

Abstract
Single-cell technologies have become essential in investigative dermatology. Despite the multitude of available datasets, a central reference atlas of normal human skin is still lacking. As part of the Human Cell Atlas (HCA) project, we have assembled a Skin Biological Network to build a Human Skin Cell Atlas (HSCA) and outline a roadmap toward that goal. We define the drivers of skin diversity to be considered, hurdles that impede comprehensive representation, and technical considerations for tissue processing and computational analysis. By outlining our goals for Atlas 1.0, we discuss how it will uncover new aspects of skin biology.
I. Why a Skin Cell Atlas?
Skin contains diverse cell lineages, including epithelial cells of the epidermis and ectodermal appendages—hair follicles, nails, sebaceous glands, and sweat glands—which exist in close association with mesenchymal cell lineages, including smooth muscle cells, adipocytes, and fibroblasts. The latter produce extracellular matrix for mechanical support and signals that guide immune and epithelial cell behavior across both spatial dimensions of the skin (e.g., epidermal differentiation at the surface), and time (e.g., cyclic growth of hair follicles). In addition to the principal skin cell types, there are less abundant cell types essential for skin function, including pigment-producing melanocytes, innate and adaptive immune cells, vascular and perivascular cells, and cells of neuroendocrine origin. Working together, these cell populations form a barrier organ—so large that no individual dataset can sample the entire skin—that plays mechano-protective, UV-shielding, antimicrobial, thermoregulatory functions, and more (Alexander et al., 2015; Donati et al., 2017; Gurtner et al., 2008; Takeo et al., 2015; Watt, 2014). To support these functions, different skin compartments are richly populated by stem cells that respond to insults by mounting reparative responses. As skin heals, such as after wounding, it restores anatomical integrity and functions by forming a scar containing new stable cell states that are distinct from unwounded cell states (Donati et al., 2017; Gurtner et al., 2008; Sun et al., 2022; Takeo et al., 2015).
Several hundred clinically distinct disorders, both monogenic and multifactorial, affect human skin (Feramisco et al., 2009). While many skin diseases are well characterized clinically, histologically, genetically, and by bulk biochemical assays, deep mechanistic understanding remains obscured in part by a lack of characterization at a single-cell resolution. Moreover, numerous distinct diseases have near-identical clinical manifestations (Feramisco et al., 2009; Lamartine, 2003), challenging correct diagnosis and resulting in ineffective therapies. Certain diseases, like psoriasis, are spatially predisposed to appear in certain body regions (Dhabale & Nagpure, 2022). Other diseases, such as facial acne, disproportionately occur at certain ages (Williams et al., 2012), and other diseases occur more commonly in females than males, such as scleroderma (Andersen & Davis, 2016). Diseases such as keloids are more common in certain ancestries (Chike-Obi et al., 2009) and require tissue injury to manifest (Tuan & Nichter, 1998). Developing greater understanding of how different skin regions, age, gender, ancestry, and the wound response affect skin cell types and cell states at the single-cell resolution will enable further understanding of skin disease mechanisms via comparison of pathological states to normal skin at multiple scales.
The Skin Biological Network set a goal to build a consensus HSCA, as part of the HCA (humancellatlas.org) (Regev et al., 2017). This effort builds on previous skin cell atlas work under the auspices of the HCA and newer CZI-supported efforts on pediatric and ancestral skin, as well as the work of individual labs that have been working independently to profile normal human skin using scRNA-seq, to develop a comprehensive atlas of diverse skin cell types across several scales (Figure 1): (a) space, capturing different body sites (Figure 1a); (b) time, capturing maturation states of the skin at the same body site (e.g., facial skin during prenatal development, childhood, adolescence, adulthood and advanced age) (Figure 1b); (c) gender scale, capturing major sexually dimorphic skin regions and changes associated with puberty (Figure 1c); (d) ancestry, capturing cutaneous anatomical features that prominently vary across ancestries, such as skin pigmentation, sweat gland and hair follicle differences (Figure 1d); (e) wound response scale, capturing new cell states of healed skin, i.e., scars, vs. unwounded skin (Figure 1e).
Figure 1. The many scales of biological variation in skin that we predict will have a significant effect on transcriptional heterogeneity.
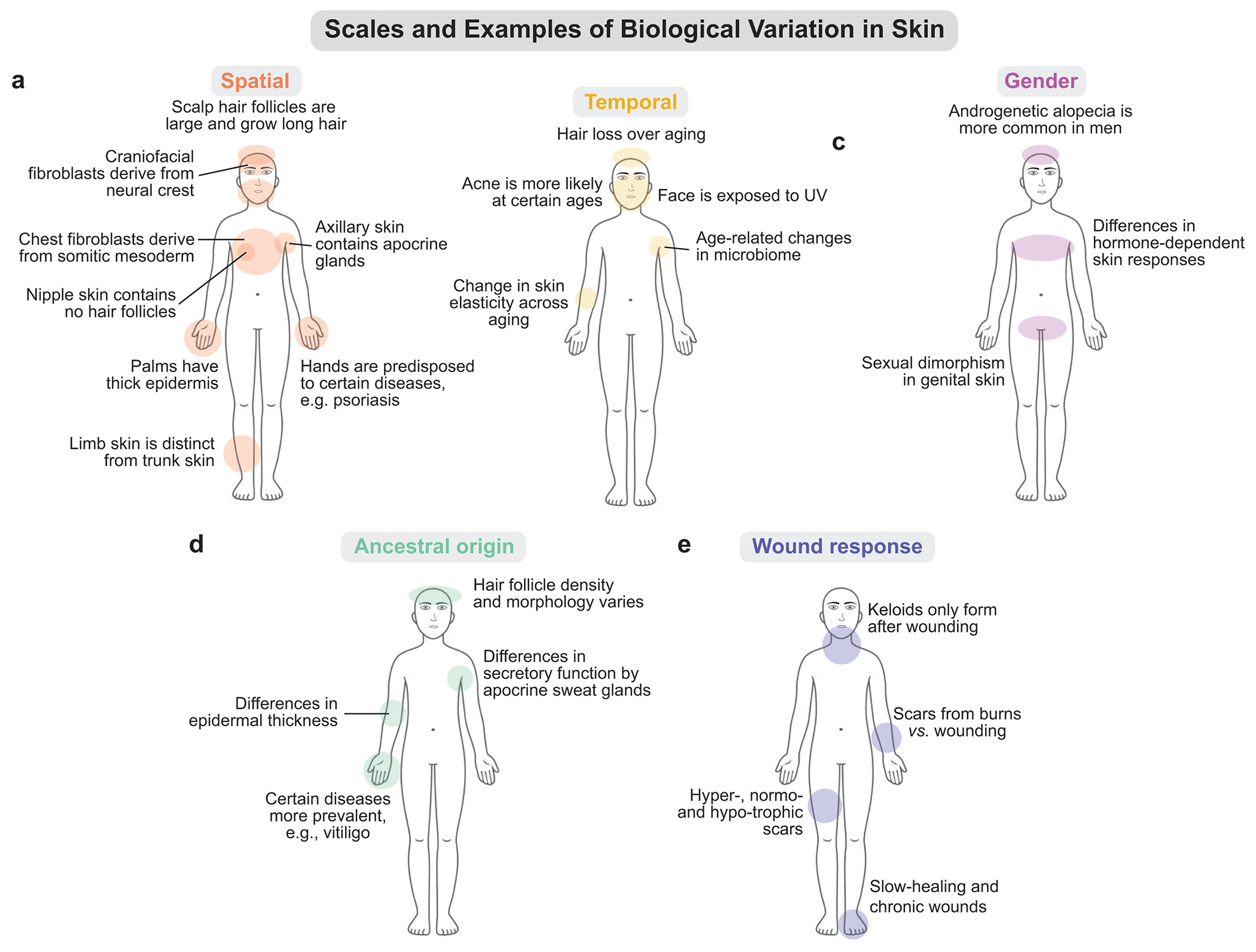
a) The spatial scale represents how skin composition and characteristics vary across anatomical regions. b) The temporal scale represents changes to skin across the human lifespan. c) The gender scale represents gender-specific differences between skin sites and function. d) The ancestral origin scale, which affects skin characteristics and proclivity to disease. e) The wound response scale, where unwounded skin is distinct from skin that is permanently altered after the innate, acute wound repair program. For each scale, we include notable examples that illustrate why these factors need to be considered when generating a high-quality single-cell skin atlas.
II. What are Key Considerations for the Human Skin Cell Atlas?
II.a. Aspects Contributing to Physiological Diversity of the Skin
To represent the full anatomical and functional diversity of skin at a single-cell level, several factors must be considered. First, skin from different body sites can have different embryonic origins. For example, dermal fibroblasts in frontal scalp skin arise from the craniofacial mesoderm, whereas fibroblasts in the chest skin originate from the somitic mesoderm (Thulabandu et al., 2018). Second, different anatomical skin sites have different dominant features. For example, eyelids have a thin epidermis, while palms and feet have significantly thicker epidermis (Sandby-Møller et al., 2003). Skin sites also vary by hair follicle size, density, and growth cycle parameters: scalp skin has large (i.e., terminal) hair follicles that produce long hairs continuously over several years, while adjacent forehead skin features diminutive (i.e., vellus) hair follicles that grow short, thin hairs (Vogt et al., 2007); and areola, lip skin, and palmoplantar skin are largely devoid of hair follicles (Stone & Wheeler, 2015; Tsai et al., 2022). Third, the skin microbiome profile varies significantly across distinct body sites and correlates with each site’s physical properties (Byrd et al., 2018); that is, whether the skin is moist (e.g., axillary or popliteal fossa skin), dry (e.g., volar forearm skin), or oily (e.g., forehead skin). Fourth, UV exposure has a prominent physiological effect on skin; its extent and history naturally differ across anatomical space and are further impacted by social dress codes and skin color (Matsumura & Ananthaswamy, 2004). For instance, sun-exposed forehead skin commonly experiences high UV exposure compared to typically clothed buttock skin. Fifth, many skin sites, particularly facial, scalp, axillary, chest and pubic skin, undergo prominent age-dependent changes (Farage et al., 2013; Haydont et al., 2019) and differ between genders (Dao & Kazin, 2007). Finally, given the inherent anatomical and physiological diversity across human populations, single-cell datasets from genetically diverse ancestries should be included to generate a comprehensive atlas. Priority should be placed on ancestral groups that are underrepresented in biomedical research, including but not limited to African, Asian, Hispanic, and Middle Eastern populations (Hirano et al., 2012; M. A. Ma et al., 2021). Further priority should be placed towards skin sites with prominent morphological differences across ancestral groups, such as scalp skin, where there is variation in hair follicle density and hair morphology, or axillary skin, where there are differences in apocrine sweat gland function (Luther et al., 2012) and propensity to disease (Kilgour et al., 2021).
II.b. Practical Considerations for Sample Collection
Sample acquisition is easier for certain body sites and states (e.g. aged vs. younger skin), and unless proactively countered, knowledge gaps in skin-wide data will persist (Figure 2). Fresh skin samples can be acquired in two main ways. The first is from discarded tissue that can be collected during certain routine surgical procedures, including: (i) blepharoplasty (upper eyelid skin), (ii) facelifts (temporal, frontal and parotid facial skin); (iii) abdominoplasty (hypogastic abdominal skin); (iv) mammoplasty (chest and areola skin); (v) hair transplantation (occipital scalp skin). As plastic surgery procedures commonly remove old scars (e.g., C-section scars during abdominoplasty), scar tissue can also be readily obtained. Second, normal skin samples can be obtained through purposeful biopsies from healthy volunteers. For certain skin sites, including trunk skin (e.g., from upper buttocks region) and the extremities (e.g., from thighs or forearms), obtaining biopsies will typically result in minimal morbidity and is well tolerated. Other skin sites, such as genital skin and axillary skin, can rarely be sampled without resulting in significant morbidity, and will require specialized arrangements.
Figure 2. Summary of current skin scRNA-seq datasets in the literature.
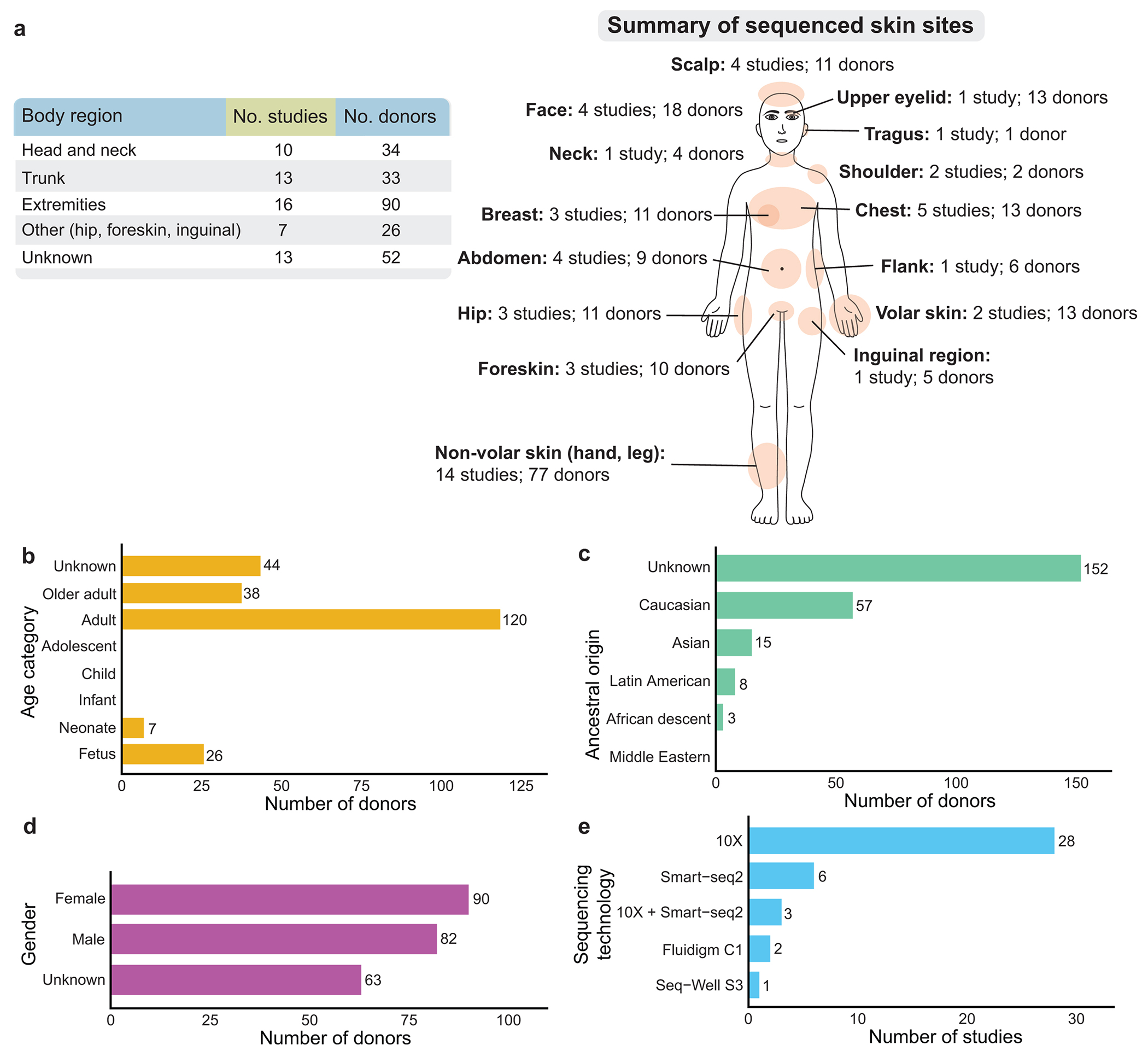
a) A summary of the number of studies and skin donors per major body region. Head and neck sites include scalp, face, upper eyelid, neck, and tragus. Trunk sites include shoulder, chest, abdomen, breast, and flank. Extremity sites include both volar skin (palm and sole) and non-volar (hand and leg) skin. b–e) The number of skin donors summarized with respect to b) age category, c) ancestral origin, d) gender, and e) sequencing technology. There are numerous gaps in the literature that can be filled when constructing new data for Atlas 1.0.
III. Distinct Challenges
III.a. Sampling Skin to Saturation
To date, predominantly fresh skin tissue has resulted in high-quality single-cell skin transcriptomes. For the reasons discussed above, many anatomical skin sites will be generally precluded from sampling, leading to under-representation of certain skin regions and their distinct anatomies and functions in Atlas 1.0. Specific efforts will need to be undertaken to minimize anticipated data gaps. One possible solution could be greater use of post-mortem tissues, albeit with the additional challenge that these tissues must be as fresh as possible to obtain high-quality data.
III.b. Global Representation of Human Skin Heterogeneity
A representative skin cell atlas must reflect the entire human population. The desire to include diverse populations and those typically underrepresented in clinical research, however, is met with multiple challenges (Mapes et al., 2020; Swartz et al., 2019) that should be addressed with directed community-engagement approaches (Borthwick et al., 2023; Holzer et al., 2014). First, low income as a barrier to participation. Indeed, low-income individuals are often excluded from clinical research, partly because individuals seen at locations that serve low-income patients are often not informed about potential opportunities for clinical trial participation, nor may they be able to afford surgery. Second, language and literacy can be a barrier to participation (Nicholson et al., 2015). Low reading literacy can make reliance on written recruitment materials ineffective. This can be partly aided through translators or by including native speakers on the research team, but cultural and language tailoring must go deeper. One should seek help from cultural and language-concordant research facilitators who can support bidirectional communication and knowledge transmission between researchers, participants, and their communities. Third, mistrust is a barrier to participation. A long history of medical and scientific exploitation has targeted and adversely affected diverse groups of people, creating perceptions of mistrust towards biomedical research. One critical way to combat mistrust is through clear communication and transparency by making a conscious effort to ensure that research materials (i.e., informed consent documents, study results, etc.) are designed in ways that promote clear understanding of the research questions, study design, participant protections, and potential community benefit (Day et al., 2020).
We note that this section specifically refers to collecting skin samples from underrepresented groups in the U.S.A., but, more broadly, researchers worldwide should be recruited to obtain these underrepresented samples. We propose to recruit participants via “deep” engagement with the local community, when the benefits of participation in such research are clearly explained and “all questions” are answered beforehand. One must work closely with community partners to understand not only why people may be interested in participating in skin research, but also to ensure that all recruitment materials and consenting documents are easy to understand and facilitate individuals making informed consents.
Participants of these studies should certainly receive monetary compensation for their time, effort, and for study-related expenses. For example, researchers must recognize that being given time off from work (e.g., sick or vacation) is a benefit that not every employer provides, so if under-resourced people are to be included in research, appropriate compensation must be provided.
III.c. Comprehensive Cell Type Analysis
When collecting representative skin data, both tissue handling and digestion protocols significantly impact the efficiency of cell isolation and viability, with uneven effects across cell types. For example, trimming adipose tissue prior to cell isolation leads to underrepresentation of cell types residing in the dermal adipose layer. Adipose minimization artifacts and loss of full-depth anagen hair follicle cell coverage also happens when skin is not biopsied at full thickness. Different skin microstructures demand different enzymatic dissociation protocols, making balanced isolation of viable cells from the epidermis, dermis, and appendages challenging. For example, dermal papilla fibroblasts from anagen scalp hair follicles are difficult to isolate because they are encased in a compact extracellular matrix and further enveloped by the epithelial matrix. While some dissociation protocols preferentially yield epithelial cells, others are better optimized for cells from the dermis. Other factors affecting cell type coverage include: (i) whether the dataset was sequenced from fresh vs. frozen tissue; (ii) using a live-cell or nuclei-based protocol; (iii) whether there was cell type enrichment prior to profiling; or (iv) whether the platform is plate- or droplet-based. Thus, each dataset included in the atlas should be accompanied by a reference and/or detailed documentation, including biological and experimental metadata and the used cell isolation method.
III.d. Balanced Batch Effect Removal
A representative skin cell atlas requires integration of samples from different body sites that exhibit both commonalities and substantial differences in micro-anatomies, functions, and gene expression signatures. For example, skin also shares many essential anatomical and functional similarities across different sites, such as the presence of an outer epidermal barrier, an elastic and tear-resistant dermis, a dynamic immune niche, or hair growth. In contrast, axillary and breast-associated skin have hormone-rich signaling environments that may result in body site-specific cell sub-clusters. Unlike laboratory mouse models, scRNA-seq data sampled from human individuals exhibit significant batch effects, even when sampled from the same anatomical site. Variation in cell composition and gene expression can occur due to differences in individual factors, such as sex, age, ancestral origin, and lifestyle factors (e.g., UV exposure, environmental pollution, and nutritional preferences), but also due to differences in technical factors, such as sampling method, dissociation protocol, or library preparation protocol. These batch effects must be accounted for and, if possible, corrected to ensure they do not mask genuine biological differences. The central challenge in constructing a skin cell atlas is that differences in gene expression patterns between samples may consist of both batch effects and meaningful biological differences. To integrate these datasets, we must remove batch effects from the data while retaining “biological variation,” i.e., to preserve both transcriptional similarities of common cell lineages from different body sites and significant axes of biological variation within a lineage, that may reflect cell type adaptation to a particular spatial niche. For example, hair-follicle keratinocytes from the scalp are expected to be more like hair follicle keratinocytes from the trunk due to their hair producing functions, rather than barrier-forming epidermal keratinocytes from the scalp. Therefore, the HSCA requires computational methodologies that strike the right balance between these two important yet diametrically opposed technical challenges of “biological conservation” and “batch correction” (Luecken et al., 2022). Additionally, it is important to integrate multiple datasets from each major axis of biological variation that we aim to represent in the atlas. For example, to show biological variation across different body sites, we must have at least two donors per site. This allows for validation that batch effects were removed (e.g., cells from different datasets per site are well mixed), while biological differences are preserved (e.g., cells sampled from different locations do not mix).
Despite the plethora of methods developed to integrate multiple scRNA-seq datasets, there is yet no “universal method” (Chazarra-Gil et al., 2021; Luecken et al., 2022; Thi et al., 2020). Some methods, e.g., fastMNN (Haghverdi et al., 2018), scANVI (Xu et al., 2021), and scGen (Lotfollahi et al., 2019), are more effective at ensuring biological conservation, at the risk of retaining some batch effects, while others, e.g., BBKNN (Polański et al., 2020), Scanorama (Hie et al., 2019), and trVAE (Lotfollahi et al., 2020), are more effective at batch correction at the risk of obscuring biological variation (Luecken et al., 2022). What is the best way to identify the most suitable method for a given dataset and intention? One strategy has been to test several methods and assess the most appropriate one using metrics that measure biological conservation and batch correction efficacy (Chazarra-Gil et al., 2021; Luecken et al., 2022), which was the adopted strategy used to construct the provisional Human Lung Cell Atlas (Sikkema et al., 2022). To confirm the robustness of cell populations identified after integration, it is important to map novel cell types and cell states that were identified in individual datasets back onto the tissue.
Constructing the atlas will require several “levels” of clustering to identify all major cell types and cell states: first, to identify the cell lineages present across all skin samples; second, to identify all cell types in a lineage; and third, to identify subtypes and cell states within each cell type. For example, the first round of clustering would identify broad immune cell populations. The second round would identify conventionally recognizable immune cell types like macrophages, T cells, or B cells. The third round of clustering, applied only to T cells, would further discriminate between major subtypes, such as CD4+, CD8+, and natural killer T cells, and possibly characterize new T cell states. Given the major variations in cell state plasticity across skin lineages, it is important to consider these different cell type resolutions. Some lineages may require new integrations at certain levels to better represent cell type heterogeneity. This multi-step integration and clustering analysis may require different batch correction methods at different levels, as has been done in previous integration studies of skin (Reynolds et al., 2021; Solé-Boldo et al., 2020; Zou et al., 2021)
IV. Toward Provisional Skin Cell Atlas
IV.a. Dataset Selection
The major goal set by the Skin Biological Network for the provisional cell atlas version 1.0, denoted as Atlas 1.0, is to generate a consensus nomenclature of cells in healthy human skin, differentiating cell types, their subtypes, and states. The atlas seeks to achieve: (i) high coverage of all major cell types (keratinocytes, fibroblasts, neural crest-derived cells, vessel-associated cells, adipose-associated cells, muscle-associated cells, immune cells); (ii) coverage of all major skin micro-structures (pilosebaceous units, sweat glands, nails, touch domes); and (iii) inclusion of men and women. Datasets that form the basis for Atlas 1.0 will naturally vary across other scales, such as age, body site origin, microbiota association and ancestry. However, these variables will not be the primary focus, since limited sample size will not permit their comprehensive coverage (Figure 2).
Rather than generating scRNA-seq data de novo, the Skin Biological Network will construct a provisional skin cell atlas using previously reported, and publicly available high-quality datasets. To date, we have identified 40 human skin scRNA-seq datasets (Figure 2a, Table S1), which provide sufficient basis to generate Atlas 1.0 and achieve the set Atlas 1.0 goals with reasonable certainty. Integrating data from these studies will produce over 500, 000 skin cells. Several candidate datasets were selected based on their high coverage of the certain cell types, such as melanocytes (Belote et al., 2021), fibroblasts (Solé-Boldo et al., 2020; Tabib et al., 2018; Wiedemann et al., 2023), keratinocytes (Cheng et al., 2018; Wang et al., 2019; Wiedemann et al., 2023), immune cells (Reynolds et al., 2021), and hair follicle cells (Takahashi et al., 2020), while all cells (including underrepresented cell types) of the individual datasets will add cumulative value to the aggregated data. In addition, integration will be performed on datasets that were generated using various cell isolation protocols and sequencing technologies, such as 10X (10X Genomics), Drop-seq (Macosko et al., 2015), Smart-seq2 (Picelli et al., 2013), and Smart-seq3 (Hagemann-Jensen et al., 2020), ensuring that outputs will be robust to major technical variables.
IV.b. Integration Strategy
Atlas 1.0 will be far from exhaustive. New scRNA-studies will continue to emerge and other biological data modalities, like epigenetic (Buenrostro et al., 2013a, 2015a), proteomic (Stoeckius et al., 2017), and spatial transcriptomics (Ståhl et al., 2016), are becoming more readily available. It will become increasingly difficult to continually reintegrate and re-annotate all datasets from scratch. Instead, newer computational approaches based on a “reference and query” strategy of integration have been proposed (Gao et al., 2021a; Hao et al., 2021; Lotfollahi et al., 2022). Methods like Azimuth (Hao et al., 2021), iNMF (Gao et al., 2021b), and scArches (Lotfollahi et al., 2022) perform integration in an iterative fashion by using the latent representation of an initial “reference” atlas, which may be constructed using a simultaneous integration approach. The result is an updated latent representation, which can then serve as the new reference atlas. This iterative strategy is particularly effective when the query datasets represent perturbations due to, for example, disease or gene mutation, and, for instance, was employed to construct the Human Lung Cell Atlas (Sikkema et al., 2022).
There are several benefits to a “sequential” integration approach over conventional “simultaneous” approaches. First, overall computation time is reduced, as integration does not need to be repeated from scratch. Second, with an initial reference atlas, subsequent integration enables more rapid identification of novel cell states present in the query dataset (in the context of the reference data), focusing on biological investigation. Third, novel computational approaches have been developed that are now able to project data from other data modalities, like single-cell ATAC-sequencing (scATAC-seq) (Buenrostro et al., 2013b, 2015b), to the transcriptomic atlas (Cao & Gao, 2022). Such projection methods require using a trained atlas reference model and thus enable building a multimodal atlas even with only few multimodal datasets. However, we note that, eventually, there will be additions so significant, addressing underrepresented or missing body sites, ancestral origins, or age categories, that a simultaneous integration approach will be needed to construct Atlas 2.0 and later versions.
With any integration task, certain axes of variation are inevitably prioritized when constructing the integrated latent embedding. As other underemphasized axes contain valuable information for certain research questions and laboratories, we will ameliorate this “subjectivity” of integration in two ways. First, we will make the individual gene expression matrices and accompanying metadata used for integration available for local analysis through, e.g., Zenodo or the Chan Zuckerberg Initiative’s CELLxGENE repository. Second, we will supplement the integrated embedding with “specialized” embeddings that emphasize specific axes, e.g., skin sites or cell lineages.
IV.c. Annotation Strategy
Annotation efforts by the Skin Biological Network aim to achieve consensus between community experts. We will follow the two-step strategy employed by the Human Lung Cell Atlas (Sikkema et al., 2022). First, we will curate original cell annotations from published datasets and generate a hierarchical reference framework that spans all appropriate annotation levels, from the broadest cell lineage classification (e.g., epithelial, mesenchymal, immune, vascular) to specific within-lineage cell types and possibly skin site-specific cell states. The framework will be a reference to annotate datasets containing unlabeled cells, help harmonize labels between datasets to guide the selection of an optimal integration method, and improve data integration via semi-supervised learning. In order to properly benchmark integration and make full of previous annotations, the original cell type annotations from published dataset must also be made consistent when constructing the reference framework. Second, we will draw upon a network of experts to generate consensus annotation and identify cell types that are prone to ambiguous or conflicting classification. This will occur in three stages: 1) providing experts with the lineage-specific integrated objects (e.g., via CELLxGENE or the Cell Annotation Platform) to pre-annotate generated clusters; 2) computationally harmonizing expert annotations and identifying disagreements; and 3) hosting an “annotation jamboree” to resolve these disagreements.
To facilitate reproducibility, we will use tools such as the Cell Annotation Platform (celltype.info) to share both our cell annotation metadata and also our rationale behind each annotation choice, e.g., marker gene expression or label transfer and tools such as protocols.io (protocos.io) to share sequencing protocols.
IV.d. Metadata Standards
For standardized and meaningful analysis of scRNA-seq data, for example, to analyze inter-individual or body site-to-site variations in cell states, it is important to annotate metadata with standard nomenclature. Current metadata accompanying published skin scRNA-seq datasets lack standardization and contain gaps (see Figure 2; Supplementary Table S1). For example, of the current identified 235 individual skin scRNA-seq datasets sampled from the 40 studies, 52 datasets do not have information about the sampled skin site (Figure 2a). Moreover, there are sites, such as dorsal skin, which are popular sites from which to sequence mouse skin, but for which there are no human samples. Data sampled from infants, children, and adolescents is also a notable age data gap (Figure 2b). One concerning data gap is that over half of the datasets (n=152) are lacking ancestry information (Figure 2c), while almost over one quarter of the datasets lack gender information (Figure 2d). Future studies should aim to address these data gaps as they have unique skin biology and disease profiles. Furthermore, most current studies use 10X-based sequencing technology (Figure 2e). Finally, the lack of detailed and standardized annotation of anatomic sites is a significant limitation of many studies. Table S1 details metadata variables that the Skin Biological Network suggests to document for each individual skin dataset (as far as regional ethical and consent policies allow). Biological variations include age, ancestral origin, gender, sex, time of day, sun exposure, smoking history, skin and systemic disease condition(s); while technical variations include cell enrichment protocols (if any), sequencing library chemistry, sequencing platform, and sequencing technology. To mitigate risk to privacy, strategies such as metadata aggregation will be used.
IV.e. Data Visualization and Web Portals
Intuitive and informative data visualization is necessary to convey scRNA-seq findings meaningfully. However, there are significant differences and limitations across current visualization methods: the linear PCA method can be confounded by technical factors; the nonlinear t-SNE method prioritizes local structure over global structure; while the nonlinear UMAP method can be distorted by cell composition. Moreover, these visualizations can only be interpreted qualitatively. For quantitative interpretation, direct visualizations of gene expression and cell state composition, which can take the form of box plots, violin plots, heatmaps, and general line graphs, are required. Tools like web-based portals are important to make visualization user-friendly.
We will make Atlas 1.0 accessible for easy data exploration and as a reference to project new disease states and annotations. Atlas 1.0 will be available on several web portals for exploration. Current web portals for skin include the Development Cell Atlas (developmental.cellatlas.io) and SkinGenes (skingenes.net). To facilitate new data annotation and projection onto the reference atlas, we will host Atlas 1.0 on other portals, such as Azimuth (azimuth.hubmapconsortium.org) or CellTypist (celltypist.org). For local analysis, the atlas will be uploaded to platforms like Zenodo. All related tools and Skin Biological Network updates will be available on the skin community landing platform (skincommunity.org).
V. Beyond Atlas 1.0
V.a. Sample Procurement
All datasets considered for such collections come with logistical and ethical limitations. For a more comprehensive atlas, sampling strategies should be expanded to include: (1) post-mortem tissues to enable a dramatic expansion of body site coverage and sampling skin sites that are otherwise challenging to procure from voluntary donors (e.g., lip skin, nail fold skin); and (2) frozen tissues to enable collection when or where fresh tissue cannot be immediately processed (e.g., hard-to-reach geographical locations or at unexpected sampling times).
V.b. Closing Data Gaps
Constructing Atlas 1.0 will lead to the identification of gaps that will set goals for Atlas 2.0. These may include: (i) anatomically distinct body sites for which there is no scRNA-seq data; (ii) different axes of variation with missing data such as age groups or ancestral groups; or (iii) missing or underrepresented cell lineages; or (iv) lack of within-donor datasets. Several members of the Skin Biological Network are generating body map datasets by sampling from multiple distinct body sites per donor.
Human skin cells have been investigated using nearly all available scRNA-seq technologies (Figure 2). While scarce in number, there are studies that have employed single-nucleus (snRNA-seq) technology, such as by Satpathy et al. (Satpathy et al., 2019), who performed an snRNA-seq and scATAC-seq study of cells from human basal cell carcinoma. Using snRNA-seq-based approaches is important for certain cell types such as lipid-containing adipocytes, for which droplet-based technologies are not feasible due to their large size and high buoyancy. Additionally, as most common scRNA-seq methods are based on short-read sequencing, long-read sequencing data will be generated using technologies such as FLASH-seq (Hahaut et al., 2022) and Smart-seq3xpress (Hagemann-Jensen et al., 2022), to capture full-length transcript isoform information and enable variant discovery of skin-specific differences between, for example, subjects of different ancestral origins (Cechova & Miga, 2023). Data with high sequencing depth and sensitivity is also needed to better capture stem cell markers and effector transcription factors that are naturally expressed at low levels, or to better represent the biology of epidermal lineages and hair follicles that undergo gradual changes in gene expression (Cockburn et al., 2022). Furthermore, other types of sequencing, such as DNA sequencing (DNA-seq), may help provide information about somatic mutations and clonal organization in, for example, sun-exposed skin, which would be particularly relevant for “same-site, different ancestral origin” studies. While current DNA-seq methods cannot be reliably used for single-cell studies, we expect newer technologies to emerge that can perform DNA-seq at the single-cell level.
Other data modalities, including scATAC-seq (Buenrostro et al., 2013a, 2015a) and CITE-seq (Stoeckius et al., 2017), can be integrated into Atlas 2.0 to better understand cell states. This multiomic approach is particularly important for lineages like skin fibroblasts, for which inferring true cell states from scRNA-seq data alone has been challenging due to their significant transcriptional state plasticity and their exhibiting characteristics of both progenitors (high proliferative potential) and specialized differentiated cells (high expression levels of specialized extracellular matrix genes). While no such multiomic study of human skin currently exists in the literature, Thompson et al. profiled neonatal fibroblasts in mice using parallel scRNA-seq and scATAC-seq (Thompson et al., 2022). The chromatin accessibility landscape revealed that despite distinct fibroblast lineages already present in neonatal mouse skin, the inferred epigenetic landscape suggested a degree of state plasticity and capacity for state transition greater than what transcriptomic data alone suggests.
Another rapidly emerging technology is spatial transcriptomics (Burgess, 2019), which can capture gene expression information while retaining spatial information in tissues, albeit, at present, at the cost of true single-cell resolution. As skin function depends on the interplay of various cell types to maintain tissue renewal and specific functions within spatially distinct niches, integrating scRNA-seq data with spatially resolved methods is highly important. There are several technologies to generate spatial data that vary by spatial resolution (single-cell vs. multicellular “spot”), tissue coverage, and transcriptome coverage. These technologies can be further divided based on RNA detection approach: some methods use in situ hybridization to profile native RNA species, capturing single-cell and even subcellular information at the cost of limited and biased gene coverage (several hundred selected genes at present), while other methods use next-generation sequencing to profile the entire transcriptome (tens of thousands of genes) at the cost of losing single-cell resolution or wide tissue coverage (Moses & Pachter, 2022). Currently, the most prominent commercially available platform is 10X Visium, which uses next-generation sequencing. Spatial information in the first-generation Visium platform is captured using micro-printed barcode-type primer spots 55µm in diameter with a center-to-center distance of 100µm. Visium has already been used in both mouse skin (Foster et al., 2021; Konieczny et al., 2022) and human skin studies (Ji et al., 2020; F. Ma et al., 2021; Schäbitz et al., 2022; Shim et al., 2022). Beyond spatial transcriptomics, scRNA-seq data can be integrated with prior imaging of skin, including with single-cell resolution multiphoton microscopy and optical coherence tomography.
VI. Utility of the Atlas
A high-quality reference skin atlas will enable numerous analyses and potential insights. First, it will allow better identification of the transcriptional heterogeneity of key cell lineages; that is, what are the invariant gene markers of cell types and what is the spectrum of within-lineage cell states and their corresponding markers? Second, consensus cell type annotation can serve as a reference map for emerging newer studies, particularly those that consider perturbations (for instance, due to disease), increasing confidence in the identification of novel cell states and markers (Hao et al., 2021; Lotfollahi et al., 2022). Third, the atlas can be used to deconvolve cell type annotation to spatial transcriptomic spot data, helping circumvent the current issue of spatial studies having reduced statistical power in analyses due to the lower number of samples (Li et al., 2022). Fourth, with comprehensive metadata and sufficient sample sizes of each skin site and cell type, well-informed statistical models can be constructed using one of the many appropriate statistical frameworks (Soneson & Robinson, 2018; Squair et al., 2021) to perform rigorous hypothesis testing. These parametric models allow for covariate specification to reduce confounding effects in statistical analyses and enable more accurate differential expression analysis of important biological factors, such as skin site, gender, sex, ancestral origin, health status, or even between individual donors. Other downstream applications of the atlas include characterizing active signaling pathways (Almet et al., 2021; Lewis et al., 2020); inferring potential gene regulatory networks of transcription factors and downstream targets that regulate epigenetic states (Pratapa et al., 2020), which will be further validated by scATAC-seq; and functional interpretation of the atlas using predefined gene sets (Buettner et al., 2017; Lotfollahi et al., 2022; Seninge et al., 2021; Zhao et al., 2021).
Atlas 1.0 can serve as a launching pad for many new scientific inquiries into human skin biology. Using statistical models, one could: (i) investigate whether there is an association between developmental origin and microanatomy of a given skin site and its functional specialization and environmental exposure, or why certain skin diseases are preferentially localized to specific body sites; (ii) examine the dependence between skin site function and cellular composition, for example, whether there are distinct mesenchymal populations that enable the elasticity and stretchability of eyelid skin or the mechanical rigidity of palmar and plantar skin; (iii) describe how stable molecular coordinates at a given body region (e.g., developmentally assigned HOX gene expression patterns) impact the transcriptional states of dermal cell lineages and, in turn, affect epithelial patterning (Chang et al., 2002; Rinn et al., 2008); (iv) analyze whether there are identifiable single cell bases for microanatomical tissue differences across ancestries; (v) determine the effect of long-term exposure to UV with respect to changes in cell lineage populations or changes in gene expression (secretome or DNA damage response); or (vi) establish how skin microbiome composition may relate to single-cell states in host tissue.
Supplementary Material
Table S1. Metadata of age, gender, anatomic site, ancestry and sequencing technology of the current skin scRNA-seq datasets. First author, PMID, URL and sample ID are included for dataset identification. Missing or unspecified information are recorded as unknown.
Table S2. Human Skin Cell Atlas Metadata Table. Tab 1: Metadata input page for authors and contributors. Note that categories with blue headings contain drop-down menus. Examples for two published studies are given. Tab 2: Explanation for each metadata heading in Tab 1. Tab 3: Drop-down menu content. Please do not modify Tab 2 and 3 content.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- Alexander CM, Kasza I, Yen CLE, Reeder SB, Hernando D, Gallo RL, Jahoda CAB, Horsley V, & MacDougald OA (2015). Dermal white adipose tissue: A new component of the thermogenic response. Journal of Lipid Research, 56(11), 2061–2069. 10.1194/jlr.R062893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almet AA, Cang Z, Jin S, & Nie Q (2021). The landscape of cell-cell communication through single-cell transcriptomics. Current Opinion in Systems Biology, 26, 12–23. 10.1016/j.coisb.2021.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen LK, & Davis MDP (2016). Sex differences in the incidence of skin and skin-related diseases in Olmsted County, Minnesota, United States, and a comparison with other rates published worldwide. In International Journal of Dermatology (Vol. 55, Issue 9, pp. 939–955). Blackwell Publishing Ltd. 10.1111/ijd.13285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belote RL, Le D, Maynard A, Lang UE, Sinclair A, Lohman BK, Planells-Palop V, Baskin L, Tward AD, Darmanis S, & Judson-Torres RL (2021). Human melanocyte development and melanoma dedifferentiation at single-cell resolution. Nature Cell Biology, 23(9), 1035–1047. 10.1038/s41556-021-00740-8 [DOI] [PubMed] [Google Scholar]
- Borthwick J, Evertsz N, & Pratt B (2023). How should communities be meaningfully engaged (if at all) when setting priorities for biomedical research? Perspectives from the biomedical research community. BMC Medical Ethics, 24(1), 6. 10.1186/s12910-022-00879-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, & Greenleaf WJ (2013a). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods, 10(12), 1213— 1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, & Greenleaf WJ (2013b). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods, 10(12), 1213— 1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, & Greenleaf WJ (2015a). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature, 523(7561), 486–490. 10.1038/nature14590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, & Greenleaf WJ (2015b). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature, 523(7561), 486–490. 10.1038/nature14590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner F, Pratanwanich N, McCarthy DJ, Marioni JC, & Stegle O (2017). f-scLVM: Scalable and versatile factor analysis for single-cell RNA-seq. Genome Biology, 18(1), 1–13. 10.1186/s13059-017-1334-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DJ (2019). Spatial transcriptomics coming of age. Nature Reviews Genetics, 20(6), 317. 10.1038/s41576-019-0129-z [DOI] [PubMed] [Google Scholar]
- Byrd AL, Belkaid Y, & Segre JA. (2018). The human skin microbiome. Nature Publishing Group, 16(3), 143–155. 10.1038/nrmicro.2017.157 [DOI] [PubMed] [Google Scholar]
- Cao ZJ, & Gao G (2022). Multi-omics single-cell data integration and regulatory inference with graph-linked embedding. Nature Biotechnology, 40(10), 1458–1466. 10.1038/s41587-022-01284-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cechova M, & Miga KH (2023). Comprehensive variant discovery in the era of complete human reference genomes. Nature Methods, 20(1), 17–19. 10.1038/s41592-022-01740-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Chi JT, Dudoit S, Bondre C, Van De Rijn M, Botstein D, & Brown PO (2002). Diversity, topographic differentiation, and positional memory in human fibroblasts. Proceedings of the National Academy of Sciences of the United States of America, 99(20), 12877–12882. 10.1073/pnas.162488599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazarra-Gil R, Van Dongen S, Kiselev VY, & Hemberg M (2021). Flexible comparison of batch correction methods for single-cell RNA-seq using BatchBench. Nucleic Acids Research, 49(7), 1–12. 10.1093/nar/gkab004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JB, Sedgewick AJ, Finnegan AI, Harirchian P, Lee J, Kwon S, Fassett MS, Golovato J, Gray M, Ghadially R, Liao W, Perez White BE, Mauro TM, Mully T, Kim EA, Sbitany H, Neuhaus IM, Grekin RC, Yu SS, … Cho RJ. (2018). Transcriptional Programming of Normal and Inflamed Human Epidermis at Single-Cell Resolution. Cell Reports, 25(4), 871–883. 10.1016/j.celrep.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chike-Obi C, Cole P, & Brissett A (2009). Keloids: Pathogenesis, Clinical Features, and Management. Seminars in Plastic Surgery, 23(03), 178–184. 10.1055/s-0029-1224797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockburn K, Annusver K, Gonzalez D, Ganesan S, May D, Kawaguchi K, Kasper M, & Greco V (2022). Gradual differentiation uncoupled from cell cycle exit generates heterogeneity in the epidermal stem cell layer. Nature Cell Biology, 142(8), B32. 10.1016/j.jid.2022.05.1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao H, & Kazin RA (2007). Gender differences in skin: A review of the literature. Gender Medicine, 4(4), 308–328. 10.1016/S1550-8579(07)80061-1 [DOI] [PubMed] [Google Scholar]
- Day S, Mathews A, Blumberg M, Vu T, Rennie S, & Tucker JD (2020). Broadening community engagement in clinical research: Designing and assessing a pilot crowdsourcing project to obtain community feedback on an HIV clinical trial. Clinical Trials, 17(3), 306–313. 10.1177/1740774520902741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabale A, & Nagpure S (2022). Types of Psoriasis and Their Effects on the Immune System. Cureus, 14(9). 10.7759/cureus.29536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati G, Rognoni E, Hiratsuka T, Liakath-Ali K, Hoste E, Kar G, Kayikci M, Russell R, Kretzschmar K, Mulder KW, Teichmann SA, & Watt FM (2017). Wounding induces dedifferentiation of epidermal Gata6 + cells and acquisition of stem cell properties. Nature Cell Biology, 19(6), 603–613. 10.1038/ncb3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farage MA, Miller KW, Elsner P, & Maibach HI (2013). Characteristics of the Aging Skin. Advances in Wound Care, 2(1), 5–10. 10.1089/wound.2011.0356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feramisco JD, Sadreyev RI, Murray ML, Grishin NV, & Tsao H (2009). Phenotypic and genotypic analyses of genetic skin disease through the online mendelian inheritance in man (OMIM) database. Journal of Investigative Dermatology, 129(11), 2628–2636. 10.1038/jid.2009.108 [DOI] [PubMed] [Google Scholar]
- Foster DS, Januszyk M, Yost KE, Chinta MS, Gulati GS, Nguyen AT, Burcham AR, Salhotra A, Ransom RC, Henn D, Chen K, Mascharak S, Tolentino K, Titan AL, Jones RE, da Silva O, Leavitt WT, Marshall CD, des Jardins-Park HE, … Longaker MT. (2021). Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proceedings of the National Academy of Sciences of the United States of America, 118(41), 1–10. 10.1073/pnas.2110025118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Liu J, Kriebel AR, Preissl S, Luo C, Castanon R, Sandoval J, Rivkin A, Nery JR, Behrens MM, Ecker JR, Ren B, & Welch JD (2021a). Iterative single-cell multi-omic integration using online learning. Nature Biotechnology, 39(8), 1000–1007. 10.1038/s41587-021-00867-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Liu J, Kriebel AR, Preissl S, Luo C, Castanon R, Sandoval J, Rivkin A, Nery JR, Behrens MM, Ecker JR, Ren B, & Welch JD (2021b). Iterative single-cell multi-omic integration using online learning. Nature Biotechnology, 39(8), 1000–1007. 10.1038/s41587-021-00867-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, & Longaker MT (2008). Wound repair and regeneration. Nature, 453(7193), 314–321. 10.1038/nature07039 [DOI] [PubMed] [Google Scholar]
- Hagemann-Jensen M, Ziegenhain C, Chen P, Ramsköld D, Hendriks GJ, Larsson AJM, Faridani OR, & Sandberg R (2020). Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nature Biotechnology, 38(6), 708–714. 10.1038/s41587-020-0497-0 [DOI] [PubMed] [Google Scholar]
- Hagemann-Jensen M, Ziegenhain C, & Sandberg R (2022). Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress. Nature Biotechnology, 40(10), 1452–1457. 10.1038/s41587-022-01311-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Lun ATL, Morgan MD, & Marioni JC (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nature Biotechnology, 36(5), 421–427. 10.1038/nbt.4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahaut V, Pavlinic D, Carbone W, Schuierer S, Balmer P, Quinodoz M, Renner M, Roma G, Cowan CS, & Picelli S (2022). Fast and highly sensitive full-length single-cell RNA sequencing using FLASH-seq. Nature Biotechnology, 40(10), 1447–1451. 10.1038/s41587-022-01312-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, … Satija R. (2021). Integrated analysis of multimodal single-cell data. Cell, 184(13), 3573–3587.e29. 10.1016/j.cell.2021.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydont V, Bernard BA, & Fortunel NO (2019). Age-related evolutions of the dermis: Clinical signs, fibroblast and extracellular matrix dynamics. Mechanisms of Ageing and Development, 177(March 2018), 150–156. 10.1016/j.mad.2018.03.006 [DOI] [PubMed] [Google Scholar]
- Hie B, Bryson B, & Berger B (2019). Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nature Biotechnology, 37(6), 685–691. 10.1038/s41587-019-0113-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano SA, Murray SB, & Harvey VM (2012). Reporting, representation, and subgroup analysis of race and ethnicity in published clinical trials of atopic dermatitis in the United States between 2000 and 2009. Pediatric Dermatology, 29(6), 749–755. 10.1111/j.1525-1470.2012.01797.x [DOI] [PubMed] [Google Scholar]
- Holzer JK, Ellis L, & Merritt MW (2014). Why we need community engagement in medical research. Journal of Investigative Medicine, 62(6), 851–855. 10.1097/JIM.0000000000000097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, Guo MG, George BM, Mollbrink A, Bergenstråhle J, Larsson L, Bai Y, Zhu B, Bhaduri A, Meyers JM, Rovira-Clavé X, Hollmig ST, Aasi SZ, Nolan GP, … Khavari PA. (2020). Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Journal of Cleaner Production, 497–514. 10.1016/j.cell.2020.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgour JM, Li S, & Sarin KY (2021). Hidradenitis suppurativa in patients of color is associated with increased disease severity and healthcare utilization: A retrospective analysis of 2 U.S. cohorts. JAAD International, 3, 42–52. 10.1016/jjdin.2021.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobak D, & Linderman GC (2021). Initialization is critical for preserving global data structure in both t-SNE and UMAP. Nature Biotechnology, 39(2), 156–157. 10.1038/s41587-020-00809-z [DOI] [PubMed] [Google Scholar]
- Konieczny P, Xing Y, Sidhu I, Subudhi I, Mansfield KP, Hsieh B, Biancur DE, Larsen SB, Cammer M, Li D, Landén NX, Loomis C, Heguy A, Tikhonova AN, Tsirigos A, & Naik S (2022). Interleukin-17 governs hypoxic adaptation of injured epithelium. Science, 9302. 10.1126/science.abg9302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamartine J (2003). Towards a new classification of ectodermal dysplasias. Clinical and Experimental Dermatology, 28(4), 351–355. 10.1046/j.1365-2230.2003.01319.x [DOI] [PubMed] [Google Scholar]
- Lewis N, Armingol E, Officer A, & Harismendy O (2020). Deciphering cell-cell interactions and communication from gene expression. Nature Reviews Genetics. 10.1038/s41576-020-00292-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Zhang W, Guo C, Xu H, Li L, Fang M, Hu Y, Zhang X, Yao X, Tang M, Liu K, Zhao X, Lin J, Cheng L, Chen F, Xue T, & Qu K (2022). Benchmarking spatial and single-cell transcriptomics integration methods for transcript distribution prediction and cell type deconvolution. Nature Methods, 19(6), 662–670. 10.1038/s41592-022-01480-9 [DOI] [PubMed] [Google Scholar]
- Lotfollahi M, Naghipourfar M, Luecken MD, Khajavi M, Büttner M, Wagenstetter M, Avsec Ž, Gayoso A, Yosef N, Interlandi M, Rybakov S, Misharin AV, & Theis FJ (2022). Mapping single-cell data to reference atlases by transfer learning. Nature Biotechnology, 40(1), 121–130. 10.1038/s41587-021-01001-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotfollahi M, Naghipourfar M, Theis FJ, & Alexander Wolf F (2020). Conditional out-of-distribution generation for unpaired data using transfer VAE. Bioinformatics, 36, I610–I617. 10.1093/bioinformatics/btaa800 [DOI] [PubMed] [Google Scholar]
- Lotfollahi M, Wolf FA, & Theis FJ (2019). scGen predicts single-cell perturbation responses. Nature Methods, 16(August). 10.1038/s41592-019-0494-8 [DOI] [PubMed] [Google Scholar]
- Luecken MD, Büttner M, Chaichoompu K, Danese A, Interlandi M, Mueller MF, Strobl DC, Zappia L, Dugas M, Colomé-Tatché M, & Theis FJ (2022). Benchmarking atlas-level data integration in single-cell genomics. Nature Methods, 19(1), 41–50. 10.1038/s41592-021-01336-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther N, Darvin ME, Sterry W, Lademann J, & Patzelt A (2012). Ethnic differences in skin physiology, hair follicle morphology and follicular penetration. Skin Pharmacology and Physiology, 25(4), 182–191. 10.1159/000337933 [DOI] [PubMed] [Google Scholar]
- Ma F, Hughes TK, Teles RMB, Andrade PR, de Andrade Silva BJ, Plazyo O, Tsoi LC, Do T, Wadsworth MH, Oulee A, Ochoa MT, Sarno EN, Luisa Iruela-Arispe M, Klechevsky E, Bryson B, Shalek AK, Bloom BR, Gudjonsson JE, Pellegrini M, & Modlin RL (2021). The cellular architecture of the antimicrobial response network in human leprosy granulomas. Nature Immunology, 22(7), 839–850. 10.1038/s41590-021-00956-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MA, Gutiérrez DE, Frausto JM, & Al-Delaimy WK (2021). Minority Representation in Clinical Trials in the United States: Trends Over the Past 25 Years. Mayo Clinic Proceedings, 96(1), 264–266. 10.1016/j.mayocp.2020.10.027 [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, & McCarroll SA (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell, 161(5), 1202–1214. 10.1016/j.cell.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapes BM, Foster CS, Kusnoor SV, Epelbaum MI, AuYoung M, Jenkins G, Lopez-Class M, Richardson-Heron D, Elmi A, Surkan K, Cronin RM, Wilkins CH, Pérez-Stable EJ, Dishman E, Denny JC, & Rutter JL (2020). Diversity and inclusion for the All of Us research program: A scoping review. PLoS ONE, 15(7), 1–14. 10.1371/journal.pone.0234962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y, & Ananthaswamy HN (2004). Toxic effects of ultraviolet radiation on the skin. Toxicology and Applied Pharmacology, 195(3), 298–308. 10.1016/j.taap.2003.08.019 [DOI] [PubMed] [Google Scholar]
- Moses L, & Pachter L (2022). Museum of spatial transcriptomics. Nature Methods, 19(5), 534–546. 10.1038/s41592-022-01409-2 [DOI] [PubMed] [Google Scholar]
- Nicholson LM, Schwirian PM, & Groner JA (2015). Recruitment and retention strategies in clinical studies with low-income and minority populations: Progress from 2004–2014. Contemporary Clinical Trials, 45, 34–40. 10.1016/j.cct.2015.07.008 [DOI] [PubMed] [Google Scholar]
- Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, & Sandberg R (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature Methods, 10(11), 1096–1100. 10.1038/nmeth.2639 [DOI] [PubMed] [Google Scholar]
- Polański K, Young MD, Miao Z, Meyer KB, Teichmann SA, Park JE, & Berger B (2020). BBKNN: Fast batch alignment of single cell transcriptomes. Bioinformatics, 36(3), 964–965. 10.1093/bioinformatics/btz625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratapa A, Jalihal AP, Law JN, Bharadwaj A, & Murali TM (2020). Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nature Methods, 17(2), 147–154. 10.1038/s41592-019-0690-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev A, Teichmann SA, & Lander ES (2017). The Human Cell Atlas. ELife. 10.7554/eLife.27041.001 [DOI] [Google Scholar]
- Reynolds G, Vegh P, Fletcher J, Poyner EFM, Stephenson E, Goh I, Botting RA, Huang N, Olabi B, Dubois A, Dixon D, Green K, Maunder D, Engelbert J, Efremova M, Polański K, Jardine L, Jones C, Ness T, … Haniffa M. (2021). Developmental cell programs are co-opted in inflammatory skin disease. Science, 371(6527). 10.1126/science.aba6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Wang JK, Allen N, Brugmann SA, Mikels AJ, Liu H, Ridky TW, Stadler HS, Nusse R, Helms JA, & Chang HY (2008). A dermal HOXtranscriptional program regulates site-specific epidermal fate. Genes and Development, 22(3), 303–307. 10.1101/gad.1610508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandby-Møller J, Poulsen T, & Wulf HC (2003). Epidermal Thickness at Different Body Sites: Relationship to Age, Gender, Pigmentation, Blood Content, Skin Type and Smoking Habits. Acta Dermato-Venereologica, 83(6), 410–413. 10.1080/00015550310015419 [DOI] [PubMed] [Google Scholar]
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, Shah P, Bell JC, Jhutty D, Nemec CM, Wang J, Wang L, Yin Y, Giresi PG, Chang ALS, … Chang HY. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nature Biotechnology, 37(8), 925–936. 10.1038/s41587-019-0206-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäbitz A, Hillig C, Mubarak M, Jargosch M, Farnoud A, Scala E, Kurzen N, Pilz AC, Bhalla N, Thomas J, Stahle M, Biedermann T, Schmidt-Weber CB, Theis F, Garzorz-Stark N, Eyerich K, Menden MP, & Eyerich S (2022). Spatial transcriptomics landscape of lesions from non-communicable inflammatory skin diseases. Nature Communications, 13(1), 1–13. 10.1038/s41467-022-35319-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seninge L, Anastopoulos I, Ding H, & Stuart J (2021). VEGA is an interpretable generative model for inferring biological network activity in single-cell transcriptomics. Nature Communications, 12(1), 1–9. 10.1038/s41467-021-26017-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J, Oh SJ, Yeo E, Park JH, Bae JH, Kim SH, Lee D, & Lee JH (2022). Integrated Analysis of Single-Cell and Spatial Transcriptomics in Keloids: Highlights on Fibrovascular Interactions in Keloid Pathogenesis. Journal of Investigative Dermatology, 142(8), 2128–2139.e11. 10.1016/jjid.2022.01.017 [DOI] [PubMed] [Google Scholar]
- Sikkema L, Strobl D, Zappia L, Madissoon E, Markov NS, Zaragosi L, Ansari M, Arguel M, Apperloo L, Bécavin C, Berg M, Chichelnitskiy E, Chung M, Collin A, Gay ACA, Hooshiar Kashani B, Jain M, Kapellos T, Kole TM, … Theis F. (2022). An integrated cell atlas of the human lung in health and disease. BioRxiv. [Google Scholar]
- Solé-Boldo L, Raddatz G, Schütz S, Mallm JP, Rippe K, Lonsdorf AS, Rodríguez-Paredes M, & Lyko F (2020). Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Communications Biology, 3(1), 1–12. 10.1038/s42003-020-0922-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, & Robinson MD (2018). Bias, robustness and scalability in single-cell differential expression analysis. Nature Methods, 15(4), 255–261. 10.1038/nmeth.4612 [DOI] [PubMed] [Google Scholar]
- Squair JW, Gautier M, Kathe C, Anderson MA, James ND, Hutson TH, Hudelle R, Qaiser T, Matson KJE, Barraud Q, Levine AJ, La Manno G, Skinnider MA, & Courtine G (2021). Confronting false discoveries in single-cell differential expression. Nature Communications, 12(1). 10.1038/s41467-021-25960-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, Mollbrink A, Linnarsson S, Codeluppi S, Borg A, Ponten F, Costea PI, Sahlén P, Mulder J, Bergmann O, … Frisén J. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science, 353(6294), 78–82. 10.1126/science.aaf2403 [DOI] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, & Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nature Methods, 14(9), 865–868. 10.1038/nmeth.4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone K, & Wheeler A (2015). A Review of Anatomy, Physiology, and Benign Pathology of the Nipple. Annals of Surgical Oncology, 22(10), 3236–3240. 10.1245/s10434-015-4760-4 [DOI] [PubMed] [Google Scholar]
- Sun X, Joost S, & Kasper M (2022). Plasticity of Epithelial Cells during Skin Wound Healing. Cold Spring Harbor Perspectives in Biology. 10.1101/cshperspect.a041232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz TH, Palermo AGS, Masur SK, & Aberg JA (2019). The Science and Value of Diversity: Closing the Gaps in Our Understanding of Inclusion and Diversity. Journal of Infectious Diseases, 220(Suppl 2), S33–S41. 10.1093/infdis/jiz174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabib T, Morse C, Wang T, Chen W, & Lafyatis R (2018). SFRP2/DPP4 and FMO1/LSP1 Define Major Fibroblast Populations in Human Skin. Journal of Investigative Dermatology, 138(4), 802–810. 10.1016/jjid.2017.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi R, Grzenda A, Allison TF, Rawnsley J, Balin SJ, Sabri S, Plath K, & Lowry WE (2020). Defining Transcriptional Signatures of Human Hair Follicle Cell States. Journal of Investigative Dermatology, 140(4), 764–773.e4. 10.1016/j.jid.2019.07.726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeo M, Lee W, & Ito M (2015). Wound healing and skin regeneration. Cold Spring Harbor Perspectives in Medicine, 5(1). 10.1101/cshperspect.a023267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi H, Tran N, Ang KS, Chevrier M, Zhang X, Yee N, Lee S, Goh M, & Chen J (2020). A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biology, 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Phan QM, Winuthayanon S, Driskell IM, & Driskell RR (2022). Parallel Single-Cell Multiomics Analysis of Neonatal Skin Reveals the Transitional Fibroblast States that Restrict Differentiation into Distinct Fates. Journal of Investigative Dermatology. 10.1016/jJid.2021.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thulabandu V, Chen D, & Atit RP (2018). Dermal fibroblast in cutaneous development and healing. Wiley Interdisciplinary Reviews: Developmental Biology, 7(2), 1–13. 10.1002/wdev.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J, Rostom M, & Garza LA (2022). Understanding and Harnessing Epithelial–Mesenchymal Interactions in the Development of Palmoplantar Identity. Journal of Investigative Dermatology, 142(2), 282–284. 10.1016/jJid.2021.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuan T, & Nichter LS (1998). The molecular basis of keloid and hypertrophic scar formation. Molecular Medicine Today, 4310(January), 19–24. [DOI] [PubMed] [Google Scholar]
- Vogt A, Hadam S, Heiderhoff M, Audring H, Lademann J, Sterry W, & Blume-Peytavi U (2007). Morphometry of human terminal and vellus hair follicles. Experimental Dermatology, 16(11), 946–950. 10.1111/j.1600-0625.2007.00602.x [DOI] [PubMed] [Google Scholar]
- Wang S, Drummond M, Guerrero-Juarez C, Tarapore E, MacLean A, Stabell A, Wu S, Gutierrez G, That B, Benavente C, Nie Q, & Atwood S (2019). Single cell transcriptomics of human epidermis reveals basal stem cell transition states. Nature Communications, 2020. 10.1101/784579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt FM (2014). Mammalian skin cell biology: At the interface between laboratory and clinic. Science, 346(6212), 937–940. [DOI] [PubMed] [Google Scholar]
- Wiedemann J, Billi AC, Bocci F, Kashgari G, Xing E, Tsoi LC, Meller L, Swindell WR, Wasikowski R, Xing X, Ma F, Gharaee-Kermani M, Kahlenberg JM, Harms PW, Maverakis E, Nie Q, Gudjonsson JE, & Andersen B (2023). Differential cell composition and split epidermal differentiation in human palm, sole, and hip skin. Cell Reports, 42(1), 111994. 10.1016/j.celrep.2023.111994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams HC, Dellavalle RP, & Garner S (2012). Acne vulgaris. The Lancet, 379(9813), 361–372. 10.1016/S0140-6736(11)60321-8 [DOI] [PubMed] [Google Scholar]
- Xu C, Lopez R, Mehlman E, Regier J, Jordan MI, & Yosef N (2021). Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models. Molecular Systems Biology, 17(1). 10.15252/msb.20209620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Cai H, Zhang Z, Tang J, & Li Y (2021). Learning interpretable cellular and gene signature embeddings from single-cell transcriptomic data. Nature Communications, 12(1), 1–15. 10.1038/s41467-021-25534-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, Long X, Zhao Q, Zheng Y, Song M, Ma S, Jing Y, Wang S, He Y, Esteban CR, Yu N, Huang J, Chan P, Chen T, Izpisua Belmonte JC, Zhang W, Qu J, & Liu GH (2021). A Single-Cell Transcriptomic Atlas of Human Skin Aging. Developmental Cell, 56(3), 383–397.e8. 10.1016/j.devcel.2020.11.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Metadata of age, gender, anatomic site, ancestry and sequencing technology of the current skin scRNA-seq datasets. First author, PMID, URL and sample ID are included for dataset identification. Missing or unspecified information are recorded as unknown.
Table S2. Human Skin Cell Atlas Metadata Table. Tab 1: Metadata input page for authors and contributors. Note that categories with blue headings contain drop-down menus. Examples for two published studies are given. Tab 2: Explanation for each metadata heading in Tab 1. Tab 3: Drop-down menu content. Please do not modify Tab 2 and 3 content.