available for many of the modified histones, thus enabling ChIP-based detection of most types of histone marks. Besides ChIP-seq, the aforementioned alternative approaches such as DamID and CUT&RUN allow for the detection of specific histone modifications throughout the genome with almost nucleotide-level resolution [62]. Cumulatively, histone tail modifications dictate the transcriptional status of genes to determine cell identity [68], define developmental transitions, such as flowering [69,70], and establish environmental stress memory, such as to recurrent drought or pathogen attack [67].
Genome-wide mapping of histone modifications enables the comparison of histone profiles of different tissues and can pinpoint key differences that distinguish divergent cell types [71]. For example, Lee et al. employed FACS in Arabidopsis to isolate normal stomatal guard cells and guard cells undergoing dedifferentiation and then compared the histone modifications associated with these two cell types [72]. ChIP-seq analysis of H3K4me3- and H3K27me3-associated genes identified several peaks of differential methylation between these two cell types. The H3K27me3 set of genes was enriched for pathways involved in transcription and regulation of postembryonic development. When Polycomb Repressive Complex 2 activity (and therefore, H3K27me3) was knocked down using a guard cell-specific miRNA, a dedifferentiation phenotype was observed in stomata. These results suggest that in stomatal guard cells, H3K27me3 distribution determines cell fate by regulating progression through the normal guard cell lineage [72]. In animal systems, the study of H3K4me3 and H3K27me3 histone marks via ChIP-seq has been implemented at single-cell resolution [73], but scChIP-seq has yet to be employed in plants [52].
In theory, native cell identity can be reprogrammed via selective modifications to the epigenome [74]. By targeting the promoters of key genes via synthetic epigenetic factors, local chromatin structure can be selectively altered to either activate or inactivate gene expression. Such regulators can be generated from a customized zinc finger DNA-binding domain, transcription activator-like effector (TALE) or dCas9 fused to a well-characterized epigenetic factor [74] and expressed in a restricted spatiotemporal pattern from a native or synthetic tissue-specific promoter. To develop such technology into a versatile molecular tool, a diverse collection of epigenetic regulators that alter DNA methylation, histone tail modifications, and/or nucleosome density would be required. Although plants are lagging behind in this arena, high-throughput studies in animals and yeast indicate that it is possible to identify epigenetic regulators capable of triggering local chromatin remodeling and altering gene expression [75,76].
Computational methods
With the advancement of computing power and the development of machine learning (ML) tools, CRE identification and syntax underlying tissue-specific gene expression can now be examined from another perspective. Furthermore, synthetic biology methods have opened up a new path for the modular testing of genetic elements from both a bottom-up and a top-down approach, but still face the same design-build-test cycle limitations as traditional molecular biology methods. To tackle these issues, databases of ‘-seq’ data and big data analysis tools should be utilized to guide experimental approaches.
CRE prediction begins with determining what genes are or are not expressed under certain stimuli and/or in a specific tissue using high-throughput methods like RNA-seq. The identification of gene expression patterns greatly benefits from a large dataset, which can be pulled from general expression data repositories such as NCBI’s Sequence Read Archive (SRA) [77] or species-specific repositories such as the Arabidopsis RNA-seq database [78] (Table 1). Once those sequences are mapped to a genome, tools like the binding site estimation suite of tools (BEST) can be used to identify the promoter region upstream of the sequences [89]. Finally, these promoter sequences can be cross-referenced with databases containing CREs, with examples of plant-specific databases like PLACE [79], PlantCARE [80], and PlantProm [81], to determine whether known or uncharacterized CREs are driving the observed tissue-specific expression pattern (Table 1). More recently developed high-throughput sequencing methods such as ChIP-seq, DAP-seq and ATAC-seq can greatly increase the accuracy of novel CRE prediction by identifying TF binding sites that may themselves either be, or contain, cis-regulatory elements [62]. PlantPAN3.0, an analysis navigator tool, provides accessibility to annotated ChIP-seq data in its PCBase database with descriptions of the type of regulatory factors involved in the experiment, the tissue, and other details about each ChIP-seq experiment [83] (Table 1).
Table 1.
List of relevant databases that may be useful when analyzing the link between tissue-specific gene expression and CREs in plants.
| Database | Scope | Type of data | Description | Established | Last Updated | Size | |
|---|---|---|---|---|---|---|---|
| NCBI Sequence Read Archive (SRA) [70] | Non-specific | NGS reads | Repository of raw, high-throughput “next generation” sequencing (NGS) data with minimal processing. Includes data related to both DNA and RNA. | 2007 | 2022 | 62+ petabases | |
| Arabidopsis RNA-seq database [71] | Species-specific: • Arabidopsis thaliana |
RNA reads | Archive that contains Arabidopsis thaliana RNA-seq libraries integrated from multiple databases and reprocessed with a standardized pipeline. | 2019 | 2019 | 20,000+ RNA-seq libraries | |
| PLACE [73] | Plant-specific | CREs | Database containing the nucleotide sequences of plant cis-acting regulatory DNA elements derived from published reports. | 1999 | 2007 | 469 entries (motifs) | |
| PlantCARE [74] | Plant-specific | CREs | Database that contains plant specific cis-acting regulatory elements, enhancers, and silencers. | 1999 | 2000 | 417 CREs | |
| PlantProm [75] | Plant-specific | CREs | Collection of RNA polymerase II proximal promoter sequences. | 2002 | 2009 | 576 annotated promoters | |
| PCBase [76] | Species-specific: • Arabidopsis thaliana • Oryza sativa • Zea mays • Solanum lycopersicum • Glycine max • Arabidopsis lyrata • Gossypium hirsutum |
ChIP-seq data | Database that utilizes plant ChIP-seq experimental data to identify TF binding sites for 7 model plants. Part of the PlantPAN3.0 navigator tool that contains 17,230 TFs and 4,703 TF binding site matrices across 78 plant species. | 2018 | 2018 | 421 processed ChIP-seq datasets | |
| RegSite [77] | Plant-specific | CREs | Database of annotated plant regulatory elements. | 2014 | 2016 | 3032 motifs | |
| PlantDHS [78] | Species-specific: • Arabidopsis thaliana • Brachypodium distachyon • Oryza sativa |
CREs and DNAse I hypersensitive sites | Utilizes histone modification, RNA-seq data, nucleosome occupancy, TF binding sites, and DNA sequencing to create a collection of DNase I hypersensitive sites for specific plant species. Data for cotton have also been collected and will be integrated into the database in the upcoming future. | 2015 | 2016 | 14.8G of processed data Coming soon: 73.9G of cotton data |
|
| AGRIS [79] | Species-specific: • Arabidopsis thaliana |
CREs and TFs | Information resource for promoter CREs , TFs and target genes. Contains three databases, AtTFDB (TFs) and AtcisDB (CREs), and AtRegNet (TF-gene interactions). | 2003 | 2019 | • 1,400 TFs • 29,388 annotated genes |
|
| plantpromoterdb [80] | Species-specific: • Arabidopsis thaliana • Oryza sativa • Physcomitrium patens • Poplar |
CREs | Database that provides promoter annotations for specific plant species. Annotations include TSS, core promoter elements, and transcriptional regulatory elements. | 2007 | 2020 | • 308 genes for Arabidopsis • 242 genes for rice |
|
| JASPAR CORE [81] | Eukaryote-specific | TF binding sites | Database of curated TF binding profiles for multiple species. Contains position frequency matrices (PFMs) to describe TF-DNA interactions. PFMs can be interpreted as motifs or TF binding profiles. | 2004 | 2022 | 1955 PFMs | |
| RGPDB [82] | Root-specific: • Zea mays • Glycine max • Sorghum bicolor |
Genes and promoters | Database containing root-specific genes and promoters for a limited set of plant species. | 2020 | 2020 | > 1200 genes |
A popular method of predicting tissue-specific gene expression and identifying respective CREs is ML, or more specifically, supervised ML. The general pipeline of a supervised ML algorithm starts with training a computer to recognize known patterns within a training set of data where the DNA features to be predicted (e.g., CREs, chromatin accessibility, DNA or histone methylation status) responsible for tissue-specific expression are known, improving upon the predictions made by this pattern recognition iteratively (Figure 2g). The ‘rules’ learned by the algorithm can then be used to identify CREs, and other features, in novel sequences. In recent years, the focus has begun to shift to deep learning, an evolution of ML, to identify DNA features through the use of artificial neural networks, i.e., enabling the machine to process information similar to a human brain [90]. For CRE prediction, the preferred approach tends to employ Convoluted Neural Networks (CNNs) which repeatedly apply self-adjusting filters to the sequence data to identify sequence characteristics with potential regulatory roles [91]. This means that if the data used to train the model contain tissue specificity information, then the algorithm will be able to predict tissue-specific CREs, methylation, DNA accessibility, etc., across different tissues. For example, Wang et al. developed a deep learning algorithm, Smart Model for Epigenetics in Plants (SMEP), which uses a CNN approach to predict multiple types of epigenomic modifications using published sequencing data [92]. The data pulled for this study were comprised of BS-seq, single-molecule real-time sequencing (SMRT-seq), ChIP-seq, and RNA-seq. The data used to train, validate, and test the model came from a number of plants, including Arabidopsis, maize, and rice. With the wide variety of plant species data included in this study, this algorithm was able to predict six types of modifications with at least 80% accuracy: 5mC, 6mA, m6a, H3K4me3, H3K27me3, and H3K9ac. SMEP exists online as a web server, making it user-friendly and available to the scientific community.
In addition to predicting epigenomic modifications, ML models can also help identify methylation features that contribute the most to accurate prediction of differential gene expression across tissues. N’Diaye et al. utilized six different ML algorithms along with a deep learning neural network to analyze methylation profiles from different tissues with a 0.81 prediction accuracy of differentially expressed genes between leaf and root tissues in wheat [93]. The algorithm highlighted that DNA methylation of the promoter, the CDS, and the exon in the context of CG methylation contribute most to the predictive power of these models [93]. In both CRE and DNA accessibility prediction in the context of tissue-specificity, ML algorithms have proved essential to solving the mystery of tissue-specific gene regulation. For additional examples and an ML-focused review on the applications of this technology in plant genomics, we refer the reader to this excellent recent review [94].
Overall, the use of computational modeling and prediction can create a positive feedback loop in identifying tissue-specific DNA elements, where experimental data guide computational prediction models and these models, in turn, guide experimental studies focused on tissue-specific gene expression, as has already been done in mammals [95]. Additional bioinformatics software such as TOMTOM, a motif comparison tool, can help compare predicted elements to CRE databases to discriminate between novel and known motifs [96]. Synthetic biology methods can then be employed to generate synthetic promoters, from the computationally predicted tissue-specific CREs, fused to genes of interest in order to create complex genetic circuits with novel expression patterns that can be both discrete and tunable (Figure 2g).
Synthetic biology approaches
Beyond the aforementioned candidate DNA element stacking to generate synthetic promoters (Figure 1e), plant biologists have not yet fully exploited the power of synthetic biology, and little progress has been made to date in understanding plant promoter architecture from the perspective of tissue specificity. Most published studies employing synthetic promoters to understand the rules of nature utilize constitutively active TF/CRE combinations. Nonetheless, some of the findings are relevant, and the technical approaches and genetic resources developed are potentially applicable to studying the mechanisms behind promoter tissue specificity.
Jores et al. generated a series of synthetic core promoters and tested them in transient expression assays in tobacco [97]. The designs of synthetic promoters (Figure 3a) were guided by the rules discovered from the self-transcribing active regulatory region sequencing (STARR-seq) analysis of thousands of Arabidopsis, maize and sorghum native core promoter fusions with barcoded GFP transiently expressed in tobacco epidermis and in maize mesophyll protoplasts [97]. Not surprisingly, the inclusion of the consensus TATA-box (Figure 3a), Initiator and Y-patch elements significantly increased reporter activity irrespective of the GC content of the rest of the core promoter sequence, with TATA having the most dramatic positive effect. In agroinfiltrated tobacco, the strongest synthetic core promoter sequences were comparable in strength to the Cauliflower Mosaic Virus 35S core promoter, but in maize protoplasts, the same promoters were much weaker than 35S. As expected, adding native CREs for TCP, NAC and/or HSF families of TFs upstream (but not downstream) of a 35S core promoter to generate synthetic proximal promoters (Figure 3a) further boosted reporter expression, with combinations of multiple CREs resulting in higher reporter activity than single or double CRE combinations [97]. Moving the location of a CRE within the proximal promoter (up to 156bp upstream of the transcription start site) did not change proximal promoter activity. Although no tissue-specific effects were directly assessed in this work, tobacco epidermis versus maize protoplast transient expression systems had different effects on promoter activities, with species-specific (tobacco versus maize), cell-specific (epidermis versus mesophyll) and condition-specific (intact cells versus protoplasts) effects potentially being responsible for the observed differences [97].
Figure 3.
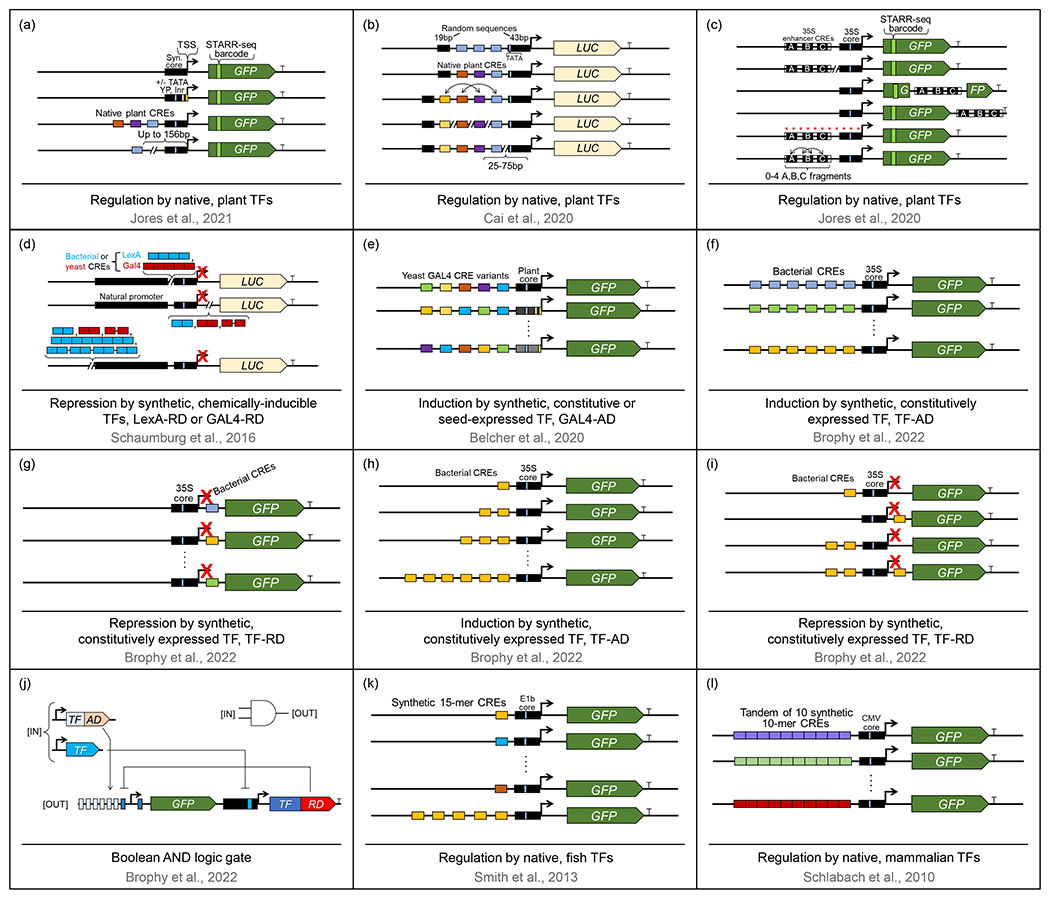
Schematic representation of genetic constructs that harbor synthetic promoters employed in the study of optimal promoter architectures. TSS – transcription start site. LexA, Gal4 – bacterial and yeast DNA binding domains, respectively. AD – transcription activation domain. RD – transcription repression domain.
Cai et al. built dozens of synthetic proximal promoters out of random combinations of three to ten CREs naturally found in constitutive plant, plant pathogen, and viral promoters, including AtACT2, AtUBQ10, AtUBC, 35S, A. tumefaciens NOS, and the Mirabilis Mosaic Virus MMV promoters [98]. The CREs were subcloned in small tandems upstream of the TATA box and sandwiched between a 19bp degenerate sequence upstream and a 43bp degenerate sequence downstream of the CRE-TATA combination (Figure 3b). The strength of a series of these “MynSyn” promoters was evaluated in transiently transformed Arabidopsis protoplasts via dual luciferase assays. Different CRE arrangements, spacing, and the distance from the TSS were tested [98]. While altering the relative positions and spacing between CREs did not have any major effects on reporter gene expression, moving CREs to more distal locations away from TATA, i.e. over 50 bp upstream (Figure 3b), weakened their effects. Four of the MynSyn promoters were also tested in B. rapa, N. benthamiana and H. vulgare protoplasts, with the promoters displaying comparable relative activities in all dicots, but little to no expression was detected in the monocot H. vulgare. Stably transformed Arabidopsis plants were also generated and MynSyn promoter activity was said to have been detected in most tissues, but no detailed tissue distribution analysis was reported [98].
Jores et al. [99] utilized STARR-seq to test the effect of the enhancer region from the proximal 35S promoter on reporter expression. The enhancers were placed individually in different locations with respect to GFP (upstream, within the coding region, or downstream, Figure 3c) and the effect of that position on GFP mRNA accumulation was examined via RNA-seq of agroinfiltrated tobacco leaves, with upstream enhancer position giving the strongest expression and internal position eliminating the effect of the enhancer [99]. Mutant versions of the 35S core promoter and 35S enhancer (all of their possible nucleotide variants) were also screened for activity via STARR-seq, with mutations in the TATA box and in previously defined A, B and C 35S enhancer regions (Figure 3c) that harbor CREs for ERF, TCP, NAC, GATA, bHLH and bZIP TF families having the most detrimental effects. Reshuffling of the enhancer A, B, and C regions and placing no, one, two or all three domains in random positions was also performed [99]. Notably, none of the combinations achieved the same level of GFP activation as the natural 35S enhancer, suggesting that the intervening sequences and/or the exact spacing between the A, B and C regions are important for the enhancer’s maximum activity [99], a key finding that may be of relevance to the rational combining of tissue-specific CREs in synthetic promoters.
Several exciting studies have implemented orthogonal transcription regulation systems in plants using synthetic activators and repressors acting upon synthetic proximal promoters harboring combinations of respective CREs. Although some of these studies did not involve tissue-specific regulation of synthetic promoters, they either illuminated critical limitations of the designs and experimental systems or shed light on the basic structural rules of promoter activity, thus providing a roadmap for constructing and testing different types of synthetic regulators and promoters in the future. Schaumburg et al. [100] employed chemically inducible synthetic TFs comprised of yeast or bacterial DNA-binding domains (DBDs) and Arabidopsis transcriptional repression domains to tune down natural constitutive promoters (35S, figwort mosaic virus (FMV) and nopaline synthase (NOS)) retrofitted with tandems of 2 to 8 copies of GAL4- and LexA-targeted CREs positioned upstream of the proximal promoter, upstream of TATA-box-containing core promoter, or downstream of the TSS (Figure 3d). Using dual Firefly and Renilla Luciferase as the readouts of input repressor levels and output target promoter activities, inferences about circuit functionality could be made for only 42 out of 128 genetic circuits tested in Arabidopsis protoplasts, indicating that many of the designs did not work as intended and suggesting that, going forward, multiple versions of each circuit component need to be tested to identify those that meet the designer’s needs [100]. For example, only some DBD-repression domain combinations were functional, and those that worked were effective with only some native promoter scaffolds. Importantly, the variability between different batches of protoplasts was often greater than the differences between different constructs. Although statistical data normalization could correct for some of the variability in the data, and TATA-proximal location for CREs (Figure 3d) was found to be the most effective for constitutive promoter repression, solid conclusions could not be drawn for many of the circuit designs, including inferring the optimal CRE spacing or the most effective TF-promoter combinations. Similar variability issues were encountered with sorghum protoplasts, highlighting the limitations of protoplast transient expression systems for these types of quantitative analyses [100].
Belcher et al. [101] chose to exploit transient assays in tobacco to test several synthetic TFs made from yeast DNA binding domains fused to viral or plant transcription activation or repression domains. These TFs targeted synthetic promoters composed of native yeast CRE variants stacked in sets of five upstream of one of 29 native plant minimal promoters driving fluorescent reporters or histochemical marker genes (Figure 3e). Importantly, tissue-specific or stimulus-regulated behavior of synthetic reporters was achieved in stable Arabidopsis transformants upon placing the artificial TFs under the control of native seed-specific (At2S3) or phosphorus-deficiency-triggered (AtPht1.1) plant promoters [101]. Furthermore, by expressing the reporters from synthetic hybrid promoters assembled from combinations of CREs recognized by multiple synthetic TFs, simple Boolean logic circuits were successfully generated [101]. These genetic devices can perform basic logical operations by integrating the input signals from several TFs to regulate the expression of the output gene of interest in a pre-defined manner, turning the target gene “on” or “off” dependent on the presence or absence of CREs and their cognate TFs. Belcher et al. built the OR (with CREs for two positive TFs) and NOR (with CREs for two negative TFs) logic gates, as well as a killer switch (with CREs for both a positive and a negative synthetic TF) [101], thus providing a clear path toward the future design of complex promoters where tissue and stimulus specificity could be combined to create novel expression patterns with the complexity typically seen in native promoters.
Brophy et al. in their groundbreaking work went one step further and successfully conferred refined patterns of gene expression to synthetic promoters in plants by placing simple genetic circuits that recapitulate basic YES, NOT, OR, NOR, AND, NAND, IMPLY and NIMPLY logic under the control of synthetic TFs driven by native promoters [102]. First, a series of ten synthetic TFs was built from each of ten different bacterial DBDs fused to a viral transcription activation domain and a nuclear localization signal (NLS). These TFs were then tested in transient assays in tobacco co-infiltrated with respective synthetic promoter constructs that stack six identical CREs for one of these synthetic transcription factors upstream of a 35S minimal promoter driving GFP (Figure 3f). A 3- to 45-fold GFP reporter activation was observed for nine out of ten synthetic TFs, with most constructs showing no cross-reactivity against non-cognate CREs [102]. The same ten bacterial DBDs were also turned into transcriptional repressors by fusion to the NLS tag alone and used in combination with a constitutively expressed 35S:GFP reporter harboring a single CRE downstream of the TSS (Figure 3g), presumably, to interfere with transcription elongation. Five out of ten of these constructs led to the reporter downregulation, with up to 13-fold repression achieved. With activator constructs, reducing the number of CREs in the target promoter lowered the GFP induction (Figure 3h). With repressor constructs, placing the CRE upstream rather than downstream of the TSS further increased the level of constitutive promoter repression achieved, and stacking of two CREs upstream of the TSS made the promoter downregulation even stronger, reaching 64-fold (as compared to just 1.5-fold repression with a single downstream CRE) (Figure 3i) [102].
Once the optimal components were determined, Brophy et al. successfully implemented the aforementioned Boolean logic circuits in tobacco transient assays [102]. Importantly, several of the designs, including the AND gate (Figure 3j), contain synthetic promoters composed of CREs for both positive and negative TFs, more closely mimicking the structure of native promoters. A set of nine logic devices was also built for stable Arabidopsis transformants, with two tissue-specific promoters, root-cap specific SOMBRERO and columella- and stele-expressed PIN4, employed to drive synthetic TFs. Finally, several versions of a simple BUFFER gate were implemented in Arabidopsis, where a lateral root cell-specific GATA23 promoter drives the expression of a synthetic positive TF that activates the stabilized dominant version of an Aux/IAA gene SLR1 via one to six CREs in its proximal promoter region. Both the control GATA23p:slr1 fusion and all buffer gates resulted in the dominant suppression of lateral root development. To tune the expression of slr1 down, point mutations were introduced into the CREs, with resulting transgenic plants showing partial lateral root suppression [102]. Thus, the strength of the output gene in logic devices can be not only turned up by stacking multiple copies of CREs, but also down by mutating CREs and thus reducing their ability to recruit their respective TFs.
The aforementioned proof-of-concept plant studies shed light on some of the foundational rules of core and proximal promoter structure and explore different ways to harness native promoters to express synthetic TFs to in turn control synthetic promoter activity. However, none of these reports leverages native tissue-specific TFs to directly control custom-built synthetic promoters made of cognate CREs in order to achieve desired patterns of activity. No systematic high-throughput efforts to build such tissue-specific promoters from scratch have been reported for plants. However, this has been done in some animal systems. In one noteworthy zebrafish study, Smith et al. compacted a library of all possible 6bp DNA sequences (4096 in total) into 184 unique synthetic 15-mer elements and cloned them upstream of a 42bp viral TATA-box-containing E1b minimal promoter driving GFP (Figure 3g) [103]. The 184 constructs were then individually tested in vivo for their ability to support GFP expression in zebrafish embryos. Interestingly, 11 of these constructs were expressed in only one of the 15 zebrafish tissues evaluated in the study [103], with four of them investigated in more detail. Three of the four 15-mers maintained their very specific expression pattern even after their minimal promoter was swapped, or when the sequences were trimmed to 9bp. Not surprisingly, concatenation of five identical copies of the 15-mer elements into tandems (Figure 3g) enhanced expression levels and maintained the original tissue-specific GFP expression patterns [103]. Likewise, concomitant expression of two constructs with different tandems resulted in additive patterns of expression. Unexpectedly, when two or three different tandems were combined into a single promoter, the tissue-specific expression of the reporters was largely abolished, highlighting the importance of relative positions (e.g., distance from the minimal promoter) and/or local sequence environment surrounding the putative regulatory elements for their proper activity [103]. Although this finding implies the possible difficulty of generating promoters with desired patterns of expression when combining individually characterized DNA elements, it also suggests that the entire 6bp-long CRE diversity can be screened via a manageable number of constructs. In plants, working with 184 constructs stably transformed into Arabidopsis may be very laborious given the need to analyze several transgenic lines for each construct. Transient expression in protoplasts, tobacco leaf epidermis, or Arabidopsis seedlings is limited to only some tissues and is impractical for the types of work aiming to identify cell-specific elements. On the other hand, multi-cellular transient transformation systems such as hairy roots [104] may be more appropriate.
The obvious limitation of the zebrafish study by Smith et al. [103] is that the 6bp putative CREs are likely to harbor only half-sites for a majority of dimeric TFs, so the 184-construct design is probably limiting. The 15-mers used for sequence compaction likely favored heterodimeric TF binding but excluded homodimer recruitment. On the other hand, increasing the size of putative CREs to accommodate possible dimers increases the number of constructs that need to be generated and screened. For example, in one animal study that aimed to test a library of all possible 10-mer DNA sequences, 52,429 100bp-long 10-copy homomeric tandems had to be evaluated [105]. These were placed upstream of a viral minimal promoter driving GFP (Figure 3h) and tested for activity in HeLa and five other types of mammalian cell lines using FACS. Although the goal of that study was to identify strong constitutive promoters, it became clear that no sequence tandem worked equivalently well in all cell types, suggesting some level of tissue specificity of all expressed synthetic promoter tandems [105]. In plants, given the scarcity of stable cell lines, such a design would probably not be practical, especially for the systematic evaluation of cell-specific expression.
Concluding remarks
The molecular underpinnings of tissue specificity in plants remain largely unexplored. Deciphering the rules of nature and key transcriptional and epigenetics mechanisms will be critical to our understanding of the fundamentals of plant biology and to our ability to harness the power of plants for developing new, more resilient, higher-yielding crop varieties. Early efforts in the area of plant synthetic biology show promise for both the discovery of the basic principles and for developing practical applications. High-throughput assays capable of linking candidate CREs with expression levels and patterns will be critical for training predictive ML models that could in turn be used to inform and accelerate the design-build-test cycle characteristic of synthetic biology approaches. Future investments in these areas are expected to provide the much-needed insights into the molecular mechanisms underlying tissue-specific gene activity.
Acknowledgement
The work in the Alonso-Stepanova lab is supported by the National Science Foundation grants 1444561 and 1940829 to JMA and ANS, and 1750006 to ANS. AEY is a recipient of the Genetics and Genomics Scholars fellowship from NC State University and of Molecular Biotechnology Training Program grant from the National Institutes of Health.
Footnotes
Conflict of interest statement
Nothing declared.
CRediT authorship contribution statement
Anna E. Yaschenko, Mario Fenech, Serina Mazzoni-Putman: Conceptualization, Writing – original draft, Visualization; Jose M. Alonso: Conceptualization, Writing – review & editing; Anna N. Stepanova: Conceptualization, Writing – original draft, Writing – review & editing, Visualization.
References and recommended reading
Papers of particular interest have been highlighted as:
* of special interest
** of outstanding interest
- [1].Riechmann JL: Transcriptional Regulation: a Genomic Overview. The Arabidopsis book 2002, 1: e0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kim J: Multifaceted Chromatin Structure and Transcription Changes in Plant Stress Response. Int. J. Mol. Sci 2021, 22: 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Klepikova AV, Kasianov AS, Gerasimov ES, Logacheva MD, Penin AA: A High Resolution Map of the Arabidopsis thaliana Developmental Transcriptome Based on RNA-seq Profiling. Plant J. 2016, 88: 1058–1070. [DOI] [PubMed] [Google Scholar]
- [4].Xu YZ, Kanagaratham C, Jancik S, Radzioch D: Promoter Deletion Analysis Using a Dual-Luciferase Reporter System. Methods Mol. Biol 2013, 977: 79–93. [DOI] [PubMed] [Google Scholar]
- [5].Rodríguez-Leal D, Lemmon ZH, Man J, Bartlett ME, Lippman ZB: Engineering Quantitative Trait Variation for Crop Improvement by Genome Editing. Cell 2017, 171: 470–480.e8. [DOI] [PubMed] [Google Scholar]; ** In vivo editing of CREs in Solanum lycopersicum CLV3 resembles the domestication process in tomato plants using guide RNA-directed gene editing by CRISPR/Cas9.
- [6].Huang D, Kosentka PZ, Liu W: Synthetic Biology Approaches in Regulation of Targeted Gene Expression. Curr. Opin. Plant Biol 2021, 63: 102036. [DOI] [PubMed] [Google Scholar]
- [7].Ali S, Kim WC: A Fruitful Decade Using Synthetic Promoters in the Improvement of Transgenic Plants. Front. Plant. Sci 2019, 10: 1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Franco-Zorrilla JM, López-Vidriero I, Carrasco JL, Godoy M, Vera P, Solano R: DNA-Binding Specificities of Plant Transcription Factors and Their Potential to Define Target Genes. Proc. Natl. Acad. Sci. U.S.A 2014, 111: 2367–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].ELMAYAN T, TEPFER M: Evaluation in Tobacco of the Organ Specificity and Strength of the rolD Promoter, Domain A of the 35S promoter and the 35S2 promoter. Transgenic Res. 1995, 4: 388–396. [DOI] [PubMed] [Google Scholar]
- [10].Mohan C, Jayanarayanan AN, Narayanan S: Construction of a Novel Synthetic Root-Specific Promoter and Its Characterization in Transgenic Tobacco Plants. 3 Biotech 2017, 7: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang R, Zhu M, Ye R, Liu Z, Zhou F, Chen H, Lin Y: Novel Green Tissue-Specific Synthetic Promoters and cis-Regulatory Elements in Rice. Sci. Rep 2015, 5: 18256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nelson T, Tausta SL, Gandotra N, Liu T: Laser Microdissection of Plant Tissue: What You See Is What You Get. Annu. Rev. Plant Biol 2006, 57: 181–201. [DOI] [PubMed] [Google Scholar]
- [13].Barcala M, Fenoll C, Escobar C: Laser Microdissection of Cells and Isolation of High-Quality RNA After Cryosectioning. Methods Mol. Biol 2021, 2170: 35–43. [DOI] [PubMed] [Google Scholar]
- [14].Olsen S, Krause K: A Rapid Preparation Procedure for Laser Microdissection-Mediated Harvest of Plant Tissues for Gene Expression Analysis. Plant Methods 2019, 15: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bonner WA, Hulett HR, Sweet RG, Herzenberg LA: Fluorescence Activated Cell Sorting. Rev. Sci. Instrum 1972, 43: 404–409. [DOI] [PubMed] [Google Scholar]
- [16].Herzenberg LA, Sweet RG, Herzenberg LA: Fluorescence-Activated Cell Sorting. Sci. Am 1976, 234: 108–117. [DOI] [PubMed] [Google Scholar]
- [17].Birnbaum K, Jung JW, Wang JY, Lambert GM, Hirst JA, Galbraith DW, Benfey PN: Cell Type-Specific Expression Profiling in Plants via Cell Sorting of Protoplasts from Fluorescent Reporter Lines. Nat. Methods 2005, 2: 615–619. [DOI] [PubMed] [Google Scholar]
- [18].Benfey PN, Galbraith DW, Lambert GM, Jung JW, Wang JY, Shasha DE, Birnbaum K: A Gene Expression Map of the Arabidopsis Root. Science 2003, 302: 1956–1960. [DOI] [PubMed] [Google Scholar]
- [19].Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. : mRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6: 377–382. [DOI] [PubMed] [Google Scholar]
- [20].Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW: Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 2015, 161: 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]; * A strategy to barcode and sequence individual cells’ mRNA and group cells with similar transcriptional profiles was developed, giving rise to high-throughput scRNA-seq.
- [21].Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. : Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161: 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]; * In a race to achieve high-throughput single-cell transcriptome profiling, this group designed an alternative strategy to that developed by Klein et al. [20] to perform high-throughput scRNA-seq.
- [22].Picelli S: Single-Cell RNA-Sequencing: The Future of Genome Biology Is Now. RNA Biol. 2017, 14: 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ryu KH, Huang L, Kang HM, Schiefelbein J: Single-Cell RNA Sequencing Resolves Molecular Relationships Among Individual Plant Cells. Plant Physiol. 2019, 179: 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Farmer A, Thibivilliers S, Ryu KH, Schiefelbein J, Libault M: Single-Nucleus RNA and ATAC Sequencing Reveals the Impact of Chromatin Accessibility on Gene Expression in Arabidopsis Roots at the Single-Cell Level. Mol. Plant 2021, 14: 372–383. [DOI] [PubMed] [Google Scholar]; ** From integrating transcriptomics and chromatin accessibility data at the single-cell level, chromatin accessibility was concluded to be a critical determinant of cell identity in Arabidopsis roots.
- [25].Nelms B, Walbot V: Defining the Developmental Program Leading to Meiosis in Maize. Science 2019, 364: 52–56. [DOI] [PubMed] [Google Scholar]
- [26].Satterlee JW, Josh S, Scanlon MJ: Plant Stem-Cell Organization and Differentiation at Single-Cell Resolution. Proc. Natl. Acad. Sci. U.S.A 2020, 117: 33689–33699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu Q, Liang Z, Feng D, Jiang S, Wang Y, Du Z, Li R, Hu G, Zhang P, Ma Y, et al. : Transcriptional Landscape of Rice Roots at the Single-Cell Resolution. Mol. Plant 2021, 14: 384–394. [DOI] [PubMed] [Google Scholar]
- [28].Zhang TQ, Chen Y, Liu Y, Lin WH, Wang JW: Single-Cell Transcriptome Atlas and Chromatin Accessibility Landscape Reveal Differentiation Trajectories in the Rice Root. Nat. Commun 2021, 12: 2053–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang Y, Huan Q, Li K, Qian W: Single-Cell Transcriptome Atlas of the Leaf and Root of Rice Seedlings. J. Genet. Genomics 2021, 48: 881–898. [DOI] [PubMed] [Google Scholar]
- [30].Schmitz RJ, Grotewold E, Stam M: Cis-Regulatory Sequences in Plants: Their Importance, Discovery, and Future Challenges. Plant Cell 2022, 34: 718–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. : Genome-Wide Location and Function of DNA Binding Proteins. Science 2000, 290: 2306–2309. [DOI] [PubMed] [Google Scholar]
- [32].Johnson DS, Mortazavi A, Myers RM, Wold B: Genome-Wide Mapping of in vivo Protein-DNA Interactions. Science 2007, 316: 1497–1502. [DOI] [PubMed] [Google Scholar]
- [33].Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. : Genome-Wide Profiles of STAT1 DNA Association Using Chromatin Immunoprecipitation and Massively Parallel Sequencing. Nat. Methods 2007, 4: 651–657. [DOI] [PubMed] [Google Scholar]
- [34].Bartlett A, O’Malley RC, Huang SC, Galli M, Nery JR, Gallavotti A, Ecker JR: Mapping Genome-Wide Transcription-Factor Binding Sites Using DAP-Seq. Nat. Protoc 2017, 12: 1659–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Steensel Bv, Henikoff S: Identification of in vivo DNA Targets of Chromatin Proteins Using Tethered Dam Methyltransferase. Nat. Biotechnol 2000, 18: 424–428. [DOI] [PubMed] [Google Scholar]
- [36].Germann S, Juul-Jensen T, Letarnec B, Gaudin V: DamI D, a New Tool for Studying Plant Chromatin Profiling in vivo, and Its Use to Identify Putative LHP1 Target Loci. Plant J. 2006, 48: 153–163. [DOI] [PubMed] [Google Scholar]
- [37].Schmid M, Durussel T, Laemmli UK: ChIC and ChEC: Genomic Mapping of Chromatin Proteins. Mol. Cell 2004, 16: 147–157. [DOI] [PubMed] [Google Scholar]
- [38].Skene PJ, Henikoff S: An Efficient Targeted Nuclease Strategy for High-Resolution Mapping of DNA Binding Sites. eLife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, Henikoff S: CUT&Tag for Efficient Epigenomic Profiling of Small Samples and Single Sells. Nat. Commun 2019, 10: 1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Leo L, Colonna Romano N: Emerging Single-Cell Technological Approaches to Investigate Chromatin Dynamics and Centromere Regulation in Human Health and Disease. Int. J. Mol. Sci 2021, 22: 8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wolter F, Puchta H: Application of CRISPR/Cas to Understand Cis- and Trans-Regulatory Elements in Plants. Methods Mol. Biol 2018, 1830: 23–40. [DOI] [PubMed] [Google Scholar]
- [42].Li T, Yang X, Yu Y, Si X, Zhai X, Zhang H, Dong W, Gao C, Xu C: Domestication of Wild Tomato is Accelerated by Genome Editing. Nat. Biotechnol 2018, 36: 1160. [DOI] [PubMed] [Google Scholar]
- [43].Wang X, Aguirre L, Rodríguez-Leal D, Hendelman A, Benoit M, Lippman ZB: Dissecting Cis-Regulatory Control of Quantitative Trait Variation in a Plant Stem Cell Circuit. Nat. Plants 2021, 7: 419–427. [DOI] [PubMed] [Google Scholar]
- [44].Swinnen G, Goossens A, Pauwels L: Lessons from Domestication: Targeting Cis-gulatory Elements for Crop Improvement. Trends Plant Sci. 2016, 21: 506–515. [DOI] [PubMed] [Google Scholar]
- [45].Lemmon ZH, Reem NT, Dalrymple J, Soyk S, Swartwood KE, Rodriguez-Leal D, Van Eck J, Lippman ZB: Rapid Improvement of Domestication Traits in an Orphan Crop by Genome Editing. Nat. Plants 2018, 4: 766–770. [DOI] [PubMed] [Google Scholar]
- [46].Lloyd JPB, Lister R: Epigenome Plasticity in Plants. Nat. Rev. Genetics 2022, 23: 55–68. [DOI] [PubMed] [Google Scholar]
- [47].Pei L, Li G, Lindsey K, Zhang X, Wang M: Plant 3D Genomics: the Exploration and Application of Chromatin Organization. New Phytol. 2021, 230: 1772–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kakoulidou I, Avramidou EV, Baránek M, Brunel-Muguet S, Farrona S, Johannes F, Kaiserli E, Lieberman-Lazarovich M, Martinelli F, Mladenov V, et al. : Epigenetics for Crop Improvement in Times of Global Change. Biology (Basel, Switzerland) 2021, 10: 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liang Z, Riaz A, Chachar S, Ding Y, Du H, Gu X: Epigenetic Modifications of mRNA and DNA in Plants. Mol. Plant 2020, 13: 14–30. [DOI] [PubMed] [Google Scholar]
- [50].Kurdyukov S, Bullock M: DNA Methylation Analysis: Choosing the Right Method. Biology (Basel, Switzerland) 2016, 5: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bochtler M, Fernandes H: DNA Adenine Methylation in Eukaryotes: Enzymatic Mark or a Form of DNA Damage? BioEssays 2021, 43: e2000243–n/a. [DOI] [PubMed] [Google Scholar]
- [52].Cole B, Bergmann D, Blaby-Haas C, Blaby IK, Bouchard KE, Brady SM, Ciobanu D, Coleman-Derr D, Leiboff S, Mortimer JC, et al. : Plant Single-Cell Solutions for Energy and the Environment. Commun. Biol 2021, 4: 962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li X, Chen L, Zhang Q, Sun Y, Li Q, Yan J: BRIF-Seq: Bisulfite-Converted Randomly Integrated Fragments Sequencing at the Single-Cell Level. Mol. Plant 2019, 12: 438–446. [DOI] [PubMed] [Google Scholar]; * The authors developed a novel approach to scBS-seq and used it to show that individual maize microspores undergo differential reprogramming of methylation.
- [54].Kawakatsu T, Stuart T, Valdes M, Breakfield N, Schmitz RJ, Nery JR, Urich MA, Han X, Lister R, Benfey PN, Ecker JR: Unique Cell-Type-Specific Patterns of DNA Methylation in the Root Meristem. Nat. Plants 2016, 2: 16058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhou M, Coruh C, Xu G, Martins LM, Bourbousse C, Lambolez A, Law JA: The CLASSY Family Controls Tissue-Specific DNA Methylation Patterns in Arabidopsis. Nat. Commun 2022, 13: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gallego-Bartolomé J, Gardiner J, Liu W, Papikian A, Ghoshal B, Kuo HY, Zhao JM, Segal DJ, Jacobsen SE: Targeted DNA Demethylation of the Arabidopsis Genome Using the Human TET1 Catalytic Domain. Proc. Natl. Acad. Sci. U.S.A 2018, 115: E2125–E2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Papikian A, Liu W, Gallego-Bartolomé J, Jacobsen SE: Site-Specific Manipulation of Arabidopsis Loci Using CRISPR-Cas9 SunTag Systems. Nat. Commun 2019, 10: 729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ghoshal B, Picard CL, Vong B, Feng S, Jacobsen SE: CRISPR-Based Targeting of DNA Methylation in Arabidopsis thaliana by a Bacterial CG-Specific DNA Methyltransferase. Proc. Natl. Acad. Sci. U.S.A 2021, 118: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors utilized CRISPR technology to specifically manipulate the methylation status of the FWA locus to rescue a late-flowering epiallele. The epigenetic change and flowering phenotype were heritable and maintained in the absence of the transgene.
- [59].Decaestecker W, Buono RA, Pfeiffer ML, Vangheluwe N, Jourquin J, Karimi M, Van Isterdael G, Beeckman T, Nowack MK, Jacobs TB: CRISPR-TSKO: A Technique for Efficient Mutagenesis in Specific Cell Types, Tissues, or Organs in Arabidopsis. Plant Cell 2019, 31: 2868–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang X, Ye L, Lyu M, Ursache R, Loytynoja A, Mahonen AP: An Inducible Genome Editing System for Plants. Nat. Plants 2020, 6: 766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chennakesavulu K, Singh H, Trivedi PK, Jain M, Yadav SR: State-of-the-Art in CRISPR Technology and Engineering Drought, Salinity, and Thermo-tolerant crop plants. Plant Cell Rep. 2021. [DOI] [PubMed] [Google Scholar]
- [62].Klein DC, Hainer SJ: Genomic Methods in Profiling DNA Accessibility and Factor Localization. Chromosome Res. 2020, 28: 69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sinha S, Satpathy AT, Zhou W, Ji H, Stratton JA, Jaffer A, Bahlis N, Morrissy S, Biernaskie JA: Profiling Chromatin Accessibility at Single-cell Resolution. Genom. Proteom. Bioinform 2021, 19: 172–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Thibivilliers SB, Anderson DK, Libault MY: Isolation of Plant Nuclei Compatible with Microfluidic Single-nucleus ATAC-sequencing. Bio Protoc. 2021, 11: e4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Dorrity MW, Alexandre CM, Hamm MO, Vigil A, Fields S, Queitsch C, Cuperus JT: The Regulatory Landscape of Arabidopsis thaliana Roots at Single-Cell Resolution. Nat. Commun 2021, 12: 3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Marand AP, Chen Z, Gallavotti A, Schmitz RJ: A Cis-Regulatory Atlas in Maize at Single-Cell Resolution. Cell 2021, 184: 3041–3055.e21. [DOI] [PubMed] [Google Scholar]; ** The authors performed scATAC-seq on six different types of maize tissue to identify cell-type specific CREs and TFs that regulate chromatin interactions.
- [67].Zhao T, Zhan Z, Jiang D: Histone Modifications and Their Regulatory Roles in Plant Development and Environmental Memory. J. Genet. Genomics 2019, 46: 467–476. [DOI] [PubMed] [Google Scholar]
- [68].Lafos M, Kroll P, Hohenstatt ML, Thorpe FL, Clarenz O, Schubert D: Dynamic Regulation of H3K27 Trimethylation during Arabidopsis Differentiation. PLoS Genet. 2011, 7: e1002040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].You Y, Sawikowska A, Neumann M, Posé D, Capovilla G, Langenecker T, Neher RA, Krajewski P, Schmid M: Temporal Dynamics of Gene Expression and Histone Marks at the Arabidopsis Shoot Meristem during Flowering. Nat. Commun 2017, 8: 15120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Deal RB, Henikoff S: Histone Variants and Modifications in Plant Gene Regulation. Curr. Opin. Plant Biol 2011, 14: 116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ikeuchi M, Iwase A, Sugimoto K: Control of Plant Cell Differentiation by Histone Modification and DNA Methylation. Curr. Opin. Plant Biol 2015, 28: 60–67. [DOI] [PubMed] [Google Scholar]
- [72].Lee LR, Wengier DL, Bergmann DC: Cell-Type-Specific Transcriptome and Histone Modification Dynamics during Cellular Reprogramming in the Arabidopsis Stomatal Lineage. Proc. Natl. Acad. Sci. U.S.A 2019, 116: 21914–21924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Grosselin K, Durand A, Marsolier J, Poitou A, Marangoni E, Nemati F, Dahmani A, Lameiras S, Reyal F, Frenoy O, et al. : High-Throughput Single-Cell ChIP-Seq Identifies Heterogeneity of Chromatin States in Breast Cancer. Nat. Genet 2019, 51: 1060–1066. [DOI] [PubMed] [Google Scholar]
- [74].Fal K, Tomkova D, Vachon G, Chaboute ME, Berr A, Carles CC: Chromatin Manipulation and Editing: Challenges, New Technologies and Their Use in Plants. Int. J. Mol. Sci 2021, 22: 10.3390/ijms22020512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, Platt RJ, Scott DA, Church GM, Zhang F: Optical Control of Mammalian Endogenous Transcription and Epigenetic States. Nature 2013, 500: 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Keung AJ, Bashor CJ, Kiriakov S, Collins JJ, Khalil AS: Using Targeted Chromatin Regulators to Engineer Combinatorial and Spatial Transcriptional Regulation. Cell 2014, 158: 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sayers EW, Bolton EE, Brister JR, Canese K, Chan J, Comeau DC, Connor R, Funk K, Kelly C, Kim S, et al. : Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50: D20–D26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zhang H, Zhang F, Yu Y, Feng L, Jia J, Liu B, Li B, Guo H, Zhai J: A Comprehensive Online Database for Exploring ~20,000 Public Arabidopsis RNA-Seq Libraries. Mol. Plant 2020, 13: 1231–1233. [DOI] [PubMed] [Google Scholar]
- [79].Higo K, Ugawa Y, Iwamoto M, Korenaga T: Plant Cis-Acting Regulatory DNA Elements (PLACE) Database: 1999. Nucleic Acids Res. 1999, 27: 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lescot M, Déhais P, Thijs G, Marchal K, Moreau Y, Van de Peer Y, Rouzé P, Rombauts S: PlantCARE, a Database of Plant Cis-Acting Regulatory Elements and a Portal to Tools for in Silico Analysis of Promoter Sequences. Nucleic Acids Res. 2002, 30: 325–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Shahmuradov IA, Gammerman AJ, Hancock JM, Bramley PM, Solovyev VV: PlantProm: a Database of Plant Promoter Sequences. Nucleic Acids Res. 2003, 31: 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chow C, Lee T, Hung Y, Li G, Tseng K, Liu Y, Kuo P, Zheng H, Chang W: PlantPAN3.0: a New and Updated Resource for Reconstructing Transcriptional Regulatory Networks from ChIP-Seq Experiments in Plants. Nucleic Acids Res. 2019, 47: D1155–D1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Solovyev VV, Shahmuradov IA, Salamov AA: Identification of Promoter Regions and Regulatory Sites. In Computational Biology of Transcription Factor Binding. Edited by Ladunga I. Humana Press; 2010: 57–83. [DOI] [PubMed] [Google Scholar]
- [84].Zhang T, Marand AP, Jiang J: PlantDHS: a Database for DNase I Hypersensitive Sites in Plants. Nucleic Acids Res. 2016, 44: D1148–D1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Yilmaz A, Mejia-Guerra M, Kurz K, Liang X, Welch L, Grotewold E: AGRIS: the Arabidopsis Gene Regulatory Information Server, an Update. Nucleic Acids Res. 2011, 39: D1118–D1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Hieno A, Naznin HA, Hyakumachi M, Sakurai T, Tokizawa M, Koyama H, Sato N, Nishiyama T, Hasebe M, Zimmer AD, et al. : ppdb: Plant Promoter Database Version 3.0. Nucleic Acids Res. 2014, 42: D1188–D1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Castro-Mondragon J, Riudavets-Puig R, Rauluseviciute I, Berhanu Lemma R, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Pérez N, et al. : JASPAR 2022: the 9th Release of the Open-Access Database of Transcription Factor Binding Profiles. Nucleic Acids Res. 2021, 50: D165–D173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Moisseyev G, Park K, Cui A, Freitas D, Rajagopal D, Konda AR, Martin-Olenski M, Mcham M, Liu K, Du Q, et al. : RGPDB: Database of Root-Associated Genes and Promoters in Maize, Soybean, and Aorghum. Database: J. Biol. Databases Curation 2020, 2020: baaa038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Che D, Jensen S, Cai L, Liu JS: BEST: Binding-Site Estimation Suite of Tools. Bioinformatics 2005, 21: 2909–2911. [DOI] [PubMed] [Google Scholar]
- [90].Zhang M, Jia C, Li F, Li C, Zhu Y, Akutsu T, Webb GI, Zou Q, Coin LJM, Song J: Critical Assessment of Computational Tools for Prokaryotic and Eukaryotic Promoter Prediction. Brief. Bioinformatics 2022, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Umarov RK, Solovyev VV: Recognition of Prokaryotic and Eukaryotic Promoters using Convolutional Deep Learning Neural Networks. PLOS One 2017, 12: e0171410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wang Y, Zhang P, Guo W, Liu H, Li X, Zhang Q, Du Z, Hu G, Han X, Pu L, et al. : A Deep Learning Approach to Automate Whole-Genome Prediction of Diverse Epigenomic Modifications in Plants. New Phytol. 2021, 232: 880–897. [DOI] [PubMed] [Google Scholar]; * Using publicly available sequencing data, the authors developed and trained a machine learning model to predict epigenetic modifications in plants with >=80% accuracy.
- [93].N’Diaye A, Byrns B, Cory AT, Nilsen KT, Walkowiak S, Sharpe A, Robinson SJ, Pozniak CJ: Machine Learning Analyses of Methylation Profiles Uncovers Tissue-Specific Gene Expression Patterns in Wheat. The Plant Genome 2020, 13: e20027. [DOI] [PubMed] [Google Scholar]; * The authors developed a deep learning algorithm to predict differentially expressed genes across tissues from methylation data with a 0.81 prediction accuracy on a scale of 0 to 1
- [94].Wang H, Cimen E, Singh N, Buckler E: Deep Learning for Plant Genomics and Crop Improvement. Curr. Opin. Plant Biol 2020, 54: 34–41. [DOI] [PubMed] [Google Scholar]
- [95].Wang H, Zhang Y, Cheng Y, Zhou Y, King DC, Taylor J, Chiaromonte F, Kasturi J, Petrykowska H, Gibb B, et al. : Experimental Validation of Predicted Mammalian Erythroid Cis-Regulatory Modules. Genome Res. 2006, 16: 1480–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS: Quantifying Similarity Between Motifs. Genome Biol. 2007, 8: R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Jores T, Tonnies J, Wrightsman T, Buckler ES, Cuperus JT, Fields S, Queitsch C: Synthetic Promoter Designs Enabled by a Comprehensive Analysis of Plant Core Promoters. Nat. Plants 2021, 7: 842–855. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** A collection of synthetic core promoters was developed and evaluated for strength by STARR-seq in transient expression assays.
- [98].Cai Y, Kallam K, Tidd H, Gendarini G, Salzman A, Patron NJ: Rational Design of Minimal Synthetic Promoters for Plants. Nucleic Acids Res. 2020, 48: 11845–11856. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Synthetic proximal promoters were constructed from combinations of native plant, bacterial, and viral CREs and optimal promoter architectures were investigated.
- [99].Jores T, Tonnies J, Dorrity MW, Cuperus JT, Fields S, Queitsch C: Identification of Plant Enhancers and Their Constituent Elements by STARR-seq in Tobacco Leaves. Plant Cell 2020, 32: 2120–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** STARR-seq was employed to study the effect of position and copy number of 35S promoter enhancer regions on gene expression.
- [100].Schaumberg KA, Antunes MS, Kassaw TK, Xu W, Zalewski CS, Medford JI, Prasad A: Quantitative Characterization of Genetic Parts and Circuits for Plant Synthetic Biology. Nat. Methods 2016, 13: 94–100. [DOI] [PubMed] [Google Scholar]
- [101].Belcher MS, Vuu KM, Zhou A, Mansoori N, Agosto Ramos A, Thompson MG, Scheller HV, Loqué D, Shih PM: Design of Orthogonal Regulatory Systems for Modulating Gene Expression in Plants. Nat. Chem. Biol 2020, 16: 857–865. [DOI] [PubMed] [Google Scholar]; ** A collection of orthogonal synthetic plant activators and repressors, along with their cognate synthetic promoters is described.
- [102].Brophy JAN, Magallon KJ, Kniazev K, Dinneny J’R: Synthetic Genetic Circuits Enable Reprogramming of Plant Roots. bioRxiv 2022. [DOI] [PubMed] [Google Scholar]; ** A small library of orthogonal synthetic transcription factors was built and tested in combination with a series of synthetic promoters in transient assays in tobacco. Several man-made genetic circuits that recapitulate Boolean logic were constructed and successfully implemented in stable Arabidopsis transformants.
- [103].Smith RP, Riesenfeld SJ, Holloway AK, Li Q, Murphy KK, Feliciano NM, Orecchia L, Oksenberg N, Pollard KS, Ahituv N: A Compact, in Vivo Screen of All 6-mers Reveals Drivers of Tissue-Specific Expression and Guides Synthetic Regulatory Element Design. Genome Biol. 2013, 14: R72. [DOI] [PMC free article] [PubMed] [Google Scholar]; * A library of 184 synthetic 15-mer DNA elements was screened in zebrafish for tissue-specific activity.
- [104].Gutierrez-Valdes N, Häkkinen ST, Lemasson C, Guillet M, Oksman-Caldentey K, Ritala A, Cardon F: Hairy Root Cultures- a Versatile Tool with Multiple Applications. Front. Plant Sci 2020, 11: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Schlabach MR, Hu JK, Li M, Elledge SJ: Synthetic Design of Strong Promoters. Proc. Natl. Acad. Sci. U.S.A 2010, 107: 2538–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Over 50,000 homomeric sequence tandems were evaluated in mammalian cell cultures by FACS for CRE-like activity in the context of synthetic proximal promoters driving GFP.