

Abstract
Background
Hypertension is a modifiable cardiovascular risk factor. Although it is established that low‐dose thiazides reduce mortality as well as cardiovascular morbidity, the dose‐related effect of thiazides in decreasing blood pressure has not been subject to a rigorous systematic review. It is not known whether individual drugs within the thiazide diuretic class differ in their blood pressure‐lowering effects and adverse effects.
Objectives
To determine the dose‐related decrease in systolic and/or diastolic blood pressure due to thiazide diuretics compared with placebo control in the treatment of patients with primary hypertension. Secondary outcomes included the dose‐related adverse events leading to patient withdrawal and adverse biochemical effects on serum potassium, uric acid, creatinine, glucose and lipids.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL 2014, Issue 1), Ovid MEDLINE (1946 to February 2014), Ovid EMBASE (1974 to February 2014) and ClinicalTrials.gov.
Selection criteria
We included double‐blind, randomized controlled trials (RCTs) comparing fixed‐dose thiazide diuretic monotherapy with placebo for a duration of 3 to 12 weeks in the treatment of adult patients with primary hypertension.
Data collection and analysis
Two authors independently screened articles, assessed trial eligibility, extracted data and determined risk of bias. We combined data for continuous variables using a mean difference (MD) and for dichotomous outcomes we calculated the relative risk ratio (RR) with 95% confidence interval (CI).
Main results
We included 60 randomized, double‐blind trials that evaluated the dose‐related trough blood pressure‐lowering efficacy of six different thiazide diuretics in 11,282 participants treated for a mean duration of eight weeks. The mean age of the participants was 55 years and baseline blood pressure was 158/99 mmHg. Adequate blood pressure‐lowering efficacy data were available for hydrochlorothiazide, chlorthalidone and indapamide. We judged 54 (90%) included trials to have unclear or high risk of bias, which impacted on our confidence in the results for some of our outcomes.
In 33 trials with a baseline blood pressure of 155/100 mmHg, hydrochlorothiazide lowered blood pressure based on dose, with doses of 6.25 mg, 12.5 mg, 25 mg and 50 mg/day lowering blood pressure compared to placebo by 4 mmHg (95% CI 2 to 6, moderate‐quality evidence)/2 mmHg (95% CI 1 to 4, moderate‐quality evidence), 6 mmHg (95% CI 5 to 7, high‐quality evidence)/3 mmHg (95% CI 3 to 4, high‐quality evidence), 8 mmHg (95% CI 7 to 9, high‐quality evidence)/3 mmHg (95% CI 3 to 4, high‐quality evidence) and 11 mmHg (95% CI 6 to 15, low‐quality evidence)/5 mmHg (95% CI 3 to 7, low‐quality evidence), respectively.
Direct comparison of doses did not show evidence of dose dependence for blood pressure‐lowering for any of the other thiazides for which RCT data were available: bendrofluazide, chlorthalidone, cyclopenthiazide, metolazone or indapamide.
In seven trials with a baseline blood pressure of 163/88 mmHg, chlorthalidone at doses of 12.5 mg to 75 mg/day reduced average blood pressure compared to placebo by 12.0 mmHg (95% CI 10 to 14, low‐quality evidence)/4 mmHg (95% CI 3 to 5, low‐quality evidence).
In 10 trials with a baseline blood pressure of 161/98 mmHg, indapamide at doses of 1.0 mg to 5.0 mg/day reduced blood pressure compared to placebo by 9 mmHg (95% CI 7 to 10, low‐quality evidence)/4 (95% CI 3 to 5, low‐quality evidence).
We judged the maximal blood pressure‐lowering effect of the different thiazides to be similar. Overall, thiazides reduced average blood pressure compared to placebo by 9 mmHg (95% CI 9 to 10, high‐quality evidence)/4 mmHg (95% CI 3 to 4, high‐quality evidence).
Thiazides as a class have a greater effect on systolic than on diastolic blood pressure, therefore thiazides lower pulse pressure by 4 mmHg to 6 mmHg, an amount that is greater than the 3 mmHg seen with angiotensin‐converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs) and renin inhibitors, and the 2 mmHg seen with non‐selective beta‐blockers. This is based on an informal indirect comparison of results observed in other Cochrane reviews on ACE inhibitors, ARBs and renin inhibitors compared with placebo, which used similar inclusion/exclusion criteria to the present review.
Thiazides reduced potassium, increased uric acid and increased total cholesterol and triglycerides. These effects were dose‐related and were least for hydrochlorothiazide. Chlorthalidone increased serum glucose but the evidence was unclear for other thiazides. There is a high risk of bias in the metabolic data. This review does not provide a good assessment of the adverse effects of these drugs because there was a high risk of bias in the reporting of withdrawals due to adverse effects.
Authors' conclusions
This systematic review shows that hydrochlorothiazide has a dose‐related blood pressure‐lowering effect. The mean blood pressure‐lowering effect over the dose range 6.25 mg, 12.5 mg, 25 mg and 50 mg/day is 4/2 mmHg, 6/3 mmHg, 8/3 mmHg and 11/5 mmHg, respectively. For other thiazide drugs, the lowest doses studied lowered blood pressure maximally and higher doses did not lower it more. Due to the greater effect on systolic than on diastolic blood pressure, thiazides lower pulse pressure by 4 mmHg to 6 mmHg. This exceeds the mean 3 mmHg pulse pressure reduction achieved by ACE inhibitors, ARBs and renin inhibitors, and the 2 mmHg pulse pressure reduction with non‐selective beta‐blockers as shown in other Cochrane reviews, which compared these antihypertensive drug classes with placebo and used similar inclusion/exclusion criteria.
Thiazides did not increase withdrawals due to adverse effects in these short‐term trials but there is a high risk of bias for that outcome. Thiazides reduced potassium, increased uric acid and increased total cholesterol and triglycerides.
Plain language summary
Thiazide diuretics for the treatment of high blood pressure
Thiazide diuretics are a class of drugs commonly recommended as first‐line treatment for raised blood pressure because they significantly reduce death, stroke and heart attacks. This class includes bendrofluazide, chlorthalidone, cyclopenthiazide, hydrochlorothiazide, indapamide and metolazone. We asked by how much does this class of drugs lower blood pressure and whether there is a difference between individual drugs within the class. We searched the available scientific literature to find all the trials that had assessed this question. The data included in this review was up to date as of February 2014.
We found 60 trials that randomly assigned 11,282 adult participants, mean age 55 years, 53% male and 47% female, with blood pressure above 140/90 mmHg (mean blood pressure 158/99 mmHg) to take one of six thiazide diuretics or placebo for an average duration of eight weeks. Most of the trials (82%) were published before the year 2000 and most were found to have a high risk of bias in the adverse effect data. Co‐morbidities were not reported in most trials. The blood pressure‐lowering effect was modest. Thiazide diuretics reduced blood pressure by 9 points in the upper number (called systolic blood pressure) and 4 points in the lower number (called diastolic blood pressure). Different thiazide drugs have similar effects in lowering blood pressure and thiazides lower systolic blood pressure more than other classes of antihypertensive drugs.
This review could not provide a valid estimate of short‐term harms from all thiazide diuretics because there was incomplete reporting of metabolic effects (serum potassium, uric acid, creatinine, glucose, total cholesterol, low‐density cholesterol and triglycerides) and the number of participants who dropped out of the trials due to adverse drug effects.
Summary of findings
Summary of findings for the main comparison. Dose‐ranging blood pressure‐lowering efficacy of hydrochlorothiazide for primary hypertension.
| Hydrochlorothiazide compared with placebo for primary hypertension | |||||
|
Patient or population: adults with primary hypertension Settings: outpatient Intervention: hydrochlorothiazide 3 to 100 mg/day Comparison: placebo | |||||
| Outcomes | Daily dose | MD (95% CI) mmHg | No of participants (studies) | Quality of the evidence (GRADE) | Comments |
| Systolic blood pressure | 3 to 6.25 mg | ‐3.6 (‐5.6 to ‐1.5) | 663 (8) | ⊕⊕⊕⊝ moderate1 |
Similar to the effect as a second‐line drug (Chen 2009). This is an indirect comparison of the effect size with a wider confidence interval compared to the Chen review, which is based on 22 trials in 3283 patients using similar inclusion/exclusion criteria with systolic blood pressure‐lowering of ‐3.7 (‐4.6 to ‐2.8) mmHg |
| 12.5 mg | ‐6.3 (‐7.2 to ‐5.3) | 2645 (22) | ⊕⊕⊕⊕ high | A narrow confidence interval based on a large sample size with a magnitude of lowering very similar to the effect as a second‐line drug (Chen 2009), which was ‐6.0 (‐6.5 to ‐5.4) mmHg | |
| 25 mg | ‐8.0 (‐9.0 to ‐7.0) | 3062 (25) | ⊕⊕⊕⊕ high | A narrow confidence interval based on a large sample size with a magnitude of lowering very similar to the effect as a second‐line drug (Chen 2009), which was ‐8.0 (‐8.7 to ‐7.3) mmHg | |
| 50 to 100 mg | ‐10.2 (‐13.1 to ‐7.3) | 315 (2) | ⊕⊕⊝⊝ low1 | The 2 included studies have a high risk of bias. The confidence interval is very wide with small a sample size providing insufficient data in both this review as well as in the Chen review comparing the effect as a second‐line drug (Chen 2009) | |
| Diastolic blood pressure | 3 to 6.25 mg | ‐2.4 (‐3.7 to ‐1.2) | 662 (8) | ⊕⊕⊕⊝ moderate1 |
Similar to the effect as a second‐line drug (Chen 2009). This is an indirect comparison of the effect size with a wider confidence interval compared to the Chen review, which is based on 23 trials in 3364 patients using similar inclusion/exclusion criteria, with diastolic blood pressure‐lowering of ‐1.7 (‐2.2 to ‐1.2) mmHg. |
| 12.5 mg | ‐3.1 (‐3.7 to ‐2.5) | 2877 (25) | ⊕⊕⊕⊕ high | Similar to the effect as a second‐line drug (Chen 2009), with a narrow confidence interval based on a large sample size with a magnitude of lowering similar to the effect as a second‐line drug (Chen 2009), which was ‐3.1 (‐3.4 to ‐2.8) mmHg | |
| 25 mg | ‐3.3(‐3.8 to ‐2.8) | 3359 (29) | ⊕⊕⊕⊕ high | Similar to the effect as a second‐line drug (Chen 2009). This is an indirect comparison of the effect size with a wider confidence interval compared to the Chen review, which is based on 42 trials in 6153 patients using similar inclusion/exclusion criteria with diastolic blood pressure‐lowering of ‐4.0 (‐4.4 to ‐3.6) mmHg | |
| 50 to 100 mg | ‐4.7 (‐6.1 to ‐3.3) | 345 (4) | ⊕⊕⊝⊝ low1 | The 4 included studies had a high risk of bias. The confidence interval is very wide with a small sample size providing insufficient data in both this review as well as in the Chen review comparing the effect as a second‐line drug (Chen 2009) | |
| GRADE Working Group grades of evidence
High quality: Further research is very unlikely to change our confidence in the estimate of effect.
Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate.
Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate.
Very low quality: We are very uncertain about the estimate. CI: confidence interval; MD: mean difference | |||||
1Downgraded due to the small number of patients and wide confidence intervals.
Summary of findings 2. Overall effects of thiazides for primary hypertension.
| Thiazide compared with placebo for primary hypertension | ||||
|
Patient or population: adults with primary hypertension Settings: outpatient Intervention: all thiazides^ Comparison: placebo | ||||
| Outcomes | MD (95% CI) mmHg | No of participants (studies) | Quality of the evidence (GRADE) | Comments |
| Systolic blood pressure | ‐9.1 (‐9.7 to ‐8.5) | 7733 (47) | ⊕⊕⊕⊕ high | At doses achieving maximal effect and above |
| Diastolic blood pressure | ‐3.6 (‐4.0 to ‐3.3) | 8064 (51) | ⊕⊕⊕⊕ high | At doses achieving maximal effect and above |
| Withdrawal due to adverse effects | RR 0.64 (95% CI 0.43 to 0.93) |
3698 (20) | ⊕⊝⊝⊝ very low | See comments 1 and 2 |
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. CI: confidence interval; RR: risk ratio | ||||
^Includes thiazide and thiazide‐like diuretics.
1Based on a high risk of selective reporting of outcome from 20 out of 40 trials meeting the inclusion criteria.
2Withdrawals due to inclusion of an increase in blood pressure as an adverse effect (AE) was the major reason for withdrawals in the placebo group.
Background
Description of the condition
Hypertension is a common and potentially serious problem. It is one of the risk factors for stroke, heart and blood vessel disease, kidney disease and early death that can most easily be reduced by treatment. Studies show a correlation between elevation of systolic or diastolic blood pressure and increased risk of stroke, heart failure, renal disease and coronary heart disease. There is considerable evidence that antihypertensive drugs reduce death, stroke and heart disease when given to people with moderate to severe hypertension (Musini 2009a; Wright 1999; Wright 2009). However, the magnitude of blood pressure reduction does not always parallel a reduction in mortality or cardiovascular morbidity. Other factors independent of blood pressure reduction may contribute to the beneficial and harmful effects of drug treatment. Nonetheless, the magnitude of blood pressure reduction is often considered an important surrogate or indicator of the likelihood that people will benefit from drug treatment.
Description of the intervention
Thiazide diuretics were developed during the 1950s, when chemists and physiologists tested derivatives of sulfonamide‐based carbonic anhydrase inhibitors, with the goal of discovering drugs that enhance the excretion of sodium with chloride rather than sodium bicarbonate. Thiazide diuretics have been widely used as pharmacological agents for the treatment of hypertension for over five decades. The members of this drug class are derived from benzothiadiazine. Drugs with a similar pharmacologic action on the kidney that do not have the thiazide chemical structure, such as indapamide, chlorthalidone and metolazone, are termed 'thiazide‐like diuretics'. Metolazone is a quinazoline. Chemically, metolazone is not a substitute for benzothiadiazine but it, and other drugs such as indapamide, act on the same co‐transporter in the kidney as thiazides. Therefore, they are appropriately grouped with thiazide diuretics despite not being thiazides themselves (Edwin 2006). In this review, we use the term 'thiazide' to encompass thiazides and 'thiazide‐like' diuretics including the following drugs: hydrochlorothiazide, chlorothiazide, buthiazide, bendroflumethiazide, hydroflumethiazide, trichlormethiazide, methyclothiazide, polythiazide, cyclothiazide, cyclopenthiazide, chlorthalidone, metolazone, quinethazone, fenquizone, clorexolone, clopamide, indapamide, diapamide, isodapamide, mefruside and xipamide. Thiazide diuretics were originally marketed and prescribed in starting doses much higher than the average starting and maximum doses that are currently used for the treatment of hypertension (Edwin 2006).
How the intervention might work
Physiological studies in people show that the early effect of diuretic therapy is to decrease the extracellular volume, plasma volume and cardiac output with relatively unchanged peripheral resistance (Edwin 2006). After several weeks of therapy cardiac output returns to normal and total peripheral resistance decreases. At the level of the kidney, thiazides inhibit reabsorption of sodium and chloride ions from the distal convoluted tubules by blocking the thiazide‐sensitive Na+Cl‐ co‐transporter (Hughes 2004). Basic research studies demonstrate mechanisms whereby the different thiazide and thiazide‐like drugs might differ in their actions and effects (Kurtz 2010). However, none of these differential effects have been demonstrated in people (Campbell 2004).
The thiazide diuretics have a unique adverse effect profile.They potentially affect blood lipids, glucose, potassium, calcium, magnesium, uric acid and chloride concentrations. Thiazides potentially increase Ca2+ concentrations by increasing calcium reabsorption at the distal tubule. This is thought to be due to lowering of the sodium concentration within the epithelial cells, and thus increase of the activity of the Na+/Ca2+‐ATPase on the basolateral membrane to pump more Ca2+ into the interstitium. Thiazides are also thought to increase the reabsorption of Ca2+ by a mechanism involving the reabsorption of sodium and calcium in the proximal tubule in response to sodium depletion. Some of this response is thought to be due to augmentation of the action of parathyroid hormone. Thiazides do not affect potassium transport directly; instead, they stimulate potassium urinary secretion indirectly. Hypokalemia results primarily from increased Na and fluid delivery to the distal tubule with an enhanced aldosterone effect. Thiazides also enhance potassium secretion by activating flow‐sensitive maxi‐K channels; these channels are molecularly distinct from the potassium secretory channels (Edwin 2006).
Why it is important to do this review
A previous review showed that treatment of primary hypertension with different antihypertensive drug classes, compared with placebo or no treatment, decreased stroke but had varied effects on coronary heart disease and all‐cause mortality (Wright 2009). Thiazide trials were classified as low‐dose (hydrochlorothiazide equivalent of less than 50 mg/day) or high‐dose (hydrochlorothiazide equivalent of > 50 mg/day) based on the starting dose in each trial. All trials used stepped care therapy with drugs from other drug classes, aiming to achieve blood pressure targets of < 140/90 mmHg. Interestingly enough, despite a similar magnitude of blood pressure reduction at one year (13/5 mmHg with first‐line, low‐dose thiazides versus 14/7 mmHg with first‐line high‐dose thiazides), there were dose‐related differences in the impact on mortality and coronary heart disease. Surprisingly, high‐dose thiazides (mean dose 90 mg/day hydrochlorothiazide equivalent) reduced stroke but not all‐cause mortality or coronary heart disease significantly, whereas low‐dose thiazides (mean dose of 24 mg/day hydrochlorothiazide equivalent) decreased all‐cause mortality, stroke and coronary heart disease. Several different drugs from the thiazide or thiazide‐like class were used in these trials, including bendrofluazide, chlorothiazide, chlorthalidone, hydrochlorothiazide, hydrochlorothiazide/amiloride, hydrochlorothiazide/triamterene, indapamide, methyclothiazide and trichlormethiazide.
Although it is now well established that first‐line, low‐dose thiazides reduce mortality as well as morbidity, the dose‐related effect of thiazides on systolic as well as diastolic blood pressure, when used as first‐line single drugs, has not been established. Using excessive doses of thiazides may increase toxicity, by causing hypokalemia, hyponatraemia or other metabolic disturbances without additional blood pressure‐lowering effect or commensurate clinical benefit. We also cannot assume that all diuretics will have the same efficacy in reducing blood pressure. The different classes of diuretics and individual drugs within each class might have differing efficacy and adverse effects. It is important to know whether the blood pressure‐lowering effect of thiazides and thiazide‐like diuretics as a class is different from other classes and to know the blood pressure‐lowering dose‐response relationship in relation to other effects of thiazides, such as their metabolic adverse effects.
The main aim of this systematic review is to quantify the dose‐response relationship of thiazides in lowering blood pressure in patients with hypertension. The information derived from this review should facilitate future reviews of head‐to‐head comparisons with other drug classes and assist clinicians in choosing a specific thiazide drug at an appropriate dose.
Objectives
Primary objective
To determine the dose‐related decrease in systolic and/or diastolic blood pressure due to thiazide diuretics compared with placebo control in the treatment of patients with primary hypertension.
Secondary objectives
To determine the dose‐related adverse events leading to patient withdrawal and adverse biochemical effects on serum potassium, uric acid, creatinine, glucose and lipids.
Methods
Criteria for considering studies for this review
Types of studies
Study design must meet the following criteria: placebo‐controlled; random allocation to thiazide diuretic group and parallel placebo group; duration of follow‐up of at least three weeks; office blood pressure measurement at baseline (following wash‐out) and at one or more time points between 3 and 12 weeks after starting treatment.
We included data from cross‐over trials if the authors reported data for the initial treatment period versus the parallel placebo control group followed by an adequate wash‐out period before crossing over to the other active treatment and if data were reported in a similar manner during the second treatment period.
Exclusion criteria
We excluded any of the following: non‐randomized trials; single‐blind as well as open‐label trials; trials using a thiazide diuretic in combination with other classes of drugs as first‐line treatment; abstracts without a complete trial report; trials reporting placebo blood pressure levels following a wash‐out period and comparing them with the treatment levels following randomization; and trials in patients with secondary causes of hypertension.
Types of participants
Adults (18 years or older) with a baseline systolic blood pressure of at least 140 mmHg or a diastolic blood pressure of at least 90 mmHg, measured in a standard way. We excluded trials in which patients had significant renal insufficiency and a documented serum creatinine level more than 1.5 times the normal range from analysis. Participants were not further restricted by age, sex, cardiovascular baseline risk or any other co‐morbid conditions.
Types of interventions
We included monotherapy with any of the following thiazide and thiazide‐like diuretics: bendroflumethiazide, buthiazide, chlorthalidone, chlorothiazide, clopamide, clorexolone, cyclopenthiazide, cyclothiazide, diapamide, fenquizone, hydrochlorothiazide, hydroflumethiazide, indapamide, isodapamide, mefruside, methyclothiazide, metolazone, polythiazide, quinethazone, trichlormethiazide and xipamide. These drugs are referred to as 'thiazide diuretics' in this review for simplicity.
We did not include data from trials in which the thiazide was titrated to a higher dose in a subset of randomized patients to achieve target blood pressure levels because dose‐response relationships cannot be analyzed if patients within each randomized group are taking different doses of the same drug.
If all the patients in the trial were given a forced titrated dose irrespective of the blood pressure recorded, then we included the trial under the highest dose given.
Potassium supplementation was allowed in patients with low serum potassium levels.
Types of outcome measures
Primary outcomes
Change in systolic and diastolic blood pressure compared with placebo. If blood pressure measurements were available at more than one time during the 24‐hour period, we used the trough measurement. We defined peak level as within 12 hours of the dose and trough level as between 12 and 24 hours. If several blood pressure measurements were available within the acceptable window, we used the weighted mean of all blood pressure measurements reported by the study authors during the 3 to 12‐week range as the best estimate of treatment effect.
Secondary outcomes
Patient withdrawals due to adverse effects compared with placebo.
Change in the concentration of serum potassium, uric acid, creatinine, glucose and lipids compared with placebo. If several measurements were available within the acceptable window, then we used the weighted mean of all measurements reported by the study authors during the 3 to 12‐week range as the best estimate of treatment effect.
Search methods for identification of studies
Electronic searches
We searched the following electronic databases for primary studies: the Cochrane Central Register of Controlled Trials (CENTRAL 2014, Issue 1), Ovid MEDLINE (1946 to February 2014), Ovid EMBASE (1974 to February 2014), ClinicalTrials.gov and reference lists of included studies.
We searched the electronic databases using a strategy combining the Cochrane Highly Sensitive Search Strategy for identifying randomized trials in MEDLINE: sensitivity‐maximizing version (2008 revision) with selected MeSH terms and free‐text terms relating to the individual thiazide drugs and hypertension. We used no language restrictions. We used the standard search strategy of the Cochrane Hypertension Review Group with additional terms related to thiazide diuretics in the above listed databases to identify relevant articles. We translated the MEDLINE search strategy (Appendix 1) into EMBASE (Appendix 2) and CENTRAL (Appendix 3) using the appropriate controlled vocabulary as applicable.
Searching other resources
We also searched Web of Science and bibliographic citations. In case of incomplete reports, we used MEDLINE to search for related papers and contacted authors to retrieve missing information. We searched the bibliographies of pertinent articles, reviews and texts for additional citations. We used previously published meta‐analyses on the dose response of thiazide diuretics, as well as narrative reviews, to help identify references to trials. We assessed trials included in the Law et al systematic review and meta‐analysis for eligibility for this review (Law 2009). Several trials included in the Law meta‐analysis do not meet the inclusion criteria for this review and the reasons for exclusion are listed under Characteristics of excluded studies. We applied no language restrictions.
Data collection and analysis
Selection of studies
We screened all potentially relevant articles and rejected articles on the initial screen if the title or abstract was not a report of a randomized, placebo‐controlled trial or if it did not meet the minimum inclusion criteria. We retrieved the full texts of the remaining articles. Two independent review authors (VM and MN) assessed the eligibility of the trials using a trial selection form. The third review author (CJ) assessed the eligibility of non‐English trials. JMW or KB resolved discrepancies. We counted trials with more than one publication only once. See Figure 1.
1.
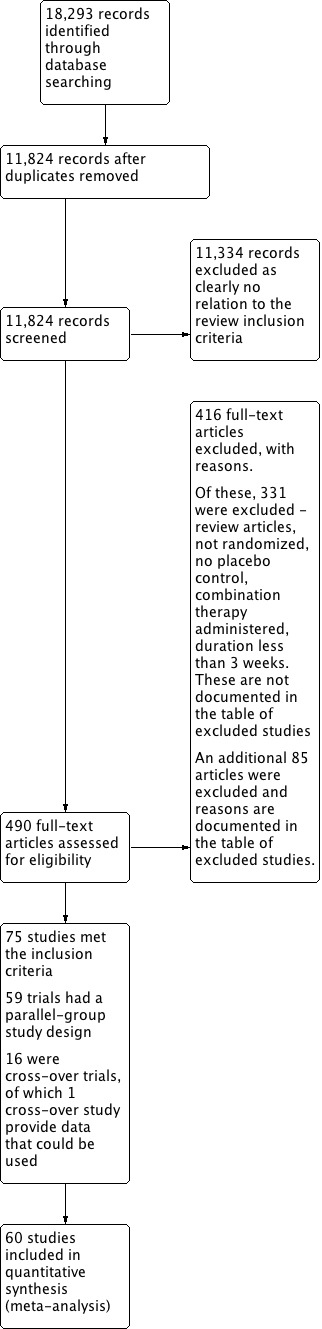
Study flow diagram.
Data extraction and management
Once it was determined that the trials met the inclusion criteria, two independent review authors (VM and MN) abstracted data for all primary and secondary outcomes using a standard form, and then cross‐checked. If data were presented numerically (tables or text) or graphically (in figures), we preferred the numeric data because of possible measurement errors when estimating from the graphs. Both review authors (VM and MN) cross‐checked all numeric calculations and graphic interpolations.
The position of the patient during blood pressure measurement may affect the blood pressure‐lowering effect. However, in order not to lose valuable data, if only one position was reported, we collected data from that position. When blood pressure measurement data were available in more than one position, sitting blood pressure was the first preference, followed by standing and supine blood pressure measurements.
We extracted data for the following outcome measures:
The trough and/or peak systolic and diastolic blood pressure at baseline following the wash‐out period.
The trough and/or peak systolic and diastolic blood pressure measurement taken between 3 and 12 weeks of the treatment period. If more than one blood pressure measurement was available then we used the weighted mean blood pressure levels.
The number of patient withdrawals due to adverse effects for each drug dose during the specified period of time the patient is taking the drug.
The blood concentrations of serum potassium, uric acid, creatinine, glucose and lipids with standard deviation (SD) at baseline as well as between 3 and 12 weeks during the treatment period. If more than one measurement was available between 3 and 12 weeks then we used the weighted mean level.
Assessment of risk of bias in included studies
Two independent review authors (VM and MN) assessed the risk of bias of the included studies to create 'Risk of bias' tables as outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Handbook 2011). We resolved any discrepancies between the review authors by discussion with a third review author (JW or KB).
We evaluated the following items: randomization; allocation concealment; blinding; incomplete outcome data reporting; selective reporting and other biases (e.g. industry sponsorship).
Measures of treatment effect
For continuous outcomes, we combined data for placebo‐corrected systolic and diastolic blood pressure reduction and for placebo‐corrected serum concentrations of potassium, uric acid, creatinine, glucose and lipids using a mean difference (MD) method, presented with 95% confidence intervals (CI). Blood pressure data are presented as systolic /diastolic blood pressure, with parentheses for 95% CI with accuracy up to one decimal point. Metabolic data are presented as mean difference with parentheses for 95% CI, with accuracy up to two decimal points.
For the dichotomous outcome withdrawals due to adverse effects, data are presented as risk ratio (RR) with 95% CI. We have not provided absolute risk difference and number needed to treat to benefit or harm due to very low‐quality evidence for the outcome 'withdrawal due to adverse effects' due to high risk of selective reporting bias.
Unit of analysis issues
For cross‐over trials that met the inclusion criteria, we used the first parallel‐group period when patients were randomized to thiazide diuretics or placebo in data analyses. For dose‐ranging trials that compared a single placebo group to several different doses, the forest plot includes the number of patients adjusted for placebo‐controlled treatment group in these trials for an accurate overall effect across all thiazide trials.
Dealing with missing data
In order to address missing data we attempted to contact the study's authors using the first author firstly then any of the co‐authors. We used the publication year 1990 as a cut‐off for verifying the authors' contact information. We did not check anything older than that. Most contacts were email addresses; the rest were telephone numbers, fax numbers or business addresses. We were not successful in obtaining additional data despite our efforts. In case missing information was not available, we included the best estimate based on the information in the same trial or from other trials using the same dose.
In case of missing SD for the change in blood pressure, we imputed the SD based on the information in the same trial or from other trials using the same dose. We used the following hierarchy (listed from high to low preference) to impute SD values:
SD calculated either from the t statistics corresponding to the exact P value reported or from the 95% CI of the mean difference between treatment groups.
SD of change in blood pressure from a different position than that of the blood pressure data used.
SD of blood pressure at the end of treatment.
SD of blood pressure at the end of treatment measured from a different position than that of the blood pressure data used.
SD of blood pressure at baseline (except if this measure is used for entry criteria) (Musini 2009b).
Mean SD of change in blood pressure from other trials using the same drug and dose.
Mean weighted SD of change available from all other trials using the same class of drug at any dose.
Assessment of heterogeneity
We calculated the heterogeneity of treatment effects between the trials using a standard Chi2 test in RevMan 5.2 (RevMan 2012). We applied the fixed‐effect model to obtain summary statistics of pooled trials, unless significant between‐study heterogeneity was present, in which case we used the random‐effects model. If a statistically significant difference compared to placebo control was still present using the random‐effects model, then we reported the fixed pooled estimate and CI because of the tendency for smaller trials, which are susceptible to publication bias, to be over‐weighted with a random‐effects analysis. We compared overall effect size using both a fixed‐effect and random‐effects model and determined that they were not significantly different from each other.
If the calculated I2 statistic value was greater than 50%, we explored the reasons for heterogeneity and the trials contributing to the heterogeneity (differences in baseline characteristics between trials and their possible impact on the magnitude of systolic and diastolic blood pressure reduction).
Data synthesis
We carried out data synthesis and analyses using the Cochrane RevMan 5.2 software (RevMan 2012). We directly compared the effect size between doses for each thiazide diuretic drug only using data from trials that randomized participants to different doses of the drug within the same trial. In case direct comparison was not possible, we did an 'adjusted indirect comparison' using the computer software developed by Wells 2009. We considered a P value less than 0.05 (P < 0.05) to be statistically significant for all comparisons.
Subgroup analysis and investigation of heterogeneity
We planned subgroup analyses at the protocol stage based on age (18 to 59 years versus 60 years or older), sex, race, comorbid conditions and severity of blood pressure at baseline. Due to a lack of available data, we could only perform subgroup analyses based on classifying trials reporting mean baseline severity of blood pressure level.
Sensitivity analysis
We intended to test the robustness of the results using sensitivity analyses (high versus low trial quality; fixed‐effect versus random‐effects model; position of blood pressure measurement; trough versus peak blood pressure measurement; reported versus imputed SD and industry versus non‐industry‐sponsored trials). Sufficient data were available to perform sensitivity analyses for overall thiazide diuretics compared to placebo including three drugs: chlorthalidone (14 RCTs were included), hydrochlorothiazide (44 RCTs were included) and indapamide (12 RCTs were included) for systolic and diastolic blood pressure‐lowering effects.
Results
Description of studies
Results of the search
The search strategy identified 18,293 citations; after de‐duplicating we screened 11,824 citations. Of these, 75 (0.7% of screened articles) were double‐blind, randomized, placebo‐controlled trials meeting the minimum inclusion criteria. Fifty‐nine RCTs were of a parallel‐group design yielding data that could be used in the evaluation of dose‐related blood pressure and metabolic effects of six thiazide diuretics (bendrofluazide, chlorthalidone, cyclopenthiazide, hydrochlorothiazide, indapamide and metolazone). See the PRISMA diagram (Figure 1).
Sixteen of the 75 RCTs were cross‐over trials, of which we excluded 15 because data were not reported for the initial parallel period. The single cross‐over trial which provided separate data for periods one and two compared indapamide 2.5 mg/day with placebo in 24 patients for a duration of eight weeks (Soltero 1989). See Characteristics of excluded studies.
Table 1: Included trials sorted according to the year of publication
| Year of publication |
Trials included in meta‐analyses |
| 1946 to 1949 | 0 |
| 1950 to 1959 | 0 |
| 1960 to 1969 | 0 |
| 1970 to 1979 | 2 |
| 1980 to 1989 | 13 |
| 1990 to 1999 | 34 |
| 2000 to 2009 | 11 |
| 2010 to 2012 | 0 |
| Overall | 60 trials |
Forty‐nine of the 60 included trials (82%) were published before the year 2000.
Included studies
Please refer to Characteristics of included studies for details of each of the 60 included trials. Studies included adult patients with systolic blood pressure entry criteria of 140 mmHg or more and/or diastolic blood pressure entry criteria of 90 mmHg or more. Co‐morbidities were not reported in most trials. Across all 60 trials, the total number of randomized patients was 11,282; mean age was 55 years; mean blood pressure was 158/99 mmHg; and mean duration of treatment was eight weeks. Of the total population, 53% of patients were male and 47% were female.
Table 2: Overall summary of the 60 trials meeting the inclusion criteria
| Thiazide drug | Dose range |
Number of trials |
Total patients randomized |
N (% males) and N (% females) |
Mean duration (weeks) |
Mean age (years) |
Baseline systolic/diastolic blood pressure mmHg |
| Bendrofluazide | 1.25 to 10.0 mg/day | 1 | 257 | 103 (40%) 154 (60%) |
12 weeks | 57 years | 165/102 mmHg |
| Chlorthalidone | 12.5 to 100 mg/day | 8* | 1265 | 581 (50.4%) 684 (49.6%) |
7 weeks | 53 years | 163/88 mmHg |
| Cyclopenthiazide | 0.05 to 0.50 mg/day | 1 | 53 | 22 (41.5%) 31 (49.6%) |
8 weeks | 57 years | 164/97 mmHg |
| Hydrochlorothiazide | 3 to 100 mg/day | 40 | 7560 | 4152 (57%) 3408 (43%) |
8 weeks | 54 years | 155/100 mmHg |
| Indapamide | 1 to 5 mg/day | 10 | 2075 | 1018 (48.4%) 1057 (51.6%) |
10 weeks | 58 years | 161/98 mmHg |
| Metolazone | 0.5 to 2.0 mg/day | 1 | 105 | 46 (43.8%) 59 (56.2%) |
6 weeks | Not reported | 150/98 mmHg |
| Overall | 60 trials | 11,282* |
5922 (53.0%) 5360 (47.0%) |
8 weeks | 55 years | 158/99 mmHg |
*The Siegel 1992 RCT has both hydrochlorothiazide 50 mg/day and chlorthalidone 50 mg/day treatment arms therefore it is counted once in the overall included trial total. Also, the 33 placebo patients in the Siegel 1992 study are counted only once in the overall total.
For bendrofluazide , one randomized, double‐blind, parallel‐group, placebo‐controlled trial met the inclusion criteria (Carlsen 1990). Two hundred and fifty‐seven patients were randomized, with a mean age of 57 years and a mean baseline blood pressure of 165/102 mmHg; the percentage of male participants was 40% and of female participants 60%. The duration of treatment was 12 weeks.
For chlorthalidone , eight randomized, double‐blind, placebo‐controlled, parallel‐group trials met the inclusion criteria. There were 1265 randomized patients, with a mean age of 53 years and a mean baseline blood pressure of 163/88 mmHg; the percentage of male participants was 50% and of female participants 50%. The mean duration of treatment was seven weeks.
For cyclopenthiazide , one randomized, double‐blind, placebo‐controlled, parallel‐group trial met the inclusion criteria (McVeigh 1988). Fifty‐three patients were randomized, with a mean age 57 years and a mean baseline blood pressure of 164/97 mmHg; the percentage of male participants was 42% and of female participants 58%. The duration of treatment was eight weeks.
Forhydrochlorothiazide , 40 randomized, double‐blind, placebo‐controlled, parallel‐group trials met the inclusion criteria. There were 7560 randomized patients, with a mean age of 54 years and a mean baseline blood pressure of 155/100 mmHg; the percentage of male participants was 55% and of female participants 45%. The mean duration of treatment was eight weeks.
Forindapamide , 10 randomized, double‐blind, placebo‐controlled, parallel‐group trials met the inclusion criteria. There were 2075 randomized patients, with a mean age of 58 years and a mean baseline blood pressure of 161/98 mmHg; the percentage of male participants was 49% and of female participants 51%. The mean duration of treatment was 10 weeks.
For metolazone , one randomized, double‐blind, placebo‐controlled, parallel‐group trial met the inclusion criteria (Curry 1986). There were 105 randomized patients; mean age was not reported and the baseline blood pressure was 150/98 mmHg; the percentage of male participants was 44% and of female participants 56%. The duration of treatment was six weeks.
Excluded studies
We excluded 86 studies. See Characteristics of excluded studies for details. Reasons for exclusion include: not a randomized trial; had no parallel, placebo‐controlled treatment arm; used combination therapy; cross‐over trial design with no wash‐out between treatment periods either reporting data before the minimum three weeks duration period or not reporting data at the end of the first parallel placebo treatment period; improper blood pressure measurement; stepped up therapy only in non‐responders (not meeting the target goal blood pressure levels) and dose was not titrated in all randomized patients. Articles were also excluded if full‐text reports were not available or data were reported in a way that could not be used for analysis.
Risk of bias in included studies
We evaluated each trial that provided data for at least one of the outcome measures using the Cochrane 'Risk of bias' tool. See Figure 2 and Figure 3.
2.
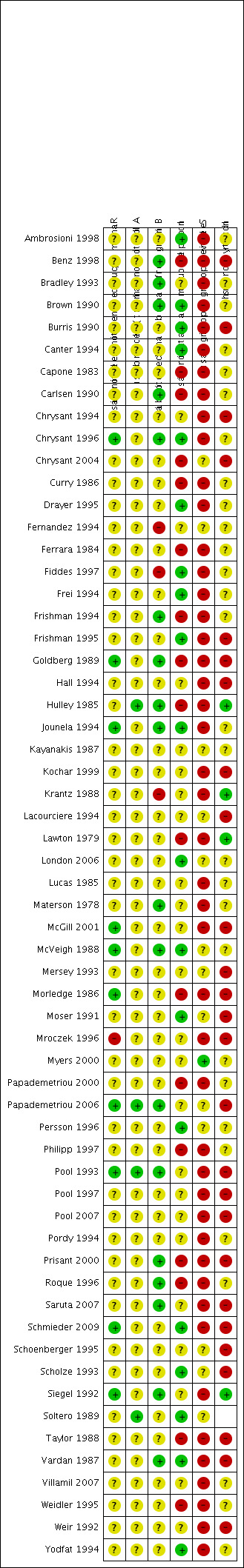
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
3.
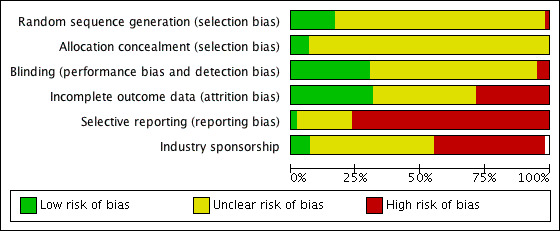
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
For details of the following seven factors evaluated for risk of bias in each individual study, see the 'Risk of bias' tables in Characteristics of included studies.
Random sequence generation (selection bias)
We determined nine of the 60 included trials (15%) to have adequate random sequence generation (Chrysant 1996; Goldberg 1989; Jounela 1994; McVeigh 1988; McGill 2001; Morledge 1986; Papademetriou 2006; Schmieder 2009; Siegel 1992). We judged 49 (82%) of the trials as at unclear risk of bias as the technique of randomization was not reported and in one trial we judged reporting of randomization as at high risk of bias (Mroczek 1996).
Allocation
We judged four of the 60 trials (7%) as at low risk of bias for allocation concealment (Hulley 1985; Papademetriou 2006; Pool 1993; Soltero 1989). The remaining 56 trials (93%) did not report on how allocation concealment was performed and therefore we judged them as at unclear risk of bias, probably resulting in high risk of selection bias.
Blinding
We judged 11 of the 60 trials (18%) as at low risk of bias since they adequately described how patients and physicians and outcome assessors were blinded (Benz 1998; Bradley 1993; Brown 1990; Carlsen 1990; Chrysant 1996; Frishman 1994; Goldberg 1989; Hulley 1985; Jounela 1994; Materson 1978; McVeigh 1988). We judged three trials as at high risk of bias (Fernandez 1994; Fiddes 1997; Krantz 1988). We judged the remaining 46 trials (77%) as at unclear risk of bias due to lack of reporting.
Incomplete outcome data
We judged 19 of the 60 trials (32%) as at low risk of bias since they adequately described total withdrawals (which were less than 20% of the total randomized patients) and how these patients were accounted for in the analysis (Ambrosioni 1998; Brown 1990; Burris 1990; Canter 1994; Chrysant 1996; Drayer 1995; Fiddes 1997; Frei 1994; Frishman 1995; Jounela 1994; London 2006; McVeigh 1988; Moser 1991; Persson 1996; Schmieder 2009; Scholze 1993; Soltero 1989; Vardan 1987; Weir 1992). We judged 17 (28%) trials as at high risk of bias (Benz 1998; Capone 1983; Carlsen 1990; Chrysant 2004; Curry 1986; Ferrara 1984; Frishman 1994; Goldberg 1989; Hulley 1985; Lawton 1979; Morledge 1986; Papademetriou 2000; Philipp 1997; Prisant 2000; Roque 1996; Taylor 1988; Weidler 1995). We judged the remaining 24 trials (40%) as at unclear risk of bias.
Selective reporting
Systolic and diastolic blood pressure data were provided in all trials and therefore they were not subject to selective reporting bias. For other outcomes, we judged one out of 60 trials (2%) to be at low risk of bias since it adequately described all outcome measures specified in the publication (Myers 2000). We judged 21 of the 60 trials (35%) as at unclear risk and the remaining 38 trials (63%) as at high risk of bias since they did not report on outcomes such as total adverse events, withdrawal due to adverse effects or metabolic data even though these were collected, according to the methods sections of the publications.
Other potential sources of bias
A factor considered as another potential sources of bias was the source of funding for each included study. We judged 14 of the 60 trials (23%) as at low risk of other bias (Ambrosioni 1998; Benz 1998; Chrysant 1994; Chrysant 2004; Frishman 1994; Frishman 1995; Hulley 1985; Kochar 1999; London 2006; McGill 2001; Schmieder 2009; Schoenberger 1995; Scholze 1993; Vardan 1987). We judged nine trials (15%) as at high risk of other bias (Bradley 1993; Brown 1990; Curry 1986; Ferrara 1984; Fiddes 1997; Frei 1994; Lucas 1985; Papademetriou 2000; Prisant 2000), and 37 trials (62%) as at unclear risk of other bias. For details, please see the 'Risk of bias' tables in Characteristics of included studies.
Of all 60 included studies, we only judged six (10%) as at low risk of bias in at least three of the six items evaluated using the 'Risk of bias' tool (Chrysant 1996; Jounela 1994; McVeigh 1988; Papademetriou 2006; Schmieder 2009; Vardan 1987). We judged the other 54 included trials (90%) as at unclear or high risk of bias. Thus, the overall evidence in this review is subject to a high risk of bias. This has to be taken into consideration in interpreting the findings. It is unclear what effects residual bias may have on our estimate of the efficacy of systolic and diastolic blood pressure reduction with thiazides, but it is likely that their harms are underestimated, including withdrawal due to adverse effects and adverse or potentially adverse metabolic changes.
Funding bias
Twenty‐eight (47%) of the 60 included studies were industry‐sponsored but in 26 of the 28 the bias would have been in favor of the other drug being tested and not in favor of the thiazide. Only five trials (8%) were sponsored by national agencies such as the National Institutes of Health, National Institute of Aging, National Heart and Lung Institute etc. and all of these were older trials and studied high doses of chlorthalidone and hydrochlorothiazide. For the remaining 26 trials (42.4%), the source of funding was not reported. See the 'Risk of bias' tables in Characteristics of included studies.
Publication bias
Publication bias is defined in this review as selective publication of studies with positive results and is another source of bias that may have skewed the results of this review. Examining the funnel plots for systolic and diastolic blood pressure in this review suggested asymmetry, indicating that there was a high risk of publication bias (Figure 4).
4.
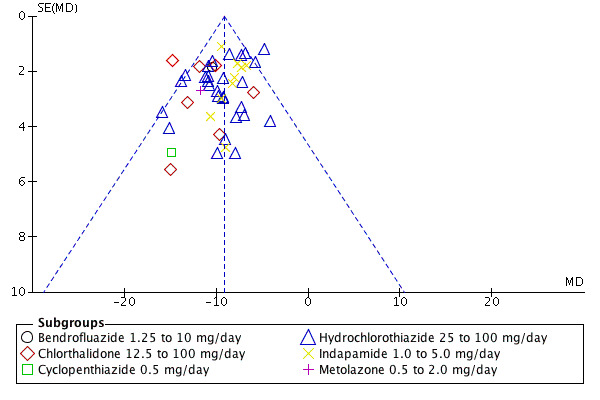
Funnel plot of comparison: 7 Thiazide versus placebo, outcome: 7.1 Systolic blood pressure.
Subgroup analyses based on risk of bias
In view of the fact that it was not possible to predict the direction of the bias in the industry‐funded trials and the trials where the source of funding was not reported, plus the higher doses used in the trials sponsored by national agencies, we did not attempt any subgroup comparisons.
Effects of interventions
Dose‐related systolic and diastolic blood pressure‐lowering
Three thiazide diuretics (bendrofluazide, cyclopenthiazide and metolazone) had only one trial each that met the minimum inclusion criteria. Therefore, there were insufficient data to present forest plots for these three drugs.
Bendrofluazide
Bendrofluazide dose ranged from 1.25 to 10 mg/day as monotherapy versus placebo control for a mean duration of treatment of 12 weeks in 257 patients in one trial (Carlsen 1990). See Analysis 1.1; Analysis 1.2.
1.1. Analysis.
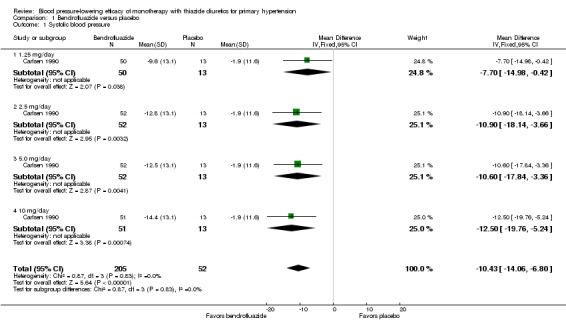
Comparison 1 Bendrofluazide versus placebo, Outcome 1 Systolic blood pressure.
1.2. Analysis.
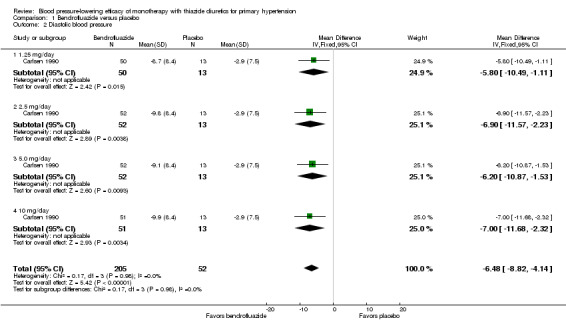
Comparison 1 Bendrofluazide versus placebo, Outcome 2 Diastolic blood pressure.
Table 3: Dose‐related systolic and diastolic blood pressure‐lowering efficacy of bendrofluazide
|
Bendrofluazide Carlsen 1990 |
Systolic blood pressure mmHg (95% CI) |
Diastolic blood pressure mmHg (95% CI) |
| 1.25 mg/day | ‐7.7 (‐15.0 to ‐0.4) | ‐5.8 (‐10.5 to ‐1.1) |
| 2.5 mg/day | ‐10.9 (‐18.1 to ‐3.7) | ‐6.9 (‐11.6 to ‐2.2) |
| 5 mg/day | ‐10.6 (‐17.8 to ‐3.4) | ‐6.2 (‐10.9 to ‐1.5) |
| 10 mg/day | ‐12.5 (‐19.8 to ‐5.2) | ‐7.0 (‐11.7 to ‐2.3) |
| Overall | ‐10.4(‐14.1 to ‐6.8) | ‐6.5 (‐8.8 to ‐4.1) |
The lowest dose of bendrofluazide that showed a statistically significant blood pressure‐lowering was 1.25 mg/day for both systolic and diastolic blood pressure. The overall placebo‐corrected systolic blood pressure‐lowering effect size across 1.25 to 10 mg/day doses was ‐10.4 mmHg (P value < 0.00001, with I2 = 0% and test for subgroup differences P value = 0.87, with I2 = 0%) and for diastolic blood pressure was ‐6.5 mmHg (P value < 0.00001, with I2 = 0% and test for subgroup differences P value = 0.98, with I2 = 0%). Direct comparison of doses in the Carlsen 1990 dose‐ranging trial showed no significant differences in systolic or diastolic blood pressure‐lowering between the different doses used.
The placebo‐corrected systolic/diastolic blood pressure‐lowering with bendrofluazide was 10.4/6.5 mmHg.
Cyclopenthiazide
Cyclopenthiazide doses ranged from 50 to 500 µg/day and were administered as monotherapy versus placebo control over eight weeks' duration in one trial (McVeigh 1988). See Analysis 2.1; Analysis 2.2.
2.1. Analysis.
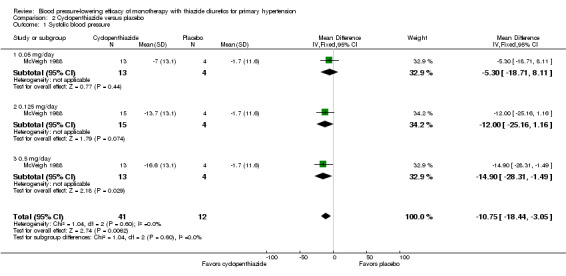
Comparison 2 Cyclopenthiazide versus placebo, Outcome 1 Systolic blood pressure.
2.2. Analysis.
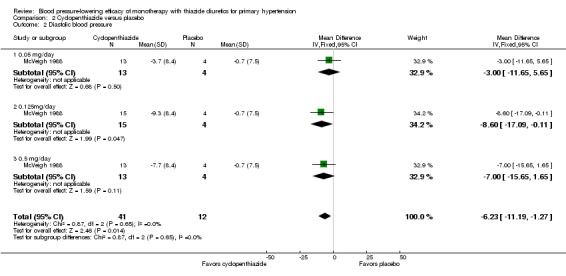
Comparison 2 Cyclopenthiazide versus placebo, Outcome 2 Diastolic blood pressure.
Table 4: Dose‐related systolic and diastolic blood pressure‐lowering efficacy of cyclopenthiazide
|
Cyclopenthiazide McVeigh 1988 |
Systolic blood pressure mmHg (95% CI) |
Diastolic blood pressure mmHg (95% CI) |
| 0.05 mg/day | ‐5.3 (‐18.7to 8.1) | ‐3.0 (‐11.7 to 5.7) |
| 0.125 mg/day | ‐12.0 (‐25.2 to 1.2) | ‐8.6 (‐17.1 to ‐0.1) |
| 0.5 mg/day | ‐14.9 (‐28.3 to ‐1.5) | ‐7.0 (‐15.7 to 1.7) |
| Overall | ‐10.8 (‐18.4 to ‐3.1) | ‐6.2 (‐11.2 to ‐1.3) |
The lowest dose of cyclopenthiazide that showed a statistically significant systolic blood pressure‐lowering was 500 µg/day. Cyclopenthiazide 0.125 mg/day showed a significant difference from placebo in lowering diastolic blood pressure. The overall placebo‐corrected systolic blood pressure‐lowering effect size across 0.05 to 0.5 mg/day doses was ‐10.8 mmHg (P value = 0.006, with I2= 0% and test for subgroup differences P value = 0.60, with I 2= 0%). For diastolic blood pressure it was ‐6.2 mmHg (P value = 0.01, with I 2 = 0% and test for subgroup differences P value = 0.65, with I 2= 0%). Direct comparison between doses showed no demonstrable dose response.
The placebo‐corrected systolic/diastolic blood pressure‐lowering with cyclopenthiazide was 10.8/6.2 mmHg.
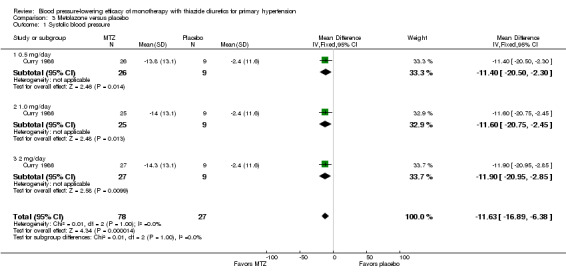
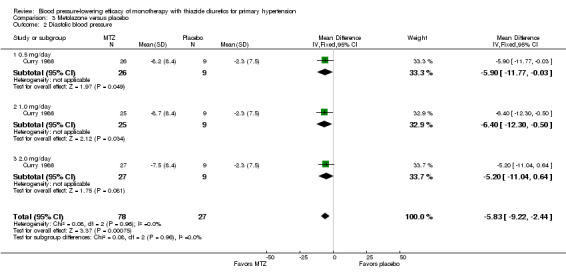
Metolazone
Metolazone doses ranged from 0.5 to 2 mg/day and were administered as monotherapy versus placebo control over six weeks' duration in one trial (Curry 1986). See Analysis 3.1; Analysis 3.2.
3.1. Analysis.
Comparison 3 Metolazone versus placebo, Outcome 1 Systolic blood pressure.
3.2. Analysis.
Comparison 3 Metolazone versus placebo, Outcome 2 Diastolic blood pressure.
Table 5: Dose‐related systolic and diastolic blood pressure‐lowering efficacy of metolazone
|
Metolazone Curry 1986 |
Systolic blood pressure mmHg (95% CI) |
Diastolic blood pressure mmHg (95% CI) |
| 0.5 mg/day | ‐11.4 (‐20.5 to ‐2.3) | ‐5.9 (‐11.8 to ‐0.0) |
| 1.0 mg/day | ‐11.6 (‐20.8 to ‐2.5) | ‐6.4 (‐12.3 to ‐0.5) |
| 2 mg/day | ‐11.9 (‐21.0 to ‐2.9) | ‐5.2 (‐11.0 to 0.6) |
| Overall | ‐11.6 (‐16.9 to ‐6.4) | ‐5.8 (‐9.2 to ‐2.4) |
The lowest dose of metolazone that showed a statistically significant systolic and diastolic blood pressure‐lowering was 0.5 mg/day. The overall placebo‐corrected systolic blood pressure‐lowering effect across all doses was ‐11.6 mmHg (P value < 0.0001, with I2= 0% and the test for subgroup differences P value = 1.00, with I2 = 0%). For diastolic blood pressure it was ‐5.8 mmHg (P value = 0.0007, with I2 = 0% and the test for subgroup differences P value = 0.96, with I2 = 0%). Direct comparison of doses did not show any significant differences in systolic or diastolic blood pressure‐lowering between the different doses used.
The placebo‐corrected systolic/diastolic blood pressure‐lowering with metolazone was 11.6/5.8 mmHg.
Three thiazides: chlorthalidone, hydrochlorothiazide and indapamide had sufficient trials to pool data in meta‐analyses and conduct subgroup and sensitivity analyses.
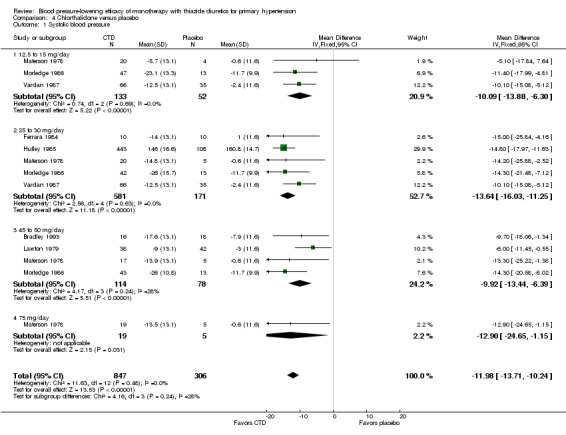
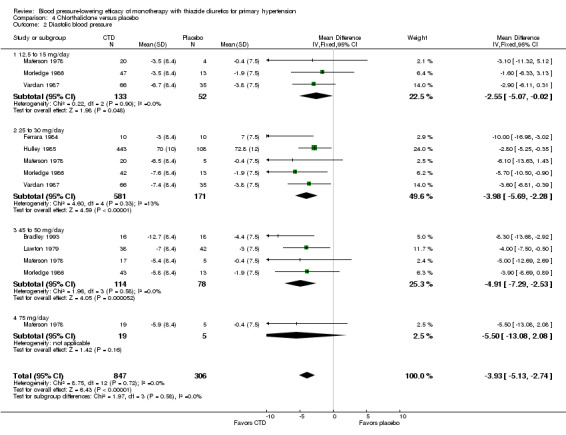
Chlorthalidone
Seven trials met the inclusion criteria, enrolling a total of 1297 patients who were treated for a mean duration of seven weeks (Bradley 1993; Ferrara 1984; Hulley 1985; Lawton 1979; Materson 1978; Morledge 1986; Vardan 1987). These trials compared chlorthalidone doses ranging from 12.5 to 75 mg/day to a placebo control. See Analysis 4.1; Analysis 4.2.
4.1. Analysis.
Comparison 4 Chlorthalidone versus placebo, Outcome 1 Systolic blood pressure.
4.2. Analysis.
Comparison 4 Chlorthalidone versus placebo, Outcome 2 Diastolic blood pressure.
Table 6: Dose‐related systolic and diastolic blood pressure‐lowering efficacy of chlorthalidone
| Chlorthalidone |
Systolic blood pressure mmHg (95% CI) |
Diastolic blood pressure mmHg (95% CI) |
| 12.5 to 15 mg/day | ‐10.1 (‐13.9 to ‐6.3) | ‐2.6 (‐5.1 to ‐0.0) |
| 25 mg/day | ‐13.6 (‐16.0 to ‐11.3) | ‐4.0 (‐5.7 to ‐2.3) |
| 50 mg/day | ‐9.9 (‐13.4 to ‐6.4) | ‐4.9 (‐7.3 to ‐2.5) |
| 75 mg/day | ‐12.9 (‐24.7 to ‐1.2) | ‐5.5 (‐13.1 to 2.1) |
| Overall | ‐12.0 (‐13.7 to ‐10.2) | ‐3.9 (‐5.1 to ‐2.7) |
The lowest dose of chlorthalidone that showed a statistically significant blood pressure‐lowering was 12.5 to 15 mg/day. Based on seven RCTs, the overall placebo‐corrected systolic blood pressure‐lowering effect for 12.5 to 75 mg/day doses was ‐12.0 mmHg (P value < 0.00001, with I 2 = 0%, test for subgroup differences P value = 0.24, with I2 = 27.8%). For diastolic blood pressure it was ‐3.9 mmHg (P value < 0.00001, with I2 = 0%, test for subgroup differences P value = 0.58, with I 2 = 0%).
Dose‐related systolic blood pressure‐lowering response of chlorthalidone by direct comparison
Two dose‐ranging trials allowed direct dose comparisons to be performed (N = 276) (Materson 1978; Morledge 1986). Chlorthalidone 25 mg/day did not lower systolic blood pressure more than 12.5 mg/day. Chlorthalidone 75 mg/day did not lower systolic blood pressure more than 50 mg/day, and 50 mg/day did not lower systolic blood pressure more than 25 mg/day or 12.5 to 15 mg/day. Therefore the maximum systolic blood pressure‐lowering dose of chlorthalidone is likely to be 12.5 mg/day, which lowered systolic blood pressure by ‐10.1 (95% CI ‐13.9 to ‐6.3) mmHg. Systolic blood pressure‐lowering efficacy at doses > 12.5 mg/day was ‐12.0 (95% CI ‐13.7 to ‐10.2) mmHg.
Dose‐related diastolic blood pressure‐lowering response of chlorthalidone by direct comparison
Two dose‐ranging trials allowed direct dose comparisons to be performed (N = 276) (Materson 1978; Morledge 1986). Both chlorthalidone 75 mg and the 50 mg/day dose did not significantly lower diastolic blood pressure compared to 12.5 mg/day. However, 25 mg/day significantly lowered diastolic blood pressure more than 12.5 mg/day by ‐2.2 (95% CI ‐4.3 to ‐0.2) mmHg. Chlorthalidone 75 mg/day dose was not significantly different from 50 mg/day dose nor was 50 mg dose significantly different from 25 mg/day. Therefore chlorthalidone 25 mg/day is likely to result in maximum diastolic blood pressure‐lowering efficacy. The maximum diastolic blood pressure‐lowering efficacy at doses > 25 mg/day was ‐4.3 (95% CI ‐5.7 to ‐3.0) mmHg.
Thus the dose to produce maximal systolic/diastolic blood pressure‐lowering with chlorthalidone is between 12.5 and 25 mg/day with an average blood pressure reduction of 12.0/3.9 mmHg.
Hydrochlorothiazide
Forty trials of hydrochlorothiazide met the inclusion criteria, with doses ranging from 3 to 100 mg/day for a mean treatment duration of eight weeks.
Since there were 15 dose‐ranging trials that compared a single placebo group to several different doses, the forest plot includes the number of patients adjusted for placebo‐controlled treatment group in these trials for an accurate overall effect across all thiazide trials (Benz 1998; Canter 1994; Chrysant 1994; Goldberg 1989; Jounela 1994; Kochar 1999; Lucas 1985; McGill 2001; Papademetriou 2000; Papademetriou 2006; Philipp 1997; Pool 1997; Pool 2007; Scholze 1993; Villamil 2007). See Analysis 5.1; Analysis 5.2.
5.1. Analysis.
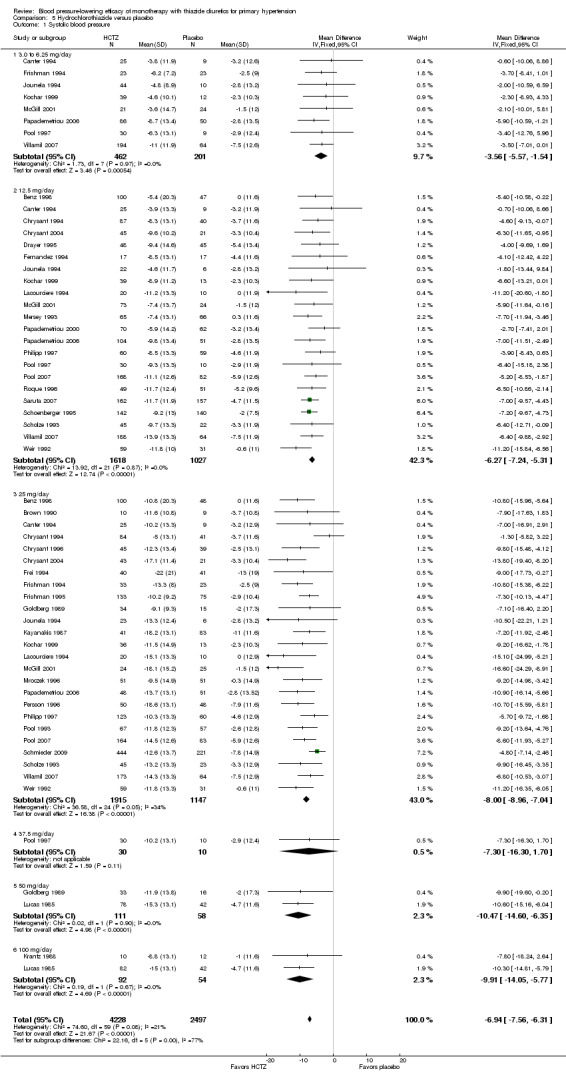
Comparison 5 Hydrochlorothiazide versus placebo, Outcome 1 Systolic blood pressure.
5.2. Analysis.
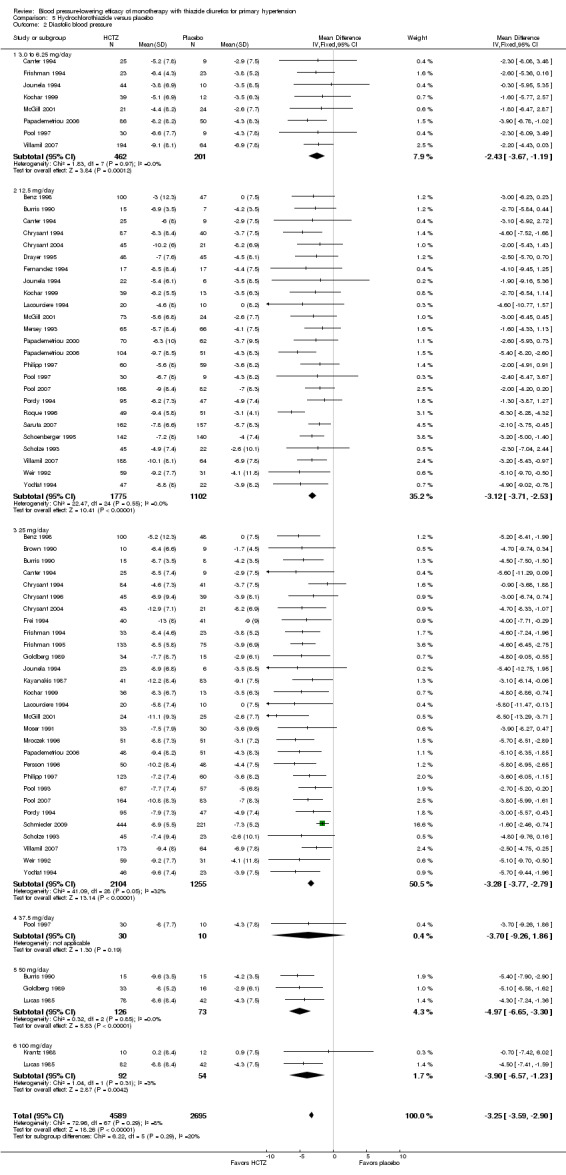
Comparison 5 Hydrochlorothiazide versus placebo, Outcome 2 Diastolic blood pressure.
Table 7: Dose‐related systolic and diastolic blood pressure‐lowering efficacy of hydrochlorothiazide
| Hydrochlorothiazide |
Systolic blood pressure mmHg (95% CI) |
Diastolic blood pressure mmHg (95% CI) |
| 3 to 6.25 mg/day | ‐3.6 (‐5.6 to ‐1.5) | ‐2.4 (‐3.7 to ‐1.2) |
| 12.5 mg/day | ‐6.3 (‐7.2 to ‐5.3) | ‐3.1 (‐3.7 to ‐2.5) |
| 25 mg/day | ‐8.0 (‐9.0 to ‐7.0) | ‐3.3 (‐3.8 to ‐2.8) |
| 37.5 mg/day | ‐7.3 (‐16.3 to 1.7) | ‐3.7 (‐9.3 to 1.9) |
| 50 mg/day | ‐10.5 (‐14.6 to ‐6.4) | ‐5.0 (‐6.7 to ‐3.3) |
| 100 mg/day | ‐9.9 (‐14.1 to ‐5.8) | ‐3.9 (‐6.6 to ‐1.2) |
| Overall | ‐6.9 (‐7.6 to ‐6.3) | ‐3.3 (‐3.6 to ‐2.9) |
The lowest dose of hydrochlorothiazide (3 to 6.25 mg/day) statistically significantly reduced both systolic and diastolic blood pressure in eight trials with 663 patients. Based on 33 trials, for a mean duration of treatment of eight weeks, the overall placebo‐corrected systolic blood pressure‐lowering effect size across 3 to 100 mg/day doses was ‐6.9 mmHg in 6725 patients (P value < 0.00001, with I2 = 21% and test for subgroup differences P value = 0.0005, with I2 = 77.4%). For diastolic blood pressure it was ‐3.3 mmHg in 7284 patients (P value < 0.00001, with I 2 = 8% and test for subgroup differences P value = 0.29, with I2 = 19.6%). The significant heterogeneity is explained by the significant differences in systolic blood pressure‐lowering between doses.
Dose‐related systolic blood pressure‐lowering response of hydrochlorothiazide by direct comparison
Hydrochlorothiazide 12.5 mg lowered systolic blood pressure more than hydrochlorothiazide 3 to 6.25 mg/day by ‐2.2 (95% CI ‐3.8 to ‐0.6) mmHg based on seven dose‐ranging trials in 920 patients (P value = 0.008; heterogeneity was not significant, with I 2 = 0%) (Canter 1994; Jounela 1994; Kochar 1999; McGill 2001; Papademetriou 2006; Pool 1997; Villamil 2007). Hydrochlorothiazide 25 mg/day lowered systolic blood pressure more than hydrochlorothiazide 12.5 mg/day by ‐2.7 (95% CI ‐3.9 to ‐1.5) mmHg based on 14 trials in 2019 patients (P value < 0.0001; heterogeneity was significant: P value = 0.007, with I 2 = 55%). Doses of 50 and 100 mg/day appeared to lower systolic blood pressure more (> 10 mmHg) but because the systolic blood pressure reduction at these doses was not statistically significantly greater than the 25 mg/day dose, hydrochlorothiazide 25 mg/day was chosen as the lowest dose with maximum systolic blood pressure‐lowering efficacy. The systolic blood pressure‐lowering efficacy at doses > 25 mg/day was ‐8.2 (95% CI ‐9.1 to ‐7.3) mmHg in 3417 patients (P value < 0.0001, and no significant heterogeneity was present, with I 2 = 25%; test of subgroup differences P value = 0.57, with I2 = 0%).
Dose‐related diastolic blood pressure‐lowering response of hydrochlorothiazide by direct comparison
Hydrochlorothiazide 12.5 mg/day significantly reduced diastolic blood pressure compared with 3 to 6.25 mg/day by ‐1.1 (95% CI ‐2.1 to ‐0.1) mmHg based on seven trials in 920 patients. Hydrochlorothiazide 25 mg significantly reduced diastolic blood pressure more than hydrochlorothiazide 12.5 mg/day by ‐1.00 (95% CI ‐1.6 to ‐0.4) mmHg based on 17 trials in 2315 patients and also compared to 3 to 6.25 mg/day by ‐1.6 (95% CI ‐2.6 to ‐0.6) mmHg based on seven trials in 917 patients.
Hydrochlorothiazide 37.5 mg/day versus 12.5 mg/day; hydrochlorothiazide 50 to 25 mg/day or hydrochlorothiazide 100 mg versus 50 mg/day were not significantly different from each other. Therefore hydrochlorothiazide 25 mg/day was chosen as the lowest dose with maximum diastolic blood pressure‐lowering efficacy. The maximum diastolic blood pressure‐lowering efficacy at doses > 25 mg/day was ‐3.4 (95% CI ‐3.9 to ‐3.0) mmHg in 3744 patients (P value < 0.0001; no significant heterogeneity was present, with I 2 = 26%; test of subgroup difference P value = 0.29, with I2 = 19.9%).
The placebo‐corrected systolic/diastolic blood pressure‐lowering with hydrochlorothiazide 3 to 100 mg/day was 6.9/3.3 mmHg.
We also plotted a weighted log dose‐response curve using individual data points from each study and the resulting curve showed a significant dose response for systolic blood pressure (slope ‐6.16 (‐8.75 to ‐3.56) and r = ‐ 0.58 but not for diastolic blood pressure slope ‐0.82 (‐3.44 to 1.79) and r = ‐0.43). See Figure 5 and Figure 6.
5.
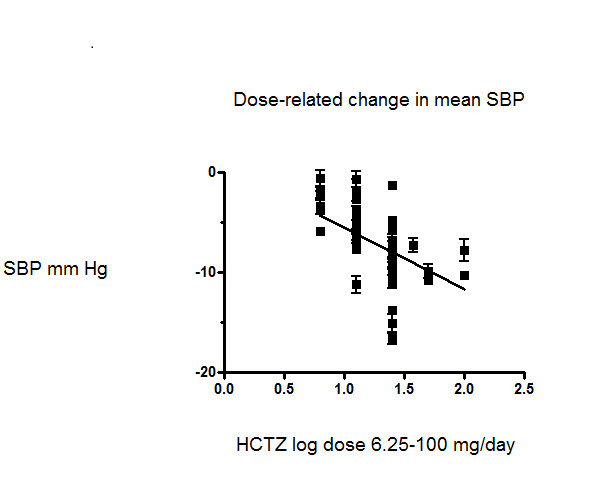
Dose‐related effect of hydrochlorothiazide on systolic blood pressure
6.
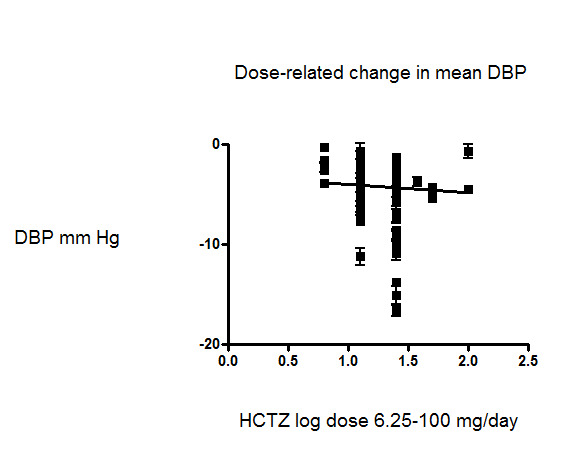
Dose‐related effect of hydrochlorothiazide on diastolic blood pressure
Indapamide
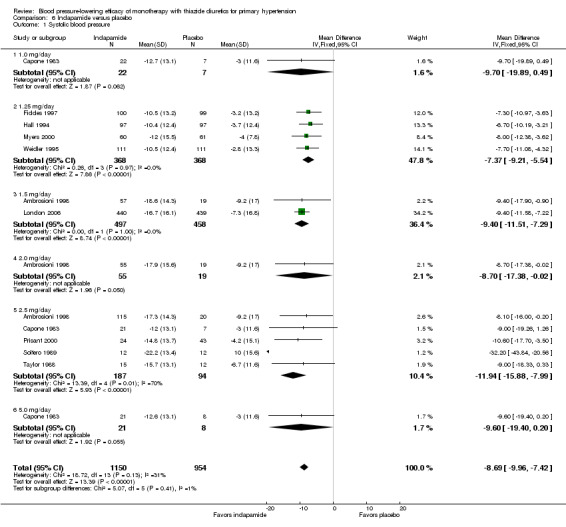
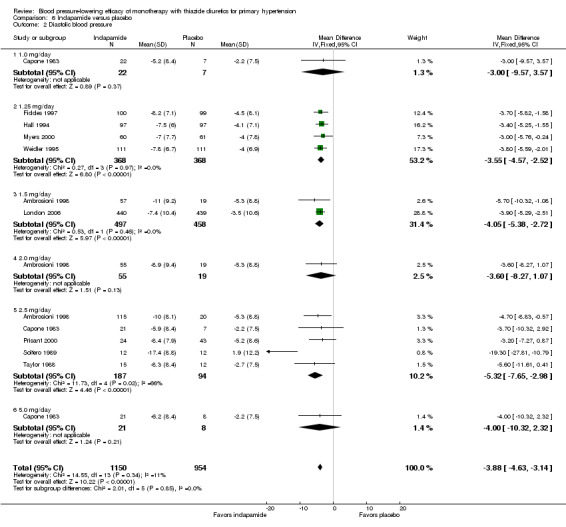
Ten trials compared indapamide at doses ranging from 1 to 5 mg/day treated for a mean duration of 10 weeks (Ambrosioni 1998; Capone 1983; Fiddes 1997; Hall 1994; London 2006; Myers 2000; Prisant 2000; Soltero 1989; Taylor 1988; Weidler 1995). See Analysis 6.1; Analysis 6.2.
6.1. Analysis.
Comparison 6 Indapamide versus placebo, Outcome 1 Systolic blood pressure.
6.2. Analysis.
Comparison 6 Indapamide versus placebo, Outcome 2 Diastolic blood pressure.
Table 8: Dose‐related systolic and diastolic blood pressure‐lowering efficacy of indapamide
| Indapamide |
Systolic blood pressure mmHg (95% CI) |
Diastolic blood pressure mmHg (95% CI) |
| 1.0 mg/day | ‐9.7 (‐19.9 to 0.5) | ‐3.0 (‐9.6 to 3.6) |
| 1.25 mg/day | ‐7.4 (‐9.2 to ‐5.5) | ‐3.6 (‐4.6 to ‐2.5) |
| 1.5 mg/day | ‐9.4 (‐11.5 to ‐7.3) | ‐4.1 (‐5.4 to ‐2.7) |
| 2.0 mg/day | ‐8.7 (‐17.4 to ‐0.0) | ‐3.6 (‐8.3 to 1.1) |
| 2.5 mg/day | ‐11.9 (‐15.9 to ‐8.0) | ‐5.3 (‐7.7 to ‐3.0) |
| 5.0 mg/day | ‐9.6 (‐19.4 to 0.2) | ‐4.0 (‐10.3 to 2.3) |
| Overall | ‐8.7 (‐10.0 to ‐7.4) | ‐3.9 (‐4.6 to ‐3.1) |
Based on the 10 trials (N = 2150 patients), the lowest dose of indapamide that statistically significantly lowered both systolic and diastolic blood pressure was 1.25 mg/day. The overall placebo‐corrected systolic blood pressure‐lowering effect across 1 to 5 mg/day doses was ‐8.7 mmHg (P value < 0.00001, with I2 = 31% and the test for subgroup differences P value = 0.41, with I2 = 1.5%). For diastolic blood pressure it was ‐3.9 mmHg (P value < 0.00001, with I2 = 11% and the test for subgroup differences P value = 0.85, with I2 = 0%).
Direct comparison of doses from one dose‐ranging trial did not show any significant differences in systolic or diastolic blood pressure between different doses used (McVeigh 1988).
The placebo‐corrected systolic/diastolic blood pressure‐lowering with indapamide was 8.4/3.8 mmHg.
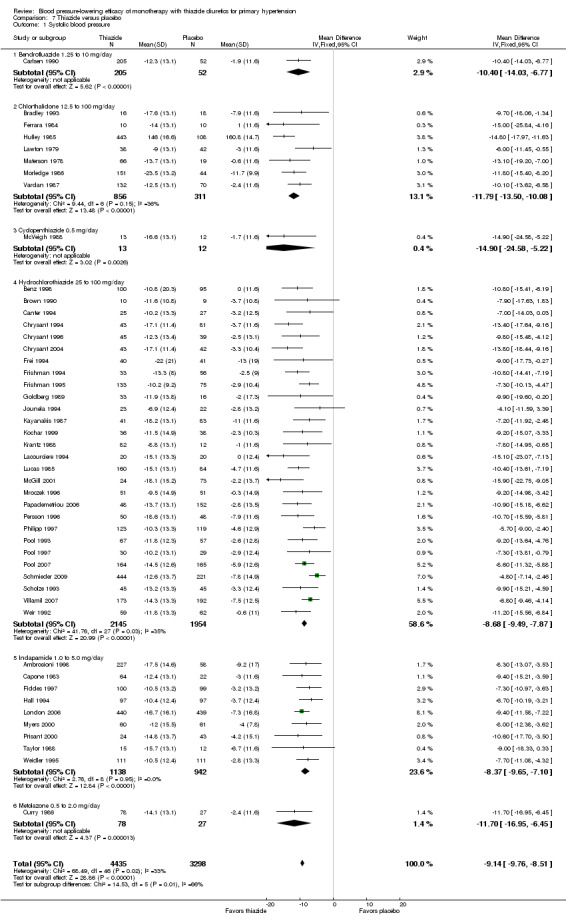
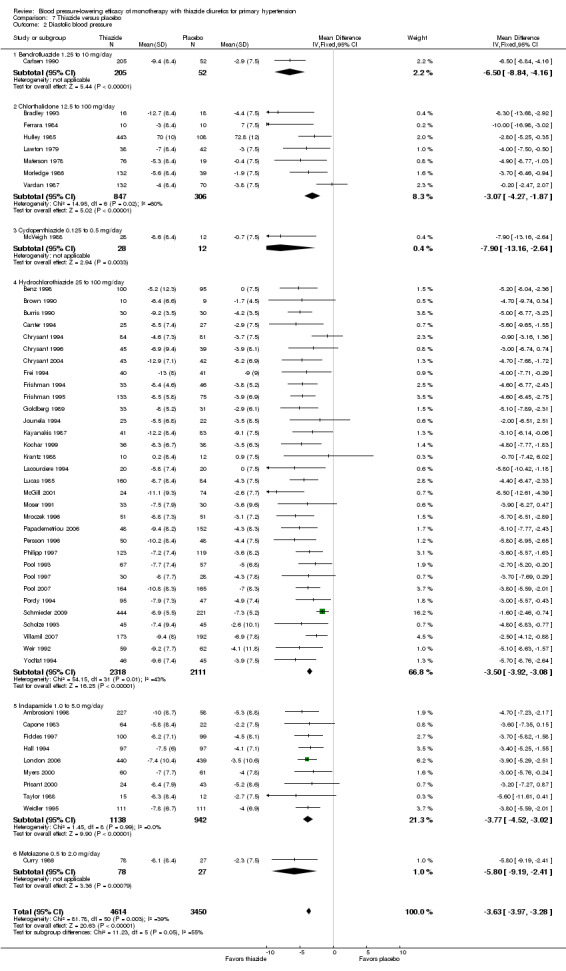
Thiazides (all six drugs)
When the lowest dose of each of the six thiazide drugs that achieved maximal systolic and diastolic blood pressure reduction and all doses above it were pooled, the overall systolic blood pressure reduction for thiazide diuretics as a class was ‐9.1 (95% CI ‐9.7to ‐8.5) mmHg (heterogeneity: Chi2 = 68.49, df = 46 (P value = 0.02); I2 = 33%; test for overall effect: Z = 28.86 (P value < 0.00001); test for subgroup differences: Chi2 = 14.53, df = 5 (P value = 0.01), I2 = 65.6% was significant). For diastolic blood pressure it was ‐3.6 (95% CI ‐4.0 to ‐3.3) mmHg (heterogeneity: Chi2 = 81.78, df = 50 (P value = 0.003); I2 = 39%; test for overall effect: Z = 20.63 (P value < 0.00001); test for subgroup differences: Chi2 = 11.23, df = 5 (P value = 0.05), I2 = 55.5%). See Analysis 7.1; Analysis 7.2.
7.1. Analysis.
Comparison 7 Thiazide versus placebo, Outcome 1 Systolic blood pressure.
7.2. Analysis.
Comparison 7 Thiazide versus placebo, Outcome 2 Diastolic blood pressure.
Subgroup analyses
Due to lack of data being reported in each trial for individual participants based on age (18 to 59 and 60 or older); sex; race (black, white and others); co‐morbid conditions; or baseline severity of hypertension (mild, moderate or severe) subgroup analyses could not be performed. However, based on trials reporting the mean baseline blood pressure levels of all included participants, we classified trials according to systolic and diastolic blood pressure and performed subgroup analyses.
Table 9: Subgroup analyses based on baseline mean systolic blood pressure
|
Based on systolic blood pressure at baseline |
# of trials |
Systolic blood pressure decrease mmHg (95% CI) |
| < 140 mmHg | 2 | ‐6.7 (‐11.0 to ‐2.3) |
| 140 to 149 mmHg | 4 | ‐10.9 (‐13.4 to ‐8.4) |
| 150 to 159 mmHg | 20 | ‐9.1 (‐10.1 to ‐8.1) |
| 160 mmHg or > | 13 | ‐10.3 (‐11.5 to ‐9.2) |
| Overall | 39 | ‐9.6 (‐10.4 to ‐8.9) |
Table 9 summarizes the finding based on baseline systolic blood pressure. Heterogeneity and subgroup differences were not significant. There were no significant differences in systolic blood pressure‐lowering based on systolic blood pressure at baseline.
Table 10: Subgroup analyses based on baseline mean systolic blood pressure
|
Based on diastolic blood pressure at baseline |
# of trials |
Diastolic blood pressure decrease mmHg (95% CI) |
| < 90 mmHg | 3 | ‐2.9 (‐4.7 to ‐1.1) |
| 90 to 99 mmHg | 14 | ‐2.8 (‐3.3 to ‐2.3) |
| 100 to 109 mmHg | 25 | ‐4.7 (‐5.2 to ‐4.1) |
| Overall | 42 | ‐3.6 (‐4.0 to ‐3.3) |
Table 10 summarizes the finding based on baseline diastolic blood pressure. Heterogeneity was significant (P value < 0.00001, with I 2 = 56%) and subgroup difference significant (P value < 0.0001, with I2 = 91.1%). There was a significantly greater magnitude of diastolic blood pressure‐lowering (by 1.8 mmHg) in trials with the highest mean diastolic blood pressure (between 100 and 109 mmHg at baseline).
Withdrawals due to adverse effects
Withdrawals due to adverse effects were reported for bendrofluazide, chlorthalidone, hydrochlorothiazide and indapamide trials. Please see Analysis 1.3; Analysis 4.3; Analysis 5.3 and Analysis 6.3
1.3. Analysis.
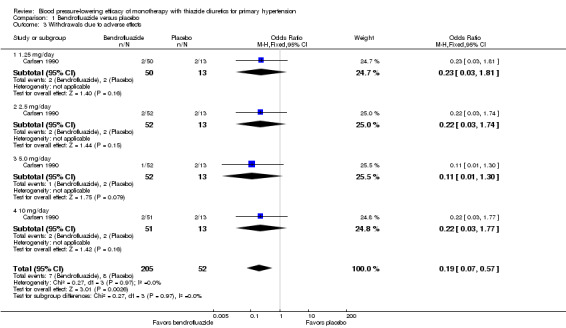
Comparison 1 Bendrofluazide versus placebo, Outcome 3 Withdrawals due to adverse effects.
4.3. Analysis.
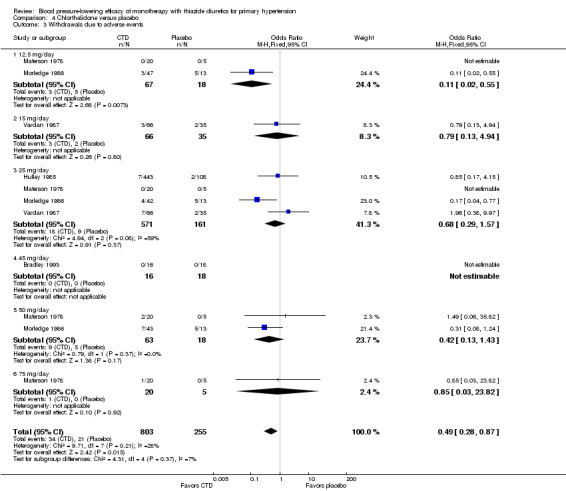
Comparison 4 Chlorthalidone versus placebo, Outcome 3 Withdrawals due to adverse events.
5.3. Analysis.
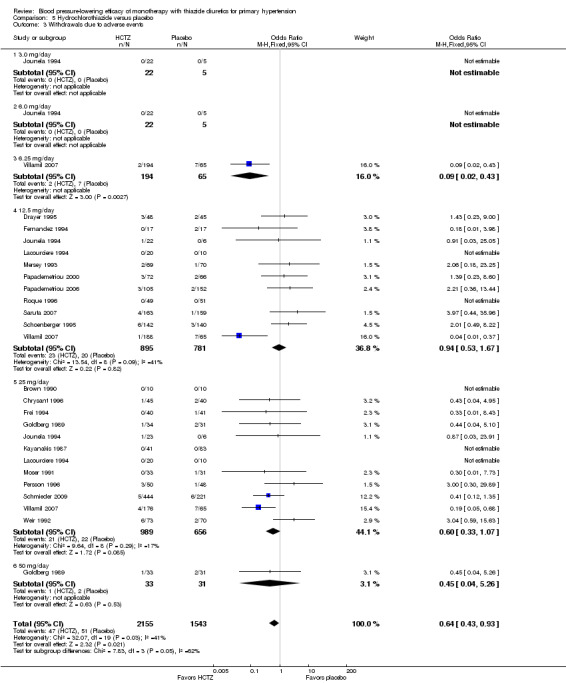
Comparison 5 Hydrochlorothiazide versus placebo, Outcome 3 Withdrawals due to adverse events.
6.3. Analysis.
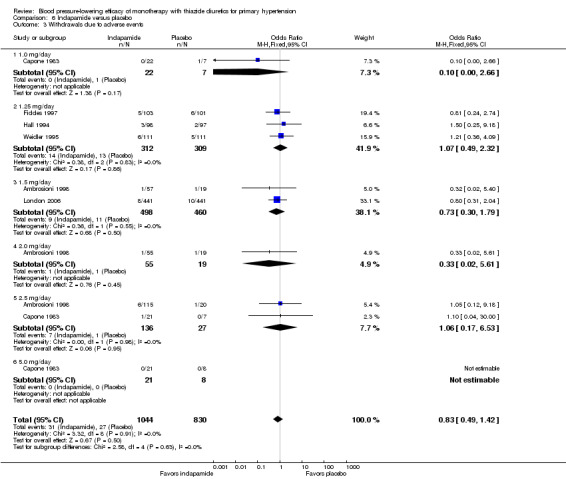
Comparison 6 Indapamide versus placebo, Outcome 3 Withdrawals due to adverse events.
There is selective outcome reporting of withdrawals due to adverse effects. This outcome was reported only in 31 of the 60 trials (52%) meeting the inclusion criteria.
Bendrofluazide resulted in significantly lower withdrawals due to adverse effects compared with placebo based on one trial in 257 patients (risk ratio (RR) 0.19, 95% CI 0.07 to 0.57) (Carlsen 1990). Chlorthalidone resulted in significantly lower withdrawals due to adverse effects compared with placebo based on five out of eight trials in 1058 patients (RR 0.49, 95% CI 0.28 to 0.87) (Bradley 1993; Hulley 1985; Materson 1978; Morledge 1986; Vardan 1987). Hydrochlorothiazide resulted in significantly lower withdrawals due to adverse effects compared with placebo based on 20 out of 40 trials in 3698 patients (RR 0.64, 95% CI 0.43 to 0.93). Indapamide did not significantly change withdrawals due to adverse effects compared with placebo based on six out of nine trials in 1874 patients (RR 0.87, 95% CI 0.52 to 1.46)) (Ambrosioni 1998; Capone 1983; Fiddes 1997; Hall 1994; London 2006; Weidler 1995). The major reason for withdrawals in the placebo group was an increase in blood pressure, which was reported as an adverse effect and therefore included in the number of patients who withdrew due to adverse effects.
The selective reporting of adverse effects across trials (e.g. data on the number of patients with serious adverse events, the nature of these events or reporting of only drug‐related adverse events) and specific reasons for withdrawal due to adverse effects are provided in detail in the 'Risk of bias' tables (in the attrition and selective reporting sections). Due to the very low quality of evidence resulting from selective reporting bias, data are not reported as absolute risk difference or as number needed to treat to benefit or harm.
Metabolic data
The reporting on metabolic data is very limited and due to the high risk of selective reporting bias strong conclusions cannot be made.
Data on serum potassium, uric acid, creatinine, glucose, total cholesterol, low‐density cholesterol and triglycerides were limited to a minority of the trials:
bendrofluazide (Carlsen 1990);
cyclopenthiazide (McVeigh 1988);
chlorthalidone(Bradley 1993; Hulley 1985; Materson 1978; Morledge 1986; Siegel 1992; Vardan 1987);
hydrochlorothiazide(Chrysant 1994; Drayer 1995; Goldberg 1989; Jounela 1994; Mersey 1993; Pool 2007; Pool 1993; Saruta 2007; Scholze 1993; Schoenberger 1995; Siegel 1992);
indapamide (Capone 1983; Fiddes 1997; Hall 1994; Soltero 1989; Taylor 1988).
Dose‐related serum potassium levels (mmol/L)
Serum potassium levels were reported for bendrofluazide, chlorthalidone, cyclopenthiazide, hydrochlorothiazide and indapamide. No data were reported for metolazone. The overall decrease in serum potassium levels for each thiazide drug is shown below.
Table 11: Serum potassium levels (mmol/L)
| Drug and dose range |
Number of trials reporting data |
MD (95% CI) mmol/L |
| Bendrofluazide 1.25 to 10 mg/day | 1 | ‐0.37 (‐0.50 to ‐0.24) |
| Cyclopenthiazide 0.05 to 0.5 mg/day | 1 | ‐0.18 (‐0.42 to 0.07) |
| Chlorthalidone 12.5 to 100 mg/day | 4 | ‐0.40 (‐0.45 to ‐0.34) |
| Hydrochlorothiazide 3 to 100 mg/day | 11 | ‐0.23 (‐0.26 to ‐0.19) |
| Indapamide 1 to 5 mg/day | 6 | ‐0.32 (‐0.38 to ‐0.26) |
| Overall^ | 23 | ‐0.25 (‐0.28 to ‐0.22) |
^Heterogeneity: Chi2 = 22.07, df = 6 (P value = 0.001), I2 = 73%; test for overall effect: Z = 10.06 (P value < 0.00001); test for subgroup differences: Chi2 = 10.06, df = 3 (P value = 0.02), I2 = 70.2%.
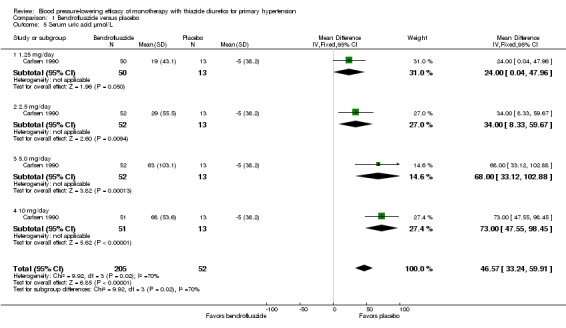
Bendrofluazide ‐ See Table 11 and Analysis 1.4. Heterogeneity between doses was not significant. The 10 mg dose lowered serum potassium significantly more than the 2.5 mg/day dose by ‐0.25 (95% CI ‐0.40 to ‐0.10) mmol/L by direct dose comparison.
1.4. Analysis.
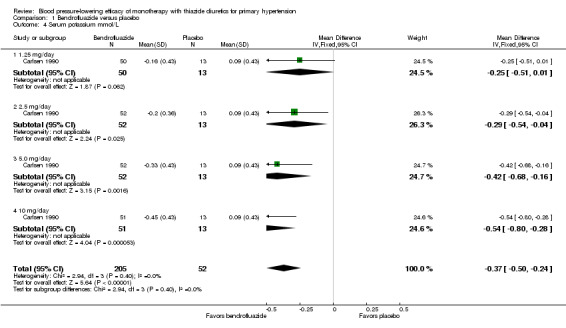
Comparison 1 Bendrofluazide versus placebo, Outcome 4 Serum potassium mmol/L.
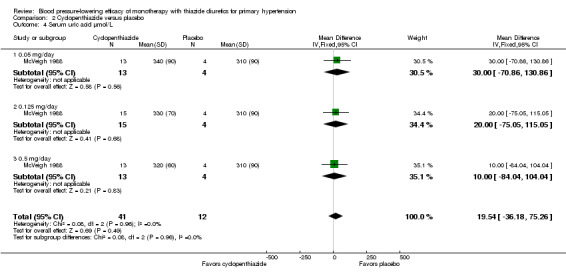
Cyclopenthiazide ‐ See Table 11 and Analysis 2.3. Heterogeneity between doses was not significant. Cyclopenthiazide 0.5 mg/day lowered serum potassium significantly more compared to 0.05 mg/day by ‐0.60 (95% CI ‐0.87 to ‐0.33) mmol/L and by ‐0.40 (95% CI ‐0.62 to ‐0.18) mmol/L compared to 0.125 mg/day by direct dose comparison.
2.3. Analysis.
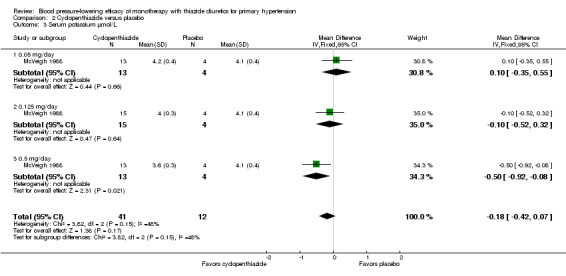
Comparison 2 Cyclopenthiazide versus placebo, Outcome 3 Serum potassium µmol/L.
Chlorthalidone ‐ See Table 11 and Analysis 4.4. Heterogeneity between doses was significant (P value < 0.0001, with I2 = 93.3%). Chlorthalidone 25 mg lowered serum potassium significantly more compared to a 12.5 to 15 mg/day dose ‐0.20 (95% CI ‐0.32 to ‐0.08) mmol/L based on 252 patients. However, doses higher than 25 mg did not differ significantly from 25 mg/day by direct dose comparison. We could not determine the cause of heterogeneity.
4.4. Analysis.
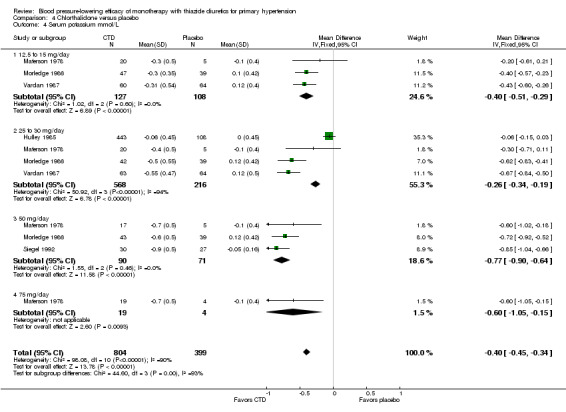
Comparison 4 Chlorthalidone versus placebo, Outcome 4 Serum potassium mmol/L.
Hydrochlorothiazide ‐ See Table 11 and Analysis 5.4. A dose‐related significant decrease in serum potassium levels was observed: ‐0.16 (95% CI ‐0.21 to ‐0.11) mmol/L at a 12.5 mg/day dose; ‐0.30 (95% CI ‐0.36 to ‐0.24) mmol/L at 25 mg/day; and ‐0.48 (95% CI ‐0.68 to ‐0.29) mmol/L at 50 mg/day. Heterogeneity between doses was significant (P value < 0.00001, with I2 = 60%).
5.4. Analysis.
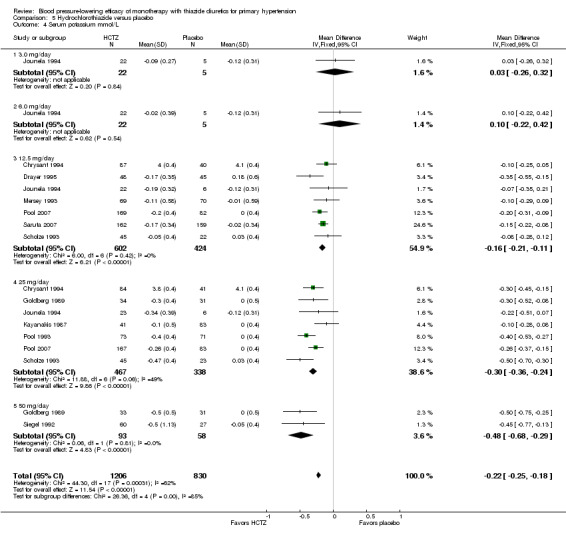
Comparison 5 Hydrochlorothiazide versus placebo, Outcome 4 Serum potassium mmol/L.
In direct dose comparisons, hydrochlorothiazide 25 mg/day lowered serum potassium significantly more compared to hydrochlorothiazide 12.5 mg/day by ‐0.15 (95% CI ‐0.22 to ‐0.09) mmol/L based on four trials in 642 patients. No significant differences were observed between other direct dose comparisons.
Indapamide ‐ See Table 11 and Analysis 6.4. Indapamide 1.25 mg/day lowered serum potassium significantly more than indapamide 1.0 mg/day by ‐0.43 (95% CI ‐0.66 to ‐0.20) mmol/L based on one trial in 47 patients. No significant differences were observed between other doses.
6.4. Analysis.
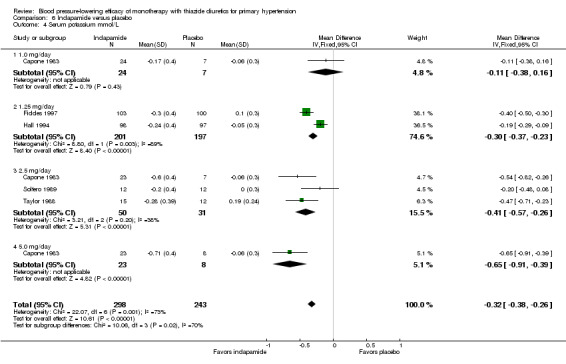
Comparison 6 Indapamide versus placebo, Outcome 4 Serum potassium mmol/L.
The overall reduction in serum potassium for all thiazide drugs compared to placebo, based on available data (based on 23 trials; N = 3868), was ‐0.25 (95% CI ‐0.28 to ‐0.22) mmol/L. See Analysis 7.3.
7.3. Analysis.
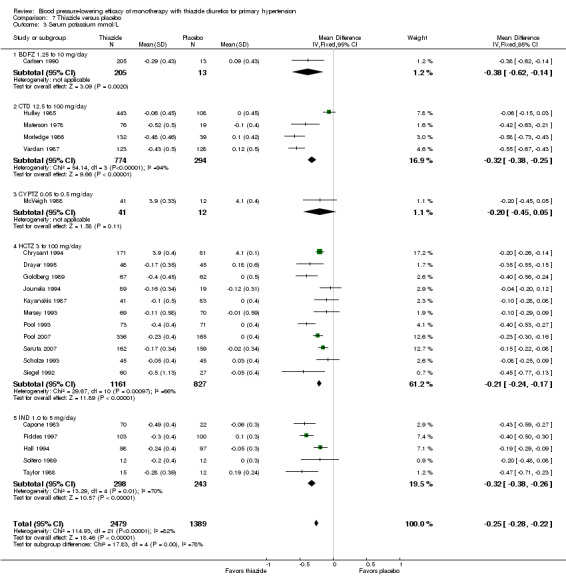
Comparison 7 Thiazide versus placebo, Outcome 3 Serum potassium mmol/L.
Indirect comparison shows significantly greater lowering of serum potassium with chlorthalidone compared to hydrochlorothiazide (‐0.10, 95% CI ‐0.17 to ‐0.30) mmol/L, but no other statistically significant difference was observed between different drugs.
Dose‐related serum uric acid levels (µmol/L)
Serum uric acid levels were reported for bendrofluazide, chlorthalidone, cyclopenthiazide, hydrochlorothiazide and indapamide. No data were reported for metolazone.
Table 12: Serum uric acid levels (µmol/L)
| Drug and dose range |
Number of trials reporting data |
MD (95% CI) µmol/L |
| Bendrofluazide 1.25 to 10.0 mg/day | 1 | 46.6 (33.2to 59.9) |
| Cyclopenthiazide 0.05 to 0.5 mg/day | 1 | 19.5 (‐36.2 to 75.3) |
| Chlorthalidone 12.5 to 100 mg/day | 2 | 64.2 (45.7 to 82.6) |
| Hydrochlorothiazide 3 to 100 mg/day | 5 | 32.9 (26.1 to 39.7) |
| Indapamide 1 to 5 mg/day | 4 | 39.8 (33.5 to 46.1) |
| Overall^ | 13 | 38.2 (34.2 to 42.2) |
^Heterogeneity: Chi2 = 23.13, df = 12 (P value = 0.03); I2 = 48%; test for overall effect: Z = 18.82 (P, 0.0001); test for subgroup differences: Chi2 = 13.76, df = 4 (P value = 0.008), I2 = 70.9%.
Bendrofluazide ‐ See Table 12 and Analysis 1.5. Bendrofluazide 5 mg/day dose showed a significant increase compared to 2.5 mg/day of 34 (95% CI 2.2to 65.8) µmol/L based on one trial in 104 patients. Bendrofluazide 10 mg/day also showed a significant increase compared to bendrofluazide 1.25 mg/day and bendrofluazide 2.5 mg/day by direct dose comparison. Bendrofluazide 10 mg was not significantly different from 5 mg/day.
1.5. Analysis.
Comparison 1 Bendrofluazide versus placebo, Outcome 5 Serum uric acid µmol/L.
Cyclopenthiazide ‐ See Table 12 and Analysis 2.4. Heterogeneity between doses was not significant (P value = 0.96, with I2 = 0%). No significant differences were observed between doses by direct dose comparison.
2.4. Analysis.
Comparison 2 Cyclopenthiazide versus placebo, Outcome 4 Serum uric acid µmol/L.
Chlorthalidone ‐ See Table 12 and Analysis 4.5. Direct comparison between doses showed no significant differences between doses.
4.5. Analysis.
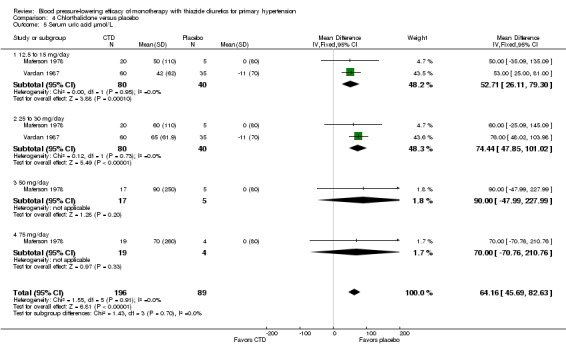
Comparison 4 Chlorthalidone versus placebo, Outcome 5 Serum uric acid µmol/L.
Hydrochlorothiazide ‐ See Table 12 and Analysis 5.5. No significant differences between doses were observed by direct dose comparison.
5.5. Analysis.
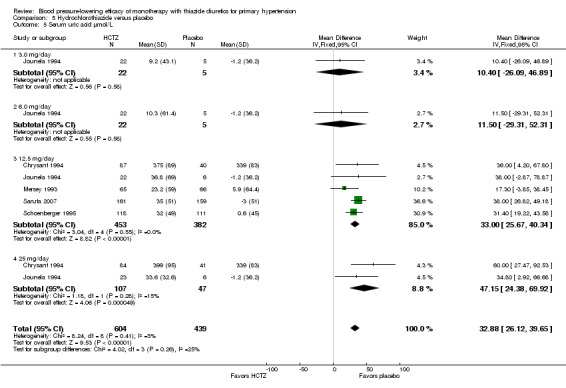
Comparison 5 Hydrochlorothiazide versus placebo, Outcome 5 Serum uric acid µmol/L.
Indapamide ‐ See Table 12 and Analysis 6.5. Direct comparison between doses showed no significant difference.
6.5. Analysis.
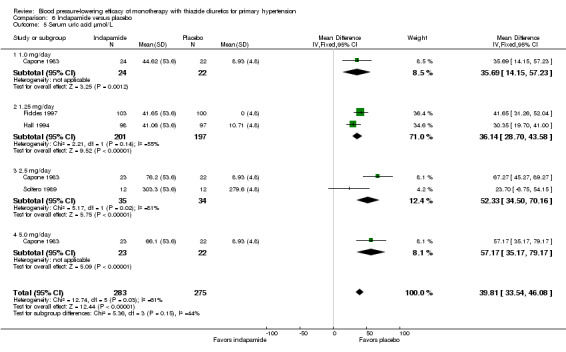
Comparison 6 Indapamide versus placebo, Outcome 5 Serum uric acid µmol/L.
The overall increase in serum uric acid for all thiazide drugs compared to placebo, based on available data, was 38.2 (34.2 to 42.2) mmol/L. See Analysis 7.4.
7.4. Analysis.
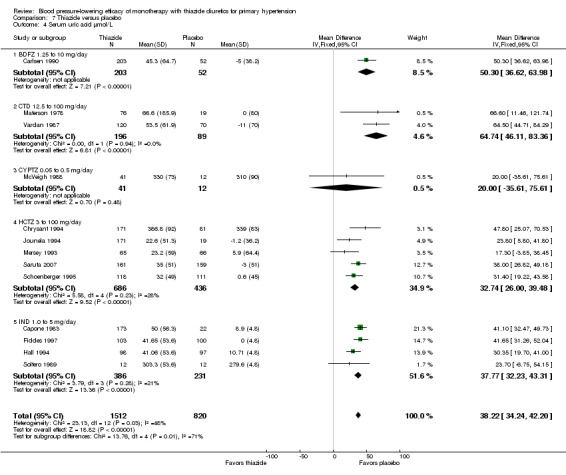
Comparison 7 Thiazide versus placebo, Outcome 4 Serum uric acid µmol/L.
Indirect comparison shows a significant increase in serum uric acid with chlorthalidone compared to hydrochlorothiazide (32.0, 95% CI 12.17 to 51.83 µmol/L) and compared to indapamide (26.4, 95% CI 6.91 to 45.89 µmol/L).
Dose‐related serum creatinine levels (µmol/L)
Serum creatinine levels were reported for bendrofluazide, hydrochlorothiazide and indapamide. Data were not reported for chlorthalidone, cyclopenthiazide and metolazone.
Table 13: Serum creatinine levels (µmol/L)
| Drug and dose range |
Number of trials reporting data |
MD (95% CI) µmol/L |
| Bendrofluazide 1.25 to 10.0 mg/day | 1 | 5.5 (1.9to 9.11) |
| Hydrochlorothiazide 3 to 100 mg/day | 3 | 0.32 (‐2.63 to 3.26) |
| Indapamide 1 to 5 mg/day | 1 | 0.00(‐2.43 to 2.43) |
| Overall^ | 5 | 1.34 (‐0.31 to 2.99) |
^Heterogeneity: Chi2 = 7.14, df = 4 (P value = 0.13); I2 = 44%; test for overall effect: Z = 1.59 (P value = 0.11); test for subgroup differences: Chi2 = 7.06, df = 2 (P value = 0.03), I2 = 71.7%.
See Analysis 7.5.
7.5. Analysis.
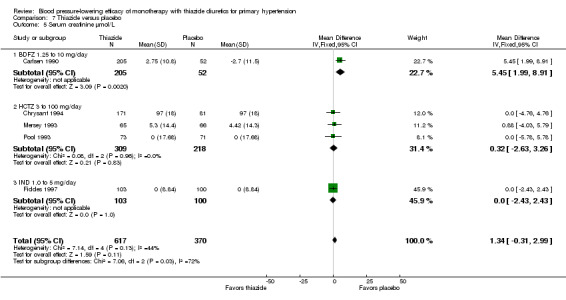
Comparison 7 Thiazide versus placebo, Outcome 5 Serum creatinine µmol/L.
Bendrofluazide is the only drug that increased creatinine (see Table 13 and Analysis 1.6). Direct comparison between doses showed no significant difference.
1.6. Analysis.
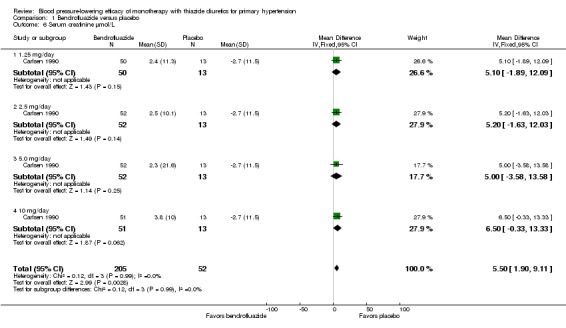
Comparison 1 Bendrofluazide versus placebo, Outcome 6 Serum creatinine µmol/L.
Dose‐related serum glucose levels (mmol/L)
Serum glucose levels were reported for bendrofluazide, chlorthalidone, hydrochlorothiazide and indapamide. Data were not reported for cyclopenthiazide and metolazone.
Table 14: Serum glucose levels (mmol/L)
| Drug and dose range |
Number of trials reporting data |
MD (95% CI) mmol/L |
| Bendrofluazide 1.25 to 10.0 mg/day | 1 | 0.13 (‐0.06 to 0.33) |
| Chlorthalidone 12.5 to 100 mg/day | 3 | 0.34 (0.12 to 0.55) |
| Hydrochlorothiazide 3 to 100 mg/day | 6 | ‐0.11 (‐0.22 to 0.01) |
| Indapamide 1 to 5 mg/day | 3 | 0.13 (‐0.11 to 0.37) |
| Overall^ | 13 | 0.03 (‐0.05to 0.12) |
^Heterogeneity: Chi2 = 20.88, df = 12 (P value = 0.05), with I2 = 43%; test for overall effect: Z = 0.81 (P value = 0.42); test for subgroup differences: Chi2 = 16.0, df = 3 (P value = 0.001), with I2 = 81.3%. See Analysis 7.6.
7.6. Analysis.
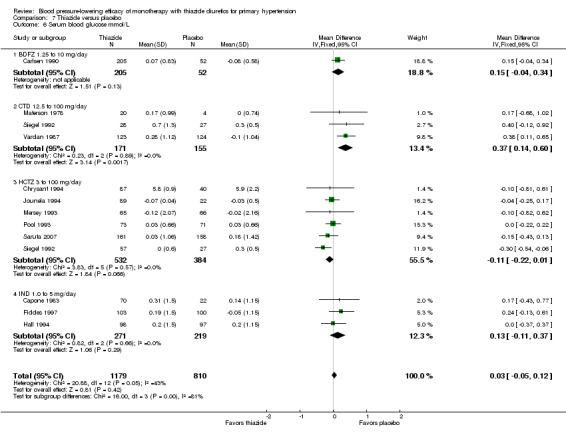
Comparison 7 Thiazide versus placebo, Outcome 6 Serum blood glucose mmol/L.
No significant difference from placebo was noted with bendrofluazide, hydrochlorothiazide and indapamide.
Chlorthalidone ‐ a statistically significant increase in serum glucose was observed with chlorthalidone 25 mg/day (0.58, 95% CI 0.23 to 0.93 mmol/L). The overall increase from 12.5 to 75 mg/day was also significant (see Table 14). Heterogeneity between doses was not significant (P value = 0.39, with I2 = 4%) (see Analysis 4.6). There were no significant differences between doses.
4.6. Analysis.
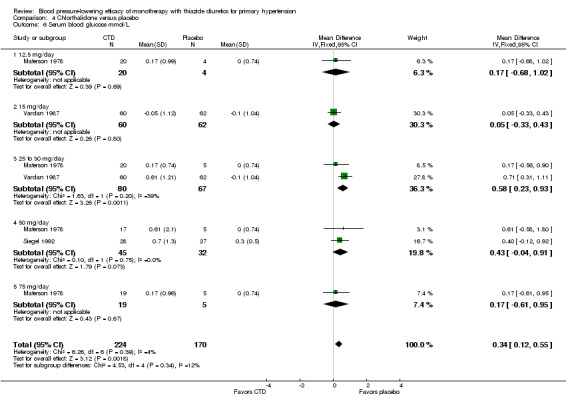
Comparison 4 Chlorthalidone versus placebo, Outcome 6 Serum blood glucose mmol/L.
Indirect comparison showed a significant increase in serum glucose with chlorthalidone compared to hydrochlorothiazide (0.45, 95% CI 0.21 to 0.69 mmol/L).
Dose‐related total cholesterol levels (mmol/L)
Total cholesterol levels were reported for bendrofluazide, chlorthalidone, cyclopenthiazide, hydrochlorothiazide and indapamide.
Table 15: Serum total cholesterol levels (mmol/L)
| Drug and dose range |
Number of trials reporting data |
MD (95% CI) mmol/L |
| Bendrofluazide 1.25 to 10.0 mg/day | 1 | 0.15 (‐0.05 to 0.35) |
| Chlorthalidone 12.5 to 100 mg/day | 2 | 0.41 (0.18 to 0.64) |
| Cyclopenthiazide 0.05 to 0.5 mg/day | 1 | 0.79 (0.36 to 1.23) |
| Hydrochlorothiazide 3 to 25 mg/day | 4 | 0.20 (0.17 to 0.22) |
| Indapamide 1 to 5 mg/day | 3 | 0.11 (0.01 to 0.21) |
| Overall^ | 11 | 0.21 (0.18 to 0.23) |
^Heterogeneity: Chi2 = 26.03, df = 10 (P value = 0.004); I2 = 62%; test for overall effect: Z = 15.59 (P value < 0.0001); test for subgroup differences: Chi2 = 14.01, df = 4 (P value = 0.007), I2 = 71.5%.
See Analysis 7.7.
7.7. Analysis.
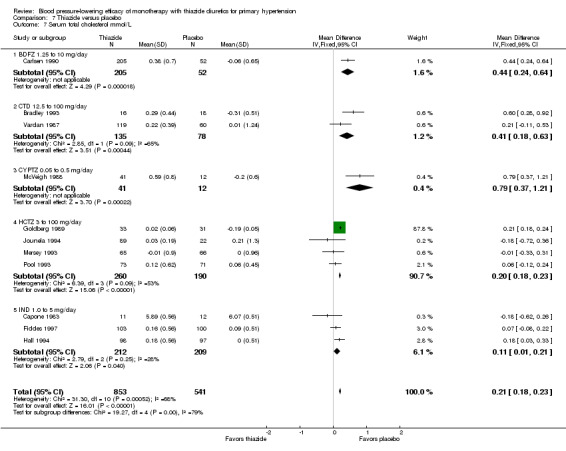
Comparison 7 Thiazide versus placebo, Outcome 7 Serum total cholesterol mmol/L.
Bendrofluazide ‐ See Table 15 and Analysis 1.8. Based on one trial in 257 patients there were no significant differences in serum total cholesterol compared to placebo or between different doses.
1.8. Analysis.
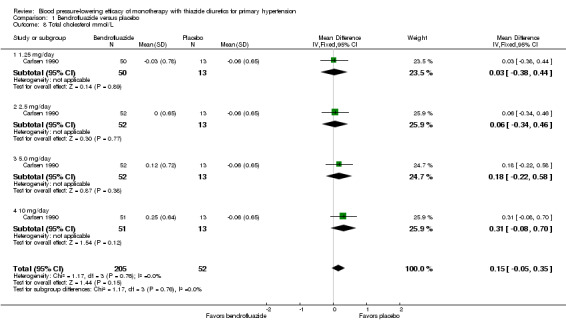
Comparison 1 Bendrofluazide versus placebo, Outcome 8 Total cholesterol mmol/L.
Chlorthalidone ‐ See Table 15 and Analysis 4.8. Based on one trial in 213 patients there was a significant increase in total serum cholesterol (0.41, 95% CI 0.18 to 0.64 mmol/L), but no difference between 25 mg versus 15 mg.
4.8. Analysis.
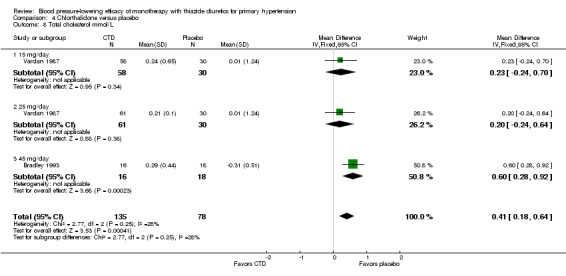
Comparison 4 Chlorthalidone versus placebo, Outcome 8 Total cholesterol mmol/L.
Cyclopenthiazide ‐ See Table 15 and Analysis 2.5. Based on one trial in 47 patients, there was a statistically significant increase in total serum cholesterol (0.79, 95% CI 0.36 to 1.23 mmol/L), but no significant difference between doses.
2.5. Analysis.
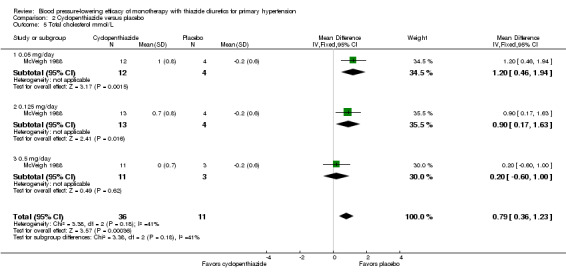
Comparison 2 Cyclopenthiazide versus placebo, Outcome 5 Total cholesterol mmol/L.
Hydrochlorothiazide ‐ See Table 15 and Analysis 5.9. Based on four trials in 450 patients there was a statistically significant increase in total serum cholesterol (0.20, 95% CI 0.17 to 0.22 mmol/L). A significant difference between doses was observed. Heterogeneity between doses was significant (P value = 0.0002, with I2 = 77%).
5.9. Analysis.
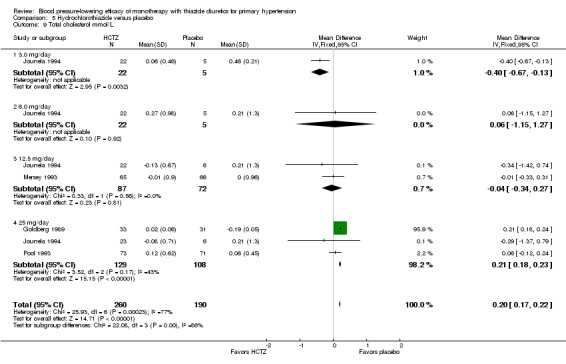
Comparison 5 Hydrochlorothiazide versus placebo, Outcome 9 Total cholesterol mmol/L.
Indapamide ‐ See Table 15 and Analysis 6.9. Based on two trials in 398 patients, there was a statistically significant increase in total serum cholesterol at a 1.25 mg/day dose (0.11, 95% CI 0.01 to 0.21 mmol/L)
6.9. Analysis.
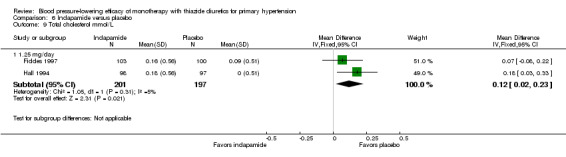
Comparison 6 Indapamide versus placebo, Outcome 9 Total cholesterol mmol/L.
Indirect comparison showed a significant increase in total cholesterol with chlorthalidone compared to indapamide (0.3, 95% CI 0.05 to 0.55 mmol/L), but not compared to hydrochlorothiazide.
Dose‐related high‐density lipoprotein (HDL) cholesterol levels (mmol/L)
HDL cholesterol levels were reported for hydrochlorothiazide, chlorthalidone and indapamide. Data were not reported for bendrofluazide, cyclopenthiazide and metolazone.
Table 16: Serum HDL cholesterol levels (mmol/L)
| Drug and dose range |
Number of trials reporting data |
MD (95% CI) mmol/L |
| Chlorthalidone 45 mg/day | 1 | ‐0.11 (‐0.22 to 0.00) |
| Hydrochlorothiazide 3 to 25 mg/day | 1 | ‐0.17 (‐0.53 to 0.19) |
| Indapamide 1 to 5 mg/day | 1 | ‐0.04 (‐0.03 to ‐0.00) |
| Overall^ | 3 | ‐0.06 (‐0.10 to ‐0.02) |
^Heterogeneity: Chi2 = 1.06, df = 2 (P value = 0.59); I2 = 0%; test for overall effect: Z = 2.76 (P value = 0.006); test for subgroup differences: Chi2 = 1.06, df = 2 (P value = 0.59); I2 = 0%.
The overall significant decrease in HDL cholesterol levels compared to placebo (‐0.06, 95% CI ‐0.10 to ‐0.02 mmol/L) was based on three trials in 348 patients. Heterogeneity and subgroup differences were not significant (see Table 16 and Analysis 7.8).
7.8. Analysis.
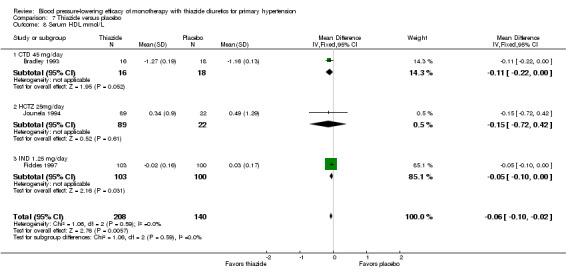
Comparison 7 Thiazide versus placebo, Outcome 8 Serum HDL mmol/L.
Dose‐related serum triglycerides levels (mmol/L)
Serum triglyceride levels were reported for bendrofluazide, chlorthalidone, cyclopenthiazide, hydrochlorothiazide and indapamide.
Table 17: Serum triglyceride levels (mmol/L)
| Drug and dose range |
Number of trials reporting data |
MD (95% CI) mmol/L |
| Bendrofluazide 1.25 to 10.0 mg/day | 1 | 0.26 (‐0.06 to 0.58) |
| Chlorthalidone 45 mg/day | 1 | 0.69 (0.05 to 1.33) |
| Cyclopenthiazide 0.05 to 0.5 mg/day | 1 | 0.20 (‐0.17 to 0.57) |
| Hydrochlorothiazide 3 to 25 mg/day | 2 | 0.09 (‐0.11 to 0.30) |
| Indapamide 1 to 5 mg/day | 1 | 0.21 (0.02 to 0.40) |
| Overall^ | 6 | 0.21 (0.08 to 0.33) |
^Heterogeneity: Chi2 = 5.63, df = 5 (P value = 0.34); I2 = 11%; test for overall effect: Z = 3.17 (P value = 0.002); test for subgroup differences: Chi2 = 3.88, df = 4 (P value = 0.42), I2 = 0%.
Bendrofluazide ‐ See Table 17 and Analysis 1.9. Based on one trial in 257 patients there was no significant difference in serum triglycerides compared to placebo. Heterogeneity between doses was not significant (P value = 0.79 and I2 = 0%).
1.9. Analysis.
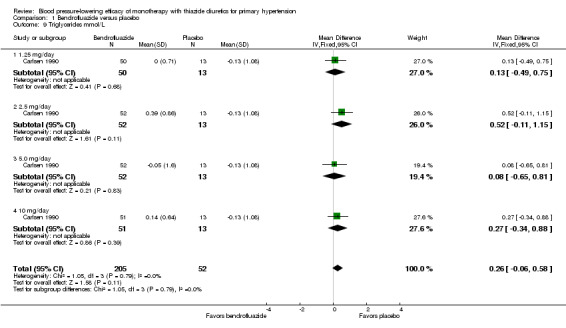
Comparison 1 Bendrofluazide versus placebo, Outcome 9 Triglycerides mmol/L.
Cyclopenthiazide ‐ See Table 17 and Analysis 2.6. Based on one trial in 48 patients there was a significant increase in serum triglycerides compared to placebo. Heterogeneity between doses was not significant (P value = 0.53 and I2 = 0%).
2.6. Analysis.
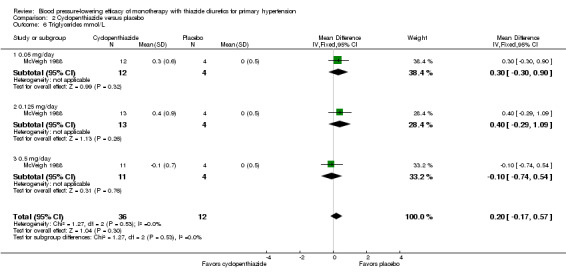
Comparison 2 Cyclopenthiazide versus placebo, Outcome 6 Triglycerides mmol/L.
Chlorthalidone ‐ See Table 17 and Analysis 4.9. Based on one trial in 36 patients there was a significant increase in serum triglycerides compared to placebo with chlorthalidone 45 mg/day.
4.9. Analysis.
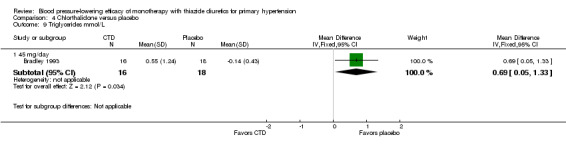
Comparison 4 Chlorthalidone versus placebo, Outcome 9 Triglycerides mmol/L.
Hydrochlorothiazide ‐ See Table 17 and Analysis 5.10. Based on two trials in 255 patients there was no significant difference in serum triglycerides compared to placebo. Heterogeneity between doses was not significant (P value = 0.75 and I2 = 0%).
5.10. Analysis.
Comparison 5 Hydrochlorothiazide versus placebo, Outcome 10 Triglycerides mmol/L.
Indapamide ‐ See Table 17 and Analysis 6.10. Based on one trial in 203 patients there was a significant increase in serum triglycerides compared to placebo with indapamide 1.25 mg/day.
6.10. Analysis.
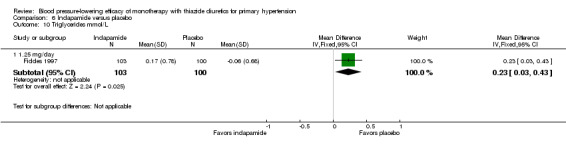
Comparison 6 Indapamide versus placebo, Outcome 10 Triglycerides mmol/L.
For the overall increase in serum triglycerides across drugs (see Table 17), heterogeneity and subgroup differences were not significant. See Analysis 7.9.
7.9. Analysis.
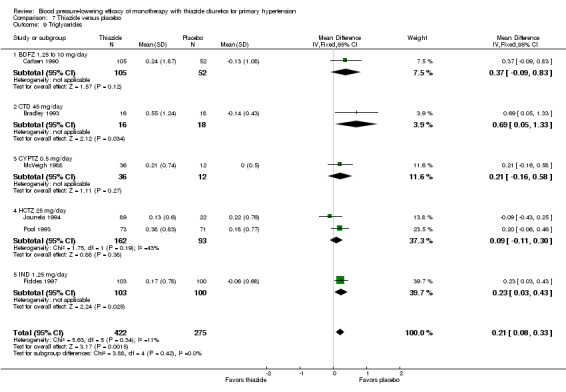
Comparison 7 Thiazide versus placebo, Outcome 9 Triglycerides.
Sensitivity analyses
Sufficient data were available to carry out sensitivity analyses for systolic and diastolic blood pressure data for thiazide diuretics overall.
1. Quality of trials
Trials of high quality versus poor quality: since only two trials had adequate randomization, allocation concealment and blinding as opposed to unclear or high risk of bias in the other 58 trials (Papademetriou 2006; Pool 1993), this analysis was not meaningful.
2. Fixed‐effect versus random‐effects model
Sensitivity analyses using a fixed‐effect versus a random‐effects model showed no significant difference in the overall effect estimate (data not shown).
3. Position in which blood pressure was measured
Robustness of the overall effect size in relation to the position in which blood pressure was measured: trials with blood pressure data measured in a sitting position versus other measurements:
Blood pressure data in a sitting position were available in 30 trials. The overall magnitude of systolic blood pressure‐lowering was ‐9.1 (95% CI ‐9.8 to ‐8.3) mmHg and diastolic blood pressure reduction was ‐3.6 (95% CI ‐4.0 to ‐3.1) mmHg. Blood pressure data in a standing position were available in 11 trials: the magnitude of systolic blood pressure‐lowering was ‐10.2 (95% CI ‐11.6 to ‐8.4) mmHg and diastolic blood pressure reduction was ‐3.7 (95% CI ‐4.7 to ‐2.7) mmHg. Blood pressure data in a supine position were available in seven trials: the magnitude of systolic blood pressure‐lowering was ‐8.7 (‐95% CI 10.2 to ‐7.2) mmHg and diastolic blood pressure reduction was ‐3.8 (95% CI ‐4.7 to ‐3.0) mmHg. No significant differences were observed based on the position in which blood pressure was measured.
4. Peak versus trough ‐ magnitude of blood pressure‐lowering
Thirty trials reported trough systolic blood pressure measurement and the magnitude of systolic blood pressure‐lowering was ‐9.1 (95% CI ‐9.8 to ‐8.4) mmHg. In the remaining trials timing of systolic blood pressure measurement was not reported.
Thirty‐three trials reported trough diastolic blood pressure measurement and the magnitude of diastolic blood pressure‐lowering was ‐3.4 (95% CI ‐3.8 to ‐3.1) mmHg. In the remaining trials the timing of diastolic blood pressure measurement was not reported.
5. Available versus imputed standard deviation
Trials with published standard deviations of blood pressure change versus imputed standard deviations showed no significant difference in the overall estimate of effect for both systolic and diastolic blood pressure (data not shown).
6. Industry versus non‐industry‐sponsored trials
Twenty‐eight trials (47%) reported industry‐sponsored funding and overall the systolic blood pressure‐lowering of thiazide diuretics was ‐8.9 (95% CI ‐9.5 to ‐8.0) mmHg, which was similar in magnitude to five non‐industry‐sponsored trials (8.3%) (‐8.6, 95% CI ‐9.5 to ‐7.6 mmHg). The remaining trials did not report source of funding.
Twenty‐four (40%) trials reported industry‐sponsored funding and overall the diastolic blood pressure‐lowering of thiazide diuretics was ‐3.2 (95% CI ‐3.7 to ‐2.8) mmHg, which was similar in magnitude to five non‐industry sponsored trials (8.3%) (‐3.8, 95% CI ‐5.5 to ‐2.1 mmHg). The remaining trials did not report source of funding.
Discussion
This review provides the best available evidence of the blood pressure‐lowering effect of thiazide monotherapy for the treatment of elevated blood pressure. The drug for which we have the most data is hydrochlorothiazide: 35 randomized controlled trials (RCTs) in 6725 patients. It is the only drug for which we have sufficient RCTs over the commonly prescribed dose range to demonstrate a clear dose‐response effect. There is a very clear dose response for systolic blood pressure over the range 6.25, 12.5 and 25 mg/day (Analysis 5.1; Analysis 5.2). The test for subgroup differences for systolic blood pressure was significant(Chi2 = 22.16, df = 5 (P value = 0.0005), I2 = 77.4%), but not for diastolic blood pressure (Chi2 = 6.22, df = 5 (P value = 0.29), I2 = 19.6%). This means that systolic blood pressure‐lowering at higher doses of hydrochlorothiazide was significantly greater than at lower doses, but diastolic blood pressure‐lowering was not significantly different between the higher or lower dose.
See also Figure 5 and Figure 6. We plotted the log dose‐response curve using individual data points from each study and the resulting curve showed a significant dose response for systolic blood pressure (slope ‐6.16 (‐8.75 to ‐3.56) and r = ‐ 0.58) but not for diastolic blood pressure (slope ‐0.82 (‐3.44 to 1.79) and r = ‐0.43). The significant dose response for hydrochlorothiazide over the dose range 6.25 to 50 mg demonstrates that for each doubling of the dose there is a 2 mmHg greater reduction in systolic blood pressure. There are not enough data at 50 and 100 mg/day but the data that are available suggest that the maximal effect is achieved with a dose of 50 mg/day and that at least 80% of the blood pressure‐lowering effect occurs with 25 mg/day. The confidence intervals for the estimates for 12.5 mg/day and 25 mg/day are narrow, demonstrating that the findings are robust and unlikely to be changed by further RCTs. Further RCTs studying the blood pressure‐lowering effect of 50 mg/day are necessary to have a better estimate of the effect for that dose.
We compared the results obtained in this review with the published review of hydrochlorothiazide given as a second‐line drug (Chen 2009) (see Table 18). The Chen 2009 review has more data on hydrochlorothiazide, from 53 RCTs in 15,129 hypertensive patients with baseline blood pressure of 156/101 mmHg, compared to the 40 RCTs in 7284 patients with baseline blood pressure of 155/100 mmHg included in this review. Results based on both of these reviews show that the magnitude of the systolic blood pressure‐lowering effect is the same whether the drug is given alone or added as a second‐line drug to another antihypertensive drug. This provides strong evidence for the dose‐response relationship and the average magnitude of effect for each dose. Since 6.25 to 50 mg/day is also the common dose range prescribed, the review provides valuable support for dose titration over this range of doses for physicians treating hypertension.
Table 18: Comparing systolic blood pressure reduction with different doses of hydrochlorothiazide
|
Hydrochlorothiazide dose mg/day |
This review Monotherapy versus placebo |
Chen 2009
review Second‐line drug versus placebo |
Weighted combined systolic blood pressure‐lowering effect from both reviews |
||||
| RCTs | # of patients | MD (95% CI) mmHg | RCTs | # of patients | MD (95% CI) mmHg | mmHg | |
| 3.0 to 6.25 | 8 | 663 | ‐3.6 (‐5.6 to ‐1.5) | 22 | 3283 | ‐3.7 (‐4.6 to ‐2.8) | 3.7 |
| 12.5 | 22 | 2645 | ‐6.3 (‐7.2 to ‐5.3) | 53 | 8482 | ‐6.0 (‐6.5 to ‐5.4) | 6.1 |
| 25 | 25 | 3062 | ‐8.0 (‐9.0 to ‐7.0) | 39 | 5799 | ‐8.0 (‐8.7 to ‐7.3) | 8.0 |
| 50 | 2 | 169 | ‐10.5 (‐14.6 to ‐6.4) | 3 | 189 | ‐17.8 (‐21.6 to ‐14.0) | 14.4 |
| 100 | 2 | 146 | ‐9.9 (‐14.1 to ‐5.8) | No data | 9.9 | ||
Reduction in systolic blood pressure shows a dose‐response relationship based on 37 RCTs in 6685 patients. It is similar whether the drug is given as monotherapy (this review) or as a second‐line drug (Chen 2009). Hydrochlorothiazide lowers systolic blood pressure by 4, 6 and 8 mmHg at 6.25, 12.5 and 25 mg/day respectively. See Figure 5.
Table 19: diastolic blood pressure reduction with different doses of hydrochlorothiazide
|
Hydrochlorothiazide dose mg/day |
This review Monotherapy versus placebo |
Chen 2009
review Second‐line drug versus placebo |
Weighted combined diastolic blood pressure‐lowering effect from both reviews |
||||
| RCTs | # of patients | MD (95% CI) mmHg | RCTs | # of patients | MD (95% CI) mmHg | mmHg | |
| 5.0 to 6.25 | 8 | 663 | ‐2.4 (‐3.7 to ‐1.2) | 23 | 3364 | ‐1.7 (‐2.2 to ‐1.2) | 1.8 |
| 12.5 | 25 | 2877 | ‐3.1 (‐3.7 to ‐2.5) | 55 | 8659 | ‐3.1 (‐3.4 to ‐2.8) | 3.1 |
| 25 | 29 | 3359 | ‐3.3 (‐3.8 to ‐2.8) | 42 | 6153 | ‐4.0 (‐4.4 to ‐3.6) | 3.8 |
| 37.5 to 50 | 3 | 239 | ‐4.5 (‐6.7 to ‐3.3) | 3 | 189 | ‐8.3 (‐10.7 to ‐6.0) | 6.4 |
| 100 | 2 | 146 | ‐3.9 (‐6.6 to ‐1.2) | ‐ | ‐ | No data | 3.9 |
Reduction in diastolic blood pressure does not show a significant dose‐response relationship based on 40 RCTs in 7284 patients (Figure 6). Based on the results of both of these reviews, there is a trend toward a dose‐response relationship, and a similar magnitude of effect whether the drug is given as monotherapy or as a second‐line drug.
The greater reduction and dose‐related reduction in systolic compared to diastolic blood pressure means that hydrochlorothiazide has a dose‐related reduction of pulse pressure by 2 to 6 mmHg over the dose range of 6.25 to 50 mg/day. At maximal doses the average reduction in pulse pressure is 5.5 mmHg. This magnitude and pattern is different from other drug classes. Angiotensin receptor blockers (ARBs), angiotensin‐converting enzyme (ACE) inhibitors and renin inhibitors lower pulse pressure by 3 mmHg on average and there is no dose‐response relationship (Heran 2009a; Heran 2009b; Musini 2008). Non‐selective beta‐blockers have little or no effect on pulse pressure: at most 2 mmHg and no dose‐response effect (Wong 2014).
The thiazide with the second most data is indapamide, which lowered blood pressure by 7.4/3.6 mmHg at the lowest dose studied (1.25 mg/day) and 9/4 mmHg for all doses combined. The lowest dose result is very similar to the results with 25 mg hydrochlorothiazide. There was no dose‐response relationship for doses of indapamide higher than 1.25 mg/day and unfortunately there were no RCTs for doses of one‐half or one‐quarter of 1.25 mg/day. These data for indapamide show that the maximal blood pressure lowering effect is achieved with the lowest doses of 1 to 2 mg/day and there is no rationale for using higher doses.
The thiazide with the third most data is chlorthalidone. Like indapamide, chlorthalidone showed no dose response over the doses studied. It appeared that the lowest dose studied(12.5 mg/day) had the maximum blood pressure‐lowering effect. On first look it appears that chlorthalidone lowers blood pressure more than hydrochlorothiazide and indapamide. However, the baseline systolic blood pressure was significantly higher and the baseline diastolic blood pressure was significantly lower in the chlorthalidone trials. When the isolated systolic blood pressure trials were deselected, the blood pressure‐lowering effect of chlorthalidone was not significantly different from the other thiazides and, furthermore, the overall blood pressure‐lowering effect of chlorthalidone was not different from hydrochlorothiazide at the maximally effective doses of 50 mg and above.
The fact that chlorthalidone is more potent than hydrochlorothiazide (12.5 mg of chlorthalidone being equivalent to 50 mg of hydrochlorothiazide) is likely due to pharmacokinetic differences between it and hydrochlorothiazide. Hydrochlorothiazide has a half‐life of 8 to 15 hours with long‐term dosing. However, several studies show that the pharmacodynamic response is much longer than predicted by the half‐life (Allen 1982; Lutterodt 1980). Chlorthalidone in comparison has a half‐life ranging from 45 to 60 hours with long‐term dosing. Interindividual variability in half‐life is large. Chlorthalidone serum concentrations after 100 mg are only twice those of a 25 mg dose, indicating a flat dose‐serum concentration curve (Carter 2004; Riess 1977; Russell 1981). However, the longer half‐life does not mean that chlorthalidone is a superior antihypertensive compound. The data in this review suggest that doses of chlorthalidone that should be prescribed are 12.5 mg/day and lower. RCTs of the blood pressure‐lowering effect of chlorthalidone at doses lower than 12.5 mg/day would be useful.
The other three thiazides had only one RCT each so no conclusions can be made about dose‐response effects or relative blood pressure‐lowering potency. From the data available there is no reason to suggest that they are any different from hydrochlorothiazide in their blood pressure‐lowering effect. Any subgroup differences between the different thiazides in blood pressure‐lowering effect are more likely due to differences in patient population (baseline blood pressure, etc.) or biases in trial conduct than to any pharmacological differences in the magnitude or pattern of the blood pressure‐lowering effect.
Harms of thiazides
The limited data on withdrawal due to adverse effects suggested a decrease in the treatment group compared to the placebo group. This does not mean that placebo is more harmful than a thiazide. The likely reason for this is that withdrawal due to an increase in blood pressure was inappropriately counted as an adverse effect. As can be seen in the table below, the proportion of RCTs reporting withdrawals due to adverse effects (32/60 (53%)) is much less than the total number reporting blood pressure data (48/60 (80%)). This suggests that the trials in which withdrawals due to adverse effects were higher for the drug than for placebo were selectively under reported and this represents a high risk of selective reporting bias. In this review, therefore, no conclusions can be drawn as to whether thiazides increase withdrawals due to adverse effects with short‐term use.
Table 20: Withdrawals due to adverse effects
| Drug and dose range |
Number of trials reporting data |
WDAEs RR (95% CI) |
| Bendrofluazide 1.25 to 10.0 mg/day | 1/1 (N = 257) | 0.19 (0.07 to 0.57) |
| Chlorthalidone 12.5 to 100 mg/day | 5/7 (N = 1058) | 0.49 (0.28 to 0.87) |
| Cyclopenthiazide 0.05 to 0.5 mg/day | 0/1 | Not reported |
| Hydrochlorothiazide 3 to 100 mg/day | 20/40 (N = 3698) | 0.64 (0.43 to 0.93) |
| Indapamide 1 to 5 mg/day | 6/10 (N = 1874) | 0.83 (0.49 to 1.42) |
| Metolazone 0.5 to 2 mg/day | 0/1 | Not reported |
Metabolic data were also only reported in a minority of trials (see Table 21 below).
The available data show a clear reduction in serum potassium, an increase in serum uric acid and an increase in serum total cholesterol and triglycerides. All of these effects are well known to occur with thiazides. New findings from this review are that the metabolic effects were greater with higher doses and were less, in general, with hydrochlorothiazides than the other thiazides. In addition, serum glucose was not increased by thiazides overall. In fact the only thiazide associated with an increase in glucose in this review was chlorthalidone. However, in this review, the high risk of selective reporting bias for the metabolic data and indirect comparison between different thiazide drugs makes drawing strong conclusions impossible.
Table 21: Metabolic data
| Drug and dose range |
Number of trials reporting data (N) |
MD with 95% CI all thiazide trials |
| Serum potassium mmol/L | 22/59 (3868) | ‐0.25 (‐0.28 to ‐0.22) |
| Serum uric acid µmol/L | 13/59 (2332) | 38.2 (34.2 to 42.2) |
| Serum creatinine µmol/L | 5/59 (987) | 1.34 (‐0.31 to 2.99) |
| Serum glucose mmol/L | 13/59 (1989) | 0.03 (‐0.05 to 0.12) |
| Serum total cholesterol mmol/L | 11/59 (431) | 0.20 (0.18 to 0.23) |
| Serum high‐density lipoprotein cholesterol mmol/L | 3/59 (348) | ‐0.06 (‐0.10 to ‐0.02) |
| Serum triglycerides mmol/L | 6/59 (697) | 0.21 (0.08 to 0.33) |
Summary of main results
The magnitude of the dose‐related systolic and diastolic blood pressure‐lowering is from a low to high quality of evidence for hydrochlorothiazide. We determined the evidence for the overall blood pressure‐lowering effect of maximal doses of thiazides to be of high quality. We judged the data on withdrawals due to adverse effects to be of very low quality since there is selective reporting bias for this outcome and because withdrawal due to an increase in blood pressure was inappropriately included as an adverse event in the placebo group.
Overall completeness and applicability of evidence
In this review we systematically searched various databases from 1946 until February 2014 and it is unlikely that any RCTs have been missed. The robustness of the blood pressure‐lowering efficacy data is validated by comparing and demonstrating the similar magnitude of blood pressure reduction in this systematic review to that by Chen 2009 on the blood pressure‐lowering efficacy of hydrochlorothiazide as second‐line therapy for primary hypertension. The Chen 2009 review has more data on hydrochlorothiazide (from 53 RCTs in 15,129 hypertensive patients with baseline blood pressure of 156/101 mmHg) compared to the 40 RCTs in 7284 patients with baseline blood pressure of 155/100 mmHg included in this review.
Quality of the evidence
We assessed the risk of bias for each of the RCTs included in this review (Figure 2). The majority of RCTs (82% of included studies) were published before the year 2000, prior to standardization of reporting of RCTs. However, it is clear that thiazides lower blood pressure, that this is dose‐related and that the magnitude is probably approximately what is reported here. Although the magnitude of systolic and diastolic blood pressure‐lowering is similar to the effect when using thiazide as a second‐line drug (Chen 2009), we downgraded the quality of evidence from high to moderate for some doses as it is an indirect comparison of the effect size with wider confidence intervals compared to the Chen review, which used similar inclusion/exclusion criteria.
There is a high risk of bias in the adverse effect data (Figure 3), therefore the available evidence for adverse events is likely not an accurate reflection of reality.
The evidence is very low quality for the metabolic data due to the high risk of selective outcome reporting bias and a weak interpretation due to multiple indirect comparisons between different thiazide drugs.
Potential biases in the review process
One limitation of this review is that it is restricted to published trials and it is possible that smaller trials are missing (publication bias). See funnel plot (Figure 4). Our judgement of an unclear to high risk of bias in most of the included trials could also have led to an overestimation of blood pressure‐lowering effect and underestimation of adverse metabolic effects. The finding in this review that withdrawals due to adverse effects are lower with a thiazide compared to placebo is unlikely to be true. The short duration of trials in this review make it good for estimating the blood pressure‐lowering effect, but not accurate for estimating long‐term benefits or harms of thiazides.
Although we have followed the method of analysis for multiple armed studies described in the Cochrane Handbook for Systematic Reviews of Interventions (section 16.5.4) (Handbook 2011), since many of the included studies had multiple dose‐ranging treatment arms being compared to the same placebo group, the resulting comparisons remain co‐related and this method only partially overcomes unit of analysis error. The approach we used to overcome this unit of analysis error was to split the shared placebo group in to two or more groups with smaller sample sizes, to include two or more reasonably independent comparisons in the meta‐analysis as if they were from different studies.
We have followed the usual convention to round up numbers if > 0.5 and round down if < 0.5, therefore the magnitude of the blood pressure‐lowering effect is reported up to one decimal point and for metabolic data it is reported up to two decimal points in the Results section of this review. However, in the Abstract, for the sake of simplicity, using the usual convention the overall systolic/diastolic blood pressure‐lowering effect in mmHg is rounded to the nearest number (mmHg) without decimal points.
Agreements and disagreements with other studies or reviews
Messerli 2011 conducted a systematic review of the blood pressure‐lowering efficacy of hydrochlorothiazide for a minimum treatment period of four weeks in randomized trials comparing hydrochlorothiazide to other antihypertensive drug classes. Both office and ambulatory blood pressure measurements were available in eight studies (488 patients in total) using hydrochlorothiazide 12.5 to 25 mg/day for a mean duration of eight weeks. The mean baseline office systolic/diastolic blood pressure was 163/98 mmHg. The reduction in office systolic blood pressure from baseline was 12/7 mmHg, whereas the reduction in ambulatory 24‐hour blood pressure was 8/4 mmHg. The Messerli review is misleading and has been used to suggest that hydrochlorothiazide lowers office blood pressure more than ambulatory blood pressure. The ambulatory blood pressure reduction observed in the Messerli review is very similar to the reduction in placebo‐corrected office systolic/diastolic blood pressure in our review(6 to 8/3 to 4 mmHg). Our review is a more robust treatment effect estimate based on a greater number of RCTs and patients compared to the Messerli 2011 review.
Law 2009 included 354 randomized, double‐blind, placebo‐controlled trials of thiazides, beta‐blockers, ACE inhibitors, ARBs and calcium channel‐blockers administered either singly or in combination. They included both parallel and cross‐over trials together in their analysis. However, they presented placebo‐adjusted reductions in systolic and diastolic blood pressure according to dose expressed as a multiple of standard (recommended) doses of the drugs. What were considered as the standard doses for thiazide drugs were not described in the review. They combined trial data for specified equivalent daily doses of different drugs as the "usual maintenance dose" in reference pharmacopoeias. When a range was given they considered the lower dose as standard dose. The Law review did not present the magnitude of blood pressure‐lowering efficacy of each of the drugs within the thiazide diuretic class at all available doses from the included trials but presented data from all thiazide drugs together as a multiple of standard doses. Therefore, unlike the Law review, our review provides evidence that there is a dose‐related effect for hydrochlorothiazide and thus combining all doses is not rational.
The overall estimated reduction in systolic/diastolic blood pressure with thiazide diuretics in this review (‐9/‐4 mmHg) is similar to the treatment effect estimate in a systematic review of loop diuretics (‐8/‐4 mmHg) (Musini 2009c). This estimate was based on nine trials in 460 patients with a baseline blood pressure of 162/103 mmHg for a mean duration of nine weeks and was likely an overestimate due to the high risk of bias in the included studies. The loop diuretics review also did not provide a good estimate of the incidence of associated harms because of the short duration of the trials and the lack of reporting of adverse effects in many of the them. This review shows that thiazides lower systolic blood pressure and pulse pressure more and diastolic blood pressure less than shown in other Cochrane reviews on ARBs (Heran 2009b), ACE inhibitors (Heran 2009a), renin inhibitors (Musini 2008) and non‐selective beta‐blockers (Wong 2014). This observation is based on an indirect comparison between different antihypertensive drug class reviews compared to placebo control using similar inclusion/exclusion criteria. Therefore the common belief that different classes of antihypertensive drugs have the same blood pressure‐lowering effect is likely to be wrong.
Authors' conclusions
Implications for practice.
This systematic review demonstrates a dose‐related blood pressure‐lowering effect of hydrochlorothiazide. The blood pressure‐lowering effect over the dose range 6.25, 12.5, 25 and 50 mg/day is 4/2, 6/3, 8/3 and 11/5 mmHg, respectively. The data for the other thiazide drugs did not show evidence of a dose response, but showed that lower doses (indapamide 1.25 to 1.5 mg/day and chlorthalidone 12.5 mg/day) achieved a probable maximal blood pressure‐lowering effect of 8 to 10/4 mmHg. As there is a greater effect on systolic blood pressure than diastolic blood pressure, thiazides lower pulse pressure by 4 to 6 mmHg. This exceeds the mean 3 mmHg pulse pressure reduction achieved by angiotensin‐converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs) and renin inhibitors and the 2 mmHg pulse pressure reduction with non‐selective beta‐blockers as shown in other Cochrane reviews, which compared these antihypertensive drug classes with placebo and used similar inclusion/exclusion criteria (Heran 2009a; Heran 2009b; Musini 2008; Wong 2014).
Thiazides did not increase withdrawals due to adverse effects in these short‐term trials, but there is a high risk of bias for that outcome. Thiazides reduced potassium, increased uric acid and increased total cholesterol and triglycerides. These effects were dose‐related and were least for hydrochlorothiazide. Chlorthalidone increased serum glucose but the other thiazides did not. There is a high risk of bias in the metabolic data.
Implications for research.
For hydrochlorothiazide, more trials are needed for doses of 50 to 100 mg/day. For thiazides other than hydrochlorothiazide, more randomized controlled trials (RCTs) are needed at lower doses to define the dose‐related blood pressure‐lowering effect. Many thiazides have no RCT data on the blood pressure‐lowering effect.
All RCTs should report withdrawals due to adverse effects and all laboratory data that are measured in the trial.
Blood pressure‐lowering effect should be reported separately by sex and race, as at the present time it is not known whether there are differences in these subgroups.
Acknowledgements
We would like to acknowledge Stephen Adams for the innumerable articles he retrieved, which possibly met the inclusion criteria of this review.
Ciprian Jauca translated non‐English articles and helped in determining trials meeting inclusion/exclusion criteria.
Devin Godfrey helped in collating baseline characteristics of the 60 included trials.
Sam Soufi helped to categorize trials to conduct subgroup and sensitivity analyses.
Appendices
Appendix 1. MEDLINE search strategy
Database: Ovid MEDLINE(R) 1946 to Present with Daily Update Search date: 19 February 2014 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 exp thiazides/ 2 exp sodium chloride symporter inhibitors/ 3 (sodium chloride adj (symporter or cotransporter? or co‐transporter?)).tw. 4 (potassium depleting adj2 diuretic?).tw. 5 (bendrofluazide? or bendroflumethiazide? or benzhydroflumethiazide? or benzthiazide? or benzydroflumethiazide? or benzothiadiazine? or buthiazide? or chlorothiazide? or cyclopenthiazide? or cyclothiazide or diucardin or diuril or enduron or esidrix or ezna or hydrochlorothiazide? or hydroflumethiazide? or methyclothiazide? or metolazone? or microzide or mykrox or naqua or naturetin or polythiazide? or renese or trichlormethiazide? or thiazide? or zaroxolyn).mp. 6 (chlortalidone? or chlorthalidone? or chlorphthalidolone? or clopamid? or clopamine? or clorexolone? or diapamide? or fenquizone? or hydromox or hygroton or indapamide? or isodapamide? or lozol or mefruside? or metindamide? or oxodoline? or phthalamudine? or quinethazone? or s‐1520 or s1520 or se‐1520 or se1520 or thalitone? or xipamide?).mp. 7 or/1‐6 8 hypertension/ 9 hypertens$.tw. 10 exp blood pressure/ 11 blood pressure.mp. 12 or/8‐11 13 randomized controlled trial.pt. 14 controlled clinical trial.pt. 15 randomized.ab. 16 placebo.ab. 17 dt.fs. 18 randomly.ab. 19 trial.ab. 20 groups.ab. 21 or/13‐20 22 animals/ not (humans/ and animals/) 23 21 not 22 24 7 and 12 and 23
Appendix 2. EMBASE search strategy
Database: EMBASE <1974 to 2014 Week 07> Search date: 19 February 2014 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 exp thiazide diuretic agent/ 2 (sodium chloride adj (symporter or cotransporter? or co‐transporter?)).tw. 3 potassium depleting diuretic?.tw. 4 (bendrofluazide? or bendroflumethiazide? or benzhydroflumethiazide? or benzthiazide? or benzydroflumethiazide? or benzothiadiazine? or buthiazide? or chlorothiazide? or cyclopenthiazide? or cyclothiazide or diucardin or diuril or enduron or esidrix or ezna or hydrochlorothiazide? or hydroflumethiazide? or methyclothiazide? or metolazone? or microzide or mykrox or naqua or naturetin or polythiazide? or renese or trichlormethiazide? or thiazide? or zaroxolyn).mp. 5 (chlortalidone? or chlorthalidone? or chlorphthalidolone? or clopamid? or clopamine? or clorexolone? or diapamide? or fenquizone? or hydromox or hygroton or indapamide? or isodapamide? or lozol or mefruside? or metindamide? or oxodoline? or phthalamudine? or quinethazone? or s‐1520 or s1520 or se‐1520 or se1520 or thalitone? or xipamide?).mp. 6 or/1‐5 7 exp hypertension/ 8 (anti‐hypertens$ or hypertens$).tw. 9 exp blood pressure/ 10 (blood pressure or bloodpressure).mp. 11 or/7‐10 12 randomized controlled trial/ 13 crossover procedure/ 14 double‐blind procedure/ 15 single‐blind procedure/ 16 random$.tw. 17 (crossover$ or cross‐over$).tw. 18 placebo$.tw. 19 (doubl$ adj blind$).tw. 20 allocat$.tw. 21 comparison.ti. 22 trial.ti. 23 or/12‐22 24 (exp animal/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans).ti.) 25 23 not 24 26 6 and 11 and 25
Appendix 3. CENTRAL search strategy
Database: Cochrane Central Register of Controlled Trials on Wiley <Issue 1, 2014> Search date: 19 February 2014 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ ID Search #1 MeSH descriptor: [Thiazides] explode all trees #2 MeSH descriptor: [Sodium Chloride Symporter Inhibitors] explode all trees #3 "sodium chloride" next (symporter or cotransporter* or co‐transporter*):ti,ab #4 "potassium depleting" near/2 diuretic*:ti,ab in Trials #5 (bendrofluazide* or bendroflumethiazide* or benzhydroflumethiazide* or benzthiazide* or benzydroflumethiazide* or benzothiadiazine* or buthiazide* or chlorothiazide* or cyclopenthiazide* or cyclothiazide or diucardin or diuril or enduron or esidrix or ezna or hydrochlorothiazide* or hydroflumethiazide* or methyclothiazide* or metolazone* or microzide or mykrox or naqua or naturetin or polythiazide* or renese or trichlormethiazide* or thiazide* or zaroxolyn):ti,ab,kw #6 (chlortalidone* or chlorthalidone* or chlorphthalidolone* or clopamid* or clopamine* or clorexolone* or diapamide* or fenquizone* or hydromox or hygroton or indapamide* or isodapamide* or lozol or mefruside* or metindamide* or oxodoline* or phthalamudine* or quinethazone* or s‐1520 or s1520 or se‐1520 or se1520 or thalitone* or xipamide*):ti,ab,kw #7 #1 or #2 or #3 or #4 or #5 or #6 #8 MeSH descriptor: [Hypertension] this term only #9 hypertens*:ti,ab #10 MeSH descriptor: [Blood Pressure] explode all trees #11 "blood pressure":ti,ab,kw #12 #8 or #9 or #10 or #11 #13 #7 and #12
Data and analyses
Comparison 1. Bendrofluazide versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Systolic blood pressure | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | ‐10.43 [‐14.06, ‐6.80] |
| 1.1 1.25 mg/day | 1 | 63 | Mean Difference (IV, Fixed, 95% CI) | ‐7.70 [‐14.98, ‐0.42] |
| 1.2 2.5 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | ‐10.9 [‐18.14, ‐3.66] |
| 1.3 5.0 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | ‐10.6 [‐17.84, ‐3.36] |
| 1.4 10 mg/day | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | ‐12.5 [‐19.76, ‐5.24] |
| 2 Diastolic blood pressure | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | ‐6.48 [‐8.82, ‐4.14] |
| 2.1 1.25 mg/day | 1 | 63 | Mean Difference (IV, Fixed, 95% CI) | ‐5.80 [‐10.49, ‐1.11] |
| 2.2 2.5 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | ‐6.9 [‐11.57, ‐2.23] |
| 2.3 5.0 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | ‐6.20 [‐10.87, ‐1.53] |
| 2.4 10 mg/day | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | ‐7.0 [‐11.68, ‐2.32] |
| 3 Withdrawals due to adverse effects | 1 | 257 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.07, 0.57] |
| 3.1 1.25 mg/day | 1 | 63 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.03, 1.81] |
| 3.2 2.5 mg/day | 1 | 65 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.03, 1.74] |
| 3.3 5.0 mg/day | 1 | 65 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 1.30] |
| 3.4 10 mg/day | 1 | 64 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.03, 1.77] |
| 4 Serum potassium mmol/L | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | ‐0.37 [‐0.50, ‐0.24] |
| 4.1 1.25 mg/day | 1 | 63 | Mean Difference (IV, Fixed, 95% CI) | ‐0.25 [‐0.51, 0.01] |
| 4.2 2.5 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | ‐0.29 [‐0.54, ‐0.04] |
| 4.3 5.0 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | ‐0.42 [‐0.68, ‐0.16] |
| 4.4 10 mg/day | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | ‐0.54 [‐0.80, ‐0.28] |
| 5 Serum uric acid µmol/L | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | 46.57 [33.24, 59.91] |
| 5.1 1.25 mg/day | 1 | 63 | Mean Difference (IV, Fixed, 95% CI) | 24.0 [0.04, 47.96] |
| 5.2 2.5 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | 34.0 [8.33, 59.67] |
| 5.3 5.0 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | 68.0 [33.12, 102.88] |
| 5.4 10 mg/day | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 73.0 [47.55, 98.45] |
| 6 Serum creatinine µmol/L | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | 5.50 [1.90, 9.11] |
| 6.1 1.25 mg/day | 1 | 63 | Mean Difference (IV, Fixed, 95% CI) | 5.1 [‐1.89, 12.09] |
| 6.2 2.5 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | 5.2 [‐1.63, 12.03] |
| 6.3 5.0 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | 5.00 [‐3.58, 13.58] |
| 6.4 10 mg/day | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 6.5 [‐0.33, 13.33] |
| 7 Serum blood glucose mmol/L | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | 0.13 [‐0.06, 0.33] |
| 7.1 1.25 mg/day | 1 | 63 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.49, 0.27] |
| 7.2 2.5 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | 0.22 [‐0.15, 0.59] |
| 7.3 5.0 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | 0.12 [‐0.25, 0.49] |
| 7.4 10 mg/day | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 0.35 [‐0.09, 0.79] |
| 8 Total cholesterol mmol/L | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | 0.15 [‐0.05, 0.35] |
| 8.1 1.25 mg/day | 1 | 63 | Mean Difference (IV, Fixed, 95% CI) | 0.03 [‐0.38, 0.44] |
| 8.2 2.5 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | 0.06 [‐0.34, 0.46] |
| 8.3 5.0 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | 0.18 [‐0.22, 0.58] |
| 8.4 10 mg/day | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 0.31 [‐0.08, 0.70] |
| 9 Triglycerides mmol/L | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | 0.26 [‐0.06, 0.58] |
| 9.1 1.25 mg/day | 1 | 63 | Mean Difference (IV, Fixed, 95% CI) | 0.13 [‐0.49, 0.75] |
| 9.2 2.5 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | 0.52 [‐0.11, 1.15] |
| 9.3 5.0 mg/day | 1 | 65 | Mean Difference (IV, Fixed, 95% CI) | 0.08 [‐0.65, 0.81] |
| 9.4 10 mg/day | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 0.27 [‐0.34, 0.88] |
1.7. Analysis.
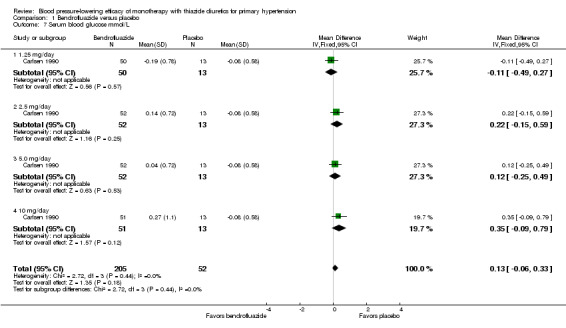
Comparison 1 Bendrofluazide versus placebo, Outcome 7 Serum blood glucose mmol/L.
Comparison 2. Cyclopenthiazide versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Systolic blood pressure | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐10.75 [‐18.44, ‐3.05] |
| 1.1 0.05 mg/day | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | ‐5.3 [‐18.71, 8.11] |
| 1.2 0.125 mg/day | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | ‐12.0 [‐25.16, 1.16] |
| 1.3 0.5 mg/day | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | ‐14.90 [‐28.31, ‐1.49] |
| 2 Diastolic blood pressure | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐6.23 [‐11.19, ‐1.27] |
| 2.1 0.05 mg/day | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | ‐3.0 [‐11.65, 5.65] |
| 2.2 0.125mg/day | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | ‐8.60 [‐17.09, ‐0.11] |
| 2.3 0.5 mg/day | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | ‐7.0 [‐15.65, 1.65] |
| 3 Serum potassium µmol/L | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐0.18 [‐0.42, 0.07] |
| 3.1 0.05 mg/day | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.35, 0.55] |
| 3.2 0.125 mg/day | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.52, 0.32] |
| 3.3 0.5 mg/day | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | ‐0.50 [‐0.92, ‐0.08] |
| 4 Serum uric acid µmol/L | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | 19.54 [‐36.18, 75.26] |
| 4.1 0.05 mg/day | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | 30.0 [‐70.86, 130.86] |
| 4.2 0.125 mg/day | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | 20.0 [‐75.05, 115.05] |
| 4.3 0.5 mg/day | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | 10.0 [‐84.04, 104.04] |
| 5 Total cholesterol mmol/L | 1 | 47 | Mean Difference (IV, Fixed, 95% CI) | 0.79 [0.36, 1.23] |
| 5.1 0.05 mg/day | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 1.2 [0.46, 1.94] |
| 5.2 0.125 mg/day | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | 0.90 [0.17, 1.63] |
| 5.3 0.5 mg/day | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 0.2 [‐0.60, 1.00] |
| 6 Triglycerides mmol/L | 1 | 48 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐0.17, 0.57] |
| 6.1 0.05 mg/day | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 0.3 [‐0.30, 0.90] |
| 6.2 0.125 mg/day | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | 0.4 [‐0.29, 1.09] |
| 6.3 0.5 mg/day | 1 | 15 | Mean Difference (IV, Fixed, 95% CI) | ‐0.1 [‐0.74, 0.54] |
Comparison 3. Metolazone versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Systolic blood pressure | 1 | 105 | Mean Difference (IV, Fixed, 95% CI) | ‐11.63 [‐16.89, ‐6.38] |
| 1.1 0.5 mg/day | 1 | 35 | Mean Difference (IV, Fixed, 95% CI) | ‐11.40 [‐20.50, ‐2.30] |
| 1.2 1.0 mg/day | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐11.6 [‐20.75, ‐2.45] |
| 1.3 2 mg/day | 1 | 36 | Mean Difference (IV, Fixed, 95% CI) | ‐11.90 [‐20.95, ‐2.85] |
| 2 Diastolic blood pressure | 1 | 105 | Mean Difference (IV, Fixed, 95% CI) | ‐5.83 [‐9.22, ‐2.44] |
| 2.1 0.5 mg/day | 1 | 35 | Mean Difference (IV, Fixed, 95% CI) | ‐5.90 [‐11.77, ‐0.03] |
| 2.2 1.0 mg/day | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐6.40 [‐12.30, ‐0.50] |
| 2.3 2.0 mg/day | 1 | 36 | Mean Difference (IV, Fixed, 95% CI) | ‐5.2 [‐11.04, 0.64] |
Comparison 4. Chlorthalidone versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Systolic blood pressure | 7 | 1153 | Mean Difference (IV, Fixed, 95% CI) | ‐11.98 [‐13.71, ‐10.24] |
| 1.1 12.5 to 15 mg/day | 3 | 185 | Mean Difference (IV, Fixed, 95% CI) | ‐10.09 [‐13.88, ‐6.30] |
| 1.2 25 to 30 mg/day | 5 | 752 | Mean Difference (IV, Fixed, 95% CI) | ‐13.64 [‐16.03, ‐11.25] |
| 1.3 45 to 50 mg/day | 4 | 192 | Mean Difference (IV, Fixed, 95% CI) | ‐9.92 [‐13.44, ‐6.39] |
| 1.4 75 mg/day | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | ‐12.9 [‐24.65, ‐1.15] |
| 2 Diastolic blood pressure | 7 | 1153 | Mean Difference (IV, Fixed, 95% CI) | ‐3.93 [‐5.13, ‐2.74] |
| 2.1 12.5 to 15 mg/day | 3 | 185 | Mean Difference (IV, Fixed, 95% CI) | ‐2.55 [‐5.07, ‐0.02] |
| 2.2 25 to 30 mg/day | 5 | 752 | Mean Difference (IV, Fixed, 95% CI) | ‐3.98 [‐5.69, ‐2.28] |
| 2.3 45 to 50 mg/day | 4 | 192 | Mean Difference (IV, Fixed, 95% CI) | ‐4.91 [‐7.29, ‐2.53] |
| 2.4 75 mg/day | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | ‐5.5 [‐13.08, 2.08] |
| 3 Withdrawals due to adverse events | 5 | 1058 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.28, 0.87] |
| 3.1 12.5 mg/day | 2 | 85 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.02, 0.55] |
| 3.2 15 mg/day | 1 | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.13, 4.94] |
| 3.3 25 mg/day | 4 | 732 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.29, 1.57] |
| 3.4 45 mg/day | 1 | 34 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.5 50 mg/day | 2 | 81 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.13, 1.43] |
| 3.6 75 mg/day | 1 | 25 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.03, 23.82] |
| 4 Serum potassium mmol/L | 5 | 1203 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐0.45, ‐0.34] |
| 4.1 12.5 to 15 mg/day | 3 | 235 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐0.51, ‐0.29] |
| 4.2 25 to 30 mg/day | 4 | 784 | Mean Difference (IV, Fixed, 95% CI) | ‐0.26 [‐0.34, ‐0.19] |
| 4.3 50 mg/day | 3 | 161 | Mean Difference (IV, Fixed, 95% CI) | ‐0.77 [‐0.90, ‐0.64] |
| 4.4 75 mg/day | 1 | 23 | Mean Difference (IV, Fixed, 95% CI) | ‐0.6 [‐1.05, ‐0.15] |
| 5 Serum uric acid µmol/L | 2 | 285 | Mean Difference (IV, Fixed, 95% CI) | 64.16 [45.69, 82.63] |
| 5.1 12.5 to 15 mg/day | 2 | 120 | Mean Difference (IV, Fixed, 95% CI) | 52.71 [26.11, 79.30] |
| 5.2 25 to 30 mg/day | 2 | 120 | Mean Difference (IV, Fixed, 95% CI) | 74.44 [47.85, 101.02] |
| 5.3 50 mg/day | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 90.0 [‐47.99, 227.99] |
| 5.4 75 mg/day | 1 | 23 | Mean Difference (IV, Fixed, 95% CI) | 70.0 [‐70.76, 210.76] |
| 6 Serum blood glucose mmol/L | 3 | 394 | Mean Difference (IV, Fixed, 95% CI) | 0.34 [0.12, 0.55] |
| 6.1 12.5 mg/day | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | 0.17 [‐0.68, 1.02] |
| 6.2 15 mg/day | 1 | 122 | Mean Difference (IV, Fixed, 95% CI) | 0.05 [‐0.33, 0.43] |
| 6.3 25 to 30 mg/day | 2 | 147 | Mean Difference (IV, Fixed, 95% CI) | 0.58 [0.23, 0.93] |
| 6.4 50 mg/day | 2 | 77 | Mean Difference (IV, Fixed, 95% CI) | 0.43 [‐0.04, 0.91] |
| 6.5 75 mg/day | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | 0.17 [‐0.61, 0.95] |
| 7 HDL cholesterol mmol/L | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 45 mg/day | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.22, 0.00] |
| 8 Total cholesterol mmol/L | 2 | 213 | Mean Difference (IV, Fixed, 95% CI) | 0.41 [0.18, 0.64] |
| 8.1 15 mg/day | 1 | 88 | Mean Difference (IV, Fixed, 95% CI) | 0.23 [‐0.24, 0.70] |
| 8.2 25 mg/day | 1 | 91 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐0.24, 0.64] |
| 8.3 45 mg/day | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.6 [0.28, 0.92] |
| 9 Triglycerides mmol/L | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1 45 mg/day | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.69 [0.05, 1.33] |
4.7. Analysis.
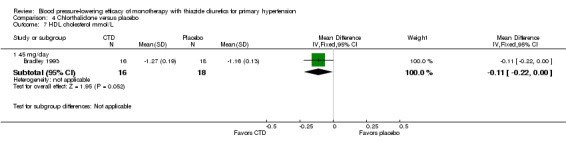
Comparison 4 Chlorthalidone versus placebo, Outcome 7 HDL cholesterol mmol/L.
Comparison 5. Hydrochlorothiazide versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Systolic blood pressure | 35 | 6725 | Mean Difference (IV, Fixed, 95% CI) | ‐6.94 [‐7.56, ‐6.31] |
| 1.1 3.0 to 6.25 mg/day | 8 | 663 | Mean Difference (IV, Fixed, 95% CI) | ‐3.56 [‐5.57, ‐1.54] |
| 1.2 12.5 mg/day | 22 | 2645 | Mean Difference (IV, Fixed, 95% CI) | ‐6.27 [‐7.24, ‐5.31] |
| 1.3 25 mg/day | 25 | 3062 | Mean Difference (IV, Fixed, 95% CI) | ‐6.00 [‐8.96, ‐7.04] |
| 1.4 37.5 mg/day | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | ‐7.30 [‐16.30, 1.70] |
| 1.5 50 mg/day | 2 | 169 | Mean Difference (IV, Fixed, 95% CI) | ‐10.47 [‐14.60, ‐6.35] |
| 1.6 100 mg/day | 2 | 146 | Mean Difference (IV, Fixed, 95% CI) | ‐9.91 [‐14.05, ‐5.77] |
| 2 Diastolic blood pressure | 39 | 7284 | Mean Difference (IV, Fixed, 95% CI) | ‐3.25 [‐3.59, ‐2.90] |
| 2.1 3.0 to 6.25 mg/day | 8 | 663 | Mean Difference (IV, Fixed, 95% CI) | ‐2.43 [‐3.67, ‐1.19] |
| 2.2 12.5 mg/day | 25 | 2877 | Mean Difference (IV, Fixed, 95% CI) | ‐3.12 [‐3.71, ‐2.53] |
| 2.3 25 mg/day | 29 | 3359 | Mean Difference (IV, Fixed, 95% CI) | ‐3.28 [‐3.77, ‐2.79] |
| 2.4 37.5 mg/day | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | ‐3.7 [‐9.26, 1.86] |
| 2.5 50 mg/day | 3 | 199 | Mean Difference (IV, Fixed, 95% CI) | ‐4.97 [‐6.65, ‐3.30] |
| 2.6 100 mg/day | 2 | 146 | Mean Difference (IV, Fixed, 95% CI) | ‐3.90 [‐6.57, ‐1.23] |
| 3 Withdrawals due to adverse events | 20 | 3698 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.43, 0.93] |
| 3.1 3.0 mg/day | 1 | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 6.0 mg/day | 1 | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.3 6.25 mg/day | 1 | 259 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.09 [0.02, 0.43] |
| 3.4 12.5 mg/day | 11 | 1676 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.53, 1.67] |
| 3.5 25 mg/day | 12 | 1645 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.33, 1.07] |
| 3.6 50 mg/day | 1 | 64 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.04, 5.26] |
| 4 Serum potassium mmol/L | 11 | 2036 | Mean Difference (IV, Fixed, 95% CI) | ‐0.22 [‐0.25, ‐0.18] |
| 4.1 3.0 mg/day | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 0.03 [‐0.26, 0.32] |
| 4.2 6.0 mg/day | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.22, 0.42] |
| 4.3 12.5 mg/day | 7 | 1026 | Mean Difference (IV, Fixed, 95% CI) | ‐0.16 [‐0.21, ‐0.11] |
| 4.4 25 mg/day | 7 | 805 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐0.36, ‐0.24] |
| 4.5 50 mg/day | 2 | 151 | Mean Difference (IV, Fixed, 95% CI) | ‐0.48 [‐0.68, ‐0.29] |
| 5 Serum uric acid µmol/L | 5 | 1043 | Mean Difference (IV, Fixed, 95% CI) | 32.88 [26.12, 39.65] |
| 5.1 3.0 mg/day | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 10.40 [‐26.09, 46.89] |
| 5.2 6.0 mg/day | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 11.5 [‐29.31, 52.31] |
| 5.3 12.5 mg/day | 5 | 835 | Mean Difference (IV, Fixed, 95% CI) | 33.00 [25.67, 40.34] |
| 5.4 25 mg/day | 2 | 154 | Mean Difference (IV, Fixed, 95% CI) | 47.15 [24.38, 69.92] |
| 6 Serum creatinine µmol/L | 3 | 527 | Mean Difference (IV, Fixed, 95% CI) | 0.32 [‐2.63, 3.26] |
| 6.1 12.5 mg/day | 2 | 258 | Mean Difference (IV, Fixed, 95% CI) | 0.57 [‐3.40, 4.55] |
| 6.2 25 mg/day | 2 | 269 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐4.38, 4.38] |
| 6.3 50 mg/day | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Serum blood glucose mmol/L | 6 | 1041 | Mean Difference (IV, Fixed, 95% CI) | ‐0.12 [‐0.24, 0.01] |
| 7.1 3.0 mg/day | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 0.07 [‐0.45, 0.59] |
| 7.2 6.0 mg/day | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | ‐0.18 [‐0.68, 0.32] |
| 7.3 12.5 mg/day | 4 | 605 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.33, 0.13] |
| 7.4 25 mg/day | 3 | 298 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐0.19, 0.20] |
| 7.5 50 mg/day | 1 | 84 | Mean Difference (IV, Fixed, 95% CI) | ‐0.5 [‐0.81, ‐0.19] |
| 8 HDL cholesterol mmol/L | 1 | 159 | Mean Difference (IV, Fixed, 95% CI) | ‐0.17 [‐0.53, 0.19] |
| 8.1 3.0 mg/day | 1 | 44 | Mean Difference (IV, Fixed, 95% CI) | ‐0.52 [‐1.15, 0.11] |
| 8.2 6.0 mg/day | 1 | 44 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐0.68, 0.70] |
| 8.3 12.5 mg/day | 1 | 32 | Mean Difference (IV, Fixed, 95% CI) | ‐0.12 [‐1.03, 0.79] |
| 8.4 25 mg/day | 1 | 39 | Mean Difference (IV, Fixed, 95% CI) | 0.04 [‐0.67, 0.75] |
| 9 Total cholesterol mmol/L | 4 | 450 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [0.17, 0.22] |
| 9.1 3.0 mg/day | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | ‐0.4 [‐0.67, ‐0.13] |
| 9.2 6.0 mg/day | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 0.06 [‐1.15, 1.27] |
| 9.3 12.5 mg/day | 2 | 159 | Mean Difference (IV, Fixed, 95% CI) | ‐0.04 [‐0.34, 0.27] |
| 9.4 25 mg/day | 3 | 237 | Mean Difference (IV, Fixed, 95% CI) | 0.21 [0.18, 0.23] |
| 10 Triglycerides mmol/L | 2 | 255 | Mean Difference (IV, Fixed, 95% CI) | 0.09 [‐0.11, 0.30] |
| 10.1 3.0 mg/day | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 0.03 [‐0.67, 0.73] |
| 10.2 6.0 mg/day | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | ‐0.17 [‐0.88, 0.54] |
| 10.3 12.5 mg/day | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.77, 0.55] |
| 10.4 25 mg/day | 2 | 173 | Mean Difference (IV, Fixed, 95% CI) | 0.16 [‐0.08, 0.40] |
5.6. Analysis.
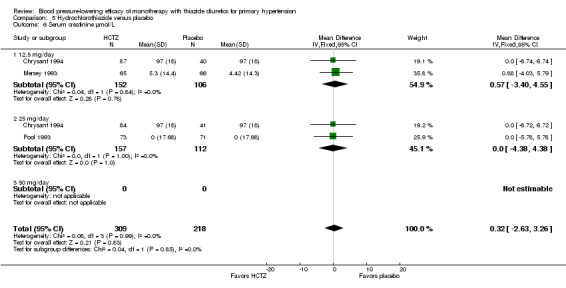
Comparison 5 Hydrochlorothiazide versus placebo, Outcome 6 Serum creatinine µmol/L.
5.7. Analysis.
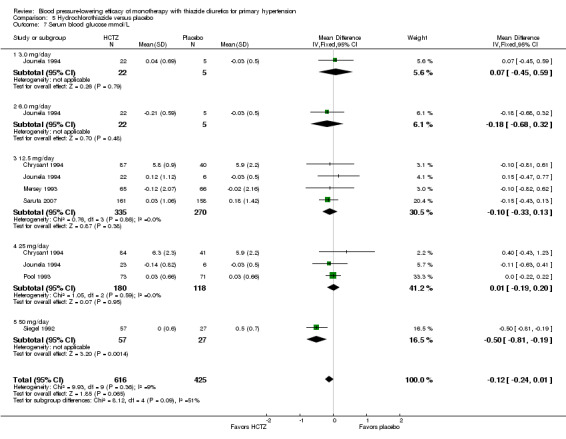
Comparison 5 Hydrochlorothiazide versus placebo, Outcome 7 Serum blood glucose mmol/L.
5.8. Analysis.
Comparison 5 Hydrochlorothiazide versus placebo, Outcome 8 HDL cholesterol mmol/L.
Comparison 6. Indapamide versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Systolic blood pressure | 10 | 2104 | Mean Difference (IV, Fixed, 95% CI) | ‐8.69 [‐9.96, ‐7.42] |
| 1.1 1.0 mg/day | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐9.7 [‐19.89, 0.49] |
| 1.2 1.25 mg/day | 4 | 736 | Mean Difference (IV, Fixed, 95% CI) | ‐7.37 [‐9.21, ‐5.54] |
| 1.3 1.5 mg/day | 2 | 955 | Mean Difference (IV, Fixed, 95% CI) | ‐9.40 [‐11.51, ‐7.29] |
| 1.4 2.0 mg/day | 1 | 74 | Mean Difference (IV, Fixed, 95% CI) | ‐8.7 [‐17.38, ‐0.02] |
| 1.5 2.5 mg/day | 5 | 281 | Mean Difference (IV, Fixed, 95% CI) | ‐11.94 [‐15.88, ‐7.99] |
| 1.6 5.0 mg/day | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐9.6 [‐19.40, 0.20] |
| 2 Diastolic blood pressure | 10 | 2104 | Mean Difference (IV, Fixed, 95% CI) | ‐3.88 [‐4.63, ‐3.14] |
| 2.1 1.0 mg/day | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐1.00 [‐9.57, 3.57] |
| 2.2 1.25 mg/day | 4 | 736 | Mean Difference (IV, Fixed, 95% CI) | ‐3.55 [‐4.57, ‐2.52] |
| 2.3 1.5 mg/day | 2 | 955 | Mean Difference (IV, Fixed, 95% CI) | ‐4.05 [‐5.38, ‐2.72] |
| 2.4 2.0 mg/day | 1 | 74 | Mean Difference (IV, Fixed, 95% CI) | ‐3.60 [‐8.27, 1.07] |
| 2.5 2.5 mg/day | 5 | 281 | Mean Difference (IV, Fixed, 95% CI) | ‐5.32 [‐7.65, ‐2.98] |
| 2.6 5.0 mg/day | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐4.0 [‐10.32, 2.32] |
| 3 Withdrawals due to adverse events | 6 | 1874 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.49, 1.42] |
| 3.1 1.0 mg/day | 1 | 29 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.00, 2.66] |
| 3.2 1.25 mg/day | 3 | 621 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.49, 2.32] |
| 3.3 1.5 mg/day | 2 | 958 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.30, 1.79] |
| 3.4 2.0 mg/day | 1 | 74 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.02, 5.61] |
| 3.5 2.5 mg/day | 2 | 163 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.17, 6.53] |
| 3.6 5.0 mg/day | 1 | 29 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Serum potassium mmol/L | 5 | 541 | Mean Difference (IV, Fixed, 95% CI) | ‐0.32 [‐0.38, ‐0.26] |
| 4.1 1.0 mg/day | 1 | 31 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.38, 0.16] |
| 4.2 1.25 mg/day | 2 | 398 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐0.37, ‐0.23] |
| 4.3 2.5 mg/day | 3 | 81 | Mean Difference (IV, Fixed, 95% CI) | ‐0.41 [‐0.57, ‐0.26] |
| 4.4 5.0 mg/day | 1 | 31 | Mean Difference (IV, Fixed, 95% CI) | ‐0.65 [‐0.91, ‐0.39] |
| 5 Serum uric acid µmol/L | 4 | 558 | Mean Difference (IV, Fixed, 95% CI) | 39.81 [33.54, 46.08] |
| 5.1 1.0 mg/day | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | 35.69 [14.15, 57.23] |
| 5.2 1.25 mg/day | 2 | 398 | Mean Difference (IV, Fixed, 95% CI) | 36.14 [28.70, 43.58] |
| 5.3 2.5 mg/day | 2 | 69 | Mean Difference (IV, Fixed, 95% CI) | 52.33 [34.50, 70.16] |
| 5.4 5.0 mg/day | 1 | 45 | Mean Difference (IV, Fixed, 95% CI) | 57.17 [35.17, 79.17] |
| 6 Serum creatinine µmol/L | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 1.25 mg/day | 1 | 203 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐2.43, 2.43] |
| 7 Serum blood glucose mmol/L | 3 | 490 | Mean Difference (IV, Fixed, 95% CI) | 0.13 [‐0.11, 0.37] |
| 7.1 1.0 mg/day | 1 | 31 | Mean Difference (IV, Fixed, 95% CI) | ‐0.08 [‐1.12, 0.96] |
| 7.2 1.25 mg/day | 2 | 398 | Mean Difference (IV, Fixed, 95% CI) | 0.12 [‐0.14, 0.38] |
| 7.3 2.5 mg/day | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | 0.34 [‐0.71, 1.39] |
| 7.4 5.0 mg/day | 1 | 31 | Mean Difference (IV, Fixed, 95% CI) | 0.25 [‐0.76, 1.26] |
| 8 HDL cholesterol mmol/L | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 1.25 mg/day | 1 | 203 | Mean Difference (IV, Fixed, 95% CI) | ‐0.05 [‐0.10, ‐0.00] |
| 9 Total cholesterol mmol/L | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1 1.25 mg/day | 2 | 398 | Mean Difference (IV, Fixed, 95% CI) | 0.12 [0.02, 0.23] |
| 10 Triglycerides mmol/L | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.1 1.25 mg/day | 1 | 203 | Mean Difference (IV, Fixed, 95% CI) | 0.23 [0.03, 0.43] |
6.6. Analysis.
Comparison 6 Indapamide versus placebo, Outcome 6 Serum creatinine µmol/L.
6.7. Analysis.
Comparison 6 Indapamide versus placebo, Outcome 7 Serum blood glucose mmol/L.
6.8. Analysis.
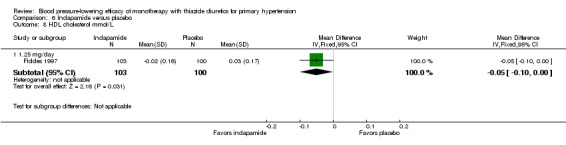
Comparison 6 Indapamide versus placebo, Outcome 8 HDL cholesterol mmol/L.
Comparison 7. Thiazide versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Systolic blood pressure | 47 | 7733 | Mean Difference (IV, Fixed, 95% CI) | ‐9.14 [‐9.76, ‐8.51] |
| 1.1 Bendrofluazide 1.25 to 10 mg/day | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | ‐10.4 [‐14.03, ‐6.77] |
| 1.2 Chlorthalidone 12.5 to 100 mg/day | 7 | 1167 | Mean Difference (IV, Fixed, 95% CI) | ‐11.79 [‐13.50, ‐10.08] |
| 1.3 Cyclopenthiazide 0.5 mg/day | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐14.90 [‐24.58, ‐5.22] |
| 1.4 Hydrochlorothiazide 25 to 100 mg/day | 28 | 4099 | Mean Difference (IV, Fixed, 95% CI) | ‐8.68 [‐9.49, ‐7.87] |
| 1.5 Indapamide 1.0 to 5.0 mg/day | 9 | 2080 | Mean Difference (IV, Fixed, 95% CI) | ‐8.37 [‐9.65, ‐7.10] |
| 1.6 Metolazone 0.5 to 2.0 mg/day | 1 | 105 | Mean Difference (IV, Fixed, 95% CI) | ‐11.7 [‐16.95, ‐6.45] |
| 2 Diastolic blood pressure | 51 | 8064 | Mean Difference (IV, Fixed, 95% CI) | ‐3.63 [‐3.97, ‐3.28] |
| 2.1 Bendrofluazide 1.25 to 10 mg/day | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | ‐6.5 [‐8.84, ‐4.16] |
| 2.2 Chlorthalidone 12.5 to 100 mg/day | 7 | 1153 | Mean Difference (IV, Fixed, 95% CI) | ‐3.07 [‐4.27, ‐1.87] |
| 2.3 Cyclopenthiazide 0.125 to 0.5 mg/day | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | ‐7.90 [‐13.16, ‐2.64] |
| 2.4 Hydrochlorothiazide 25 to 100 mg/day | 32 | 4429 | Mean Difference (IV, Fixed, 95% CI) | ‐3.50 [‐3.92, ‐3.08] |
| 2.5 Indapamide 1.0 to 5.0 mg/day | 9 | 2080 | Mean Difference (IV, Fixed, 95% CI) | ‐3.77 [‐4.52, ‐3.02] |
| 2.6 Metolazone 0.5 to 2.0 mg/day | 1 | 105 | Mean Difference (IV, Fixed, 95% CI) | ‐5.8 [‐9.19, ‐2.41] |
| 3 Serum potassium mmol/L | 22 | 3868 | Mean Difference (IV, Fixed, 95% CI) | ‐0.25 [‐0.28, ‐0.22] |
| 3.1 BDFZ 1.25 to 10 mg/day | 1 | 218 | Mean Difference (IV, Fixed, 95% CI) | ‐0.38 [‐0.62, ‐0.14] |
| 3.2 CTD 12.5 to 100 mg/day | 4 | 1068 | Mean Difference (IV, Fixed, 95% CI) | ‐0.32 [‐0.38, ‐0.25] |
| 3.3 CYPTZ 0.05 to 0.5 mg/day | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐0.45, 0.05] |
| 3.4 HCTZ 3 to 100 mg/day | 11 | 1988 | Mean Difference (IV, Fixed, 95% CI) | ‐0.21 [‐0.24, ‐0.17] |
| 3.5 IND 1.0 to 5 mg/day | 5 | 541 | Mean Difference (IV, Fixed, 95% CI) | ‐0.32 [‐0.38, ‐0.26] |
| 4 Serum uric acid µmol/L | 13 | 2332 | Mean Difference (IV, Fixed, 95% CI) | 38.22 [34.24, 42.20] |
| 4.1 BDFZ 1.25 to 10 mg/day | 1 | 255 | Mean Difference (IV, Fixed, 95% CI) | 50.30 [36.62, 63.98] |
| 4.2 CTD 12.5 to 100 mg/day | 2 | 285 | Mean Difference (IV, Fixed, 95% CI) | 64.74 [46.11, 83.36] |
| 4.3 CYPTZ 0.05 to 0.5 mg/day | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | 20.0 [‐35.61, 75.61] |
| 4.4 HCTZ 3 to 100 mg/day | 5 | 1122 | Mean Difference (IV, Fixed, 95% CI) | 32.74 [26.00, 39.48] |
| 4.5 IND 1.0 to 5 mg/day | 4 | 617 | Mean Difference (IV, Fixed, 95% CI) | 37.77 [32.23, 43.31] |
| 5 Serum creatinine µmol/L | 5 | 987 | Mean Difference (IV, Fixed, 95% CI) | 1.34 [‐0.31, 2.99] |
| 5.1 BDFZ 1.25 to 10 mg/day | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | 5.45 [1.99, 8.91] |
| 5.2 HCTZ 3 to 100 mg/day | 3 | 527 | Mean Difference (IV, Fixed, 95% CI) | 0.32 [‐2.63, 3.26] |
| 5.3 IND 1.0 to 5 mg/day | 1 | 203 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐2.43, 2.43] |
| 6 Serum blood glucose mmol/L | 12 | 1989 | Mean Difference (IV, Fixed, 95% CI) | 0.03 [‐0.05, 0.12] |
| 6.1 BDFZ 1.25 to 10 mg/day | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | 0.15 [‐0.04, 0.34] |
| 6.2 CTD 12.5 to 100 mg/day | 3 | 326 | Mean Difference (IV, Fixed, 95% CI) | 0.37 [0.14, 0.60] |
| 6.3 HCTZ 3 to 100 mg/day | 6 | 916 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.22, 0.01] |
| 6.4 IND 1.0 to 5 mg/day | 3 | 490 | Mean Difference (IV, Fixed, 95% CI) | 0.13 [‐0.11, 0.37] |
| 7 Serum total cholesterol mmol/L | 11 | 1394 | Mean Difference (IV, Fixed, 95% CI) | 0.21 [0.18, 0.23] |
| 7.1 BDFZ 1.25 to 10 mg/day | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | 0.44 [0.24, 0.64] |
| 7.2 CTD 12.5 to 100 mg/day | 2 | 213 | Mean Difference (IV, Fixed, 95% CI) | 0.41 [0.18, 0.63] |
| 7.3 CYPTZ 0.05 to 0.5 mg/day | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | 0.79 [0.37, 1.21] |
| 7.4 HCTZ 3 to 100 mg/day | 4 | 450 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [0.18, 0.23] |
| 7.5 IND 1.0 to 5 mg/day | 3 | 421 | Mean Difference (IV, Fixed, 95% CI) | 0.11 [0.01, 0.21] |
| 8 Serum HDL mmol/L | 3 | 348 | Mean Difference (IV, Fixed, 95% CI) | ‐0.06 [‐0.10, ‐0.02] |
| 8.1 CTD 45 mg/day | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.22, 0.00] |
| 8.2 HCTZ 25mg/day | 1 | 111 | Mean Difference (IV, Fixed, 95% CI) | ‐0.15 [‐0.72, 0.42] |
| 8.3 IND 1.25 mg/day | 1 | 203 | Mean Difference (IV, Fixed, 95% CI) | ‐0.05 [‐0.10, ‐0.00] |
| 9 Triglycerides | 6 | 697 | Mean Difference (IV, Fixed, 95% CI) | 0.21 [0.08, 0.33] |
| 9.1 BDFZ 1.25 to 10 mg/day | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | 0.37 [‐0.09, 0.83] |
| 9.2 CTD 45 mg/day | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.69 [0.05, 1.33] |
| 9.3 CYPTZ 0.5 mg/day | 1 | 48 | Mean Difference (IV, Fixed, 95% CI) | 0.21 [‐0.16, 0.58] |
| 9.4 HCTZ 25 mg/day | 2 | 255 | Mean Difference (IV, Fixed, 95% CI) | 0.09 [‐0.11, 0.30] |
| 9.5 IND 1.25 mg/day | 1 | 203 | Mean Difference (IV, Fixed, 95% CI) | 0.23 [0.03, 0.43] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ambrosioni 1998.
| Methods | Randomized, double‐blind, placebo‐controlled trial (dose‐finding). Wash‐out period = 1 month. Multicenter; France, UK, Italy, Belgium, Spain | |
| Participants | DBP 95 to 114 mmHg. Mean age 54 years. Males 51.5%. Baseline BP was 164.5/101.7 mmHg in the treatment group and 164.4/102.5 mmHg in the control group. Pulse pressure = 62.8 in the treatment group and 61.9 in the control group | |
| Interventions | Indapamide IR 2.5 mg/d (N = 59), indapamide SR 1.5 mg (N = 57), 2.0 mg (N = 55) or 2.5 mg/d (N = 56), or placebo (N = 58) Trial duration = 2 months. IR = immediate release and SR = sustained release |
|
| Outcomes | Change from the baseline in trough supine and standing DBP and SBP; BP response rate; serum biochemistry | |
| Notes | A sample size calculation was provided based on 200 patients to detect a 10 mmHg difference in supine DBP between placebo and treatment groups. Baseline characteristics did not differ between treatment groups. No placebo data for metabolic changes
Indapamide 2.5 SR and 2.5 IR results added and presented as weighted mean changes in SBP, DBP and withdrawal due to adverse events (WDAE). Results were pooled from 2 identical studies. Original publication also included an equivalence study comparing indapamide 1.5 SR to indapamide 2.5 IR (no placebo arm; therefore not included in this review) Additional publications: Mallion JM et al. Journal of Cardiovascular Pharmacology 1998; 32(4): 673‐8ambulatory BP was measured in a subset of patients from the original trial) and Leonetti G. Drugs 2000;, 59 (Suppl 2):27‐38 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...hypertensive patients were randomly allocated to parallel groups..." (line 15 under "Patients and Methods‐Study objective and design" p.1678). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "...hypertensive patients were randomly allocated to parallel groups (inclusion visit month 0) for one of the above treatments on a double‐blind basis for 2 months..." (line 15 under "Patients and Methods‐Study objective and design" p.1678). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Efficacy and safety analysis was based on an ITT with LOCF (last observation carried forward) technique Exclusions: 79/364 (22%) patients were excluded from study during the 1‐ month, single‐blind, placebo run‐in period prior to randomization Attrition: 17/285 (6%) patients withdrew from the study. Reasons were, in the IND 2.5 IR group: 1 patient ‐ adverse events; IND 1.5 SR group: 1 ‐ major protocol violation, 1 ‐ causes unrelated to treatment, and 1 ‐ adverse events; IND 2.0 SR group: 1 ‐ major protocol violation and 1 ‐ adverse events; IND 2.5 SR group: 1 ‐ causes unrelated to treatment and 5 ‐ adverse events; Placebo group: 2 ‐ major protocol violation, 1 ‐ severe hypertension, 1 ‐ causes unrelated to treatment and 1 ‐ adverse events WDAEs: 9/285 (3%) patients withdrew due to adverse events for the following reasons: 1 patient in the IND 2.5 IR group for "serum potassium of 2.8 mmol/L"; 1 patient for a "skin allergy" in the IND 1.5 SR group; 1 patient for "skin allergy" in the IND 2.0 SR group; in the IND 2.5 SR group: 1 ‐ "serum potassium of 2.8 mmol/L", 2 ‐ "dizziness", 1 ‐ "asthenia" and 1 ‐ "gout"; and in the placebo group 1 patient for a "headache" |
| Selective reporting (reporting bias) | High risk | Baseline standing BP was measured but not reported. Serum biochemical data were reported, but not mentioned in the methods section of the study SAEs were not clearly documented but were inferred based on the authors' statement that "no serious treatment‐related adverse event was observed..." (line 3 under "Safety" p.1680) Placebo and indapamide 2.0 and 2.5 mg treatment arms were not shown for biochemical data (IND 2.5 IR and 1.5 SR only) Study authors did not comment on all‐cause mortality and total AEs were not reported |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Benz 1998.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 2 to 4 weeks; conducted in USA | |
| Participants | DBP 95 to 115 mmHg. Mean age 52 years. Males 57%. Baseline BP was 152.8/101.5 mmHg in the treatment group and 152.7/101.4 mmHg in the control group. Pulse pressure = 51.3 | |
| Interventions | Valsartan 80 mg (N = 99) or 160 mg/d (N = 99), valsartan 80 mg or 160 mg/d + HCTZ 12.5 mg or 25 mg/d (all combined, N = 379), HCTZ 12.5 mg (N = 100) or 25 mg/d (N = 100), or placebo (N = 94) Treatment duration = 8 weeks | |
| Outcomes | Change from the placebo in trough mean sitting SBP and DBP; pulse rate, body weight, ECG, serum chemistry, hematology and urinalysis. Withdrawals due to adverse events were not given in each treatment group | |
| Notes | A sample size calculation was provided based on 85 patients per treatment group to detect a difference in mean sitting DBP of 4 mmHg (standard deviation = ± 8 mmHg) between treatments at a power of 90%. Baseline patient demographics, measurements and medical history were similar across all treatment groups (P value = NS). Standard deviation (SD) of change in BP not given (95% CI given) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The study was ... randomized..." (line 2 under "Study Design" p.862). "Eligible patients were randomized into one of nine double‐blind treatment groups..." (line 10 under "Study Design" p.862). Technique for sequence generation not stated explicitly by study authors |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "....a randomized, double‐blind, multiple dose, placebo controlled, multifactorial, parallel trial" (line 2 under "Study Design" p.862). "Study drugs were packaged in double‐dummy fashion to maintain blinding, with each patient taking two capsules per day at 8 am." (line 3 from bottom under "Study Design" p.862). Both low and high doses of HCTZ and placebo were supplied as identical capsules |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "The primary efficacy analysis was an ITT analysis..." (line 7 under "Statistical analysis" p.862). Exclusions: number of patients excluded from study after the single‐blind placebo run‐in period before randomization was not reported Attrition: there were 79/871 (9%) withdrawals due to: 41 ‐ AEs, 9 ‐ unsatisfactory therapeutic effect, 7 ‐ did not meet protocol criteria, 2 ‐ non‐compliance, 15 ‐ withdrew consent, 5 ‐ lost to follow‐up WDAEs: 41/871 (4.7%) of patients withdrew due to an adverse event (description of AE not given). Note that data from each treatment group were pooled and therefore it could not be ascertained how many patients receiving HCTZ monotherapy or placebo were withdrawn |
| Selective reporting (reporting bias) | High risk | Baseline age, height and weight were measured but not reported. Only differences in change in mean sitting DBP or SBP (HCTZ ‐ placebo) were shown at endpoint, not the actual mean change for each individual treatment group (see Table 2, p.864). Standard deviation of change in BP not given. Study authors did not comment on mortality. Serious adverse events: 1 patient from the HCTZ 12.5 mg group, 2 from the HCTZ 25 mg group and 1 from placebo group (reasons not given). Adverse events regardless of relationship to study drug were reported for all treatment groups combined only (464/867), not for individual groups; whereas, drug‐related AEs were reported for each treatment group (PLB = 17/93, HCTZ 12.5 = 23/100, HCTZ 25 = 18/100) |
| Industry sponsorship | High risk | Sponsored by Novartis |
Bradley 1993.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 2 to 8 weeks (depending on previous use of anti‐hypertensive medication). Conducted in USA | |
| Participants | DBP 90 to 104 mmHg. Non‐smoking men. Mean age 51 years. Males 100%. Baseline BP was 140/92 mmHg in the treatment group and 145/91 mmHg in the control group | |
| Interventions | Chlorthalidone 45 mg/d (N = 16) or placebo (N = 18) Trial duration = 12 weeks | |
| Outcomes | Change from baseline in sitting SBP and DBP; LDL‐3, LDL‐1, LDL‐2, TG, VLDL, TC, HDL‐C, HDL‐2, HDL‐3, pulse rate, body weight; serum biochemistry (i.e. serum potassium, glucose and creatinine) and insulin. WDAEs | |
| Notes | A sample size calculation was based on change in LDL‐3; refer to Discussion p.639. BP was a secondary outcome measure of the study, and therefore statistical powering was based on LDL 3, the primary endpoint. No SD for BP data. Withdrawals due to adverse events were none. Patients were all non‐smoking, hypertensive males with mild hypertension. This inclusion criteria differ compared to studies with no restrictions on smoking and/or gender. A small sample size of 34 patients. There were no statistically significant differences (P value = NS) across treatment groups in patients' lipid levels, BP, glucose, insulin or body weight at baseline | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...we conducted a randomized, double‐blind, placebo‐controlled, 12 week clinical trial..." (line 7 from bottom of p.636). "Participants were randomly assigned to treatment with 45 mg/day of chlorthalidone or placebo." (line 21 under "Design" p.637). Comment: no further information given on how patients were randomized |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "A double‐blind, placebo‐controlled, parallel group, 12 week trial was conducted." (line 1 under "Design" p.637). "Participants randomized to the chlorthalidone group took three 15 mg tablets once daily while those randomized to placebo took three identical placebo tablets." (line 28 under "Design" p.637) Comment: no further information given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not known whether efficacy analysis was based on an ITT or per‐protocol technique Exclusions: no patients were excluded from study after the wash‐out period prior to randomization Attrition: t here was 1/18 (5.6%) withdrawals from the placebo group due to "symptomatic ulcerative colitis" WDAEs: it is presumed that the 1 patient withdrawal for "symptomatic ulcerative colitis" was considered to be an adverse event, however this is not stated explicitly by the study authors |
| Selective reporting (reporting bias) | High risk | Variability (i.e. SD) of mean change from baseline in BP was not given. Mortality, SAEs and AEs were not reported. Pulse, serum potassium, creatinine, insulin and glucose levels were measured at baseline and endpoint, however the change in mean ± SD at endpoint was not given. Variability for change in weight at endpoint was not given |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Brown 1990.
| Methods | Randomized, double‐blind, placebo‐controlled trial. Wash‐out period = 2 weeks. 2 centers: conducted in UK and France | |
| Participants | Supine DBP 95 to 115 mmHg. Patients 18 to 70 years. Mean age 58 years. Males 47.5%. Baseline supine and erect BP was 184/105 mmHg and 183/108 in the treatment group and 174/103 and 172/106 in the control group | |
| Interventions | Perindopril 4 mg/d (N = 10), perindopril 4 mg/d + HCTZ 25 mg/d (N = 10), HCTZ 25 mg (N = 10) or placebo (N = 10) Treatment duration = 4 weeks DB | |
| Outcomes | Change from baseline in trough mean supine and erect SBP and DBP; ECG, hematology and serum biochemistry; plasma renin and ACE activity, plasma aldosterone; perindopril/perindoprilat assay | |
| Notes | Sample size calculation was not provided. Small sample size of 10 patients per group. The study authors did not state whether there were statistically significant differences across treatment groups in the baseline patient demographics and characteristics. However, the reviewers determined the P value between HCTZ and placebo groups to be statistically significant (P value < 0.05) for supine SBP and DBP and for erect SBP. Baseline patient demographics such as age, sex ratio and body weight were not reported. Only standard error of the mean (SEM) of change in BP was given. Metabolic data were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "All 40 patients selected were then randomised to treatment and received either placebo (N = 10), perindopril 4 mg/day (N = 10), hydrochlorothiazide 25 mg/day (N = 10), or perindopril 4 mg/day and hydrochlorothiazide 25 mg/day (N = 10) for the next 4 weeks." (line 11 under "Study Design" p.328) Comment: no further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "In order to ensure double‐blind conditions, each patient received once daily, two tablets which were identical in appearance." (line 15 under "Study Design" p.328). "The double‐blind code was then broken by a nurse who had no part in the clinical study and those patients taking perindopril (10 taking perindopril alone and 10 taking the combination of perindopril and hydrochlorothiazide) continued to take their treatment daily for a further 3 or 4 days." (line 16 from bottom of p.328) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Not known whether efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol technique Exclusions: no patients were excluded from the study during the single‐blind placebo run‐in period prior to randomization Attrition: no patients withdrew from the study, however, there was one patient (1/10 = 10%) from the placebo group who was excluded from analysis because of "agitation which prevented BP measurements under stable conditions" WDAEs: not stated, but presumed to be none |
| Selective reporting (reporting bias) | High risk | Variability in baseline patient demographics and characteristics was not given. SEM (standard error of the mean) of BP was provided at baseline and SEM of change in BP at endpoint. Weight and heart rate were measured but not reported at the end of the study. Metabolic data were not given. Mortality, SAEs and total AEs were not documented. There was no systematic way of reporting AEs, only case by case reports |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Burris 1990.
| Methods | Randomized, double‐blind, placebo‐controlled trial. Wash‐out period 4 to 6 weeks. Multicenter, conducted in USA | |
| Participants | Supine DBP 95 to 110 mmHg. Patients 18 to 70 years. Mean age 52 years. Males 62%. Baseline supine BP was 151.6/99.4 mmHg. The majority of patients were male (62%) with an average weight of 94 kg, indicating that they were obese | |
| Interventions | Diltiazem SR 60 mg (N = 15), 90 mg (N = 15), 120 mg (N = 15) or 180 mg bid (N = 15), diltiazem 60 mg, 90 mg, 120 mg or 180 mg bid + HCTZ 6.25 mg, 12.5 mg or 25 mg bid (all combined, N = 180), HCTZ 6.25 mg (N = 15), 12.5 mg (N = 15) or 25 mg bid (N = 15), or placebo (N = 15) Treatment duration = 6 weeks | |
| Outcomes | Change from baseline in trough mean supine DBP (primary) and SBP (secondary); heart rate, ECG, serum biochemistry, liver function | |
| Notes | A sample size calculation was provided based on 15 patients per treatment group to detect a difference in DBP reduction of 4 mmHg (SD ± 7.5 mmHg) between treatments (combination therapy versus HCTZ monotherapy) at a power of 94%. The study authors stated that there were no statistically significant differences across treatment groups in the baseline patient demographics and characteristics. SAEs and total AEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This was a randomised, double‐blind, factorial‐design, placebo‐controlled, parallel‐group, multicenter study..." (line 1 under "Methods‐Study Design" p.1508). "...qualifying patients were randomised to a 6‐week double‐blind treatment phase..." (line 19 under "Methods‐Study Design" p.1508) Comment: no further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "This was a randomised, double‐blind, factorial‐design, placebo‐controlled, parallel‐group, multicenter study..." (line 1 under "Methods‐Study Design" p.1508). "...qualifying patients were randomised to a 6‐week double‐blind treatment phase..." (line 19 under "Methods‐Study Design" p.1508) Comment: no further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The efficacy analysis was based on an intention‐to‐treat technique Exclusions: 127/424 (30%) of patients were excluded from the study during the single‐blind placebo run‐in period prior to randomization Attrition: 36/297 (12%) of patients withdrew from the study for the following reasons: 10 patients for "intolerable side effects from medication", 7 for "inadequate BP control", 19 for "administrative reasons". Data were pooled, therefore it could not be determined from which treatment groups the patients originated WDAEs: 2% of patients from all HCTZ groups (6.25, 12.5 and 25 mg) combined. Reasons were not specified |
| Selective reporting (reporting bias) | High risk | SBP, heart rate, ECG, body weight, hematology, liver function and serum biochemical values (except for serum glucose and cholesterol levels) were measured, but not reported at endpoint of the study. Variability in DBP was not given. Effects of HCTZ 6.25, 12.5 and 25 mg on DBP at time points between week 0 and 6 were not shown. Mortality: none; SAEs: not given; and total AEs were not reported only those related to treatment. Reporting of AEs was incomplete |
| Industry sponsorship | High risk | The study was supported by grants and statistical services provided by Marion Laboratories |
Canter 1994.
| Methods | Randomized, double‐blind, placebo‐controlled trial. Wash‐out period = 2 to 4 weeks. Multicenter, conducted in USA and Europe | |
| Participants | Sitting DBP 100 to 115 mmHg. Patients ≥ 18 years. Mean age 53 years. Males 63%. Baseline sitting BP was 162/105 mmHg | |
| Interventions | Quinapril 2.5 mg, 10 mg or 40 mg/d (all combined, N = 86), quinapril 2.4 mg, 10 mg or 40 mg/d + HCTZ 6.25 mg, 12.5 mg or 25 mg/d (all combined, N = 259), HCTZ 6.25 mg, 12.5 mg or 25 mg/d (all combined, N = 88) or placebo (N = 27) Treatment duration = 8 weeks | |
| Outcomes | Change from baseline in trough mean sitting DBP (primary) and SBP (secondary); response rate; serum potassium | |
| Notes | A sample size calculation was provided based on 25 patients per treatment group to detect a difference in mean DBP of 3.5 mmHg (SD ± 7 mmHg) between treatments (combination therapy versus HCTZ or quinapril monotherapy) at a power of 80%. "No clinically important differences between treatment groups were found for any of the baseline demographic parameters." (line 3 under "Results‐Patient characteristics and disposition" p.158). Patient demographics/characteristics were given for all randomized patients combined (not separately for each treatment arm). Therefore statistical differences could not be determined. Standard deviations for BP change was not reported There were no mortalities during the study. SAEs defined as "hospitalizations" occurred in 1 patient from the HCTZ 25 mg group only (due to prostate resection) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "..qualifying patients were randomised to an eight week double‐blind phase with one of 16 parallel treatments: placebo, one of three doses of quinapril (2.5, 10 or 40 mg once daily), one of three doses of HCTZ (6.25, 12.5 or 25 mg once daily) or one of nine possible corresponding combinations of quinapril and HCTZ doses." (line 11 under "Patients and methods‐Patient selection and study design" p.156) Comment: no further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "..qualifying patients were randomised to an eight week double‐blind phase with one of 16 parallel treatments..." (line 11 under "Patients and methods‐Patient selection and study design" p.156). "All patients received fixed doses throughout the double‐blind phase..." (line 17 under "Patients and methods‐Patient selection and study design" p.156). No further information on blinding was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The analysis in the study was based on an ITT technique with the LOCF (last observation carried forward) Exclusions: number of patients excluded from study during the single‐blind placebo run‐in period before randomization was not reported Attrition: 41/460 (9%) patients withdrew from the study for the following reasons: 14 ‐ adverse events, 15 ‐ lack of efficacy and 12 ‐ non‐compliance and surgery. The number of patients who withdrew from each treatment group was not given WDAEs: 14/460 (3%); specific reasons were not given |
| Selective reporting (reporting bias) | High risk | Standard deviations (SD) were not reported for baseline patient demographics/characteristics, BP or serum potassium levels. Heart rate and laboratory parameters were measured but not reported. Total AEs were not reported on. AEs associated with treatment in all HCTZ groups combined = 15% of patients, and in the placebo group = 19%. The nature of each AE was included in a table (see table 2 p.160) if it occurred in at least 6 or more patients |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Capone 1983.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 6 weeks. Multicenter, conducted in USA | |
| Participants | DBP 95 to 114 mmHg. Mean age 52 years. Males 66%. Baseline BP was 152/102.8 mmHg in the treatment group and 153/103.8 in the control group | |
| Interventions | Indapamide 1.0 mg (N = 24), 2.5 mg (N = 23) or 5.0 mg/d (N = 23) or placebo (N = 22) Trial duration = 8 weeks (+ 2 weeks of single‐blind follow‐up) | |
| Outcomes | Standing and supine SBP and DBP (taken at 2‐week intervals); response rate; pulse rate, ECG, body weight, serum biochemistry and urinalysis | |
| Notes | Primary efficacy analysis not stated by study authors. A sample size calculation was not provided. Baseline patient demographics, measurements and other variables were similar across all treatment groups (P value = NS). BP data in graph form only (see Fig. 2 and 4, p.310‐311) with no SD. No SD for metabolic data and number of subjects not provided. Mortalities, SAEs and WDAEs were not explicitly reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The study was conducted according to a randomised, double‐blind, parallel design with four groups receiving once‐daily dosages of placebo or 1mg, 2.5 mg, or 5 mg indapamide. Equal numbers of patients were randomly assigned to each of the four treatment groups." (line 1 under "Methods" p.307). Technique for sequence generation not stated explicitly by study authors |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "The study was....double‐blind..." (line 1 under "Methods" p.307). No further information on blinding was given |
| Incomplete outcome data (attrition bias) All outcomes | High risk | It is not known whether study was based on an ITT or per‐protocol technique
Exclusions: number of patients excluded from study after the single‐blind placebo run‐in period and before randomization was not reported
Attrition: there were 4/92 (4.3%) patient withdrawals for dizziness (IND 2.5), pancreatitis (IND 1.0), lack of therapeutic response (PLB), and dizziness + lack of response (PLB). In addition, 5/92 (5.4%) patients were excluded from the evaluation of the efficacy of indapamide due to various reasons including poor compliance and headaches (IND 1.0 mg group), increased hypertension (PLB), a stomach lesion (IND 5.0), concomitant use of phenytoin (IND 2.5 mg) and rash (IND 5.0 mg), however, it is not known whether any of these 5 patients were amongst the 4 patients who eventually withdrew WDAEs were not stated explicitly by study authors (but based on information in the results section of the study it was presumed to be: 2 patients from the IND 1.0 mg group, 1 from IND 2.5 mg group, 2 from IND 5.0 and 2 from the placebo group) |
| Selective reporting (reporting bias) | High risk | Baseline patient demographics and characteristics were not reported. Weight and pulse were measured but not reported. BP data were graphed in figures only. No standard deviations (SD) for BP or metabolic data were given. Study authors did not comment on all‐cause mortality, and SAEs and WDAEs were not clearly stated. AEs, regardless of relationship to study drug were not reported. Only drug‐related AEs were reported; for each treatment group there were: PLB = 1/22 (4.5%), IND 1.0 = 4/24 (17%), IND 2.5 = 6/23 (26%) and IND 5.0 = 6/23 (26%) |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Carlsen 1990.
| Methods | Randomized, double‐blind, placebo‐controlled dose‐ranging trial (parallel arms). Wash‐out period = 6 weeks. Conducted in New Zealand and Denmark | |
| Participants | DBP 100 to 120 mmHg. Mean age 57.4 years. Males 40%. Baseline BP was 165.2/104 mmHg in the treatment group and 161.9/101.8 in the control group | |
| Interventions | Bendrofluazide 1.25 mg (N = 50), 2.5 mg (N = 52), 5 (N = 52) or 10 mg/d (N = 51), or placebo (N = 52) Treatment duration = 12 weeks | |
| Outcomes | Sitting SBP and DBP (taken at 4, 10 and 12 weeks); BP response rate; heart rate; serum biochemistry | |
| Notes | A sample size calculation was provided based on 50 patients per treatment group to detect a difference in BP of 5 mmHg (SD ± 7 mmHg) between treatments (power level not given). Patients who took less than 80% of their tablets (placebo) during the 6‐week placebo run‐in period were excluded from entering the study. After taking placebo for 6 weeks only patients with DBP between 100 and 120 mmHg were included in the study. The 10 mg bendrofluazide dose was chosen based only on 1 prior trial by the Medical Research Council. The study's authors stated that "there were no large differences" between treatment groups in patients' baseline demographics and characteristics (Table 1 p.976) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "[Patients] were randomly allocated in blocks of 10 on a double‐blind basis to receive placebo or bendrofluazide at a dose of 1.25, 2.5, 5, or 10 mg a day." (line 1 under "Study Design" p.975). No further detail provided |
| Allocation concealment (selection bias) | Unclear risk | Not stated explicitly by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "[Patients] were randomly allocated in blocks of 10 on a double‐blind basis..." (line 1 under "Study Design" p.975). "Placebo and active tablets were identical in appearance and taste. All patients received four tablets daily, two in the morning and two at lunch. Those receiving fewer than four active tablets daily were given the active tablets in the morning." (line 15 from bottom under "Study Design" p.975) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not known whether efficacy analysis was based on an ITT or per‐protocol technique
Exclusions: number of patients excluded from study after the placebo run‐in period and before randomization was not reported Attrition: total withdrawals and reasons for withdrawing were not given WDAEs: 9/257 (3.5%) of patients withdrew due to adverse events (2 in placebo, 1.25, 2.5 and 10 mg group, and 1 in 5 mg group), however the precise reasons were not given |
| Selective reporting (reporting bias) | High risk | Sodium levels and pulse were measured both at beginning and end of study but not reported on. Variability in baseline patient demographics and characteristics was not given. Change in weight from baseline was not reported. Mortalities and SAEs were not documented. Total AEs: BDFZ 1.25 (8 patients), 2.5 (14 patients), 5 (12 patients), 10 mg/d (24 patients) and placebo (9 patients) |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Chrysant 1994.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Multicenter, conducted in USA | |
| Participants | DBP 100 to 114 mmHg. Mean age 53.5 years. Males 58.2%. Baseline BP was 155/103 mmHg in both the treatment groups as well as the control group | |
| Interventions | Lisinopril 10 mg/d (N = 85), lisinopril 10 mg/d + HCTZ 12.5 mg or 25 mg/d (all combined, N = 168), HCTZ 12.5 mg (N = 87), or 25 mg/d (N = 84), or placebo (N = 81) Trial duration = 8 weeks | |
| Outcomes | Change from the baseline in trough mean sitting and standing SBP and DBP; ECG, heart rate, serum biochemistry, hematology and urinalysis | |
| Notes | Sample size calculation was provided ‐ study had a power of 95% to detect a change in sitting DBP of 5 mmHg between treatment groups. The study authors stated that "there were no significant differences between the various treatment groups" in baseline patient demographics and characteristics (i.e. age, body weight, BP and heart rate). BP data in graphical form only. Mean change from the placebo group with 95% CIs available. WDAEs, mortalities, SAEs and total AEs not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...patients...were randomised into the double‐blind phase of the study..." (line 21 under "Patients and Methods" p.738). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "This was a multicenter, double‐blind, parallel, placebo‐controlled study." (line 1 under "Patients and Methods" p.738). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Efficacy analysis was based on an ITT technique Exclusions: 244/749 (33%) patients were excluded from study after the 4‐week single‐blind, placebo run‐in period prior to randomization for "having BP lower than those required for inclusion." Attrition: 38/505 (7.5%) patients withdrew from the study, 8 from the HCTZ 12.5 mg group, 7 from the HCTZ 25 mg group and 10 from the placebo group. The reasons for withdrawing were not given WDAEs: not given |
| Selective reporting (reporting bias) | High risk | Gender, medical history and variability (i.e. SD) in baseline patient demographics and characteristics were not given. BP data were graphed only and variability was expressed as SEM (standard error of the mean), not SD. Blood cell counts and ECG were measured but not reported; weight and heart rate were measured and reported at baseline, but not at endpoint. Mortalities, SAEs and total AEs were not reported. Only 5 of the most commonly reported adverse of events were given. Results from a subgroup analysis of BP‐lowering effects in black and elderly patients were shown but the analysis was not mentioned a priori (i.e. was not included in the methods section of the study); refer to Table 3 p.740 |
| Industry sponsorship | High risk | Supported by a grant from ICI Pharmaceuticals Group |
Chrysant 1996.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 1 to 4 weeks. Multicenter, conducted in USA | |
| Participants | Sitting DBP 95 to 114 mmHg. Mean age: HCTZ 25 mg = 53.2; placebo = 53.8 years. Males: HCTZ 25 mg = 53% and placebo = 55% | |
| Interventions | Benazepril 20 mg/d (N = 42), benazepril 5 mg, 10 mg or 20 mg/d + HCTZ 6.25 mg, 12.5 mg or 25 mg/d (all combined, N = 207), HCTZ 25 mg/d (N = 45) or placebo (N = 40) Treatment duration = 6 weeks | |
| Outcomes | Change from the baseline in trough mean sitting DBP and SBP; pulse rate, serum potassium, body weight | |
| Notes | Study was adequately powered (73% and 87% power to detect a 5 and 6 mmHg difference in BP, respectively between any pair of treatments). The authors of the study did not state whether there were statistically significant differences across treatment groups in baseline demographics and characteristics. SD for BP data were not given (SEM only). Baseline BP was not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "...patients were assigned to receive placebo or one of seven active treatment regimens according to a computer‐generated randomisation table." (line 4 under "Design" p.18) |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "The study was a 6‐week, randomised, double‐blind, placebo‐controlled, parallel‐group multicenter trial." (line 1 under "Design" p.18). "Study medication was provided as tablets of identical appearance and capsules of identical appearance..." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Efficacy analysis was based on an ITT technique Exclusions: 73/407 (18%) of patients were excluded from the study during the single‐blind, placebo run‐in period prior to randomization, the majority of whom failed "to meet the Sitting DBP entry criterion." Attrition: 33/334 (10%) of patients withdrew from the study for the following reasons: 12 ‐ lack of efficacy, 11 ‐ adverse events, 5 ‐ lost to follow‐up, 3 ‐ consent withdrawal and 2 ‐ protocol violations WDAEs: 11/334 (3.3%) of patients (across all 8 treatment arms) withdrew due to adverse events (termed "adverse experiences"). This included 1 patient (1/45) receiving HCTZ 25 mg (1/45 = 2.2%) and 2 patients in the placebo group (2/40 = 5%); however the precise reasons were not given. Note: Data on total withdrawals were pooled (not presented as separate treatment arms) |
| Selective reporting (reporting bias) | High risk | Weight, heart rate, hematology and serum chemistry (except for potassium levels), urinalysis and ECG were measured, but not reported. BP data were provided as graphs only; variability in BP was expressed as SEM (standard error of the mean); baseline BP was not given. Variability in baseline patient demographics and characteristics was not given. Medical history of patients at baseline was not reported. SAEs: 4/334 (1.2%) across all treatment groups; authors did not account for all 4 patients, e.g. reasons were given for 2 patients only (hypotension and syncope). Note that data for SAEs were combined, not presented separately across all treatment groups. Total AEs were not reported; only AEs possibly or probably related to study drug were. Except for potassium levels, the following were measured but not reported on: weight, serum hematology, glucose, lipids, electrolytes, ECG and urinalysis. Mortalities were not mentioned. A post hoc subgroup analysis of black versus non‐black patients and patients < 65 and ≥ 65 years of age was performed and the results presented in 2 graphs; yet there was no mention of it in the methods section of the study |
| Industry sponsorship | Unclear risk | Sponsor was not reported |
Chrysant 2004.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). 3 x 4 factorial design. Wash‐out period = 4 weeks. Multicenter, conducted in USA | |
| Participants | Sitting DBP ≥ 100 and ≤ 115 mmHg. Mean age: HCTZ 12.5 mg = 54.1; HCTZ 25 mg = 54.7; placebo = 54.0 years. Males: HCTZ 12.5 mg = 55.6%; HCTZ 25 mg = 49%; placebo = 64.3%. Baseline mean sitting SBP/DBP ranged from 151.9 to 156.6 mmHg/102.6 to 104.4 mmHg | |
| Interventions | Olmesartan medoxomil 10 mg (N = 39), 20 mg (N = 41) or 40 mg/d (N = 45), olmesartan medoxomil 10 mg, 20 mg or 40 mg/d + HCTZ 12.5 mg or 25 mg/d (all combined, N = 247), HCTZ 12.5 mg (N = 45) or 25 mg/d (N = 43), or placebo (N = 42) Trial duration = 8 weeks | |
| Outcomes | Change from baseline in trough mean sitting and standing DBP and SBP; BP response rate; heart rate, serum biochemistry, hematology and urinalysis | |
| Notes | Sample size calculation was based on change (magnitude not given) from baseline in sitting DBP at week 8 (90% power). SD for BP data were not given. Biochemical data not given. Mortalities and total AEs were not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This was a randomised, double‐blind, factorial design study..." (line 1 under "Methods‐Study Population" p.253) "...eligible patients (N = 502) were randomised to one of 12 treatment groups..." (line 2 under "Methods‐Study Design" p.253). No further information given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "...eligible patients (N = 502) were randomised to one of 12 treatment groups for 8 weeks of double‐blind treatment with placebo, olmesartan medoxomil monotherapy, HCTZ monotherapy, or olmesartan medoxomil/HCTZ combination therapy..." (line 2 under "Methods‐Study Design" p.253). No further information given |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Efficacy analysis was based on an ITT technique with the LOCF (last observation carried forward) Exclusions: 863 patients were screened and 750 of these were enrolled. 248/750 (33%) of patients were excluded from the study during the single‐blind, placebo run‐in period prior to randomization. The reasons for these exclusions were not given Attrition: 51/502 (10%) patients withdrew from the study; the reasons for withdrawing were not given. Data were combined, not presented separately for each treatment group WDAEs: the study reported WDAEs only for those patients receiving one or both of the study drugs (not placebo), which was about 2%. The precise reasons for these withdrawals and from which treatment group the patients originated were not given |
| Selective reporting (reporting bias) | Unclear risk | Baseline and endpoint body weight, heart rate, height, hematology, serum chemistry and urinalysis were measured but actual values were not reported; any changes at endpoint were noted qualitatively. Variability in BP data was not given. Variability in baseline patient demographics and characteristics was also not given. Baseline standing BP was measured, but not reported. Medical history was measured but not reported with the baseline patient demographics and characteristics. Mortalities were not mentioned SAEs: 1 patient in the placebo group with "unstable angina"; no other SAEs were reported. Total AEs were not reported; only the most commonly reported AEs were mentioned |
| Industry sponsorship | High risk | Supported by a grant from Sankyo Pharma Inc. |
Curry 1986.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period ≤ 4 weeks. Multicenter, conducted in USA | |
| Participants | Sitting DBP 90 to 110 mmHg. Age range: 30 to 71 years. Males 43%. 59% Black race. Baseline BP 146 to 154/97 in the 3 treatment groups and 151/99 in the control group | |
| Interventions | Metolazone 0.5 mg (N = 26), 1 mg (N = 25), 2 mg/d (N = 27) or placebo (N = 27) Treatment duration = 6 weeks | |
| Outcomes | Sitting and standing SBP and DBP (at 2‐week intervals); response rate; ECG, hematology, serum biochemistry and urinalysis | |
| Notes | A sample size calculation was not provided. Anomalously high DBP response (‐31 mmHg) from 1 patient in the placebo group increased the mean and variability in that group substantially enough to lessen the magnitude of statistical difference compared to active treatment (analysis was done including this outlier patient). There were no significant differences between treatment groups in baseline demographics. Medical history was not included in baseline characteristics. All AE data grouped together. Detailed reporting of biochemical data restricted to potassium levels only. Mortalities, SAEs and total AEs were not reported. Publication consisted of 2 studies (the positive‐controlled study did not include a placebo arm, therefore this study will not be discussed further in this review) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "In the placebo‐controlled study, patients were randomly assigned to receive single daily doses of placebo, or 0.5, 1.0, or 2.0 mg of...metolazone." (line 3 under "Drugs" p.49). Technique for sequence generation not stated explicitly by study authors |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "...a double‐blind, parallel clinical [study]" (line 17 from top under "Introduction" p.48). No further information given of how the study was blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not known whether efficacy analysis was based on an ITT or per‐protocol technique
Exclusions: 43/148 (29%) of patients were excluded from the study during the placebo run‐in phase prior to randomization Attrition: 7/105 (6.7%) of patients withdrew from the study. Reasons for withdrawals included: 1 ‐ voluntary withdrawal, 4 ‐ increased blood pressure and 2 ‐ missed visit WDAEs: 0/105 (0%) of patients withdrew due to adverse events (termed "adverse experiences"), however the precise reasons were not given. Note: data from each treatment group were pooled (not presented separately) |
| Selective reporting (reporting bias) | High risk | Except for potassium levels, the following were measured but not reported on: serum hematology, glucose, lipids, electrolytes, ECG and urinalysis. Mean and SD for baseline patient demographics and characteristics were not given. Mortalities, SAEs and total AEs were not documented (see line 1 under "Adverse Experiences" p.55) |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Drayer 1995.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Multicenter, conducted in USA | |
| Participants | Sitting DBP 95 to 114 mmHg. Mean age 56 years. Males 62%. Baseline DBP was 99.9 mmHg in the HCTZ 12.5 mg group and 100.2 mmHg in the placebo group. Pulse pressure was not reported | |
| Interventions | Moexipril 3.75 mg (N = 49), 7.5 mg (N = 42), 15 mg (N = 47) or 30 mg/d (N = 45), moexipril 3.75 mg, 7.5 mg or 15 mg/d + HCTZ 12.5 mg/d (all combined, N = 137), HCTZ 12.5 mg/d (N = 48), or placebo (N = 45) Trial duration = 8 weeks | |
| Outcomes | Change from baseline in mean sitting DBP and SBP after 8 weeks; serum biochemistry, plasma renin activity and aldosterone levels | |
| Notes | Sample size calculation was not provided. Study authors did not state whether there were statistically significant differences across treatment groups in baseline patient demographics and characteristics. BP data in graph form only | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This was a randomised, double‐blind, multicenter, placebo‐controlled, parallel group study to evaluate the efficacy and safety of moexipril, alone or in combination with a low‐dose diuretic." (line 1 under "Methods" p.526) "The second [phase] was a double‐blind treatment period during which patients were randomly allocated to one of nine separate study groups." (line 5 under "Protocol" p.526). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "This was a randomised, double‐blind, multicenter, placebo‐controlled, parallel group study to evaluate the efficacy and safety of moexipril, alone or in combination with a low‐dose diuretic." (line 1 under "Methods" p.526). "The second [phase] was a double‐blind treatment period during which patients were randomly allocated to one of nine separate study groups." (line 5 under "Protocol" p.526) Comment: no further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The primary efficacy analysis was presumed to be based on an ITT technique (from information given in the Results section of the study, line 3 from bottom of p.527) Exclusions: 90/503 (18%) of patients were excluded from the study during the single‐blind placebo run‐in period prior to randomization Attrition: 22/413 (5%) of patients withdrew from the study: 14 due to "adverse experiences", 2 due to "lack of therapeutic response" and the 6 other patients for "other reasons" WDAEs: 3/48 (6%) and 2/45 (4.4%) of patients from the HCTZ and placebo groups, respectively, withdrew due to adverse events. The specific reasons were not given |
| Selective reporting (reporting bias) | High risk | Heart rate and results for serum biochemistry, except for potassium levels, were not given (i.e. actual values were not shown at baseline or endpoint). Variability in baseline patient demographics and characteristics was not shown; medical history was also not included. BP data (mean change ± SEM) were presented in graph form only. Mortalities and SAEs were not stated explicitly. Total AEs were reported (HCTZ = 46%, placebo = 47%); a detailed listing of only the "principal adverse experiences" was provided |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Fernandez 1994.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out = 4 weeks. Multicenter, conducted in Mexico | |
| Participants | DBP 95 to 110 mmHg. Mean age 54.7 years. All patients were of "mestizo" race. Males 34.3%. Baseline BP was 149.9/100.6 mmHg in the treatment group and 146.6/100.3 mmHg in the control group | |
| Interventions | Fosinopril 20 mg/d (N = 16), fosinopril 20 mg/d + HCTZ 12.5 mg/d (N = 17), HCTZ 12.5 mg/d (N = 17) or placebo (N = 17). Treatment duration = 8 weeks | |
| Outcomes | Change from the baseline in trough mean sitting SBP and DBP at 0, 2, 4, 6 and 8 weeks; BP response rate; ECG, hematology, urinalysis and serum biochemistry | |
| Notes | A sample size calculation was not provided. Study authors stated that there were no significant differences between treatment groups in baseline general demographics, BP and heart rate. Gender was not balanced between HCTZ and placebo groups. Biochemical data were measured, but values were not shown. Mortalities and SAEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomised in a double‐blind, placebo‐controlled fashion in four parallel groups..." (line 12 from bottom p.I‐207) Comment: no further information given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | High risk | "Those continuing to period B received randomised, double‐blind, placebo‐controlled treatment for 8 weeks." (line 3 from top under "Methods" p.I‐208). No further information given on how study was blinded, however, it is suspected that blinding was broken for 2 reasons as stated by the authors: 1) "Fosinopril plus hydrochlorothiazide combination therapy consisted of individual distinct tablets of each drug," (line 7 from bottom p.I‐207) and 2) "The double‐blind procedure was not tested at the end of the study." (line 4 from bottom under "Methods" p.I‐208) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Primary efficacy analysis was based on an ITT technique Exclusions: number of patients excluded from the study during the single‐blind placebo run‐in period was not reported Attrition: 3/67 (4.5%) of patients withdrew from the study: 1 receiving fosinopril (an ACE inhibitor) for an acute gout attack, 1 receiving placebo for persisting dizziness and headache, 1 receiving placebo for unknown reasons WDAEs: information not given (it is presumed that the dizziness and headache experienced by the patient on placebo could be counted as an AE). Note: fosinopril, an ACE inhibitor, is outside the scope of this review and will not be discussed further |
| Selective reporting (reporting bias) | Unclear risk | Heart rate, body mass index, hematology, serum chemistry (including lipids, glucose, electrolytes, metabolites) and urinalysis were measured both at baseline and at end of study but not reported on accordingly. Mortalities were not stated explicitly. SAEs: none. Total AEs: 8/17 (47%) of patients on HCTZ and 5/17 (29%) on placebo |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Ferrara 1984.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 2 weeks. Conducted in Italy | |
| Participants | Mild to moderate hypertension. Values not mentioned. Mean age 45.5 years. % of males not given. Mean baseline SBP/DBP was 161/107 mmHg in the treatment group and 151/104 in the placebo group. Pulse pressure 54/47 respectively | |
| Interventions | Slow‐release nifedipine 20 mg/d, chlorthalidone 25 mg/d or placebo (all combined, N = 30 patients randomized) Trial duration = 8 weeks | |
| Outcomes | Standing and supine SBP and DBP; heart rate; left ventricular mass and function; systolic time intervals | |
| Notes | A sample size calculation was not provided. Study authors did not state whether there were statistically significant differences between treatment groups in baseline patient demographics and characteristics. The majority of patients were overweight (average BMI = 27) at baseline. A separate group of 10 patients (group A) who did not enter a wash‐out period at the beginning of the study due to adequate BP control on other antihypertensives were not included in this review. Total withdrawals (TW), WDAEs, mortalities, SAEs and total AEs were not reported. Biochemical data not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...patients...were randomly allocated to chlorthalidone 25 mg/day (Group B), slow release nifedipine 20 mg/day (Group C) or placebo (Group D)." (line 9 under "Summary" p.525). No further information was given for how patients were randomized to treatment |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "The study was performed according to a double blind model since echocardiograms were numerically coded and read by two blinded observers." (line 3 from bottom of p.526). As well as the statement by the study authors above, the title of study included the word "double‐blind"; no further information was given, however |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Whether primary efficacy analysis was based on an ITT or per‐protocol technique was not reported. Exclusions: the number of patients excluded from the study during the run‐in period prior to randomization was not given Attrition: total number of withdrawals and their reasons were not given WDAEs: information not given |
| Selective reporting (reporting bias) | High risk | Gender, weight and medical history of patients were not given at baseline. Biochemical data including hematology, serum chemistry, urinalysis and ECG were not reported. For BP data, statistical significance was calculated within groups but not between groups. Number of patients randomized to each treatment arm was not reported and it is not known how many were included in the efficacy and safety analysis. Mortalities, SAEs and total AEs were not reported |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Fiddes 1997.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel groups). Wash‐out period = 4 weeks. Multicenter, conducted in USA | |
| Participants | DBP 95 to 114 mmHg. Mean age 69.7 years. Males 55%. Baseline BP was 159.3/98.8 mmHg in the treatment group and 160.3/99.8 in the control group | |
| Interventions | Indapamide 1.25 mg/d (N = 103) or placebo (N = 101) Treatment duration = 8 weeks | |
| Outcomes | Mean change from the baseline in standing and supine DBP and SBP (at 2‐week intervals); response rate, heart rate, body weight, ECG, serum biochemistry, hematology and urinalysis | |
| Notes | Sample size calculation was not provided. The study authors did not state whether there were statistically significant differences between indapamide and placebo treatment groups in baseline patient demographics and characteristics. The minimum age of 65 years for patients to be included in the study was much older than most other studies assessed in this review. This elderly population may present with a different profile of underlying diseases not seen in young adult patients of 18 years of age or older. Patients with low serum potassium during the trial were given potassium supplements. For BP and biochemical data the number of subjects changes over the duration of treatment | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This was a multicentre, randomised study..." (line 1 under "Design" p.240) "[Patients] were randomised to receive indapamide 1.25 mg or placebo..." (line 5 from top of p.240). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | High risk | "This was a multicentre, randomised study consisting of two periods: a single‐blind placebo washout period and a double‐blind treatment period." (line 1 under "Design" p.240) "Eligible patients were...entered into an 8‐week double‐blind treatment period." (line 4 from top of p.240). The investigator at his/her discretion gave hypokalemic patients potassium supplementation which could have broken the blinding No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Efficacy and safety analysis was based on an "all‐treated patient population" (line 1 under "Statistical Analysis" p.240) Exclusions: the number of patients excluded from the study during the placebo run‐in period prior to randomization was not given Attrition: 11/103 (11%) and 16/101 (16%) of patients from the indapamide and placebo groups, respectively, withdrew from the study. Reasons for withdrawals included (IND versus PLB): 4 versus 6 patients ‐ adverse events, 1 versus 6 ‐ ineffective therapy, 1 versus 2 ‐ protocol deviations, 1 versus 1 ‐ withdrawn consent, 2 versus 0 ‐ lost to follow‐up and 2 versus 1 ‐ other reasons WDAEs: 5/103 (5%) patients receiving indapamide and 6/101 (6%) receiving placebo withdrew due to adverse events; precise reasons were not given. Note: 11 WDAEs is contrary to the number (n = 10) the study accounted for on line 9 under "Results" p.241 |
| Selective reporting (reporting bias) | High risk | Variability was not included in baseline patient demographics and characteristics or in baseline mean BP readings. Body weight was reported at baseline, but not at endpoint. Heart rate and ECG were measured but not reported. Reporting of biochemical data was based on a selection of measured parameters, therefore some data like hematology and urinalysis were missing. Baseline standing BP was measured but not reported and the variability in mean change in standing BP at endpoint was not reported. Medical history of patients was not given. Mortalities: 1 patient in the indapamide group died from arteriosclerosis and 1 patient in the placebo group died from a myocardial infarction). SAEs: 2/103 (1.9%) patients on indapamide and 5/101 (5%) patients on placebo. Total AEs: 32/103 (31%) patients on indapamide and 38/101 (38%) patients on placebo. A list of AEs (with description) occurring in at least 2% of patients was provided |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Frei 1994.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out (placebo run‐in) period = 4 weeks. Multicenter, Germany | |
| Participants | DBP 95 to 114 mmHg and SBP ≤ 240 mmHg. Mean age: 55.1 years. Males: 50%. Baseline BP was 166/103 mmHg in the HCTZ 25 mg treatment group and 166/102 in the placebo group. Pulse pressure was not reported | |
| Interventions | Moxonidine 0.4 mg/d (N = 38), moxonidine 0.4 mg/d + hydrochlorothiazide 25 mg/d (N = 42), hydrochlorothiazide (HCTZ) 25 mg/d (N = 40) or placebo (N = 41) Treatment duration = 8 weeks | |
| Outcomes | Mean change from the baseline in sitting DBP and SBP; responder rates; blood and urine lab tests | |
| Notes | Sample size calculation was not provided. Patients were permitted to take medication for diseases unrelated to hypertension which is contrary to what most other studies investigating the BP‐lowering effects of thiazides allow. Most other studies either restrict or ban and monitor any use of other medications during the study. Details were not provided. Patients with more serious SBP levels as high as 240 mmHg were permitted to enter the study. The study authors did not state whether there were statistically significant differences between HCTZ and placebo treatment groups in baseline patient demographics and characteristics. Biochemical data measured but not reported. Total AEs not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The study was designed as a multicenter, double‐blind, placebo‐controlled, parallel‐group, prospectively randomised study..." (line 23 under "Subjects and Methods" p.S26). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "The study was designed as a multicenter, double‐blind, placebo‐controlled, parallel‐group, prospectively randomised study..." (line 23 under "Subjects and Methods" p.S26) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Efficacy analysis was based on an intention‐to‐treat (ITT) technique Exclusions: 14/177 (8%) of patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: 3 patients (3/163 = 1.8%) were not included in the efficacy analysis of the ITT population: 2 patients did not take study medications following randomization and 1 patient was without a post‐baseline measurement. It is not known from which treatment groups these patients came WDAEs: 4/161 (2.5%) of patients withdrew due to adverse events, 1 patient from the placebo group for headache and the remaining 3 from other treatment groups other than the HCTZ 25 mg group (note: safety data were based on an ITT group of 161 patients) |
| Selective reporting (reporting bias) | High risk | Baseline patient demographics and characteristics did not include weight or vital signs and variability was omitted. Biochemical data were measured but not reported. Mortalities and SAEs were not explicitly stated by study authors. Total AEs were not reported, only the most commonly occurring AEs were included (but no systematic approach to reporting them was taken) |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Frishman 1994.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel groups). 3 x 4 factorial design. Wash‐out period = 4 to 6 weeks. Multicenter, USA | |
| Participants | Sitting DBP 95 to 115mmHg. Mean age: 53 years. Males: 71%. Race: 71% non‐black. Baseline sitting BP was 151/101 mmHg. Sitting heart rate = 76 bpm | |
| Interventions | Bisoprolol 2.5 mg, 10 mg or 40 mg/d (all combined, N = 197), bisoprolol 2.5 mg, 10 mg or 40 mg/d + HCTZ 6.25 mg or 25 mg/d (all combined, N = 190), HCTZ 6.25 mg (N = 29) or 25 mg/d (N = 33), or placebo (N = 63) Duration = 12 weeks + 2‐week taper period | |
| Outcomes | Mean change from the baseline in trough sitting DBP and SBP at 3 to 4 weeks; analyses of the effects of age, sex, race and baseline DBP on change in BP; serum biochemistry | |
| Notes | A sample size calculation was provided based on 60 patients in the bisoprolol monotherapy group and 30 patients in all others groups to detect a difference of 3.6 to 5.1 mmHg and 5.1 to 7.3 mmHg, respectively, with 80% power. The study authors stated that there were no significant differences across treatment groups in the baseline patient characteristics. SD for BP data not given. BP data at endpoint (i.e. 12 weeks) not reported; midpoint only | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The study reported on here was 12‐week, randomised, double‐blind, placebo‐controlled, multicenter 3x4 factorial trial..." (line 1 under "Methods‐Study Design" p.1462). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Patients whose mean sitting diastolic blood pressure was stable and between 95 and 115 mmHg (inclusive) qualified for randomisation to one of 12 double‐blind treatment groups. To maintain blinding, matching placebo tablets were provided for both bisoprolol and hydrochlorothiazide." (line 27 under "Methods‐Study Design" p.1462) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | It is not known whether efficacy analysis was based on an intention‐to‐treat or per‐protocol technique Exclusions: 208/720 (29%) patients were excluded from the study during the single‐blind placebo run‐in period prior to randomization. The specific reasons for these exclusions were not given Attrition: 109/512 (21%) patients withdrew from the study; the specific reasons were not given WDAEs: 41/512 (8%) patients withdrew due to "a laboratory abnormality or adverse experience". This included 9 patients (or 14%) from the placebo group and a range of between 2% and 10% in each of the HCTZ 6.25 mg and HCTZ 25 mg treatment groups (actual % was not given). The specific reasons were not given. Note that the data above were pooled, not presented as separate for each treatment group |
| Selective reporting (reporting bias) | High risk | Baseline BP in each of the treatment groups was not reported (data were combined). Although the duration of the study was 12 weeks, the primary efficacy outcome (change from baseline in sitting DBP) was reported on at weeks 3 to 4 only. Reductions in DBP between 3 to 4 and 12 weeks were mentioned as being "similar" (no further information was given). All‐cause mortality and SAEs were not clearly documented. Total AEs were not reported; only AEs occurring in at least 2% of patients and either dose‐related or somehow related to active treatment were listed. Weight, heart rate, ECG, serum chemistry including glucose, lipids and calcium were measured but actual values were not reported on at endpoint |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Frishman 1995.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 to 6 weeks. Multicenter, conducted in USA | |
| Participants | Sitting DBP 95 to 115 mmHg. Mean age: not given. % patients < 60 years: HCTZ 25 mg group = 61% versus placebo = 69%. Males: HCTZ 25 mg = 62% versus placebo = 64%. Non‐black race: HCTZ 25 mg = 84% versus placebo = 80%. Baseline sitting BP was 151/101 mmHg in HCTZ 25 mg group and 152/100 in placebo group. Sitting heart rate = 76 versus 75 |
|
| Interventions | Bisoprolol 5 mg/d (N = 158), bisoprolol 5 mg/d + HCTZ 6.25 mg/d (N = 160), HCTZ 25 mg/d (N = 148) or placebo (N = 81) Trial duration = 4 weeks | |
| Outcomes | Mean change from baseline in trough sitting DBP and SBP at 3 and 4 weeks; analyses of the effects of age, sex, race, smoking status and baseline sitting DBP on change in BP; response rates; serum biochemistry | |
| Notes | A sample size calculation was provided based on 120 patients in each treatment group and 60 patients in the placebo group to detect a difference of at least 2.5 mmHg between active treatments and 3.1 mmHg between active treatment and placebo with 80% power. The study authors stated that there were no statistically significant differences across treatment groups in the baseline patient demographics and characteristics. SD for BP data (change from baseline) not given; it was reported as SE | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This was a multicenter, randomised, placebo‐controlled, parallel‐group study..." (line 1 under "Methods‐Study Design" p.183). Eligible patients were randomized to treatment (see line 9 under "Methods‐Study Design" p.183). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "This was a multicenter, randomised, placebo‐controlled, parallel‐group study...followed by a 4‐week double‐blind treatment period..." (line 1 under "Methods‐Study Design" p.183). Eligible patients entered the 4‐week, double‐blind treatment period (line 20 from top of p.183). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Efficacy analysis was based on an ITT technique Exclusions: the number of patients who were excluded from the study during the single‐blind, placebo run‐in period prior to randomization was not given Attrition: 38/547 (7%) patients were not included in the BP analysis. It is not known how many patients withdrew from the study and for what reasons WDAEs: 9/547 (1.6%) patients withdrew due to "clinical adverse experiences" for the following reasons: 7 ‐ "related to underlying medical conditions or concomitant illness" and 1 ‐ "bradycardia" and 1 ‐ "impotence". 2 other patients withdrew due to "laboratory findings" (not clearly explained). Note that the WDAE data were pooled, not presented as separate for each treatment group |
| Selective reporting (reporting bias) | High risk | Heart rate was measured but not reported at the end of the study. ECG, a vital sign, was not reported. Variability in metabolic data was not reported. Glucose levels were measured but not reported All‐cause mortality and SAEs were not documented. Total AEs were not reported; drug‐related "adverse experiences" were |
| Industry sponsorship | High risk | Supported by a grant from American Cyanamid Company |
Goldberg 1989.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel groups). 3 x 4 factorial design. Wash‐out period = up to 4 weeks on placebo followed by another 4 weeks "qualification phase". Multicenter, conducted in USA | |
| Participants | DBP 95 to 110 mmHg. Mean age 53.6 years. Males 52%. Baseline BP was 151.0/99.9 mmHg. Heart rate 74.1 bpm | |
| Interventions | Pinacidil 12.5 mg (N = 30), 25 mg (N = 34) or 37.5 mg bid (N = 32), pinacidil 12.5 mg, 25 mg or 37.5 mg bid + HCTZ 12.5 mg or 25 mg bid (N = 190, all combinations), HCTZ 12.5 mg (N = 34) or 25 mg bid (N = 33) or placebo (N = 31) Duration of treatment = 8 weeks | |
| Outcomes | Change from the baseline in trough mean supine and standing SBP and DBP; heart rate, body weight; hematology, urinalysis, serum biochemistry, pinacidil and HCTZ plasma concentrations | |
| Notes | Sample size calculation was not provided. The study authors stated that there were no significant differences across treatment groups in the baseline patient characteristics (line 3 under "Results" p.213). Supine BP in graphical form. Mortalities, SAEs and total AEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "...a randomised, double‐blind, 4 x 3 factorial, modified fixed‐dose multicenter trial." (see abstract p.208) "...patients were randomly allocated to one of 12 treatments in blocks of 12." (line 15 from top of p.209, left column) Comment: no further information was given. |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "...a randomised, double‐blind, 4 x 3 factorial, modified fixed‐dose multicenter trial." (see abstract p.208) "At entry into the study, a placebo was prescribed for all patients (one capsule bid, identical in appearance to eventual double‐blind capsules)." (line 8 under "Study design p.209). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Efficacy and safety analysis was based on an ITT technique Exclusions: 205/589 (35%) of patients were excluded from study during the placebo run‐in period before randomization Attrition: 87/384 (22.7%) of patients withdrew early from the study of which 9.1% withdrew for "failure to report, protocol violation, or physician/patient decision." The number of withdrawals from each treatment group was not given WDAEs: HCTZ 12.5 mg bid = 1/34 (2.9%) for "other reasons", HCTZ 25 mg bid = 1/33 (3%) for "other reasons" and placebo = 2/31 (6.5%) 1 ‐ weight gain, 1 ‐ "other reasons" |
| Selective reporting (reporting bias) | High risk | Standing SBP and DBP were measured but not reported. Supine SBP and DBP is shown in graphical form only (no standard deviations shown in graph). All‐cause mortality, SAEs and total AEs were not documented |
| Industry sponsorship | High risk | Supported by Eli Lilly and Company |
Hall 1994.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Conducted in USA | |
| Participants | DBP 95 to 110 mmHg. Mean age 50.2 years. Males 40%. Baseline BP was 150.2/100.1 mmHg in the treatment group and 149.8/99.6 in the control group | |
| Interventions | Indapamide 1.25 mg daily (N = 98) or placebo (N = 97) Trial duration = 8 weeks | |
| Outcomes | Change from the baseline in trough mean sitting and standing SBP and DBP (at 2, 4, 6 and 8 weeks); response rate; heart rate, ECG, hematology, urinalysis, serum biochemistry, body weight, vital signs | |
| Notes | A sample size calculation was not provided. Baseline patient characteristics did not include medical history. The study authors did not state whether there were statistically significant differences between indapamide and placebo groups with regard to baseline patient demographics and characteristics. Standard error (SE) given for BP data. SD or SE not given for biochemical data | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients who met the entry criteria were randomised at the end of a four week single‐blind placebo wash‐out period to either 1.25 mg indapamide or placebo..." (line 1 under "Design" p.572). No further information about sequence generation was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "Three of the [indapamide] patients received potassium supplements concomitant with double‐blind medication." (line 8 p.575). This could have broken the blinding. Blinding not explicitly stated by study authors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not known whether efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol technique, however sitting SBP results were based on the "all‐treated population..." (line 22 from top p.573) Exclusions: number of patients excluded from the study during the placebo run‐in period prior to randomization was not given Attrition: 16/98 (16%) and 7/97 (7%) of patients from the indapamide and placebo groups, respectively, withdrew from the study. Reasons for withdrawals included (IND versus PLB): 8 ‐protocol violations, 3 ‐ treatment failures, 3 ‐ clinical adverse experiences, 1 ‐ withdrawal of consent and 1 ‐ lost to follow‐up versus 2 ‐ clinical adverse experiences, 2 ‐ lost to follow‐up, 1 ‐ protocol violation, 1 ‐ treatment failure and 1 ‐ other reasons WDAEs: withdrawals due to "clinical adverse experiences" included: IND group, 3/98 (3.1%) versus placebo group, 2/97 (2.1%) for the following reasons: (rash + abnormal ECG), (dizziness) and (dizziness + headache + nausea + vomiting + photophobia + hypertension) versus (breast carcinoma) and (headache) |
| Selective reporting (reporting bias) | High risk | Baseline sitting BP did not include SD or SE; change in sitting BP was reported at endpoint with SE. Variability was not reported for baseline patient characteristics or biochemical data. Baseline standing BP was measured but not reported. Change in heart rate, serum sodium and chloride levels, hematology, urinalysis, vital signs, weight and ECG were measured but not reported at endpoint. Mortalities and SAEs were not clearly documented. Total AEs: 54/98 (55%) patients in indapamide group and 46/97 (47%) in the placebo group. A list of AEs (with description of event) occurring in at least 3% of patients was included |
| Industry sponsorship | High risk | Received grants from General Research Support of the School of Public Health, University of Minnesota and from Merck Sharp & Dohme |
Hulley 1985.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms) ‐ a pilot study of the SHEP program‐isolated systolic hypertension in the elderly. Wash‐out period = 8 weeks. Multicenter, conducted in USA | |
| Participants | SBP 160 to 219 mmHg. DBP < 90 mmHg. Age > 70 years: 61% of patients. Males 36%. Baseline BP was 172/75 in the treatment group and 174/77 in the control group. The study included elderly (60 years or older) patients with isolated systolic hypertension only (i.e. SBP 160 to 219 mmHg and DBP < 90 mmHg) | |
| Interventions | Chlorthalidone 25 mg daily (N = 443) or placebo (N = 108) for first 4 weeks. A step‐up protocol was used wherein poor response (i.e. BP goal not reached) after 4 weeks led to a doubling of drug dosage from 1 to 2 capsules of chlorthalidone per d; poor response in patients receiving placebo led to a simulated randomization with a doubling of placebo capsules. If the goal BP was not reached after 12 additional weeks, patients on chlorthalidone were re‐randomized to step II drugs including reserpine, metoprolol, or hydralazine | |
| Outcomes | Trough mean sitting SBP and DBP; serum biochemistry including potassium, uric acid, creatinine, glucose and cholesterol levels | |
| Notes | A sample size calculation was provided based on a sample of 500 patients to detect a mean difference in SBP of 6 mmHg at 90% power. BP results were taken from the first 4 weeks of fixed monotherapy. The study authors stated that the baseline patient demographics, characteristics and medical history were "reasonably well distributed" across the 2 groups except for angina and carotid bruit which were statistically more common in the placebo group (P value < 0.05). BP data of the 1st 4 weeks was used from figure 1 on page 916 (SD not given). Change in serum potassium level given but no SD Additional publications: Smith WM et al. Drugs 1986; 31(Suppl 4):154‐64; Hulley SB et al. J Am Ger Society 1986 [data from first 3 months], SHEP. JAMA 1991; 265(24):3255‐64 and Kostis JB et al. JAMA 1997;278(3):2126. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Elderly men and women who met eligibility criteria at 4 baseline examinations were randomised in a double‐blind fashion to chlorthalidone or matching placebo." (line 1 under "Methods" p.914). "We used an adaptive randomisation procedure that varied treatment assignment probabilities by 10% in one or the other direction in order to balance the step I study groups within race, sex, age and baseline systolic BP strata." (line 15 from top of p.914, right column). No further information was given |
| Allocation concealment (selection bias) | Low risk | "Each randomisation was carried out by telephone between the clinic staff and the coordinating centre data manager, who checked that eligibility criteria were met before assigning the participant to chlorthalidone or placebo." (line 9 from top of p.914, right column) |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Elderly men and women who met eligibility criteria at 4 baseline examinations were randomised in a double‐blind fashion to chlorthalidone or matching placebo." (line 1 under "Methods" p.914). "Upon randomisation into the study, participants entered the step‐up protocol and received 25 mg/day of chlorthalidone or placebo (supplied as identical capsules by USV Pharmaceutical Corp.) (line 1 under "Methods‐Blood pressure treatment, p.914) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The analysis of efficacy was based on an intention‐to‐treat (ITT) technique
Exclusions: 27,199 patients were screened; 2130 of these were included in the first baseline clinic visit. 1579/2130 (74%) of patients were excluded from the study during the 3 baseline clinic visits following screening, but prior to randomization Attrition: total number of withdrawals were not clearly documented WDAEs: not given. 7 patients receiving chlorthalidone (3 ‐ dizziness on standing, 1 ‐ syncope on standing, 1 ‐ rash, 1 ‐ hyperglycemia and 1 ‐ sexual dysfunction) and 2 patients receiving placebo (1 ‐ asthma and 1 ‐ escaping BP) had their medications terminated due to drug‐related events; the time point was not given, therefore these data were not useful (note that only the 1st 4 weeks out of the 12‐month trial were useable) |
| Selective reporting (reporting bias) | High risk | BP variability was not given. BP data from the placebo group were in graph form only (Fig. 1, p.916). Except for serum potassium levels, all other biochemical data were reported at baseline and 12 months only, not at 4 weeks (therefore not useable). Mortalities, SAEs and total AEs were not reported. Only more severe AEs characterized as "troublesome" or "intolerable" were reported in the study (AEs determined to be "not troublesome" were not mentioned) |
| Industry sponsorship | Low risk | Supported by grants from the National Heart, Lung and Blood Institute, the National Institute of Aging and the National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland |
Jounela 1994.
| Methods | Randomized, double‐blind, placebo‐controlled dose‐ranging trial (parallel arms). Wash‐out period = 4 weeks. Multicenter, conducted in Scandinavia (Finland, Sweden, Norway and Denmark) | |
| Participants | DBP 95 to 115 mmHg. Mean age 48.5 years. Males 39.6%. Baseline BP was 152.8/99.3 mmHg in the treatment group and 152.5/99.8 mmHg in the control group | |
| Interventions | HCTZ 3 mg (N = 22), 6 mg (N = 22), 12.5 mg (N = 22), 25 mg/d (N = 23) or placebo (N = 22) Treatment duration = 6 weeks | |
| Outcomes | Change from the baseline in mean standing and supine DBP and SBP; heart rate; serum biochemistry, hematology, urinalysis, ECG; plasma renin activity | |
| Notes | A sample size calculation was not provided. Baseline patient characteristics did not include medical history. Study authors stated that baseline patient characteristics were similar across treatment groups. Standing BP change from baseline available but no SD given. Supine BP with SD given. Mortality data were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients...were randomly allocated on a double‐blind basis to receive placebo or hydrochlorothiazide (HCTZ) at a dose of 3, 6, 12.5, or 25 mg once daily." (line 1 under "Study Design" p.231). "Random allocation was performed using randomisation tables." (line 5 under "Study Design" p.231) |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Patients...were randomly allocated on a double‐blind basis to receive placebo or hydrochlorothiazide..." (line 1 under "Study Design" p.231) "Placebo and active tablets were identical in appearance and taste." (line 6 under "Study Design" p.231) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Not known whether efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol technique Exclusions: no patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: 3/111 (3%) patients withdrew from the study for the following reasons: 1 ‐ headache, palpitation and vertigo (12.5 mg HCTZ group), 1 ‐ palpitation (25 mg HCTZ group) and 1 ‐ lost to follow‐up (group not specified) WDAEs: HCTZ 12.5 mg group: 1 patient due to headache, palpitation and vertigo; HCTZ 25 mg group: 1 patient due to palpitation |
| Selective reporting (reporting bias) | High risk | ECG, heart rate, urinalysis, serum creatinine levels, hemoglobin, hematocrit, WBC and platelets were measured but their values (mean ± SD) not reported at baseline or endpoint. Variability was not given for baseline patient characteristics. Standing BP was not reported at baseline and its variability not reported at endpoint. Mortalities were not documented. SAEs: no patients had what study authors reported to be "serious clinical adverse experiences". Total AEs: in HCTZ 3, 6, 12.5 and 25 mg/d and placebo groups there were 2 (9%), 4 (18%), 3 (13.6%), 4 (17%) and 1 (4.5%) patients who experienced AEs |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Kayanakis 1987.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 2 weeks. Multicenter, conducted in France | |
| Participants | SBP 160 to 200 mmHg. DBP 95 to 120 mmHg. Mean age 53.5 years. Males 54.8%. Baseline BP was 176.6/103.2 mmHg in the treatment group and 172/102.5 mmHg in the control group | |
| Interventions | Captopril 50 mg/d (N = 43), captopril 50 mg/d + HCTZ 25 mg/d (N = 45), HCTZ 25 mg daily (N = 43) or placebo (N = 86) Treatment duration = 8 weeks | |
| Outcomes | Trough standing and supine SBP and DBP; response rate; heart rate, hematology, urinalysis, body weight, serum biochemistry | |
| Notes | A sample size calculation was not provided. Although the study's focus was on mild to moderate essential hypertension, patients with relatively severe BP as high as 200/120 mmHg were included (line 2 under "Patients" p.89S). Study authors stated that the baseline patient demographics and characteristics were similar across treatment groups. BP and SD in graph form only (Fig. 1 and 2 p.91S). Biochemical data restricted to serum potassium from patients in the HCTZ group. Mortalities and SAEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The study was divided into two phases: a placebo run‐in period of 2 week's duration and an active treatment period of 8 weeks duration when the patients were randomised in four groups: captopril 50 mg, hydrochlorothiazide 25 mg combination once daily, captopril 50 mg alone once daily, hydrochlorothiazide 25 mg alone once daily or placebo once daily." (line 1 under "Methods‐Trial Design" p.90S). No further information given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "A double‐blind design was used for the 8 weeks of active treatment." (line 9 under "Methods‐Trial Design" p.90S). Comment: no further information given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not known whether efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol technique Exclusions: 4/221 (1.8%) of patients were excluded from the study during the placebo run‐in period prior to randomization (line 1 under "Results‐Patient Population" p.90S) Attrition: 2/43 (4.7%) and 3/86 (3.5%) of patients from the HCTZ and placebo groups, respectively, withdrew from the study. Reasons for withdrawals included (HCTZ versus PLB): 1 ‐ inefficacy and 1 ‐ lost to follow‐up versus 2 ‐ inefficacy and 1 ‐ lost to follow‐up WDAEs: 0/211 (0%) of patients withdrew due to adverse events |
| Selective reporting (reporting bias) | Unclear risk | Baseline patient characteristics did not include medical history. Standing BP, heart rate and weight were measured but not reported at the end of the study. BP data were graphed only. Except for potassium levels, changes in mean ± SD for serum biochemistry, hematology and urinalysis were not shown at endpoint. Mortalities and SAEs were not clearly documented. Total AEs: 9/43 (20%) patients in HCTZ group and 14/86 (16%) patients on placebo. The most commonly occurring AE, GI upset was experienced by 8 patients receiving HCTZ and 10 patients receiving placebo |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Kochar 1999.
| Methods | Randomized, double‐blind, placebo‐controlled trial. 4 x 4 factorial design. Wash‐out period = 4 to 5 weeks. Multicenter, conducted in USA | |
| Participants | Supine, seated or standing DBP ≥ 95 mmHg (and seated DBP < 110 mmHg). Mean age 55 years. Males: 65%. Baseline seated BP was 151/100 mmHg | |
| Interventions | Irbesartan 37.5 mg, 100 mg or 300 mg/d (all combined, N = 126), irbesartan 37.5 mg, 100 mg or 300 mg/d + HCTZ 6.25 mg, 12.5 mg or 25 mg/d (all combined, N = 390), HCTZ 6.25 mg, 12.5 mg or 25 mg/d (all combined, N = 123) or placebo (N = 44) Treatment duration = 8 weeks | |
| Outcomes | Mean change from baseline in trough DBP measured at 8 weeks; heart rate; serum biochemistry and ECG. WDAE given | |
| Notes | A sample size calculation based on 40 patients in each treatment group to detect a difference of 1.3 mmHg from the true mean in sitting DBP was provided at 95% power. The study authors stated that there were no statistically significant differences between treatment groups in baseline patient demographics and characteristics. Baseline BP was not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This randomised, double‐blind, placebo‐controlled study was conducted at 46 sites in the United States." (line 1 under "Materials and Methods‐Study Design" p.798). "To be eligible for randomisation, patients had to have a mean seated DBP at both weeks 3 and 4 between 95 and 100 mmHg and demonstrate good compliance." (line 5 under "Materials and Methods‐Study Design" p.798). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "This randomised, double‐blind, placebo‐controlled study was conducted at 46 sites in the United States." (line 1 under "Materials and Methods‐Study Design" p.798). "All patients were instructed to take three capsules (irbesartan or matching placebo) and one tablet (HCTZ or matching placebo) once daily between 6 AM and 10 AM for 8 weeks." (line 1 from top of p.798). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Efficacy analysis was not mentioned (was it based on an intention‐to‐treat (ITT) or per‐protocol technique?). It was stated that 680 patients were included in the efficacy analysis Exclusions: 443/1126 (39%) patients were excluded from the study during the single‐blind run‐in period prior to randomization. Specific reasons for each excluded patient were not given Attrition: 52/683 (7.6%) patients withdrew from the study for the following reasons: 22 ‐ "adverse events", 9 ‐ "patient request", 10 ‐ "poor BP control", 4 ‐ "loss to follow‐up", 7 ‐ for various reasons, including "poor compliance", "use of prohibited medications" and "administrative issues". It is not known from which treatment groups these patients withdrew WDAEs: 22/683 (3.2%) patients withdrew due to adverse events. WDAE data were expressed in percentages only and were pooled, not presented as separate according to each treatment group (i.e. HCTZ 6.25/12.5/25 mg = 4.1%; placebo = 4.5%). The most common reasons for the adverse events were given and were expressed in percentages, not in terms of numbers of patients |
| Selective reporting (reporting bias) | High risk | Baseline patient medical history was not given. Glucose, lipids and blood urea nitrogen levels; heart rate, ECG and urinalysis were measured but actual values were not reported on at the study's endpoint. Standing BP was measured, but not reported. Subgroup analysis of elderly versus young, black versus white and male versus female patients was reported, but not mentioned in the methods section of the study. Variability in the mean change of serum potassium and uric acid levels was not shown in the graphs at endpoint. Mortalities: none (0%). SAEs: 8/683 (1.2%) patients; specific reasons were not given. For the reporting of AEs, the HCTZ 6.25, 12.5 and 25 mg treatment groups were combined. Total AEs were not reported, only the "most common treatment‐emergent adverse events" were |
| Industry sponsorship | High risk | Sponsored by Bristol‐Myers Squibb Pharmaceutical Research Institute |
Krantz 1988.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 6 weeks. Conducted in USA | |
| Participants | DBP 90 to 108 mmHg. Mean age 45.2 years. All patients were male. Baseline BP was 138.2/89 mmHg in the treatment group and 136.3/86.8 mmHg in the placebo group | |
| Interventions | Atenolol 25 mg bid (N = 12), propranolol 40 mg bid (N = 12), HCTZ 25 mg bid (N = 10) or placebo (N = 12). After 2 weeks on atenolol, propranolol or HCTZ non‐responsive patients had their dosages doubled. All patients were determined to either have not achieved BP response and/or exceeded exercise‐induced increases in heart rate, therefore, this review will compare the HCTZ 50 mg bid dosage versus placebo Treatment duration = 2 weeks (low‐dose) + 4 weeks (high‐dose) | |
| Outcomes | Sitting (resting) DBP and SBP measured at 4 weeks; heart rate, ECG; psychological and behavioral testing | |
| Notes | A sample size calculation was not provided. Dosage of HCTZ was doubled from 25 to 50 mg bid for all patients receiving HCTZ after 2 weeks of treatment. Study authors did not report whether there were statistically significant differences in baseline patient demographics and characteristics across treatment groups. Resting BP measurements were taken while patient was sitting. Baseline BP was not given. Peak or trough BP not mentioned. WDAEs, mortalities, SAEs and total AEs were not reported. Biochemical data not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "In a double‐blind study, mild hypertensive's were assigned randomly to receive either propranolol, atenolol, placebo, or a diuretic (hydrochlorothiazide) for 6 weeks." (line 1 under "Methods‐Overview" p.617). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | High risk | "In a double‐blind study, mild hypertensive's were assigned randomly to receive either propranolol, atenolol, placebo, or a diuretic (hydrochlorothiazide) for 6 weeks." (line 1 under "Methods‐Overview" p.617) "...subjects were randomised into one of four treatments following double‐blind procedures." (line 6 from top of p.617). "...only the treating physician (JDL or EF), who did not conduct any of the behavioural or psychophysiological testing, was aware of individual patients' drug assignments." (line 8 from top of p.617) This did not imply that the treating physician did not perform BP readings and, therefore, had knowledge of which patients may be receiving treatment as opposed to a placebo pill "Initial drug dosages were 25 mg b.i.d. for hydrochlorothiazide and for atenolol and 40 mg b.i.d. for propranolol, taken in visually identical white capsules." (line 14 from top of p.617). After the second week of treatment, drug dosages were doubled for all patients, but it is not known if the placebo group also received 2 times as many capsules |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It was not stated whether efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol technique Exclusions: the number of patients excluded from the study during the drug wash‐out period prior to randomization was not given Attrition: 5 patients withdrew from the study for the following reasons: 1 ‐ "untoward side effects", 4 ‐ "errors in conducting the protocol" or "life events requiring withdrawal". Although the study authors stated that the patients were about equally distributed between treatment groups, the actual number of dropouts between groups was not reported WDAEs: not given |
| Selective reporting (reporting bias) | High risk | Except for mean age, baseline patient demographics and characteristics were not documented. Variability was not included in the mean age of patients at baseline. Biochemical including hematology, serum chemistry and urinalysis data were not reported in the study. Psychological and symptom questionnaires were conducted during the study, but their results were not reported. Mortalities, SAEs and total AEs were not reported |
| Industry sponsorship | Low risk | Supported by NIH grant HL31514 and USUHS protocol R07233 |
Lacourciere 1994.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). 3 x 4 factorial design. Wash‐out period = 4 weeks. Conducted in Canada | |
| Participants | DBP 95 to 110 mmHg. Mean age not given. % of males not given. Baseline BP was 158/101 mmHg in both the treatment as well as the control group | |
| Interventions | Nebivolol 1 mg (N = 20), 5 (N = 20) or 10 mg/d (N = 20), nebivolol 1 mg, 5 mg or 10 mg/d + HCTZ 12.5 mg or 25 mg/d (N = 120, all combinations), HCTZ 12.5 mg (N = 20) or 25 mg/d (N = 20) or placebo (N = 20) Treatment duration = 12 weeks | |
| Outcomes | Change from the baseline in mean trough sitting SBP and DBP; heart rate, body weight and serum biochemistry (i.e. lipids profile). WDAE data available | |
| Notes | A sample size calculation was not provided. In addition to BP taken in the clinical setting, ambulatory BP, using a non‐invasive device was monitored to address "white‐coat hypertension". The latter was considered to be a research tool primarily and not yet clinically useful in the diagnosis and management of hypertension (see additional publication ‐ line 2 from top of p.143). After only 4 weeks of DB therapy (out of a total of 12 weeks), unresponsive patients who were withdrawn from the study were still considered fully evaluable in the efficacy analysis. (see additional publication ‐ line 19 from top of p.138) Study authors stated that sex, age, clinic and ambulatory BP and body mass index did not differ significantly across treatment groups. Metabolic data available as % change from baseline and not as actual values. Mortalities, SAEs and total AEs were not stated clearly Additional publication: Lacourciere Y et al. Am J Hypertens 1994;7:137‐45; ambulatory BP was measured using a 24 hour, non‐invasive BP device. Incidence of AEs (listed as WHO terms) was included in duplicate publication only |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients...were randomly assigned to period B which consisted of a 12 week double‐blind treatment." (line 8 under "Study Design" p.284) |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "Patients...were randomly assigned to period B which consisted of a 12 week double‐blind treatment." (line 8 under "Study Design" p.284) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Primary efficacy analysis was based on an intention‐to‐treat (ITT) technique (see additional publication ‐ p.139)
Exclusions: 73/313 (23%) of patients were excluded from the study during the single‐blind placebo run‐in period prior to randomization (see duplicate publication ‐ p.139)
Attrition: 14/240 (5.8%) patients withdrew from the study. Reasons for withdrawals included, in the placebo group, 6 patients for inadequate response; in the HCTZ 12.5 mg group 1 patient was lost to follow‐up and in the HCTZ 25 mg group there were no withdrawals. The remaining patients who withdrew were receiving other (non‐thiazide) drugs (see additional publication ‐ p.139) WDAEs: 0/240 (0%) of patients withdrew due to an adverse event from the HCTZ 12.5 mg, HCTZ 25 mg or placebo groups (see additional publication ‐ p.139) |
| Selective reporting (reporting bias) | Unclear risk | Baseline BP was not given. Variability in mean change from baseline in BP was not given. Baseline patient demographics and characteristics were measured but not reported. Vital signs, laboratory values and ECG were measured but not reported. Total AEs were reported in the duplicate publication, if present, in at least 3 patients receiving active treatment which is equivalent to 3/40 = 7.5% of patients. This cut‐off percentage is much higher than the minimum of 3% chosen to present AE data in most other studies on use of thiazides for BP‐lowering. AEs were pooled using data from the HCTZ 12.5mg and HCTZ 25 mg treatment groups. Mortalities, SAEs and total AEs were not stated clearly |
| Industry sponsorship | High risk | Supported in part by a grant from Janssen Research Foundation and by Le Centre Hospitalier de l'Universite Lavale |
Lawton 1979.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Conducted in USA | |
| Participants | DBP 90 to 105 mmHg. Mean age 37 years. Males 71%. Baseline BP was 135/93 mmHg in the treatment group and 137/93 mmHg in the control group | |
| Interventions | Chlorthalidone 50 mg/d (N = 42) or placebo (N = 42) Trial duration = 4 weeks | |
| Outcomes | Sitting SBP and DBP; urinalysis (includes urinary sodium, creatinine, catecholamines and norepinephrine), plasma renin activity and serum dopamine β‐hydroxylase enzyme | |
| Notes | A sample size calculation was not provided. The average age of patients included in the study was younger than in most other studies under this review. The study authors stated that there were no statistically significant differences across treatment groups in the baseline patient demographics and characteristics, except for age (average age was 34.5 in chlorthalidone group and 38.9 in placebo group). Baseline BP following randomization was not reported. BP data with SD at 1 month available. Total withdrawals, WDAEs, mortalities, SAEs, total AEs and biochemical data were not reported. Study included a separate group of 116 normotensive patients whose dopamine β‐hydroxylase levels were monitored (not included in the review) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...patients were randomly assigned to receive either active treatment with chlorthalidone 50 mg qam or placebo." (line 32 from top of p.1064). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "The study was conducted in a double‐blind manner." (line 35 from top of p.1064). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Primary efficacy analysis is not known (was it based on an intention‐to‐treat (ITT) or per‐protocol technique). Exclusions: the number of patients excluded from the "before treatment" period of the study prior to randomization was not given Attrition: total patient withdrawals and their reasons were not given WDAEs: not given |
| Selective reporting (reporting bias) | High risk | Mean age was the only of the baseline patient demographics and characteristics reported by the study authors. Metabolic data including hematology, serum chemistry, lipids and ECG were not documented (urine sodium and creatinine were included). Mortalities, SAEs and total AEs were not documented |
| Industry sponsorship | Low risk | Supported by Research and Development Division, Veterans Administration Hospital and by VA‐NIH Mild Hypertension Cooperative Study #8 |
London 2006.
| Methods | Randomized, double‐blind, placebo‐controlled trial. Wash‐out period = 4 weeks. Multicenter; conducted in France, Germany and Spain | |
| Participants | SBP/DBP 150 to <180/95 to < 110 mmHg or SBP/DBP 160 to < 180/< 90. Mean age 59 years. Males 50.7%. Baseline BP was 164/96.5 mmHg in the IND SR treatment group and 165/9 mmHg in the control group | |
| Interventions | Candesartan 8 mg/d (N = 435), amlodipine 5 mg/d (N = 445), indapamide SR 1.5 mg/d (N = 441), or placebo (N = 441) Treatment duration = 12 weeks | |
| Outcomes | Mean trough supine SBP and DBP; pulse pressure; response rate; automated ambulatory BP over a 24‐hour period; heart rate, body weight, hematology, serum biochemistry, hepatic and renal function | |
| Notes | A sample size calculation (based on unknown number of patients in each treatment group) was provided to detect a difference of 3 mmHg at 83% power. The study authors stated that the baseline patient demographics and characteristics (including their cardiovascular risk factors) were similar across all treatment groups (P value = NS). Biochemical data not given (except for serum potassium in IND SR group only). A subset of patients with isolated systolic hypertension was also evaluated, but not included in this review | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This was a multicenter, multinational, randomised, double‐blind, placebo‐controlled study with four parallel treatment arms." (line 1 under "Study Design" p.114). "...patients were randomised to receive either placebo, indapamide (1.5 mg) SR (sustained release), candesartan (8 mg), or amlodipine (5 mg)..." (line 4 under "Study Design" p.114). No further information given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "This was a multicenter, multinational, randomised, double‐blind, placebo‐controlled study with four parallel treatment arms." (line 1 under "Study Design" p.114). No further information given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Primary efficacy analysis was based on the an intention‐to‐treat (ITT) technique Exclusions: 608/2370 (25.7%) of patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: 14/441 (3.2%) patients from the IND SR group and 36/441 (8.2%) of patients on placebo, respectively, withdrew from the study. Reasons included (IND SR versus placebo) protocol violation: 3 versus 8; lack of efficacy: 1 versus 14; non‐medical reason: 2 versus 4 and adverse events: 8 versus 10 WDAEs: 8/441 (1.8%) of patients from the IND SR group and 10/441 (2.3%) of patients from the placebo group withdrew due to adverse events; the precise reasons were not given |
| Selective reporting (reporting bias) | Unclear risk | Weight, heart rate and laboratory parameters (including hematology, glucose levels, lipids profile and hepatic and renal function) were measured but not reported at baseline or endpoint. Mortalities: 1 patient from myocardial infarction in IND SR group; 0 in placebo group. SAEs: none. Total AEs were not reported |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Lucas 1985.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Conducted in USA | |
| Participants | DBP 100 to 115 mmHg. Mean age 50 years. Males 65%. Baseline BP not given | |
| Interventions | HCTZ 25 mg bid (N = 78), HCTZ 50 mg bid (N = 82) or placebo (N = 84) Treatment duration = 4 weeks. This period was followed by 4 weeks of add‐on therapy with bevantolol 200 mg bid (therefore only the first 4 weeks were included in this review) | |
| Outcomes | Mean change from baseline in trough sitting DBP at 4 weeks; BP response rate; heart rate, body weight, hematology, urinalysis, serum biochemistry | |
| Notes | A sample size calculation was not provided. "At each clinic visit, four blood pressure readings were obtained at a time as close as possible to 12 hours after the previous dose." (line 5 from bottom on p.51). The study authors did not state whether baseline patient demographics and characteristics were statistically different or similar across treatment groups. No SD for BP or biochemical data. Biochemical data as % and not actual values. Mortalities, SAEs and total AEs were not reported. Total withdrawals and WDAEs were shown as combined data | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Subjects were then randomly allocated to one of three treatment groups and received either 50 mg/day of hydrochlorothiazide (25 mg twice daily), 100 mg/day of hydrochlorothiazide (50 mg twice daily), or placebo for four weeks." (line 4 under "Methods" p.51). No further information given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "...patients entering the double‐blind phase..." (line 4 from bottom of p.51). No further information given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Primary efficacy analysis whether based on an intention‐to‐treat (ITT) or per‐protocol technique is not reported Exclusions: it is not known whether any patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: 16/244 (6.6%) patients withdrew from the monotherapy phase of the study. Reasons for withdrawals were not given WDAEs: 7/244 (3%) patients withdrew due to adverse events; the precise reasons were not given. Data on total withdrawals and WDAEs (above) were pooled rather than shown separately for each treatment group |
| Selective reporting (reporting bias) | High risk | Weight, general physical condition, hematology and urinalysis results were measured but not reported at baseline or endpoint. Serum biochemical levels including lipids were expressed as percentages rather than as mean ± SD changes from baseline. Variability in BP and heart rate data were not given. Mortalities, SAEs and total AEs were not documented |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Materson 1978.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Multicenter, conducted in USA | |
| Participants | Standing DBP 90 to 109 mmHg. Mean age 53.6 years. Males 58%. Baseline BP was 146.1/97.2 mmHg in the treatment group and 145.3/95.8 mmHg in the control group | |
| Interventions | Chlorthalidone 12.5 mg (N = 20), 25 mg (N = 20), 50 mg (N = 20), 75 mg/d (N = 20) or placebo (N = 20) Treatment duration = 12 weeks | |
| Outcomes | Mean standing and supine SBP and DBP (Korotkoff sounds 4 and 5 reported for DBP) at baseline and end of treatment; BP response rate; ECG; serum biochemistry, urinalysis and body weight | |
| Notes | A sample size calculation was not provided. Patients were excluded based on a disability, geographic location, poor motivation, psychosis and other restrictive criteria which surpassed those of similar trials investigating the BP lowering of thiazides. The mean BP at endpoint was calculated by taking the average of BP readings at the 3 last treatment visits. Normally endpoint measurements are taken from the last evaluable time point in the study. The study authors stated that the baseline patient demographics and characteristics (i.e. BP, body weight and serum biochemical values) were similar across treatment groups (P value = NS). Korotkoff phase 5 was used for determining DBP in this review. Mortalities and SAEs not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...patients were randomly and in a double‐blind fashion placed on one of the following 5 regimens of medication..." (line 7 from bottom of p.193). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "...patients were randomly and in a double‐blind fashion placed on one of the following 5 regimens of medication taken once daily replacing an identical‐appearing placebo used in the placebo run‐in period..." (line 7 from bottom of p.193) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It is not known if primary efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol technique Exclusions: the study did not report how many, if any patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: a total of 6/100 (6%) patients withdrew from the study, this included 3/20 (15%) from the chlorthalidone 50 mg group: 2 patients for "adverse reactions" and 1 for "treatment failure"; 1/20 (5%) from the chlorthalidone 75 mg group for "adverse reactions"; and 2/20 (10%) patients from the placebo group for "treatment failure" and "prolonged hospitalization for low back pain" WDAEs: 3/100 (3%) patients withdrew due to what study authors referred to as "adverse reactions" (presumed to be adverse events); 2 from the chlorthalidone 50 mg group for "rash" and "severe headaches" and 1 from the chlorthalidone 75 mg for "orthostatic hypotension" |
| Selective reporting (reporting bias) | High risk | Heart rate, ECG and urinalysis were measured but not reported on in the study. Variability was not included in baseline patient demographics and characteristics; medical history of patients was not reported. BP and biochemical (i.e. serum potassium, uric acid and glucose) data were expressed as baseline and endpoint averages, not as change from baseline; in addition, SE (standard error of mean), not SD, was given. Baseline weight and serum chloride levels were measured, but the study only reported the mean change from baseline. Mortalities and SAEs were not stated explicitly. Total AEs: in the chlorthalidone 12.5, 25, 50 and 75 mg groups: 3 (15%), 6 (30%), 7 (35%) and 7 (35%) patients; and in the placebo group: 3 patients (15%) |
| Industry sponsorship | Unclear risk | Sponsor not reported |
McGill 2001.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). 4 x 5 factorial design. Wash‐out period = 4 weeks Multicenter, conducted in USA | |
| Participants | Supine DBP 90 to 114 mmHg. Mean age 53 years. Males 60.3%. Baseline BP was 154.0/100.7 mmHg | |
| Interventions | Telmisartan 20 mg, 40 mg, 80 mg or 160 mg/d (all combined, N = 209), telmisartan 20 mg, 40 mg, 80 mg or 160 mg/d + HCTZ 6.25 mg, 12.5 mg or 25 mg/d (all combined, N = 414), HCTZ 6.25 mg, 12.5 mg or 25 mg/d (all combined, N = 121); or placebo (N = 74) Treatment duration = 8 weeks | |
| Outcomes | Change from baseline in mean trough supine and standing DBP and SBP; BP response rate; heart rate; vital signs, ECG, plasma renin activity and serum biochemistry | |
| Notes | A sample size calculation based on 75 patients in each key treatment group to detect a difference of at least 4 mmHg for each comparison (combination versus monotherapy) was provided at a power of 86%. The study authors stated that the baseline patient demographics and characteristic were comparable across treatment groups. Primary efficacy analysis did not include 6.25 and 25 mg doses of HCTZ. SD not used (SE was) Additional publications: McGill. Blood Pressure Monitoring 2001; 6 (Suppl 1): S3‐S13. Reported data were identical. Subgroup analysis of black patients included in Littlejohn III. Blood Pressure Monitoring 2001; 6 (Suppl 1): S15‐S21 and McGill. Clinical Cardiology 2001;24:66‐72 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "This was a multicenter, randomised, double‐blind, placebo‐controlled, parallel‐group study...". "Randomization was according to enrolment order and a computer‐generated list..." (line 17 under "Study Design" p.836) |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "[An] 8‐week, double‐blind, double‐dummy comparison of telmisartan monotherapy, HCTZ monotherapy, telmisartan/HCTZ combination therapy and placebo." (line 5 under "Study Design" p.836). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The primary efficacy analysis was based on an intention‐to‐treat (ITT) technique with the LOCF (last observation carried forward) Exclusions: 475/1293 (37%) patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: 69/818 (8.4%) patients withdrew from the study for the following reasons: 24 patients for an "adverse event", 18 ‐ "lack of efficacy", 7 ‐ "noncompliance", 6 ‐ "lost to follow up", 6 ‐ "withdrawal of consent" and 8 ‐ "other reasons". Note that data were pooled, which made it impossible to ascertain from which treatment groups the patients originated. WDAEs: 24/818 (3%) patients withdrew due to adverse events. The study authors stated that 3 of the 24 patients were receiving HCTZ monotherapy or placebo, but it was not mentioned from which groups the patients originated. The precise reasons were not given |
| Selective reporting (reporting bias) | High risk | Vital signs, ECG and serum chloride, uric acid, BUN and glucose levels were measured but not reported at endpoint. Serum potassium levels and PRA (plasma renin activity) were expressed as mean change from baseline and did not include baseline measurements or variability. Variability in some of the baseline patient demographics and characteristics was not given; moreover, the data for the patients in the HCTZ groups were combined into one. Primary efficacy analysis did not include 6.25 and 25 mg doses of HCTZ. SE (standard error of the mean) was given for BP data, not SD. Results for standing DBP and SBP as well as heart rate were not reported. Trough‐peak ratios were not clearly stated for all treatment groups. Mortalities were not mentioned. SAEs: 1 patient in the HCTZ 6.25 mg group had a "uterine fibroid", 1 patient in the HCTZ 12.5 mg group had "syncope" and 1 patient on placebo had "oesophageal ulceration". Total AEs were reported in 50% of patients (all HCTZ groups combined) and in 42% of patients on placebo |
| Industry sponsorship | High risk | Supported by a restricted grant from Boehringer Ingelheim Pharmaceuticals, Inc |
McVeigh 1988.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Conducted in Belfast | |
| Participants | DBP 90 to 110 mmHg. Mean age 57 years. Males 41.5%. Baseline BP was 166.7/97 mmHg in the treatment group and 157/94 mmHg in the control group | |
| Interventions | Cyclopenthiazide 50 μg/d (N = 13), 125 μg/d (N = 15), 500 μg/d (N = 13) or placebo (N = 12) Trial duration = 8 weeks | |
| Outcomes | Sitting SBP and DBP (at weeks 2, 4, 6 and 8 intervals); ECG, serum biochemistry, plasma renin activity, urinalysis and body weight | |
| Notes | A sample size calculation based on 6 or more patients per treatment group to detect a 10 mmHg (± 5 mmHg) difference in DBP was provided at a power of 80%. There was a small sample size of 12 to 15 patients in each treatment group. The study authors stated that the baseline patient age and body weight were similar across treatment groups. Mortalities and SAEs not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "...patients were randomly allocated in a double‐blind fashion to one of four regimens of treatment incorporating 50μg, 125μg and 500μg cyclopenthiazide or a placebo that looked identical." (line 17 from bottom of p.96). "Randomisation was achieved with a balanced block design." (line 20 from bottom of p.96) |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "...patients were randomly allocated in a double‐blind fashion to one of four regimens of treatment incorporating 50μg, 125μg and 500 μg cyclopenthiazide or a placebo that looked identical." (line 17 from bottom of p.96) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | It was not shown whether the primary efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol technique Exclusions: 30/83 (36%) patients were excluded from the study during the placebo run‐in period prior to randomization. 22 of these patients became normotensive (DBP < 90 mmHg), 3 were unable to tolerate the placebo, 2 were hospitalized for low back pain, 1 developed "unacceptable" ankle edema, 1 had a SBP exceeding 240 mmHg and 1 had DBP exceeding 110 mmHg Attrition: no patients withdrew from the study WDAEs: none |
| Selective reporting (reporting bias) | Unclear risk | Mean body weight was reported at baseline only. Serum levels of creatinine and magnesium were measured but actual values were not shown. BP data were presented as mean ± SD at both baseline and endpoint. Medical history was not included in the baseline patient characteristics. Mortalities and SAEs were not mentioned. Total AEs: cyclopenthiazide 50, 125 and 500 μg/d groups ‐ 13, 13 and 8 patients; placebo group ‐ 9 patients |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Mersey 1993.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Placebo run‐in period = 4 to 6 weeks Multicenter, conducted in USA | |
| Participants | Seated DBP 92 to 109 mmHg. Mean age (HCTZ 12.5 mg versus placebo): 52.1 versus 50.7 years. Males (HCTZ 12.5 mg versus placebo): 63% versus 59%. Baseline BP was 143.5/97.3 mmHg in the treatment group and 142.8/97.6 mmHg in the placebo group | |
| Interventions | Captopril 25 mg/d (N = 68), captopril 25 mg/d + HCTZ 12.5 mg/d (N = 69), captopril 50 mg/d + HCTZ 25 mg/d (N = 69), HCTZ 12.5 mg/d (N = 69) or placebo (N = 70) Trial duration = 8 weeks | |
| Outcomes | Change from baseline in mean trough seated, standing and supine DBP and SBP; BP response rate, heart rate; serum biochemistry (glucose, potassium, cholesterol, uric acid, BUN and creatinine levels) | |
| Notes | A sample size calculation was not provided. Study authors stated that baseline patient demographics and characteristics were similar across treatment groups. Variability in BP data was not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Qualified patients were randomly assigned in a double‐blind manner to one of five treatment groups..." (line 12 under "Patients and Methods‐Patients and Treatment" p.503). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "Qualified patients were randomly assigned in a double‐blind manner to one of five treatment groups..." (line 12 under "Patients and Methods‐Patients and Treatment" p.503) Comment: no further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It is not known if the primary efficacy analysis was based on the per‐protocol or an intention‐to‐treat (ITT) population of patients Exclusions: 152/497 (31%) of patients were excluded from the study during the single‐blind placebo run‐in period prior to randomization Attrition: total withdrawals were reported separately based on study phase. 23/345 (7%) of patients withdrew from the study following randomization but prior to collection of efficacy data. The reasons were: 9 ‐ "loss to follow‐up", 6 ‐ "adverse drug experiences", 4 ‐ "patient request", 1 ‐ "concomitant illness", 1 ‐ "noncompliance", 1 ‐ "prohibited medication", 1 ‐ seated DBP < 92 mmHg after the lead‐in period". 26/345 (7.5%) more patients withdrew before the end of the study for the following reasons: 8 ‐ "adverse drug experiences", 3 ‐ "loss to follow‐up", 5 ‐ "specific requests", 3 ‐ "abnormal test results", 2 ‐ "prohibited medication", 2 ‐ "seated DBP ≥ 115 mmHg, 3 ‐ "other reasons". Therefore, total number of withdrawals during the study was 49/345 (14%) of patients. Note that the data were pooled, making it impossible to determine from which treatment group the patients originated WDAEs: the number of patients who withdrew due to any adverse events is not known; data were presented in terms of "adverse drug experiences" of which there were 14/345 (4%). Three more patients were withdrawn due to "abnormal lab test results"; if taken into account (presuming abnormal laboratory result = an adverse event), there were a total 17/345 (4.9%) WDAEs for the following reasons: in the HCTZ 12.5 mg group, 1 patient with "tightness and fluttering in chest" and 1 ‐ "proteinuria"; in the placebo group, 1 ‐ "chest pain". The remaining 14 patients with WDAEs were receiving non‐thiazide monotherapy or thiazide + non‐thiazide combo therapy (outside the scope of this systematic review). |
| Selective reporting (reporting bias) | Unclear risk | Hematology results were not shown; other biochemical data included the mean change and SE (standard error of the mean). Heart rate and change from baseline in standing and supine DBP and SBP were measured but actual values were not reported on at endpoint. Mean changes in seated DBP and SBP were represented in 2 graphs, however, variability in BP was not included in either. Variability was not given for baseline patient demographics and characteristics. There were no mortalities during the study. SAEs were not clearly stated. Total number of patients with any AEs were not reported, but a list of only the most frequently reported AEs was shown |
| Industry sponsorship | High risk | Funded by a grant from Bristol‐Myers Squibb Company |
Morledge 1986.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Multicenter, USA | |
| Participants | SBP ≥ 160 mmHg (patients with isolated systolic hypertension). Mean age 73 years. Males 38.5%. Baseline BP was 176/84 in the treatment group and the control group | |
| Interventions | Chlorthalidone 12.5 mg/d (N = 47), 25 mg/d (N = 43), 50 mg/d (N = 47) or placebo (N = 39) Trial duration = 12 weeks | |
| Outcomes | Sitting and standing SBP and DBP (average of 6 readings at 1, 2, 4, 6, 8, 10 and 12 weeks); BP response rate; pulse rate, ECG, body weight; hematology, urinalysis and serum biochemistry | |
| Notes | A sample size calculation was not provided. Study authors stated that baseline patient demographics and characteristics were comparable across treatment groups (P value > 0.10), except in the case of concomitant illness (P value < 0.05). Number of participants unknown for BP and biochemical data; data on BP from figure only (no standard deviation). Patients received potassium supplements at the discretion of the physician | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "...patients were randomly assigned, according to a computer‐generated code, to one of four treatment groups: chlorthalidone 12.5, 25.0, or 50.0 mg or placebo." (line 5 from top of p.200) |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "During the double‐blind treatment period..." (line 17 from top of p.200). Some of the patients received potassium supplements as required which could have broken the blinding. No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | High risk | It is not known if the primary efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol population of patients. The study authors did state however, that "patients who failed to respond satisfactorily to their assigned medication could be dropped from the study any time after the first week of treatment, but were included in the statistical analyses of safety and efficacy." (line 12 from top of p.200). The total number of patients presumed to be randomized to treatment was 176 Exclusions: it is not known if any patients were excluded from the study during the wash‐out period prior to randomization Attrition: 36/176 (20%) patients withdrew from the study. 17/176 (10%) of these patients withdrew due to "treatment failures (i.e. unsatisfactory response to treatment)": 6 (13%), 1 (2%), 2 (5%) and 8 (21%) in the chlorthalidone 12.5, 25.0, 50 mg and placebo groups, respectively. It is not known for what reasons the other 19 patients withdrew WDAEs: 19/176 (11%) of patients withdrew due to adverse events (referred to as "adverse reactions"). 3 (6%), 4 (9%), 7 (15%) and 5 (13%) patients from the chlorthalidone 12.5, 25.0, 50.0 mg and placebo groups, respectively. The precise reasons were not given |
| Selective reporting (reporting bias) | High risk | Body weight, pulse rate and ECG were measured but actual values were not shown at endpoint. Results for serum chemistry parameters (except for potassium and uric acid levels), hematology and urinalysis were not shown. Mean BP (from baseline to endpoint) was presented in graph form only (Fig 1 and 2, p.201; no values were shown) and variability was not given Mortalities: 1 patient from the chlorthalidone 50.0 mg group died of "ventricular fibrillation". SAEs: not stated explicitly by the study's authors. The total number of patients with AEs (referred to as "adverse reactions") was shown (chlorthalidone 12.5, 25.0, 50.0 mg and placebo: 22, 23, 31 and 21 patients), but a complete listing of descriptions for the AEs was not; only the 5 most frequently occurring AEs were mentioned in the study (data were pooled) |
| Industry sponsorship | High risk | Supported by a grant from USV Laboratories |
Moser 1991.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 2 to 4 weeks. Multicenter, conducted in USA | |
| Participants | Sitting DBP 95 to 114 mmHg. Mean age 50 years. Males 66%. Baseline BP range was 151 to 154/101 to 102 mmHg | |
| Interventions | Benazepril 2 mg (N = 34), 5 mg (N = 38), 10 mg (N = 34) or 20 mg/d (N = 36), hydrochlorothiazide 25 mg/d (N = 33) or placebo (N = 31) Trial duration = 4 weeks (followed by a 2‐week open‐label HCTZ phase in non‐responders; not to be discussed here) |
|
| Outcomes | Mean trough sitting, standing and supine DBP and SBP (at 1‐week intervals from weeks 1 to 4); response rate; pulse rate; ECG and on a periodic basis plasma renin activity, hematology, urinalysis and serum biochemistry | |
| Notes | A sample size calculation was not provided. Study authors stated that there were no statistically significant differences (P value = NS) in baseline patient demographics and characteristics across treatment groups. Mortalities and SAEs not given. Subgroup analysis in black versus non‐black patients | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...patients were randomised to placebo, 25 mg, hydrochlorothiazide, or 2, 5, 10, or 20 mg benazepril once daily for a 4‐week double‐blind treatment period." (line 5 from top of p.323). No further information was given. |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "...patients were randomised to placebo, 25 mg, hydrochlorothiazide, or 2, 5, 10, or 20 mg benazepril once daily for a 4‐week double‐blind treatment period." (line 5 from top of p.323). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The primary efficacy analysis was based on an intention‐to‐treat (ITT) technique Exclusions: 87/293 (30%) of patients were excluded from the study during the single‐blind placebo run‐in period prior to randomization Attrition: 2/33 (6%) versus 5/31 (16%) of patients from the HCTZ and placebo groups, respectively withdrew from the study due to 1 ‐ "lost to follow up" and 1 ‐ "protocol violation", versus 2 ‐ "lost to follow up", 2 ‐ "lack of efficacy" and 1 ‐ "adverse experience" WDAEs: 1 patient from the placebo group withdrew due to an "adverse experience" for "lymphoma" |
| Selective reporting (reporting bias) | Unclear risk | Standing and supine BP, ECG, serum chemistry and urinalysis were measured, but actual values were not reported on at the end of the study. Mortalities and SAEs were not given. Total AEs (referred to in the study as "adverse experiences") were reported in terms of percentages of patients: HCTZ, 55%; placebo, 45%. Only the most frequently occurring AE (i.e. headache) was listed |
| Industry sponsorship | High risk | Supported by a grant from Ciba‐Geigy Pharmaceuticals |
Mroczek 1996.
| Methods | Randomized, double‐blind placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Multicenter, conducted in USA (and Germany) | |
| Participants | Sitting DBP 95 to 114 mmHg. Mean age 56 years. Males 66%. Baseline BP was 154/102 in the treatment group and 154/101 in the control group | |
| Interventions | Moexipril 7.5 mg (N = 51) or 15 mg/d (N = 47), HCTZ 25 mg/d (N = 51) or placebo (N = 51) Trial duration = 12 weeks | |
| Outcomes | Change from baseline in mean trough sitting and standing DBP and SBP (at 2‐week intervals); pulse rate; plasma renin activity and aldosterone; serum biochemistry and other laboratory tests (not specified) | |
| Notes | A sample size calculation was not provided.The study authors stated that the baseline patient demographics and characteristics including BP, pulse rates and duration of hypertension, were comparable across treatment groups (P ≥ 0.38). WDAEs, mortalities and SAEs not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | "This investigation was a randomised, double‐blind, multicenter, placebo‐controlled, parallel‐group study. After a 4‐week, single‐blind placebo period, patients who fulfilled the entrance requirements were randomised to receive moexipril 7.5 mg or 15 mg, HCTZ 25 mg, or placebo once a day for 12 weeks. (line 11 under "Patients and Methods" p.80). "Patients with mild hypertension (SiDBP 95 to 104 mmHg) were randomised according to a schedule in ascending order, whereas patients with moderate hypertension (SiDBP 105 to 114 mmHg) were randomised according to a schedule in descending order." (line 1 from top of p.81) |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "This investigation was a randomised, double‐blind, multicenter, placebo‐controlled, parallel‐group study". (line 11 under "Patients and Methods" p.80). "To enter the double‐blind period, the patients needed an SiDBP between 95 and 114 mmHg at the last two consecutive placebo follow‐up visits..." (line 1 from bottom of p.80). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Efficacy analysis was based on an intention‐to‐treat (ITT) technique Exclusions: 38/238 (16%) of patients were excluded from the study during the placebo run‐in period prior to randomization. Specific reasons for each patient were not given, however the authors did state that "inadequate therapeutic response was the most frequent reason..." (refer to line 3 under "Results" p.82) Attrition: 23/200 (11.5%) of patients withdrew from the study, 7/51 (14%) and 6/51 (12%) from the HCTZ and placebo groups, respectively. The specific reasons for withdrawing were not given, except that "inadequate therapeutic response" caused 2 patients to discontinue from the HCTZ group and 4 from the placebo group; the other 7 patients were not accounted for WDAEs: not given |
| Selective reporting (reporting bias) | High risk | Variability was not shown for the baseline patient demographics and characteristics and a medical history at baseline was not included in the study. Standing BP readings between 0 and 12‐week time points were measured, but not reported on (only endpoint changes were given). Pulse, serum biochemistry, plasma renin activity (PRA) and aldosterone were measured, but actual values were not reported on at endpoint. The study authors commented on PRA, aldosterone and serum potassium levels in the discussion section of the study only. Mortalities and SAEs were not documented. Total AEs: 53% (27/51) of patients in the HCTZ group and 61% (31/51) in the placebo group. Other AEs: the 2 most commonly occurring AEs were upper respiratory infection and headache |
| Industry sponsorship | High risk | Supported by a grant from Schwarz Pharma AG |
Myers 2000.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out = 4 weeks (single‐blind placebo run‐in). Multicenter; conducted in Europe and Canada | |
| Participants | In Europe: supine DBP 95 to < 114 mmHg; in Canada: 95 to < 109 mmHg. Mean age 56 years. Males 54% Baseline supine (standing) BP was 161/101 (160/103) in the treatment group and 164/102 (166/104) in the control group | |
| Interventions | Perindopril 2 mg, 4 mg or 8 mg/d + indapamide 0.625 mg, 1.25 mg or 2.5 mg/d (N = 317, all combined), indapamide 1.25 mg daily (N = 60) or placebo (N = 61) Trial duration = 8 weeks | |
| Outcomes | Change from baseline in mean trough supine and standing DBP and SBP; 24‐hour ambulatory BP, response rate; heart rate; serum biochemistry | |
| Notes | A sample size calculation was not provided. Markedly obese patients (BMI > 32) were excluded from the study. Patients received potassium supplementation if their serum potassium levels fell below 3.4 mmol/L. The study authors stated that there were no significant differences in the baseline patient characteristics across treatment groups. Total AEs not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...patients were randomly allocated to an 8‐week treatment period with either a perindopril/indapamide combination or placebo being administered using a double‐blind, parallel‐group study design." (line 9 under "Methods‐Patient population and inclusion criteria" p.318). "The study was a multinational, randomised, double‐blind comparison of perindopril and indapamide versus placebo using a seven‐way parallel‐group study design." (line 1 under "Study Design" p.318) |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "...patients were randomly allocated to an 8‐week treatment period with either a perindopril/indapamide combination or placebo being administered using a double‐blind, parallel‐group study design." (line 9 under "Methods‐Patient population and inclusion criteria" p.318) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Efficacy and safety analysis was based on an intention‐to‐treat technique with the last observation carried forward (LOCF) Exclusions: 58/496 (12%) of patients were excluded from the study during the placebo run‐in period prior to randomization. Specific reasons included: 12 patients for "patient choice", 19 ‐ supine DBP < 95 mmHg, 9 ‐ side effects, 6 ‐ supine DBP < 114 (Europe) or > 109 (Canada); 4 ‐ abnormal laboratory parameter, 3 ‐ investigator decision and 3 ‐ protocol violation. Note that 2 patients (i.e. their reasons for being excluded from the study) were not accounted for Attrition: 17/438 (3.9%) of patients withdrew from the study, 12 for adverse events, 4 for lack of efficacy and 1 for non‐medical reasons. The treatment groups from which these 17 patients withdrew were not mentioned WDAEs: 12/438 (2.7%) of patients withdrew due to AEs (dizziness, headache and nausea). The treatment groups from which these patients withdrew was not given |
| Selective reporting (reporting bias) | Low risk | Heart rate was measured, but not reported. Several serum biochemical levels, including sodium, uric acid, creatinine, glucose and cholesterol were reported in the results but not mentioned in the methods section. Variability in baseline patient characteristics and BP data was expressed in terms of standard error of the mean (SEM), rather than as standard deviation (SD). Mortalities: none. SAEs: 1 patient with myocardial infarction and 1 with angina reported as adverse events but that did not lead to death were mentioned, however there is no indication whether these patients were hospitalized or were forced to withdraw from the study. Total AEs: not reported. The incidence of 1 specific AE, cough, was reported (in the indapamide 1.25 mg group: 5%; in the placebo group: 0%) |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Papademetriou 2000.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 1 week wash‐out + single‐blind placebo run‐in of 4 to 5 weeks. Multicenter, USA | |
| Participants | DBP 95 to 114 mmHg. Mean age 52 years. Males 56%; blacks 21%. Average weight: males, 213 lb and females, 184 lb Baseline BP was 152/100 |
|
| Interventions | Candesartan 32 mg/d (N = 73), candesartan 32 mg/d + HCTZ 12.5 mg/d (N = 64), HCTZ 12.5 mg/d (N = 72) or placebo (N = 66) Trial duration = 8 weeks | |
| Outcomes | Change from baseline in trough sitting and standing DBP and SBP (at weeks 2, 4 and 8); peak SBP and DBP; BP response rate; heart rate; serum potassium levels and other laboratory parameters (not specified). Subgroup analyses were performed (black versus non‐blacks) | |
| Notes | A sample size calculation was not provided. The study included patients who had an average body weight of 213 and 184 lb. for men and women. Excess weight and obesity is associated with other diseases, which could have biased the BP and safety results. The medical history of patients at baseline was not included in the study. It was not stated whether there were statistically significant differences in baseline patient demographics and characteristics across treatment groups. BP data contained SEM (standard error of the mean), not SD. Total withdrawals and total AEs not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "[Patients] were randomly assigned to one of the following 4 groups: placebo, hydrochlorothiazide (HCTZ) 12.5 mg daily, candesartan 32 mg daily, or a combination of candesartan and HCTZ." (line 1 from top of p.373) |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "This was a multicenter, double‐blind, placebo‐controlled study of patients with essential hypertension." (line 1 under "Methods" p.373). "The duration of the double‐blind treatment phase was 8 weeks." (line 3 from top of p.373) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Primary efficacy analysis was based on an intention‐to‐treat (ITT) technique with the LOCF (last observation carried forward) Exclusions: not given Attrition: not given WDAEs: a total of 10/275 (3.6%) patients withdrew due to adverse events (5 from non‐thiazide treatment groups), 3/72 (4.2%) patients receiving HCTZ 12.5 mg and 2/66 (3%) patients on placebo. The precise reasons were not given |
| Selective reporting (reporting bias) | High risk | Body weight (except at baseline), heart rate and serum chemistry (except for potassium) were measured but actual values were not given. ECG was not reported. Mortalities: none. SAEs: 1 patient in the HCTZ group and 4 patients in the placebo group. Total AEs were not reported. The most commonly reported AEs were upper respiratory infections and headache (treatment groups not specified) |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Papademetriou 2006.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Factorial design. Wash‐out period = 4 to 5 weeks. Multicenter, USA | |
| Participants | Sitting DBP 95 to 114 mmHg and SBP < 180 mmHg. Mean age 53 years. Males ˜50%. Mean body mass index > 30: 57% of patients. Baseline BP was 151/100 | |
| Interventions | ER‐metoprolol 25 mg/ (N = 89), 50 mg (N = 94), 100mg (N = 96) or 200 mg/d (N = 52), ER‐metoprolol 25 mg, 50 mg, 100 mg or 200 mg/d + hydrochlorothiazide 6.25 mg/d or 25 mg/d (all combined, N = 849), hydrochlorothiazide 6.25 mg (N = 86), 12.5 mg (N = 105), 25 mg daily (N = 48) or placebo (N = 152) Trial duration = 8 weeks (followed by a 2‐week drug taper period) | |
| Outcomes | Change from baseline in trough sitting and standing DBP and SBP; peak BP; BP response rate; ECG, heart rate, hematology, urinalysis and serum biochemistry. Subgroup analyses were performed (i.e. based on age, sex and race) | |
| Notes | A sample size calculation was provided and was based on 1485 patients to detect a difference in standard deviation of 8 mmHg (sitting DBP) at 80% power. The majority of patients in the study (57%) were overweight with a body mass index (BMI) over 30, a characteristic that is not reflected in most other trials included in this review. The study authors stated that baseline patient characteristics were well balanced across treatment groups, however the reviewers determined there to be statistically significant differences (P value < 0.05) in terms of mean age, incidence of diabetes mellitus and BP goals above JNC‐7 at baseline. Total AEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "This was a multicenter (in the USA), randomised, double‐blind, placebo‐controlled, parallel group, unbalanced factorial study..." (line 1 under "Study Design" p.1218) "...eligible patients were randomly allocated to one of the 17 treatment groups..." (line 4 from bottom of p.1218). "A central, computer‐generated randomisation schedule using an interactive voice response system allocated patient to treatment groups within the study centre. As certain treatment groups were of greater importance for pair wise comparisons, patients were assigned in an allocation ratio of 1 (indicating least important), 2 (more important), or 3 (most important)". (line 1 under "Randomization, Blinding and Study Treatments" p.1218) |
| Allocation concealment (selection bias) | Low risk | "A central, computer‐generated randomisation schedule using an interactive voice response system allocated patient to treatment groups within the study centre." (line 1 under "Randomization, Blinding and Study Treatments" p.1218) |
| Blinding (performance bias and detection bias) All outcomes | Low risk | The dose of ER‐metoprolol was doubled after 1 week of therapy, however the investigators stated that it was a "blinded escalation to the assigned dose". (line 1 from top of p.1218) "To blind study treatments, HCTZ in identically appearing tablets of 6.25, 12.5, or 25 mg, or matching placebo and ER‐metoprolol tablets of 25 or 100 mg or matching placebo, were blister‐card packaged according to a double‐dummy design." (line 7 under "Randomization, Blinding and Study Treatments p.1218) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Primary efficacy analysis was based on an intention‐to‐treat (ITT) technique with the LOCF (last observation carried forward) Exclusions: 1260/2831 (44.5%) of patients were excluded from the study during the placebo run‐in period prior to randomization. Some of the reasons included patients failing eligibility criteria (814) and those withdrawing consent (262) Attrition: 176/1571 (11%) of patients withdrew from the study for the following reasons: 46 ‐ "adverse event", 51 ‐ "lack of therapeutic response", 45 ‐ "patient consent withdrawn", 19 ‐ "loss to follow‐up" and 15 ‐ "other reasons". Note that since the data were combined, it was impossible to ascertain from which treatment groups the withdrawn patients originated (there were 17 treatment groups in total, some with patients receiving non‐thiazide mono‐ and combo‐therapies) WDAEs: a total of 46/1571 (2.9%) patients withdrew due to adverse events (5 from non‐thiazide treatment groups), 3/72 (4.2%) patients receiving HCTZ 12.5 mg and 2/66 (3%) patients on placebo. The precise reasons were not given |
| Selective reporting (reporting bias) | Unclear risk | The study was a 4 x 5 factorial design which means there were 20 possible treatment groups to assign patients, however the authors stated that 3 of the groups (combo‐therapy: ER‐metoprolol/HCTZ: 200/6.25, 25/25 and 50/25 mg) were not investigated (see line 5 under "Study Design" p.1218). Results for serum chemistry (except for potassium and uric acid levels), hematology, lipids and urinalysis tests and ECG were measured but actual values were not reported at the study's endpoint. Standing BP data were measured, but not reported on. Peak:trough BP was measured but not reported for each separate treatment group. Subgroup analyses of sex (male versus female) age (< 65 years versus ≥ 65 years) and ethnicity (African American versus other) were documented in the results section of the study only (actual values were not shown for sex and age differences); but not included in the methods. Mortalities: none. SAEs: 5 patients had a "coronary artery disease manifestation" e.g. myocardial infarction, but it was not stated from which treatment group the patients originated. Total AEs were not reported. Only the single most commonly occurring AE (headache) was reported in the study |
| Industry sponsorship | High risk | Supported by AstraZeneca LP |
Persson 1996.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Multicenter; conducted in Germany and Sweden | |
| Participants | DBP 95 to 114 mmHg. Mean age 70 years. Males 57%. Baseline BP was 171/102 in the treatment group and 172/103 in the control group | |
| Interventions | Moexipril 7.5 mg (N = 50) or 15 mg/d (N = 53), HCTZ 25 mg/d (N = 50) or placebo (N = 48) Trial duration = 8 weeks | |
| Outcomes | Change from the baseline in mean trough sitting and standing SBP and DBP (at 2, 4, 6 and 8 weeks); BP response rate; trough:peak BP ratios; pulse rate; ECG, urinalysis, hematology, body weight, serum biochemistry; plasma renin activity (PRA) and aldosterone. Subgroup analysis was performed based on mild versus moderate hypertension | |
| Notes | A sample size calculation was not provided. This study included elderly patients only (65 to 80 years of age). Elderly patients present with a different set of problems, some of which are more concomitant illnesses and metabolic differences, so results of this study may differ. Study authors stated that age and DBP of patients at baseline were not significantly different (P value = NS) across treatment groups. Biochemical data (except for PRA and aldosterone) not available. Mortalities not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel study..." (line 1 under "Patients and Methods" p.259) "...patients were randomised to treatment with moexipril 7.5 mg, moexipril 15 mg, HCTZ or placebo for 8 weeks." (line 8 under "Protocol" p.260). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel study..." (line 1 under "Patients and Methods" p.259). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Primary efficacy analysis was based on an intention‐to‐treat (ITT) technique Exclusions: 88/289 (30%) of patients were excluded from the study during the single‐blind, placebo run‐in period prior to randomization. The specific reasons were not given Attrition: a total of 17/201 (8.4%) of patients withdrew from the study for the following reasons: 6 ‐ "withdrawn consent, protocol violations, or failure to meet criteria for continuation"; 4 ‐ "inadequate therapeutic response (1 patient receiving moexipril (group was not given: 7.5 mg or 15 mg?) and 3 on placebo); and 7 ‐ "adverse experiences". Note that data were pooled, not presented as separate according to each treatment group WDAEs: 7/201 (3.5%) of patients withdrew due to "adverse experiences". The reasons included, 3/50 (6%) of patients in the HCTZ treatment group: 1 ‐ "malignancy", 1 ‐ atrial fibrillation" and 1 ‐ "serum creatinine increased"; and 1/48 (2.1%) patient in the placebo group for "myocardial infarction". (Note: the other 3 patients were receiving a non‐thiazide drug) |
| Selective reporting (reporting bias) | Unclear risk | Variability of the mean in baseline patient demographics and characteristics and in results for serum biochemistry (except aldosterone and plasma renin activity), heart rate, ECG and urinalysis were not given. SEM (standard error of the mean) was included in BP data and in PRA and aldosterone; instead of SD Trough: peak ratios were reported in the results section of the study, but not documented in the methods as an outcome to be measured. Mortalities: not given. SAEs: 2 patients in the HCTZ treatment group, 1 each for "haemorrhoid bleeding" and "second‐degree heart block". No patients in the placebo group had SAEs. Total AEs: 28% (14/50) of patients in the HCTZ group and 35% (17/48) of patients on placebo. The authors listed only the most commonly occurring AEs, "headache" and "respiratory symptoms" |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Philipp 1997.
| Methods | Randomized, double‐blind, placebo‐controlled trial. Factorial design. Wash‐out period = 2 to 6 weeks. Multicenter, conducted in Germany | |
| Participants | DBP 95 to 110 mmHg. Mean age 55.1 years | |
| Interventions | Candesartan 2 mg, 4 mg, 8 mg or 16 mg/d, candesartan 2 mg, 4 mg, 8 mg or 16 mg/d + HCTZ 12.5 mg or 25 mg/d, HCTZ 12.5 mg (N = 60) and 25 mg/d (N = 123) or placebo (N = 119). N = 1096 randomized Trial duration = 8 weeks | |
| Outcomes | Change from the baseline in mean sitting DBP and SBP (measured using an automated BP device); BP response rate; heart rate, ECG, urinalysis and blood tests | |
| Notes | A sample size calculation was not provided. Study authors stated that baseline patient demographics and characteristics, including medical history and previous treatment with antihypertensives were "well matched" across treatment groups. Baseline BP was not given. Variability in BP not given. Mortalities, SAEs and total AEs were not given. Biochemical data measured but not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...1096 patients were randomised to once‐daily oral treatment with candesartan cilexetil 2, 4, 8, or 16 mg, HCTZ 12.5 or 25 mg, combination therapy with both agents at these respective doses, or placebo." (line 7 under "Patients and Methods" p.S67). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "[Patients] were recruited to this double‐blind, factorial design study..." (line 3 under "Patients and Methods" p.S67). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Primary efficacy analysis was based on an intention‐to‐treat (ITT) technique Exclusions: 210/1306 (16%) of patients were excluded from the study during the wash‐out and placebo run‐in periods prior to randomization. The specific reasons were not given Attrition: the number of patients who withdrew from the study was not given nor how their data were analyzed has been provided WDAEs: not clearly documented. It was stated that "2.4% of patients withdrew from the study due to adverse occurrences." (line 7 from top of p.S68), but no further information was given |
| Selective reporting (reporting bias) | High risk | Heart rate, ECG, serum chemistry and urinalysis were measured, but not given. Except for mean age, all other baseline patient demographics and characteristics were undocumented. Variability was not included in BP data. Baseline BP was not reported. Mortalities, SAEs and total AEs were not given |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Pool 1993.
| Methods | Randomized, double‐blind, placebo‐controlled trial. Wash‐out period = 4 to 6 weeks. Multicenter, USA | |
| Participants | Mean age 53 years. Males 67%. Supine DBP 95 to 110 mmHg. Supine SBP 153.4 + 2.2 (SEM) mmHg and supine DBP 100.2 + 0.5 (SEM) mmHg | |
| Interventions | Diltiazem SR 120 mg q12h (N = 73), diltiazem SR 120 mg q12h + HCTZ 12.5 mg q12h (N = 77), HCTZ 12.5 mg q12h (N = 74), or placebo (N = 74) Trial duration = 6 weeks | |
| Outcomes | Change from the baseline in mean trough supine and standing DBP and SBP; 12‐hour BP monitoring; heart rate; ECG; hematology and serum biochemistry. Subgroup analyses were performed (based on age, race, weight, sex and smoking status) | |
| Notes | A sample size calculation was not provided. Both male and female patients were, on average overweight or obese which has several implications including a higher incidence of concomitant illnesses compared to other studies reviewed here. 114 of the study's patients selected from certain clinics underwent BP monitoring while in clinic both at baseline and at endpoint to determine residual drug effects every 2 hours for up to 12 hours post‐dose. Study authors stated that the baseline patient demographics and characteristics were not significantly different across treatment groups. WDAEs, mortalities and SAEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Eligible patients were allocated to one of four treatment groups, as noted above, using a blinded, blocked randomisation schedule." (line 1 under "Randomization and Dose Selection" p.489) |
| Allocation concealment (selection bias) | Low risk | "Eligible patients were allocated to one of four treatment groups, as noted above, using a blinded, blocked randomisation schedule. (line 1 under "Randomization and Dose Selection" p.489) |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "In this multicenter, randomised, double‐blind, placebo‐controlled, parallel group trial, DTZ SR‐HCTZ 120 mg‐12.5 mg (Cardizide), DTZ SR 120 mg (Cardizem SR), HCTZ 12.5 mg, or placebo was administered every 12 hours." (line 1 under "Methods‐design" p.488). "All drugs and placebo were identical in appearance." (line 3 under "Randomization and Dose Selection" p.489) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Primary efficacy analysis was based on an intention‐to‐treat (ITT) technique Exclusions: 206/504 (41%) of patients were excluded from the study during the single‐blind, placebo run‐in period prior to randomization Attrition: 44/298 (15%) of patients withdrew from the study. 10 patients withdrew for "protocol violations", but the specific reasons were not given for the other 34 patients. Data were pooled from all treatment groups, therefore it could not be determined from which groups the withdrawing patients originated WDAEs: not given |
| Selective reporting (reporting bias) | High risk | Medical history was not included in the baseline patient demographics and characteristics. Heart rate and ECG were measured, but not reported on at the study's endpoint. Multivariate analyses comparing BP effects based on age, sex, weight, ethnicity and smoking status were conducted without any details included in the methods section of the study. Variability in residual BP and HR effects (i.e. 0 to 12 hours post‐dose) was not reported. Mortalities and SAEs were not documented. All adverse events were not reported. Treatment‐related adverse events: 40.5% (30/74) of patients in the HCTZ group and 27% (20/74) of patients in the placebo group. Only the 8 most commonly reported AEs "possibly" related to the study drug were listed |
| Industry sponsorship | High risk | Funded by a research grant from Marion Merrell Dow Inc. |
Pool 1997.
| Methods | Randomized, double‐blind, placebo‐controlled trial. Modified 4 x 4 factorial design. Wash‐out period = 4 to 5 weeks. Multicenter, USA | |
| Participants | Mean age 51.5 years. Males 61%. Body weight: 90.5; body mass index: 30.6. Seated DBP 95 to 110 mmHg. Seated SBP 149.5 + 15.7 mmHg and seated DBP 100.1 + 4.0 mmHg | |
| Interventions | Fosinopril 2.5 mg (N = 33), 10 mg (N = 30) or 40 mg/d (N = 32), fosinopril 2.5 mg, 10 mg, 20 mg or 40 mg/d + HCTZ 5 mg, 12.5 mg or 37.5 mg/d (all combined, N = 325), HCTZ 5 mg (N = 32), 12.5 mg (N = 32) or 37.5 mg/d (N = 32), or placebo (N = 32) Trial duration = 8 weeks | |
| Outcomes | Change from the baseline in mean trough sitting, standing and supine DBP and SBP; response rate; heart rate; hematology, urinalysis, serum biochemistry | |
| Notes | A sample size calculation was not provided. Patients in the study were overweight/obese with an average BMI of 31. Study authors stated that except for seated DBP, the baseline patient demographics and characteristics were not significantly different across treatment groups (P value = NS). Variability in BP not given. Biochemical data measured but not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The...study was a modified 4 x 4 factorial, randomised, double‐blind, parallel group trial of 17 different doses of the combination of Fos and HCTZ, comparing each of the combinations with the individual components and placebo." (line 1 under "Study Design" p.118). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "The...study was a modified 4 x 4 factorial, randomised, double‐blind, parallel group trial..." (line 1 under "Study Design" p.118). "Subjects took their first randomised, double‐blind medication under supervision in the clinic..." (line 20 under "Study Design" p.118). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Primary efficacy analysis was based on both an intention‐to‐treat (ITT) and per‐protocol technique, however the study included data from the ITT population only Exclusions: 159/709 (22.4%) of patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: 45/550 (8%) of patients withdrew from the study; the specific reasons were not given. Data were pooled from all treatment groups, therefore it could not be determined from which groups the patients originated WDAEs: 19/550 (3.5%) of patients withdrew due to adverse events; this included 1% of patients from the HCTZ 5, 12.5 and 37.5 mg groups combined and 3.1% of patients from the placebo group. The specific reasons were not given |
| Selective reporting (reporting bias) | High risk | Parameters for biochemistry, hematology and urinalysis were measured, but not reported on. Variability in BP data was not reported. Serum biochemistry, hematology and urinalysis results were measured but not reported. Mortalities and total AEs were not documented. There were 5 SAEs; specific reasons were not given |
| Industry sponsorship | High risk | Supported by grant from Bristol‐Myers Squibb Company |
Pool 2007.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 2 weeks, followed by a 2 to 4‐week single‐blind, placebo run‐in. Multicenter, conducted in USA and Canada | |
| Participants | Mean age, all HCTZ groups combined (˜53 years); placebo (52 years). Males, all HCTZ groups (62%); placebo (49%). Body mass index, HCTZ group (32); placebo (31). Sitting DBP 90 to < 110 mmHg after wash‐out. SBP 151.2 + 12.7 mmHg and DBP 99.1 + 3.9 mmHg in HCTZ group and SBP = 150.4 + 12.7 mmHg and DBP = 99.1 + 3.7 in placebo group | |
| Interventions | Valsartan 160 mg (N = 166) or 320 mg/d (N = 170), valsartan 160 mg or 320 mg/d + HCTZ 12.5 mg or 25 mg/d (all combined, N = 505), HCTZ 12.5 mg (N = 169), 25 mg/d (N = 167) or placebo (N = 169) Trial duration = 8 weeks | |
| Outcomes | Change from the baseline to endpoint in mean trough seated DBP and SBP; response rate; pulse rate; hematology, serum biochemistry | |
| Notes | A sample size calculation was provided (number of patients per treatment group not specified) to detect a difference of 3 mmHg in seated DBP (SD ± 8 mmHg) at 90% power. Patients in the study were overweight/obese with an average BMI of 31 to 32. Study authors stated that except for gender (P value = 0.03), the baseline patient demographics and characteristics, including BP were not significantly different across treatment groups (P value = NS). SE (standard error of mean) was given for BP, not SD. Biochemical data incomplete. SAEs and total AEs were not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...this study was an 8‐week, multicenter, randomised, double‐blind, placebo‐controlled, parallel‐group trial..." (line 1 under "Patients and Methods" p.62). "...patients who met the study inclusion criteria were randomised (visit 3, week 0) in a double‐blind fashion to 1 of 8 treatment groups: VAL monotherapy at a dose of 160 or 320 mg; HCTZ monotherapy at 12.5 or 25 mg; VAL/HCTZ 160/12.5, 320/12.5, or 320/25 mg; or placebo." (line 7 under "Study Design" p.62). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "...this study was an 8‐week, multicenter, randomised, double‐blind, placebo‐controlled, parallel‐group trial..." (line 1 under "Patients and Methods" p.62). No further detail provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Primary efficacy analysis was based on an intention‐to‐treat (ITT) technique with the LOCF (last observation carried forward) Exclusions: the number of patients excluded from the study during the placebo run‐in period prior to randomization was not given Attrition: 185/1346 (14%) of patients withdrew from the study. Data on total withdrawals was incomplete. Study authors selectively reported the specific reasons i.e. "the most common reasons for discontinuations were unsatisfactory therapeutic effect (4.4%), AEs (3.1%) and withdrawn consent (2.2%)." (see line 8 under "Results" p.65). Note that data were pooled from all treatment groups, therefore it could not be determined from which groups the patients originated WDAEs: 3.1% of patients withdrew due to adverse events. The specific reasons were not given and no details were given as to which treatment groups the patients originated. The study authors were selective in their reporting of WDAEs, i.e. in terms of a range of percentages only |
| Selective reporting (reporting bias) | High risk | Pulse rate, hematology, serum biochemistry (except for potassium levels) were measured, but not reported. BP data included SE (standard error of the mean), not SD. Variability in BP data and in serum potassium levels were not reported. Mortalities: 1 patient receiving valsartan 320 mg died (reason was not given). No patients from HCTZ or placebo groups died. SAEs: not given. Total AEs were not reported; study authors were selective in their reporting of AEs, i.e. in terms of a range of percentages and a listing of the 3 most commonly occurring AEs only |
| Industry sponsorship | High risk | Funded by Novartis Pharmaceuticals Corporation |
Pordy 1994.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). 4 x 3 factorial design. Wash‐out period = 4 weeks. Multicenter, USA | |
| Participants | Mean age 54 years. 60% males. Sitting DBP 95 to 115 mmHg. Baseline DBP = 100.3 mmHg | |
| Interventions | Cilazapril 0.5 mg, 5 mg or 10 mg/d (all combined, N = 288), cilazapril 0.5 mg, 5 mg or 10 mg/d + HCTZ 12.5 mg or 25 mg/d (all combined, N = 579), HCTZ 12.5 mg or 25 mg/d (all combined, N = 198), or placebo (N = 97) Trial duration = 4 weeks | |
| Outcomes | Change from baseline in trough mean sitting DBP; response rate; peak BP and trough:peak ratios; heart rate, ECG, body weight, hematology, serum biochemistry, urinalysis | |
| Notes | A sample size calculation was not provided. Study authors stated that there were no "overt differences" (P value = NS) in baseline patient demographics and characteristics across treatment groups. Total AEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This double‐blind, randomised, placebo‐controlled, multicenter study compared the effects of three doses of the ACE inhibitor, cilazapril (CLZ) and two doses of HCTZ, alone and in combination." (line 21 from top of p.312). "...patients [were] randomly assigned in blocks of 12 to one of 12 treatment groups." (line 1 under "Materials and Methods‐Study Design" p.312) |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "...period II (active treatment) consisted of a 4‐week, double‐blind, placebo‐controlled comparison of three doses of CLZ and two doses of HCTZ, alone and in combination." (line 4 from top of p.312). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The primary efficacy analysis was based on an intention‐to‐treat (ITT) technique Exclusions: 856/2018 (42%) of patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: 74/1088 (6.8%) of patients withdrew from the study, 13/198 (6.6%) from the HCTZ 12.5 and 25 mg groups combined and 11/97 (11%) from the placebo group (the other 50 patients were receiving non‐thiazides). The reasons for withdrawing were given, but with the data pooled, it was not possible to ascertain from which treatment groups the patients originated WDAEs: 33/1088 (3%) of patients withdrew due to adverse events, 5 (2.5%) from the HCTZ 12.5 and 25 mg group and 3 (3%) from the placebo group; the other 25 patients were receiving non‐thiazides. No specific reasons were given |
| Selective reporting (reporting bias) | High risk | Heart rate, body weight, ECG and laboratory results including serum biochemistry, hematology, lipids and urinalysis were measured but actual values were not reported on at either baseline or endpoint. BP data were expressed in terms of mean ± SEM (standard error of the mean). Mortalities: 1 death from the CLZ + HCTZ combo therapy group. None from HCTZ or placebo groups. SAEs: 30 patients in total, including 6 from the HCTZ 12.5 and 25 mg groups, combined. Total AEs were not reported. Only AEs related in some way to the medication were reported |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Prisant 2000.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Multicenter, USA | |
| Participants | Supine DBP 95 to 114 mmHg. Mean age 51.6 years. Males 60%. Baseline BP was 156.7/101.8 mmHg in the indapamide 2.5 mg group and 153.4/100.5 mmHg in the placebo group | |
| Interventions | Diltiazem XR 120 mg, 180 mg, 240 mg, or 360 mg/d (all combined, N = 187), diltiazem XR 120 mg, 180 mg or 240 mg/d + indapamide 1.25 mg or 2.5 mg/d (all combined, N = 75), indapamide 1.25 mg (N not reported) or 2.5 mg/d (N = 24), or placebo (N = 43) Trial duration = 6 weeks | |
| Outcomes | Change from baseline in mean trough supine SBP and DBP; 24‐hour ambulatory BP (using an automated monitoring device); ECG, hematology, urinalysis, serum biochemistry | |
| Notes | A sample size calculation was not provided. The small sample in the indapamide treatment group could have biased the results. Forced‐titration schedule for all patients assigned to receive non‐thiazides in the 1st week of DB treatment. Female patients were in the majority in the placebo group (60%), but in the minority in the indapamide 2.5 mg group (25%). Study authors stated that baseline patient demographics of age, race and gender were "evenly distributed" across treatment groups. Study does not report on the patient group receiving 1.25 mg/d indapamide due to large variability relative to effect. Total withdrawals and total AEs not given. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This was a randomised, double‐blind, placebo‐controlled, parallel‐group, multicenter study in patients with mild to moderate hypertension. After 4 weeks on single‐blind placebo treatment, qualifying patients were randomly assigned to double‐blind medication." (line 1 under "Methods‐Study Design" p.178). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Blinding was achieved by placing diltiazem XR tablets and indapamide powder in capsules, which were identical in appearance, size, shape and colour to those of their respective placebos. Each patient took three capsules of medication once a day in the morning." (line 11 under "Methods‐Study Design" p.178) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | It was not stated whether the primary efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol technique Exclusions: the number of patients excluded from the study during the placebo run‐in period prior to randomization was not given. However, because of the high variability in the BP effect in patients receiving indapamide 1.25 mg/day, this treatment arm was excluded from the analysis of results (presumed to be an N = 67 patients) Attrition: not given WDAEs: 29 (7.3%) patients withdrew due to adverse events (referred to by the authors as "adverse clinical events"). The specific reasons were not given and no details were given as to which treatment groups the patients originated from |
| Selective reporting (reporting bias) | High risk | Serum chemistry (except potassium and uric acid levels), hematology, urinalysis and ECG were measured, but actual values were not reported on at endpoint. Statistical analysis of the effects of indapamide on ambulatory 24‐hour BP changes was not performed. Study authors removed patients in the indapamide 1.25 mg treatment group from analysis due to large variability effect in that group. Variability in BP data was not given. Mortalities: None. SAEs: 17 patients (treatment group not specified and reasons not given). Total AEs not reported. There was no detailed list of each AE patients experienced. |
| Industry sponsorship | High risk | study was sponsored by manufacturer |
Roque 1996.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period ≥ 1 week. Multicenter, Argentina | |
| Participants | DBP 95 to 115 mmHg. No black patients. Mean age 62.5 years. Males 41%. Baseline BP was 160.2/98.8 mmHg in the treatment group and 163.4/99.1 mmHg in the control group | |
| Interventions | HCTZ 12.5 mg/d (N = 49) or placebo (N = 51) Trial duration = 8 weeks | |
| Outcomes | Change from baseline in mean supine SBP and DBP (at 4 and 8 weeks); BP response rate; heart rate. Subgroup analyses were performed (based on sex, age, severity of hypertension) | |
| Notes | A sample size calculation was provided based on 100 patients (50 per treatment group) to detect a difference of 4 mmHg (SD ± 7 mmHg) between groups at a power 80%. Patients were enrolled without regard to previous anti‐hypertensive medication (sensitivity to prior drugs was most likely a case of the study authors not stating it, but it was possibly overlooked) (refer to inclusion criteria p.285). Study authors stated that baseline patient demographics and characteristics were not significantly different across treatment groups. The majority of patients were female with mild hypertension (in both HCTZ and placebo groups). Total withdrawals, WDAEs, biochemical data, mortalities, SAEs and total AEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | This was a..."prospective, multicenter, randomised, double‐blind, placebo‐controlled trial conducted in Argentina." (line 2 under "Patients and Methods" p.285). No further information given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Coded medications were prepared by the Pharmaceutical Development Center at the University Medical School of South Carolina. All qualified patients were randomised to receive either a 12.5 mg HCTZ tablet or a matched placebo capsule once daily in the morning." No further information given |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Primary efficacy analysis was based on the per‐protocol population of patients as well as the an intention‐to‐treat (ITT) population with LOCF (last observation carried forward) Exclusions: 18/118 (15%) of patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: total withdrawals not stated explicitly WDAEs: not given |
| Selective reporting (reporting bias) | High risk | Heart rate and AEs were measured but not reported at the end of the study. For AEs, the authors stated "no side effects attributable to either study medication were reported." (p.289). Serum biochemistry, hematology, urinalysis and ECG were not reported. Mortalities, SAEs and total AEs were not given |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Saruta 2007.
| Methods | Randomized, double‐blind, placebo‐controlled trial. Wash‐out period = 4 weeks. Multicenter; Japan, USA and Peru | |
| Participants | Sitting DBP 95 to 115 mmHg. Mean age 55 years. Males 59%. Baseline BP was 155/99.8 mmHg in the HCTZ 12.5 mg group and 153/100 mmHg in the placebo group | |
| Interventions | Losartan 50 mg/d (N = 160), losartan 25 mg or 50 mg/d + HCTZ 6.25 mg or 12.5 mg/d (all combined, N = 472), HCTZ 12.5 mg/d (N = 163) or placebo (N = 159) Trial duration = 8 weeks | |
| Outcomes | Change from the baseline in mean trough sitting, standing and supine DBP and SBP; heart rate, ECG, body weight, laboratory tests (including serum biochemistry and urinalysis). Subgroup analyses were performed (based on age and severity of hypertension) | |
| Notes | A sample size calculation was not provided. Study authors stated that there were no significant differences in the baseline patient demographics and characteristics across treatment groups. Patients were of Japanese descent. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This randomised, placebo‐controlled, double‐blind, parallel‐group study..." (line 1 under "Study Procedures" p.730). "..those [patients] who continued to fulfil the eligibility criteria were randomised into 1 of 6 treatment arms..." (line 20 from bottom of p.731). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "If eligible, patients were given 5 placebo tablets matched to each of losartan 50 mg plus HCTZ 12.5 mg, losartan 50 mg plus HCTZ 6.25 mg, losartan 25 mg plus HCTZ 6.25 mg, losartan 50 mg and HCTZ 12.5 once daily for 4 weeks." (line 9 from top of p.731) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The primary efficacy analysis was based on a "full‐analysis set" with the LOCF (last observation carried forward) Exclusions: it was not stated whether any patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: the total number of withdrawals was not given, except that there were 7 patients excluded from both efficacy and safety analyses in whom "previous medication for essential hypertension was discontinued or tapered before obtaining written consent" WDAEs: 4/163 (2.5%) patients receiving HCTZ12.5 and 1/159 (0.6%) patients receiving placebo withdrew due to adverse events; the specific reasons were not given |
| Selective reporting (reporting bias) | High risk | Trough standing BP, heart rate, ECG and body weight were measured, but not reported in the study. BP data at 2, 4 and 6 week time points were graphed. Results for the subgroup analyses comparing BP effects in terms of age and severity of hypertension were not shown. Serum biochemistry (except for uric acid, potassium and glucose levels) was not reported. Mortalities: none. SAEs: 9 patients (reasons were not given; and treatment groups were not specified). Total AEs were not clearly documented; they were reported separately as: "clinical" and "laboratory" AEs. By combining these 2 groups, it was then determined that 107/163 (65.6%) and 103/159 (64.8%) patients in the HCTZ and placebo groups, respectively, experienced AEs. A description of the 6 most commonly occurring "clinical" AEs in at least 2% of patients were reported. In addition, 5 of the most commonly occurring "laboratory" AEs in at least 4% of patients were reported |
| Industry sponsorship | High risk | Sponsored by Banyu Pharmaceutical Co. |
Schmieder 2009.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 2 weeks followed by a single‐blind placebo run‐in of 2 to 5 weeks. Multicenter; Belgium, Finland, Germany, Italy, the Netherlands and Spain | |
| Participants | DBP 90 to < 110 mmHg. Mean age in HCTZ group: 56 years. Males 56%, white 99%, obese 34% in HCTZ group Baseline BP in HCTZ group was 154.3/99.0 mmHg (PLB arm demographics/characteristics not mentioned) | |
| Interventions | Aliskiren 150 mg/d forced titrated at week 3 to 300 mg/d (N = 459), HCTZ 12.5 mg/d forced titrated at week 3 to 25 mg/d (N = 444) or placebo (N = 221) Duration 12 weeks (full trial duration including extension phase was 52 weeks). After the first 6 weeks patients in the placebo arm were re‐assigned to aliskiren or HCTZ and after 12 weeks, combination therapy was introduced with aliskiren 300 mg/d + amlodipine 5 mg or 10 mg/d, or HCTZ 25 mg/d + amlodipine 5 mg or 10 mg/d | |
| Outcomes | Change from the baseline to endpoint in mean trough sitting DBP and SBP; response rate; ECG, hematology, urinalysis and serum biochemistry. Subgroup analysis was performed (based on age) | |
| Notes | A sample size calculation was provided based on approximately 440 patients in each active treatment group to detect a non‐inferiority margin of 2 mmHg between aliskiren and HCTZ regimens (SD ± 8 mmHg) at a power of 95%. The study authors stated that there were no statistically significant differences in the baseline patient demographics and characteristics between the HCTZ and aliskiren (not classified as a thiazide) treatment groups. The study did not make comparisons between HCTZ and placebo groups. Baseline patient demographics and characteristics were not reported in the placebo group. Patients randomized to receive the placebo were on it for 6 weeks before being switched to active treatment, therefore only BP data of up to 6 weeks can be used in this review, including that from the HCTZ group. Another important note: after 3 weeks on HCTZ 12.5 mg, patients were given a forced titration to 25 mg |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "...[a] randomised, double‐blind, parallel‐group, active‐controlled, dose‐titration study was performed..." (line 1 under "Study Design" p.418). "Randomization by centre was performed by the interactive system that automates the random assignment of patients to randomisation numbers." (line 19 from top of p.418, right column) |
| Allocation concealment (selection bias) | Unclear risk | "Randomization data were kept strictly confidential until the time of unblinding." (line 22 from top of p.418, right column). No further information was given |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "...[a] randomised, double‐blind, parallel‐group, active‐controlled, dose‐titration study was performed..." (line 1 under "Study Design" p.418). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The primary efficacy analysis was based on an intention‐to‐treat (ITT) technique with the LOCF (last observation carried forward) Exclusions: 151/1275 (12%) of patients were excluded from the study during the placebo run‐in period prior to randomization (specific reasons were given) Attrition: 67/1124 (6%) of patients withdrew from the study after 6 weeks of DB therapy; 26/444 (5.9%) and 21/221 (9.5%) of patients from the HCTZ and placebo groups, respectively. The main reasons for withdrawing: AEs (see below) and withdrawal of consent (HCTZ: 11 patients; placebo: 6 patients) WDAEs: 5/444 (1%) of patients in the HCTZ group and 6/221 (2.7%) of patients from the placebo group withdrew due to adverse events |
| Selective reporting (reporting bias) | High risk | Baseline demographics and characteristics were not reported in patients on the placebo. Results from physical exams, hematology, biochemistry and urine samples were not reported at baseline and only BUN and serum potassium and creatinine levels (patients were grouped according to certain threshold levels; actual mean values were not given) were reported at the study's endpoint. Mortalities: none. SAEs: Only those considered by the investigator to be related to the study medications were counted; they included 1 from the HCTZ group (moderate hypokalemia) and 1 from the placebo group (myocardial infarction). Total AEs: 24.5% of patients in HCTZ group and 28.5% in the placebo group experienced AEs. A listing of other AEs and laboratory results from patients in the placebo group were not reported |
| Industry sponsorship | High risk | Supported by Novartis Pharmaceuticals |
Schoenberger 1995.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Multicenter, USA | |
| Participants | DBP 95 to 115 mmHg. Mean age 53.5 years. Males 60%. Baseline BP was 152.2/100.9 in the treatment group and 152.3/101.3 in the control group | |
| Interventions | Losartan 50 mg/d (N = 139), losartan 50 mg/d + HCTZ 6.25 mg or 12.5 mg/d (all combined, N = 282), HCTZ 12.5 mg/d (N = 142) or placebo (N = 140) Trial duration = 12 weeks | |
| Outcomes | Change from the baseline in trough mean sitting and standing SBP and DBP; BP response rate; heart rate and laboratory tests (not specified). Subgroup analyses were performed (black versus non‐black patients; severity of hypertension) | |
| Notes | A sample size calculation was provided based on 703 patients in total to detect a difference of 3 to 5 mmHg between treatment groups at a power of 95%. Regarding baseline patient demographics and characteristics, the study authors stated that "there were no statistically significant differences between treatment groups" (P value = NS). No SD given for BP data. Biochemical data not available Additional publication: MacKay JH et al. Arch Intern Med 1996;156: 278‐85. Additional data on AEs and serum uric acid levels |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The study was randomly allocated..." (line 3 from top of p.S44, right column). The authors stated that the sequence generation design was randomized block (see line 12 under "Statistical Methods" in duplicate publication); but no further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "The study was randomly allocated, in parallel and placebo‐controlled and double‐blinded." (line 3 from top of p.S44, right column). No further information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It is not known if primary efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol technique
Exclusions: it was not reported whether any patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: a total of 99/703 (14%) patients withdrew from the study; this included 20/142 (14%) from the HCTZ 12.5 mg group: 6 patients for "clinical adverse experiences", 1 ‐ "other adverse experience", 5 ‐ "therapy ineffective" and 8 ‐ "other"; and 26/140 (19%) from the placebo group, 3 patients for "clinical adverse experiences", 14 ‐ "therapy ineffective" and 9 ‐ "other". WDAEs: 6/142 (4.2%) and 3/140 (2.1%) patients from the HCTZ and placebo groups, respectively, withdrew due to what the authors referred to as "adverse experiences"; the precise reasons were not given |
| Selective reporting (reporting bias) | Unclear risk | Heart rate, body weight, ECG, hematology, serum chemistry (except for uric acid and potassium levels ‐ see duplicate publication) were measured but not reported at the end of the study. Variability was not included in the baseline patient demographics and characteristics. Peak sitting DBP was reported in the results, yet the intention to measure it was not clearly stated in the methods section. Mortalities: none. SAEs: 1 patient in the placebo group with "subarachnoid hemorrhage". Total AEs: 52% (74/142) of patients from the HCTZ group and 52% (73/140) from the placebo group. The most commonly reported clinical AEs (≥ 4%) were reported in the secondary publication |
| Industry sponsorship | High risk | Supported by Merck Research Laboratories |
Scholze 1993.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). 4 x 3 factorial design. Wash‐out period = 2 to 4 weeks. Multicenter, conducted in Germany | |
| Participants | DBP 100 to 115 mmHg. Median age 48.2 years (range 21 to 68). Males 56.6%. Baseline BP range was 157.4 to 163.9/105.9 to 108.1 mmHg across the 12 treatment groups | |
| Interventions | Ramipril 2.5 mg, 5 mg or 10 mg/d, ramipril 2.5 mg, 5 mg or 10 mg/d + HCTZ 12.5 mg or 25 mg/d, HCTZ 12.5 mg or 25 mg/d, or placebo. N = 534 randomized (42 to 48 patients per treatment group) Trial duration = 6 weeks | |
| Outcomes | Change from the baseline in trough mean standing and supine SBP and DBP (at weeks 1, 2, 4 and 6); pulse rate; biochemical data including serum potassium and uric acid; hematology and urinalysis (values not shown) | |
| Notes | A sample size calculation of 40 patients per treatment group was required to detect a difference ranging from 3 to 6 mmHg between groups depending on monotherapy versus placebo or combination therapy versus monotherapy comparisons at a power of 37% to 99%. The study authors stated that "overall, the treatment groups were balanced" with respect to baseline patient demographics and characteristics and baseline BP. Baseline data were incomplete, therefore we could not calculate P values. Body weight, hematology, urinalysis and serum biochemistry (except for potassium levels) were measured but actual values were not reported on. Mortalities, SAEs and total AEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This study was a randomised placebo‐controlled, parallel‐group, Multicenter trial with a 4 x 3 factorial design (2.5, 5 and 10 mg ramipril; 12.5 and 25 mg HCT; all combinations of ramipril and HCT; placebo)." (line 1 under "Methods‐Design" p.218). "Randomization was stratified by centre." (line 9 under "Methods‐Design" p.218). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "A 2‐ or 4‐week, single‐blind, placebo run‐in phase was followed by a 6‐week, double‐blind, active treatment phase." (line 5 under "Methods‐Design" p.218). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Efficacy analysis was based on an intention‐to‐treat (ITT) technique Exclusions: 47/581 (8%) of patients were excluded from the study before randomization i.e. during or after the run‐in period Attrition: a total of 17/534 (3.2%) of patients withdrew from the study early due to the following reasons: 5 ‐ adverse events alone, 6 ‐ adverse events + elevated BP, 2 ‐ adverse events + low BP, 3 ‐ persistent elevation of BP and 1 ‐ alcohol abuse. Four of the 17 patients were receiving combination therapy; it is not known which treatment groups the remaining 13 patients were from WDAEs: 13/534 (2.4%) patients withdrew due to adverse events: 5 ‐ adverse events alone, 6 ‐ adverse events + elevated BP, 2 ‐ adverse events + low BP. Of these 13 patients, 1 (0.19%) was receiving HCTZ and 1 placebo, however it is not known what the adverse event was that caused them to withdraw |
| Selective reporting (reporting bias) | Unclear risk | Body weight, hematology, urinalysis and serum biochemistry (except for potassium levels) were measured at baseline and endpoint but actual values were not reported on. BP was measured but not reported at weeks 1, 2 and 4. Variability was not given for serum potassium levels or baseline patient demographics and characteristics. Mortalities, SAEs and total AEs were not reported. Treatment‐related (possibly or probably related) AEs: 6.8% of patients, all HCTZ groups combined; and 9.1% in the placebo group |
| Industry sponsorship | High risk | Supported by Hoechst AG |
Siegel 1992.
| Methods | Randomized, double‐blind, placebo‐controlled trial. Wash‐out period = 1 month | |
| Participants | DBP 95 to 105 mmHg. Mean age range: 58.1 to 62.2 years. All male patients with abnormal resting ECG (e.g. arrhythmias) | |
| Interventions | HCTZ 50 mg/d + potassium 40 mmol/d (N = 32), HCTZ 50 mg/d + potassium 40 mmol/d and magnesium 400 mg/d (N = 35), HCTZ 50 mg/d + triamterene 100 mg/d (N = 32), HCTZ 50 mg/d (N = 66), chlorthalidone 50 mg/d (N = 34) or placebo (N = 33) Trial duration = 8 weeks | |
| Outcomes | 24‐hour Holter monitoring (for arrhythmias). Serum and intracellular potassium and magnesium levels. Data on glucose and insulin levels available from duplicate publication | |
| Notes | A sample size calculation was not provided. All of the patients were males with resting electrocardiographic abnormalities (stratified by the presence or absence of left ventricular hypertrophy). It is not known how many patients were randomized to each treatment group; a range of 42 to 48 per group was given. The study authors stated that there were no statistically significant differences (P value = NS) in the baseline patient demographics and characteristics across all treatment groups. No data on DBP or SBP. WDAEs, mortalities, SAEs and total AEs were not reported Additional publication: Siegel et al. Hypertension 1994;23(part 1): 688‐94; measurement of glucose and insulin levels only |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Participants were then assigned to a study medication using a randomised block design, stratified by the presence or absence of left ventricular hypertrophy on ECG." (line 15 from top of p.1084, middle column). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Treatment assignment was blinded from participants, clinicians and laboratory staff by having another member of the staff dispense and count the blindly labelled medications, which were identically packaged." (line 19 from top of p.1084, middle column) Comment: no further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The primary efficacy analysis was based on an intention‐to‐treat (ITT) technique Exclusions: It is not known if there were any patients who were excluded from the study during the placebo run‐in period prior to randomization Attrition: 21/233 (9%) of patients withdrew from the study (data were not available for each treatment group). Reasons for withdrawing were not specified WDAEs: not given |
| Selective reporting (reporting bias) | High risk | BP was not measured. Except for serum potassium, glucose and insulin levels, biochemical data were not reported on. Mortalities, SAEs and total AEs were not reported |
| Industry sponsorship | Low risk | Supported by grant HL‐36821 and by National Heart, Lung and Blood Institute Preventive Cardiology Academic Award HL‐02081 |
Soltero 1989.
| Methods | Randomized, double‐blind, placebo‐controlled cross‐over trial. Wash‐out period = 15 days. Conducted in Venezuela | |
| Participants | Mild to moderate BP (cut‐offs not defined). Patients either never received antihypertensive treatment before or in the 3 months prior to entering the trial. Mean age 48 years (range: 20 to 70). Males 33%. Baseline sitting (standing) BP was 147.2/103.7 (149.5/102.1) mmHg in the treatment group and 140.0/100.6 (142.9/100.8) mmHg in the control group | |
| Interventions | Indapamide 2.5 mg/d or placebo. N = 24 patients randomized. Each treatment period = 8 weeks (2 periods in all with a 4‐week wash‐out period in between) | |
| Outcomes | Trough mean sitting and standing SBP and DBP; biochemical data including serum potassium, sodium, uric acid and lipids; heart rate; body weight | |
| Notes | A sample size calculation was not provided. Total withdrawals and WDAEs reported for periods I and II combined. Mortalities and SAEs were not reported and data for total AEs not useable. BP variability was given as standard error of the mean (SEM), not standard deviation | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patients were randomised to group A (indapamide followed by placebo) or group B (placebo followed by indapamide), following a 15‐day washout period." (line 12 under "Patients and Methods" p.164). No further information was provided |
| Allocation concealment (selection bias) | Low risk | "The codes of the active drug and the placebo were kept at the laboratory supplying the tablets." (line 15 under "Patients and Methods" p.164) |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "The study was conducted according to a double‐blind, crossover, placebo‐controlled methodology..." (line 11 under "Patients and Methods" p.164). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | It is not known if the study was based on an intention‐to‐treat (ITT) or per‐protocol technique Attrition: a total of 3/24 patients (12.5%) withdrew from the study early; 2 patients while receiving indapamide due to severe faintness and 1 patient while receiving placebo due to severe hypertension WDAEs: 3/24 (12.5%) |
| Selective reporting (reporting bias) | Unclear risk | Variability was not given for heart rate, biochemical data or body weight. BP, biochemical data and vital signs were presented separately (not combined) for each group of patients (group A and group B) as well as for the indapamide and placebo treatment phase. Therefore, both indapamide and placebo represent 2 data sets. Mortalities and SAEs were not reported. Total AEs were presented for group A and B separately, however there is no information indicating which treatment (IND or PLB) the patient was receiving at the time the adverse event was reported |
Taylor 1988.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 6 weeks. Johannesburg, South Africa | |
| Participants | DBP 95 to 115 mmHg. Mean age 61 years. Males 7%. Baseline standing BP was 157/96 in the treatment group and 158/96 in the control group | |
| Interventions | Indapamide 2.5 mg/d or placebo. N = 35 randomized Trial duration = 8 weeks (followed by another 8 weeks of magnesium chloride added onto IND or PLB in patients with low serum potassium levels; not discussed in this review) | |
| Outcomes | Mean standing and supine SBP and DBP at weeks 4, 8, 12 and 16; heart rate; body weight, serum biochemistry; RBC Na+, K+ and Mg2+ | |
| Notes | Incomplete reporting of total withdrawals. WDAEs, mortalities, SAEs and total AEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "....patients were randomly allocated, in double‐blind fashion, to indapamide 2.5 mg daily or matching placebo." (line 2 under "Trial Design" p.274). No further information given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No further information given on how blinding was achieved. "After 7 weeks both groups received magnesium chloride presented as enteric‐coated slow‐release 535 mg tablets, taken in two doses, 3 at breakfast and 3 at dinner, for a further 8 weeks." (line 6 under "Trial Design" p.274). It was further stated that, "Although magnesium chloride was not given in double‐blind form it appears to have had no additive effect, either alone or with indapamide, on the blood pressure in these elderly patients with mild hypertension." (line 6 from bottom p.276). Comment: adding another active medication at the midpoint of the trial could have compromised patient blinding. Some patients, those with low serum potassium levels, were administered supplemental potassium medication |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The study authors did not state whether the primary efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol technique Exclusions: it is not known if any patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: 8/35 (23%) of patients withdrew from the study, 2 ‐ "tachycardia", 4 ‐ "developed diastolic pressure > 115 mmHg (1 ‐ indapamide, 3 ‐ placebo) and 2 ‐ "left for domestic reasons." WDAEs: not given |
| Selective reporting (reporting bias) | High risk | Heart rate, serum creatinine, calcium, uric acid and hematology were measured but not reported on at the end of the study. Results were expressed as mean ± SEM. Mortalities, SAEs and total AEs were not reported |
| Industry sponsorship | High risk | Supported by Servier Laboratories SA (Pty) Ltd and the South African Medical Research Council |
Vardan 1987.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 2 weeks. USA | |
| Participants | DBP 91 to 104 mmHg. Mean age not given. Males 65.7%. Baseline BP was 142.9/97.3 mmHg in the treatment group and 144.9/96.8 mmHg in the control group | |
| Interventions | Chlorthalidone 15 mg (N = 71) or 25 mg/d (N = 75) or placebo (N = 76) Trial duration = 12 weeks | |
| Outcomes | Mean standing and supine SBP and DBP (at weeks 2, 4, 8 and 12); pulse rate; body weight, ECG, hematology, urinalysis, serum biochemistry; liver function tests | |
| Notes | A sample size calculation was not provided. The study included elderly (60 years or older) patients with isolated systolic hypertension only (i.e. SBP 160 to 219 mmHg and DBP < 90 mmHg). A step‐up protocol was used wherein poor response (i.e. BP goal not reached) after 4 weeks led to a doubling of drug dosage from 1 to 2 capsules of chlorthalidone per day; poor response in patients receiving placebo led to a simulated randomization with a doubling of placebo capsules. Therefore with increasing and/or changing medication, BP results were taken from the first 4 weeks of the study only. The study authors stated that there were no statistically significant differences (P value = NS) across treatment groups in baseline SBP and DBP. Gender and race were available for all patients combined into one group therefore P values could not be calculated. We determined that baseline biochemical levels were not statistically significant between treatment groups. Mean ± SEM for BP data at 4, 8 and 12 weeks useful. Not all patients received potassium supplementation. Mortalities and SAEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patients were then randomly allocated in double‐blind fashion to receive the 15‐mg or 25‐mg chlorthalidone preparation or the placebo..." (left column, bottom line of p.485). No further information given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "The patients were then randomly allocated in double‐blind fashion to receive the 15‐mg or 25‐mg chlorthalidone preparation or the placebo in identical tablet forms to be taken once a day throughout the trial period of 12 weeks." (left column, bottom line of p.485) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The study authors did not state whether the primary efficacy analysis was based on an intention‐to‐treat (ITT) or per‐protocol technique
Exclusions: 27,199 patients were screened; 2130 of these were included in the first baseline clinic visit. 1579/2130 (74%) of patients were excluded from the study during the 3 baseline clinic visits following screening, but prior to randomization Attrition: 5/71 (7%) and 9/75 (12%) patients receiving chlorthalidone 15 mg and 25 mg doses, respectively, and 6/76 (8%) patients receiving placebo withdrew from the study. Reasons included: 12 patients for "unacceptable side effects", 5 ‐ "further elevation in blood pressure" and 3 ‐ "protocol violations" WDAEs: 12 patients withdrew due to adverse events (referred in the study as "unacceptable side effects"). The specific reasons in the chlorthalidone 15 mg group were 3/71 (4%) patients: 1 ‐ "rash, drowsiness, depression, weakness, sleep disturbances", 1 ‐ "nausea" and 1 ‐ "rash with pruritus"; in the chlorthalidone 25 mg group were 7/75 (9%) patients: 1 ‐ "rash with pruritus", 1 ‐ "painful eyes, headache, cramps, nausea", 1 ‐ "light‐headedness, diplopia", 1 ‐ "coryza, pruritus, flu symptoms", 1 ‐ "palpitations, light‐headedness", 1 ‐ "headache" and 1 ‐ "hypokalemia". In the placebo group were 3/76 (4%) patients: 1 ‐ "headache, edema, tinnitus" and 1 ‐ "rash, periorbital edema, choking, swollen, ulcerated uvula" |
| Selective reporting (reporting bias) | High risk | ECG, hematology, urinalysis and liver function were measured, but not reported on in the study. Pulse rate was measured, but not reported. Baseline for body weight was not shown. Endpoint BP was expressed as mean ± SE (standard error of the mean), not as change from baseline to endpoint. SE was given for biochemical data, not SD. Reporting of baseline patient demographics and characteristics was incomplete, i.e. mean age and medical history were not shown. Mortalities and SAEs were not documented Total AEs: 22.5% (16/71), 32% (24/75) and 19.7% (15/76) of patients from the CTD 15 mg and 25 mg and placebo groups, respectively. The 3 to 4 most commonly reported AEs in each treatment group were mentioned |
| Industry sponsorship | High risk | Supported by Boehringer Ingelheim Pharmaceuticals Inc. |
Villamil 2007.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Multi‐factorial. Wash‐out period = 1 week followed by a single‐blind, placebo run‐in of 2 to 4 weeks. Multicenter, USA | |
| Participants | Sitting DBP 95 to < 110 mmHg. Mean age: HCTZ (all doses) 55.2 years; placebo 54.4 years. Males 55%. Baseline BP was 153.8/99.2 mmHg in the HCTZ treatment group (all doses) and 152.7/99.3 mmHg in the placebo group | |
| Interventions | Aliskiren 75 mg (N = 184), 150 mg (N = 185) or 300 mg/d (N = 183), aliskiren 75 mg, 150 mg or 300 mg/d + HCTZ 6.25 mg, 12.5 mg or 25 mg/d (all combined, N = 1471), HCTZ 6.25 mg (N = 194), 12.5 mg (N = 188) or 25 mg/d (N = 176) or placebo (N = 195) Trial duration = 8 weeks | |
| Outcomes | Change from the baseline in mean trough sitting DBP and SBP (at 2‐week intervals); response rate; heart rate, plasma renin activity and renin concentrations; serum potassium levels | |
| Notes | A sample size calculation was provided based on 161 patients per treatment group to detect a difference of 3.2 mmHg (SD ± 8 mmHg) in DBP between combination therapy and both respective monotherapy treatment groups at a power of 90%. Comparison between aliskiren monotherapy and placebo groups also calculated. The study authors stated that the baseline patient demographics and characteristics were similar across treatment groups. Total withdrawals and SAEs not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This was an 8‐week, multicenter, randomised, double‐blind, placebo‐controlled, multifactorial, parallel‐group trial." (line 1 under "Study Design" p.218). No further information given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "Eligible patients were randomised to receive double‐blind treatment with placebo, aliskiren monotherapy (75, 150 or 300 mg), HCTZ monotherapy (6.25, 12.5 or 25 mg), or a combination of aliskerin and HCTZ (every dose combination except aliskerin/HCTZ 300/6.25 mg) in a factorial design." (line 11 under "Study Design" p.218). No further information given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The primary and secondary efficacy analysis was based on an intention‐to‐treat (ITT) technique Exclusions: 414/3190 (13%) of patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: total withdrawals were not given WDAEs: 1%, 0.5% and 2.3% of patients in the HCTZ 6.25, 12.5 and 25 mg treatment groups, respectively, and 3.6% of patients in the placebo group withdrew due to adverse events. The specific reasons were not given |
| Selective reporting (reporting bias) | High risk | Dose‐response surface analyses graphs for BP were not shown. Heart rate was measured, but not reported in the study. Variability was not included in the baseline patient demographics and characteristics. SE was given for DBP data, not SD. Variability in SBP was not shown. Other than the reporting of results for PRA and serum potassium levels, biochemical data were incomplete. Mortalities: no patients died while receiving HCTZ monotherapy or placebo. SAEs were not clearly documented. The study authors only provided a general range of 0% to 2.6% in the incidence of SAEs across all treatment groups. Total AEs were: 39%, 42% and 42% of patients in the HCTZ 6.25, 12.5 and 25 mg treatment groups, respectively, and 44% of patients on placebo. Treatment‐related AEs were also reported |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Weidler 1995.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Multicenter, USA | |
| Participants | DBP 95 to 110 mmHg. Mean age 60.9 years. Males 48%. Baseline BP was 150.7/98.9 mmHg in the treatment group and 152.5/98.7 mmHg in the control group | |
| Interventions | Indapamide 1.25 mg/d (N = 111) or placebo (N = 111) Trial duration = 8 weeks | |
| Outcomes | Change from the baseline in mean trough sitting and standing SBP and DBP (at weeks 2, 4, 6 and 8); BP response rate; heart rate; ECG, vital signs, body weight, hematology, urinalysis, serum biochemistry | |
| Notes | A sample size calculation was not provided. Only patients older than 50 years of age were included in the study. The study authors did not state whether there were any statistically significant differences in baseline patient demographics and characteristics across treatment groups. The number of participants changes for BP over time. Mortalities and SAEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients who met the entry criteria were randomised at the end of a 4‐week single‐blind placebo washout period to either 1.25 mg of indapamide or placebo in a double‐blind manner." (line 1 under "Design" p.46). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by the study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "Patients who met the entry criteria were randomised at the end of a 4‐week single‐blind placebo washout period to either 1.25 mg of indapamide or placebo in a double‐blind manner." (line 1 under "Design" p.46). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The primary efficacy analysis was based on the "treated patient" population Exclusions: 414/3190 (13%) of patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: 30/111 (27%) of patients in the indapamide (IND) group and 24/111 (21.6%) of patients on placebo withdrew from the study for the following reasons: (IND; placebo) 20;12 ‐ "protocol violations", 6;5 ‐ "clinical adverse events", 1;2 ‐ "lack of efficacy", 1;1 ‐ "lost to follow‐up" and 2;1 ‐ "withdrawal of consent"; 0;1 ‐ "laboratory adverse event"; 0;2 ‐ "other reasons" WDAEs: 6/111 (5.4%) of patients in the indapamide group withdrew due to adverse events including 1 patient each for "headache and dizziness", "headache and abnormal vision", "hyperthyroidism", "dizziness and tinnitus", "atrial fibrillation" and "dizziness". 5/111 of patients on placebo (4.5%) withdrew due to AEs including 1 patient each for "bone fracture and skin disorder", "edema", "depression", "hypotension, dizziness and asthenia" and "dizziness" |
| Selective reporting (reporting bias) | High risk | Variability in baseline patient demographics and characteristics was not given. Results for serum biochemistry (except for potassium, uric acid and BUN levels), hematology, lipids, urinalysis, heart rate and ECG were measured, but actual values were not shown in the study. SE (standard error of the mean) was given for mean change from baseline in BP, not SD. Mortalities and SAEs were not documented. Total AEs: 41% (46/111) of patients from the indapamide group and 40% (44/111) from the placebo group. AEs were listed by body system in at least 3% of patients in either treatment group |
| Industry sponsorship | Unclear risk | Sponsor not reported |
Weir 1992.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 to 6 weeks. Multicenter, USA | |
| Participants | Supine DBP 95 to 110 mmHg. Mean age 53.5 years. Males 65%. Baseline BP 152.6/99.8 mmHg in HCTZ group and 152.7/99.5 mmHg in placebo group | |
| Interventions | Diltiazem SR 60 mg, 90 mg or 120 mg bid (all combined, N = 72), diltiazem SR 60 mg, 90 mg or 120 mg bid + HCTZ 6.25 mg or 12.5 mg bid (all combined, N = 75), HCTZ 6.25 mg or 12.5 mg bid (all combined, N = 76), or placebo (N = 75) Trial duration = 12 weeks (3 consecutive 4‐week treatment periods; 1st 8 weeks of data available) | |
| Outcomes | Change from baseline in mean trough standing and supine DBP and SBP (at weeks 4, 8 and 12); BP response rate; heart rate, ECG and serum biochemistry | |
| Notes | A sample size calculation was not provided. Patients in the 6.25 mg indapamide group were subjected to forced titration to 12.5 mg indapamide 8 weeks into DB treatment. Therefore, data after 8 weeks were excluded from the review. The study authors stated that the baseline patient demographics and characteristics (i.e. age, race, smoking status, height and body weight) were similar across treatment groups except for gender. All patients receiving the HCTZ 6.25 mg bid dose were force‐titrated to HCTZ 12.5 mg bid after 8 weeks. Mortalities, SAES and total AEs were not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This was a randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study comparing various doses of DTZ SR/HCTZ combination therapy with DTZ SR and HCTZ monotherapies and placebo... " (line 1 under "Methods‐Study Design" p.134). "...qualifying patients were randomised to a 12‐week double‐blind treatment phase..." (line 13 under "Methods‐Study Design" p.134). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "...qualifying patients were randomised to a 12‐week double‐blind treatment phase..." (line 13 under "Methods‐Study Design" p.134). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The primary efficacy analysis was based on an intention‐to‐treat (ITT) technique with the LOCF (last observation carried forward) Exclusions: 115/413 (28%) of patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: 44/298 (15%) of patients withdrew from the study. Reasons given included "intolerable side effects" and "inability to control BP", however these 2 reasons did not account for all patients withdrawing. Specific reasons were not given and from which group the patients originated is not known WDAEs: if assumed to be equivalent to "intolerable side effects", there were 6 patients from the HCTZ group and 2 from the placebo group who withdrew due to adverse events. Specific reasons were not given |
| Selective reporting (reporting bias) | High risk | ECG was not reported at the study's endpoint. BP data were available in graph form only. SE (not SD) was included in mean BP change. Mortalities, SAEs, total AEs were not documented. Treatment‐related AEs were reported |
| Industry sponsorship | High risk | Funded by research grant from Marion Merrell Dow Inc. |
Yodfat 1994.
| Methods | Randomized, double‐blind, placebo‐controlled trial (parallel arms). Wash‐out period = 4 weeks. Multicenter; Israel and Italy | |
| Participants | Sitting DBP > 100 mmHg. Mean age 53 years. 65% males. Baseline BP was not given | |
| Interventions | Cilazapril 2.5 mg or 5 mg/d (all combined, N = 94), cilazapril 1.25 mg, 2.5 mg or 5 mg/d + HCTZ 6.25 mg, 12.5 mg or 25 mg/d (all combined, N = 142), HCTZ 12.5 mg or 25 mg/d (all combined, N = 95), or placebo (N = 46) Trial duration = 8 weeks | |
| Outcomes | Change from baseline in mean trough sitting DBP and SBP (at weeks 2, 6 and 8); peak BP; response rate; pulse rate, ECG, hematology, urinalysis and serum biochemistry | |
| Notes | A sample size calculation was not provided. The study authors did not state whether there were statistically significant differences between the treatment groups in terms of the baseline patient demographics and characteristics. We determined there to be no statistically significant differences (P value = NS) in mean age, body weight, height or gender. Mortalities and total AEs not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "This report presents the results of a parallel‐group, placebo‐controlled, randomised study..." (line 12 from top of p.118). "All patients who entered period II were randomly assigned to a double‐blind, fixed dose, active treatment period of eight weeks." (line 6 (right column) from top of p.118). No further information was given |
| Allocation concealment (selection bias) | Unclear risk | Not stated by study authors |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "All patients who entered period II were randomly assigned to a double‐blind, fixed dose, active treatment period of eight weeks." (line 6 (right column) from top of p.118). No further information was given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The primary efficacy analysis was based on an intention‐to‐treat (ITT) technique Exclusions: 161/538 (30%) of patients were excluded from the study during the placebo run‐in period prior to randomization Attrition: 14/377 (3.7%) of patients withdrew from the study; the specific reasons and the treatment groups from which the patients originated were not given WDAEs: 7/377 (1.9%) of patients withdrew due to "adverse reactions", including 1 from the HCTZ group, however it is not known which dosage, 12.5 or 25 mg, the patient was receiving (the other 6 patients were receiving cilazapril + HCTZ combo therapy). The specific reasons were not given |
| Selective reporting (reporting bias) | High risk | Heart rate, body weight, ECG, hematology, serum biochemistry and urinalysis results were measured but actual values were not reported at the study's endpoint. Data for the HCTZ 12.5 mg and 25 mg treatment groups were pooled. BP measurements were not shown beyond week 4 of the 8‐week study. All DBP data were in graph form only and variability in the mean was not given. SBP was measured, but not shown Baseline patient demographics and characteristics did not include medical history. Mortalities: not given. SAEs: 3 patients receiving HCTZ (12.5 and 25 mg groups combined) for "aggravated hypertension", "AV‐block" and "angina". Total AEs were not reported; treatment‐related AEs were. Only the 5 most commonly occurring AEs in patients receiving combination therapy (i.e. cilazapril + HCTZ) were listed |
| Industry sponsorship | Unclear risk | Sponsor not reported |
ACE: angiotensin‐converting enzyme AE: adverse effect BDFZ: bendrofluazide bid: twice a day BMI: body mass index BP: blood pressure BUN: blood urea nitrogen CI: confidence interval CLZ: cilazapril CTD: chlorthalidone d: day DB: double‐blind DBP: diastolic blood pressure ECG: electrocardiography ER: extended release GI: gastrointestinal HCTZ: hydrochlorothiazide HDL: high‐density lipoprotein HR: heart rate IND: indapamide IR: immediate release ITT: intention‐to‐treat LDL: low‐density lipoprotein LOCF: last observation carried forward NS: non‐significant PLB: placebo PRA: plasma renin activity q12h: every 12 hours RBC: red blood cells SAE: serious adverse event SBP: systolic blood pressure SD: standard deviation SE: standard error SEM: standard error of the mean SR: sustained release VAL: valsartan WBC: white blood cells WDAE: withdrawal due to adverse events XR: extended release
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Amery 1978 | EPWHE study. Combination of 25 to 50 mg HCTZ with triamterene. Exclusion criteria met |
| Anavekar 1979 | Randomized, double‐blind, placebo‐controlled trial (a cross‐over trial). There is no parallel placebo treatment arm |
| Bateman 1979 | Randomized, double‐blind, placebo‐controlled trial. Meets criteria but data not available. Also there was no wash‐out period in between treatment periods. 1st 4 weeks' data useful but not available. Chlorthalidone 25 mg versus placebo |
| Batterman 1966 | Publication includes 110 trials in 62 patients which used both randomized and double‐blind methods. Blood pressure measurements were not taken under resting condition but determined as soon as the patients were seated |
| Blaufox 1992 | There is no mention of whether this RCT was double‐blinded |
| Boike 1982 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Borghi 1993 | No mention if trial was blinded. Data available only from 6 months onwards. BP and biochemical data from hypertensive patients were not reported separately from those of normotensive patients |
| Carretta 1988a | A single‐blinded, cross‐over trial. Indapamide 2.5 mg versus placebo for 3 months. There was no wash‐out between cross‐over treatment so data from 1st 3 months useful but not available |
| Carretta 1988b | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Chalmers 1976 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Chalmers 1982 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Chalmers 1986 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Christiansen 1981 | Randomized, double‐blind, placebo‐controlled trial. Bendroflumethiazide 5 mg/day versus placebo. Patients were all healthy, postmenopausal women (selection of patients was not based on blood pressure, except if > 170/105 mmHg) |
| Consoli 1985 | Foreign language (Italian). No placebo treatment arm |
| Cranston 1962 | Although it is a double‐blind RCT in 10 patients randomized to chlorthalidone 50 mg/day or placebo, data are available at week 27 and not between 3 and 12 weeks |
| Crowe 1987 | There was a placebo wash‐out period prior to randomization but no parallel placebo treatment arm |
| Datta 1989 | Randomized, double‐blind, placebo‐controlled trial. BP data reported at baseline and 24 weeks. Data requested but not available for other time points in between. The author is deceased |
| Davis 1993 | Subgroup of TAIM study. The TAIM study is a double‐blind RCT. Patients were randomized to placebo, chlorthalidone 25 mg/day or atenolol 50 mg/day. However, the dose was titrated only in non‐responders from 2 weeks onwards |
| Dean 1971 | Although randomized patients to HCTZ and placebo there is no mention of a wash‐out period before randomization |
| Durel 1992 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Eames 2005 | Randomized, double‐blind, placebo‐controlled trial. Bendrofluazide 2.5 mg/d versus placebo. Study duration was 7 days |
| Elliot 1991 | Randomized, double‐blind, placebo‐controlled trial (a cross‐over trial). Indapamide 1.25 mg, HCTZ 25 mg/day or placebo for 4 weeks. No wash‐out between treatments. 1st 4 weeks' data not available |
| Erwteman 1984 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Fagard 1976 | No mention if trial was randomized. In addition to patients with essential hypertension there were 7 with renovascular hypertension and 5 with renal parenchymal hypertension, however separate data for these 3 groups of patients were not available |
| Fernandez 1980 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Flack 2001 | Multicentre randomized, double‐blind, placebo‐controlled trial. Combination therapy consisting of hydrochlorothiazide and losartan versus placebo. HCTZ monotherapy treatment arm absent |
| Gall 1992 | Randomized, double‐blind, placebo‐controlled trial (a cross‐over trial). No wash‐out phase between cross‐over. 12.5 mg/day in 1st 4 weeks after which the dose was doubled in non‐responders only. Therefore, only 1st 4 weeks' data useful but not available |
| Galloway 1974 | Randomized, double‐blind, placebo‐controlled trial. Meets criteria but data not available. Cross‐over without a wash‐out period in between treatment. 1st 4 weeks' data useful but not available. Bendrofluazide 2.5 mg or placebo for 4 weeks. DBP recorded by phase IV muffle sound |
| Gerber 1985 | This is a single‐blinded RCT and is therefore excluded |
| Gleerup 1996 | Randomized, double‐blind, placebo‐controlled trial (a cross‐over trial). HCTZ 24 mg versus placebo. Since no wash‐out period before cross‐over, data from 1st 4 weeks useful but not available |
| Goldman 1980 | VA study. Randomized, double‐blind, placebo‐controlled trial. Baseline inclusion 85 to 105 mmHg; since mean DBP > 90 mmHg, inclusion was justified. Chlorthalidone 50 mg/day versus placebo. Step 1 data useful. At step 2, dose was doubled in non‐responders only. Data given at the end of 1 year and not the required 3 to 12‐week window |
| Grimm 1981 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Grimm 2002 | Randomized, double‐blind, placebo‐controlled trial. Chlorthalidone 15 mg/day versus placebo. 15 mg dose of CTD was doubled to 30 mg/day in non‐responders only. Also, BP data was not reported separately for the 2 CTD doses |
| Hobbs 1964 | Randomized, double‐blind placebo controlled trial that meets the criteria but provides only mean BP data. Data for SBP and DBP were not provided |
| Horvath 1979 | Bendrofluazide versus placebo in 18 patients with a treatment period of 8 weeks' duration. No wash‐out period. The 1st 8 weeks' data useful but not available. Also BP recorded as mean arterial pressure |
| Jackson 1986 | Randomized, double‐blind, placebo‐controlled trial (a cross‐over trial). HCTZ + amiloride combination was used therefore the trial was excluded. There was no thiazide monotherapy arm |
| Jain 1985 | All patients were started on chlorthalidone 25 mg/day open‐label for 2 weeks and then randomized to added therapy with either guanfacine or clonidine. There was no parallel placebo arm |
| Johnson 1986 | Randomized, double‐blind, placebo‐controlled trial. A cross‐over trial without a wash‐out period between treatment. Data needed from the 1st 4 weeks but not available. HCTZ 100 mg/day versus placebo |
| Jueng 1987 | After 1 week of double‐blind therapy, dose of HCTZ was doubled (from 25 mg to 50 mg) in patients not responding to treatment (defined as a diastolic BP > 90 mmHg). Inclusion criteria not met |
| Koskelainen 1985 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Kuramoto 1981 | Randomized, double‐blind, placebo‐controlled trial. Meets criteria but data not available. 35/44 patients were treated with thiazide monotherapy (i.e. trichlormethiazide 1 to 4 mg), however the exact number of patients in each arm is not known. Data between weeks 3 and 12 are not reported |
| Kuramoto 1985 | Japanese language trial. No parallel placebo arm, therefore excluded |
| Lechi 1982 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Lutterodt 1980 | Randomized, double‐blind, placebo‐controlled trial. Meets criteria but data not available. A cross‐over trial without a wash‐out period in between treatment. 1st 12 weeks' data useful but not available. HCTZ 50 mg or placebo. N = 27 of which 16 were eliminated so more than 50% withdrawals. The remaining 11 patients completed the trial. Mean arterial pressure given. SBP or DBP not given |
| Materson 1993 | Randomized, double‐blind, placebo‐controlled trial. Inclusion DBP 95 to 109 mmHg. HCTZ 12.5 mg up to 50 mg/day. Dose titrated every 2 weeks until DBP < 90 mmHg was achieved or maximal pre‐determined dose reached (data, however, were not available in the 1st 4 weeks) |
| Maus 1978 | Foreign language trial (French). No placebo arm |
| Maximilian 1970 | Foreign language trial (Romanian) |
| McCorvey 1993 | Randomized, double‐blind, placebo‐controlled trial (cross‐over trial). No adequate drug wash‐out prior to randomization of patients to either enalapril, propranolol, HCTZ or placebo |
| Merrill 1987 | No placebo arm of study |
| Milliez 1975 | Randomized, double‐blind, placebo‐controlled trial (cross‐over design). Meets criteria but data not available. No wash‐out period in between treatment periods. 1st 6 weeks data useful but not available. Indapamide 5 mg or chlorothiazide 500 mg or placebo for 1 month. WDAE not given |
| Morgan 2001 | Randomized, placebo‐controlled trial. Trial not blinded |
| Moser 1986 | Trial not randomized |
| MRC Working Party 1983 | Randomized, double‐blind, placebo‐controlled trial. Meets criteria but data not available. Bendrofluazide 5 or 10 mg/day versus placebo. Also the number of patients taking 5 mg or 10 mg not specified |
| Muiesan 1987 | Randomized, placebo‐controlled trial. Single‐blinded, stepped‐up dosage for patients who did not respond after 4 weeks of double‐blind active treatment. Only patients with a DBP < 100 mmHg were permitted to enter a subsequent phase of double‐blind treatment |
| Myers 1982 | Randomized, double‐blind, placebo‐controlled trial. Meets criteria but data not available. Diuretic treatment arms combined together. No wash‐out period prior to double‐blind treatment |
| Myers 1983 | Randomized, double‐blind, placebo‐controlled trial (cross‐over trial). No wash‐out period prior to randomization of patients to double‐blind treatment and no drug wash‐out between treatment periods. It meets the criteria but data from 1st 6 weeks not available. HCTZ 50 mg versus placebo |
| Okun 1978 | Randomized, double‐blind, placebo‐controlled trial with 4 weeks wash‐out period. Ticrynafen 250 mg, HCTZ 50 mg or placebo for 6 weeks. Dose titrated after 2 weeks in patients whose BP did not decrease by more than 10 mmHg |
| Okun 1979 | Randomized, double‐blind, placebo‐controlled trial. Tienilic acid 250 mg, HCTZ 50 mg versus placebo. 2 weeks after randomization dose doubled in patients whose BP did not decrease by 10 mmHg. Does not meet inclusion criteria |
| PATS Col. Group 1995 | Randomized, double‐blind, placebo‐controlled trial. Indapamide 2.5 mg versus placebo. Patients included hypertensive and normotensive with history of a minor or major stroke (cut‐off values for baseline BP were not specified) |
| Petersen 1996 | Randomized, double‐blind, placebo‐controlled trial (a cross‐over trial). HCTZ 6 mg versus placebo. Since no wash‐out period before cross‐over, data from 1st 4 weeks useful but not available |
| Reisin 1997 | Randomized, double‐blind, placebo‐controlled trial where data from first 4 weeks after randomization, before dose was titrated in non‐responders could have been included. Author (Reisin) was contacted by email. He lost the data from this study in hurricane Katrina |
| Russel 1968 | Meets criteria but data not available. Randomized, double‐blind, placebo‐controlled trial cross‐over without wash‐out period in between treatment. 1st 6 weeks' data useful but not available. HCTZ 200 mg versus placebo |
| Safar 1994 | Although it meets the inclusion criteria, data are available only in poster format. Details of the study methodology not available and unable to grade quality of evidence |
| Salvetti 1969 | Randomized, double‐blind, placebo‐controlled trial (cross‐over design). Wash‐out phase between double‐blind treatment not mentioned. Inclusion DBP 101 to 114 mmHg. Chlorthalidone 25 mg/day or placebo for 1 month. Data for 1 month not available |
| Salvetti 1989 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Samson 1965 | Randomized, double‐blind, placebo‐controlled trial. Cross‐over design (3 treatment periods in each patient: chlorthalidone 100 mg/d, polythiazide 4 mg/d, placebo; duration of each period was 2 months ‐ no data between 3 and 12 weeks available). Also, there was no placebo run‐in period |
| Schaller 1985 | This trial, although double‐blind, does not mention whether patients were randomized to indapamide or placebo |
| Shahinfar 1999 | Double‐blind randomized controlled trial. There was no wash‐out period prior to randomization ‐ all patients received HCTZ |
| Shimizu 1977 | A foreign language RCT (Japanese). Non‐blinded in methodology, therefore excluded from this review |
| Siegel 1990 | Patients were withdrawn from diuretic treatment before entering study. Outcome is left ventricular hypertrophy (LVH) on ECG. No data on SBP, DBP or WDAEs |
| Stein 1992 | Dose‐ranging trial in 19 black patients. Each dose of 6.25 mg, 12.5 mg, 25 mg and 50 mg of hydrochlorothiazide or placebo given randomly for 6 weeks. No wash‐out between treatment periods. Data from 1st 6 weeks useful but not available |
| Stornello 1990 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| TOMHS 1991 | Randomized, double‐blind, placebo‐controlled trial. TOMHS (the Treatment Of Mild Hypertension Research) Group, USA. Trial excluded because dose of chlorthalidone was doubled from 15 mg to 30 mg daily in non‐responding patients only (defined as a diastolic BP ≥ 95 mmHg on 3 successive visits or ≥ 105 mmHg at a single visit). Data from the titrated group were not available separately Duplicate publications (e.g. Grimm Jr. JAMA,1996) |
| Valmin 1975 | Randomized, double‐blind, placebo‐controlled trial (cross‐over design). Meets criteria but data not available. No wash‐out period in between treatment periods. 1st 4 weeks' data useful but not available. Furosemide 12.5 mg, 25 mg, 40 mg, or HCTZ 25 mg or placebo for 4 weeks |
| Wassertheil‐Smoller 1992 | TAIM study is a double‐blind RCT with patients randomized to placebo, chlorthalidone 25 mg/day or atenolol 50 mg/day. However, dose was titrated in non‐responders only from 2 weeks onwards |
| Weber 1977 | Randomized, double‐blind, placebo‐controlled trial (cross‐over trial). Meets criteria but data not available. No wash‐out in between treatment periods. 1st 4 weeks' data useful but not available. Xipamide 20 mg, 40 mg or placebo |
| Webster 1980 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Weinberger 1983 | Randomized, placebo‐controlled trial (Study B). Blinding of trial not mentioned nor referenced as blinded in PubMed (MeSH terms) |
| Wiggam 1999 | A double‐blind, cross‐over study in which data are not reported at the end of the first treatment period (parallel placebo control part of the trial) |
| Wilcox 1978 | Randomized, double‐blind, placebo‐controlled trial (cross‐over trial). Does not meet inclusion criteria as treatment below the minimum 3 weeks' duration. Bendrofluazide 5 mg or 10 mg versus placebo. Each dose given for only 2 weeks |
| Wing 1982 | Meets criteria but data not available. Randomized, double‐blind, placebo‐controlled trial, cross‐over study with no wash‐out period in between treatment. 1st 5 weeks' data valid but not available |
| Wing 1997 | Randomized, double‐blind, placebo‐controlled trial (cross‐over trial). No wash‐out period before crossing over treatment. After 1st 2 weeks of double‐blind treatment HCTZ 25 mg titrated to achieve SBP < or = 160 mmHg in non‐responders only |
| Wing 1998 | Randomized, double‐blind trial (cross‐over trial). Not placebo‐controlled. No wash‐out between treatment periods. Last 2 weeks of data provided for each 6‐week phase of treatment. No thiazide monotherapy arm; combination therapy of indapamide plus perindopril only |
| Zachariah 1993 Study #1 | This double‐blind placebo‐controlled RCT meets the inclusion criteria but since the number of patients randomized to each treatment group has not been reported it had to be excluded in the data analysis |
| Zachariah 1993 Study #2 | This double‐blind placebo‐controlled RCT meets the inclusion criteria but since the number of patients randomized to each treatment group has not been reported it had to be excluded in the data analysis |
BP: blood pressure CTD: chlorthalidone d: day DBP: diastolic blood pressure ECG: electrocardiogram HCTZ: hydrochlorothiazide RCT: randomized controlled trial SBP: systolic blood pressure WDAE: withdrawals due to adverse effects
Differences between protocol and review
The inclusion criteria for patients with hypertension in the protocol was defined as those with systolic and/or diastolic blood pressure of 160/90 mmHg or more, which was corrected to include the generally accepted standard definition of hypertension as systolic and/or diastolic blood pressure of 140/90 mmHg or more.
The search methods section in the protocol was limited to searching of MEDLINE, EMBASE and CENTRAL until June 2008, using the standard search strategy of the Cochrane Hypertension Group, with additional terms used to identify the relevant articles. In this completed review, the search of electronic databases also included ClinicalTrials.gov, Web of Science and bibliographic citations. In case of incomplete reports, we used MEDLINE to search for related papers. We searched bibliographies of pertinent articles, reviews and texts for additional citations. We used previously published meta‐analyses on the dose response of thiazide diuretics, as well as narrative reviews, to help identify references to trials. We searched electronic databases using a strategy combining the Cochrane Highly Sensitive Search Strategy for identifying randomized trials in MEDLINE: sensitivity‐maximizing version (2008 revision) with selected MeSH terms and free‐text terms relating to the individual thiazide drugs and hypertension. We used no language restrictions. We updated the search from June 2008 until February 2014.
In order to address missing data we attempted to contact the study's authors using the first author firstly then any of the co‐authors. Since using the address and telephone contact details of authors of trials before 1990 was proving to be very difficult, we decided to use the publication year 1990 as a cut‐off for verifying the authors' contact information.
The hierarchy of imputing standard deviation data in this review has minor differences from that stated in the protocol and was standardized to match other Cochrane reviews measuring the blood pressure‐lowering efficacy of other antihypertensive drug classes (ACE inhibitors, angiotensin receptor blockers) using similar inclusion/exclusion criteria.
To reflect the content of the review better the title was changed from 'Blood pressure lowering efficacy of thiazide diuretics for primary hypertension' to 'Blood pressure‐lowering efficacy of monotherapy with thiazide diuretics for primary hypertension'.
Contributions of authors
James Wright and Vijaya Musini formulated the idea for the review and developed the basis for the protocol.
Vijaya Musini took the lead role in searching, identifying and assessing studies.
Vijaya Musini and Mark Nazer independently assessed each trial against the inclusion criteria and carried out data abstraction and analyses.
James Wright and Ken Bassett confirmed the accuracy of the data and were the review authors who settled any discrepancies in the inclusion criteria or data abstraction.
Vijaya Musini wrote the first draft of this review. All authors contributed to the interpretation of the findings and to the final draft of the review.
Sources of support
Internal sources
University of British Columbia, Department of Anesthesiology, Pharmacology & Therapeutics, Canada.
External sources
-
Canadian Institutes of Health Research, Canada.
Grant to the Cochrane Hypertension Review Group
Declarations of interest
Vijaya Musini: none known.
Mark nazer: none known.
Ken Bassett: none known.
James Wright: none known.
New
References
References to studies included in this review
Ambrosioni 1998 {published data only}
- Ambrosioni E, Safar M, Degaute JP, Malin PL, MacMahon M, Pujol DR, et al. Low‐dose antihypertensive therapy with 1.5 mg sustained‐release indapamide: results of randomised double‐blind controlled studies. Journal of Hypertension 1998;16:1677‐84. [DOI] [PubMed] [Google Scholar]
- Asmar R, Guez D, Malbezin M, Brault Y, Cordoue A, Barrandon S, et al. Therapeutic benefit of a low dosage of indapamide: result of a double‐blind, placebo controlled European study [Benefice therapeutique d'une faible dose d'indapamide: resultats d'une etude europeenne controlee en double aveugle contre placebo]. Archives des Maladies du Coeur et des Vaisseaux 1995;88:1083‐7. [PubMed] [Google Scholar]
- Guez D, Mallion J‐M, Degaute J‐P, Malini P‐L, Baldwin R, Rodriguez‐Pujol D, et al. Treatment of hypertension with indapamide 1.5 mg sustained‐release form: synthesis of results [Traitement de l'hypetension arterielle par l'indapamide 1,5 mg comprime enrobe a liberation prolongee: synthese des resultats]. Archives des Maladies du Coeur et des Vaisseaux 1996;89(IV):17‐25. [PubMed] [Google Scholar]
- Leonetti G. Clinical positioning of indapamide sustained release 1.5mg in management protocols for hypertension. Drugs 2000;59(Suppl 2):27‐38. [DOI] [PubMed] [Google Scholar]
- Mallion J‐M, Asmar R, Boutelant S, Guez D. Twenty‐four hour antihypertensive efficacy of indapamide, 1.5‐mg sustained release: results of two randomized double‐blind controlled studies. Journal of Cardiovascular Pharmacology 1998;32(4):673‐8. [DOI] [PubMed] [Google Scholar]
Benz 1998 {published data only}
- Benz JR, Black HR, Graff A, Reed A, Fitzsimmons S, Shi Y. Valsartan and hydrochlorothiazide in patients with essential hypertension. A multiple dose, double‐blind, placebo controlled trial comparing combination therapy with monotherapy. Journal of Human Hypertension 1998;12:861‐6. [DOI] [PubMed] [Google Scholar]
Bradley 1993 {published data only}
- Bradley K, Flack JM, Belcher J, Elmer P, Miller P, Grimm R Jr. Chlorthalidone attenuates the reduction in total cholesterol and small, dense LDL cholesterol subclass associated with weight loss. American Journal of Hypertension 1993;6(7 Pt 1):636‐9. [DOI] [PubMed] [Google Scholar]
Brown 1990 {published data only}
- Brown CL, Backhouse CI, Grippat JC, Santoni JP. The effect of perindopril and hydrochlorothiazide alone and in combination on blood pressure and on the renin‐angiotensin system in hypertensive subjects. European Journal of Clinical Pharmacology 1990;39(4):327‐32. [DOI] [PubMed] [Google Scholar]
Burris 1990 {published data only}
- Burris JF, Weir MR, Oparil S, Wever M, Cady WJ, Stewart WH. An assessment of diltiazem and hydrochlorothiazide in hypertension. Application of factorial trial design to a multicenter clinical trial of combination therapy. JAMA 1990;263(11):1507‐12. [PubMed] [Google Scholar]
Canter 1994 {published data only}
- Canter D, Frank GJ, Knapp LE, Phelps M, Quade M, Texter M. Quinapril and hydrochlorothiazide combination for control of hypertension:assessment by factorial design. Quinapril Investigator Group [see comments]. Journal of Human Hypertension 1994;8(3):155‐62. [PubMed] [Google Scholar]
Capone 1983 {published data only}
- Capone P, Vukovich RA, Neiss ES, Bolton S, Reeves RL. Multicenter dose‐response study of the effect of indapamide in the treatment of patients with mild to moderate hypertension. Clinical Therapeutics 1983;5(3):305‐16. [PubMed] [Google Scholar]
Carlsen 1990 {published data only}
- Carlsen JE, Kober L, Torp‐Pedersen C, Johansen P. Relation between dose of bendrofluazide, antihypertensive effect, and adverse biochemical effects [see comments]. BMJ 1990;300(6730):975‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsen JE, Kober L, Torp‐Pedersen CT, Johansen P. The optimal dose of bendroflumathiazide in hypertension. A randomized double‐blind dose‐response study [Optimal dosis af bendroflumetiazid ved hypertension. En randomiseret dobbeltblind dosis‐responsunderogelse]. Ugeskrift for Laeger 1990;152(42):3072‐5. [PubMed] [Google Scholar]
Chrysant 1994 {published data only}
- Chrysant SG, The Lisinopril‐Hydrochlorothiazide Group. Antihypertensive effectiveness of low‐dose lisinopril‐hydrochlorothiazide combination. A large multicenter study. Archives of Internal Medicine 1994;154(7):737‐43. [PubMed] [Google Scholar]
Chrysant 1996 {published data only}
- Chrysant SG, Fagan T, Glazer R, Kriegman A. Effects of benazepril and hydrochlorothiazide, given alone and in low‐ and high‐dose combinations, on blood pressure in patients with hypertension. Archives of Family Medicine 1996;5(1):17‐24. [DOI] [PubMed] [Google Scholar]
Chrysant 2004 {published data only}
- Chrysant SG, Weber MA, Wang AC, Hinman DJ. Evaluation of antihypertensive therapy with the combination of olmesartan medoxomil and hydrochlorothiazide. American Journal of Hypertension 2004;17(3):252‐9. [DOI] [PubMed] [Google Scholar]
Curry 1986 {published data only}
- Curry CL, Janda SM, Harris R, MacKay JH, Nugent CA, Ryan J, et al. Clinical studies of a new, low‐dose formulation of metolazone for the treatment of hypertension. Clinical Therapeutics 1986;9(1):47‐62. [PubMed] [Google Scholar]
Drayer 1995 {published data only}
- Drayer JI, Stimpel M, Fox A, Weber M. The antihypertensive properties of the angiotensin‐converting enzyme inhibitor moexipril given alone or in combination with a low dose of a diuretic. American Journal of Therapeutics 1995;2(8):525‐31. [DOI] [PubMed] [Google Scholar]
Fernandez 1994 {published data only}
- Fernandez M, Madero R, Gonzalez D, Camacho P, Villalpando J, Arriaga J. Combined versus single effect of fosinopril and hydrochlorothiazide in hypertensive patients. Hypertension 1994;23(Suppl 1):I207‐10. [DOI] [PubMed] [Google Scholar]
Ferrara 1984 {published data only}
- Ferrara LA, Simone G, Mancini M, Fasano ML, Pasanisi F, Vallone G. Changes in left ventricular mass during a double‐blind study with chlorthalidone and slow‐release nifedipine. European Journal of Clinical Pharmacology 1984;27(5):525‐8. [DOI] [PubMed] [Google Scholar]
Fiddes 1997 {published data only}
- Fiddes R, Blumenthal J, Dawson JE, Dyckman E, John Hammond PG, Harris S, et al. Evaluation of indapamide 1.25 mg once daily in elderly patients with mild to moderate hypertension. Journal of Human Hypertension 1997;11(4):239‐44. [DOI] [PubMed] [Google Scholar]
Frei 1994 {published data only}
- Frei M, Küster L, Gardosch von Krosigk PP, Koch HF, Küppers H. Moxonidine and hydrochlorothiazide in combination: a synergistic antihypertensive effect. Journal of Cardiovascular Pharmacology 1994;24(Suppl 1):S25‐8. [DOI] [PubMed] [Google Scholar]
Frishman 1994 {published data only}
- Frishman WH, Bryzinski BS, Coulson LR, DeQuattro VL, Vlachakis ND, Mroczek WJ, et al. A multifactorial trial design to assess combination therapy in hypertension. Treatment with bisoprolol and hydrochlorothiazide. Archives of Internal Medicine 1994;154(13):1461‐8. [PubMed] [Google Scholar]
Frishman 1995 {published data only}
- Frishman WH, Burris JF, Mroczek WJ, Weir MR, Alemayehu D, Simon JS, et al. First‐line therapy option with low‐dose bisoprolol fumarate and low‐dose hydrochlorothiazide in patients with stage I and stage II systemic hypertension. Journal of Clinical Pharmacology 1995;35(2):182‐8. [DOI] [PubMed] [Google Scholar]
Goldberg 1989 {published data only}
- Goldberg MR, Rockhold FW, Offen WW, Dornseif BE. Dose‐effect and concentration‐effect relationships of pinacidil and hydrochlorothiazide in hypertension. Clinical Pharmacology and Therapeutics 1989;46(2):208‐18. [DOI] [PubMed] [Google Scholar]
Hall 1994 {published data only}
- Hall WD, Weber MA, Ferdinand K, Flamenbaum W, Marbury T, Jain AK, et al. Lower dose diuretic therapy in the treatment of patients with mild to moderate hypertension. Journal of Human Hypertension 1994;8(8):571‐5. [PubMed] [Google Scholar]
Hulley 1985 {published data only}
- Black DM, Brand RJ, Greenlick M, Hughes G, Smith J. Compliance to treatment for hypertension in elderly patients: The SHEP trial. Journal of Gerontology 1987;42(5):552‐7. [DOI] [PubMed] [Google Scholar]
- Hulley SB, Feigal D, Ireland C, Kuller LH, Smith WM. Systolic hypertension in the elderly program (SHEP). The first three months. Journal of the American Geriatrics Society 1986;34(2):101‐5. [DOI] [PubMed] [Google Scholar]
- Hulley SB, Furberg CD, Gurland B, McDonald R, Perry HM, Schnaper HW, et al. Systolic Hypertension in the Elderly Program (SHEP): Antihypertensive efficacy of chlorthalidone. American Journal of Cardiology 1985;56(15):913‐1038. [DOI] [PubMed] [Google Scholar]
- Kostis JB, Davis BR, Cutler J, Grimm RH, Berge KG, Cohen JD, et al. Prevention of heart failure by antihypertensive drug treatment in older persons with isolated systolic hypertension. SHEP Cooperative Research Group. JAMA 1997;278(3):212‐6. [PubMed] [Google Scholar]
- Moye LA, Davis BR, Hawkins CM, Probstfield JL. Conclusions and implications of the systolic hypertension in the elderly program. Clinical and Experimental Hypertension 1993;15(6):911‐24. [DOI] [PubMed] [Google Scholar]
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the systolic hypertension in the elderly program (SHEP). JAMA 1991;265(24):3255‐64. [PubMed] [Google Scholar]
- Smith WM, Feigal DW, Furberg CD, Greenlick M, Kuller L, Perry HM, et al. Use of diuretics in treatment of hypertension in the elderly. Drugs 1986;31(Suppl 4):154‐64. [DOI] [PubMed] [Google Scholar]
Jounela 1994 {published data only}
- Jounela AJ, Lilja M, Lumme J, Morlin C, Hoyem A, Wessel‐Aas T, et al. Relation between low dose of hydrochlorothiazide, antihypertensive effect and adverse effects. Blood Pressure 1994;3(4):231‐5. [DOI] [PubMed] [Google Scholar]
Kayanakis 1987 {published data only}
- Kayanakis JG, Baulac L. Comparative study of once‐daily administration of captopril 50 mg, hydrochlorothiazide 25 mg and their combination in mild to moderate hypertension. British Journal of Clinical Pharmacology 1987;23(Suppl 1):89S‐92S. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kochar 1999 {published data only}
- Kochar M, Guthrie R, Triscari J, Kassler‐Taub K, Reeves RA. Matrix study of irbesartan with hydrochlorothiazide in mild‐to‐moderate hypertension. American Journal of Hypertension 1999;12(8 Pt 1):797‐805. [DOI] [PubMed] [Google Scholar]
Krantz 1988 {published data only}
- Krantz DS, Contrada R, Durel LA, Hill DR, Friedler E. Comparative effects of two beta‐blockers on cardiovascular reactivity and type A behaviour in hypertensives. Psychosomatic Medicine 1988;50:615‐26. [DOI] [PubMed] [Google Scholar]
Lacourciere 1994 {published data only}
- Lacourciere Y, Arnott W. Placebo‐controlled comparison of the effects of nebivolol and low‐dose hydrochlorothiazide as monotherapies and in combination on blood pressure and lipid profile in hypertensive patients. Journal of Human Hypertension 1994;8(4):283‐8. [PubMed] [Google Scholar]
- Lacourciere Y, Lefebvre J, Poirier L, Archambault F, Arnott W. Treatment of ambulatory hypertensives with nebivolol or hydrochlorothiazide alone and in combination. A randomized, double‐blind, placebo‐controlled factorial‐design trial. American Journal of Hypertension 1994;7(2):137‐45. [DOI] [PubMed] [Google Scholar]
Lawton 1979 {published data only}
- Lawton WJ, Fitz A, Grant C, Witte DL. Dopamine beta‐hydroxylase and plasma renin activity in patients with low‐, normal‐, and high‐renin essential hypertension. Circulation 1979;59(5):1063‐9. [DOI] [PubMed] [Google Scholar]
London 2006 {published data only}
- London G, Schmieder R, Calvo C, Asmar R. Indapamide SR versus candesartan and amlodipine in hypertension: the X‐CELLENT study. American Journal of Hypertension 2006;19:113‐21. [DOI] [PubMed] [Google Scholar]
Lucas 1985 {published data only}
- Lucas CP, Morledge JH, Tessman DK. Comparison of hydrochlorothiazide and hydrochlorothiazide plus bevantolol in hypertension. Clinical Therapeutics 1985;8(1):49‐60. [PubMed] [Google Scholar]
Materson 1978 {published data only}
- Materson BJ, Oster JR, Ulrich FM, Bolton SM, et al. Dose response to chlorthalidone in patients with mild hypertension. Efficacy of a lower dose. Clinical Pharmacology and Therapeutics 1978;24(2):192‐8. [DOI] [PubMed] [Google Scholar]
McGill 2001 {published data only}
- Littlejohn III TW. Clinical considerations in hypertensive black patients: the role of angiotensin II receptor blockers in combination with a diuretic. Blood Pressure Monitoring 2001;6(Suppl 1):S15‐21. [Google Scholar]
- McGill JB. Angiotensin II receptor antagonist plus a thiazide diuretic is more efficacious for treating hypertension than either drug alone. Blood Pressure Monitoring 2001;6 (supplement 1):S3‐13. [Google Scholar]
- McGill JB, Reilly PA. Combination treatment with telmisartan and hydrochlorothiazide in black patients with mild to moderate hypertension. Clinical Cardiology 2001;24:66‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill JB, Reilly PA. Telmisartan plus hydrochlorothiazide versus telmisartan or hydrochlorothiazide monotherapy in patients with mild to moderate hypertension: a multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group trial. Clinical Therapeutics 2001;23(6):833‐50. [DOI] [PubMed] [Google Scholar]
McVeigh 1988 {published data only}
- McVeigh G, Galloway D, Johnston D. The case for low dose diuretics in hypertension: comparison of low and conventional doses of cyclopenthiazide. British Medical Journal 1988;297(6641):95‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mersey 1993 {published data only}
- Mersey J, D'Hemecourt P, Blaze K. Once‐daily fixed combination of captopril and hydrochlorothiazide as first line therapy for mild to moderate hypertension. Current Therapeutic Research and Clinical Experiment 1993;53(5):502‐12. [Google Scholar]
Morledge 1986 {published data only}
- Morledge JH, Ettinger B, Aranda J, McBarron F, Barra P, Gorwit J, et al. Isolated systolic hypertension in the elderly. A placebo‐controlled, dose‐response evaluation of chlorthalidone. Journal of the American Geriatrics Society 1986;34(3):199‐206. [DOI] [PubMed] [Google Scholar]
Moser 1991 {published data only}
- Moser M, Abraham PA, Bennett WM, Brachfeld N, Goodman RP, McKenney JM, et al. The effects of benazepril, a new angiotensin‐converting enzyme inhibitor, in mild to moderate essential hypertension: a multicenter study. Clinical Pharmacology and Therapeutics 1991;49(3):322‐9. [DOI] [PubMed] [Google Scholar]
Mroczek 1996 {published data only}
- Mroczek WJ, Stimpel M. A double‐blind evaluation of moexipril versus hydrochlorothiazide in hypertension. Advances in Therapy 1996;13(2):79‐87. [Google Scholar]
Myers 2000 {published data only}
- Myers MG, Asmar R, Leenen FHH, Safar M. Fixed low‐dose combination therapy in hypertension ‐ a dose response study of perindopril and indapamide. Journal of Hypertension 2000;18(3):317‐25. [DOI] [PubMed] [Google Scholar]
Papademetriou 2000 {published data only}
- Papademetriou V, Reif M, Henry D, et al. Combination therapy with candesartan cilexetil and hydrochlorothiazide in patients with systemic hypertension. Journal of Clinical Hypertension 2000;2:372‐8. [Google Scholar]
Papademetriou 2006 {published data only}
- Papademetriou V, Hainer JW, Sugg J, Munzer D, ATTACH Study Group. Factorial antihypertensive study of an extended‐release metoprolol and hydrochlorothiazide combination. American Journal of Hypertension 2006;19(12):1217‐25. [DOI] [PubMed] [Google Scholar]
Persson 1996 {published data only}
- Persson B, Stimpel M. Evaluation of the antihypertensive efficacy and tolerability of moexipril, a new ACE inhibitor, compared to hydrochlorothiazide in elderly patients. European Journal of Clinical Pharmacology 1996;50:259‐64. [DOI] [PubMed] [Google Scholar]
Philipp 1997 {published data only}
- Philipp T, Letzel H, Arens HJ. Dose‐finding study of candesartan cilexetil plus hydrochlorothiazide in patients with mild to moderate hypertension. Journal of Human Hypertension 1997;11(Suppl 2):S67‐8. [PubMed] [Google Scholar]
Pool 1993 {published data only}
- Pool PE, Applegate WB, Woehler T, Sandall P, Cady WJ. A randomized, controlled trial comparing diltiazem, hydrochlorothiazide, and their combination in the therapy of essential hypertension. Pharmacotherapy 1993;13(5):487‐93. [PubMed] [Google Scholar]
Pool 1997 {published data only}
- Pool JL, Cushman WC, Saini RK, Nwachuku CE, Battikha JP. Use of the factorial design and quadratic response surface models to evaluate the fosinopril and hydrochlorothiazide combination therapy in hypertension. American Journal of Hypertension 1997;10(1):117‐23. [DOI] [PubMed] [Google Scholar]
Pool 2007 {published data only}
- Pool JL, Glazer R, Weinberger M, Alvarado R, Huang J, Graff A. Comparison of valsartan/hydrochlorothiazide combination therapy at doses up to 320/25 mg versus monotherapy: a double‐blind, placebo‐controlled study followed by long‐term combination therapy in hypertensive adults. Clinical Therapeutics 2007;29(1):61‐73. [DOI] [PubMed] [Google Scholar]
Pordy 1994 {published data only}
- Pordy RC and the Cilazapril/Hydrochlorothiazide 4x3 Factorial Design Study Group. Cilazapril plus hydrochlorothiazide: improved efficacy without reduced safety in mild to moderate hypertension. A double‐blind placebo‐controlled multicenter study of factorial design. Cardiology 1994;85:311‐22. [DOI] [PubMed] [Google Scholar]
Prisant 2000 {published data only}
- Prisant LM. Ambulatory blood pressure profiles in patients treated with once‐daily diltiazem extended‐release or indapamide alone or in combination. American Journal of Therapeutics 2000;7:177‐84. [DOI] [PubMed] [Google Scholar]
Roque 1996 {published data only}
- Roque F, Mon G, Cravero C, Courreges J, et al. Antihypertensive efficacy of hydrochlorothiazide 12.5 mg in patients with mild to moderate hypertension. Advances in Therapy 1996;13(5):284‐91. [Google Scholar]
Saruta 2007 {published data only}
- Saruta T, Ogihara T, Matsuoka H, Suzuki H, Toki M, Hirayama Y, et al. Antihypertensive efficacy and safety of fixed‐dose combination therapy with losartan plus hydrochlorothiazide in Japanese patients with essential hypertension. Hypertension Research 2007;30(8):729‐39. [DOI] [PubMed] [Google Scholar]
Schmieder 2009 {published data only}
- Schmieder RE, Philipp T, Guerediaga J, Gorostidi M, Smith B, Weissbach N, et al. Long‐term antihypertensive efficacy and safety of the oral direct renin inhibitor aliskiren. A 12‐month randomized, double‐blind comparator trial with hydrochlorothiazide. Circulation 2009;119:417‐25. [DOI] [PubMed] [Google Scholar]
Schoenberger 1995 {published data only}
- MacKay JH, Arcuri KE, Goldberg AI, Snapinn SM, Sweet CS. Losartan and low‐dose hydrochlolorthiazide in patients with essential hypertension. Archives of Internal Medicine 1996;156:278‐85. [PubMed] [Google Scholar]
- Schoenberger JA, Bauer J, Barden LP, Brown R, Byyny R, Cohen J, et al. Losartan with hydrochlorothiazide in the treatment of hypertension. Journal of Hypertension 1995;13(Suppl 1):S43‐7. [PubMed] [Google Scholar]
Scholze 1993 {published data only}
- Scholze J, Breitstadt A, Cairns V, Bauer B, Bender N, Priestley C, et al. Short report: ramipril and hydrochlorothiazide combination therapy in hypertension: a clinical trial of factorial design. The East Germany Collaborative Trial Group. Journal of Hypertension 1993;11(2):217‐21. [DOI] [PubMed] [Google Scholar]
Siegel 1992 {published data only}
- Siegel D, Hulley SB, Black DM, Cheitlin MD, Sebastian A, Seeley DG, et al. Diuretics, serum, and intracellular electrolyte levels, and ventricular arrhythmias in hypertensive men. JAMA 1992;267(8):1083‐9. [PubMed] [Google Scholar]
- Siegel D, Saliba P, Haffner S. Glucose and insulin levels during diuretic therapy in hypertensive men. Hypertension 1994;23(6 Pt 1):688‐94. [DOI] [PubMed] [Google Scholar]
Soltero 1989 {published data only}
- Soltero I, Fuenmayor I, Colmenares A, et al. Action of indapamide on lipid profiles: a double blind cross over study. Current Therapeutic Research 1989;46(1):163‐72. [Google Scholar]
Taylor 1988 {published data only}
- Taylor DR, Constable J, Sonnekus M, Milne FJ. Effect of indapamide on serum and red cell cations, with and without magnesium supplementation, in subjects with mild hypertension. South African Medical Journal 1988;74(6):273‐6. [PubMed] [Google Scholar]
Vardan 1987 {published data only}
- Vardan S, Mehrotra KG, Mookherjee S, Willsey GA, Gens JD, Green DE. Efficacy and reduced metabolic side effects of a 15‐mg chlorthalidone formulation in the treatment of mild hypertension. A multicenter study. JAMA 1987;258(4):484‐8. [DOI] [PubMed] [Google Scholar]
Villamil 2007 {published data only}
- Villamil A, Chrysant SG, Calhoun D, Schober B, Hsu H, Matrisciano‐Dimichino L, et al. Renin inhibition with aliskiren provides additive antihypertensive efficacy when used in combination with hydrochlorothiazide. Journal of Hypertension 2007;25:217‐26. [DOI] [PubMed] [Google Scholar]
Weidler 1995 {published data only}
- Weidler D, Jallad NS, Curry C, Ferdinand K, Jain AK, Schnaper HW, et al. Efficacious response with lower dose indapamide therapy in the treatment of elderly patients with mild to moderate hypertension. Journal of Clinical Pharmacology 1995;35:45‐51. [DOI] [PubMed] [Google Scholar]
Weir 1992 {published data only}
- Weir MR, Weber MA, Punzi HA, Serfer HM, Rosenblatt S, Cady WJ. A dose escalation trial comparing the combination of diltiazem SR and hydrochlorothiazide with the monotherapies in patients with essential hypertension. Journal of Human Hypertension 1992;6(2):133‐8. [PubMed] [Google Scholar]
Yodfat 1994 {published data only}
- Yodfat Y, Zimilchman R and the Israeli‐Italian Cilazapril/Hydrochlorothiazide Research Study Group. Dose‐finding and dose justification of once‐daily cilazapril in combination with hydrochlorothiazide in non‐obese patients with mild‐to‐moderate essential hypertension. Journal of Drug Development 1994;6(3):117‐21. [Google Scholar]
References to studies excluded from this review
Amery 1978 {published data only}
- Amery A, Bulpitt, Schaepdryver A, Fagard R, Hellemans J, Mutsers A, et al. Glucose intolerance during diuretic therapy. Results of trial by the European Working Party on Hypertension in the Elderly. Lancet 1978;311(8066):681‐3. [DOI] [PubMed] [Google Scholar]
Anavekar 1979 {published data only}
- Anavekar SN, Ludbrooke A, Louis WJ, Doyle AE. Evaluation of indapamide in the treatment of hypertension. Journal of Cardiovascular Pharmacology 1979;1(4):389‐94. [DOI] [PubMed] [Google Scholar]
Bateman 1979 {published data only}
- Bateman D, Dean CR, Mucklow JC, Bulpitt CJ, Dollery CT. Atenolol and chlorthalidone combination for hypertension. British Journal of Clinical Pharmacology 1979;7:357‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Batterman 1966 {published data only}
- Batterman RC. Hypertensive treatment with veratrum alkaloids and thiazides alone and in combination. The Angiology Research Foundation Inc. 1966;3(1):1‐11. [PubMed] [Google Scholar]
Blaufox 1992 {published data only}
- Blaufox MD, Lee HB, Davis B, Oberman A, Wassertheil‐Smoller S, Langford H. Renin predicts diastolic blood pressure response to nonpharmacologic and pharmacologic therapy. JAMA 1992;267:1221‐5. [PubMed] [Google Scholar]
Boike 1982 {published data only}
- Boike SC, Durley Y, Cubberley RB. Atenolol and chlorthalidone administered alone and in combination for essential hypertension. Clinical Pharmacy 1982;1:449‐53. [PubMed] [Google Scholar]
Borghi 1993 {published data only}
- Borghi L, Meschi T, Guerra A, Novarini A. Randomized prospective study of a non thiazide diuretic, indapamide, in preventing calcium stone recurrences. Journal of Cardiovascular Pharmacology 1993;22(Suppl 6):S78‐86. [PubMed] [Google Scholar]
Carretta 1988a {published data only}
- Carretta R, Fabris B, Bardelli M, Muiesan S, Fischetti F, Vran F, et al. Arterial compliance and baroreceptor sensitivity after chronic treatment with indapamide. Journal of Human Hypertension 1988;2:171‐5. [PubMed] [Google Scholar]
Carretta 1988b {published data only}
- Carretta R, Fabris B, Fischetti F, Muiesan S, Bardelli M, Melchior C, et al. Platelet alpha2‐adrenoceptor modifications induced by long‐term treatment with indapamide in essential hypertension. American Journal of Medicine 1988;84(Suppl 1B):31‐5. [PubMed] [Google Scholar]
Chalmers 1976 {published data only}
- Chalmers J, Tiller D, Horvath J, Bune A. Effects of timolol and hydrochlorothiazide on blood‐pressure and plasma renin activity. Double‐blind factorial trial. Lancet 1976;2(7981):328‐31. [DOI] [PubMed] [Google Scholar]
- Chalmers JP, Horvath JS, Korner PI, Tiller DJ, Bune AJ, England JD, et al. Quantitative effects of timolol and hydrochlorothiazide on blood pressure, heart rate and plasma renin activity: results of a double‐blind factorial trial in patients with essential hypertension. Clinical Sciences and Molecular Medicine 1976;51:517S‐19S. [DOI] [PubMed] [Google Scholar]
- Chalmers JP, Korner PI, Tiller DJ, Bune AJ, Steiner JD, West MJ, et al. Double‐blind factorial trial of prindolol and hydrochlorothiazide in hypertension. The Medical Journal of Australia 1976;1:650‐3. [PubMed] [Google Scholar]
Chalmers 1982 {published data only}
- Chalmers JP, Wing LMH, Grygiel MJ, Graham JR, Bune AJ. Effects of once daily indapamide and pindolol on blood pressure, plasma aldosterone concentration and plasma renin activity in a general practice setting. European Journal of Clinical Pharmacology 1982;22:191‐6. [DOI] [PubMed] [Google Scholar]
Chalmers 1986 {published data only}
- Chalmers JP, Morris JM, Wing LMH, West MJ, Bune AJC, Elliott JM. Effects of enalapril and hydrochlorothiazide on blood pressure, renin‐angiotensin system, and atrial natriuretic factor in essential hypertension: a double‐blind factorial cross‐over study. Australian New Zealand Journal of Medicine 1986;16:475‐80. [DOI] [PubMed] [Google Scholar]
Christiansen 1981 {published data only}
- Christiansen C, Christensen MS, Hagen C, Stocklund K‐E, Transbol I. Effects of natural estrogen/gestagen and thiazide on coronary risk factors in normal postmenopausal women. A 2‐year double‐blind placebo study. Acta Obstetricia et Gynecologica Scandinavica 1981;60:407‐12. [DOI] [PubMed] [Google Scholar]
Consoli 1985 {published data only}
- Consoli G, Martino G. Evaluation of the hypotensive efficacy in a double‐blind study of xipamide versus chlorthalidone [Valutazione dellefficacia ipotensiva in uno studio a doppio cieco della xipamide verso il clortalidone]. La Clinica Terapeutica 1985;113(5):379‐83. [PubMed] [Google Scholar]
Cranston 1962 {published data only}
- Cranston WI, Juel‐Jensen BE. The effects of spironolactone and chlorthalidone on arterial pressure. The Lancet 1962;June 2:1161‐4. [DOI] [PubMed] [Google Scholar]
Crowe 1987 {published data only}
- Crowe PF, Finnegan OC, Rusell CJMcL, Varma MPS. A double‐blind study of inderex in the treatment of essential hypertension. British Journal of Clinical Practice 1987;41(10):967‐70. [PubMed] [Google Scholar]
Datta 1989 {published data only}
- Datta SR. A double‐blind comparison of indapamide with placebo in the treatment of elderly hypertensives. Clinical Trials Journal 1989;26(5):356‐62. [Google Scholar]
Davis 1993 {published data only}
- Davis BR, Blaufox MD, Oberman A, Wassertheil‐Smoller S, Zimbaldi N, Cutler JA, et al. Reduction in long‐term antihypertensive medication requirements. Effects of weight reduction by dietary intervention in overweight persons with mild hypertension. Archives of Internal Medicine 1993;153(15):1773‐82. [DOI] [PubMed] [Google Scholar]
Dean 1971 {published data only}
- Dean G, Louw S, Hersch C, Kirsten HO, Brereton DN, Finnemore L, et al. A double‐blind trial in hypertension comparing Baycaron (FBA 1500), hydrochlorothiazide and placebo. South African Medical Journal 1971;45(12):323. [PubMed] [Google Scholar]
Durel 1992 {published data only}
- Durel LA, Hayashi PJ, Weidler DJ, Schneiderman N. Effectiveness of antihypertensive medications in office and ambulatory settings: a placebo‐controlled comparison of atenolol, metoprolol, chlorthalidone, verapamil, and an atenolol‐chlorathalidone combination. Journal of Clinical Pharmacology 1992;32:564‐70. [DOI] [PubMed] [Google Scholar]
Eames 2005 {published data only}
- Eames PJ, Robinson TG, Panerai RB, Potter JF. Bendrofluazide fails to reduce elevated blood pressure levels in the immediate post‐stroke period. Cerebrovascular Diseases 2005;19:253‐9. [DOI] [PubMed] [Google Scholar]
Elliot 1991 {published data only}
- Elliot WJ, Weber RP, Murphy MB. A double‐blind randomised, placebo‐controlled comparison of the metabolic effects of low‐dose hydrochlorothiazide and indapamide. Journal of Clinical Pharmacology 1991;31:751‐7. [DOI] [PubMed] [Google Scholar]
Erwteman 1984 {published data only}
- Erwteman TM, Nagelkerke N, Lubsen J, Koster M, Dunning AJ. β Blockade, diuretics, and salt restriction for the management of mild hypertension: a randomised double blind trial. British Medical Journal 1984;289:406‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fagard 1976 {published data only}
- Fagard R, Amery A, Plaen J‐F, Lijnen P, Missotten A. Relative value of beta blockers and thiazides for initiating antihypertensive therapy. Acta Cardiologica 1976;31(5):411‐26. [PubMed] [Google Scholar]
Fernandez 1980 {published data only}
- Fernandez PG, Zachariah PK, Bryant DG, Missan SS. Antihypertensive efficacy of alpha‐methyldopa, chlorothiazide and supres‐150 (alpha methyldopa‐chlorothiazide). Canadian Medical Association Journal 1980;123:284‐7. [PMC free article] [PubMed] [Google Scholar]
Flack 2001 {published data only}
- Flack JM, Saunders E, Gradman A, Kraus WE, Lester M, Pratt JH, et al. Antihypertensive efficacy and safety of losartan alone and in combination with hydrochlorothiazide in adult African Americans with mild to moderate hypertension. Clinical Therapeutics 2001;23(8):1193‐208. [DOI] [PubMed] [Google Scholar]
Gall 1992 {published data only}
- Gall MA, Rossing P, Skott P, Hommel E, Mathiesen ER, Geradel LU, et al. Placebo‐controlled comparison of captopril, metoprolol, and hydrochlorothiazide therapy in non‐insulin dependent diabetic patients with primary hypertension. American Journal of Hypertension 1992;5(5 Pt 1):257‐65. [DOI] [PubMed] [Google Scholar]
Galloway 1974 {published data only}
- Galloway DB, Beattie AG, Petrie JC. Practolol and bendrofluazide in treatment of hypertension. British Heart Journal 1974;36:867‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gerber 1985 {published data only}
- Gerber A, Weidmann P, Bianchetti MG, Ferrier C, Laederach K, Mordasini R, et al. Serum lipoproteins during treatment with the antihypertensive agent indapamide. Hypertension 1985;7(6 Suppl II):II164‐9. [DOI] [PubMed] [Google Scholar]
Gleerup 1996 {published data only}
- Gleerup G, Petersen JR, Mehelsen J, Winther K. Effect of spirapril and hydrochlorothiazide on platelet function and euglobulin clot lysis time in patients with mild hypertension. Angiology 1996;47(10):951‐6. [DOI] [PubMed] [Google Scholar]
Goldman 1980 {published data only}
- Goldman AI, Steele BW, Schnaper HW, Fitz AE, Frohlich ED, Perry HM Jr. Serum lipoprotein levels during chlorthalidone therapy. A Veterans Administration‐National Heart, Lung, and Blood Institute Cooperative Study on Antihypertensive Therapy: mild hypertension. JAMA 1980;244(15):1691‐5. [DOI] [PubMed] [Google Scholar]
Grimm 1981 {published data only}
- Grimm RH, Leon AS, Hunninghake DB, Lenz K, Hannan P, Blackburn H. Effect of thiazide diuretics on plasma lipids and lipoproteins in mildly hypertensive patients. Annals of Internal Medicine 1981;1(94):7‐11. [DOI] [PubMed] [Google Scholar]
Grimm 2002 {published data only}
- Grimm Jr. RH, Black H, Rowen R, Lewin A, Shi H, Ghadanfar M and the Amlodipine study group. Amlodipine versus chlorthalidone versus placebo in the treatment of stage I isolated systolic hypertension. American Journal of Hypertension 2002;15(1 Part 1):31‐6. [DOI] [PubMed] [Google Scholar]
Hobbs 1964 {published data only}
- Hobbs LF. Essential hypertension. Management with reserpine‐thiazide combination and the separate components (report of a double‐blind study). Western Medicine ‐ the Medical Journal of the West 1964;5:316‐9. [PubMed] [Google Scholar]
Horvath 1979 {published data only}
- Horvath JS, Caterson RJ, Collett P, Duggin GG, Kelly DH, Tiller DJ. Labetolol and bendrofluazide: comparison of their antihypertensive effects. Medical Journal of Australia 1979;1:626‐62. [DOI] [PubMed] [Google Scholar]
Jackson 1986 {published data only}
- Jackson G, Rowland M, Adam G, MacFarlane E, Jackson PG. Placebo controlled double‐blind randomised cross‐over trial of atenolol, hydrochlorothiazide and amiloride, and the combination (Kalten) in patients over 60 years of age. British Journal of Clinical Practice 1986;40(6):230‐4. [PubMed] [Google Scholar]
Jain 1985 {published data only}
- Jain AK, Hiremath A, Michael R, Ryan JR, McMahon FG. Clonidine and guanfacine in hypertension. Clinical Pharmacology and Therapeutics 1985;37(3):271‐6. [DOI] [PubMed] [Google Scholar]
Johnson 1986 {published data only}
- Johnson BF, Saunders R, Hickler R, Marwaha R, Johnson J. The effect of thiazide diuretics upon plasma lipoproteins. Journal of Hypertension 1986;4:235‐9. [DOI] [PubMed] [Google Scholar]
Jueng 1987 {published data only}
- Jueng C, Halperin AK, Hashimoto F, Callender K. Nifedipine GITS and hydrochlorothiazide in essential hypertension. Journal of Clinical Hypertension 1987;3:695‐703. [PubMed] [Google Scholar]
Koskelainen 1985 {published data only}
- Koskelainen J, Turpeinen T, Lehto H, et al. Metabolic effects of hydrochlorothiazide and hydrochlorothiazide‐amiloride and trichlormethiazide‐triamterene combinations. Current Therapeutic Research 1985;37(3):554‐65. [Google Scholar]
Kuramoto 1981 {published data only}
- Kuramoto K, Matsushita S, Kuwajima I, Murakami M. Prospective study on the treatment of mild hypertension in the aged. Japanese Heart Journal 1981;22(1):75‐85. [DOI] [PubMed] [Google Scholar]
Kuramoto 1985 {published data only}
- Kuramoto K, Masuyama Y. Comparison of the clinical usefulness of diuretics in elderly and younger essential hypertension‐double blind trial using tripamide. Nippon Ronen Igakkai Zasshi (Japanese Journal of Geriatrics) 1985;22(4):346‐53. [DOI] [PubMed] [Google Scholar]
Lechi 1982 {published data only}
- Lechi A, Pomari S, Berto R, Buniotto P, Parrinello A, Marini F, et al. Clinical evaluation of labetalol alone and combined with chlorthalidone in essential hypertension: a double‐blind multicentre controlled study. European Journal of Clinical Pharmacology 1982;22:289‐93. [DOI] [PubMed] [Google Scholar]
Lutterodt 1980 {published data only}
- Lutterodt A, Nattel S, Mclead PJ. Duration of antihypertensive effect of a single daily dose of hydrochlorothiazide. Clinical Pharmacology and Therapeutics 1980;27(3):324‐7. [DOI] [PubMed] [Google Scholar]
Materson 1993 {published data only}
- Materson BJ, Reda DJ, Cushman WC, Henderson WG. Results of the combination antihypertensive therapy after failure of each of the components. Journal of Human Hypertension 1995;9:791‐6. [PubMed] [Google Scholar]
- Materson BJ, Reda DJ, Cushman WC, Massie BM, Freis ED, Kochar MS, et al. Single‐drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. New England Journal of Medicine 1993;328(13):914‐21. [DOI] [PubMed] [Google Scholar]
Maus 1978 {published data only}
- Maus Y, Rorive G. Comparison using the double‐blind method of the hypotensive action of indapamide and that of another salidiuretic agent, chlorthalidone [Comparison selon la method du double insu de laction hypotensive de lindapamide avec celle dun autre salidiuretique la chlorthalidone]. Revue Medicale de Liege 1978;33(10):365‐8. [PubMed] [Google Scholar]
Maximilian 1970 {published data only}
- Maximilian V, Gavrila F, Zeana C, Barbulescu I, Dimitriu CG. Effectiveness of a compound preparation (DCR 515) in the treatment of arterial hypertension (double‐blind test) [Eficacitatea unui preparat compus (DCR 515) in tratamentul hipertensiunii arteriale (test dublu orb)]. Medicina Interna 1970;22(3):297‐305. [PubMed] [Google Scholar]
McCorvey 1993 {published data only}
- McCorvey E Jr, Wright JT Jr, Culbert JP, McKenny M, Proctor JD, Annet MP. Effect of hydrochlorothiazide, enalapril, and propranolol on quality of life and cognitive and motor function in hypertensive patients. Clinical Pharmacy 1993;12:300‐5. [PubMed] [Google Scholar]
Merrill 1987 {published data only}
- Merrill BA, Byyny RL, Carr A, Dauer AD, Kazilionis JE, Lester FM, et al. Lisinopril/HCTZ in essential hypertension. Pharmacology and Therapeutics 1987;41(2):227 [Abstract]. [Google Scholar]
Milliez 1975 {published data only}
- Milliez P, Tehaedakoff P. Antihypertensive activity of a new agent, indapamide: a double‐blind study. Current Medical Research Opinion 1975;3:9‐15. [DOI] [PubMed] [Google Scholar]
Morgan 2001 {published data only}
- Morgan TO, Anderson AIE, MacInnis RJ. ACE inhibitors, beta‐blockers, calcium blockers, and diuretics for the control of systolic hypertension. American Journal of Hypertension 2001;14(3):241‐7. [DOI] [PubMed] [Google Scholar]
Moser 1986 {published data only}
- Moser M. Low dose diuretic therapy for hypertension. Clinical Therapeutics 1986;8(5):554‐62. [PubMed] [Google Scholar]
MRC Working Party 1983 {published data only}
- MRC Working Party on Mild Hypertension. Ventricular extrasystoles during thiazide treatment: sub‐study of MRC mild hypertension trial. British Medical Journal 1983;287:1249‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Muiesan 1987 {published data only}
- Muiesan G, Agabiti‐Rosei E, Buoninconti R, Cagli V, Carotti A, Corea L, et al. Antihypertensive efficacy and tolerability of captopril in the elderly: comparison with hydrochlorothiazide and placebo in a multicentre, double‐blind study. Journal of Hypertension 1987;5(Suppl 5):S599‐602. [PubMed] [Google Scholar]
Myers 1982 {published data only}
- Myers MG, Wein ME, Fisher RH, Gryfe CI, Shulman HS. Unnecessary diuretic therapy in the elderly. Age and Aging 1982;11:213‐21. [DOI] [PubMed] [Google Scholar]
Myers 1983 {published data only}
- Myers MG. Effects of atenolol and hydrochlorothiazide on blood pressure and plasma catecholamines in essential hypertension. Hypertension 1983;5:591‐6. [DOI] [PubMed] [Google Scholar]
Okun 1978 {published data only}
- Okun R, Beg M. Tricynafen and hydrochlorothiazide in hypertension. Clinical Pharmacology and Therapeutics 1978;23(6):703‐11. [DOI] [PubMed] [Google Scholar]
Okun 1979 {published data only}
- Okun R, Beg MA. A double‐blind study of tienilic acid with two year follow‐up of patients with mild to moderate essential hypertension. Postgraduate Medical Journal 1979;55(Suppl 3):103‐9. [PubMed] [Google Scholar]
PATS Col. Group 1995 {published data only}
- PATS Collaborating Group. Epidemiology Survey. Post‐stroke antihypertensive treatment study: a preliminary result. Chinese Medical Journal 1995;108(9):710‐7. [PubMed] [Google Scholar]
Petersen 1996 {published data only}
- Petersen JR, Drabaek H, Gleerup G, Mehlsen J, Petersen LJ, Winther K. ACE inhibitor with spirapril improves diastolic function at rest independent of vasodilation during treatment with spirapril in mild to moderate hypertension. Angiology 1996;47(3):233‐40. [DOI] [PubMed] [Google Scholar]
Reisin 1997 {published data only}
- Reisin E, Weir MR, Falkner B, Hutchinson HG, Anzalone DA, Tuck ML. Lisinopril versus hydrochlorothiazide in obese hypertensive patients: a multicenter placebo‐controlled trial. Treatment in obese patients with hypertension (TROPHY) study group. Hypertension 1997;30(1 Pt 1):140‐5. [DOI] [PubMed] [Google Scholar]
- Weir MR, Reisin E, Falkner B, Hutchinson HG, Sha L, Tuck ML. Nocturnal reduction of blood pressure and the antihypertensive response to a diuretic or angiotensin converting enzyme inhibitor in obese hypertensive patients. TROPHY study group. American Journal of Hypertension 1998;11(8 Pt 1):914‐20. [DOI] [PubMed] [Google Scholar]
Russel 1968 {published data only}
- Russel RP, Lindeman RD, Prescott LF. Metabolic and hypotensive effect of ethacrynic acid. Comparative study with hydrochlorothiazide. JAMA 1968;205(1):81‐5. [DOI] [PubMed] [Google Scholar]
Safar 1994 {published data only}
- Safar M, Zanchetti A, Sever PS, et al. Perindopril and indapamide as a combination in the treatment of mild to moderate hypertension ‐ a double‐blind randomized placebo‐controlled European multicenter study [POSTER]. American Journal of Hypertension 1994;7(4):43A. [Google Scholar]
Salvetti 1969 {published data only}
- Salvetti A, Magagna A, Innocenti P, Ponzanelli F, Caiganelli A, et al. Chlorthalidone does not increase the hypotensive effect of nifedipine in essential hypertensives: a crossover multicenter study. Journal of Hypertension 1969;7(Suppl 6):S250‐1. [DOI] [PubMed] [Google Scholar]
Salvetti 1989 {published data only}
- Salvetti A, Magagna A, Innocenti P, Cagianelli A, Cipriani M, Gandolfi E, et al. Chlorthalidone does not increase the hypotensive effect of nifedipine in essential hypertensives: a cross over multicentre study. Journal of Hypertension 1989;7(Suppl 6):S250‐1. [DOI] [PubMed] [Google Scholar]
- Salvetti A, Magagna A, Innocenti P, Ponzanelli F, Caiganelli A, et al. The combination of chlorthalidone with nifedipine dose does not exert an additive antihypertensive effect in essential hypertensives: A crossover multicenter study. Journal of Cardiovascular Pharmacology 1991;17:332‐5. [DOI] [PubMed] [Google Scholar]
Samson 1965 {published data only}
- Samson WE. Clinical evaluation of polythiazide in hypertension and congestive heart failure: a comparative double‐blind study. American Journal of the Medical Sciences 1965;249:571‐5. [PubMed] [Google Scholar]
Schaller 1985 {published data only}
- Schaller M‐D, Waeber B, Brunner HR. Double‐blind comparison of indapamide with a placebo in hypertensive patients treated by practicing physicians. Clinical and Experimental Theory and Practice 1985;A7(7):985‐94. [DOI] [PubMed] [Google Scholar]
Shahinfar 1999 {published data only}
- Shahinfar S, Simpson RL, Carides AD, Thiyagarajan B, Nakagawa Y, Umans JG, et al. Safety of losartan in hypertensive patients with thiazide‐induced hyperuricemia. Kidney International 1999;56:1879‐85. [DOI] [PubMed] [Google Scholar]
Shimizu 1977 {published data only}
- Shimizu N, Miyashita H, Koide K, Inoue N, Ogasawara M. Comparative study of anti‐aldosterone agents and hypotensive diuretics in essential hypertension by a double bind test. Horumon To Rinsho 1977;25(2):167‐74. [PubMed] [Google Scholar]
Siegel 1990 {published data only}
- Siegel D, Cheitlin MD, Black DM, Seeley D, Hearst N, Hulley SB. Risk of ventricular arrhythmias in hypertensive men with left ventricular hypertrophy. American Journal of Cardiology 1990;65(11):742‐7. [DOI] [PubMed] [Google Scholar]
Stein 1992 {published data only}
- Stein CM, Neill P, Kusemarnuriwo T. Antihypertensive effects of low doses of hydrochlorothiazide in hypertensive black Zimbabweans. International Journal of Cardiology 1992;37:231‐5. [DOI] [PubMed] [Google Scholar]
Stornello 1990 {published data only}
- Stornello M, Valvo EV, Scapellato L. Effect of sustained‐release nicardipine, chlorthalidone, and the two drugs combined in patients with mild to moderate hypertension. Current Therapeutic Research 1990;47(2):405‐11. [Google Scholar]
TOMHS 1991 {published data only}
- Lewis CE, Grandits GA, Flack J, McDonald, Elmer PJ. Efficacy and tolerance of antihypertensive treatment in men and women with stage 1 diastolic hypertension. Archives of Internal Medicine 1996;156(4):377‐85. [PubMed] [Google Scholar]
- Liebson PR, Grandits GA, Dianzumba S, Prineas RJ, Grimm Jr. RH, Neaton JD, et al. Comparison of five antihypertensive mono therapies and placebo for change in left ventricular mass in patients receiving nutritional‐hygienic therapy in the treatment of mild hypertension study (TOMHS). Circulation 1995;91(3):698‐706. [DOI] [PubMed] [Google Scholar]
- The Treatment of Mild Hypertension Research Group (TOMHS). The treatment of mild hypertension study. A randomized, placebo‐controlled trial of a nutritional‐hygienic regiment along with various drug monotherapies. Archives of Internal Medicine 1991;151:1413‐23. [DOI] [PubMed] [Google Scholar]
Valmin 1975 {published data only}
- Valmin K, Hansen T. Treatment of benign essential hypertension. Comparison of furosemide and hydrochlorothiazide. European Journal of Clinical Pharmacology 1975;8:393‐401. [DOI] [PubMed] [Google Scholar]
Wassertheil‐Smoller 1992 {published data only}
- Wassertheil‐Smoller S, Oberman A, Blaufox M, et al. The trial of antihypertensive medications (TAIM) study. Final results with regards to blood pressure, cardiovascular risk and quality of life. American Journal of Hypertension 1992;5:37‐44. [DOI] [PubMed] [Google Scholar]
Weber 1977 {published data only}
- Weber JCP, Bird H, Cosh J, Davies PS, Dixon J, Lister J, et al. Once daily treatment of mild to moderate hypertension with xipamid: controlled study. British Journal of Pharmacology 1977;4:283‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Webster 1980 {published data only}
- Webster J, Dollery CT, Hensby CN. Circulating prostacyclin concentrations may be increased by bendrofluazide in patients with essential hypertension. Clinical Science 1980;59:125‐8S. [DOI] [PubMed] [Google Scholar]
- Webster J, Dollery CT, Hensby CN, Friedman LA. Antihypertensive action of bendroflumethiazide: increased prostacyclin production?. Clinical Pharmacology and Therapeutics 1980;28(6):751‐8. [DOI] [PubMed] [Google Scholar]
Weinberger 1983 {published data only}
- Weinberger MH. Influence of an angiotensin converting‐enzyme inhibitor on diuretic‐induced metabolic effects in hypertension. Hypertension 1983;5(5 Suppl III):III132‐III138. [DOI] [PubMed] [Google Scholar]
Wiggam 1999 {published data only}
- Wiggam MI, Bell PM, Sheridan B, Walmsley A, Atkinson AB. Low dose bendrofluazide (1.25 mg) effectively lowers blood pressure over 24 hours. Results of a randomised, double‐blind, placebo‐controlled cross over study. American Journal of Hypertension 1999;12:528‐31. [DOI] [PubMed] [Google Scholar]
Wilcox 1978 {published data only}
- Wilcox RG. Randomised study of six beta‐blockers and a thiazide diuretic in essential hypertension. British Medical Journal 1978;2(6134):383‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wing 1982 {published data only}
- Wing LMH, West MJ, Graham JR, Chalmers JP. Long‐acting and short‐acting diuretics in mild essential hypertension. Clinical and Experimental Hypertension ‐ Theory and Practice 1982;A4(8):1429‐41. [DOI] [PubMed] [Google Scholar]
Wing 1997 {published data only}
- Wing LMH, Arnolda LF, Harvey PJ, Upton J, et al. Lacidipine, hydrochlorothiazide and their combination in systolic hypertension in the elderly. Journal of Hypertension 1997;15:1503‐10. [DOI] [PubMed] [Google Scholar]
Wing 1998 {published data only}
- Wing LMH, Arnolda LF, Harvey PJ, Upton J, Molloy D, Gabb GM, et al. Low‐dose diuretic and/or dietary sodium restriction when blood pressure is resistant to ACE inhibitor. Blood Pressure 1998;7:299‐307. [DOI] [PubMed] [Google Scholar]
Zachariah 1993 Study #1 {published data only}
- Zachariah PK, Messerli FH, Mroczek W. Low‐dose bisoprolol/hydrochlorothiazide: an option in first‐line, antihypertensive treatment. Clinical Therapeutics 1993;15(5):779‐87. [PubMed] [Google Scholar]
Zachariah 1993 Study #2 {published data only}
- Zachariah PK, Messerli FH, Mroczek W. Low‐dose bisoprolol/hydrochlorothiazide: an option in first‐line, antihypertensive treatment. Clinical Therapeutics 1993;15(5):779‐87. [PubMed] [Google Scholar]
Additional references
Allen 1982
- Allen JH, McKenney JM, Stratton MA, Link K. Antihypertensive effect of hydrochlorothiazide administered once or twice daily. Clinical Pharmacology 1982;1:239–43. [PubMed] [Google Scholar]
Campbell 2004
- Campbell B, Brackman F. Cardiovascular protective properties of indapamide. American Journal of Cardiology 2004;65(17):H11‐27. [DOI] [PubMed] [Google Scholar]
Carter 2004
- Carter BL, Ernst ME, Cohen JD. Hydrochlorothiazide versus chlorthalidone evidence supporting their interchangeability. Hypertension 2004;43:4‐9. [DOI] [PubMed] [Google Scholar]
Chen 2009
- Chen JMH, Heran BS, Wright JM. Blood pressure lowering efficacy of diuretics as second‐line therapy for primary hypertension. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD007187.pub2] [DOI] [PubMed] [Google Scholar]
Edwin 2006
- Edwin JK. Diuretics. In: Brunton Lawrence L, et al. editor(s). Goodman & Gillman's The Pharmacological basis of Therapeutics. 11th Edition. New York: McGraw Hill, 2006. [Google Scholar]
Handbook 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Heran 2009a
- Heran BS, Wong MMY, Heran IK, Wright JM. Blood pressure lowering efficacy of angiotensin receptor blockers for primary hypertension. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD003822.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Heran 2009b
- Heran BS, Wong MMY, Heran IK, Wright JM. Blood pressure lowering efficacy of angiotensin converting enzyme(ACE) inhibitors for primary hypertension. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD003823.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hughes 2004
- Hughes AD. How do thiazide and thiazide like diuretics lower blood pressure?. Journal of the Renin‐angiotensin‐aldosterone System 2004;5(4):155‐60. [DOI] [PubMed] [Google Scholar]
Kurtz 2010
- Kurtz TW. Chlorthalidone: don't call it "thiazide‐like" anymore. Hypertension 2010;56:335‐7. [DOI] [PubMed] [Google Scholar]
Law 2009
- Law MR, Morris J, Wald NJ. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: meta‐analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ 2009;338:b1665doi:10.1136/bmj.b1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Messerli 2011
- Messerli FH, Makani H, Benjo A, Romero J, Alviar C, Bangalore S. Antihypertensive efficacy of hydrochlorothiazide as evaluated by ambulatory blood pressure monitoring: a meta‐analysis of randomized trials. Journal of American College of Cardiology 2011;57:590‐600. [DOI: 10.1016/j.jacc.2010.07.053] [DOI] [PubMed] [Google Scholar]
Musini 2008
- Musini VM, Fortin PM, Bassett K, Wright JM. Blood pressure lowering efficacy of renin inhibitors for primary hypertension. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD007066.pub2] [DOI] [PubMed] [Google Scholar]
Musini 2009a
- Musini VM, Tejani AM, Bassett K, Wright JM. Pharmacotherapy for hypertension in the elderly. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD000028.pub2] [DOI] [PubMed] [Google Scholar]
Musini 2009b
- Musini VM, Wright JM. Factors affecting blood pressure variability: lessons learned from two systematic reviews of randomized controlled trials. PloS One 2009;4(5):e5673. [DOI: 10.1371/journal.pone.0005673] [DOI] [PMC free article] [PubMed] [Google Scholar]
Musini 2009c
- Musini VM, Wright JM, Bassett K, Jauca CD. Blood pressure lowering efficacy of loop diuretics for primary hypertension. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD003825.pub2] [DOI] [PubMed] [Google Scholar]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2012.
Riess 1977
- Riess W, Dubach UC, Burckhardt D, Theobald W, Vuillard P, ZimmerliM. Pharmacokinetic studies with chlorthalidone (Hygroton) in man. European Journal of Clinical Pharmacology 1977;12:375–82. [DOI] [PubMed] [Google Scholar]
Russell 1981
- Russell JG, Mayhew SR, Humphries IS. Chlorthalidone in mild hypertension:dose response relationship. European Journal of Clinical Pharmacology 1981;20:407–11. [DOI] [PubMed] [Google Scholar]
Wells 2009 [Computer program]
- Wells GA, Sultan SA, Chen L, Khan M, Coyle D. Indirect treatment comparison. Version 1. Canadian Agency for Drugs and Technology in Health, 2009.
Wong 2014
- Wong GWK, Wright JM. Blood pressure lowering efficacy of nonselective beta‐blockers for primary hypertension. Cochrane Database of Systematic Reviews 2014, Issue 2. [DOI: 10.1002/14651858.CD007452.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wright 2009
- Wright JM, Musini VM. First‐line drugs for hypertension. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD001841.pub2] [DOI] [PubMed] [Google Scholar]
Wright 1999
- Wright JM, Cheng‐Han Lee, Chambers KC. Systematic review of antihypertensive therapies: does the evidence assist in choosing a first‐line drug?. Canadian Medical Association Journal 1999;161(1):25‐32. [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Musini 2000
- Musini V. A systematic review of the blood pressure lowering efficacy of thiazide and loop diuretics in the treatment of primary hypertension (Masters Thesis). Available at http://hdl.handle.net/2429/10427. University of British Columbia, 2000. [Google Scholar]