

Abstract
The secretome from mesenchymal stem cells (MSCs) has recently gained attention for new therapeutics. However, clinical application requires in vitro cell manufacturing to attain enough cells. Unfortunately, this process often drives MSCs into a senescent state that drastically changes cellular secretion activities. Antioxidants are used to reverse and prevent the propagation of senescence; however, their activity is short-lived. Polymer-stabilized crystallization of antioxidants has been shown to improve bioactivity, but the broad crystal size distribution (CSD) significantly increases the efficacy variation. Efforts were made to crystalize drugs in microdroplets to narrow the CSD, but the fraction of drops containing at least one crystal can be as low as 20%. To this end, this study demonstrates that in-drop thermal cycling of hyaluronic acid-modified antioxidant crystals, named microcrystal assembly for senescence control (MASC), can drive the fraction of microdrops containing crystals to >86% while achieving significantly narrower CSDs (13±3μm) than in bulk (35±11μm). Therefore, this approach considerably improves the practicality of CSD-control in drops. In addition to exhibiting uniform release, MASC made with antioxidizing N-acetylcysteine extended the release time by 40%. MASC further improves the restoration of reactive oxygen species homeostasis in MSCs, thus minimizing cellular senescence and preserving desired secretion activities. We propose that MASC is broadly useful to controlling senescence of a wide array of therapeutic cells during biomanufacturing.
Keywords: biomanufacturing, drop-microfluidics, exosome, hyaluronic acid-dopamine, mesenchymal stem cells
Graphical Abstract
Adult stem cells are being extensively tested for various therapeutics because of their secretion activities, however cellular senescence complicates mesenchymal stem cell (MSC) manufacturing. In this study we engineer a microcrystal assembly for senescence control (MASC) that maintains reactive oxygen species homeostasis through highly uniform release. Conditioning of MSCs with MASC protects against senescence and preserves therapeutic secretion activities.
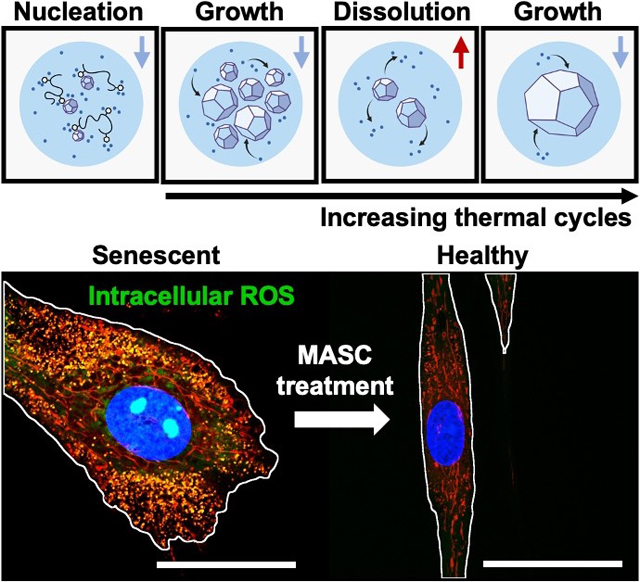
1. Introduction
Mesenchymal stem cells (MSCs) have long been studied for new generations of medicine due to their secretion of various therapeutic proteins and extracellular vesicles;[1–3] however in vitro expansion required to attain enough cells for clinical applications often leads to cell heterogeneity.[3,4] In turn, changes in the cellular secretome and loss of therapeutic efficacy are observed.[3,4] MSC heterogeneity is further exacerbated by cellular senescence, which is heavily influenced by the oxidative state of the cell.[5–7] Moreover, senescent MSCs produce reactive oxygen species (ROS) and spread senescence to neighboring cells, compounding adverse effects on the secretome bio-efficacy.[8,9]
Many high throughput label-free approaches exist to separate senescent cells from primary MSC samples prior to expansion including density gradient centrifugation[10] and microfluidic mechanical[11] or hydrodynamic[12–14] filtration. Nevertheless, these size-dependent techniques miss quiescent cells that can transition into senescent cells without proper intervention. Therefore, hydrophilic antioxidants, such as -acetylcysteine (NAC) and ascorbic acid, are added to cell culture media to reverse quiescence and prevent senescence spreading through ROS scavenging.[15–18] However, most hydrophilic antioxidants are metabolized within 6 hours in media, leaving <5% bioactive.[19,20] The increased and more frequent dosing for compensation leads to antioxidant stress, DNA damage in proliferating MSCs, and large efficacy variation.[18] In addition, even micromolar fluctuations in the ROS levels in MSCs can lead to substantial changes in intracellular transcription and translation of therapeutic paracrine factors,[21–23] driving the need for prolonged and sustained antioxidant release.
Recently, crystallization of antioxidants with stabilizing polymers have illustrated improved release kinetics and efficacy.[19,20] MSC-secretome manufacturing can benefit from these emerging systems by achieving ROS homeostasis, through the careful design and fabrication of an antioxidant crystal platform. In pharmaceutical manufacturing, crystallization is an important step in active pharmaceutical ingredient (API) assembly, and control of the crystal size distribution (CSD) has major implications on API bio-efficacy. Although CSD control operations such as milling are conventional, sensitive drug systems require alternative approaches.[19,24–30] These crystal systems include APIs prone to polymorph changes, sensitive to temperature degradation, or, as in our case, interfaced with polymeric excipients.[19,27–31] As such, crystallization using droplet-microfluidics has been discussed as an alternative CSD control tool.[32–36]
Compared to crystallization in batch, drop-microfluidic techniques have the benefit of precise compartmentalization. Therefore, drug loading in drops is highly uniform and, moreover, provides nucleation control by avoiding reactor-induced nucleation. A major drawback, however, is that over reasonable time scales, the fraction of microdroplets containing at least one hydrophilic API crystal can be as low as 20%, challenging both high-throughput operation and material recovery.[37,38]
Previously, a handful of approaches have been studied to improve in-drop nucleation such as API seed loading and solvent extraction.[36,39,40] However, CSD control is sacrificed with these approaches, compromising the unique benefit of the in-drop approach. We hypothesize that loading polymeric excipients, with strong thermodynamic interactions with the API surfaces, in microdroplets will increase the fraction of drops containing crystals due to increased heterogeneous nucleation and stabilization of nuclei by physical adsorption. Furthermore, taking advantage of drop confinement, sequential in-drop thermal cycles, can be used to yield single crystals per drop and refine the CSD (Figures 1a–c and Figure S1a).[40,41]
Figure 1.

(a) CAD image of drop generator geometry. (b) Image of drop generation at microfluidic junction. (c) Scheme of polymer induced nucleation and sequential in-drop thermal cycles. (d) Scheme of MASC scavenging ROS and preserving the healthy MSC phenotype.
As a model API-polymeric excipient system, we fabricated antioxidizing, NAC crystals stabilized by hyaluronate-dopamine (HA-dopa) polymers. Previously, we demonstrated that dopamine conjugation to hyaluronate could interface with NAC crystals and is necessary to provide extended stability and release through aggregation.[20] We anticipate that a reduction in the CSD will further extend NAC release and minimize variation through more uniform aggregation. This microcrystal assembly for senescence control (MASC) can then actively scavenge ROS, alleviate oxidative stress, and reduce MSC heterogeneity in order to improve the therapeutic efficacy of the MSC secretome (Figure 1d). In order to explore the ability of MASC to protect against MSC senescence and remediate normal paracrine signaling, we established an ROS-induced acute senescence model. We first characterized the MSC oxidative state post MASC treatment and determined the degree of cellular senescence through quantification of -galactosidase-positive cells and p16 gene expression. To validate preserved paracrine signaling, we monitored exosome secretion and VEGF, IGF, and IL-10 gene expression.
2. Results and Discussion
2.1. HA-dopa induced nucleation of -acetylcysteine in drops
First, to study if HA-dopa loading in drops can improve the fraction of drops containing crystals, we quantified the number of crystals per drop as a function of drop content (no polymer or HA-dopa) (Figure 2a). During thermal cycling, the fraction of drops containing no crystals () was less for drops with HA-dopa. After 7 thermal cycles and a 24hr hold time to reach complete crystallization, 86.3% of drops containing HA-dopa had at least 1 crystal (), a 4-fold improvement than previously reported efficiencies for similar cooling crystallization timescales (Figure S1b).[40,42,43] Furthermore, the time for 50% of drops to contain a crystal is 10-fold faster for HA-dopa loaded drops, illustrating the drastic effect of HA-dopa on early nucleation kinetics and considerably improving the practicality of crystallization using droplet microfluidics.
Figure 2. HA-dopa induced NAC nucleation in drops.

(a) Number of crystals per drop with successive thermal cycle events. 500 drops were characterized for each population and error bars represent one standard deviation. (b) ln() over 4,000min (only when drops are at 4°C). Dark and light solid lines depict fits to the Pound and LaMer expression for the mean and one standard deviation respectively. (c) Fitted homogeneous nucleation rate (), heterogeneous nucleation rate (), and number of active centers () within drops. *p < 0.05, **p < 0.01, ***p < 0.001 as determined by Wilcoxon rank sum test. Scale bar equals 25μm.
The high non-linearity of ln() suggests that more than one type of nucleation is occurring (Figure 2b). Therefore, we modeled the primary nucleation with a probabilistic model proposed by Pound and LaMer.[44,45] With this model, we decoupled homogeneous () from heterogeneous () nucleation rates and predicted the mean number of active nucleation centers () (Equation 1).
| (1) |
Heterogeneous nucleation was dominant in both drop conditions, evident by being over an order of magnitude greater than . Nonetheless, and were greatly increased in drops containing HA-dopa (Figure 2c). As such, the observed increase in nucleation kinetics in HA-dopa loaded drops is attributed to the promotion of heterogeneous nucleation and nuclei stabilization facilitated by polymer adsorption.
2.2. Crystal size distribution control with in-drop thermal cycling
We then compared bulk and microfluidic fabrication approaches to illustrate the extent that controlling CSDs minimize NAC release variation. For NAC crystallization, we chose the supersaturation condition (364mg/mL at 45°C) that provided a large nucleation driving force and prevented crystal-induced drop deformation (Figure S2). In order to illustrate the ability to fabricate crystals small enough for various delivery routes (~10μm), we generated 25μm-diameter drops that were collected in PCR tubes. The collected drops were then thermal cycled 5-times between 65°C to 5°C, followed by a 24hr hold time at 4°C.[46–48] In principle, during thermal cycling, heating events fully dissolve small crystals in the crystal population. As such, during the next cooling step, growth is preferred on the larger crystals that survived the aforementioned heating step over the formation of nuclei (Figure 3). Crystallization without HA-dopa was also explored (Figure S3a). To emphasize process compatibility with other hydrophilic APIs, we crystallized and thermal cycled ascorbic acid in-drop (Figure S3b). Separately, bulk-assembled NAC crystals (denoted as “bulk”) with HA-dopa were processed with the same thermal cycling conditions. The histograms quantifying the major axis diameter of NAC crystals illustrate two significant differences between the fabrication approaches: (1) droplet confinement reduced the average crystal diameter (13.1μm compared to 34.9μm in bulk), and (2) thermal cycling in drops narrowed the CSD (7.4μm to 2.9μm). In contrast, bulk thermal cycling did not reduce the CSD, likely due to uncontrollable secondary nucleation.
Figure 3. Thermal cycling approach to CSD control.

(a) Representative images of in-drop thermal cycling. (b) Histograms and representative SEM images for NAC crystals fabricated and thermal cycled in bulk or 25μm-diameter drops. All crystals were prepared in the presence of 2.5wt% HA-dopa. represents the average major axis diameter of the crystal population and one standard deviation. A minimum of 2,000 crystals were characterized for each population.
2.3. -acetylcysteine crystal dissolution
The dissolution profile of NAC crystals and the release variation were quantified (Figure 4). Surprisingly, despite the smaller size, microfluidic-assembled NAC crystals (denoted as “MASC”) extended the release of NAC by 40% compared to bulk NAC crystals. For our HA-dopa NAC crystal system, reducing the CSD considerably enhances the stabilizing effect of HA-dopa on the NAC crystals. Furthermore, the release variation, quantified as the ratio of the coefficient of variation () was greater than 1.0 for all but one time point and increased with time, illustrating faster propagation of release variation for the crystal populations with a broader CSD.
Figure 4. NAC crystal dissolution.

(a) NAC dissolution profile of bulk crystals and MASC assembled with either no polymer or HA-dopa. All crystal populations were thermal cycled 5 times. Error bars represent one standard deviation (n=5). (b) Change in release time between bulk- and microfluidic-assembled crystals (). (c) release as a function of time.
2.4. Efficacy of MASC to protect against MSC senescence
2.4.1. Characterization of MSC oxidative state
As alluded to before, minimal variation in antioxidant release can improve the therapeutic efficacy by maintaining cellular homeostasis. As such, we tested whether MASC is more effective in reverting ROS-driven senescence in human adipose-derived MSCs and preserving cellular paracrine secretion activity compared to bulk NAC crystals. Senescent MSCs were created through H2O2 exposure for 5 days (Figure 5a). Then, MSCs were exposed to either antioxidant-free media (denoted as “H2O2 exposure”), bulk NAC crystals, or MASC for up to 6 days. To confirm that the H2O2 exposure condition did not induced cell death, we conducted a LIVE/DEAD assay (Figure S5). We found no statistical difference in cell death between cells never exposed to H2O2 or exposed to 25μM H2O2 daily for 5 days. In order to confirm that MASC is biocompatible, we screened a range of MASC concentrations and the effect on MSC metabolic function (Figure S6). We observe no statistical difference in metabolic function at a concentration of 200μM and lower when compared to MSCs never exposed to MASC.
Figure 5. Oxidative state of MSCs post MASC treatment.

(a) Timeline for MSC study. (b) Confocal images of intracellular ROS. All images were taken after 6 days of treatment. (c) Quantified intracellular ROS levels relative to healthy MSCs (n=5) (Figure S7a). (d) Colocalization of intracellular ROS and mitochondria. Quantified in Figure S7b–d. *p < 0.05, **p < 0.01, ***p < 0.001, # no statistical significance with healthy MSCs, as determined by a student’s t-test. Scale bar equals 50μm.
To illustrate the antioxidant activity of the crystals, the MSC oxidative state was monitored through live-cell fluorescence staining of intracellular ROS levels (Figures 5b–c and Figure S7a). The mean intracellular ROS levels were over 1.8-times greater in the cell population exposed to H2O2 compared to normal cells. Furthermore, senescent cells showed increased green-fluorescent puncta, indicating intracellular oxidative stress levels in the mitochondria as marked by co-localization with a fluorescent mitochondrial stain (Figure 5d and Figures S7b–d). This observation is consistent with the literature characterization of senescent cells and matches the current hypothesis that ROS-mediated cellular senescence starts with mitochondrial dysfunction and ROS overproduction.[5–9] When the cells were treated with either bulk NAC crystals or MASC, the mean intracellular green intensity decreased. While MASC restored the intracellular ROS levels to that of normal cells by day 4, cells treated with bulk crystals exhibited a 24% higher ROS level than normal cells.
2.4.2. Characterization of MSC senescence and paracrine signaling
The degree of senescence was also characterized with two well-established markers: (1) the fraction of -galactosidase-positive cells (Figure 6a and Figure S8) and (2) the gene expression level of tumor suppression protein, p16 (Figure 6b).[6,7] Cells incubated with either NAC crystals exhibited a substantial decrease in the fraction of -galactosidase-positive cells and p16 gene expression. Interestingly, MASC greatly reduced the number of senescent cells compared to treatment with bulk crystals. After 6 days of treatment with MASC, the fraction of -galactosidase-positive cells was reduced from 90% to 21%, and the p16 gene expression was reduced from 1.93 to 1.16, suggesting that control of the oxidative state can protect against cellular senescence. Treatment with bulk NAC crystals for 6 days only reduced the fraction of -galactosidase-positive cells to 33.9% and the p16 gene expression to 1.30.
Figure 6. MASC efficacy in treating MSC senescence.

(a) Fraction of cells positively stained for -galactosidase (n=5). (b) p16 gene expression normalized to the healthy MSC p16 expression (n=5). (c) Exosome release relative to baseline exosome release in healthy MSCs (n=5). (d) VEGF, IGF, and IL-10 gene expression following 6 days of treatment normalized to the expression in healthy MSCs (n=5). (e) for bulk crystal and MASC treatment. (f) as a function of time for all time-dependent in vitro studies. *p < 0.05, **p < 0.01, ***p < 0.001, # no statistical significance with healthy MSCs, as determined by a student’s t-test.
We characterized the secretome of the MSCs to gauge their therapeutic potential by monitoring the exosome secretion levels (Figure 6c) and paracrine gene expression for vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF), and interleukin-10 (IL-10) relative to healthy MSCs (Figure 6d). MSCs exposed to H2O2 showed low exosome secretion, but NAC crystal treatment led to a significant recovery in exosome secretion following 6 days of treatment. In particular, cells treated with MASC restored exosome secretion levels equal to those never exposed to H2O2. A similar trend was observed for the gene expressions of VEGF, IGF, and IL-10. MASC treatment enabled cells to retain healthy baseline expression compared to NAC-free media and bulk NAC crystal treatment.
2.4.3. Variation in MSC phenotypic expression
Finally, we observed whether the in each experiment was smaller for MASC () than bulk NAC crystals (). Plotting for all in vitro experiments allowed us to find two key trends (Figure 6e–f): (1) A decrease in release variation leads to a decrease in the variation of cell state and (2) increases faster than over time. This characterization helps to emphasize that drugs designed to control cellular homeostasis require predictable and uniform release but also have major implications in long-term treatment of an injury or disease. Heterogeneity in cell state negatively influences cellular homeostasis and can change the direction of disease pathogenesis.
2.4.4. NAC efficacy based only on uniform release
To confirm that the improved efficacy from MASC is linked to the CSD and release variation and not just the release rates, we engineered a NAC crystal (denoted as “broad CSD”) and repeated the MSC efficacy studies (Figure 7a–c). Briefly, the crystal assembly followed the same protocol previously described for fabricating bulk NAC crystals, except an additional crystal growth step was included. Following the 5 thermocycles and 12-hour hold at 4°C, 200μL of supersaturated NAC was added dropwise into the crystal suspension. This approach promotes crystal growth rather than the formation of new nuclei. The resulting broad CSD crystals show a similar release profile to MASC but have a larger size distribution and release variation than MASC. While the measured therapeutic differences between the broad CSD crystals and MASC are reduced, MASC still better preserves the healthy MSC state than the broad CSD crystals (Figure 7d–h). Notably, the oxidative stress levels following MASC treatment are still lower and less variable, leading to cell function most similar to the healthy MSC population. Therefore, we propose that the improved efficacy of MASC results from both the prolonged and uniform release of NAC from crystals with narrow CSDs.
Figure 7: Comparison of MASC and broad CSD crystals release and efficacy.

(a) Histograms and representative SEM images of broad CSD crystals and MASC. All crystals were prepared in the presence of 2.5wt% HA-dopa. represents the average major axis diameter of the associated NAC crystal population and one standard deviation error. A minimum of 2,000 crystals were characterized for each population. (b) NAC dissolution profile of broad CSD crystals and MASC. Error bars represent one standard deviation (n=5). (c) release as a function of time. (d) Confocal (oxidative state) and brightfield (-galactosidase) images of senescent MSCs treated with NAC crystals with a broad CSD (Figure 7a – blue histogram) or MASC (Figure 7a – green histogram) for 6 days. 20x objective confocal images depict nuclear counterstain (DAPI – blue) and intracellular ROS (CellROX – green). Optical images where blue stained cells depict -galactosidase-positive MSCs. (e) Quantification of intracellular ROS in MSCs from confocal images (n=5) (f) Fraction of MSCs positively stained for -galactosidase following treatment with NAC crystals with a broad CSD (blue) or MASC (green). Normalized gene expression for p16 following treatment with NAC crystals with a broad CSD (blue) or MASC (green). (n=5) (g) VEGF, IGF, and IL-10-encoding mRNA expression in MSCs following treatment with NAC crystals with a broad CSD (blue) or MASC (green). (n=5) (h) Coefficient of variation for broad CSD crystals () and MASC (). *p < 0.05, **p < 0.01, ***p < 0.001 as determined by Wilcoxon rank sum test.
Overall, translation of cell-derived secretome-based therapeutics into the clinical setting is heavily setback by functional cell heterogeneity.[49] Cell heterogeneity can be caused by donor-to-donor variability as well as cell manufacturing approaches.[49,50] As discussed in this study, the oxidative state of MSCs has major implications on phenotypic expression and needs to be controlled when manufacturing MSC-derived therapeutics.[3] To date, most clinical protocols require extensive MSC expansion. Therefore, easy to implement and relatively cheap approaches to minimizing cell heterogeneity are most actively pursued.[3] Namely, ‘rejuvenating’ media is used to preserve cellular homeostasis. Nonetheless, the antioxidant additives lose their activity prior to 48 hours when media is conventionally changed. Our MASC system tackles this approach by extending antioxidant release. More interestingly, the MASC system minimizes burst release which can drive cells into antioxidative stress, lead to DNA damage, and promote cellular senescence. Suppressing variation in cell phenotypic expression allows for more predictable MSC secretion activities and, therefore, leads to reliable biologics manufacturing. Furthermore, this system can be directly applied to the manufacturing patient-derived therapeutics which add benefit of minimizing the probability of a host response.
3. Conclusion
In summary, by including polymeric excipients, we have achieved crystallization in >86% of microdrops, 4 times larger than previously reported values with similar crystallization timescales, while being able to finely control CSDs via in-drop thermal cycling. The resulting HA-dopa-stabilized NAC crystals, named MASC, show significantly extended and uniform release profiles. They minimize ROS-triggered senescence, as confirmed by senescent biomarkers, exosome secretion, and paracrine factor-encoding gene expression. We envision that the in-drop thermal cycling process can be extended to control the CSDs of various hydrophilic API crystals with desired functions, such as stimulus-responsive dissolution or tissue-targeting transport.[19] Moreover, APIs that modulate cellular environments sensitive to micromolar biochemical fluctuations would experience improved efficacy.
4. Experimental Section/Methods
Materials:
N-acetyl-L-cysteine (NAC) (>99%), N-(3-Dimethylaminopropyl)-N′-ethyl carbodiimide hydrochloride (EDC) (98%), N-Hydroxysulfosuccinimide sodium salt (sulfo-NHS) (>98%), 4-Morpholineethanesulfonic acid monohydrate (MES) (>99%), and dopamine hydrochloride (DA) were purchased from Sigma-Aldrich. Hyaluronic acid (HA, Mw ≈ 620 – 1200 kg/mol) was purchased from Kikkoman. For drop making polydimethylsiloxane (PDMS) and Novec™ 7500 (HFE 7500) were purchased from Fisher Scientific, an aquapel applicator was purchased from Aquapel glass treatment, and 008-FluoroSurfactant (RAN surfactant) was purchased from RAN Biotechnologies. SU-8 2025 and 2050 were purchased from Kayaku Advanced Materials. Human adipose-derived mesenchymal stem cells (MSCs) were obtained from Essent Biologics and cultured according to supplier’s instruction. The senescence cells histochemical staining kit (CS0030) was purchased from Sigma-Aldrich. The LIVE/DEAD™ Cell Imaging Kit (R37601) was purchased from ThermoFisher. 4′,6-diamidino-2-phenylindole (DAPI), CellROX™ Green Reagent (C10444), and MitoTracker™ Red FM (M22425) were purchased from ThermoFisher. Total Exosome Isolation Reagent (4478359) was purchased from ThermoFisher. RNeasy Plus Mini Kit (74134 and 74136) was purchased from Qiagen. iScript™ cDNA Synthesis Kit (1708890) was purchased from BioRAD. Forward and reverse primers for p16, VEGF, IGF, and IL-10 were custom designed and ordered from Integrated DNA Technologies. iTaq Universal SYBR Green Supermix (1725121) was purchased from BioRAD.
Fabrication of single emulsion drop generator:
SU-8 2025 was used to fabricate microfluidic devices with 30μm channel heights and SU-8 2050 was used to fabricate devices with 50μm channel heights. Silicon wafers were cleaned with IPA, DI water, and acetone and dried with N2 at 65°C prior to spin coating of SU-8. According to the supplier’s recommendations, SU-8 was spin-coated onto the Si wafers, soft baked, hard baked, and exposed to UV. Photomasks of device features were designed in AutoCAD and produced by CADart. Following exposure, the wafers underwent a post-exposure bake and development with Kayaku Advanced Materials SU-8 developer according to the supplier’s recommendations.
A 1:10 weight ratio of PDMS monomer and curing agent was poured into the SU-8 stamps and degassed for several hours. The PDMS was transferred to an oven at 65°C and cured overnight. The PDMS was removed and bound to glass following plasma treatment of the surface of the glass slide and PDMS. The device was transferred to an oven at 65°C to allow the bond to set. To increase the hydrophobicity of the PDMS channels, aquapel was injected into the device and dried with N2 prior to drop making.
Synthesis of hyaluronate-dopamine (HA-dopa):
HA was dissolved in a 0.1M solution of MES (pH = 5) for 12 hours at room temperature. Once the polymer was fully dissolved, 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-Hydroxysulfosuccinimide (sulfo-NHS) were dissolved in the solution for 30 min. EDC and sulfo-NHS were both added in excess at a 0.625:1 molar ratio to sodium glucuronate of HA. Dopamine hydrochloride was added at the appropriate molar ratio. The mixture was dialyzed against DI water using a dialysis tube (MWCO = 3.5 kDa) for 2 days and then lyophilized for 3 days before storage at 4°C until future use. As we have previously reported, the degree of substitution of dopamine (DSdopa) was controlled by fixing the molar ratio between dopamine to sodium glucuronate of HA at 8:9.[20] This ratio leads to ~20% of the carboxylic acids on HA being conjugated with dopamine.
Bulk fabrication of crystals:
50mg (5% w/w of NAC) of polymer additives (HA or HA-dopa) were dissolved in DI water (10mL) for 6 hours at 45°C to ensure the complete dissolution. Then, 1g of NAC was added into the polymer solution and stirred at 45°C until all the solids were dissolved (6 hours). The solution was cooled to 4°C at a rate of 1°C/min and heated to 65°C at a rate of 1°C/min for anywhere between 0 and 5 thermocycles and finally cooled to 4°C where the temperature was held for an additional 12 hours all under constant stirring. The obtained crystal solution was filtered through a polyvinylidene fluoride (PVDF) membrane (HVLP04700, pore size: 0.45μm, Millipore) to collect NAC crystals. The crystals were gently washed 2 – 3 times with DI water to remove the unbound polymers. Then, the samples were dried under vacuum at room temperature for 2 days. NAC crystals were stored at room temperature under N2 until used for future experiments. The morphology of NAC crystals was examined by an optical microscope (Leica DMIL) and an environmental scanning electron microscope (ESEM, Quanta FEG 450, FEI) at 7kV acceleration voltage.
In order to illustrate that efficacy differences between the bulk-assembled NAC crystals and microfluidic-assembled NAC crystals are coupled to the release variation, we engineered a second bulk-assembled crystal (“Broad CSD crystal”). The bulk-assembly followed the same protocol previously described, except an additional crystal growth step was employed. Following the 5 thermocycles and 12-hour hold at 4°C, 200μL of supersaturated NAC (20mg of NAC was dissolved in 200μL at RT and slowly cooled to 4°C – 0.1°C/min) was added to the crystal suspension. Rather than the formation of new nuclei, crystal growth occurred on the current crystal population. This procedure resulted in larger crystals that dissolve slower but have the same CSD as the first bulk-assembled crystals (Figure 7a–b). Again, the release variation was higher for this crystal population than for the microfluidic crystals (Figure 7c). The obtained crystal solution was filtered through a polyvinylidene fluoride (PVDF) membrane (HVLP04700, pore size: 0.45μm, Millipore) to collect NAC crystals. The crystals were gently washed 2 – 3 times with DI water to remove the unbound polymers. Then, the samples were dried under vacuum at room temperature for 2 days. NAC crystals were stored at room temperature under N2 until used for future experiments. The morphology of NAC crystals was examined by an optical microscope (Leica DMIL) and an environmental scanning electron microscope (ESEM, Quanta FEG 450, FEI) at 7kV acceleration voltage.
Microfluidic assembly of crystals:
364mg of NAC and 18.2mg (5% w/w of NAC) of polymer additives (HA or HA-dopa) were dissolved in DI water (1mL) for 6 hours at 55°C to ensure complete dissolution. Using a heated glass syringe (heated with a conductive fabric (8.4V) to 55°C), the NAC solution was injected into the microfluidic device (Q=100μL/hr) while HFE 7500 with 2wt% RAN surfactant was injected into the second inlet of the microfluidic device (Q=200μL/hr) to generate drops with a diameter of 25μm. Although the NAC saturation temperature was 45°C, the operating temperature of 55°C was chosen to prevent crystallization prior to drop generation as the chip itself was not heated. The drop diameter could be changed for subsequent experiments by changing the width of the aqueous-organic (drop-making) junction or by changing the ratio of the aqueous and organic phase flow rates. Drops were collected in PCR tubes and topped with a mineral oil layer to minimize evaporation and shrinking of the aqueous drops.
Using a thermocycler (BIO-RAD T100 thermocycler), the temperature of the drops was brought to 80°C for 1hr to ensure any crystals/nuclei dissolved. Then drops were cycled (up to 7 times) between 65°C and 5°C and held at 4°C for 24hrs after thermal cycling. For details on heating and cooling rates and hold times, refer to Figure S1c. Following crystallization, the drops were washed with 10% perfluorooctanoic acid in HFE7500 to break drops. The obtained crystal solution was filtered through a polyvinylidene fluoride (PVDF) membrane (HVLP04700, pore size: 0.45μm, Millipore) to collect NAC crystals. The crystals were gently washed 2 – 3 times with DI water to remove the unbound polymers. Then, the samples were dried under vacuum at room temperature for 2 days. NAC crystals were stored at room temperature under N2 until used for future experiments. The morphology of NAC crystals was examined by an optical microscope (Leica DMIL) and an environmental scanning electron microscope (ESEM, Quanta FEG 450, FEI) at 7kV acceleration voltage.
Post crystallization processing:
During the thermocycling, drop merging occurred resulting in crystals with significantly larger major axis diameters. As such, we employed density gradient centrifugation to separate drop size (Figure S4a). Following thermocycling, drop suspensions were lightly centrifuged (800rpm) for 45s. For analysis of crystal diameters, only the bottom 1/3 of drops were collected and washed with 10% perfluorooctanoic acid to harvest the NAC crystals.
While the drop diameter can be used to control crystal size, another approach is to add heating steps post-thermal cycling to partially dissolve crystals but preserve the CSD (Figure S4b–c). This approach helps to maximize the crystallization efficiency by having a larger driving force for crystallization (45°C → 4°C compared to 45°C → 35°C). Following 5 thermocycles in 25μm diameter drops as previously described, the temperature of the drops was raised to either RT or 35°C and held at this temperature for 6 hours. The drops were then washed with HFE containing 10% perfluorooctanoic acid to harvest the NAC crystals. The obtained crystal solution was filtered through a PVDF membrane (HVLP04700, pore size: 0.45μm, Millipore) to collect NAC crystals. The crystals were gently washed 2 – 3 times with DI water to remove the unbound polymers. Then, the samples were dried under vacuum at room temperature for 2 days. NAC crystals were stored at room temperature under N2 until used for future experiments. The morphology of NAC crystals was examined by an optical microscope (Leica DMIL) and an environmental scanning electron microscope (ESEM, Quanta FEG 450, FEI) at 7kV acceleration voltage.
Characterization of NAC dissolution:
50mg of NAC crystals were placed in a dialysis tube (MWCO = 500 – 1,000 Da) with phosphate-buffered saline (PBS, 1mL). The dialysis tube was placed in the 499 mL of PBS media and incubated at 37°C under continuous shaking at 100rpm. At the designated time points, dissolved NAC was collected from the incubation media and determined by reading the absorbance at the wavelength of 260 nm using the microplate spectrophotometer (Infinite 200 PRO, Tecan).
Human adipose-derived mesenchymal stem cells (MSCs) culture and treatment:
In order to put MSCs into a senescent state, cells were exposed to H2O2 to disrupt mitochondrial functions and upregulate intracellular ROS levels. First, MSCs (passage 2) were seeded at 25% confluency on 0.1% gelatin-coated glass bottom dishes and allowed to proliferate to 50% confluency over 24 hours. After 24 hours, the FBS concentration was reduced from 10 to 5% to slow proliferation, and 25μM of H2O2 was added to the media daily for 5 days.
To confirm that the H2O2 treatment did not cause cell death, a LIVE/DEAD assay was performed according to the supplier’s recommendations. 2μM calcein AM (marker for live cells – green fluorescence) and 4μM EthD-1 solution (marker for dead cells – red fluorescence) were added directly to the cells following H2O2 exposure and washing with PBS and allowed to incubate for 45min. Finally, the cells were imaged using a confocal microscope (Zeiss LSM 700), and the intensity was quantified using ImageJ software (NIH) (Figure S6a). The assay was used to assess MSCs either cultured with H2O2-free media or exposed to 25μM H2O2 daily for 5 days (Figure S6b).
To confirm the biocompatibility and toxicity of MASC, a commercially available MTT assay kit (11465007001, Roche) was used to measure the metabolic activity of MSCs exposed to MASC at a concentration range of 50μM-500μM. The assay was performed with a few modifications to the manufacturer’s directions. MSCs were seeded into wells in a 96-well plate at a density of 10,000 cells per well. When the cells reached 50% confluency, the FBS concentration was reduced from 10 to 5% to slow proliferation. MSCs were exposed to MASC every 2 days alongside the media change for 6 days. After the exposure period, 90μL of MASC-free media and 10μL of MTT solution was used to replace media. The cells were incubated at 37°C for 3 hours. The formazan crystals formed as result of metabolic cell function were dissolved with 65μL of DMSO. The absorbance of MTT was measure using a microplate spectrophotometer (Infinite 200 Pro, Tecan) at the wavelengths 550 and 650nm. The relative metabolic activity was quantified as [(A550-A650)MASC/(A550-A650)Control]. Five samples (n=5) were prepared for each condition.
Following H2O2 exposure, the MSCs were treated with either NAC-free media, bulk-assembled NAC crystals (10ppm), or microfluidic-assembled NAC crystals (10ppm) every 2 days for 6 days. NAC was added every two days as an illustration that the crystals can be used as a media supplement and only needs to be applied during conventional media changes (every 2 days). The cell state and behavior were monitored over the 6 treatment days and were described in detail in later methods sections.
Intracellular reactive oxygen species (ROS) levels in MSCs:
MSCs were seeded 0.1% gelatin-coated 18mm cover glass at 10000 cells/dish exposed to H2O2 as described before. After the H2O2 exposure, the cells were treated with either NAC-free media, bulk-assembled NAC crystals (10ppm), or microfluidic-assembled NAC crystals (10ppm) every 2 days for 6 days. Following treatment, cells were washed 3 times with 1x PBS. After 2, 4, or 6 days, the intracellular oxidative stress was evaluated using the CellROX green reagent (ThermoFisher), following the manufacturer’s instruction. Briefly, the MSCs were incubated with 5μM of CellROX reagent for 30 min, and the cells were washed with 1x PBS three times. To identify the mitochondria, cells were stained with 300nM of MitoTracker Red (ThermoFisher) for 45 min. The cells were washed with 1xPBS three times. The cells were fixed with a 1:1 acetone:methanol solution for 15 min. For the nuclear counterstain, cells were stained with 500 nM of 4′,6-diamidino-2- phenylindole (DAPI). Finally, the cells were imaged using a confocal microscope (Zeiss LSM 700), and the intensity was quantified using ImageJ software (NIH). 5 samples (n = 5) were prepared for each condition.
Senescence characterization in MSCs:
The histological kit for -galactosidase detection was used to determine if a cell is in a senescent state. MSCs were exposed to H2O2 as previously described for 5 days, then washed with 1x PBS 5 times to remove any extracellular H2O2. The cells were then treated with either NAC-free media or NAC crystals according to our previously described protocol. Following treatment, the cells were fixed with the supplied fixation buffer for 7min at room temperature and washed with 1x PBS 3 times. According to the supplier’s recommendations, the staining mixture was prepared and used to cover the cells. The cells were incubated in the staining mixture at 37°C without CO2 for 8 hours. The CO2 levels were minimized to ensure no fluctuations in the pH levels. Cells were washed with 1x PBS 3 times and imaged with an optical microscope (Leica DMIL). 5 samples (n = 5) were prepared for each condition.
As a second marker for senescence, the gene expression for the tumor suppression protein, p16, was quantified via qRT-PCR. MSCs were exposed to 25μM H2O2 daily for 5 days and then treated with either NAC-free media, bulk-assembled NAC crystals (10ppm), or microfluidic-assembled NAC crystals (10ppm) every 2 days for 6 days. Following treatment, cells were washed 3 times with 1x PBS, detached with trypsin, and centrifuged at 1,250rpm for 5min. The supernatant was discarded, and cells were lysed and homogenized in the supplied RLT Plus buffer. Following the supplier’s recommendations, the cell debris and DNA were isolated from the RNA through centrifugation with the supplied spin columns and filters. The harvested RNA was then synthesized into cDNA using the iScript™ cDNA Synthesis Kit. Following the supplier’s recommendations, 4μL of the iScript Reaction Mix, 1μL of iScript Reverse Transcriptase, and 15μL of total RNA in nuclease-free water (0.05μg/μL) were added to dome-capped PCR tubes and thermal cycled (BIO-RAD T100 thermocycler) according to the supplier’s recommendations. Using iTaq Universal SYBR Green Supermix according to the supplier’s recommendations, the forward and reverse primers for p16 (designed and ordered from Integrated DNA Technologies) were mixed with the synthesized cDNA in a PCR tube via vortexing and added to a PCR plate. qRT-PCR was used to quantify ( method) the expression of p16 and was normalized to GAPDH gene expression levels. 5 samples (n = 5) were prepared for each condition.
Quantification of exosome release and paracrine gene expression in MSCs:
The exosomes secreted from the MSCs (~30–120nm) were isolated from the cell media daily using Total Exosome Isolation Reagent (ThermoFisher). As previously described, MSCs were exposed to 25μM H2O2 daily for 5 days and then treated with either NAC-free media, bulk-assembled NAC crystals (10ppm), or microfluidic-assembled NAC crystals (10ppm) every 2 days for 6 days. At the end of each day, 50% of the cell media was harvested and centrifuged at 2000 x g for 30 min to remove any cell debris or crystals. The supernatant was transferred to a separate centrifuge tube and mixed with the supplied reagent (1:2 v/v), vortexed, and incubated at 4°C overnight. After incubation, the sample was centrifuged at 10000 x g for 60 min at 4°C. The supernatant was discarded, and the exosomes were resuspended in 1x PBS. The concentration of the exosomes was calculated using nanoparticle tracking analysis (NanoSight NS300).
Expression levels of mRNAs encoding paracrine factors including VEGF, IGF, and IL-10, were quantified via qRT-PCR. MSCs were exposed to 25μM H2O2 daily for 5 days and then treated with either NAC-free media, bulk-assembled NAC crystals (10ppm), or microfluidic-assembled NAC crystals (10ppm) every 2 days for 6 days. Following treatment, cells were washed 3 times with 1x PBS, detached with trypsin and centrifuged at 1,250rpm for 5min. The supernatant was discarded, and cells were lysed and homogenized in the supplied RLT Plus buffer. Following the supplier’s recommendations, the cell debris and DNA were isolated from the RNA through centrifugation with the supplied spin columns and filters. The harvested RNA was then synthesized into cDNA using the iScript™ cDNA Synthesis Kit. Following the supplier’s recommendations, 4μL of the iScript Reaction Mix, 1μL of iScript Reverse Transcriptase, and 15μL of total RNA in nuclease-free water (0.05μg/μL) were added to dome-capped PCR tubes and thermal cycled (BIO-RAD T100 thermocycler) according to the supplier’s recommendations. Using iTaq Universal SYBR Green Supermix according to the supplier’s recommendations, the forward and reverse primers for p16 (designed and ordered from Integrated DNA Technologies) were mixed with the synthesized cDNA in a PCR tube via vortexing and added to a PCR plate. qRT-PCR was used to quantify the expression ( method) of VEGF, IGF, and IL-10, and was normalized to GAPDH gene expression levels. 5 samples (n = 5) were prepared for each condition.
Coefficient of variation calculation and statistical analysis:
The coefficient of variation () was calculated as the standard deviation over the mean. The CV was used as a metric to determine the variability of each sample condition over the time course of an experiment. The CV was calculated for all experiments involving either the bulk-assembled NAC crystals or the microfluidic-assembled NAC crystals.
To determine the colocalization of the mitochondria and intracellular ROS, the built-in tool (Coloc2) in ImageJ (NIH) was used to determine the Pearson’s coefficient, a metric for co-localization that takes into account relative pixel intensity (Figure S7b–d). 10 samples (n = 10) were prepared for each condition.
Averaged data are presented as mean ± one standard deviation. Statistical significance between pairs of experimental populations is determined by a Wilcoxon rank sum test or a nonpaired student t-test. *p < 0.05, **p < 0.01 and ***p < 0.001.
Supplementary Material
Acknowledgements
Funding Sources
This work was supported partly by the National Science Foundation Research Training Grant (NRT-MBM 1735252), National Science Foundation (NSF-CBET 1932192), National Research Foundation (NRF-2017M3A7B6052455), Alzheimer Association (2019-AARG-NTF-644507), and the National Institute of Health (NIH 1R01AI160671-A1).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Thermal cycle conditions; nucleation efficiency in drops; crystal size distribution histograms; post-thermal cycle processing; LIVE/DEAD assay; metabolic activity assay; intracellular ROS characterization; mitochondria and intracellular ROS co-localization; -galactosidase histochemical stain (PDF)
No competing financial interests have been declared.
References
- [1].Wechsler ME, Rao VV, Borelli AN, Anseth KS, Adv. Healtc. Mater 2021, 10, 2001948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Santamaria G, Brandi E, Vitola PL, Grandi F, Ferrara G, Pischiutta F, Vegliante G, Zanier ER, Re F, Uccelli A, Forloni G, Kerlero de Rosbo N, Balducci C, Cell Death Differ. 2021, 28, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yin JQ, Zhu J, Ankrum JA, Nat. Biomed. Eng 2019, 3, 90–104 [DOI] [PubMed] [Google Scholar]
- [4].Jung S, Panchalingam KM, Wuerth RD, Rosenberg L, Behie LA, Biotechnol. Appl. Biochem 2012, 59, 106. [DOI] [PubMed] [Google Scholar]
- [5].Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G, Cell 2013, 153, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liu J, Ding Y, Liu Z, Liang X, Front. Cell Dev. Biol 2020, 8, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Di Micco R, Krizhanovsky V, Baker D, di Fagagna F, Nat. Revs. Mol. Cell Biol 2021, 22, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li Y, Wu Q, Wang Y, Li L, Bu H, Bao J, Int. J. Mol. Med 2017, 39, 775. [DOI] [PubMed] [Google Scholar]
- [9].Ye G, Xie Z, Zeng H, Wang P, Li J, Zheng G, Wang S, Cao Q, Li M, Liu W, Cen S, Li Z, Wu Y, Ye Z, Shen H, Cell Death Dis. 2020, 11, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kovacovicova K, Vinciguerra M, Cell Prolif. 2019, 52, e12674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen Y, Mao P, Snijders AM, Wang D, Aging Cell 2018, 17, e12722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yin L, Wu Y, Yang Z, Tee CA, Denslin V, Lai Z, Lim CT, Lee EH, Han J, Lab Chip 2018, 18, 878. [DOI] [PubMed] [Google Scholar]
- [13].Poon Z, Lee WC, Guan G, Nyan LM, Lim CT, Han J, Van Vliet KJ, Stem Cells Transl. Med 2015, 4, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lee WC, Shi H, Poon Z, Nyan LM, Kaushik T, Shivashankar GV, Chan JKY, Lim CT, Han J, Van Vilet KJ, PNAS 2014, 11, E4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang M, Teng S, Ma C, Yu Y, Wang P, Yi C, Cytotechnology 2018, 70, 1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liao N, Shi Y, Zhang C, Zheng Y, Wang Y, Zhao B, Zeng Y, Liu X, Liu J, Stem Cell Res. Ther 2019, 10, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Park SY, Jeong AJ, Kim GY, Jo A, Lee JE, Leem SH, Yoon JH, Ye SK, Chung JW, J. Microbiol. Biotechnol 2017, 27, 1877. [DOI] [PubMed] [Google Scholar]
- [18].Weng Z, Wang Y, Ouchi T, Liu H, Qiao X, Wu C, Zhao Z, Li L, Li B, Stem Cells Transl. Med 2022, 11, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kim BS, Leong J, Yu SJ, Cho Y, Park CG, Kim DH, Ko E, Im SG, Lee J, Kim YJ, Kong H, Small 2019, 15, 1900765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Miller R, Kim Y, Park CG, Torres C, Kim BS, Lee J, Flaherty D, Han HS, Kim YJ, Kong H, ACS Appl. Mater. Interfaces 2022, 14, 39759 [DOI] [PubMed] [Google Scholar]
- [21].Chuang YC, Su WH, Lei HY, Lin YS, Liu HS, Chang CP, Yeh TM, PLoS One 2012, 7, e37613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rashedi I, Talele N, Wang XH, Hinz B, Radisic M, Keating A, PLoS One 2017, 12, e0187348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang YH, Tao YC, Wu DB, Wang ML, Tang H, Chen EQ, Stem Cell Res. Ther 2021, 12, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tung HH, Org. Process Res. Dev 2013, 17, 445 [Google Scholar]
- [25].Rasenack N, Müller BW, Pharm. Dev. Technol 2004, 9, 1. [DOI] [PubMed] [Google Scholar]
- [26].Brown CJ, McGlone T, Yerdelen S, Srirambhatla V, Mabbott F, Gurung R, Briuglia ML, Ahmed B, Polyzois H, McGinty J, Perciballi F, Fysikopoulos D, MacFhionnghaile P, Siddique H, Raval V, Harrington TS, Vassileiou AD, Robertson M, Prasad E, Johnston A, Johnston B, Nordon A, Srai JS, Halbert G, ter Horst JH, Price CJ, Rielly CD, Sefcik J, Florence AJ, Mol. Syst. Des. Eng 2018, 3, 518 [Google Scholar]
- [27].Taylor LS, Braun DE, Steed JW, Cryst. Growth and Des 2021, 21, 1375 [Google Scholar]
- [28].Puhl S, Li L, Meinel L, O. Mol. Pharm 2014, 11, 2372. [DOI] [PubMed] [Google Scholar]
- [29].Chen D, Singh D, Sirkar KK, Pfeffer R, Langmuir 2015, 31, 432. [DOI] [PubMed] [Google Scholar]
- [30].Shpigel T, Taguri GC, Lewitus DY, J. Appl. Polym. Sci 2019, 136, 47227 [Google Scholar]
- [31].Besenhard MO, Neugebauer P, Ho CD, Khinast JG, Cryst. Growth and Des 2015, 15, 1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Leng J, Salmon JB, Lab Chip 2009, 9, 24. [DOI] [PubMed] [Google Scholar]
- [33].Shi HH, Xiao Y, Ferguson S, Huang X, Wang N, Hao HX, Lab Chip 2017, 17, 2167. [DOI] [PubMed] [Google Scholar]
- [34].Tona RM, McDonald TAO, Akhavein N, Larkin JD, Lai D, Lab Chip 2019, 19, 2127. [DOI] [PubMed] [Google Scholar]
- [35].Pan LJ, Tu JW, Ma HT, Yang YJ, Tian ZQ, Pang DW, Zhang ZL, Z L Lab Chip 2018, 18, 41. [DOI] [PubMed] [Google Scholar]
- [36].Tona RM, McDonald TAO, Akhavein N, Larkin JD, Lai D, Lab Chip 2019, 19, 2127. [DOI] [PubMed] [Google Scholar]
- [37].Devos C, van Gerven T, Kuhn S, S. Cryst. Growth and Des 2021, 21, 2541 [Google Scholar]
- [38].Goh L, Chen K, Bhamidi V, He G, Kee NCS, Kenis PJA, Zukoski CF, Braatz RD, Cryst. Growth and Des 2010, 10, 2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li S, Zeng M, Gaule T, McPherson MJ, Meldrum FC, Small 2017, 13, 1702154. [DOI] [PubMed] [Google Scholar]
- [40].Garg N, Tona R, Martin R, Martin-Soladana PM, Ward G, Douillet N, Lai D, Lab Chip 2020, 20, 1815. [DOI] [PubMed] [Google Scholar]
- [41].Shan G, Igarashi K, Noda H, Ooshima H, J. Chem. Eng 2002, 85, 161 [Google Scholar]
- [42].Kwon S, Lakerveld R, Ind. Eng. Chem. Res 2022, 61, 11108 [Google Scholar]
- [43].Kwon S, Thomas KM, Lakerveld R, Chem. Eng. Process 2022, 171, 108751 [Google Scholar]
- [44].Pound GM, LaMer VK, J. Am. Chem. Soc 1952, 74, 2323 [Google Scholar]
- [45].Teychené S, Biscans B, Chem. Eng. Sci 2012. 77, 242 [Google Scholar]
- [46].Bragagni M, Gil-Alegre ME, Mura P, Cirri M, Ghelardini C, Mannelli L, L. Int. J. Pharm 2018, 547, 24. [DOI] [PubMed] [Google Scholar]
- [47].Jang JH, Shea LD, J. Controlled Release 2006, 112, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Brannon-Peppas L, Int. J. of Pharm 1995, 116, 1 [Google Scholar]
- [49].Marklein RA, Lam J, Guvendiren M, Sung KE, Bauer SR, Trends in Biotechnol. 2018, 36, 105. [DOI] [PubMed] [Google Scholar]
- [50].Phinney DG, J. Cell. Biochem 2012, 113, 2806. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.