

Abstract
Recent advances in sequencing technologies and collaborative efforts have led to substantial progress in identifying the genetic causes of amyotrophic lateral sclerosis (ALS). This momentum has, in turn, fostered the development of putative molecular therapies. In this Review, we outline the current genetic knowledge, emphasizing recent discoveries and emerging concepts such as the implication of distinct types of mutation, variability in mutated genes in diverse genetic ancestries and gene–environment interactions. We also propose a high-level model to synthesize the interdependent effects of genetics, environmental and lifestyle factors, and ageing into a unified theory of ALS. Furthermore, we summarize the current status of therapies developed on the basis of genetic knowledge established for ALS over the past 30 years, and we discuss how developing treatments for ALS will advance our understanding of targeting other neurological diseases.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that primarily affects motor neurons, leading to progressive muscle weakness and death from respiratory failure within 2–5 years of symptom onset1. The word ‘amyotrophic’ is derived from Greek, referring to muscle loss due to lack of nourishment, whereas ‘lateral sclerosis’ refers to scarring or hardening of the lateral column of the spinal cord. The term ‘amyotrophic lateral sclerosis’ was introduced by Jean-Martin Charcot, who described the clinical syndrome 150 years ago2. Consequently, ALS is also referred to as ‘Charcot’s disease’ in France, whereas it is known as ‘motor neuron disease’ in the UK. In China, it is called ‘jian dong ren’, meaning ‘gradually frozen men’. In the USA, the disease is recognized as Lou Gehrig’s disease after a famous baseball player who died of ALS in 1941. ALS and frontotemporal dementia (FTD) are part of a spectrum, and cognitive dysfunction is a part of the ALS disease course that was previously under-recognized.
ALS is relatively rare, with a worldwide incidence of ~2 per 100,000 person-years3,4. Despite this rarity, more than half a million individuals have died of ALS in the 80 years since Lou Gehrig gave his famous ‘luckiest man’ speech in Yankee stadium5. The number of affected individuals is soaring, mainly because of population ageing, and ~400,000 people will live with ALS globally by 2040 (ref. 6). The high fatality rate and the severe physical burden associated with ALS have further enhanced worldwide awareness of the disease, especially after the public attention attracted by the Ice Bucket Challenge7–10.
Approximately 10% of patients with ALS have a family history of the disease, and more than 30 ALS-associated genes have been identified in these familial ALS cases so far1,11. Although the remaining 90% of cases are apparently sporadic, pathogenic mutations identified in familial ALS were also described in many sporadic cases12. Familial aggregation studies, twin studies and heritability studies based on genome-wide association studies (GWAS) confirmed a substantial genetic component (61% by twin studies and 21% by GWAS) in ALS13–15.
Pathogenic variants in the most common ALS-associated genes, superoxide dismutase 1 (SOD1), TAR DNA-binding protein (TARDBP), fused in sarcoma (FUS) and chromosome 9 open reading frame 72 (C9orf72), account for approximately 60% of familial cases and about 10% of sporadic ALS. This equates to one in six ALS cases explained by these four genes12 (Table 1). More recently, whole-exome sequencing and whole-genome sequencing have identified several other ALS-causing genes to the point that we now know the underlying genetic mutation in about three-quarters of familial and one-fifth of sporadic ALS cases in populations of European ancestries16 (Table 1). In this Review, we summarize what is known about the genetic basis of ALS and describe how new genomic methodologies and collaborative efforts aim to unravel the remaining unsolved portion of the disease. We also discuss the genetic overlap between ALS and other conditions, ALS studies in populations of diverse genetic ancestry, gene–environment interactions and how genomic discoveries are the starting point for targeted drug development and personalized medicine17.
Table 1 |.
Summary of ALS-associated genes
| Year | Locus | Gene | Inheritance | Familial (%)a | Sporadic (%)a | Disease-associated mechanism | Other associated phenotypesb | Refs. |
|---|---|---|---|---|---|---|---|---|
| 1993 | 21q22.11 | SOD1 | Autosomal dominant, autosomal recessive, de novo | 12 | 1–2 | Oxidative stress, excitotoxicity, mitochondrial dysfunction, axonal transport disruption | Frontotemporal dementia, spastic tetraplegia and axial hypotonia | 19 |
| 1994 | 22q12.2 | NEFH | Autosomal dominant | Unknown | Unknown | Axonal transport disruption | Axonal Charcot-Marie-Tooth disease type 2CC | 212 |
| 2001 | 2q33.1 | ALS2 | Autosomal recessive | Unknown | Unknown | Vesicular trafficking defects | Juvenile primary lateral sclerosis, infantile hereditary spastic paraplegia | 213 |
| 2003 | 2p13.1 | DCTN1 | Autosomal dominant | Unknown | Unknown | Axonal transport disruption | Distal hereditary motor neuropathy type VIIB, Perry syndrome | 214 |
| 2004 | 20q13.32 | VAPB | Autosomal dominant | Unknown | Unknown | Proteostasis defects | Finkel-type spinal muscular atrophy | 215 |
| 2004 | 9q34.13 | SETX | Autosomal dominant | Unknown | Unknown | Altered ribostasis | Autosomal recessive spinocerebellar ataxia type 1 | 216 |
| 2006 | 3p11.2 | CHMP2B | Autosomal dominant | Unknown | Unknown | Proteostasis defects, vesicular trafficking defects | Frontotemporal dementia | 217,218 |
| 2008 | 1p36.22 | TARDBP | Autosomal dominant, autosomal recessive, de novo | 4 | 1 | Altered ribostasis, nucleocytoplasmic transport defects | Frontotemporal dementia | 38,39 |
| 2009 | 16p11.2 | FUS | Autosomal dominant, autosomal recessive, de novo | 4 | 1 | Altered ribostasis, nucleocytoplasmic transport defects | Frontotemporal dementia, essential tremor | 46,47 |
| 2010 | 9p13.3 | VCP | Autosomal dominant, de novo | 1 | 1 | Proteostasis defects | Frontotemporal dementia, Charcot-Marie-Tooth disease type 2Y, inclusion body myopathy with early-onset Paget disease | 144 |
| 2010 | 15q21.1 | SPG11 | Autosomal recessive | Unknown | Unknown | DNA damage | Hereditary spastic paraplegia, Charcot-Marie-Tooth disease type 2X | 219 |
| 2010 | 10p13 | OPTN | Autosomal dominant, autosomal recessive | <1 | <1 | Autophagy, inflammation | Adult-onset primary open-angle glaucoma | 220 |
| 2011 | Xp11.21 | UBQLN2 | X-linked dominant | <1 | <1 | Proteostasis defects | None | 221 |
| 2011 | 5q35.3 | SQSTM1 | Autosomal dominant | 1 | <1 | Autophagy, inflammation | Frontotemporal dementia, distal myopathy, childhood-onset neurodegeneration with ataxia, dystonia and gaze palsy, Paget disease of bone-3 | 222 |
| 2011 | 9p21.2 | C9orf72 | Autosomal dominant | 40 | 7 | Autophagy, global RNA alterations, intracellular trafficking defects, nucleocytoplasmic transport defects, proteostasis defects | Frontotemporal dementia | 55,56 |
| 2012 | 17p13.2 | PFN1 | Autosomal dominant | <1 | <1 | Impaired axonal growth and cytoskeletal organization | None | 223 |
| 2013 | 7p15.2 | HNRNPA2B1 | Autosomal dominant | Unknown | Unknown | Altered ribostasis | Inclusion body myositis with early-onset Paget disease with or without frontotemporal dementia 2, multisystem proteinopathy | 224 |
| 2013 | 12q13.13 | HNRNPA1 | Autosomal dominant, de novo | Unknown | Unknown | Altered ribostasis | Inclusion body myositis with early-onset Paget disease with or without frontotemporal dementia 3, multisystem proteinopathy | 224 |
| 2014 | 2q35 | TUBA4A | Autosomal dominant | <1 | <1 | Impaired axonal growth and cytoskeletal organization | Frontotemporal dementia | 225 |
| 2014 | 5q31.2 | MATR3 | Autosomal dominant | <1 | <1 | Altered ribostasis | Distal myopathy with vocal cord and pharyngeal weakness | 226 |
| 2014 | 22q11.23 | CHCHD10 | Autosomal dominant | <1 | <1 | Mitochondrial dysfunction | Frontotemporal dementia, spinal muscular atrophy (Jokela type), isolated mitochondrial myopathy | 227 |
| 2015 | 12q14.2 | TBK1 | Autosomal dominant | <1 | <1 | Autophagy, inflammation | Frontotemporal dementia | 112 |
| 2016 | 4q33 | NEK1 | Not established | 2 | 2 | DNA damage, impaired cytoskeletal organization and cell cycle | Short-rib thoracic dysplasia 6 with or without polydactylism | 112,114 |
| 2016 | 16p13.3 | CCNF | Autosomal dominant | 4 | 2 | Proteostasis defects | Frontotemporal dementia | 228 |
| 2016 | 21q22.3 | CFAP410 | Not established | Unknown | Unknown | Impaired cytoskeletal organization | Axial spondylometaphyseal dysplasia, retinal dystrophy with macular staphyloma | 107 |
| 2017 | 10q22.3 | ANXA11 | Autosomal dominant | Unknown | Unknown | Dysregulation of calcium homeostasis and stress granule dynamics | Inclusion body myopathy and brain white matter abnormalities | 229 |
| 2018 | 12q13.3 | KIF5A | Autosomal dominant | <1 | <1 | Impaired cytoskeletal organization and axonal transport | Charcot-Marie-Tooth type 2, hereditary spastic paraplegia | 72,73 |
| 2018 | 10q24.31 | ERLIN1 | Autosomal recessive | Unknown | Unknown | Dysregulation of inositol 1,4,5-trisphosphate intracellular ion channels | Hereditary spastic paraplegia | 78 |
| 2019 | 3p21.1 | GLT8D1 | Autosomal dominant | Unknown | Unknown | Impaired ganglioside synthesis | None | 230 |
| 2019 | 17q21.2 | DNAJC7 | Not established | Unknown | Unknown | Not established | None | 84 |
| 2021 | 4p16.3 | HTT | Autosomal dominant | Unknown | Unknown | Not established/nucleocytoplasmic transport defects | Huntington disease, Lopes-Maciel-Rodan syndrome | 91 |
| 2022 | 9q22.31 | SPTLC1 | De novo | Unknown | Unknown | Disruption of sphingolipid metabolism | Hereditary sensory and autonomic neuropathy type 1A | 97,98 |
Genes are listed chronologically based on their year of discovery.
Percentage of amyotrophic lateral sclerosis (ALS) cases explained by mutations in the corresponding disease-causing genes.
Phenotypes associated with the genes extracted from the Online Mendelian Inheritance in Man database.
Mendelian genes causing ALS
The four most common ALS genes
SOD1.
Thirty years ago, one of the first successful applications of linkage analysis in neurological disease identified a chromosome 21q locus segregating with the disease in 23 families with ALS cases18. Mutation screening and segregation analysis subsequently identified pathogenic variants in SOD1 as the causative gene19. This ground-breaking discovery showed for the first time that a genetic mutation could underlie ALS and that modern genetic approaches could reliably detect causative mutations. More than 200 SOD1 mutations have been reported, accounting for approximately 12% of familial and 1–2% of sporadic ALS in populations of European ancestries20,21. Most disease-causing SOD1 variants are missense mutations20. Although most of these mutations are inherited in an autosomal dominant manner, Asp91Ala, which is frequent in the Scandinavian population, is associated with a recessive form of ALS22–24.
The clinical phenotype of ALS is variable across the various SOD1 mutations but can also be remarkably stereotypical for a particular disease-causing variant. For example, the Ala5Val substitution, which is one of the most common SOD1 mutations among North American patients with ALS, leads to a rapidly progressive, dominant form of the disease25. Similarly, the Gly73Ser and Ser106Leu mutations present with a more severe phenotype than many other SOD1 mutations (for example, Gly38Arg and Gly94Cys)26–31. By contrast, patients homozygous for the Asp91Ala allele progress much more slowly, often developing urinary incontinence and sensory abnormalities in the late stages of their illness22.
Mutant SOD1 protein has a toxic gain of function. It seems to cause neuronal cell death via several mechanisms, including excitotoxicity, oxidative stress, non-cell-autonomous toxicity of neuroglia, mitochondrial dysfunction and axonal transport disruption32. Misfolded wild-type SOD1 was also suggested to be involved in sporadic ALS33. These mechanisms provided essential insights into the pathogenesis of ALS, showing how wild-type and mutant SOD1 could drive the disease process independently. Building on these observations, SOD1-related ALS was among the first neurological diseases for which molecular therapy development was attempted to block the production of the misfolded protein34–36.
TARDBP.
Another milestone in ALS research was the 2006 discovery by V. Lee, J. Trojanowski and colleagues of the nuclear depletion and cytoplasmic accumulation of TAR DNA-binding protein (TDP43) in motor neurons of patients with ALS37. Two subsequent studies identified mutations in the C terminus-encoding region of the gene TARDBP as a cause of autosomal dominant ALS38,39. The prevalence of TARDBP mutations in ALS is lower than that of mutations in SOD1, accounting for only 4% of familial and 1% of sporadic cases12. Regardless of the underlying pathogenic variants, abnormal cytoplasmic TDP43 inclusions are found in the brain and spinal cord in 97% of ALS cases40. This widespread pathological feature has become a hallmark of ALS and FTD, a rare and progressive disease resulting from neurodegeneration in the frontal and temporal lobes of the brain. Subsequent research has shown that TDP43 can be deposited in other neurological conditions (for example, FTD and limbic-predominant age-related TDP43 encephalopathy (LATE))41. The C-terminal domain of TDP43, also known as the glycine-rich region, has prion sequence similarity42. As for FUS and SOD1, prion-like motifs in TDP43 have been implicated in cell-to-cell transmission that may induce misfolding and aggregation of normal counterparts43–45. The role of TDP43 as an RNA-binding protein also implicated RNA processing as a central process in the motor neuron degeneration observed in ALS40,42.
FUS.
A year after the discovery of TARDBP and its involvement in ALS as a dysregulated RNA-binding protein, pathogenic variants were detected in a similar RNA-binding protein encoded by FUS46,47. Disease-causing variants are clustered in the region of the gene that encodes the C terminus of the protein, which functions as a nuclear localization signal, and they account for 4% of familial and 1% of sporadic ALS12,44. Although mutations in FUS are predominantly associated with autosomal dominant ALS, de novo mutations have also been reported46,48. In contrast to patients carrying mutated forms of other ALS-associated genes, some patients with FUS mutations may present at a young age and with an aggressive disease course49–51.
The identification of FUS provided additional evidence, along with TARDBP, that points to RNA metabolism disruption as a putative pathogenic mechanism in ALS. Reminiscent of TARDBP, FUS mutations are associated with a similar nuclear to cytoplasmic mislocalization and the formation of cytoplasmic inclusions of the mutated protein, highlighting the role of these cellular processes in ALS pathogenesis46,47,50,52. Recent research has focused on the role of stress granules in the pathogenesis of TARDBP- and FUS-related ALS53,54, although the pathogenic role of these phenomena remains unclear.
C9orf72.
The next breakthrough in ALS research was the discovery of a hexanucleotide (GGGGCC) repeat expansion in the C9orf72 gene in 2011 (refs. 55,56). Since then, it has become clear that C9orf72 repeat expansion is the most common genetic cause of ALS and FTD, accounting for ~40% of familial and ~7% of sporadic ALS in populations of European ancestries and an equivalent proportion of FTD cases57. Wild-type alleles carry fewer than 30 repeats, whereas pathogenic alleles have been reported to range between 700 and 1,600 repeats55. Expanded repeats are detected using a repeat-primed PCR; accurate sizing of the expansion relies on Southern blotting55,56,58, although initial studies suggest that long-read sequencing may also be helpful59. Studies examining repeat size and clinical heterogeneity among patients with C9orf72 expansion have yielded contradictory results60, with some reporting a negative correlation between expansion size and age at onset61, whereas others found a positive correlation62,63.
Multiple mechanisms have been proposed to explain C9orf72-related neurodegeneration. Expanded repeats may cause a toxic gain of function that involves disruption of RNA metabolism similar to TARDBP- and FUS-mediated pathogenesis. For example, the expansion-containing RNAs sequester important cellular factors into nuclear RNA foci64. In addition, a non-canonical mechanism of translation initiation, known as repeat-associated non-AUG-initiated (RAN) translation, was shown to produce dipeptide repeats from multiple reading frames in C9orf72-positive cases12,65,66. These dipeptides interfere with cytoskeletal dynamics and axonal transport by interacting with kinesin motor proteins, such as KIF5A, which has also been implicated in ALS67 (see below). A loss-of-function mechanism because of C9orf72 haploinsufficiency may likewise trigger neurodegeneration in ALS68. Although considerable in vitro and in vivo evidence supports each of these mechanisms, additional studies are required to learn precisely how the C9orf72 repeat expansion causes neuronal death. An intriguing possibility is that multiple pathological processes operate in tandem to trigger the neurodegenerative process. Indeed, the C9orf72 expansion has also been reported in other neurological disorders, including Alzheimer disease, Parkinson disease, progressive muscular atrophy and Huntington disease-like syndrome69,70.
Recent gene discoveries in ALS
Mutations in several other genes have been described as less frequent causes of ALS, including VCP, PFN1, MATR3, CHCHD10, TBK1 and NEK1 (ref. 16) (Fig. 1). Here, we discuss five ALS genes identified after 2018 using whole-exome and whole-genome sequencing, each providing novel insights into disease pathogenesis and a novel starting point for therapeutic development.
Fig. 1 |. Proportion of familial and sporadic ALS cases attributed to mutations in the corresponding disease-causing genes.
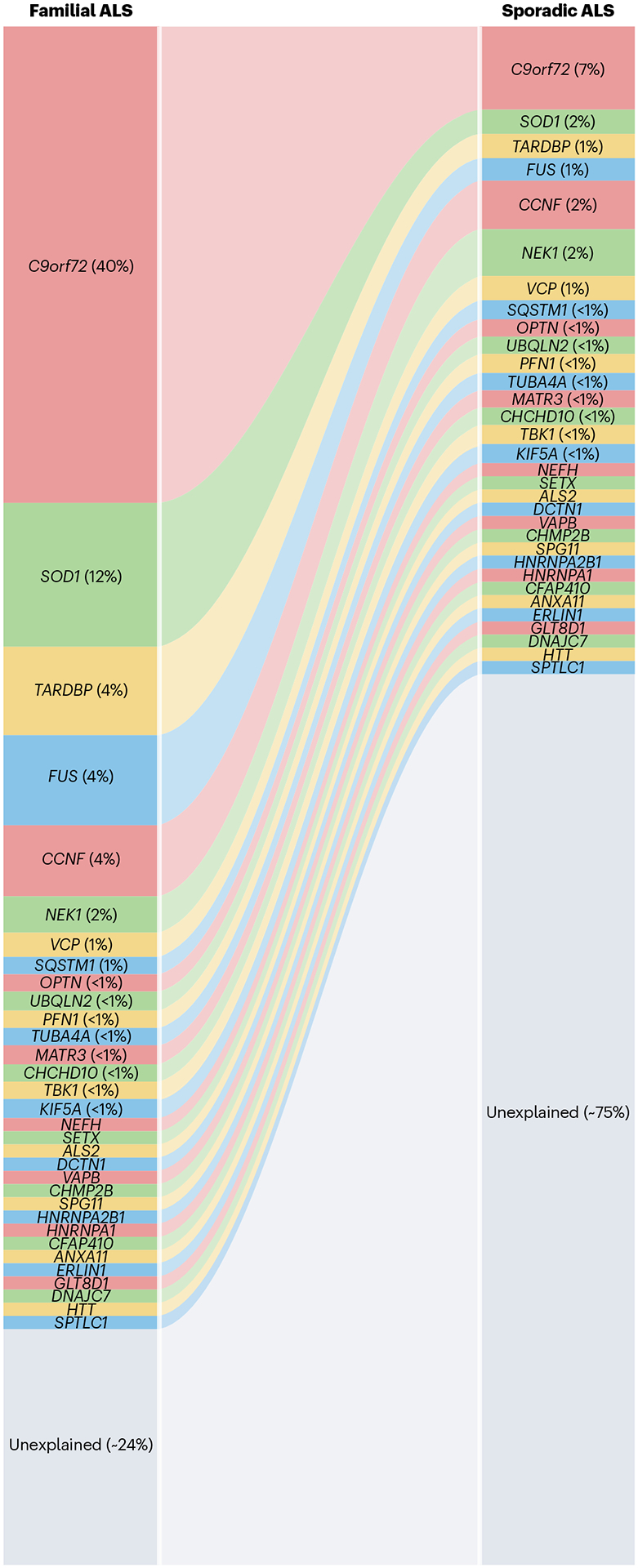
Mutations in the known amyotrophic lateral sclerosis (ALS) genes explain approximately 76% of familial and 25% of sporadic ALS231. The four most common ALS-associated genes, C9orf72, SOD1, TARDBP and FUS comprise 60% of familial and 11% of sporadic ALS. The proportion attributed to recently identified or rarely implicated genes has not been established.
Kinesin family member 5A (KIF5A).
Although the KIF5A locus had been suggested as a candidate genetic risk factor71, it was identified as a definitive ALS gene based on a genome-wide study of common and rare variants in 2018 (refs. 72,73). The ALS-related mutations result in the skipping of exon 27, resulting in a protein with a novel 39 amino acid residue C-terminal sequence. The same mutations have also been shown to cause FTD74. These mutations lead to a toxic gain of function involving abolished auto-inhibition of the kinesin activity, resulting in hyperactive axonal transport75. Discovering mutations in KIF5A and several other motor protein and cytoskeleton-related genes, such as DCTN1, PFN1, SPAST and TUBA4A, highlighted the pivotal role of the cytoskeleton in ALS pathogenesis76. It also suggests that therapeutic approaches that enhance cytoskeletal integrity or maintain axonal transport machinery may benefit patients with ALS77.
Endoplasmic reticulum lipid raft associated protein 1 (ERLIN1).
ERLIN1 was identified as a causative gene for an early-onset, autosomal recessive form of ALS based on a whole-exome sequencing analysis of a large, consanguineous Turkish family78. Many affected family members manifested symptoms in early life (before 25 years) and displayed a slowly progressive form of ALS. ERLIN1 mutations were first reported in three families with hereditary spastic paraplegia, and since then, only one additional mutation has been described in a Chinese hereditary spastic paraplegia pedigree, indicating a rare role of ERLIN1 in neuromuscular diseases79,80.
ERLIN1 encodes endoplasmic reticulum lipid raft associated protein (ERLIN1), which is part of a protein complex implicated in degrading inositol 1,4,5-trisphosphate intracellular receptor (IP3R) ion channels. Dysregulation of IP3Rs disrupts calcium release from the neuronal endoplasmic reticulum, leading to synaptic loss and dysfunction81. Aberrant activity of IP3Rs was noted in Huntington disease82 and ITPR1-related spinocerebellar ataxia83, again showing that mutations in a single gene may be associated with diverse neurological conditions (that is, pleiotropy).
DnaJ heat shock protein (Hsp40) family member C7 (DNAJC7).
Rare protein-truncating and damaging missense variants in DNAJC7 were enriched in patients with ALS in an exome-wide association analysis84. Further exploration in Asian cohorts identified potentially deleterious DNAJC7 variants in sporadic cases of ALS85–88. However, pathogenic DNAJC7 mutations co-segregating with the disease in an ALS pedigree have not been reported to date, which would confirm mutations in this gene as disease-causing. Nonetheless, functional evidence supports involvement of the gene in ALS. DNAJC7 encodes a member of the Hsp40 heat shock protein family, and other members of the same family act as molecular chaperones involved in the folding and clearance of misfolded proteins89. For example, Hsp90 contributes to the elimination of misfolded TDP43, and DNAJC7 in mice may regulate the degradation of ALS-associated proteins, such as SOD1, TDP43 and FUS90. Protein misfolding and aggregation are a central theme in ALS, and mitigating misfolding and promoting clearance by modulating the DNAJC7 function presents an attractive therapeutic target. Such an intervention would be broadly useful for other ALS-related proteinopathies.
Huntingtin (HTT).
Pathogenic CAG repeat expansions in the first exon of the HTT gene were recently identified as a rare cause of pure ALS/FTD phenotypes based on large whole-genome sequence datasets91. Although infrequent, the repeat expansion was present in multiple patient cohorts91, and recent work has confirmed the original findings92,93. None of the patients displayed chorea, abnormal involuntary movements that are a core symptom of Huntington disease, during their illness, implying a pure ALS/FTD phenotype in these cases. The post-mortem findings of two patients with ALS with HTT repeat expansions revealed the classic motor neuron loss and TDP43 pathology of ALS/FTD, ruling out mimic syndromes as an explanation. The neostriatal atrophy that pathologically defines Huntington disease was also absent in these cases91. Interestingly, mutant huntingtin protein disrupts the nucleocytoplasmic transport94, a process also implicated in the pathogenic C9orf72 repeat expansions95. Although HTT expansions are rare in ALS/FTD, this finding again emphasizes the molecular overlap across distinct neurodegenerative clinical entities. It highlights the power of genomics to define and classify them molecularly. Clinically, it opens opportunities to bridge to disease-modifying interventions, such as antisense oligonucleotide (ASO) therapies designed to lower the amount of mutant proteins in the brain (see below)96.
Serine palmitoyltransferase long chain base subunit 1 (SPTLC1).
Recently, de novo mutations in the SPTLC1 gene were identified as a cause of juvenile ALS, a rare form of ALS that occurs before the age of 25 years97,98. Mutations in this gene cause autosomal dominant hereditary sensory autonomic neuropathy, type 1A (HSAN1A), by disrupting an essential enzyme complex in the sphingolipid synthesis pathway99. These data broaden the phenotypes associated with SPTLC1 mutations to include juvenile ALS and implicate disruption of sphingolipid metabolism in motor neuron disease. Cell-based assays of serine palmitoyltransferase activity confirmed that the recurrent de novo Ala20Ser mutation altered the function of the encoded enzyme, leading to increased aberrant utilization of alanine and glycine as substrates97,100. Notably, nutritional supplementation with serine is a current therapeutic strategy for HSAN1A101 and may be a viable option among juvenile patients with ALS, although further work is needed to confirm this97.
Other aspects of ALS genetics
Genome-wide association studies in ALS
As for other neurological diseases, GWAS have been a major contributor to our knowledge of the genetic basis of ALS102. The first ALS GWAS was published in 2007 and did not identify any significant genetic loci103. Since then, much larger GWAS projects have revealed additional risk loci for ALS. In 2009, an association analysis for sporadic ALS first implicated the UNC13A locus104. The association signal in chromosome 9p21 and SOD1 reached genome-wide significance in a Finnish cohort of familial ALS105,106. These efforts paved the way for identifying the C9orf72 repeat expansion in 2011 (refs. 12,55,56). Further GWAS identified and replicated variants in C9orf72, CFAP410, KIF5A, MOBP, SARM1, SOD1, TBK1, TNIP1 and UNC13A72,107–110 (Fig. 2). Initial studies comprised individuals with European genetic ancestry, whereas recent multi-ethnic meta-analyses contained Asian cohorts105,107,108,111. Notably, GWAS and familial studies worked synergistically to unravel the genetic architecture of ALS with many Mendelian genes involved in ALS, such as SOD1, NEK1, C9orf72 and TBK1, also showing association in GWAS19,55,71,105,106,112–114.
Fig. 2 |. Thirty years of gene discovery in ALS and FTD.

The first Mendelian gene for amyotrophic lateral sclerosis (ALS) was identified in 1993 (ref. 19), and the first genome-wide association study (GWAS) for ALS was published in 2007 (ref. 103). Further GWAS and gene hunting studies identified several significant loci (P < 5 × 10−8) and genes associated with ALS, frontotemporal dementia (FTD) or ALS/FTD. Summary-level associations were extracted from the EBI-GWAS website102.
Pathway and multi-omic analyses in ALS
Genetic data have implicated various pathways and cellular mechanisms in the pathogenesis of familial ALS, such as protein homeostasis, cytoskeleton alterations and RNA metabolism16. More recently, our knowledge of the biological processes involved in the sporadic form of the disease has also improved; enrichment analysis based on large-scale datasets identified genes and pathways that can be targeted for therapeutic intervention. These include neuronal projection, membrane trafficking and signal transduction mediated by ribonucleotides115. Detailed dissection of these pathways identified six differentially expressed genes associated with increased ALS risk (ATG16L2, ACSL5, MAP1LC3A, MAPKAPK3, PLXNB2 and SCFD1)115. Autophagy was also central to C9orf72-related neurodegeneration115, an observation consistent with the encoded protein being an integral part of the autophagy initiation complex116,117.
Genetic data can also identify the cell types involved in ALS, often yielding surprising insights and new avenues for research. For instance, an approach combining GWAS data and single-nucleus RNA sequencing data identified cortical GABAergic interneurons and oligodendrocytes as associated with ALS risk115. Nearly 20 years ago, autopsy studies implicated the same class of interneurons118. The confluence of genetic and pathological data suggests that damage to GABAergic interneurons is an early event in ALS. Similarly, this data-driven approach demonstrates that oligodendrocytes contribute to the disease pathogenesis and are worthy of additional research.
Various omics-based approaches have provided insights into mechanisms of ALS pathogenesis. Spatial transcriptomics is an emerging technique that maps gene expression in individual cell types by generating spatially resolved RNA sequencing profiles from tissues of interest. Using this cutting-edge approach, Maniatis and colleagues119 identified TYROBP–TREM2-mediated signalling as an early mechanism associated with microglial gene expression in ALS mice. More recently, large-scale transcriptomics analysis of human post-mortem ALS spinal cords identified new candidate genes with cell-type-specific functions; microglia were assigned as a putative cell type for ACSL5 and oligodendrocytes for FNBP1, SH3RF1 and NFASC120. Transcriptome-wide analysis incorporating clinical data revealed that expression of microglial gene modules correlated with disease duration, highlighting the role of glial activation in ALS120. A multi-omic analysis of human induced pluripotent stem cell-derived spinal motor neurons identified altered lipid metabolism pathways in mutant SOD1 and C9orf72 spinal motor neuron populations121. Additional metabolomics profiling confirmed the upregulation of lipid metabolism and identified elevated arachidonic acid levels in spinal motor neurons, pinpointing a potential therapeutic target121.
Although these studies are relatively new, they highlight the application of genetic data to predict functional outcomes. Future large-scale studies will shape future drug development as personalized medicine takes centre stage.
Whole-genome sequencing and other mutation types in ALS
Various other mutation types have been implicated in ALS, highlighting the diverse genetic architecture of the disease. Repeat expansions, structural alterations, non-coding mutations, de novo variation and activation of human endogenous retroviruses (HERVs) fill the contours of the ALS genetics puzzle. Our ability to detect these distinct types of mutation has been driven by advances in next-generation sequencing technologies and the evolution of more powerful bioinformatics tools.
The discovery of the C9orf72 repeat expansions implicated this type of structural variation in ALS55. This realization, and the availability of reliable in silico repeat detection tools, paved the way for the subsequent identification of HTT repeat expansion as a cause of ALS91. In addition, repeat expansions in ATXN1, ATXN2 and NIPA1 have now been associated with an increased risk of developing ALS122–126. Other structural variants have been identified occasionally in patients with ALS, including an insertion in ERRB4 and an inversion in VCP127.
Non-coding variants within the introns and untranslated regions of genes may account for a considerable proportion of the missing heritability of ALS128. Previously, a linkage analysis combined with targeted sequencing in a large family identified an intronic variant that introduces a pseudoexon into the SOD1 transcript, leading to truncation of the protein129. More recently, next-generation sequencing analysis showed an increased burden of untranslated region variants in SOD1, TARDBP, FUS, OPTN, VCP and UBQLN2 (ref. 130). Variants in the 3′ untranslated region of the IL18RAP gene may protect against developing ALS131. Intronic mutations leading to aberrant splicing of SOD1 and KIF5A have also been described in large ALS families, confirming the pathogenic potential of this often-overlooked part of the genome74,129,132,133. Additionally, aberrant transcript splicing has long been recognized as a core feature of ALS due to the loss of the normal function of TDP43 of repressing non-conserved cryptic exons, leading to nonsense-mediated decay of the associated transcripts134,135. The UNC13A risk factor can act in a similar manner by potentiating cryptic exon inclusion136.
Although similar molecular mechanisms are involved in familial and sporadic ALS, the disease-causing mutations identified in familial cases to date explain only a small proportion of sporadic cases. This may be due to incomplete penetrance in the families, lack of DNA from other relatives, small family structure, or lack of the proband knowing clinical information about another. In addition, spontaneous de novo mutations have been described in the FUS, SOD1 and VCP genes, explaining at least some sporadic cases48,137,138. Parent–offspring trio sequencing identified spontaneous variants in SPTLC1 as the underlying cause in sporadic juvenile ALS cases97,98. This research suggests that de novo mutations may be more crucial in the pathogenesis of ALS than previously recognized.
A limitation of the current genetic studies is that they are constrained by the number of nucleotides sequenced in each read (that is, ~200 bp on Illumina platforms). However, nearly 40% of the genome is made of highly repetitive retroviral elements, and short reads derived from these regions are challenging to align to reference genomes and are thus often discarded from analyses. Long-read sequencers, such as the Pacific Biosciences (PacBio) or Oxford Nanopore technologies, are needed for this purpose. These retrovirus-derived genomic regions may have a critical role in the pathophysiology of ALS, and it is crucial to determine whether mutations in these regions contribute to ALS. For example, autopsy studies show that reactivation of HERVs, particularly the HERV-K subtype HML-2, is associated with ALS and may influence the clinical course of the disease139. Reverse transcriptase, a marker of endogenous retroviral activity, has been detected in the blood and cerebrospinal fluid (CSF) of patients with ALS, suggesting retroviral involvement in ALS140. Moreover, the HML-2 envelope triggers an ALS-like syndrome in transgenic mice141. Antiretroviral therapies that inhibit HERV-K in patients with ALS are currently in clinical trials142. Despite these compelling data, the information provided by long-read sequencing will be required to confirm the role of HERVs in the pathogenesis of ALS because of their repetitive nature and typical length (~7 kb).
As the size and diversity of cohorts increase, so will the ability to resolve the genetic architecture of ALS, providing further insight into the cellular processes that underlie motor neuron death. Making these data readily available to researchers is equally essential and will maintain the momentum of gene discovery in ALS by lowering the costs associated with genomic research. Several online platforms, such as Answer ALS, ALS Compute, New York Genome Center ALS Consortium, Project MinE and the database of genotypes and phenotypes (dbGaP), are already sharing genetic data and enabling future research.
Overlap between ALS and other diseases
A central theme emerging from genetic studies is the overlap of ALS with other diseases and metabolic traits. An early hint of this came with the discovery of VCP mutations in families with ALS that also manifested FTD143,144. Mutations in this gene had been implicated as a cause of an unusual syndrome comprising inclusion body myositis, Paget disease of the bone and FTD. The rarity of VCP mutations led to the belief that the co-occurrence of ALS and FTD in the same patient was an outlier phenomenon. It was not until the revelation of C9orf72 as a common cause of both ALS and FTD that the importance of the phenotypic overlap was appreciated55. Since then, several other genes causing both ALS and FTD, such as CHCHD10, KIF5A, FUS, HTT, SPTLC1, SQSTM1, TARDBP and TBK1 (refs. 12,69), have been identified, strengthening their convergence into a single spectrum. It is now recognized that up to 60% of patients with ALS develop cognitive impairment during their illness145. Conversely, up to one-third of patients with FTD develop motor dysfunction127.
Genetic evidence also suggests that energy metabolism is dysregulated in ALS. Older epidemiological studies had reported high levels of cholesterol and other lipids in the central nervous system (CNS) and circulation in ALS97,146–156. However, these results were based on small numbers of patients, and several other studies observed no difference in blood lipid levels between patients with ALS and controls156 or reported a negative association157–159. A possible explanation for excess cholesterol in the CNS of patients with ALS is that the cholesterol is released after neuronal death and is not removed properly147,159. More recently, Mendelian randomization studies based on genetic data from sizeable patient cohorts confirmed a causal link between blood LDL-cholesterol levels and ALS109,160,161. Furthermore, a metabolomics analysis identified recurrent dysregulation in pathways related to lipid metabolism in independent ALS cohorts162.
Mendelian randomization studies also suggested that type 2 diabetes is protective against ALS163–166. These data are consistent with population-based observational studies showing that a high body mass index and type 2 diabetes are associated with a lower risk of ALS167,168. These observations suggest that metabolic disturbance may be a fundamental component of the ALS syndrome, similar to what has been observed in more common neurodegenerative diseases169.
Aside from the phenotypic overlap, there is also considerable evidence of pleiotropy, with mutations in the same gene causing two distinct neurological diseases (but not in the same patient). For example, mutations in the N-terminal domain-encoding region of KIF5A are a known cause of Charcot–Marie–Tooth type 2 and hereditary spastic paraplegia170,171. Another example of genetic intersection is the SPTLC1 gene: the disease-causing mutations result in diverse pathologies leading to either ALS or a form of peripheral neuropathy known as hereditary sensory autonomic neuropathy type 1 (refs. 97,99,101). Expanded repeats in the genes associated with hereditary cerebellar ataxia (ATXN1 and ATXN2) and hereditary spastic paraplegia (NIPA1) are also risk factors for ALS122–126. Genetic data show that ALS is a complex and nuanced disease with phenotypic and aetiological overlap with other neurological and systemic conditions.
Diverse populations and rare ALS subtypes
The incidence of ALS may be lower among populations of African, Asian and Hispanic ancestries than among populations of European ancestries3,172. It is unclear whether this rate disparity is due to differences in epidemiological study design or in population demographics, environmental factors or genetic predisposition3. There is compelling evidence that the genetic architecture of ALS varies with geography173. The pathogenic repeat expansion of C9orf72 is a widespread cause of the disease in populations of European ancestries because of a founder effect but is diminishingly rare in populations of Asian ancestries174 (Fig. 3). By contrast, mutations in SOD1 and FUS are more common in Japanese patients with ALS175.
Fig. 3 |. Frequency of causal mutations in the four most common ALS genes in diverse populations.
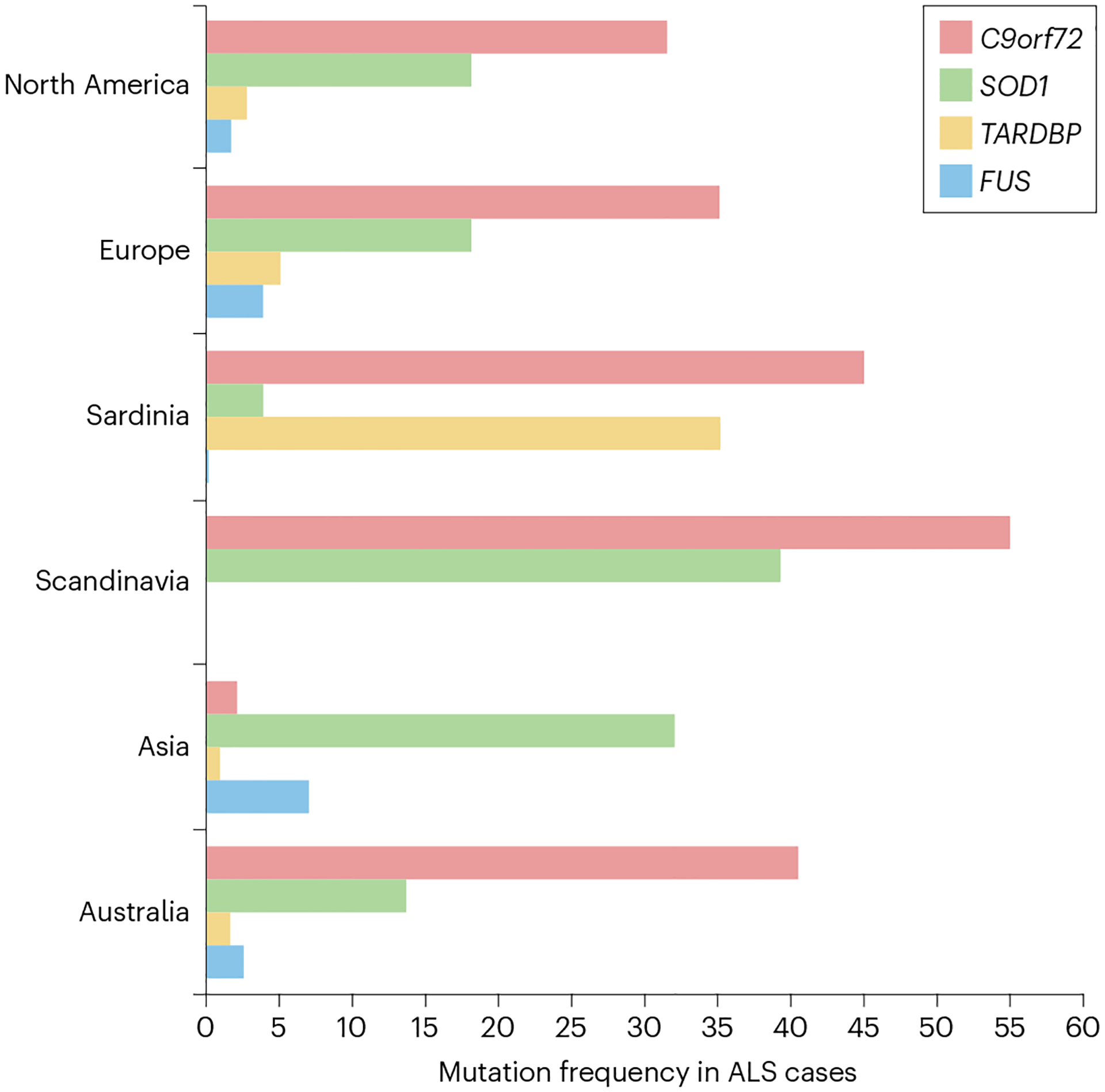
The incidence of amyotrophic lateral sclerosis (ALS) varies in distinct populations. Although the C9orf72 repeat expansion is rare in Asia, it is the most frequent cause of ALS in Scandinavian populations. Similarly, a founder mutation in TARDBP explains approximately 35% of the disease in Sardinia.The frequency of familial cases explained by the mutated genes in different geographical regions is obtained from references173,232,233; the regions are North America (Canada and USA); Europe (Italy, Germany, Spain, Switzerland, France, Belgium, UK, Netherlands and Slovenia); Sardinia; Scandinavia (Sweden and Finland); Asia (Japan, Korea, China, India and Iran); and Australia.
Isolated populations with a high incidence of ALS have been closely studied to understand the disease176,177. For example, nearly one-third of ALS cases in the island population of Sardinia are due to a founder mutation in TARDBP (Ala383Thr)178, and the Asp91Ala mutation in SOD1 is more frequent in the Scandinavian population23,24 (Fig. 3). Likewise, a VAPB mutation (Phe56Ser) accounts for 44% of familial ALS in Portuguese–Brazilian populations179. Investigating unique ALS variants also offers meaningful opportunities to understand ALS. For example, exome sequencing of families with Brown–Vialetto–Van Laere syndrome (ALS plus deafness and blindness) identified mutations in a series of genes encoding riboflavin receptors180,181. This finding led to high-dose vitamin B2 supplementation as an effective treatment that halts progression in this rare form of ALS180.
Gene–environment interactions
Epidemiological studies showed that several environmental and lifestyle factors, such as agricultural and industrial chemicals, certain occupations, cigarette smoking and heavy exercise, may be involved in ALS160. However, these studies may have been hampered by myriad confounding variables and the difficulty of establishing causal relationships based on observational data. Mendelian randomization overcomes this limitation by using genetic elements to explore the causal relationship between exposure and outcome. This analytical technique relies on genotypes that remain constant over a lifetime and are randomized during game-togenesis. Because of this, it provides a reliable identification of causal associations by reducing confounding and excluding reverse causality161,182. To date, Mendelian randomization studies point to alcohol consumption, educational attainment, physical exercise, dyslipidaemia and smoking as ALS risk factors109,160,161,166,183. Epigenetic mechanisms, such as DNA methylation, microRNA (miRNA) dysregulation, chromatin remodelling and histone modifications, have been shown to operate in ALS184–189 and are suspected of mediating the effects of these environmental factors on disease pathogenesis. How these exposures alter the cell’s epigenetic profile to bring about ALS is unknown.
These environmental and lifestyle exposures are common in the population, yet ALS is rare. Gene–environment interactions may explain this incongruity; extrinsic or genetic factors alone would not lead to motor neuron degeneration. Instead, only those exposed individuals with an underlying genetic predisposition would be at risk of disease190. Gene–environment interactions may also be especially relevant in late-onset, rare diseases whose sporadic occurrence appears stochastic. In reality, it hints at a much more complicated theory of the disease160.
Similarly, infections can also unmask or precipitate ALS. In particular, patients with HIV or HTLV-I infection can occasionally develop ALS. When ALS occurs in patients with HIV, it is often rapidly progressive; however, if treated with antiretroviral drugs early in the syndrome, the symptoms can be halted or reversed in some cases191.
A model of ALS that integrates genetics, environment and ageing
Various models have been proposed to explain the occurrence of ALS. Although each schema has merit, they typically postulate that disease-causing events occur sequentially or do not account for the interactions between the various factors that drive cell death11,192,193. For example, the multistep model of ALS portrays the path to the disease as a series of steps that occur over time from genetic and environmental insults193. Aside from the obvious difficulty in calculating the number of steps involved in the disease process, it implies that these events occur separately. Instead, numerous factors impinge on motor neurons in a complex and interdependent manner to cause neurodegeneration.
Here, we propose an updated model whereby disease-causing mutations, genetic modifiers, ageing and environmental factors come together synergistically to cause disease. Under this paradigm, ALS risk can be visualized as a series of overlapping concentric rings (Fig. 4). At the centre are ALS and the long-term health of the motor neurons. The first ring represents the principal disease-causing mutation, such as in C9orf72 or SOD1, or polygenic risk in the case of sporadic ALS. The second ring represents genetic variation influencing phenotypic manifestations of the disease, such as onset age, site of symptom onset and progression rate. These ancillary genetic effects can be monogenic or polygenic. The third ring symbolizes environmental and lifestyle factors that affect a patient’s risk of developing ALS. These include cigarette smoking, hyperlipidaemia, excessive exercise and military service160. The outer ring denotes the ageing process, which is still one of the most substantial risk factors for ALS.
Fig. 4 |. A model of ALS that integrates genetics, environment and ageing.
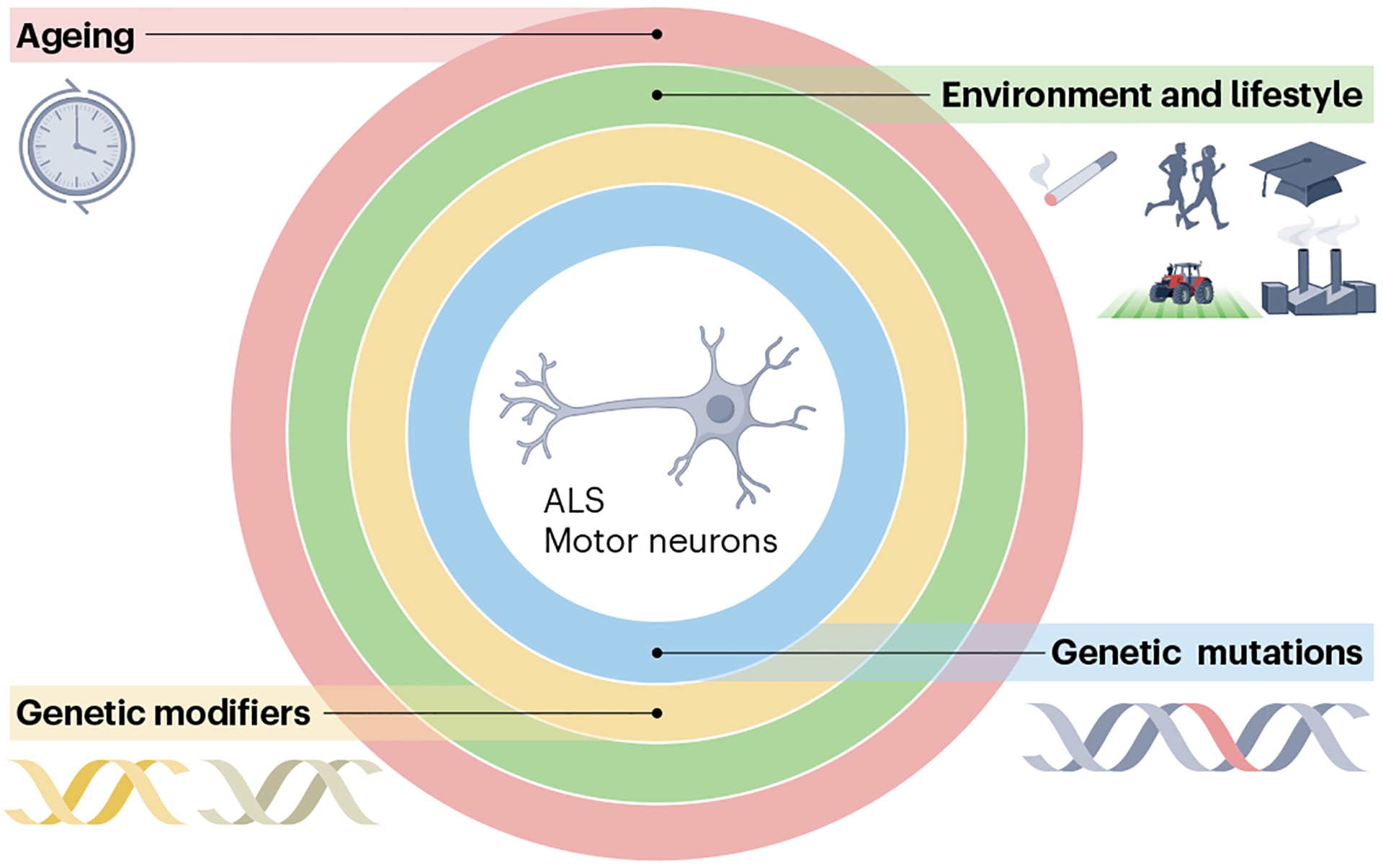
Amyotrophic lateral sclerosis (ALS) may occur owing to multiple overlapping factors, including a primary disease-causing mutation, a genetic predisposition that influences disease manifestation, environmental and lifestyle factors, and ageing. Together, they comprise a multifaceted, complex system, in which the integrity of each element is vital to maintain the integrity of the motor neurons.
Our model conveys the essential concept of the underlying factors operating collectively. This captures the interdependent forces that lead to neurodegeneration, as well as the view that each has a role in disease occurrence. Such multiform interactions between genetic, environmental and lifestyle factors are now recognized as central drivers of neurodegeneration194. For instance, several studies have shown that an individual could be genetically predisposed to cigarette smoking, which then increases the risk of developing ALS161,166. Gene–environment interactions may be particularly relevant in the pathogenesis of rare, sporadic diseases that occur in later life, such as ALS160.
The model is reminiscent of Pritchard’s omnigenic archetype proposed for complex diseases, while also incorporating salient clinical aspects of ALS195. Indeed, this multidimensional model applies to almost all neurodegenerative diseases, such as Alzheimer disease and Parkinson disease. Instead of offering a mechanistic view of cellular pathophysiology, a perspective that shifts with each discovery, it provides a holistic and enduring compendium into how geneticists think about diseases. In this way, our prototype is a helpful guide for designing future genetic and environmental studies by emphasizing the need to incorporate diverse factors into the analysis.
Our ring model also has implications for ALS treatment. The complex ecosystem may explain treatment failures in molecular therapies targeting monogenic forms of ALS; addressing ancillary factors may be necessary to slow disease progression once symptoms emerge. Applying molecular therapies to patients with ALS will test this hypothesis in the next few years. These first-in-human clinical trials may also establish a better understanding of how the components interact to cause motor neuron degeneration.
Molecular therapies in ALS
Current treatment options for ALS are limited, with only three drugs currently on the market. Riluzole, a small molecule that targets voltage-dependent Na+ channels to reduce motor neuron excitotoxicity, was approved by the FDA in 1995. Edaravone, a small-molecule antioxidant, gained FDA approval in 2017. More recently, the FDA approved a combination of sodium phenylbutyrate and ursodoxicoltaurine (Albrioza) that is aimed at ameliorating endoplasmic reticulum stress and mitochondrial dysfunction.
Although these drugs represent progress in the ALS field, they have only a modest effect on slowing disease progression196,197. Unsurprisingly, efforts continue towards developing drugs with greater efficacy and longer-lasting impact. To this end, molecular therapy approaches have gained traction in ALS. The advancements in our understanding of ALS genomics described above coincide with the first oligonucleotide and gene therapies hitting the markets, most notably embodied by the success of nusinersen and onasemnogene abeparvovec-xioi for treating spinal muscular atrophy198.
Molecular therapies come in various modalities that offer the ability to selectively silence, upregulate, edit or introduce a gene or transcript to correct the underlying cause of disease either temporarily or permanently (Box 1). Various molecular therapy modalities can potentially treat ALS depending on the nature of the causal mutation. Diseases caused by a gain-of-function mutation may be mitigated by silencing the mutant gene or transcript. Alternatively, upregulation or introduction of a gene or transcript may correct disease caused by a loss-of-function mutation. An early example of gene therapy attempting to treat ALS by replenishing a supporting protein is Engensis (also known as VM202), a non-viral gene therapy that uses a plasmid to deliver the hepatocyte growth factor gene directly to nerves and muscle199. A phase I/II clinical trial (Clinicaltrials.gov identifier NCT02039401) showed that intramuscular injections of the therapy were generally well-tolerated and may slow disease progression.
Box 1. Therapeutic modalities to treat ALS.
Molecular therapies include DNA or RNA moieties that are introduced into cells for therapeutic benefit. Molecular therapy that silence the expression of the causal gene can potentially treat diseases that result from gain-of-function mutations. Various modalities can accomplish gene silencing, the most promising of which are oligonucleotide therapeutics, with more than a dozen of this class having gained approval from the FDA. Antisense oligonucleotides (ASOs) are single-stranded oligonucleotides, typically 14–25 nucleotides in length. ASOs are used to alter splicing patterns of genes (for example, nusinersen) or directly decrease the expression level of a transcript. In that instance, the binding of the ASO to a complementary strand sequence in the target mRNA leads to recruitment of RNase H and subsequent degradation of the target mRNA, thereby preventing the translation of the aberrant mRNA. At least five ASOs are in clinical trials for amyotrophic lateral sclerosis (ALS) and more are in development (Table 2).
Small interfering RNAs (siRNAs) and short hairpin RNAs (shRNAs), which function as precursors to siRNAs, are oligonucleotide therapies that use RNA interference (RNAi). RNAi involves recruitment of the RNA-induced silencing complex (RISC), leading to cleavage of the target mRNA, silencing the expression of the target gene. At least five RNAi drugs have been approved, predominantly for rare diseases. Ongoing research shows promise for bringing RNAi-based ALS treatments to the clinic in the future234.
Gene therapy can treat diseases arising from loss-of-function mutations by introducing or upregulating the wild-type gene or transcript. Gene delivery using viral vectors such as adeno-associated virus and lentivirus, or non-viral vectors, to deliver DNA to the patient’s cells to produce a functional protein, is being extensively tested in the clinic235. Engensis, a therapy that uses a plasmid to deliver the hepatocyte growth factor gene (HGF), is in a clinical trial for ALS. A newer approach that may lead to successful therapies aims to increase the translation of mRNA from the wild-type allele in disease that results from a mutation in one allele. Small activating RNAs (saRNAs) are also an emerging class of RNA therapeutics designed for RNA activation236. Other novel RNA-based approaches to upregulate gene expression are under investigation, such as targeted augmentation of nuclear gene output (TANGO) from Stoke Therapeutics237.
Regardless of the nature or outcome of the mutation, there is also great potential for genome editing to correct a mutation directly at the DNA level. Gene editing with CRISPR–Cas9, zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) correct mutations at the DNA level. CRISPR–Cas-based gene editing first reached the clinical stage for sickle cell disease, and various CRISPR-based therapies are in clinical trials for other blood disorders, cancers, genetic blindness and more238–240. ZFN- and TALEN-based therapies have not yet reached this stage for ALS, although the potential for their use is exciting.
Here, we focus on therapies directed at RNA, namely ASOs, as most agents in clinical trials for ALS fall into this class. In that regard, ALS has become an archetypal neurological disorder to study these new methodologies. In comparison with Alzheimer disease and Parkinson disease, the end point in ALS clinical trials is easier to measure and is reached more quickly because of the rapidly progressive nature of the disease. To date, therapeutic development has focused on dampening the toxic gain-of-function effects of the common mutant ALS genes SOD1, FUS and C9orf72. The rationale of these efforts is that removing the aberrant RNA transcripts, for example, with ASOs, will stop the disease process. Although no disease-modifying ALS treatment has yet been approved, several potential therapies are in clinical trials (Table 2).
Table 2 |.
Current clinical trials of molecular therapies for ALS
| Treatment | Modality | Target | Phase | Sponsor | clinicaltrials.gov identifier |
|---|---|---|---|---|---|
| ION363 (jacifusen) | ASO | FUS | III | Ionis Pharmaceuticals, Inc. | NCT04768972 |
| BIIB067 (tofersen) | ASO | SOD1 | III | Biogen | NCT04856982 |
| WVE-004 | ASO | C9orf72 | I (ALS), II (FTD) | Wave Life Sciences Ltd | NCT04931862 |
| BIIB105 (ION541) | ASO | ATXN2 | I | Biogen | NCT04494256 |
| BIIB078 (IONIS-C9Rx) | ASO | C9orf72 | I | Biogen | NCT04288856 |
| Engensis (VM202) | Non-viral plasmid gene delivery | HGF addition | II | Helixmith Co. Ltd | NCT05176093, NCT04632225, NCT02039401 |
ALS, amyotrophic lateral sclerosis; ASO, antisense oligonucleotide.
ION363, informally known as jacifusen after the first patient to receive the drug, is an ASO that targets FUS mRNA to reduce FUS expression200. ION363 was first administered through compassionate-use protocols to a patient with life-threatening ALS linked to the Pro525Leu mutation, and it reduced total and mutant FUS in brainstem tissue from the patient. However, the ASO has potentially broader applicability, and, as of 2021, ION363 has entered a phase III clinical trial to evaluate its efficacy in a larger number of patients with FUS-related ALS. Early indications are that the ASO silences FUS expression, although the effect on clinical outcome remains unclear200.
BIIB067, known as tofersen, is an ASO that inhibits the production of the SOD1 protein. Although tofersen reduced SOD1 levels in CSF, it failed to meet its primary end point in a phase III clinical trial but did show encouraging results in some secondary end points201,202. Maximizing the beneficial effect of tofersen may entail starting the treatment as early as possible17. Future efforts will build on this lesson, not just with tofersen, but across the gamut of molecular therapies for neurological diseases. This trend is already emerging. Post-hoc analysis of the phase III study of tominersen showed a beneficial effect among patients with Huntington disease who received treatment early in their illness203. Recruitment is underway for a phase III trial of tofersen in pre-symptomatic carriers of SOD1 mutation with elevated neurofilament, a CSF biomarker of neuronal death and a harbinger of symptom onset204.
Early reports suggested that the ASO afinersen, developed to suppress the expression of C9orf72 transcripts containing the repeat expansion, may be tolerated at low doses, and it reduced levels of toxic dipeptides within the CSF, a marker of the repeat expansion205. However, in a recent upset in the field, another C9orf72-targeting ASO BIIB078 (formerly IONIS-C9Rx) failed to show clinical benefit in a phase I trial, even trending toward greater decline at a high dose. Nonetheless, efforts to treat C9orf72-ALS continue with the C9orf72-targeting ASO WVE-004 currently in phase I trials for ALS and a phase II trial for FTD.
BIIB105 (formerly ION541) is an ASO designed to degrade ATXN2 mRNA, and it has entered clinical trials for patients with ALS carrying pathogenic CAG repeat expansion in this gene. This therapy is also being tested in patients with ALS without CAG expansions based on the premise that ATXN2 interacts with TDP43, and modulating the gene will prevent TDP43 cytoplasmic accumulation206. Even partial success in this early-stage clinical trial would represent proof of principle in neurodegenerative disorders by demonstrating the benefit of modifying an entire pathway involved in a disease.
Next steps in developing molecular therapies for ALS
Several issues must be addressed to realize the full potential of ASOs and other DNA- and RNA-based therapies as treatments for ALS, and care should be taken to incorporate lessons from previous clinical trials in the ALS space over the past 30 years. These include determining the best time to start treatment, optimizing drug delivery, ensuring adequate target engagement and discerning how to conduct clinical trials involving only a handful of patients. First-in-human dosing of new molecular therapies in ALS is typically done in clinical trials in symptomatic patients or under compassionate-use protocols in individuals in whom disease has progressed to a debilitating and life-threatening stage by the time they receive the first dose. On a molecular level, the regular pool of ~500,000 motor neurons is depleted in the later stages of the disease, decreasing the targets on which an experimental treatment can exert an effect. As the efficacy and safety profile of oligonucleotide therapeutics becomes established, we will see more clinical trials testing these therapies in patients at earlier stages of the disease or even pre-symptomatically.
Direct injection into the CSF results in a favourable distribution of oligonucleotides in the CNS207. However, repeat injections may obstruct CSF pathways leading to hydrocephalus208. Future improvements in delivery may improve the biodistribution and pharmacokinetics of ASOs for neurological diseases209. For example, lipid nanoparticles and other nanomedicines are increasingly being used to enhance the delivery of drugs to target tissue. Newer work has focused on conjugating ligands such as cholesterol moieties to therapeutic molecules, enabling them to cross the blood–brain barrier and potentially opening the intravenous route of administration for CNS diseases210. There are even early indications that the oral administration of ASOs is feasible, which would enormously reduce the burden of long-term treatment in chronic diseases211.
Molecular therapy development to date has focused mainly on the more-common genes responsible for ALS. As oligonucleotide therapeutics become established, we will see more clinical trials testing these therapies in rarer genetic forms of the disease. Establishing the efficacy of ASOs and other individualized therapies will rely on our ability to robustly quantify clinical outcomes in a few or even a single patient. Each human study is an opportunity to learn about the effectiveness and safety of this relatively new class of therapeutics and to understand the cellular mechanisms that drive ALS. Conducting small clinical trials applies across the rare disease spectrum, not just to ALS. Because of this, collaboration and data sharing to aggregate experiences, such as the N=1 Collaborative network, will support this paradigm shift in rare and ultra-rare clinical trial design.
The future of ALS genetics and ASO therapy
Our understanding of ALS has considerably improved over the past three decades, driven mainly by identifying the genetic causes of this fatal neurodegenerative disease. At the same time, we now have several elegant examples of clinical proof of concept for oligonucleotide therapies and other DNA- and RNA-based therapeutic modalities, leading to the emergence of personalized drug development platforms. ASOs are well suited to such platform development as they are rapidly customizable, relatively simple and inexpensive to manufacture and demonstrate a growing safety and efficacy record. As we learn more about the genetics of ALS, and whole-genome and long-read sequencing is applied more broadly in the clinic, there will be more and more opportunities to develop personalized treatments for familial ALS. Over time, these therapeutic approaches may also be extended to the sporadic form of the disease, where targeting multiple genes in a single patient may be necessary.
The current high cost of developing personalized therapies is preventing widespread access. However, the future optimization of therapeutic development platforms will lower the cost and shorten their development time, thereby improving access to new therapies that go straight from the gene to the patient. We envisage a future time when pharmacies in all hospitals are equipped with nucleic acid synthesizers that can be instantly activated to produce safe and effective therapies personalized to the patients admitted to the neurology wards. ALS remains at the forefront of this bold new campaign, offering real hope to patients with neurodegenerative diseases and their families.
Acknowledgements
This work was supported by the NIH Intramural Research Program, the US National Institute on Aging (NIA) grant Z01-AG000949-02, the US National Institute of Neurological Disorders and Stroke (NINDS) grant NS03130 and the US National Center for Advancing Translational Sciences (NCATS).
Glossary
- De novo mutations
Mutations not inherited from either parent and present for the first time in a family.
- End point
The targeted outcome of a clinical trial to determine the efficacy and safety of the therapy. The amyotrophic lateral sclerosis functional rating — revised (ALSFRS-R) score is the commonly used primary end point for ALS clinical trials.
- Enrichment analysis
A statistical approach to identify over-represented or differentially expressed genes or biological pathways contributing to a phenotype.
- Familial aggregation studies
Methods to assess the relative risk of a trait in affected family members compared with unaffected family members. Familial aggregation is the tendency for a trait to occur or cluster in multiple family members beyond what would be expected by chance
- Founder effect
The reduction in genetic diversity when a newly established population is descendent from a small number of ancestors.
- Genome-wide association Studies
(GWAS). Studies in which genetic variants across the genome are genotyped and tested for association with a trait.
- Haploinsufficiency
A situation in which a diploid organism has only one copy of a gene when both copies are required for proper function.
- Hydrocephalus
Abnormal cerebrospinal fluid in the ventricles of the brain
- Linkage analysis
An approach that examines the cosegregation of the chromosomal region (or regions) with traits of interest, usually designed to localize disease-causing genes in co-segregating segments.
- Mendelian randomization
The use of genetic variation to address the causal effect of one trait on another, minimizing the issues of confounding and reverse causality.
- Mimic syndromes
A diverse group of conditions, the clinical features of which may resemble amyotrophic lateral sclerosis
- Missing heritability
The discrepancy between the heritability explained by trait-associated genetic variants from genome-wide association studies and the amount of total heritability from familial aggregation or twin studies
- Prion-like motifs
Unstructured protein domains that can undergo conformational switches in response to environmental stress. Their phase transition activity renders RNA-binding proteins prone to misfolding in neurodegenerative diseases.
- Segregation analysis
A study that examines the carrier status of a genetic variant in the affected and unaffected members of a family. It is used to validate whether a specific genetic variant is causal for the disease in a family and the established mode of inheritance based on the pedigreestructure.
- Twin studies
Studies that compare the resemblance of monozygotic (identical) and dizygotic (non-identical) twins. An increased concordance rate in monozygotic twins compared with dizygotic twins provides evidence of a genetic component.
- Whole-exome sequencing
Targeted sequencing method that selectively determines exome sequences, which are highly enriched for protein-coding regions.
- Whole-genome sequencing
Sequencing method that examines the entire genome of an individual
Footnotes
Competing interests
B.J.T. holds a patent on the diagnostic and therapeutic applications of the pathogenic repeat expansion in C9orf72. The other authors declare no competing interests.
References
- 1.Rowland LP & Shneider NA Amyotrophic lateral sclerosis. N. Engl. J. Med 344, 1688–1700 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Kumar DR, Aslinia F, Yale SH & Mazza JJ Jean-Martin Charcot: the father of neurology. Clin. Med. Res 9, 46–49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chio A et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41, 118–130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu L et al. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J. Neurol 267, 944–953 (2020). [DOI] [PubMed] [Google Scholar]
- 5.U.S. Food and Drug Administration. Meeting of the Peripheral and Central Nervous System Drugs Advisory Committee Meeting [online], https://www.youtube.com/watch?v=AVVZMMvDOUg (2022).
- 6.Arthur KC et al. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun 7, 12408 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Achtert K & Kerkemeyer L The economic burden of amyotrophic lateral sclerosis: a systematic review. Eur. J. Health Econ 22, 1151–1166 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Chio A, Gauthier A, Calvo A, Ghiglione P & Mutani R Caregiver burden and patients’ perception of being a burden in ALS. Neurology 64, 1780–1782 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Wicks P The ALS Ice Bucket Challenge – can a splash of water reinvigorate a field? Amyotroph. Lateral Scler. Frontotemporal Degener 15, 479–480 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Sohn E Fundraising: the Ice Bucket Challenge delivers. Nature 550, S113–S114 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Goutman SA et al. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 21, 465–479 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Renton AE, Chio A & Traynor BJ State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci 17, 17–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keller MF et al. Genome-wide analysis of the heritability of amyotrophic lateral sclerosis. JAMA Neurol. 71, 1123–1134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Chalabi A et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 81, 1324–1326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang F et al. Familial aggregation of amyotrophic lateral sclerosis. Ann. Neurol 66, 94–99 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chia R, Chio A & Traynor BJ Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez ER, Borschel WF & Traynor BJ New antisense oligonucleotide therapies reach first base in ALS. Nat. Med 28, 25–27 (2022). [DOI] [PubMed] [Google Scholar]
- 18.Siddique T et al. Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N. Engl. J. Med 324, 1381–1384 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Rosen DR et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 (1993). [DOI] [PubMed] [Google Scholar]; This is the first study to identify SOD1 mutations as the genetic cause of familial ALS.
- 20.Abel O, Powell JF, Andersen PM & Al-Chalabi A ALSoD: a user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat 33, 1345–1351 (2012). [DOI] [PubMed] [Google Scholar]
- 21.McCann EP et al. Evidence for polygenic and oligogenic basis of Australian sporadic amyotrophic lateral sclerosis. J. Med. Genet 58, 87–95 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Andersen PM et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain 119, 1153–1172 (1996). [DOI] [PubMed] [Google Scholar]
- 23.Parton MJ et al. D90A-SOD1 mediated amyotrophic lateral sclerosis: a single founder for all cases with evidence for a Cis-acting disease modifier in the recessive haplotype. Hum. Mutat 20, 473 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Feneberg E, Turner MR, Ansorge O & Talbot K Amyotrophic lateral sclerosis with a heterozygous D91A SOD1 variant and classical ALS-TDP neuropathology. Neurology 95, 595–596 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Cudkowicz ME et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann. Neurol 41, 210–221 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Berdynski M et al. Recurrent G41S mutation in Cu/Zn superoxide dismutase gene (SOD1) causing familial amyotrophic lateral sclerosis in a large Polish family. Amyotroph. Lateral Scler 13, 132–136 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Berdynski M et al. SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci. Rep 12, 103 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuzma-Kozakiewicz M et al. Putative founder effect in the Polish, Iranian and United States populations for the L144S SOD1 mutation associated with slowly uniform phenotype of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener 22, 80–85 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Bernard E et al. Clinical and molecular landscape of ALS patients with SOD1 mutations: novel pathogenic variants and novel phenotypes. A single ALS center study. Int. J. Mol. Sci 21, 6807 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gamez J et al. Mutational analysis of the Cu/Zn superoxide dismutase gene in a Catalan ALS population: should all sporadic ALS cases also be screened for SOD1. J. Neurol. Sci 247, 21–28 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Hou L et al. Screening of SOD1, FUS and TARDBP genes in patients with amyotrophic lateral sclerosis in central-southern China. Sci. Rep 6, 32478 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayashi Y, Homma K & Ichijo H SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv. Biol. Regul 60, 95–104 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Bosco DA et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci 13, 1396–1403 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raoul C et al. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat. Med 11, 423–428 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Ralph GS et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med 11, 429–433 (2005). [DOI] [PubMed] [Google Scholar]; The first study to show the therapeutic efficacy of RNAi in ALS.
- 36.Smith RA et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest 116, 2290–2296 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neumann M et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006). [DOI] [PubMed] [Google Scholar]; This study was the first to identify cytoplasmic accumulation and nuclear depletion of TDP43 in motor neurons of patients with ALS.
- 38.Kabashi E et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet 40, 572–574 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Sreedharan J et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; Kabashi et al. (2008) and Sreedharan et al. (2008) were the first reports of pathogenic TARDBP mutations in ALS.
- 40.Turner MR et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Besser LM, Teylan MA & Nelson PT Limbic predominant age-related TDP-43 encephalopathy (LATE): clinical and neuropathological associations. J. Neuropathol. Exp. Neurol 79, 305–313 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suk TR & Rousseaux MWC The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener 15, 45 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nonaka T et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Nomura T et al. Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J. Biol. Chem 289, 1192–1202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chia R et al. Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS ONE 5, e10627 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kwiatkowski TJ Jr et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Vance C et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; Kwiatkowski et al. (2009) and Vance et al. (2009) were the first reports of pathogenic FUS mutations in ALS.
- 48.Chio A et al. A de novo missense mutation of the FUS gene in a “true” sporadic ALS case. Neurobiol. Aging 32, 553 e523–553 e556 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki N et al. FALS with FUS mutation in Japan, with early onset, rapid progress and basophilic inclusion. J. Hum. Genet 55, 252–254 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Lattante S, Rouleau GA & Kabashi E TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum. Mutat 34, 812–826 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Hubers A et al. De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol. Aging 36, 3117 e3111–3117 e3116 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Kim SH, Shanware NP, Bowler MJ & Tibbetts RS Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J. Biol. Chem 285, 34097–34105 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khalfallah Y et al. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci. Rep 8, 7551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bosco DA et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet 19, 4160–4175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Renton AE et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeJesus-Hernandez M et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; Renton et al. (2011) and DeJesus-Hernandez et al. (2011) were the first studies that identified GGGGCC repeat expansion in C9orf72 in ALS and FTD.
- 57.Majounie E et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study was among the first studies to evaluate the genetics of ALS and FTD globally and across populations of different ancestries.
- 58.Dols-Icardo O et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet 23, 749–754 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Ebbert MTW et al. Long-read sequencing across the C9orf72 ‘GGGGCC’ repeat expansion: implications for clinical use and genetic discovery efforts in human disease. Mol. Neurodegener 13, 46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van der Ende EL et al. Unravelling the clinical spectrum and the role of repeat length in C9ORF72 repeat expansions. J. Neurol. Neurosurg. Psychiatry 92, 502–509 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gijselinck I et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry 21, 1112–1124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nordin A et al. Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum. Mol. Genet 24, 3133–3142 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Beck J et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet 92, 345–353 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu Z et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl Acad. Sci. USA 110, 7778–7783 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ash PE et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balendra R & Isaacs AM C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol 14, 544–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fumagalli L et al. C9orf72-derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. Sci. Adv 7, eabg3013 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi Y et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med 24, 313–325 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abramzon YA, Fratta P, Traynor BJ & Chia R The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front. Neurosci 14, 42 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Majounie E et al. Repeat expansion in C9ORF72 in Alzheimer’s disease. N. Engl. J. Med 366, 283–284 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kenna KP et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet 48, 1037–1042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nicolas A et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283 e1266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brenner D et al. Hot-spot KIF5A mutations cause familial ALS. Brain 141, 688–697 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Nicolas et al. (2018) and Brenner et al. (2018) were the first reports of pathogenic KIF5A mutations in ALS.
- 74.Saez-Atienzar S et al. Identification of a pathogenic intronic KIF5A mutation in an ALS–FTD kindred. Neurology 95, 1015–1018 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baron DM et al. ALS-associated KIF5A mutations abolish autoinhibition resulting in a toxic gain of function. Cell Rep. 39, 110598 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Castellanos-Montiel MJ, Chaineau M & Durcan TM The neglected genes of ALS: cytoskeletal dynamics impact synaptic degeneration in ALS. Front. Cell Neurosci 14, 594975 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wenting G, Laura F & Ludo Van Den B in Amyotrophic Lateral Sclerosis (ed. Hegde LM) (IntechOpen, 2020). [Google Scholar]
- 78.Tunca C et al. ERLIN1 mutations cause teenage-onset slowly progressive ALS in a large Turkish pedigree. Eur. J. Hum. Genet 26, 745–748 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhu ZY et al. A novel homozygous mutation in ERLIN1 gene causing spastic paraplegia 62 and literature review. Eur. J. Med. Genet 65, 104608 (2022). [DOI] [PubMed] [Google Scholar]
- 80.Novarino G et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343, 506–511 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Egorova PA & Bezprozvanny IB Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J. 285, 3547–3565 (2018). [DOI] [PubMed] [Google Scholar]
- 82.Higo T et al. Mechanism of ER stress-induced brain damage by IP3 receptor. Neuron 68, 865–878 (2010). [DOI] [PubMed] [Google Scholar]
- 83.Casey JP et al. A novel gain-of-function mutation in the ITPR1 suppressor domain causes spinocerebellar ataxia with altered Ca2+ signal patterns. J. Neurol 264, 1444–1453 (2017). [DOI] [PubMed] [Google Scholar]
- 84.Farhan SMK et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nat. Neurosci 22, 1966–1974 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jih KY, Tsai PC, Tsai YS, Liao YC & Lee YC Rapid progressive ALS in a patient with a DNAJC7 loss-of-function mutation. Neurol. Genet 6, e503 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.He J et al. Validation of the pathogenic role of rare DNAJC7 variants in Chinese patients with amyotrophic lateral sclerosis. Neurobiol. Aging 106, 314 e311–314 e316 (2021). [DOI] [PubMed] [Google Scholar]
- 87.Wang M et al. A novel potentially pathogenic rare variant in the DNAJC7 gene identified in amyotrophic lateral sclerosis patients from mainland China. Front. Genet 11, 821 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tohnai G et al. Mutation screening of the DNAJC7 gene in Japanese patients with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 113, 131–136 (2022). [DOI] [PubMed] [Google Scholar]
- 89.Dilliott AA et al. DnaJC7 in amyotrophic lateral sclerosis. Int. J. Mol. Sci 23, 4076 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jinwal UK et al. Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J. Biol. Chem 287, 24814–24820 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dewan R et al. Pathogenic huntingtin repeat expansions in patients with frontotemporal dementia and amyotrophic lateral sclerosis. Neuron 109, 448–460 e444 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hickman RA et al. Amyotrophic lateral sclerosis is over-represented in two Huntington’s disease brain bank cohorts: further evidence to support genetic pleiotropy of pathogenic HTT gene expansion. Acta Neuropathol. 143, 105–108 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dewan R et al. CAG somatic instability in a Huntington disease expansion carrier presenting with a progressive supranuclear palsy-like phenotype. Mov. Disord 37, 1555–1557 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grima JC et al. Mutant huntingtin disrupts the nuclear pore complex. Neuron 94, 93–107 e106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Freibaum BD et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tabrizi SJ et al. Targeting huntingtin expression in patients with Huntington’s disease. N. Engl. J. Med 380, 2307–2316 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Johnson JO et al. Association of variants in the SPTLC1 gene with juvenile amyotrophic lateral sclerosis. JAMA Neurol. 78, 1236–1248 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mohassel P et al. Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis. Nat. Med 27, 1197–1204 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bejaoui K et al. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat. Genet 27, 261–262 (2001). [DOI] [PubMed] [Google Scholar]
- 100.Penno A et al. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J. Biol. Chem 285, 11178–11187 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garofalo K et al. Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. J. Clin. Invest 121, 4735–4745 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schymick JC et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 6, 322–328 (2007). [DOI] [PubMed] [Google Scholar]
- 104.van Es MA et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet 41, 1083–1087 (2009). [DOI] [PubMed] [Google Scholar]
- 105.Laaksovirta H et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 9, 978–985 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shatunov A et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 9, 986–994 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.van Rheenen W et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet 48, 1043–1048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Benyamin B et al. Cross-ethnic meta-analysis identifies association of the GPX3–TNIP1 locus with amyotrophic lateral sclerosis. Nat. Commun 8, 611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.van Rheenen W et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet 53, 1636–1648 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fogh I et al. A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet 23, 2220–2231 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nakamura R et al. A multi-ethnic meta-analysis identifies novel genes, including ACSL5, associated with amyotrophic lateral sclerosis. Commun. Biol 3, 526 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cirulli ET et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Freischmidt A et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci 18, 631–636 (2015). [DOI] [PubMed] [Google Scholar]
- 114.Brenner D et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 139, e28 (2016). [DOI] [PubMed] [Google Scholar]
- 115.Saez-Atienzar S et al. Genetic analysis of amyotrophic lateral sclerosis identifies contributing pathways and cell types. Sci. Adv 7, eabd9036 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ho WY et al. The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy 15, 827–842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang M et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv 2, e1601167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maekawa S et al. Cortical selective vulnerability in motor neuron disease: a morphometric study. Brain 127, 1237–1251 (2004). [DOI] [PubMed] [Google Scholar]
- 119.Maniatis S et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 364, 89–93 (2019). [DOI] [PubMed] [Google Scholar]
- 120.Humphrey J et al. Integrative transcriptomic analysis of the amyotrophic lateral sclerosis spinal cord implicates glial activation and suggests new risk genes. Nat. Neurosci 26, 150–162 (2023). [DOI] [PubMed] [Google Scholar]
- 121.Lee H et al. Multi-omic analysis of selectively vulnerable motor neuron subtypes implicates altered lipid metabolism in ALS. Nat. Neurosci 24, 1673–1685 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tazelaar GHP et al. ATXN1 repeat expansions confer risk for amyotrophic lateral sclerosis and contribute to TDP-43 mislocalization. Brain Commun. 2, fcaa064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Elden AC et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Daoud H et al. Association of long ATXN2 CAG repeat sizes with increased risk of amyotrophic lateral sclerosis. Arch. Neurol 68, 739–742 (2011). [DOI] [PubMed] [Google Scholar]
- 125.Glass JD et al. ATXN2 intermediate expansions in amyotrophic lateral sclerosis. Brain 145, 2671–2676 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Blauw HM et al. NIPA1 polyalanine repeat expansions are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet 21, 2497–2502 (2012). [DOI] [PubMed] [Google Scholar]
- 127.Al Khleifat A et al. Structural variation analysis of 6,500 whole genome sequences in amyotrophic lateral sclerosis. NPJ Genom. Med 7, 8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ross JP, Dion PA & Rouleau GA in Spectrums of Amyotrophic Lateral Sclerosis (eds Shaw CA & Morrice JR) 17–34 (Wiley, 2021). [Google Scholar]
- 129.Valdmanis PN et al. A mutation that creates a pseudoexon in SOD1 causes familial ALS. Ann. Hum. Genet 73, 652–657 (2009). [DOI] [PubMed] [Google Scholar]
- 130.Morgan S et al. A comprehensive analysis of rare genetic variation in amyotrophic lateral sclerosis in the UK. Brain 140, 1611–1618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Eitan C et al. Whole-genome sequencing reveals that variants in the Interleukin 18 Receptor Accessory Protein 3’UTR protect against ALS. Nat. Neurosci 25, 433–445 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Birve A et al. A novel SOD1 splice site mutation associated with familial ALS revealed by SOD activity analysis. Hum. Mol. Genet 19, 4201–4206 (2010). [DOI] [PubMed] [Google Scholar]
- 133.Kawata A et al. Aberrant splicing of human Cu/Zn superoxide dismutase (SOD1) RNA transcripts. Neuroreport 11, 2649–2653 (2000). [DOI] [PubMed] [Google Scholar]
- 134.Jeong YH et al. Tdp-43 cryptic exons are highly variable between cell types. Mol. Neurodegener 12, 13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ling JP, Pletnikova O, Troncoso JC & Wong PC TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Brown AL et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Alexander MD et al. “True” sporadic ALS associated with a novel SOD-1 mutation. Ann. Neurol 52, 680–683 (2002). [DOI] [PubMed] [Google Scholar]
- 138.Muller K et al. De novo mutations in SOD1 are a cause of ALS. J. Neurol. Neurosurg. Psychiatry 93, 201–206 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Garcia-Montojo M, Doucet-O’Hare T, Henderson L & Nath A Human endogenous retrovirus-K (HML-2): a comprehensive review. Crit. Rev. Microbiol 44, 715–738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.MacGowan DJ et al. A controlled study of reverse transcriptase in serum and CSF of HIV-negative patients with ALS. Neurology 68, 1944–1946 (2007). [DOI] [PubMed] [Google Scholar]
- 141.Li W et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med 7, 307ra153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Garcia-Montojo M et al. Inhibition of HERV-K (HML-2) in amyotrophic lateral sclerosis patients on antiretroviral therapy. J. Neurol. Sci 423, 117358 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Watts GD et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet 36, 377–381 (2004). [DOI] [PubMed] [Google Scholar]
- 144.Johnson JO et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chio A et al. Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology 93, e984–e994 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dupuis L et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 70, 1004–1009 (2008). [DOI] [PubMed] [Google Scholar]
- 147.Abdel-Khalik J et al. Defective cholesterol metabolism in amyotrophic lateral sclerosis. J. Lipid Res 58, 267–278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dodge JC et al. Neutral lipid cacostasis contributes to disease pathogenesis in amyotrophic lateral sclerosis. J. Neurosci 40, 9137–9147 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Andres-Benito P et al. Lipid alterations in human frontal cortex in ALS-FTLD-TDP43 proteinopathy spectrum are partly related to peroxisome impairment. Neuropathol. Appl. Neurobiol 47, 544–563 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ikeda K, Hirayama T, Takazawa T, Kawabe K & Iwasaki Y Relationships between disease progression and serum levels of lipid, urate, creatinine and ferritin in Japanese patients with amyotrophic lateral sclerosis: a cross-sectional study. Intern. Med 51, 1501–1508 (2012). [DOI] [PubMed] [Google Scholar]
- 151.Thompson AG, Talbot K & Turner MR Higher blood high density lipoprotein and apolipoprotein A1 levels are associated with reduced risk of developing amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 93, 75–81 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cutler RG, Pedersen WA, Camandola S, Rothstein JD & Mattson MP Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol 52, 448–457 (2002). [DOI] [PubMed] [Google Scholar]
- 153.Theofilopoulos S et al. Cholestenoic acids regulate motor neuron survival via liver X receptors. J. Clin. Invest 124, 4829–4842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dodge JC et al. Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc. Natl Acad. Sci. USA 112, 8100–8105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.D’Amico E et al. Metabolic abnormalities, dietary risk factors and nutritional management in amyotrophic lateral sclerosis. Nutrients 13, 2273 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chio A et al. Lower serum lipid levels are related to respiratory impairment in patients with ALS. Neurology 73, 1681–1685 (2009). [DOI] [PubMed] [Google Scholar]
- 157.Yang JW et al. Hypolipidemia in patients with amyotrophic lateral sclerosis: a possible gender difference? J. Clin. Neurol 9, 125–129 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sutedja NA et al. Beneficial vascular risk profile is associated with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 82, 638–642 (2011). [DOI] [PubMed] [Google Scholar]
- 159.Hartmann H, Ho WY, Chang JC & Ling SC Cholesterol dyshomeostasis in amyotrophic lateral sclerosis: cause, consequence, or epiphenomenon? FEBS J. 289, 7688–7709 (2022). [DOI] [PubMed] [Google Scholar]
- 160.Vasta R, Chia R, Traynor BJ & Chio A Unraveling the complex interplay between genes, environment, and climate in ALS. EBioMedicine 75, 103795 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bandres-Ciga S et al. Shared polygenic risk and causal inferences in amyotrophic lateral sclerosis. Ann. Neurol 85, 470–481 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Goutman SA et al. Metabolomics identifies shared lipid pathways in independent amyotrophic lateral sclerosis cohorts. Brain 145, 4425–4439 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zeng P, Wang T, Zheng J & Zhou X Causal association of type 2 diabetes with amyotrophic lateral sclerosis: new evidence from Mendelian randomization using GWAS summary statistics. BMC Med. 17, 225 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhang L, Tang L, Huang T & Fan D Association between type 2 diabetes and amyotrophic lateral sclerosis. Sci. Rep 12, 2544 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen H et al. Type 2 diabetes mellitus and amyotrophic lateral sclerosis: genetic overlap, causality, and mediation. J. Clin. Endocrinol. Metab 106, e4497–e4508 (2021). [DOI] [PubMed] [Google Scholar]
- 166.Julian TH et al. A review of Mendelian randomization in amyotrophic lateral sclerosis. Brain 145, 832–842 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.D’Ovidio F et al. The role of pre-morbid diabetes on developing amyotrophic lateral sclerosis. Eur. J. Neurol 25, 164–170 (2018). [DOI] [PubMed] [Google Scholar]
- 168.Nakken O, Meyer HE, Stigum H & Holmoy T High BMI is associated with low ALS risk: a population-based study. Neurology 93, e424–e432 (2019). [DOI] [PubMed] [Google Scholar]
- 169.Vasta R, D’Ovidio F, Logroscino G & Chio A The links between diabetes mellitus and amyotrophic lateral sclerosis. Neurol. Sci 42, 1377–1387 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Reid E et al. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am. J. Hum. Genet 71, 1189–1194 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Crimella C et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot–Marie-Tooth type 2. Clin. Genet 82, 157–164 (2012). [DOI] [PubMed] [Google Scholar]
- 172.Cronin S, Hardiman O & Traynor BJ Ethnic variation in the incidence of ALS: a systematic review. Neurology 68, 1002–1007 (2007). [DOI] [PubMed] [Google Scholar]
- 173.Zou ZY et al. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 88, 540–549 (2017). [DOI] [PubMed] [Google Scholar]
- 174.Pliner HA, Mann DM & Traynor BJ Searching for Grendel: origin and global spread of the C9ORF72 repeat expansion. Acta Neuropathol. 127, 391–396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Suzuki N, Nishiyama A, Warita H & Aoki M Genetics of amyotrophic lateral sclerosis: seeking therapeutic targets in the era of gene therapy. J. Hum. Genet 68, 131–152 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dombroski BA et al. C9orf72 hexanucleotide repeat expansion and Guam amyotrophic lateral sclerosis-Parkinsonism-dementia complex. JAMA Neurol. 70, 742–745 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Logroscino G & Piccininni M Amyotrophic lateral sclerosis descriptive epidemiology: the origin of geographic difference. Neuroepidemiology 52, 93–103 (2019). [DOI] [PubMed] [Google Scholar]
- 178.Chio A et al. Large proportion of amyotrophic lateral sclerosis cases in Sardinia due to a single founder mutation of the TARDBP gene. Arch. Neurol 68, 594–598 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chadi G et al. Genetic analysis of patients with familial and sporadic amyotrophic lateral sclerosis in a Brazilian Research Center. Amyotroph. Lateral Scler. Frontotemporal Degener 18, 249–255 (2017). [DOI] [PubMed] [Google Scholar]
- 180.Johnson JO et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain 135, 2875–2882 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Green P et al. Brown–Vialetto–Van Laere syndrome, a ponto-bulbar palsy with deafness, is caused by mutations in c20orf54. Am. J. Hum. Genet 86, 485–489 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bandres-Ciga S, Noyce AJ & Traynor BJ Mendelian randomization — a journey from obscurity to center stage with a few potholes along the way. JAMA Neurol. 77, 7–8 (2020). [DOI] [PubMed] [Google Scholar]
- 183.Pingault JB et al. Using genetic data to strengthen causal inference in observational research. Nat. Rev. Genet 19, 566–580 (2018). [DOI] [PubMed] [Google Scholar]
- 184.Ebbert MTW, Lank RJ & Belzil VV An epigenetic spin ALS FTD. Adv. Neurobiol 20, 1–29 (2018). [DOI] [PubMed] [Google Scholar]
- 185.Bennett SA, Tanaz R, Cobos SN & Torrente MP Epigenetics in amyotrophic lateral sclerosis: a role for histone post-translational modifications in neurodegenerative disease. Transl. Res 204, 19–30 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Dolinar A, Ravnik-Glavac M & Glavac D Epigenetic mechanisms in amyotrophic lateral sclerosis: a short review. Mech. Ageing Dev 174, 103–110 (2018). [DOI] [PubMed] [Google Scholar]
- 187.Hop PJ et al. Genome-wide study of DNA methylation shows alterations in metabolic, inflammatory, and cholesterol pathways in ALS. Sci. Transl. Med 14, eabj0264 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.McMillan CT et al. C9orf72 promoter hypermethylation is neuroprotective: neuroimaging and neuropathologic evidence. Neurology 84, 1622–1630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Butovsky O et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol 77, 75–99 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Rothman KJ Causes. Am. J. Epidemiol 104, 587–592 (1976). [DOI] [PubMed] [Google Scholar]
- 191.Alfahad T & Nath A Retroviruses and amyotrophic lateral sclerosis. Antivir. Res 99, 180–187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Van Damme P, Robberecht W & Van Den Bosch L Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis. Model. Mech 10, 537–549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Al-Chalabi A et al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. 13, 1108–1113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Traynor BJ & Singleton AB Nature versus nurture: death of a dogma, and the road ahead. Neuron 68, 196–200 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Boyle EA, Li YI & Pritchard JK An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bensimon G, Lacomblez L & Meininger V A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med 330, 585–591 (1994). [DOI] [PubMed] [Google Scholar]
- 197.Writing G & Edaravone, A. L. S. S. G. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 16, 505–512 (2017). [DOI] [PubMed] [Google Scholar]
- 198.Finkel RS et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med 377, 1723–1732 (2017). [DOI] [PubMed] [Google Scholar]
- 199.Sufit RL, Ajroud-Driss S, Casey P & Kessler JA Open label study to assess the safety of VM202 in subjects with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener 18, 269–278 (2017). [DOI] [PubMed] [Google Scholar]
- 200.Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P & Shneider NA Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat. Med 28, 104–116 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Mullard A ALS antisense drug falters in phase III. Nat. Rev. Drug Discov 20, 883–885 (2021). [DOI] [PubMed] [Google Scholar]
- 202.Biogen Investor Relations. New 12-Month Tofersen Data Presented at ENCALS Meeting Show Clinically Meaningful Benefit in People With SOD1-ALS [Online], https://investors.biogen.com/news-releases/news-release-details/new-12-month-tofersen-data-presented-encals-meeting-show (2022).
- 203.Tabrizi SJ et al. Potential disease-modifying therapies for Huntington’s disease: lessons learned and future opportunities. Lancet Neurol. 21, 645–658 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Benatar M et al. Design of a randomized, placebo-controlled, phase 3 trial of tofersen initiated in clinically presymptomatic SOD1 variant carriers: the ATLAS study. Neurotherapeutics 19, 1248–1258 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tran H et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med 28, 117–124 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Becker LA et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sullivan JM et al. Convective forces increase rostral delivery of intrathecal radiotracers and antisense oligonucleotides in the cynomolgus monkey nervous system. J. Transl. Med 18, 309 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Belov V et al. Large-volume intrathecal administrations: impact on CSF pressure and safety implications. Front. Neurosci 15, 604197 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Roberts TC, Langer R & Wood MJA Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov 19, 673–694 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Nagata T et al. Cholesterol-functionalized DNA/RNA heteroduplexes cross the blood–brain barrier and knock down genes in the rodent CNS. Nat. Biotechnol 39, 1529–1536 (2021). [DOI] [PubMed] [Google Scholar]
- 211.Gennemark P et al. An oral antisense oligonucleotide for PCSK9 inhibition. Sci. Transl. Med 13, eabe9117 (2021). [DOI] [PubMed] [Google Scholar]
- 212.Figlewicz DA et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet 3, 1757–1761 (1994). [DOI] [PubMed] [Google Scholar]
- 213.Yang Y et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet 29, 160–165 (2001). [DOI] [PubMed] [Google Scholar]
- 214.Puls I et al. Mutant dynactin in motor neuron disease. Nat. Genet 33, 455–456 (2003). [DOI] [PubMed] [Google Scholar]
- 215.Nishimura AL et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet 75, 822–831 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chen YZ et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet 74, 1128–1135 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Parkinson N et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67, 1074–1077 (2006). [DOI] [PubMed] [Google Scholar]
- 218.Skibinski G et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet 37, 806–808 (2005). [DOI] [PubMed] [Google Scholar]
- 219.Orlacchio A et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 133, 591–598 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Maruyama H et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226 (2010). [DOI] [PubMed] [Google Scholar]
- 221.Deng HX et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Fecto F et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol 68, 1440–1446 (2011). [DOI] [PubMed] [Google Scholar]
- 223.Wu CH et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kim HJ et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Smith BN et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Johnson JO et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci 17, 664–666 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Bannwarth S et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137, 2329–2345 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Williams KL et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun 7, 11253 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Smith BN et al. Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci. Transl. Med 9, eaad9157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Cooper-Knock J et al. Mutations in the glycosyltransferase domain of GLT8D1 are associated with familial amyotrophic lateral sclerosis. Cell Rep. 26, 2298–2306 e2295 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Grassano M et al. Systematic evaluation of genetic mutations in ALS: a population-based study. J. Neurol. Neurosurg. Psychiatry 93, 1190–1193 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.McCann EP et al. The genotype-phenotype landscape of familial amyotrophic lateral sclerosis in Australia. Clin. Genet 92, 259–266 (2017). [DOI] [PubMed] [Google Scholar]
- 233.Smith BN et al. The C9ORF72 expansion mutation is a common cause of ALS+/− FTD in Europe and has a single founder. Eur. J. Hum. Genet 21, 102–108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wittrup A & Lieberman J Knocking down disease: a progress report on siRNA therapeutics. Nat. Rev. Genet 16, 543–552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Shahryari A et al. Development and clinical translation of approved gene therapy products for genetic disorders. Front. Genet 10, 868 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kwok A, Raulf N & Habib N Developing small activating RNA as a therapeutic: current challenges and promises. Ther. Deliv 10, 151–164 (2019). [DOI] [PubMed] [Google Scholar]
- 237.Wengert ER et al. Targeted augmentation of nuclear gene output (TANGO) of Scn1a rescues parvalbumin interneuron excitability and reduces seizures in a mouse model of Dravet Syndrome. Brain Res. 1775, 147743 (2022). [DOI] [PubMed] [Google Scholar]
- 238.Sanders R FDA approves first test of CRISPR to correct genetic defect causing sickle cell disease [online], https://news.berkeley.edu/2021/03/30/fda-approves-first-test-ofcrispr-to-correct-genetic-defect-causing-sickle-cell-disease/ (2021).
- 239.Frangoul H et al. CRISPR–Cas9 gene editing for sickle cell disease and beta-thalassemia. N. Engl. J. Med 384, 252–260 (2021). [DOI] [PubMed] [Google Scholar]
- 240.No authors listed. FDA approves hereditary blindness gene therapy. Nat. Biotechnol 36, 6 (2018). [DOI] [PubMed] [Google Scholar]
Related links
- Answer ALS: https://www.answerals.org/
- Clinical trial information for Engensis (VM202): https://alsnewstoday.com/vm202/
- dbGaP: https://www.ncbi.nlm.nih.gov/gap/
- N=1 Collaborative network: https://www.n1collaborative.org/
- New York Genome Center ALS Consortium: https://www.nygenome.org/als-consortium/
- Online Mendelian Inheritance in Man: https://www.omim.org/
- Project MinE: https://www.projectmine.com/
- Safety study of VM202 to treat amyotrophic lateral sclerosis: https://clinicaltrials.gov/ct2/show/NCT02039401