

Abstract
Background
Allogeneic haematopoietic stem cell transplantation (allo‐HSCT) is an established treatment option for many malignant and non‐malignant disorders. In the past two decades, peripheral blood stem cells replaced bone marrow as stem cell source due to faster engraftment and practicability. Previous meta‐analyses analysed patients treated from 1990 to 2002 and demonstrated no impact of the stem cell source on overall survival, but a greater risk for graft‐versus‐host disease (GvHD) in peripheral blood transplants. As transplant indications and conditioning regimens continue to change, whether the choice of the stem cell source has an impact on transplant outcomes remains to be determined.
Objectives
To assess the effect of bone marrow versus peripheral blood stem cell transplantation in adult patients with haematological malignancies with regard to overall survival, incidence of relapse and non‐relapse mortality, disease‐free survival, transplant‐related mortality, incidence of GvHD and time to engraftment.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2014, Issue 1), MEDLINE (from 1948 to February 2014), trial registries and conference proceedings. The search was conducted in October 2011 and was last updated in February 2014. We did not apply any language restrictions.
Selection criteria
We included randomised controlled trials (RCTs) comparing bone marrow and peripheral blood allogeneic stem cell transplantation in adults with haematological malignancies.
Data collection and analysis
Two review authors screened abstracts and extracted and analysed data independently. We contacted study authors for additional information. We used the standard methodological procedures expected by The Cochrane Collaboration.
Main results
We included nine RCTs that met the pre‐defined selection criteria, involving a total of 1521 participants. Quality of data reporting was heterogeneous among the studies. Overall, the risk of bias in the included studies was low.
For the primary outcome overall survival, our analysis demonstrated comparable results between bone marrow transplantation (BMT) and peripheral blood stem cell transplantation (PBSCT) (six studies, 1330 participants; hazard ratio (HR) 1.07; 95% CI 0.91 to 1.25; P value = 0.43; high‐quality evidence).
Disease‐free survival (six studies, 1225 participants; HR 1.04; 95% CI 0.89 to 1.21; P value = 0.6; moderate‐quality of evidence) and non‐relapse or transplant‐related mortality (three studies, 758 participants; HR 0.98; 95% CI 0.76 to 1.28; P = 0.91; high‐quality evidence) were also comparable between transplantation arms.
In the related‐donor setting, data from two of eight studies with 211 participants (21%) indicated a higher relapse incidence in participants transplanted with bone marrow stem cells rather than peripheral blood stem cells (HR 2.73; 95% CI 1.47 to 5.08; P value = 0.001). There was no clear evidence of a difference in relapse incidence between transplantation groups in unrelated donors (HR 1.07; 95% CI 0.78 to 1.47; P value = 0.66). The difference between the donor‐related and ‐unrelated subgroups (P‐value = 0.008) was considered to be statistically significant.
BMT was associated with lower rates of overall and extensive chronic GvHD than PBSCT (overall chronic GvHD: four studies, 1121 participants; HR 0.72; 95% CI 0.61 to 0.85; P value = 0.0001, extensive chronic GvHD: four studies, 765 participants; HR 0.69; 95% CI 0.54 to 0.9; P value = 0.006; moderate‐quality evidence for both outcomes). The incidence of acute GvHD grades II to IV was not lower (six studies, 1330 participants; HR 1.03; 95% CI 0.89 to 1.21; P value = 0.67; moderate‐quality evidence), but there was a trend for a lower incidence of grades III and IV acute GvHD with BMT than with PBSCT (three studies, 925 participants; HR 0.75; 95% CI 0.55 to 1.02; P value = 0.07; moderate‐quality evidence).
Times to neutrophil and platelet engraftment were longer with BMT than with PBSCT (neutrophil: five studies, 662 participants; HR 1.96; 95% CI 1.64 to 2.35; P value < 0.00001; platelet: four studies, 333 participants; HR 2.17; 95% CI 1.69 to 2.78; P value < 0.00001).
Authors' conclusions
This systematic review found high‐quality evidence that overall survival following allo‐HSCT using the current clinical standard stem cell source ‐ peripheral blood stem cells ‐ was similar to that following allo‐HSCT using bone marrow stem cells in adults with haematological malignancies. We found moderate‐quality evidence that PBSCT was associated with faster engraftment of neutrophils and platelets, but a higher risk of GvHD (in terms of more overall and extensive chronic GvHD). There was an imprecise effect on relapse and on severe (grades III to IV) acute GvHD. Quality of life, which is severely affected by GvHD, was not evaluated.
Against the background of transplantation practices that have clearly changed over the past 10 to 15 years, our aim was to provide current data on the best stem cell source for allo‐HSCT, by including the results of recently conducted trials. Our review includes participants recruited up to 2009, a proportion of whom were older, had received reduced‐intensity conditioning regimens or had been transplanted with stem cells from unrelated donors. However, only one, large, study included relatively recently treated participants. Nevertheless, our findings are comparable to those of previous meta‐analyses suggesting that our results hold true for today's practice.
Plain language summary
Which is the most suitable source of donor blood‐forming (stem) cells for transplanting into adults with blood cancers?
Stem cell transplantation
Stem cell transplantation is an important treatment option for individuals with blood cancers (haematological malignancies). During the procedure, blood‐forming (stem) cells, derived from the bone marrow, peripheral blood or umbilical cord blood of a healthy donor, are transplanted into a person with a blood cancer. The aim is to replenish the recipient's body with healthy cells after treatment with conditioning regimens such as chemotherapy or radiation (or both). Peripheral blood stem cells and bone marrow stem cells are the standard stem cell sources used in adults. The most successful transplantations occur when stem cells are transplanted from a healthy donor whose tissue is genetically compatible with that of the recipient (matched related donor). If no matched donor can be identified, it is possible to transplant cells from a matched unrelated donor or from donors carrying certain mismatches. In principle, the higher the degree of genetic mismatch, the higher the risk of severe transplant‐related complications, especially graft‐versus‐host disease (GvHD), in which a donor's white blood cells (T cells) attack the recipient's healthy tissues.
Peripheral blood versus bone marrow stem cells
Peripheral blood stem cells are collected after the donor has received a drug that acts to mobilise stem cells from the bone marrow to the peripheral blood. Bone marrow stem cell donation involves the removal of stem cells from the pelvic bone of the donor under general anaesthesia. Donor convenience as well as logistic reasons favour peripheral blood stem cell donation.
This review addresses the question of which stem cell source ‐ bone marrow or peripheral blood ‐ is the most suitable for individuals undergoing stem cell transplantation.
Clinical results from several studies have been published comparing the use of bone marrow stem cells and peripheral blood stem cells in individuals with haematological malignancies. In most of these studies, the rates at which stem cells received during transplantation start to grow and make new blood cells (known as engraftment) have been shown to be faster following the transplantation of peripheral blood stem cells (PBSCT) than following transplantation of bone marrow stem cells (BMT) platelets. Some studies have reported PBSCT to be associated with a higher risk of developing GvHD than BMT. GvHD is associated with a lower risk of relapse, reflecting the capability of the immune response to simultaneously attack the malignant cells (Graft versus Malignacy effect). On the other hand, GvHD can be an important driver of transplant‐related mortality and morbidity. Disease‐free and overall survival have usually been reported not to differ between PBSCT and BMT. A systematic review from 2005, based on data from individual recipients, could not identify a preferred stem cell source and was largely based on data from the late 1990s. Since then, transplant indications and strategies, as well as supportive care measures, have changed substantially.
Results of this meta‐analysis
In this systemic review we included nine randomised controlled trials involving 1521 participants. Key inclusion criteria were adults undergoing stem cell transplantation for a blood cancer using either bone marrow stem cells or peripheral stem cells as a stem cell source. Participants were treated between 1994 and 2009. The evidence is current to February 2014.
In summary, we found overall and disease‐free survival to be comparable for both PBSCT and BMT. Recipients of bone marrow stem cells from related donors were more likely to relapse than recipients of peripheral blood stem cells from related donors, but this difference was not seen in the recipients of bone marrow stem cells from unrelated donors. The incidence of acute GvHD following PBSCT and BMT was comparable; however, there was a tendency to more severe GvHD with PBSCT. PBSCT was associated with higher rates of chronic GvHD. The time to engraftment was significantly shorter with PBSCT than with BMT. The quality of the evidence was considered moderate to high.
Conclusion
Against the background of altering clinical strategies these results confirm that the current practice of using peripheral blood rather than bone marrow as a source of stem cells for stem cell transplantation in adults with haematological malignancies is not deleterious with respect to overall survival.
Summary of findings
Summary of findings for the main comparison. BMT compared to PBSCT for haematological malignancies.
| BMT compared with PBSCT for haematological malignancies | ||||||
| Participant or population: participants with haematological malignancies Settings: Intervention: BMT Comparison: PBSCT | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| PBSCT | BMT | |||||
| Mortality Follow‐up: median 24 months | Moderate | HR 1.07 (0.91 to 1.25) | 1330 (6 studies) | ⊕⊕⊕⊕ high |
According to GRADE/summary of findings different reporting, mortality has to be calculated instead of OS | |
| 350 per 1000 | 369 per 1000 (324 to 416) | |||||
| Progress, relapse or death Follow‐up: median 24 months | Moderate | HR 1.04 (0.89 to 1.21) | 1225 (6 studies) | ⊕⊕⊕⊝ moderate1 | According to GRADE/summary of findings different reporting, progress, relapse or death has to be calculated instead of DFS | |
| 550 per 1000 | 564 per 1000 (509 to 619) | |||||
| Non relapse or transplant related mortality Follow‐up: median 24 months | Moderate | HR 0.98 (0.76 to 1.28) | 758 (3 studies) | ⊕⊕⊕⊕ high |
||
| 200 per 1000 | 196 per 1000 (156 to 248) | |||||
| Extensive chronic GvHD Follow‐up: median 24 months | Moderate | HR 0.69 (0.54 to 0.9) | 765 (4 studies) | ⊕⊕⊕⊝ moderate1 | ||
| 150 per 1000 | 106 per 1000 (84 to 136) | |||||
| Acute GvHD III to IV Follow‐up: median 100 days | Moderate | HR 0.75 (0.55 to 1.02) | 925 (3 studies) | ⊕⊕⊕⊝ moderate1 | ||
| 250 per 1000 | 194 per 1000 (146 to 254) | |||||
| Chronic GvHD overall Follow‐up: median 24 months | Moderate | HR 0.72 (0.61 to 0.85) | 1121 (4 studies) | ⊕⊕⊕⊝ moderate1 | ||
| 500 per 1000 | 393 per 1000 (345 to 445) | |||||
| Acute GvHD II to IV Follow‐up: median 100 days | Moderate | HR 1.03 (0.89 to 1.21) | 1330 (6 studies) | ⊕⊕⊕⊝ moderate1 | ||
| 800 per 1000 | 809 per 1000 (761 to 857) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Trials not blinded
BMT, bone marrow transplantation
CI, confidence interval
GRADE
GvHD, graft‐versus‐host disease
HR, hazard ratio
PBSCT, peripheral blood stem cell transplantation
Background
Description of the condition
Allogeneic haematopoietic stem cell transplantation
Allogeneic haematopoietic stem cell transplantation (allo‐HSCT) is an important and potentially curative treatment modality for many malignant haematological disorders, such as relapsed and refractory acute or chronic leukaemia, lymphoma and multiple myeloma (Appelbaum 2007). It is also used in non‐malignant haematological bone marrow disorders, such as aplastic anaemia. Since the first allo‐HSCT was performed in the 1960s, numbers have increased steadily, especially in the past few years. Approximately 13,000 allogeneic HSCTs were performed in Europe in 2009 (Baldomero 2011).
Following a conditioning regimen consisting of chemo‐ or radiotherapy, or both, the individual receives stem cells from an unrelated or related donor to replace their own haematopoietic system. Stem cells can either be derived from bone marrow, peripheral blood or umbilical cord blood, resulting in different modalities of transplantation. Donors are closely matched at defined human leukocyte antigen (HLA) class I and class II loci to prevent graft rejection and graft‐versus‐host disease (GvHD) (Petersdorf 2001).
Different strategies of conditioning regimens can be applied. Classic myeloablative conditioning consists of high‐dose chemotherapy with or without total body irradiation. Reduced‐intensity conditioning, introduced about 10 years ago, is less toxic, thus facilitating allo‐HSCT in individuals at advanced age or with comorbidity. Reduced‐intensity conditioning transplants are associated with less‐acute GvHD and fewer infectious complications, but an increased risk of relapse during follow up. Acute and chronic GvHD are the main causes of transplant‐related morbidity and mortality, as well as conditioning regimen toxicity, graft rejection or failure, and infections (Jenq 2010; Pollack 2009).
Graft‐versus‐host disease
GvHD is caused by allo‐reactive donor T cells attacking the recipients' tissues. GvHD can be acute or chronic. According to the classic definition, acute GvHD occurs up to day +100 post‐transplantation (Glucksberg 1974), reflecting the time course of GvHD development after myeloablative conditioning. More recently, with the introduction of reduced‐intensity conditioning and donor lymphocyte infusions, acute and chronic GvHD are mainly distinguished on the basis of clinical features, as acute GvHD can occur at different time points later in the post‐transplant period.
The pathophysiology of acute GvHD is commonly described as comprising three phases. In the first phase, (intensive) conditioning induces damage in host tissues, which causes a pro‐inflammatory milieu and the activation of antigen‐presenting cells. Second, donor T cells are activated by stimulatory cytokines in response to antigen‐presenting cells. Finally, expanded cytotoxic T cells and other cytolytic cells cause further damage in combination with inflammatory cytokines (Ferrera 2009; Paczesny 2009).
Acute GvHD affects mainly the skin, liver and gut. Approximately 40% of individuals experience acute GvHD after allo‐HSCT, but this ranges from 10% to 80% depending on risk factors. Although mortality due to infection or toxicity has decreased significantly within the past 15 years (Gratwohl 2007), GvHD still accounts for at least 25% of transplant‐related deaths.
The main risk factor for the development of acute GvHD is HLA incompatibility, but gender mismatch (male recipient/female donor), prior alloimmunisation, age, conditioning regimen and cytomegalovirus (CMV) serostatus of the donor and recipient are also risk factors. Due to the higher T‐cell content in peripheral blood stem cell grafts, this type of stem cell source is considered a risk factor for GvHD.
The pathophysiology of chronic GvHD is poorly understood. Allo‐reactive T and B cells, and antibody formation are suspected to cause chronic tissue inflammation. Chronic GvHD commonly occurs as a transition from acute GvHD (progressive onset). In 20% to 30% of individuals, chronic GvHD develops without prior GvHD (de novo) or after acute GvHD has resolved (quiescent). The symptoms of chronic GvHD resemble multisystemic autoimmune diseases, such as systemic sclerosis or Sjögren syndrome. Clinical features mainly include changes in the skin, mouth and eyes, but GvHD may also affect the lung and other inner organs. Staging of chronic GvHD into mild, moderate and severe disease is based on the number of organs involved and the severity of organ manifestation according to National Institutes of Health (NIH) consensus criteria (Filipovich 2005). Chronic GvHD is the primary cause of late morbidity and non‐relapse mortality in transplant survivors. Mild chronic GvHD correlates with increased long‐term relapse‐free survival, reflecting the graft versus leukaemia effect (Ferrera 2009). On average, 50% of HSCT recipients develop chronic GvHD, but this ranges from 30% to 80% depending on risk factors (Horwitz 2006). The most important risk factor is previous acute GvHD. Individuals experiencing mild chronic GvHD have a 10‐year survival of 80%; survival is less than 5% in those who develop severe chronic GvHD.
In order to prevent GvHD after allo‐HSCT an immunosuppressive drug therapy is essential as prophylaxis. Immunosuppressive drugs that are widely used include calcineurin inhibitors, such as cyclosporine and tacrolimus, and mammalian target of rapamycin (mTor) inhibitors, such as sirolimus, mycophenolate, methotrexate and antithymocyte globulin. First‐line therapy for both acute and chronic GvHD occurring under immunosuppressive medication is corticosteroids. The prognosis of steroid‐refractory GvHD is poor and there is no standard second‐ or third‐line therapy.
Description of the intervention
Following a conditioning regimen, donor haematopoietic stem cells are infused into the recipient intravenously over two to three hours. As previously mentioned, stems cells derived from bone marrow, peripheral blood or umbilical cord blood can be used. Bone marrow has been the classic source, but granulocyte‐colony stimulating factor (G‐CSF) mobilised peripheral blood stem cells have increasingly been used since the 1990s and have now replaced bone marrow as the main stem‐cell source. The Center for International Blood and Marrow Transplant Research reported that, in the period from 2007 to 2011 about 70% to 80% of adult allogeneic transplant recipients received peripheral blood stem cells (Pasquini 2013). Currently, in Europe, the number is estimated to be even higher (Gratwohl 2013).
Cord blood stem cells are collected from umbilical cords post‐partum and then stored frozen. The small volume and the total cell number limits the use of cord blood to children and individuals with low body weight (Gluckman 2006). For the donor, cord blood represents a harmless and easily accessible source of stem cells that is not associated with any risk. The donation of bone marrow requires general anaesthesia so that stem cells can be obtained from the pelvic bone. The most common adverse events associated with bone marrow donation are tiredness, collection‐site pain, back pain and nausea (Confer 2009;Siddiq 2009). Donor convenience as well as logistic reasons favour peripheral blood stem cell donation over bone marrow donation and have boosted the use of this stem‐cell source. Peripheral blood stem cells are collected using a continuous‐flow cell separation device after injecting the donor with G‐CSF for five days (de Fabritiis 2001). Myalgia, headache and malaise are the most common adverse events observed in peripheral blood stem cell donors after receiving G‐CSF (Confer 2009;Siddiq 2009). It has been suggested that G‐CSF stimulation might cause leukaemia or lymphoma in peripheral blood stem cell donors. The National Marrow Donor Program has followed up 4000 donors for between one and nine years. Twenty cases of cancer at various sites have been reported, but there have been no instances of leukaemia or lymphoma (Confer 2007).
Studies comparing these two common stem cell sources ‐ bone marrow and peripheral blood ‐ have shown peripheral blood stem cell transplantation (PBSCT) to be associated with faster engraftment of neutrophils, platelets and red blood cells than bone marrow transplantation (BMT), resulting in lower infection rates and lower requirements for supplemental blood compounds (Stem Cell Trialists' Collaborative Group 2005, Stem Cell Trialists' Collaborative Group 2006). Due to the higher T‐cell content of peripheral blood stem cells, PBSCT has been associated with faster immune recovery and stronger graft‐versus‐leukaemia reactions. On the downside, higher rates of grades III to IV acute GvHD, as well as chronic GvHD, have been reported for PBSCT than for BMT (Eapen 2007). In conditions not requiring graft‐versus leukaemia reactions, bone marrow is usually chosen as the stem‐cell source due to reportedly lower GvHD rates (Bensinger 2012).
How the intervention might work
It has yet to be determined whether the change to peripheral blood stem cells as the standard source of stem cells could have an adverse impact on outcomes in individuals undergoing allo‐HSCT. GvHD is known to be the strongest risk factor for non‐relapse mortality and morbidity post‐transplant. Studies suggest that bone marrow stem cells carry less risk of both acute and chronic GvHD. The duration of neutropenia and requirements for blood products are thought to be higher in BMT, but improvements in anti‐infectious prophylaxis/treatment and supportive care could outweigh this disadvantage. The impact of a supposedly weaker graft‐versus‐leukaemia effect with BMT than with PBSCT is not clearly defined. BMT could be the better choice for improving overall survival in allo‐HSCT.
Why it is important to do this review
A large individual participant data meta‐analysis comparing BMT and PBSCT in individuals with haematological malignancies was published in 2005 (Stem Cell Trialists' Collaborative Group 2005, Stem Cell Trialists' Collaborative Group 2006), analysing data from 1990 to 2002. In this meta‐analysis, 75% of data were generated by individuals with early disease (chronic myeloid leukaemia in chronic phase, acute myeloid leukaemia, acute lymphoblastic leukaemia in first complete remission and early myelodysplastic syndrome), and chronic myeloid leukaemia was the most frequent diagnosis (40% of all participants treated).
Indications for allo‐HSCT have altered in the past nine years. In 2007, nearly 45% of the individuals who underwent allo‐HSCT in Europe had acute lymphoid or myeloid leukaemia and only approximately 5% had chronic myeloid leukaemia (Gratwohl 2009). One reason for this development was the introduction of the drug imatinib into the treatment of individuals with chronic myeloid leukaemia (CML) in chronic phase in 2002, which had previously been the main indication for allogeneic transplantation in adults.
Furthermore, individuals with co‐morbidities and those 70 years of age and older are now eligible to undergo allo‐HSCT, following the introduction of reduced‐intensity or non‐myeloablative conditioning regimens, which have resulted in a decrease in regimen‐related morbidity and mortality. Due to laboratory improvements, such as more precise HLA typing, and improvements in recipient care, the use of unrelated donors and HLA‐mismatched HSCT has also increased. In 2006, more than one‐third of allogeneic transplants performed worldwide used unrelated donors (Gratwohl 2013).
These recent developments require an updated review of the clinical data to assess the impact of using either bone marrow or peripheral blood cells as a stem cell source on survival, relapse and GvHD under current clinical conditions. A previous meta‐analysis, published in 2011 (Chang 2012), was based on the same dataset as the Stem Cell Trialists' analysis in 2005 (Stem Cell Trialists' Collaborative Group 2005, Stem Cell Trialists' Collaborative Group 2006) and therefore did not reflect the described changes in the transplant setting.
Controversy remains regarding the appropriate stem cell source (Bensinger 2012;Pidala 2009a), as BMT in some studies has been associated with lower acute and chronic GvHD rates than PBSCT, potentially translating into an improvement in overall survival. This review will aim to update the findings of previous meta‐analyses by including recently published studies.
Objectives
To compare allo‐HSCT using stem cells derived from bone marrow or peripheral blood in adults with haematological malignancies with regard to overall survival, incidence of relapse and non‐relapse mortality, disease‐free survival, transplant‐related mortality, incidence of graft‐versus‐host disease (GvHD) and time to engraftment.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled studies. We included full text and abstract publications, and unpublished data, if sufficient information was available.
Types of participants
Adults of both sexes with haematological malignancies who received a bone marrow or peripheral blood allo‐HSCT. Although we defined adult age as an inclusion criterion, we did not mean to exclude studies involving a mixed population or a broad distribution of age. We excluded studies with a focus on paediatric participants, as the immune reconstitution and underlying disease characteristics of children differ from those in adults.
Types of interventions
Experimental intervention
Bone marrow allo‐HSCT for haematological malignancies
Control intervention
Peripheral blood allo‐HSCT for haematological malignancies
Types of outcome measures
Primary outcomes
Overall survival
Secondary outcomes
Incidence of relapse and non‐relapse mortality
Disease‐free survival
Transplant‐related mortality
Incidence of overall chronic GvHD
Incidence of extensive chronic GvHD
Incidence of acute GvHD grades II to IV
Incidence of acute GvHD grades III to IV
Time to platelet engraftment
Time to neutrophil engraftment
Quality of life
Search methods for identification of studies
Electronic searches
We adapted search strategies (Appendix 1; Appendix 2) from those suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Lefebvre 2011). We did not apply any language restrictions in order to reduce potential language bias.
We searched the following databases of medical literature:
Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2014, Issue 1);
MEDLINE (from 1948 to February 2014).
Searching other resources
We searched for abstracts in the conference proceedings of annual meetings of the following societies (electronically from 2000 to 2010 and manually from 2011 to 2013):
American Society of Hematology;
European Hematology Association;
European Group of Bone Marrow Transplantation;
American Society of Bone Marrow Transplantation.
We electronically searched the following database of ongoing trials:
meta‐register of Controlled Trials (http://www.controlled‐trials.com/mrct/).
We also handsearched references
of all identified trials and relevant review articles.
Data collection and analysis
Selection of studies
Two review authors (UH and MA) independently selected studies from the titles and abstracts of those identified from the above sources. After the first review of all titles and abstracts, we rejected all studies that were clearly ineligible. We assessed selected studies with regard to study design and compliance with inclusion criteria using an eligibility form (Higgins 2011a).
The eligibility form contained the following questions.
Was the study described as randomised?
Did the study compare the use of peripheral blood stem cells with bone marrow stem cells in allo‐HSCT?
In case of doubt, we included an analysis of the full text and reached a decision though discussion (preferably including studies rather than losing relevant data). According to Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) guidelines, we include a flow diagram showing the numbers of identified records, excluded articles and included studies (Figure 1) (Moher 2009).
1.
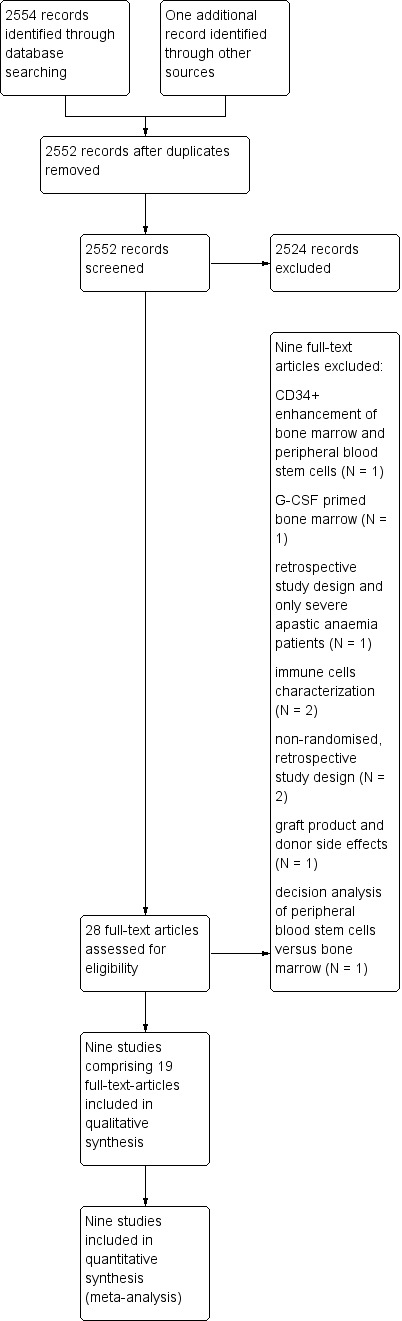
Study flow diagram.
Data extraction and management
Two review authors (UH and MA) independently extracted data according to the Cochrane Handbook for Systematic Reviews of Interventions using a standardised data extraction form containing the following items (Higgins 2011a).
General information:
author, title, source, publication date, country, language, duplicate publications;
Quality assessments:
sequence generation, allocation concealment, blinding (participants, personnel, outcome assessors), incomplete outcome data, selective outcome reporting, other sources of bias;
Study characteristics:
trial design, aims, setting and dates, source of participants, inclusion/exclusion criteria, comparability of groups, subgroup analyses, statistical methods, power calculations, treatment cross‐overs, compliance with assigned treatment, length of follow up, time point of randomisation;
Participant characteristics
age, gender, ethnicity, number of participants recruited/allocated/evaluated, participants lost to follow up, diagnosis and stage of disease, histological subtype, additional diagnosis, HLA compatibility/incompatibility;
Interventions:
setting, type, type and intensity of conditioning regimen, type of transplantation source, dose of G‐CSF, duration of follow up;
Outcomes:
overall survival, incidence of relapse and non‐relapse mortality, transplant‐related mortality, incidence of acute and chronic GvHD, disease‐free survival, time to platelet and neutrophil engraftment.
Assessment of risk of bias in included studies
Two review authors (UH and MA) assessed the methodological quality and risk of bias in each included study using the following criteria, according to the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b):
sequence generation;
allocation concealment;
blinding (participants, personnel, outcome measures);
incomplete outcome data;
selective outcome reporting;
concurrent participation in different clinicals trials;
early termination of the trial.
Judgements of the review authors for each criterion were based on a three‐point scale (low risk of bias; high risk of bias; unclear risk of bias); the review authors also provided a summary description.
Measures of treatment effect
For binary outcomes, we calculated risk ratios (RRs) with 95% confidence intervals (CIs) for each trial if they were available. We calculated continuous outcomes as standardised mean differences. For time‐to‐event outcomes, we extracted the hazard ratios (HRs). In trials where the HR was not reported, the HR for each endpoint was calculated from the observed minus expected number of events and variance. If we were unable to extract data for time‐to‐event outcomes directly, then we calculated the summary estimates (HRs and CIs) using the methods by Parmar and Tierney (Parmar 1998; Tierney 2007).
Dealing with missing data
There were some potential sources of missing data that we needed to take into account: at outcome level, at summary data level, at individual level and at study‐level (e.g. for subgroup analyses (Higgins 2011c)). First, it was important to differentiate between 'missing at random' and 'not missing at random'. We contacted original investigators to request missing outcome data. We received additional data for two studies (Anasetti 2012; Mielcarek 2011). If data were still missing, we made explicit assumptions of any methods used (e.g. that the data were assumed missing at random or that missing values were assumed to have a particular value, such as a poor outcome).
Assessment of heterogeneity
We assessed the heterogeneity of treatment effects between trials using the Chi2 test with a significance level set at a P value less than 0.1. We used the I2 statistic to quantify possible heterogeneity (I2 > 30% moderate heterogeneity, I2 > 75% considerable heterogeneity) (Deeks 2011).
We planned to explore potential causes of heterogeneity by sensitivity and subgroup analyses using meta‐regression, but with a total number of included studies less than 10 it was inappropriate to perform meta‐regression.
Assessment of reporting biases
In meta‐analyses including at least 10 trials, we planned to explore potential publication bias by generating a funnel plot and testing it statistically using a linear regression test (Sterne 2011). We considered a P value of less than 0.1 significant for this test. As we included only nine trials in the meta‐analysis, we did not perform a funnel plot analysis.
Data synthesis
We performed analyses according to the recommendations of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011). We used aggregated data for analysis. For statistical analysis, we entered data into The Cochrane Collaboration's statistical software, Review Manager (Review Manager 2011). One review author (MA) entered the data and a second review author (UH) checked them for accuracy. We performed meta‐analyses using a fixed‐effect model (e.g. the generic inverse variance method for survival data outcomes and Mantel‐Haenszel method for dichotomous data outcomes). If appropriate, we calculated the number needed to treat for an additional beneficial outcome and the number needed to treat for an additional harmful outcome. We created 'Summary of findings' tables, as suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011). Our prioritised endpoints in the 'Summary of findings' tables were overall survival, disease‐free survival, incidence of relapse, transplant‐related mortality and incidence of chronic GvHD.
Subgroup analysis and investigation of heterogeneity
We planned subgroup analyses on the following characteristics during development of the review protocol:
underlying disease;
age (18 to 60 years, adults ≥ 60 years). We chose this cut‐off point for age because of the increased risk of non‐relapse mortality which usually occurs in elderly individuals (participants aged > 60 years are usually defined as elderly in studies);
type of donor (matched unrelated donor, matched related donor, mismatched unrelated donor, mismatched related donor, age, sex, CMV status, blood group);
type of conditioning regimen (myeloablative versus reduced intensity).
Based on the data reported we performed such analyses only for the subgroups 'related donor' versus 'unrelated donor'.
We discussed possible reasons for any heterogeneity according to the prespecified subgroups.
Sensitivity analysis
We planned sensitivity analyses on the following items during development of the review protocol:
size of the trial;
funding of the trial;
the level of loss to follow up;
quality components, including full text publications/abstracts, preliminary results versus mature results.
The included studies were homogeneous with regard to the reporting quality and quality of the trials. Size differed, but mature data from all studies were published in peer‐reviewed journals. There was no third‐party funding suspected to have influenced the studies. Hence, we performed no sensitivity analyses.
Results
Description of studies
We summarise study details in 'Characteristics of included studies' and 'Characteristics of excluded studies' tables.
Results of the search
We identified a total of 2554 publications using the computerised search strategy. We handsearched references of all identified trials, relevant review articles and current treatment guidelines and found one more randomised controlled trial. We excluded two duplicates. Of the remaining 2552 records, we excluded 2524 publications and retrieved 28 full‐text articles for further assessment. We excluded nine articles and included 19 publications reporting on a total of nine randomised controlled trials (see Figure 1).
Included studies
Included studies
Nine randomised controlled studies were included (Anasetti 2012; Couban 2002; Friedrichs 2010; Heldal 2003; Mahmoud 1999; Mielcarek 2011; Mohty 2002; Powles 2002; Vigorito 2001) in this review. All studies were published in English. Outcome data for 1521 participants were evaluated. Participants were treated in 122 centres and recruitment periods ranged from 1994 to 2009, with a maximum median follow up of more than 12 years (see 'Characteristics of included studies' table).
Standard GvHD prophylaxis
For GvHD prophylaxis, the calcineurin inhibitors, cyclosporine or tacrolimus, were used in combination with methotrexate in all studies. Cyclosporine dosing was comparable in seven studies (Anasetti 2012; Couban 2002; Friedrichs 2010; Heldal 2003; Mahmoud 1999; Mohty 2002; Powles 2002). In five studies (Anasetti 2012; Couban 2002; Heldal 2003; Mahmoud 1999; Powles 2002) methotrexate was administered on days +1, +3, +6 and +11, whereas Friedrichs 2010 and Mohty 2002 omitted the day +11 administration. Mielcarek 2011 reported no exact data for cyclosporine and methotrexate dosing. Vigorito 2001 also provided no exact data regarding dosage, and three participants in the intervention arm received prednisone instead of methotrexate. Anasetti 2012 reported that most participants received tacrolimus instead of cyclosporine. Details regarding GvHD prophylaxis are summarised in Table 2.
1. Standard GvHD prophylaxis.
| Study ID | Cyclosporine | Methotrexate |
| Anasetti 2012 | From day ‐1 until day +180, first intravenously and then oral intake (data according to study protocol); most participants (66% in BMT group and 72% in PBSCT group) received tacrolimus instead of cyclosporine | Intravenously 15 mg/m² on day +1 and 10 mg/m² on days +3, +6, +11 |
| Couban 2002; Mahmoud 1999; Powles 2002 | From day ‐1 until day +180, first intravenously and then oral intake | Intravenously 15 mg/m² on day +1 and 10 mg/m² on days +3, +6, +11 |
| Heldal 2003 | From day ‐1 until day +180, first intravenously and then oral intake | Intravenously 15 mg/m² on days +1, +3, +6, +11 |
| Friedrichs 2010 | From day ‐1 until day +180, first intravenously and then oral intake | Intravenously 15 mg/m² on day +1 and 10 mg/m² on days +3, +6 |
| Mohty 2002 | From day ‐1 until day +180, first intravenously and then oral intake | Intravenously 15 mg/m² on days +1, +3, +6 |
| Mielcarek 2011 | Administered, but dose not reported | Administered, but dose not reported |
| Vigorito 2001 | Administered, but dose not reported | Administered, but dose not reported, three participants received prednisone instead of methotrexate in the intervention arm |
BMT, bone marrow transplantation
GvHD, graft‐versus‐host disease
PBSCT, peripheral blood stem cell transplantation
Conditioning regimen
In all studies participants were treated with different myeloablative conditioning regimens, depending on the underlying disease. In six of nine studies (Friedrichs 2010; Mahmoud 1999; Mielcarek 2011; Mohty 2002; Powles 2002; Vigorito 2001) participants were treated with myeloablative conditioning regimens either with total body irradiation (TBI), mostly in combination with busulphan and cyclophosphamide, or without TBI (Couban 2002; Heldal 2003). Anasetti 2012 used both myeloablative (in nearly 80% of participants) and non‐myeloablative conditioning regimens. In the same study, antithymocyte globulins were used for GvHD prophylaxis as part of the conditioning regimen in 25% and 28% of the participants transplanted with bone marrow or peripheral stem cells, respectively. A comparison of conditioning regimens is presented in Table 3.
2. Conditioning regimen.
| Study ID | Conditioning regimen intervention arm | Conditioning regimen control arm |
| Anasetti 2012 | Myeloablative: N = 223 (80%) with cyclophosphamide (120 mg/kg) + TBI (12 Gy) N = 133 or cyclophosphamide (120 mg/kg) + busulphan (14 mg/kg orally or 11.2 mg/kg intravenously) N = 90; non‐myeloablative regimen: N = 55 (20%) with fludarabine (120 mg/m2 of body‐surface area) + busulphan (250 mg/m2 or 8 mg/kg) + antithymocyte globulin N = 39 or fludarabine (120 mg/m2) + melphalan (140 mg/m2) N = 16 | Myeloablative: N = 208 (76%) with cyclophosphamide (120 mg/kg) + TBI (12 Gy) N = 133 or cyclophosphamide (120 mg/kg) + busulphan (14 mg/kg orally or 11.2 mg/kg intravenously N = 75; non‐myeloablative: N = 65 (24%) with fludarabine (120 mg/m2 of body‐surface area) + busulphan (250 mg/m2 or 8 mg/kg) + antithymocyte globulin N = 40 or fludarabine (120 mg/m2) + melphalan (140 mg/m2) N = 25 |
| Couban 2002 | Myeloablative: busulphan (1 mg/kg orally every 6 hours for 16 doses, day ‐7 to day ‐4) followed by cyclophosphamide (60 mg/kg intravenously for 2 days, day ‐3 and ‐2) N = 118 (100%) | Myeloablative: busulphan (1 mg/kg orally every 6 hours for 16 doses, day ‐7 to day ‐4) followed by cyclophosphamide (60 mg/kg intravenously for 2 days, day ‐3 and ‐2) N = 109 (100%) |
| Friedrichs 2010 | Myeloablative: TBI (single dose or fractioned) + cyclophosphamide N = 96 (58%) or + etoposide N = 2 (1%) in standard doses or + cyclophosphamide + etoposide N = 8 (5%) or + melphalan N = 1 or + etoposide + melphalan N = 1 or busulphan + cyclophosphamide N = 54 (33%) and + etoposide N = 3 (1%) or busulphan + melphalan N = 1 | Myeloablative: TBI (single dose or fractioned) + cyclophosphamide N = 101 (62%) or + etoposide N = 1 in standard doses or + cyclophosphamide + etoposide N = 3 (2%) or busulphan + cyclophosphamide N = 55 (34%), and + etoposide N = 2 (1%) or busulphan + melphalan N = 1 |
| Heldal 2003 | Myeloablative: busulphan (16 mg/kg) and cyclophosphamide (120 mg/kg) and intrathecal methotrexate (12 mg/kg) for recipients with acute myeloid leukaemia M4/5 or acute lymphoblastic leukaemia on days ‐8 and ‐4 and four times after transplantation N = 30 (100%) | Myeloablative: busulphan (16 mg/kg) and cyclophosphamide (120 mg/kg) and intrathecal methotrexate (12 mg/kg) for recipients with acute myeloid leukaemia M4/5 or acute lymphoblastic leukaemia on days ‐8 and ‐4 and four times after transplantation N = 30 (100%) |
| Mahmoud 1999 | Only TBI (10 Gy fractioned over 4 consecutive days) + cyclophosphamide (60 mg/kg/day for 2 consecutive days) N = 15 (100%) | TBI (10 Gy fractioned over 4 consecutive days) + cyclophosphamide (60 mg/kg/day for 2 consecutive days) N = 14 (93%) or busulphan (4 mg/kg/day for 4 consecutive days) + cyclophosphamide (60 mg/kg/day for 2 consecutive days) N = 1 (7%) |
| Mielcarek 2011 | Myeloablative: TBI (total dose 12 to 13.5 Gy) + busulphan N = 13 (14%) or + cyclophosphamide N = 30 (33%) or + etoposide N = 7 (8%) or busulphan + cyclophosphamide N = 40 (44%), busulphan + thiotepa N = 1 (1%) | Myeloablative: TBI (total dose 12 to 13.5 Gy) + busulphan N = 12 (15%) or + cyclophosphamide N = 24 (30%) or + etoposide N = 13 (16%) or + busulphan + cyclophosphamide N = 3 (4%) or busulphan + cyclophosphamide N = 29 (35%) |
| Mohty 2002 | Myeloablative: TBI (median dose of 12 Gy and a median fraction of 6 fractions) + cyclophosphamide (120 mg/kg) N = 39 (74%) or + cyclophosphamide (120 mg/kg) + etoposide (60 mg/kg) N = 7 (13%) or + cytarabine + melphalan N = 1 (2%) or busulphan (16 mg/kg) + cyclophosphamide (200 mg/kg) N = 6 (11%) | Myeloablative: TBI (median dose of 12 Gy and a median fraction of 6 fractions) + cyclophosphamide (120 mg/kg) N = 34 (71%) or + cyclophosphamide (120 mg/kg) + etoposide (60 mg/kg) N = 4 (8%) or + cytarabine + melphalan N = 1 (2%) or busulphan (16 mg/kg) + cyclophosphamide (200 mg/kg) N = 9 (19%) |
| Powles 2002 | Myeloablative: TBI + melphalan N = 12 (63%) or + etoposide N = 1 (5%) or busulphan + cyclophosphamide N = 6 (32%) | Myeloablative: TBI + melphalan N = 12 (60%) or + etoposide N = 1 (5%) or busulphan + cyclophosphamide N = 7 (35%) |
| Vigorito 2001 | Myeloablative: busulphan (16 mg/kg) + cyclophosphamide (120 mg/kg) N = 16 (85%), busulphan (16 mg/kg) + cyclophosphamide (120 mg/kg) + etoposide (40 mg/kg) N = 3 (15%) | Myeloablative: TBI + cyclophosphamide (120 mg/kg) N = 1 (6%) or busulphan (16 mg/kg) + cyclophosphamide (120 mg/kg) N = 17 (94%) |
TBI, total body irradiation
G‐CSF (filgrastim) after allo‐HSCT
In two of nine studies (Friedrichs 2010; Mahmoud 1999), all participants received G‐CSF after transplantation at a dose of 10 μg/kg/day from day +1 to neutrophil recovery, which could influence the time to engraftment.
Participant characteristics
Age
Median participant age for all included studies ranged from 21 to 45 years (age range over all studies was 10 to 65 years). Four studies (Friedrichs 2010; Mielcarek 2011; Mohty 2002; Powles 2002) stated age over 55 years as an exclusion criterion. Four more studies (Couban 2002; Heldal 2003; Mahmoud 1999; Vigorito 2001) accepted participants aged up to 65 years of age. Anasetti 2012 included participants under 66 years of age. Vigorito 2001 included participants over the age of 10 years, Mielcarek 2011, over the age of 12 years and Couban 2002, over the age of 16 years. Six studies (Anasetti 2012; Friedrichs 2010; Heldal 2003; Mahmoud 1999; Mohty 2002; Powles 2002) stated age over 18 years as an inclusion criterion. Details regarding participant age are summarised in Table 4.
3. Participants characteristics.
| Study ID |
Median age (range) BMT group |
Median age (range) PBSCT group |
Recipient male/ donor female |
Underlying disease BMT group |
Underlying disease PBSCT group |
Early‐stage disease | Advanced‐stage disease | Graft characteristics |
| Anasetti 2012 | > 40 years 159 participants (57%) | > 40 years 159 participants (58%) | Not reported | AML: N = 130 ALL: N = 61 CML: N = 29 MDS: N = 52 CMML: N = 4 MF: N = 2 |
AML: N = 131 ALL: N = 56 CML: N = 37 MDS: N = 41 CMML: N = 4 MF: N = 4 |
BMT: 22% PBSCT: 23% |
BMT: 78% PBSCT: 77% |
Up to 3 mismatches allowed: BMT 20%, PBSCT 24% with mismatched donor, 87.5% were single mismatch grafts |
| Couban 2002 | 44 years (19 to 64) | 45 years (19 to 64) | not reported | CML in chronic or accelerated phase, AML in first or subsequent remission, MDS | CML in chronic or accelerated phase, AML in first or subsequent remission, MDS | BMT: 74% PBSCT: 72% |
BMT: 26% PBSCT: 28% |
One antigen mismatched allowed, no detailed data provided |
| Friedrichs 2010 | 37 years (19 to 55) |
37 years (19 to 58) |
BMT: 22% PBSCT: 20% |
De‐novo AML or ALL in first or 2nd complete remission, CL in first chronic or accelerated phase or MDS | De‐novo AML or ALL in first or 2nd complete remission, CL in first chronic or accelerated phase or MDS | Not reported | Not reported | HLA‐matched related or unrelated donor |
| Heldal 2003 | 45 years (18 to 55) |
39 years (15 to 52) |
BMT: 13% PBSCT: 29% |
CML in chronic phase, AML in complete remission and early relapse, ALL in complete remission and early relapse, MDS, primary MF | CML in chronic phase, AML in complete remission and early relapse, ALL in complete remission and early relapse, MDS, primary MFs | Not reported | Not reported | Single mismatch donor allowed: BMT 3%, PBSCT 17% mismatched |
| Mahmoud 1999 | 21.8 years | 23 years | Not reported | AML: N = 6 ALL: N = 5 CML: N = 3 SAA: N = 1 |
AML: N = 3 ALL: N = 3 CML: N = 4 SAA: N = 3 MDS: N = 2 |
Not reported | Not reported | HLA‐matched related or unrelated donor |
| Mielcarek 2011 | 42 years (12 to 55) |
42 years (15 to 55) |
BMT: 24% PBSCT: 35% |
Any haematologic cancer that can be treated by transplantation | Any haematologic cancer that can be treated by transplantation | BMT: 56% PBSCT: 51% |
BMT: 44% PBSCT: 49% |
HLA‐matched related or unrelated donor |
| Mohty 2002 | 36.5 years | 37.3 years | BMT: 25% PBSCT: 31% |
Acute leukaemia in first or 2nd complete remission,CML in first chronic phase | Acute leukaemia in first or 2nd complete remission, CML in first chronic phase | Not reported | Not reported | HLA‐matched related or unrelated donor |
| Powles 2002 | 37 years (22 to 51) |
34 years (24 to 51) |
Not reported | AML: N = 3 ALL: N = 4 CML: N = 6 CLL: N = 1 NHL: N = 1 MM: N = 1 MDS: N = 1 ABL: N = 2 |
AML: N = 5 ALL: N = 3 CML: N = 6 CLL: N = 1 MDS: N = 1 MM: N = 1 ABL: N = 3 |
BMT: 63% PBSCT: 45% |
BMT: 37% PBSCT: 55% |
HLA‐matched related or unrelated donor |
| Vigorito 2001 | 35 years (17 to 56) |
29.5 years (9 to 51) |
Not reported | Haematologic malignancies as primary disease | Haematologic malignancies as primary disease | BMT: 68% PBSCT: 72% |
BMT: 32% PBSCT: 28% |
HLA‐matched related or unrelated donor |
ABL = acute biphenotypic leukaemia, AML = acute myeloid leukaemia, ALL = acute lymphoid leukaemia, BMT, bone marrow transplantation; CML = chronic myeloid leukaemia, CMML = chronic myelomonocytic leukaemia, CLL = chronic lymphoid leukaemia, MDS = myelodysplastic syndrome, MF = myelofibrosis, MM = multiple myeloma, NHL = non‐Hodgkin lymphoma, PBSCT, peripheral blood stem cell transplantation; SAA = severe aplastic anaemia
Underlying disease
The underlying diseases in all studies included acute leukaemia and chronic myeloid leukaemia. Couban 2002, Heldal 2003 and Friedrichs 2010 additionally included participants with myelodysplastic syndrome. Anasetti 2012 and Heldal 2003 also included participants with primary myelofibrosis. Three studies (Mahmoud 1999; Mielcarek 2011; Powles 2002) included participants with any haematological malignancy that can be treated using allo‐HSCT. Details are summarised in Table 4.
Stage of disease
In four of nine studies (Friedrichs 2010; Heldal 2003; Mahmoud 1999; Mohty 2002) the stage of disease was not reported (34% of the data‐set). In all the other five studies (Anasetti 2012; Couban 2002; Mielcarek 2011; Powles 2002; Vigorito 2001), with a total of 1001 participants (66%), similar percentages of participants with early and advanced stages of disease were seen in the intervention and control arms of each study. Comparing these studies, it was noticed that in two studies (Couban 2002; Vigorito 2001), 70% of participants had early stage disease and 30% had advanced disease, whereas in Mielcarek 2011 and Powles 2002, this split was nearer 50%. The most recent study of Anasetti 2012 reported that a majority (nearly 80%) of participants in both arms had advanced‐stage disease. Details about stage of disease are presented in Table 4.
Recipient/donor gender (male/female)
Five studies (Anasetti 2012; Couban 2002; Mahmoud 1999; Powles 2002; Vigorito 2001), involving a total 859 participants (56%), did not report data on the adverse combination 'male recipient with a female donor', which is known to be a risk factor for GvHD. In the remaining four studies (Heldal 2003; Friedrichs 2010; Mielcarek 2011; Mohty 2002), involving a total 662 participants (44%), between 13% and 35% of male recipients had a female donor, similarly distributed between the intervention and control arms. Details about recipient/donor gender are presented in Table 4.
Graft characteristics
Of all 1521 participants, 775 (51%) were randomised to BMT and 776 (49%) underwent PBSCT. The majority of participants were transplanted with a HLA‐matched related or unrelated donor. Studies by Couban 2002 and Heldal 2003 allowed up to one antigen mismatch. Couban 2002 did not provide data on the number of participants transplanted with mismatched grafts. Heldal 2003 reported that 3% of participants in the BMT arm as opposed to 17% in the PBSCT arm received a single‐mismatched graft. Anasetti 2012 documented that 20% and 24% of participants in the BMT and PBSCT arms, respectively, received a graft with up to three mismatches (87.5% were single mismatches). Details regarding graft characteristics are presented in Table 4.
Summaries of individual trials
All studies evaluated the transplantation of bone marrow stem cells as compared with peripheral blood stem cells from related or unrelated donors in participants with haematological malignancies.
Anasetti 2012 evaluated 526 participants between March 2004 and September 2009 in 48 transplantation centres in the USA and Canada. Only participants with unrelated donors were recruited. Median follow up was three years. Median age was not reported, but 57% and 58% of participants were aged between 41 and 66 years in the bone marrow and control groups, respectively. Participants with high‐risk disease were equally distributed (28%) in both groups. Participants predominantly received a myeloablative conditioning regimen, but 20% of the bone marrow group and 24% of the controls group received non‐myeloablative conditioning. GvHD prophylaxis consisting of tacrolimus and methotrexate was administered in 66% of participants in the intervention arm and 74% in the control arm. Most of the remaining participants received cyclosporine and methotrexate (24% of the bone marrow group and 22% of the control group). It was noted that 24% and 20% of participants in the bone marrow and peripheral blood stem cell groups received a graft with up to three mismatches (87.5% were single mismatches).
Couban 2002 recruited 227 participants between 1996 and 2000 from eight BMT centres in Canada and New Zealand, and reported data with a median follow up of 2.7 years. Median participant age was 44 years in the BMT group and 45 years in the PBSCT group. In the BMT and PBSCT groups, 74% and 72% of participants, respectively, had early‐stage disease. Participants received a myeloablative conditioning regimen with busulphan followed by cyclophosphamide without TBI. Standard GvHD prophylaxis consisted of cyclosporine and methotrexate on days +1, +3, +6 and +11.
Friedrichs 2010 presented long‐term data from a study previously published in 2002 and 2005 (Schmitz 2002; Schmitz 2005). The median follow up was 10,8 years. The recruitment period was between 1995 and 1999 from 42 transplantation centres in Europe and Australia, and 329 participants were evaluated. Median age was 37 years in both groups and gender mismatch (male recipient/female donor) occurred in 22% (36 pairs) of bone marrow transplants and 20% (33 pairs) of peripheral blood transplants. In both the BMT and PBSCT groups, 65% of participants (N = 108 / N = 105, respectively) received TBI, mostly in combination with cyclophosphamide, and 35% (N = 58/ N = 58, respectively) received cyclophosphamide in combination with busulphan. GvHD prophylaxis consisted of cyclosporine and methotrexate on days +1, +3 and +6. All participants received filgrastim until day +28 or until neutrophil recovery.
Heldal 2003 designed a single‐centre study in Norway evaluating 60 participants who had been recruited between 1994 and 1999. Median follow up was five years. Median age was 45 years in the BMT group and 39 years in the PBSCT group. Gender mismatch (male recipient/female donor) was reported in 13% (N = 4) of the BMT group and 30% (N = 9) of the PBSCT group. As a conditioning regimen, all participants received busulphan and cyclophosphamide. Standard GvHD prophylaxis included cyclosporine and methotrexate on days +1, +3, +6 and +11.
Mahmoud 1999 recruited 30 participants between1995 and 1997 from a single institution in Egypt. In the BMT group, the median age was 21.8 years as compared with 23 years in the PBSCT group. All participants received TBI followed by cyclophosphamide, except one in the PBSCT group, who received busulphan and cyclophosphamide. Standard GvHD prophylaxis included cyclosporine and methotrexate on days +1, +3, +6 and +11, and all participants received filgrastim until neutrophil recovery.
Mielcarek 2011 presented the long‐term outcomes of a US multi‐centre study first published in 2001 (Bensinger 2001), with a median follow up of 12.2 years. The recruitment period was between 1996 and 1999 from three Medical Centres in the USA. They evaluated 172 participants with a median age of 42 years in both groups. Participants were between 12 and 55 years old. Gender mismatch (male recipient/female donor) was documented in 24% of participants in the BMT group and 35% in the PBSCT group. In the BMT group, 56% of participants had a less‐advanced stage of disease compared with 51% of the control group. In the BMT group, 55% of participants received TBI in combination with cyclophosphamide (33%) or other chemotherapy (22%), 44% received busulphan in combination with cyclophosphamide without TBI. In the PBSCT arm, 64% of participants received TBI in combination with cyclophosphamide or other chemotherapy, and 36% received busulphan with cyclophosphamide. All participants received cyclosporine and methotrexate as GvHD prophylaxis.
Mohty et al (Blaise 2000; Mohty 2002) evaluated 101 participants from 17 centres in France between 1996 and 1998, and reported the data with a median follow up of 3.75 years. The median age was 36.5 years in the BMT group and 37.3 years in the PBSCT group. Gender mismatch (male recipient/female donor) was documented in 25% of the BMT group and 31% of the PBSCT group. In the BMT arm, 89% of participants received TBI in combination with cyclophosphamide or other chemotherapy, 11% had chronic myeloid leukaemia and received busulphan instead of TBI. In the PBSCT arm, 19% of participants with chronic myeloid leukaemia received busulphan and cyclophosphamide. The remaining participants (81%) received TBI with cyclophosphamide or other chemotherapy. GvHD prophylaxis included cyclosporine and methotrexate on days +1, +3 and +6.
Powles 2002 evaluated 39 participants from a single institution study in the UK. Recruitment time was between 1995 and 1997. Median follow up was 2.75 years. Median age was 37 years in the BMT group and 34 years in the PBSCT group. Sixty‐three per cent of participants in the BMT group had low‐risk disease compared with 45% of the PBSCT group. In the intervention arm, 32% of patients received busulphan and cyclophosphamide and 68% received TBI plus melphalan or etoposide. In the control arm 35% of the patients received busulphan and cyclophosphamide whereas 65% of participants received TBI plus melphalan or etoposide. Standard GvHD prophylaxis consisted of cyclosporine and methotrexate on days +1, +3, +6 and +11.
Vigorito et al (Vigorito 2001; Vigorito 1998) recruited 37 participants to a single‐centre trial in Brazil between 1995 and 1999. Median follow up was 6.7 years. Participants aged 10 to 60 years were included; median age was 35 years in the BMT group and 29.5 years in the PBSCT group. Early‐stage disease was documented in 68% of the BMT arm and in 72% of the PBSCT group. Nearly all participants received busulphan and cyclophosphamide as a conditioning regimen. Only one (6%) in the PBSCT group received TBI plus cyclophosphamide. GvHD prophylaxis included cyclosporine and methotrexate in most cases, but 16% of participants in the BMT group received methotrexate and prednisone.
Statistical evaluation of time‐to‐event outcome data The statistical evaluation of time‐to‐event data in stem cell transplantation was not reported accurately in all trials. According to the statistical guidelines recommended by the European Group Blood and Marrow Transplantation (EBMT) (Labopin 2003), time‐to event outcomes with competing risks should be compared using the cumulative incidence estimator. Using this method, only participants at risk for a specific outcome are evaluated (i.e. participants who die without experiencing the event of interest are censored). It is recommended that outcomes without competing events are analysed using Kaplan‐Meier methodology. A detailed summary of the statistical outcomes of this review, based on the EBMT guidelines (Labopin 2003), is summarised in Table 5.
4. Statistical Outcomes.
| Outcome | Relevant events | Censored cases | Competing events |
| Overall survival | Death regardless of cause | Participants alive at last contact | None |
| Incidence of relapse | Relapse | Participants alive without relapse at last contact | Death without evidence of relapse |
| Disease‐free survival | Time to relapse or death from any cause, which ever comes first | Participants alive without any disease at last contact | Death without evidence of disease |
| Non‐relapse or transplant‐related mortality | Time to deaths without relapse/recurrence. Deaths from any cause without prior progression. | — | Events related to the disease such as relapse or progression |
| Chronic GvHD | Chronic GvHD | Participants alive with no episode of chronic GvHD at last follow up | Death without chronic GvHD |
| Acute GvHD | Acute GvHD | Participants alive with no occurrence of acute GvHD at 100 days | Death without acute GvHD within 100 days |
| Engraftment | Persistent blood cells count above predefined level | Participants alive with no recovery at last follow up | Death before recovery |
GvHD, graft‐versus‐host disease
Modified table of statistical outcomes according to the EBMT statistical guidelines (Labopin 2009). Cumulative Incidence curves were not used for this meta‐analysis.
Couban 2002 and Mielcarek 2011 reported overall survival data using the Kaplan‐Meier method. All other endpoints were computed according to the method described by Kalbfleisch and Prentice (Kalbfleisch 1980). Mahmoud 1999 and Mohty 2002 reported using Kaplan‐Meier methodology, but did not state for which time‐to‐event outcomes. Friedrichs 2010 used Kaplan‐Meier methodology for the outcomes overall survival and disease‐free survival. They reported no other time‐to‐event outcomes. Powles 2002 reported using Kaplan‐Meier methodology for the outcomes overall survival and relapse. Heldal 2003 visualised competing risk outcomes, such as chronic GvHD and relapse, with cumulative incidence curves, and survival data with Kaplan‐Meier survival curves. Vigorito 2001 reported using the Kaplan‐Meier method for the outcomes engraftment, acute and chronic GvHD, overall survival and disease‐free survival.
Subgroup analysis
Based on the data reported we performed analyses only for the subgroups 'related donor' versus 'unrelated donor'.
Excluded studies
Nine studies were excluded for the following reasons. One study was excluded because bone marrow and peripheral blood stem cells were enriched for CD34‐positive stem cells (Cornelissen 2003), thereby dramatically reducing the number of T cells in the grafts, which account for the characteristics of PBSCT. Another study used G‐CSF to prime bone marrow and peripheral blood stem cells (Morton 2001). This approach also alters the graft composition and must be considered an experimental and rarely evaluated approach. Another study had a retrospective study design and involved only participants with severe aplastic anaemia (Bacigalupo 2012). Two studies focused on the characterisation of immune cells (Robinet 2003; Storek 2001). Two other studies were non‐randomised and had a retrospective study design (Champlin 2000; Kirschbaum 2012). One study focused on the differences between graft product and donor side effects after stem cell donation (Favre 2003). Finally, in one study the authors analysed and discussed decision guidelines for peripheral blood versus bone marrow stem cells for allo‐HSCT (Pidala 2009).
Risk of bias in included studies
Overall risk of bias was judged to be moderate in the included nine studies. The judgement is graphically summarised in Figure 2. For detailed information, please see the 'Risk of bias' sections in 'Characteristics of included studies tables.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
There was insufficient information regarding allocation in three of the nine studies (Mahmoud 1999; Powles 2002; Vigorito 2001). Allocation was considered to be associated with a low risk of bias in six studies (Anasetti 2012; Couban 2002; Friedrichs 2010; Heldal 2003; Mielcarek 2011; Mohty 2002).
Blinding
The information regarding the assessment of blinding was insufficient. In eight studies (Anasetti 2012; Couban 2002; Friedrichs 2010; Heldal 2003; Mahmoud 1999; Mielcarek 2011; Mohty 2002; Vigorito 2001) blinding was not reported, reflecting a high risk of bias. However, we assume that in this context the outcome and the outcome measurement are not likely to be influenced by lack of blinding. The study by Powles 2002 was described as double‐blinded.
Incomplete outcome data
Two studies excluded participants after randomisation and mentioned no reasons (Heldal 2003; Mohty 2002). Anasetti 2012 et al declared that five percent of the patients randomly assigned to the bone marrow group and 4% of those randomly assigned to the peripheral‐blood group did not undergo transplantation but were included in the intention‐to‐treat analysis (primary reason in 84% of cases was relapse of cancer). In one study information was insufficient to permit judgement (Mahmoud 1999).
Selective reporting
Study protocols were available for two studies (Anasetti 2012; Friedrichs 2010). The protocol for the EBMT study (Friedrichs 2010) is available on www.clinicaltrials.gov (ClinicalTrials.gov identifier: NCT01020175) and the BMT CTN (Bone Marrow Transplantation Clinical Trials Network) trial protocol is available on www.cibmtr.org and www.clinicaltrials.gov (NCT00075816).
There was no protocol information available for the remaining seven studies, but it was clear that the published reports included all expected outcomes, including those that were prespecified.
Other potential sources of bias
Only one study had another potential source of bias. After comparison with the randomised study by Bensinger 2001 and after an interim analysis undertaken using a Pocock stopping boundary, the study by Couban 2002 was terminated early. All other studies appeared to be free of other sources of bias.
Effects of interventions
See: Table 1
Overall survival
Kaplan‐Meier plots or hazard ratios (HRs) for overall survival were available in six of nine studies; 1330 participants were analysed. The pooled HR was 1.07 (95% CI 0.91 to 1.25; P value = 0.43, Figure 3). There was no statistically significant heterogeneity among the six trials (Chi² = 7.54, df = 5 (P value = 0.18); I² = 34%). Based on the data available there was no evidence that transplantation with bone marrow stem cells instead of peripheral blood stem cells improved overall survival. Survival data were evaluated using the generic inverse variance method.
3.
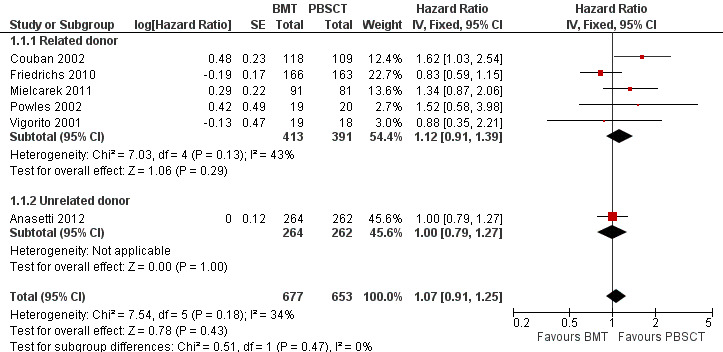
Forest plot of comparison: 1 BMT vs PBSCT, outcome: 1.1 Overall survival.
Four of the six studies (Anasetti 2012; Couban 2002; Friedrichs 2010; Mielcarek 2011) reported HRs and 95% CIs for this outcome. For the remaining two studies (Powles 2002; Vigorito 2001), we estimated the HRs from the published Kaplan‐Meier survival plots using the method suggested by Tierney et al (Parmar 1998; Tierney 2007).
Mohty 2002 reported a Kaplan‐Meier survival plot with no P‐value. The two‐year probabilities for PBSCT and BMT were 67% and 65%, respectively, and did not differ significantly (P‐value not reported).
Analysis of the related donor (HR 1.12; 95% CI 0.91 to 1.39; P value = 0.29) and unrelated donor subgroups (HR 1.00; 95% CI 0.79 to 1.27; P value = 1.00) did not reveal significant differences in overall survival between BMT and PBSCT in either subgroup (test for subgroup differences: Chi² = 0.51, df = 1 (P value = 0.47), I² = 0%, Figure 3).
Two studies (Heldal 2003; Mahmoud 1999) reported neither HRs nor survival curves and could not therefore be evaluated. Heldal 2003 noted a better outcome for PBSCT compared with BMT with regard to overall survival, but this result was not statistically significant (P value = 0.617). The respective figures or Kaplan‐Meier survival plots were not provided.
Disease‐free survival
Kaplan‐Meier plots or HRs for disease‐free survival were available in six of nine studies; 1225 participants were analysed. The pooled HR was 1.04 (95% CI 0.89 to 1.21; P value = 0.60, Figure 4). There was no statistically significant heterogeneity among the trials (Chi² = 4.66; df = 5 (P value = 0.46); I² = 0%). Based on the data available there was no evidence that BMT improved disease‐free survival compared with PBSCT.
4.
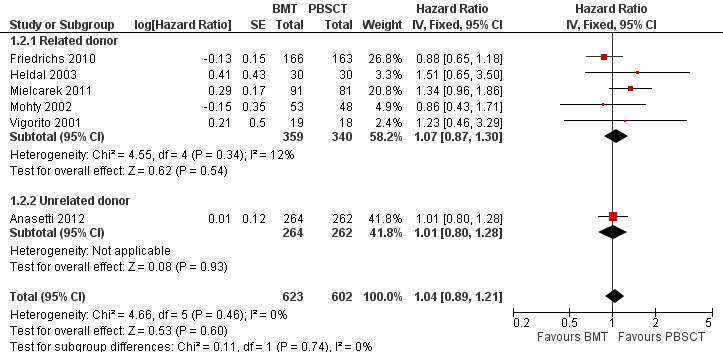
Forest plot of comparison: 1 BMT vs PBSCT, outcome: 1.2 Disease free survival.
Three of the six studies (Anasetti 2012; Friedrichs 2010; Mielcarek 2011) reported HRs and 95% CIs for this outcome. The other three studies (Heldal 2003; Mohty 2002; Vigorito 2001) provided a Kaplan‐Meier survival plot from which we estimated the HR using the method suggested by Tierney et al (Parmar 1998; Tierney 2007).
Analysis of the related donor (HR 1.07; 95% CI 0.87 to 1.30; P value = 0.54) and unrelated donor subgroups (HR 1.01; 95% CI 0.80 to 1.28; P value = 0.93) found no significant differences in disease‐free survival between BMT or PBSCT in either subgroup (test for subgroup differences: Chi² = 0.11, df = 1 (P value = 0.74); I² = 0%).
Three studies (Couban 2002; Mahmoud 1999; Powles 2002) reported neither the HR nor survival curves and could therefore not be evaluated.
Incidence of relapse
Kaplan‐Meier plots or HRs for the incidence of relapse were available in three of nine studies; 737 participants were analysed. The pooled HR was 1.3 (95% CI 0.98 to 1.72; P value = 0.07, Figure 5. There was statistically significant heterogeneity among the trials (Chi² = 8.82, df = 2 (P value = 0.01); I² = 77%). Based on the data available there was no significant difference between the BMT and PBSCT groups with respect to this outcome. Relapse data were evaluated using the generic inverse variance method.
5.
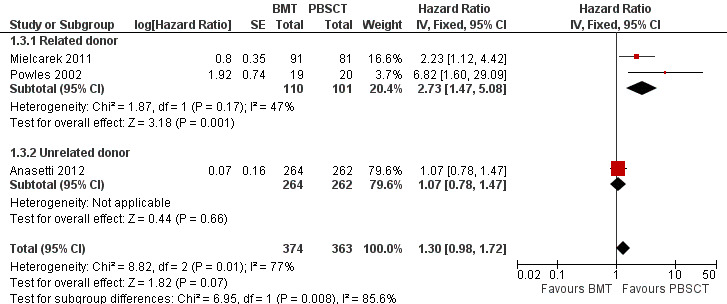
Forest plot of comparison: 1 BMT vs PBSCT, outcome: 1.3 Incidence of relapse.
Two of these three studies (Anasetti 2012; Mielcarek 2011) reported HRs and 95% CIs for this outcome. Powles 2002 provided a Kaplan‐Meier survival plot from which we estimated the HR using the method suggested by Tierney et al (Parmar 1998; Tierney 2007).
Subgroup analysis demonstrated a statistically significant advantage for the PBSCT group with regard to the incidence of relapse in recipients with related donors (HR 2.73; 95% CI 1.47 to 5.08; P value = 0.001, Figure 5); no such difference was apparent in PBSCT recipients with unrelated donors (HR 1.07; 95% CI 0.78 to 1.47; P value = 0.66; test for subgroup differences: Chi² = 6.95, df = 1 (P value = 0.008), I² = 85.6%). The difference between subgroups (P value = 0.008) was considered to be statistically significant. This may be one reason for the high level of heterogeneity detected between the studies with regard to this endpoint (I² = 85.6%). Other reasons included differences in the numbers of evaluable participants in each study (Anasetti N = 529, Mielcarek N = 172, Powles N = 39) and the difference in CIs.
Four studies (Friedrichs 2010; Mahmoud 1999; Mohty 2002; Vigorito 2001) reported neither the HR nor survival curves and could not therefore be evaluated. Couban 2002 and Heldal 2003 reported the cumulative incidence of relapse, which could not be analysed by the Tierney method.
Non‐relapse or transplant‐related mortality
Kaplan‐Meier plots or HRs for non‐relapse mortality were available in three of nine studies; 758 participants were analysed. The pooled HR was 0.98 (95% CI 0.76 to 1.28; P value = 0.91). There was no statistically significant heterogeneity among the trials (Chi² = 1.02, df = 2 (P value = 0.60); I² = 0%). Based on the data available there was no evidence that BMT improved transplant‐related mortality compared with PBSCT.
Two of the three studies (Anasetti 2012; Mielcarek 2011) reported the HRs and 95% CIs for this outcome. Heldal 2003 provided a Kaplan‐Meier survival plot from which we estimated the HR using the method suggested by Tierney et al (Parmar 1998; Tierney 2007).
Analysis of the related donor (HR 1.04; 95% CI 0.65 to 1.66; P value = 0.87) and unrelated donor subgroups (HR 0.96; 95% CI 0.70 to 1.31; P value = 0.8) did not reveal significant differences in transplant‐related mortality between BMT or PBSCT in either subgroup (test for subgroup differences: Chi² = 0.08, df = 1 (P value = 0.78), I² = 0%).
Couban 2002 reported the cumulative incidence of non‐relapse mortality, which could not be analysed by the Tierney method.
Mohty 2002 reported that 11 of 53 participants in the BMT group and 12 of 48 participants in the PBSCT group died of non‐relapse causes related to treatment (P value = not significant). Powles 2002) reported that 13 of 39 participants died of non‐relapse causes related to the treatment a median 57 days (range 15 to 733 days) after transplantation (6 of 19 in the BMT group and 7 of 20 in the PBSCT group; P value = 0.8). Vigorito 2001 reported that 7 of 19 participants in the BMT group and 5 of 18 participants in the PBSCT group died of non‐relapse causes related to treatment (P value not reported). For these studies, HRs could not be calculated.
The remaining two studies (Friedrichs 2010; Mahmoud 1999) did not report transplant‐related mortality as outcome.
Incidence of overall chronic GvHD
Four of nine studies, involving a total of 1121 participants, reported HRs or Kaplan‐Meier plots for overall chronic GvHD. The pooled HR was 0.72 (95% CI 0.61 to 0.85; P value = 0.0001; Figure 6). There was no statistically significant heterogeneity among the trials (Chi² = 3.68, df = 3 (P value = 0.30); I² = 19%). Based on the data available there was evidence for a lower incidence of overall chronic GvHD in the BMT group compared with the PBSCT group.
6.
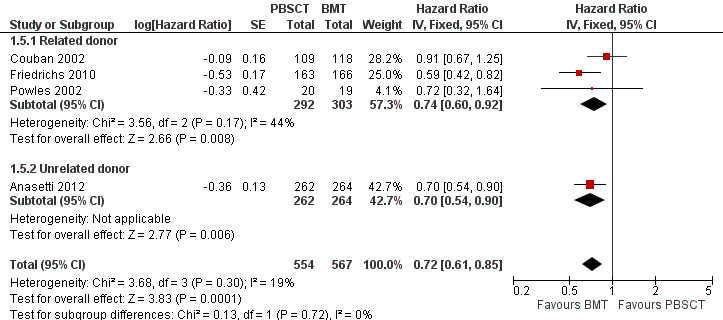
Forest plot of comparison: 1 BMT vs PBSCT, outcome: 1.5 Incidence of overall chronic GvHD.
Three studies (Anasetti 2012; Couban 2002; Friedrichs 2010) reported HRs and 95% CIs for this outcome. For one study (Powles 2002), we estimated the HR from a Kaplan‐Meier plot using the method suggested by Tierney et al (Parmar 1998; Tierney 2007).
Analysis of the related donor (HR 0.74; 95% CI 0.60 to 1.92; P value = 0.008) and unrelated donor subgroups (HR 0.7; 95% CI 0.54 to 0.90; P value = 0.006) showed significant differences in overall chronic GvHD in favour of BMT in both subgroups (Figure 6). Results for related und unrelated donor subgroups were not different with regard to overall chronic GvHD (test for subgroup differences: Chi² = 0.13, df = 1 (P value = 0.72), I² = 0%).
Three studies reported results for this outcome that were not suitable for inclusion in our analysis. Heldal 2003 reported a cumulative incidence plot, with an 11% incidence of chronic GvHD in the BMT arm and 57.7% in PBSCT arm. Mohty 2002 showed a cumulative incidence plot with an overall incidence of chronic GvHD of 36% in the BMT group and 65% in the PBSCT group after three years. Vigorito 2001 reported an incidence of overall chronic GvHD of 53.3% after BMT and 71.4% after PBSCT. The other studies did not report overall chronic GvHD as an outcome.
Incidence of extensive chronic GvHD
Kaplan‐Meier plots or HRs for extensive chronic GvHD were available in four of nine studies; 765 participants were analysed. The pooled HR was 0.69 (95% CI 0.54 to 0.90; P value = 0.006). There was no statistically significant heterogeneity among the trials (Chi² = 4.06; df = 3 (P value = 0.25); I² = 26%). Based on the data available there was evidence for a reduced incidence of extensive chronic GvHD in the BMT group compared with the PBSCT group.
Three studies (Couban 2002; Friedrichs 2010; Mielcarek 2011) reported HRs and 95% CIs for this outcome. For Vigorito 2001, we estimated the HR from a Kaplan‐Meier plot using the method suggested by Tierney et al (Parmar 1998; Tierney 2007).
Two studies reported cumulative incidences for this outcome, which could not be in included in our analyses. Heldal 2003 reported a significant difference regarding extensive chronic GvHD in favour of the BMT group (13.3% BMT versus 38.5% PBSCT; P value = 0.034). Mohty 2002 reported a benefit in the BMT group after 3 years (17% BMT versus 44% PBSCT; P value = not reported).
Incidence of acute GvHD grades II to IV
In six of nine studies, Kaplan‐Meier plots or HRs for acute GvHD grades II to IV were available; 1330 participants were analysed. At day 100 after transplantation, the pooled HR was 1.03 (95% CI 0.89 to 1.21; P value = 0.67). There was no statistically significant heterogeneity among the trials (Chi² = 7.36; df = 5 (P value = 0.20); I² = 32%). Based on the data available there was no evidence for a lower incidence of acute GvHD grades II to IV in the BMT group compared with the PBSCT group.
Four of the six studies (Anasetti 2012; Couban 2002; Friedrichs 2010; Mielcarek 2011) reported HRs and 95% CIs for this outcome. For the remaining two studies (Powles 2002; Vigorito 2001), we estimated the HR from a Kaplan‐Meier plot using the method suggested by Tierney et al (Parmar 1998; Tierney 2007).
Analyses of the related donor (five studies with 804 participants; HR 1.08; 95% CI 0.88 to 1.33; P value = 0.47) and unrelated donor subgroups (one study with 526 participants; HR 0.98; 95% CI 0.77 to 1.24; P value = 0.87) did not reveal significant differences in the incidence of acute GvHD grades II to IV between BMT and PBSCT in either subgroup (test for subgroup differences: Chi² = 0.36, df = 1 (P value = 0.55); I² = 0%).
Incidence of acute GvHD grades III to IV
In three of nine studies Kaplan‐Meier plots or HRs for acute GvHD grades III to IV were available; 925 participants were analysed. The pooled HR was 0.75 (95% CI 0.55 to 1.02; P value = 0.07). There was no statistically significant heterogeneity among the trials (Chi² = 0.18; df = 2 (P value = 0.91); I² = 0%).
All three studies included (Anasetti 2012; Couban 2002; Mielcarek 2011) reported HRs and 95% CIs for this outcome.
Analysis of the related donor (two studies with 399 participants; HR 0.71; 95% CI 0.44 to 1.13; P value = 0.15) and unrelated donor subgroups (one study with 526 participants; HR 0.79; 95% CI 0.52 to 1.19; P value = 0.25) did not reveal significant differences in the incidence of acute GvHD grades III to IV between BMT and PBSCT in either subgroup (test for subgroup differences: Chi² = 0.11, df = 1 (P value = 0.74); I² = 0%).
Time to neutrophil engraftment
Five of nine studies reported HRs or Kaplan‐Meier plots for this outcome; 662 participants were analysed. The pooled HR was 1.96 (95% CI 1.64 to 2.35; P value < 0.00001, Figure 7). There was no statistically significant heterogeneity among the trials (Chi² = 1.22; df = 4 (P value = 0.88); I² = 0%). Based on the data available neutrophil engraftment was faster in the PBSCT group than in the BMT group (mean 16.4 days (range 11 to 35 days) versus mean 19.9 days (range 13 to 68 days, respectively).
7.
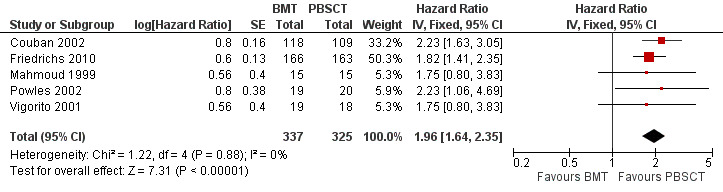
Forest plot of comparison: 1 BMT vs PBSCT, outcome: 1.9 Time to neutrophil engraftment.
Two of the five studies (Couban 2002; Friedrichs 2010) reported HRs and 95% CIs for this outcome. For the other three studies (Mahmoud 1999; Powles 2002; Vigorito 2001), we estimated the HR from a Kaplan‐Meier plot using the method suggested by Tierney et al (Parmar 1998; Tierney 2007).
One study (Mielcarek 2011, reported in Bensinger 2001) indicated significantly faster neutrophil engraftment in the PBSCT group than in the BMT group (16 days (range 11 to 29) versus 21 days (13 to 36); P value < 0.001), but the data could not be transformed into HRs.
Time to platelet engraftment
Four of nine studies reported HRs or Kaplan‐Meier plots for this outcome; 333 participants were analysed. The pooled HR was 2.17 (95% CI 1.69 to 2.78; P value < 0.00001, Figure 8). There was a moderate, non‐significant heterogeneity among the trials (Chi² = 6.46; df = 3 (P value = 0.09); I² = 54%). Based on the data available platelet engraftment was significantly faster in the PBSCT group than in the BMT group (mean 13 days (0 to 100) versus mean 19 days (0 to 100), respectively).
8.
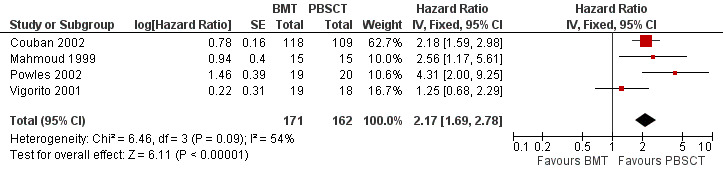
Forest plot of comparison: 1 BMT vs PBSCT, outcome: 1.10 Time to platelet engraftment.
Couban 2002 reported HRs and 95% CIs for this outcome. For the remaining three studies (Mahmoud 1999; Powles 2002; Vigorito 2001), we estimated the HR from a Kaplan‐Meier plot using the method suggested by Tierney et al (Parmar 1998; Tierney 2007).
One study (Mielcarek 2011, reported in Bensinger 2001) indicated significantly faster platelet engraftment in the PBSCT group than in the BMT group (13 days (5 to 41) versus 19 days (7 to 74), respectively; P value < 0.001), but the data could not be transformed into HRs.
Discussion
Summary of main results
In this Cochrane analysis we intended to evaluate of the most suitable stem cell source for allo‐HSCT in adults with hematological malignancies.
This systematic review included nine clinical studies evaluating the effect of BMT versus PBSCT in a total of 1521 participants. We were able to carry out subgroup analyses of recipients with unrelated and related donors for the outcomes overall and disease‐free survival, incidence of relapse, non‐relapse or transplant‐related mortality, overall chronic GvHD, and acute GvHD grades II to IV and III to IV.
Our meta‐analyses led to the following main conclusions.
There is no evidence that BMT is superior to PBSCT with regard to overall or disease‐free survival.
There is evidence for less overall and extensive chronic GvHD in individuals transplanted with bone marrow stem cells than in those transplanted with peripheral blood stem cells.
There is no evidence that the use of bone marrow stem cells in comparison with peripheral blood stem cells reduces non‐relapse mortality.
There is no evidence that the use of bone marrow stem cells in comparison with peripheral blood stem cells reduces the incidence of both grades II to IV and III to IV acute GvHD.
There is evidence that time to engraftment is faster when peripheral blood stem cells are used rather than when bone marrow stem cells are used.
There is evidence for an adverse impact of bone marrow stem cells in comparison to peripheral blood stem cells with respect to relapse incidence in the subgroup 'related donors'.
Overall survival
Overall survival after allo‐HSCT is influenced by several factors. First, direct conditioning and treatment‐related toxicity affect this endpoint. Furthermore, the balance between beneficial immune responses directed against infection and the tumour (engraftment, immune recovery and the graft‐versus‐tumour effect) and detrimental immune responses towards the recipient's healthy tissues (i.e. GvHD) is crucial (Appelbaum 2007). Acute and chronic GvHD are known to be the most important drivers of non‐relapse mortality and morbidity (Ferrera 2009). It has been speculated that the use of bone marrow stem cells, as opposed to peripheral blood stem cells, would decrease GvHD rates, thereby reducing non‐relapse mortality and translating into improved overall survival. Our meta‐analysis, however, in agreement with previous studies, found no significant difference between BMT and PBSCT with regard to overall survival. Non‐relapse mortality also did not differ between the two procedures. Although our analysis did demonstrated a significant reduction in the rates of chronic GvHD with BMT than with PBSCT, this advantage could partially be outweighed by a higher risk of relapse in this group, as demonstrated by the statistically significant increase in relapse in the BMT group in the related setting. Only one study (Couban 2002) has reported a statistically significant overall survival advantage for individuals transplanted with peripheral blood stem cells compared with those transplanted with bone marrow stem cells. Couban et al stated that this result was probably due to a reduction in transplant‐related early death and non‐relapse mortality in the PBSCT group. Participants in this study were treated between 1996 and 2000. Since then, supportive care and antimicrobial drug treatment have improved. It is likely that a shorter time to engraftment and immune recovery may have less impact in the current setting.
Disease‐free survival
Data from six studies could be included in the analysis of disease‐free survival. There was no significant difference between arms. Subgroup analysis of relapse incidence demonstrated a benefit for PBSCT in related donors. However, this did not translate into a benefit in disease‐free survival in either the donor‐related or ‐unrelated subgroup suggesting that, in this case, the survival benefit was counterbalanced by higher non‐relapse mortality (e.g. through an increase in GvHD). It has been recently reported that, in the myeloablative setting, GvHD has a negative impact on treatment‐related mortality, whereas with reduced‐intensity conditioning, GvHD‐related graft‐versus‐leukaemia effects might be required (Weisdorf 2012). Most of the participants included in this meta‐analysis received myeloablative conditioning.
Relapse incidence
Only three studies could be evaluated for the incidence of relapse. The pooled incidence of relapse did not differ between arms. As mentioned previously, there was a significant advantage for PBSCT with regard to a reduced incidence of relapse in participants receiving related donor transplants (HR 2.73; 95% CI 1.47 to 5.08; P value = 0.001). The difference between subgroups (P‐value = 0.008) was considered to be statistically significant in this context. This result implies that the smaller the genetic disparity (as in the related setting) the more important are the strength and speed of T‐cell recovery in order to mount efficient antitumour reactions. It has always been speculated that the higher numbers of T cells in the peripheral blood stem cell product could account for reduced relapse rates. Interestingly, the large study by Anasetti et al (Anasetti 2012) found no such correlation in the unrelated setting, whereas the downside of the graft‐versus‐tumour effect (i.e. chronic GvHD) was increased with PBSCT in both donor‐related and ‐unrelated subgroups.
Acute GvHD
Acute GvHD is clinically divided into grades I to IV. The more relevant grades with respect to long‐term outcomes are grades III and IV; grades I and II are usually clinically manageable (Ferrera 2009; Goldberg 2013). The higher T‐cell content in peripheral blood‐derived grafts leads to earlier immune recovery and therefore could induce more acute GvHD. In our analysis we could include data from six studies for the outcome grades II to IV acute GvHD. Here, no association with either treatment arm could be found. There was also no difference between related and unrelated donors. In the analysis of the clinically more relevant GvHD grades III to IV, only three studies could be included. Pooled data show a non‐significant trend in favour of bone marrow grafts (HR 0.75; 95% CI 0.55 to 1.02; P value = 0.07).
Chronic GvHD
Chronic GvHD accounts for a significant proportion of morbidity and mortality in the course following allo‐HSCT (Goldberg 2013; Kuzmina 2012), and often requires long‐term immunosuppression, including the administration of steroids. Chronic GvHD and its treatment substantially decrease the quality of life of surviving stem cell transplant recipients (Pidala 2009b; Wood 2013). However, mild chronic GvHD is considered to have a positive impact on survival as a consequence of a reduction in relapse incidence (Ringden 2009). We could include four studies in each of the analyses of overall chronic GvHD and extensive chronic GvHD. Both analyses demonstrated a statistically significant reduction in chronic GvHD in participants transplanted with bone marrow stem cells compared with those who received peripheral blood stem cells. These results were driven by two large studies (Anasetti 2012; Friedrichs 2010) that reported significant reductions in chronic GvHD in participants undergoing BMT. Although the other trials included in the meta‐analysis did not report a significant difference between the BMT and PBSCT groups, they did note a non‐significant advantage for BMT.
Non‐relapse mortality/transplant‐related mortality
The incidence of non‐relapse or transplant‐related mortality reflects the intensity of the conditioning regimen, the potency of GvHD prophylaxis, and the quality of donor selection and supportive care (Appelbaum 2007). Some of these aspects have improved in the clinical setup in recent years. In particular, the implementation of reduced‐intensity transplants has decreased early mortality. However, these developments have facilitated transplants in older individuals, and in those with co‐morbidities and high‐risk disease such as secondary or refractory leukaemia. In our analysis, non‐relapse mortality did not differ between treatment arms. It could be argued that the disadvantages of BMT, such as longer times to engraftment and immune recovery, are balanced by an advantage with respect to GvHD induction. Importantly, there was also no difference in non‐relapse mortality in Anasetti 2012, in which about 20% to 25% of participants received reduced‐intensity conditioning and 28% of participants had high‐risk disease.
Time to engraftment of neutrophils and platelets
Time to both neutrophil and platelet recovery was significantly shorter with PBSCT than with BMT. The higher T‐cell content of the graft facilitates the engraftment of foreign stem cells. Some studies also demonstrated less need for blood products, such as red blood cells and platelets, which was not an endpoint in this study. The shorter duration of neutropenia could lead to fewer infections and a lower use of antibiotics, faster recovery of mucosal and other lesions, as well as shorter hospitalisation times. However, none of the studies investigated hospitalisation or the need for supportive drugs.
Overall completeness and applicability of evidence
The results of this meta‐analysis should not be interpreted without considering the impact of the following factors.
The studies that were analysed range over a period of 15 years (1994 to 2009) during which transplant strategies and indications have changed substantially.
Supportive care and medical treatment have also changed considerably and this could have influenced overall outcomes.
Donors were related in eight studies (Couban 2002; Friedrichs 2010; Heldal 2003; Mahmoud 1999; Mielcarek 2011; Mohty 2002; Powles 2002; Vigorito 2001) whereas unrelated donors were exclusively included in Anasetti 2012.
HLA typing methods have changed and the older results of a six of six allele full match could have been a mismatch if higher‐resolution methods had been performed (e.g. 10 of 10 alleles). Couban 2002 and Heldal 2003 allowed up to one antigen mismatch. Anasetti 2012 included participants with up to three antigen mismatches.
Not all endpoints from all studies could be analysed for this review due to the reporting of cumulative incidence curves.
The statistical consideration of competing risks was not taken into account in the same way in the analyses of outcomes in different studies.
The conditioning regimen was myeloablative in all but the Anasetti 2012 study.
Study populations were not homogeneous and participants had different haematological diseases (in some studies the details were not reported). This is an important issue with regard to the interpretation of the findings for the outcome incidence of relapse as some malignancies are more vulnerable to a graft‐versus‐leukaemia effect than others. For a more detailed overview, a comparison of studies is given in the 'Included studies' section of this review.
The nine included studies, comprising 1521 participants, may not have been adequately powered to detect small differences, especially for outcomes with few events.
Quality of the evidence
Overall, the quality of evidence, including the nine included trials, was moderate. All the included trials were published as full text and reported as randomised studies. All but one of the studies (Powles 2002) had an open‐label design, which could have led to performance or detection biases for most of the evaluated endpoints. As death is an endpoint not susceptible to judgement by an outcome assessor, we considered the quality of the outcomes overall survival/mortality and non‐relapse/transplant‐related mortality to be high. There was no important uncertainty on the directness of the data.
Potential biases in the review process
To account for potential biases arising in the preparation of this review, important steps in the review were performed independently by two review authors. We did not apply any language restrictions in performing a comprehensive search strategy. Study authors were contacted for additional information or to resolve uncertainties. All studies included in the review were randomised controlled trials. We did not perform all the subgroup analyses that were planned in the protocol (see 'Subgroup analysis and investigation of heterogeneity' section). As individual participant data were not available, only subgroups that involved the comparison of whole trial results could be formed. Therefore, we decided to compare outcomes in the subgroups we considered to be the most important: unrelated and related donors.
In meta‐analyses involving at least 10 trials we would have tested for publication bias by generating a funnel plot and statistically testing data using a linear regression test. As we included nine trials only, we did not perform these analyses. However, as we handsearched the conference proceedings of the main BMT and haematology conferences, we minimised the chance for publication bias. If new studies report relevant data, we will perform subgroup analyses in an update of this systematic review.
The criteria we defined for undertaking a sensitivity analysis dud not differ among the included trials, with the exception of size of the trial. Only full text publications presenting mature results were included.
Agreements and disagreements with other studies or reviews
Our results are in line with those of previously published meta‐analyses (Stem Cell Trialists' Collaborative Group 2005, Chang 2012). Both these analyses evaluated only individuals with matched related donors undergoing myeloablative conditioning. The Stem Cell Trialists' analysis was based on individual participant data from 1111 participants included in nine individual trials (Stem Cell Trialists' Collaborative Group 2005). In this analysis, PBSCT led to faster engraftment of neutrophils (odds ratio (OR) 0.31; 95% CI 0.25 to 0.38; P value < 0.00001) and platelets (OR 0.52; 95% CI 0.44 to 0.61; P value < 0.00001) than BMT. PBSCT was also associated with a significant increase in the development of grades III to IV acute GvHD (OR 1.39; 95% CI 1.03 to 1.88; P value = 0.03), overall chronic GvHD (68% versus 52% at 3 years; OR 1.92; 95% CI 1.47 to 2.49; P value < 0.000001) and extensive chronic GvHD (47% versus 31% at 3 years; OR 1.89; 95% CI 1.47 to 2.42; P value < 0.000001) versus BMT. PBSCT was associated with a decrease in relapse (21% versus 27% at 3 years; OR 0.71; 95% CI 0.54 to 0.93; P value = 0.01) in both late‐stage‐ (33% versus 51% at 3 years; OR 0.59; 95% CI 0.38 to 0.93; P value = 0.02) and early‐stage–disease participants (16% versus 20% at 3 years; OR 0.69; 95% CI 0.49 to 0.98; P value = 0.04). Non‐relapse mortality did not differ between groups. Overall and disease‐free survival were significantly improved for PBSCT in participants with late‐stage disease only (overall survival 46% versus 31% at 3 years; OR 0.64; 95% CI 0.46 to 0.90; P value = 0.01; disease‐free survival: 41% versus 27% at 3 years; OR 0.63 95% CI 0.45 to 0.87; P value = 0.01). There was no such difference in participants with early disease (overall survival 65% versus 64%; OR 0.97; 95% CI 0.75 to 1.25; P value = 0.8; disease‐free survival: OR = 0.85; 95% CI 0.67 to 1.08; P value = 0.2). Findings for engraftment, GvHD, non‐relapse mortality and relapse were similar, taking into account that only related donors were included in the study. Overall and disease‐free survival in the pooled data‐set also did not differ between groups. Our data were insufficient to carry out subgroup analyses of late‐stage versus early‐stage disease.
Although more recent, the second review was based mainly on the same population included in the Stem Cell Trialists' review (Stem Cell Trialists' Collaborative Group 2005), The only updated data included were those published by Friedrichs 2010 as an update of the EBMT study by Schmitz and colleagues (Schmitz 2005), which was included in the Stem Cell Trialists' meta‐analysis. In this analysis, PBSCT was associated with faster neutrophil (HR 2.08; 95% CI 1.80 to 2.42; P value < 0.00001) and platelet (HR 2.77; 95% CI 1.78 to 4.30; P value < 0.00001) recoveries, a significant increase in the development of grades II to IV (HR 0.75; 95% CI 0.63 to 0.90; P value = 0.002) and III to IV (HR 0.63; 95% CI 0.47 to 0.84; P value = 0.001) acute GvHD, and overall (HR 0.70; 95% CI 0.59 to 0.83; P value < 0.0001) and extensive (HR 0.60; 95% CI 0.39 to 0.91; P value = 0.002) chronic GvHD compared with BMT. BMT was associated with a higher incidence of relapse (HR 1.91; 95% CI 1.34 to 2.74; P value = 0.0004). Comparable treatment‐related mortality (HR 1.08; 95% CI 0.56 to 2.10; P value = 0.81), disease‐free survival (HR 1.04; 95% CI 0.83 to 1.30; P value = 0.73) and overall survival (HR 1.06; 95% CI 0.81 to 1.39; P value = 0.65) were demonstrated for both treatment arms.
Recently, two large retrospective registry studies from the EBMT have been published (Nagler 2012;Ringden 2012). In one study (Ringden 2012), PBSCT (N = 1502) and BMT (N = 760) were compared in adults with acute myeloid leukaemia with unrelated donors after myeloablative conditioning. Recovery of neutrophils and platelets was faster with PBSCT than with BMT (P value < 0.0001). The incidence of acute GvHD was similar between arms. Again, PBSCT was associated with higher levels of chronic GVHD than BMT (HR = 1.29; P value = 0.02). There were no significant differences between the BMT and PBSCT groups in non‐relapse mortality, relapse incidence and leukaemia‐free survival amongst participants with acute myeloid leukaemia in remission. In participants with advanced leukaemia, non‐relapse mortality was lower (HR = 0.61; P value = 0.02) and leukaemia‐free survival was longer (HR = 0.67; P value = 0.002) with PBSCT than with BMT. At 3 years, leukaemia‐free survival in all participants, regardless of remission status, was 41% in both treatment groups. The second study (Nagler 2012) analysed the same endpoints against a background of reduced‐intensity conditioning in 508 participants receiving peripheral blood transplants and 94 receiving bone marrow grafts. The incidence of acute GvHD grades III and IV was significantly higher in the PBSCT group (27% versus 12% in the BMT group (P value < 0.002). Chronic GvHD was also higher in the PBSCT group (43% versus 35% in the BMT group, respectively; P value = 0.04). The 2‐year probabilities of leukaemia‐free survival were 46% in the PBSCT group versus 43% in the BMT group (P value = NS), whereas relapse incidence was significantly higher in the BMT group than in the PBSCT group: 46% versus 32%, respectively (P value = 0.014). Non‐relapse mortality was significantly higher in participants with peripheral blood transplants: 28% versus 13%, respectively (P value = 0.004). In multivariate analysis, use of peripheral blood stem cells was associated with a higher incidence of acute GvHD (grades II to IV; HR = 2.33; P value = 0.06), higher non‐relapse mortality (HR = 2.3; P value = 0.015) and a lower incidence of relapse than use of bone marrow stem cells (HR 0.61; P value = 0.02), with no statistical difference in leukaemia‐free survival between the two groups (P value = 0.88).
Authors' conclusions
Implications for practice.
There is high‐quality evidence that overall survival following allo‐HSCT using the current clinical standard stem cell source ‐ peripheral blood stem cells ‐ was similar to that following allo‐HSCT using bone marrow stem cells in adults with haematological malignancies.
Data are insufficient to state that this conclusion is valid for recipients with unrelated donors and those transplanted against the background of current transplant settings. For peripheral blood transplants there was an imprecise effect on relapse incidence in the pooled data‐set. In the related donor setting, an advantage with respect to this endpoint was found implying that participants at high risk of relapse might benefit from PBSCT. Conversely, in participants lacking a matched donor or those with a condition that does not require a graft‐versus‐malignancy effect, BMT might be more suitable as it is associated with less GvHD.
Implications for research.
Further research is needed with regard to the stem cell source under current transplant strategies. Only one study (Anasetti 2012) has been carried out in the era of reduced‐intensity transplants, now the most commonly used conditioning strategy. Even in this study, only about a quarter of participants received the reduced‐intensity option. More and more mismatched and unrelated transplants are being performed. The number of participants included in our analysis is too small to draw conclusions regarding this donor type, but hypothetically, GvHD rates are likely to increase under these conditions and differences between stem cell sources could be more pronounced. In addition, the numbers of older recipients and those with comorbidity are increasing. A clinical trial in participants older than 60 years is warranted as the medical needs, underlying conditions and immune recovery of older individuals differ from those in younger recipients (Brunner 2013; Sorror 2011). Quality of life has been paid too little consideration in transplantation trials in the past. Only one of the nine studies included in our analysis (Vigorito 2001, reported in De Souza 2002) investigated quality of life. In this study, bone marrow transplants were reported to be beneficial with regard to pain/discomfort, mobility and daily living activities. Recent data underline the importance of quality of life in long‐term survivors (Norkin 2012), and this endpoint should be evaluated in future trials.
Acknowledgements
We would like to thank the Cochrane Haematological Malignancies Group editors, affiliated consumers and members for critical advice, support and encouragement: Sabine Kluge, Ina Monsef and Andreas Engert.
We thank Dr Anasetti and the Blood and Marrow Transplant Clinical Trials Network (BMT CTN) for additional information on the BMT CTN 0201 study: “Support for the BMT CTN 0201 study was provided by grant #U10HL069294 from the National Heart, Lung, and Blood Institute and the National Cancer Institute, the Department of the Navy, Office of Naval Research, and the National Marrow Donor Program. Enrollment support was provided by DKMS Germany. Any views, opinions, findings, conclusions or recommendations expressed in this material are those of the authors and do not reflect the views or the official policy or position of the above mentioned parties.”
We would also like to thank Dr Marco Mielcarek for his prompt answers to our queries.
Appendices
Appendix 1. MEDLINE (Ovid)
MEDLINE/Ovid (January 1948 to October 2011)
| # | Search term |
| 1 | exp stem cell transplantation/ |
| 2 | hematopoietic stem cell transplantation/ |
| 3 | bone marrow transplantation/ |
| 4 | peripheral blood stem cell transplantation/ |
| 5 | (bone marrow adj2 (transplant$ or graft$ or trasplant$ or rescue$)).tw,kf,ot. |
| 6 | (stem cell$ or stem‐cell$).tw,kf,ot. |
| 7 | "progenitor cell$".tw,kf,ot. |
| 8 | (SCT or BMT or PBSC or PSCT or BMCT or BM or SCT).tw,kf,ot. |
| 9 | or/1‐8 |
| 10 | TRANSPLANTATION CONDITIONING/ |
| 11 | myeloablat$.tw,kf,ot. |
| 12 | reduced intens$.tw,kf,ot. |
| 13 | (nonmyeloablat$ or non‐myeloablat$).tw,kf,ot. |
| 14 | (mini‐tra?splant$ or minitra?splant$).tw,kf,ot. |
| 15 | or/10‐14 |
| 16 | (allotransplant$ or allo‐transplant$).tw,kf,ot. |
| 17 | (allotrasplant$ or allo‐trasplant$).tw,kf,ot. |
| 18 | (allogen$ or allo‐gen$).tw,kf,ot. |
| 19 | ((allogen$ or allo‐gen$) adj5 (transplant$ or trasplant$ or graft$ or rescue$)).tw,kf,ot. |
| 20 | (homograft$ or homo‐graft$).tw,kf,ot. |
| 21 | homolog$.tw,kf,ot. |
| 22 | (homotransplant$ or homo‐transplant$).tw,kf,ot. |
| 23 | (homotrasplant$ or homo‐trasplant$).tw,kf,ot. |
| 24 | or/16‐23 |
| 25 | randomized controlled trial.pt. |
| 26 | controlled clinical trial.pt. |
| 27 | randomized.ab. |
| 28 | placebo.ab. |
| 29 | clinical trials as topic.sh. |
| 30 | randomly.ab. |
| 31 | trial.ti. |
| 32 | or/25‐31 |
| 33 | humans.sh. |
| 34 | 32 and 33 |
| 35 | 9 or 15 |
| 36 | 35 and 24 |
| 37 | 36 and 34 |
key: tw: text word, kf: keyword heading word, nm: name of substance, ot: original title, pt: publication type, ab: abstract; fs: floating subheading; sh: medical subject heading word, sh=medical subject heading
MEDLINE/Ovid (28.06.2013 to 06.02.2014)
| # | Searches | Results |
| 1 | exp STEM CELL TRANSPLANTATION/ | 51093 |
| 2 | HEMATOPOIETIC STEM CELL TRANSPLANTATION/ | 26692 |
| 3 | BONE MARROW TRANSPLANTATION/ | 40678 |
| 4 | PERIPHERAL BLOOD STEM CELL TRANSPLANTATION/ | 2800 |
| 5 | (bone marrow adj2 (transplant$ or graft$ or trasplant$ or rescue$)).tw,kf,ot. | 30859 |
| 6 | (stem cell$ or stem‐cell$).tw,kf,ot. | 126472 |
| 7 | "progenitor cell$".tw,kf,ot. | 38083 |
| 8 | (SCT or BMT or PBSC or PSCT or BMCT or BM or SCT).tw,kf,ot. | 30803 |
| 9 | or/1‐8 | 213808 |
| 10 | TRANSPLANTATION CONDITIONING/ | 7023 |
| 11 | myeloablat$.tw,kf,ot. | 3819 |
| 12 | reduced intens$.tw,kf,ot. | 2414 |
| 13 | (nonmyeloablat$ or non‐myeloablat$).tw,kf,ot. | 1941 |
| 14 | (mini‐tra?splant$ or minitra?splant$).tw,kf,ot. | 73 |
| 15 | or/10‐14 | 10979 |
| 16 | (allotransplant$ or allo‐transplant$).tw,kf,ot. | 3896 |
| 17 | (allotrasplant$ or allo‐trasplant$).tw,kf,ot. | 5 |
| 18 | (allogen$ or allo‐gen$).tw,kf,ot. | 48486 |
| 19 | ((allogen$ or allo‐gen$) adj5 (transplant$ or trasplant$ or graft$ or rescue$)).tw,kf,ot. | 24945 |
| 20 | (homograft$ or homo‐graft$).tw,kf,ot. | 5191 |
| 21 | homolog*.tw,kf,ot. | 241223 |
| 22 | (homotransplant$ or homo‐transplant$).tw,kf,ot. | 1512 |
| 23 | (homotrasplant$ or homo‐trasplant$).tw,kf,ot. | 18 |
| 24 | or/16‐23 | 298807 |
| 25 | randomized controlled trial.pt. | 360225 |
| 26 | controlled clinical trial.pt. | 86967 |
| 27 | randomized.ab. | 261342 |
| 28 | placebo.ab. | 141439 |
| 29 | clinical trials as topic.sh. | 166817 |
| 30 | randomly.ab. | 186764 |
| 31 | trial.ti. | 111877 |
| 32 | or/25‐31 | 829699 |
| 33 | humans.sh. | 13109787 |
| 34 | 32 and 33 | 745450 |
| 35 | 9 or 15 | 215072 |
| 36 | 35 and 24 | 29146 |
| 37 | 36 and 34 | 1911 |
| 38 | limit 37 to ed=20110622‐20130628 | 211 |
| 39 | limit 37 to ed=20110622‐20130628 | 211 |
| 40 | limit 37 to ed=20130628‐20140206 | 72 |
key: tw: text word, kf: keyword heading word, nm: name of substance, ot: original title, pt: publication type, ab: abstract; fs: floating subheading; sh: medical subject heading word, sh=medical subject heading
Appendix 2. CENTRAL (The Cochrane Library)
Cochrane Central Register of Controlled Trials (Cochrane Library 2011, Issue 3)
| # | Search term |
| #1 | MeSH descriptor Stem Cell Transplantation explode all trees |
| #2 | MeSH descriptor Hematopoietic Stem Cell Transplantation explode all trees |
| #3 | MeSH descriptor Bone Marrow Transplantation explode all trees |
| #4 | MeSH descriptor Peripheral Blood Stem Cell Transplantation explode all trees |
| #5 | (bone marrow adj2 transplant*) or (bone marrow adj2 graft*) or (bone marrow adj2 trasplant*) or (bone marrow adj2 rescue*):ti |
| #6 | (stem cell* or stem‐cell*) |
| #7 | "progenitor cell" |
| #8 | (SCT or BMT or PBSC or PSCT or BMCT or BM or SCT) |
| #9 | (#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8) |
| #10 | MeSH descriptor Transplantation Conditioning explode all trees |
| #11 | myeloablat* |
| #12 | reduced intens* |
| #13 | (nonmyeloablat* or non‐myeloablat*) |
| #14 | (mini‐transplant* or minitransplant* or mini‐trasplant* or minitrasplant*) |
| #15 | (#10 OR #11 OR #12 OR #13 OR #14) |
| #16 | (allotransplant* or allo‐transplant*) |
| #17 | (allotrasplant* or allo‐trasplant*) |
| #18 | (allogen* or allo‐gen*) |
| #19 | (allogen* near/2 transplant*) or (allogen* near/2 trasplant*) or (allogen* near/2 graft*) or (allogen* near/2rescue*) |
| #20 | (allo‐gen* near/2 transplant*) or (allo‐gen* near/2 trasplant*) or (allo‐gen* near/2 graft*) or (allo‐gen* near/2rescue*) |
| #21 | (homograft* or homo‐graft*) |
| #22 | homolog* |
| #23 | (homotransplant* or homo‐transplant*) |
| #24 | (homotrasplant* or homo‐trasplant*) |
| #25 | (#16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24) |
| #26 | (#9 OR #15) |
| #27 | (#25 AND #26) |
| #28 | "accession number" near pubmed |
| #29 | (#27 AND NOT #28) |
Search Name: BMT vs PBSC bei allo Transplantation Update.2
Last Saved: 06/02/2014 22:10:42.444
Description: update 28.06.2013 ‐ 06.02.2014
| ID | Search |
| #1 | MeSH descriptor: [Stem Cell Transplantation] explode all trees |
| #2 | MeSH descriptor: [Hematopoietic Stem Cell Transplantation] explode all trees |
| #3 | MeSH descriptor: [Bone Marrow Transplantation] explode all trees |
| #4 | MeSH descriptor: [Peripheral Blood Stem Cell Transplantation] explode all trees |
| #5 | (bone marrow adj2 transplant*) OR (bone marrow adj2 graft*) OR (bone marrow adj2 trasplant*) OR (bone marrow adj2 rescue*):ti |
| #6 | (stem cell* or stem‐cell*) |
| #7 | "progenitor cell" |
| #8 | (SCT or BMT or PBSC or PSCT or BMCT or BM or SCT) |
| #9 | #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 |
| #10 | MeSH descriptor: [Transplantation Conditioning] explode all trees |
| #11 | myeloablat* |
| #12 | reduced intens* |
| #13 | (nonmyeloablat* or non‐myeloablat*) |
| #14 | (mini‐transplant* or minitransplant* or mini‐trasplant* or minitrasplant*) |
| #15 | #10 or #11 or #12 or #13 or #14 |
| #16 | (allotransplant* or allo‐transplant*) |
| #17 | (allotrasplant* or allo‐trasplant*) |
| #18 | (allogen* or allo‐gen*) |
| #19 | allogen* near/2 transplant* OR allogen* near/2 trasplant* OR allogen* near/2 graft* OR allogen* near/2rescue* |
| #20 | allo‐gen* near/2 transplant* OR allo‐gen* near/2 trasplant* OR allo‐gen* near/2 graft* OR allo‐gen* near/2rescue* |
| #21 | (homograft* or homo‐graft*) |
| #22 | homolog* |
| #23 | (homotransplant* or homo‐transplant*) |
| #24 | (homotrasplant* or homo‐trasplant*) |
| #25 | #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 |
| #26 | #9 or #15 |
| #27 | #25 and #26 |
| #28 | #27 from 2011 to 2013, in Trials |
| #29 | #27 from 2013 to 2014, in Trials |
Data and analyses
Comparison 1. BMT vs PBSCT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 6 | 1330 | Hazard Ratio (Fixed, 95% CI) | 1.07 [0.91, 1.25] |
| 1.1 Related donor | 5 | 804 | Hazard Ratio (Fixed, 95% CI) | 1.12 [0.91, 1.39] |
| 1.2 Unrelated donor | 1 | 526 | Hazard Ratio (Fixed, 95% CI) | 1.0 [0.79, 1.27] |
| 2 Disease free survival | 6 | 1225 | Hazard Ratio (Fixed, 95% CI) | 1.04 [0.89, 1.21] |
| 2.1 Related donor | 5 | 699 | Hazard Ratio (Fixed, 95% CI) | 1.07 [0.87, 1.30] |
| 2.2 Unrelated donor | 1 | 526 | Hazard Ratio (Fixed, 95% CI) | 1.01 [0.80, 1.28] |
| 3 Incidence of relapse | 3 | 737 | Hazard Ratio (Fixed, 95% CI) | 1.30 [0.98, 1.72] |
| 3.1 Related donor | 2 | 211 | Hazard Ratio (Fixed, 95% CI) | 2.73 [1.47, 5.08] |
| 3.2 Unrelated donor | 1 | 526 | Hazard Ratio (Fixed, 95% CI) | 1.07 [0.78, 1.47] |
| 4 Transplant related mortality | 3 | 758 | Hazard Ratio (Fixed, 95% CI) | 0.98 [0.76, 1.28] |
| 4.1 Related donor | 2 | 232 | Hazard Ratio (Fixed, 95% CI) | 1.04 [0.65, 1.66] |
| 4.2 Unrelated donor | 1 | 526 | Hazard Ratio (Fixed, 95% CI) | 0.96 [0.70, 1.31] |
| 5 Incidence of overall chronic GvHD | 4 | 1121 | Hazard Ratio (Fixed, 95% CI) | 0.72 [0.61, 0.85] |
| 5.1 Related donor | 3 | 595 | Hazard Ratio (Fixed, 95% CI) | 0.74 [0.60, 0.92] |
| 5.2 Unrelated donor | 1 | 526 | Hazard Ratio (Fixed, 95% CI) | 0.70 [0.54, 0.90] |
| 6 Incidence of extensive chronic GvHD | 4 | 765 | Hazard Ratio (Fixed, 95% CI) | 0.69 [0.54, 0.90] |
| 7 Incidence of acute GvHD grade II‐IV | 6 | 1330 | Hazard Ratio (Fixed, 95% CI) | 1.03 [0.89, 1.21] |
| 7.1 Related donor | 5 | 804 | Hazard Ratio (Fixed, 95% CI) | 1.08 [0.88, 1.33] |
| 7.2 Unrelated donor | 1 | 526 | Hazard Ratio (Fixed, 95% CI) | 0.98 [0.77, 1.24] |
| 8 Incidence of acute GvHD grade III‐IV | 3 | 925 | Hazard Ratio (Fixed, 95% CI) | 0.75 [0.55, 1.02] |
| 8.1 Related donor | 2 | 399 | Hazard Ratio (Fixed, 95% CI) | 0.71 [0.44, 1.13] |
| 8.2 Unrelated donor | 1 | 526 | Hazard Ratio (Fixed, 95% CI) | 0.79 [0.52, 1.19] |
| 9 Time to neutrophil engraftment | 5 | 662 | Hazard Ratio (Fixed, 95% CI) | 1.96 [1.64, 2.35] |
| 10 Time to platelet engraftment | 4 | 333 | Hazard Ratio (Fixed, 95% CI) | 2.17 [1.69, 2.78] |
1.1. Analysis.
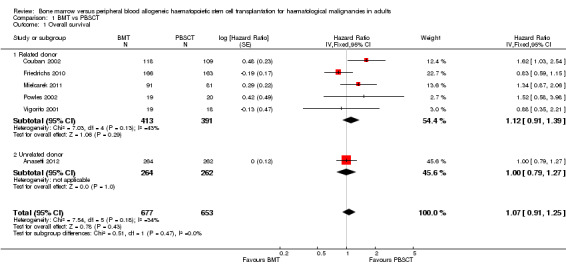
Comparison 1 BMT vs PBSCT, Outcome 1 Overall survival.
1.2. Analysis.
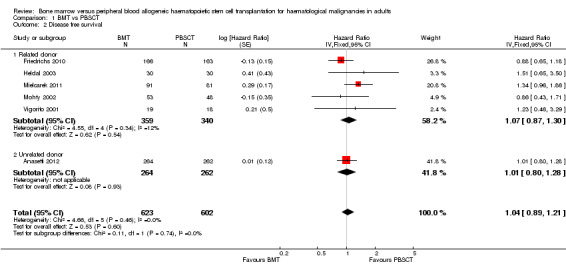
Comparison 1 BMT vs PBSCT, Outcome 2 Disease free survival.
1.3. Analysis.
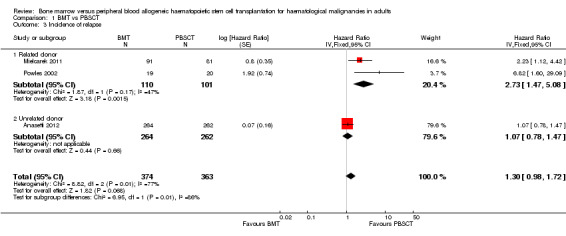
Comparison 1 BMT vs PBSCT, Outcome 3 Incidence of relapse.
1.4. Analysis.
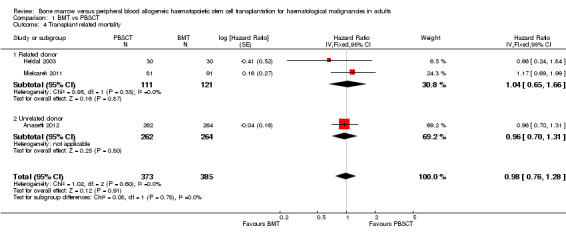
Comparison 1 BMT vs PBSCT, Outcome 4 Transplant related mortality.
1.5. Analysis.
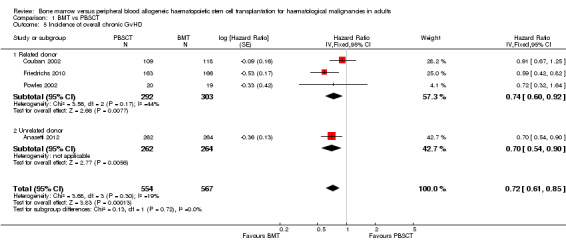
Comparison 1 BMT vs PBSCT, Outcome 5 Incidence of overall chronic GvHD.
1.6. Analysis.
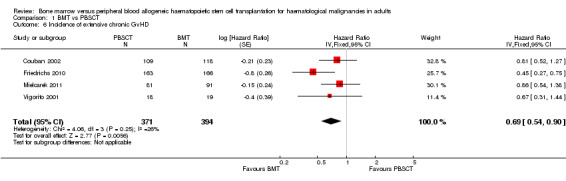
Comparison 1 BMT vs PBSCT, Outcome 6 Incidence of extensive chronic GvHD.
1.7. Analysis.
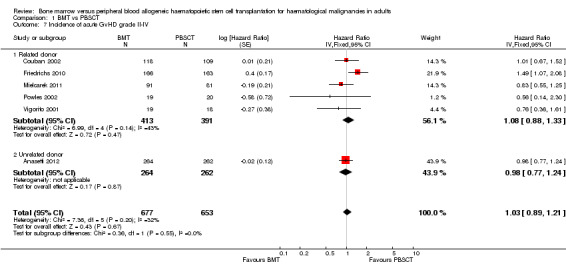
Comparison 1 BMT vs PBSCT, Outcome 7 Incidence of acute GvHD grade II‐IV.
1.8. Analysis.
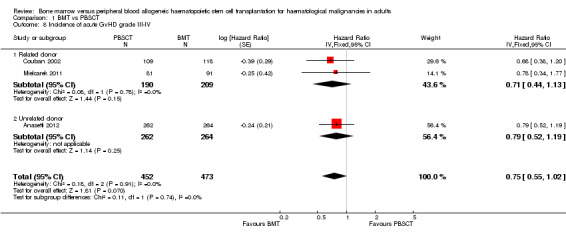
Comparison 1 BMT vs PBSCT, Outcome 8 Incidence of acute GvHD grade III‐IV.
1.9. Analysis.

Comparison 1 BMT vs PBSCT, Outcome 9 Time to neutrophil engraftment.
1.10. Analysis.
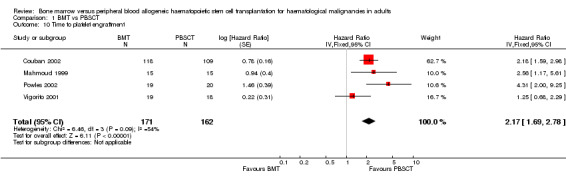
Comparison 1 BMT vs PBSCT, Outcome 10 Time to platelet engraftment.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Anasetti 2012.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Method of randomisation not mentioned, but we assume that central randomisation and allocation was performed using a computer random number generator. |
| Allocation concealment (selection bias) | Low risk | "Randomisation was performed in a 1:1 ratio, with the use of random block sizes, and was stratified according to transplantation centre and disease risk." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "No blinding was attempted." However, we assume that the outcome and the outcome measurement are not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Five percent of the patients randomly assigned to the bone marrow group and 4% of those randomly assigned to the peripheral‐blood group did not undergo transplantation but were included in the intention‐to‐treat analysis." (primary reason in 84% of cases were relapse of cancer) |
| Selective reporting (reporting bias) | Low risk | study protocol available in ClinicalTrials.gov number NCT00075816 |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Couban 2002.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Method of randomisation not mentioned, but we assume that central randomisation and allocation was performed using a computer random number generator. |
| Allocation concealment (selection bias) | Low risk | "Eligible patient‐donor pairs were randomised centrally in permuted blocks of 4. Pairs were stratified before randomisation by disease (CML, AML, or MDS) and centre." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding reported, but we assume that the outcome and the outcome measurement are not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | One patient was lost to follow‐up within 1 week of randomisation. No other patients were lost or excluded after randomisation. |
| Selective reporting (reporting bias) | Low risk | The study protocol was not available but it was clear that the published reports included all expected outcomes, including those that were pre‐specified. |
| Other bias | High risk | "Following publication of the preliminary results of the randomised study by Bensinger et al, an interim analysis was undertaken using a Pocock stopping boundary, and accrual to the study was stopped in February 2000." |
Friedrichs 2010.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The central randomisation centre (International Institiute for Drug Development, Brussels, Belgium) used minimisation to achieve the best balance of treatment allocations within centres and groups, defined by patient characteristics." |
| Allocation concealment (selection bias) | Low risk | " Stratification criteria were the following: (1) if donor is female and has ever been pregnant; (2) sex mismatch between donor and recipient; (3) recipient diagnosis of CML. The randomised treatment was communicated by fax to the investigator after the patient was registered." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding reported, but we assume that the outcome and the outcome measurement are not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 21 patients excluded: 7 due to relapse, 5 due to withdrawal of consent, 4 ineligibility, 1 aspergillus, 1 Klinefelter syndrome, 2 recipient death, 1 donor failure to mobilize |
| Selective reporting (reporting bias) | Low risk | study protocol available in ClinicalTrials.gov number NCT 01020175 |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Heldal 2003.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Method of randomisation not mentioned, but we assume that central randomisation and allocation was performed using a computer random number generator. |
| Allocation concealment (selection bias) | Low risk | "The 61 patients were stratified according to age, but not to HLA mismatch, stage of disease or other prognostic factors. Subsequently, they were randomised on blocks of six, three for each type of cell harvest." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding reported, but we assume that the outcome and the outcome measurement are not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | One patient excluded after randomisation with no reason mentioned. |
| Selective reporting (reporting bias) | Low risk | The study protocol was not available but it was clear that the published reports included all expected outcomes, including those that were pre‐specified. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Mahmoud 1999.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Method of randomisation not mentioned, but we assume that central randomisation and allocation was performed using a computer random number generator. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding reported, but we assume that the outcome and the outcome measurement are not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Low risk | The study protocol was not available but it was clear that the published reports included all expected outcomes, including those that were pre‐specified. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Mielcarek 2011.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Method of randomisation not mentioned, but we assume that central randomisation and allocation was performed using a computer random number generator |
| Allocation concealment (selection bias) | Low risk | "After random assignment to transplantation with peripheral‐blood cells or bone marrow, the patients were stratified according to treatment centre, age (<30 or >30 years), and stage of cancer (less advanced or more advanced). Within these strata, assignments were balanced in blocks of random size" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding reported, but we assume that the outcome and the outcome measurement are not likely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Shortly after randomisation but before the beginning of treatment, three patients were found to be ineligible and were given alternative therapy; the results for these three patients were excluded from further analysis" |
| Selective reporting (reporting bias) | Low risk | The study protocol was not available but it was clear that the published reports included all expected outcomes, including those that were prespecified |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Mohty 2002.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "However, to minimise the bias introduced by eventual exclusion resulting from adverse events occurring between randomisation and transplantation, the data manager randomised each patient using a minimization method that allowed the more likely randomisation of future patients to one arm if previous patients failed to receive a transplant in this arm" |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding reported, but we assume that the outcome and the outcome measurement are not likely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "One hundred eleven patients with leukaemia in the early stages and with HLA‐matched sibling donors were randomised in this study. One hundred one underwent transplantation" |
| Selective reporting (reporting bias) | Low risk | The study protocol was not available but it was clear that the published reports included all expected outcomes, including those that were prespecified |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Powles 2002.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Method of randomisation not mentioned, but we assume that central randomisation and allocation was performed using a computer random number generator |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The study was double‐blinded to eliminate the subjective aspects to assessment and care that could potentially affect the outcome of such a comparison." "Masking depended on the harvested stem cells being frozen in bags that made the source indistinguishable when frozen, thawed, and re‐infused" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The study was double‐blinded to eliminate the subjective aspects to assessment and care that could potentially affect the outcome of such a comparison." "Masking depended on the harvested stem cells being frozen in bags that made the source indistinguishable when frozen, thawed, and re‐infused" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Two participants excluded: one due to fulminant relapse, one donor withdrew after enrolment but before donating |
| Selective reporting (reporting bias) | Low risk | "The study protocol was approved by the institutional review board (scientific and ethics committees) of the Royal Marsden Hospital, and all patients and donors provided informed consent" |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Vigorito 2001.
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Method of randomisation not mentioned, but we assume that central randomisation and allocation was performed using a computer random number generator. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding reported, but we assume that the outcome and the outcome measurement are not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Three patients were excluded from the analysis: two in the PBPC group, where one refused and the other had no HLA‐identical sibling , and one in the BM group because they did not have a haematological malignancy." |
| Selective reporting (reporting bias) | Low risk | The study protocol was not available but it was clear that the published reports included all expected outcomes, including those that were pre‐specified. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
AML
BM, bone marrow
BMT, bone marrow transplantation
CML
GvHD, graft‐versus‐host disease
HLA, human leukocyte antigen
MDS, myelodysplastic syndrome
PBPC
PBSCT, peripheral blood stem cell transplantation
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bacigalupo 2012 | Retrospective study design, participants with severe aplastic anaemia only |
| Champlin 2000 | Non‐randomised, retrospective study design |
| Cornelissen 2003 | CD34+ purification of bone marrow and peripheral blood stem cells |
| Favre 2003 | focused on the differences between graft product and donor side effects after HSCT |
| Kirschbaum 2012 | Non‐randomised, retrospective study design |
| Morton 2001 | Both stem cell sources primed by granulocyte‐colony‐stimulating factor; differs from clinical standards |
| Pidala 2009 | Not a clinical study; decision analysis of peripheral blood versus bone marrow hematopoietic stem cells for allogeneic hematopoietic cell transplantation. |
| Robinet 2003 | Focuses on the characterisation of immune cells |
| Storek 2001 | Focuses on the characterisation of immune cells |
Differences between protocol and review
Because outcomes were not reported with respect to participant age below or above 18 years, it was not possible to extract data that corresponded to adults only. Even though we defined adult age as an inclusion criterion, we did not mean to exclude studies involving a mixed population or a broad distribution of age. We planned to exclude studies with a focus on paediatric participants as their immune reconstitution and underlying disease characteristics differ from those of adults. Three studies included participants younger than 18 years of age (Couban 2002; Mielcarek 2011; Vigorito 2001). We did not foresee that these studies would report their results irrespective of participant age. It can be assumed that only a small minority of participants were younger than 18 years of age. We therefore decided to evaluate studies irrespective of participant age. We also wanted to analyse differences in participants aged over of less than 60 years. However, based on the data available, this analysis could not be done.
Subgroup investigation and sensitivity analyses were not performed as planned in the protocol mainly due to a lack of comparable data or missing individual participant data. Hence, we performed subgroup analyses only with regard to type of donor: related versus unrelated. Sensitivity analyses were not performed as all trials were published as full text publications in peer‐reviewed journals and presented mature results with good reporting quality.
We did not analyse the endpoint 'quality of life' because no study reported this endpoint.
Contributions of authors
Udo Holtick: drafting of the protocol, abstract screening, data extraction, data analysis and interpretation, drafting of the review, handsearching, quality assessment (RoB);
Melanie Albrecht: drafting of the protocol, abstract screening, data extraction, data analysis and interpretation, drafting of the review, handsearching, quality assessment (RoB);
Jens M Chemnitz: clinical expertise;
Sebastian Theurich: clinical expertise;
Nicole Skoetz: administrative support, statistical and methodological advice, communication between authors, proofreading, update screening, preparation of 'Summary of Findings' table;
Christof Scheid: clinical expertise, content input;
Michael von Bergwelt‐Baildon: clinical expertise, content input.
Sources of support
Internal sources
-
Department of Internal Medicine, Stem Cell Transplantation Program, Germany.
University Hospital of Cologne
External sources
No sources of support supplied
Declarations of interest
The authors have no conflicts of interest to declare.
a authors have contributed equally (first author)
b authors have contributed equally (first author)
New
References
References to studies included in this review
Anasetti 2012 {published data only}
- Anasetti C, Logan BR, Lee SJ, Waller EK, Weisdorf DJ, Wingard JR, et al. Peripheral‐blood stem cells versus bone marrow from unrelated donors. The New England Journal of Medicine 2012;367(16):1487‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Couban 2002 {published data only}
- Couban S, Simpson DR, Barnett MJ, Bredeson C, Hubesch L, Howson‐Jan K, et al. A randomized multicenter comparison of bone marrow and peripheral blood in recipients of matched sibling allogeneic transplants for myeloid malignancies. Blood 2002;100(5):1525‐31. [DOI] [PubMed] [Google Scholar]
Friedrichs 2010 {published data only}
- Friedrichs B, Tichelli A, Bacigalupo A, Russell NH, Ruutu T, Shapira MY, et al. Long‐term outcome and late effects in patients transplanted with mobilised blood or bone marrow: a randomised trial. Lancet Oncology 2010;11(4):331‐8. [DOI] [PubMed] [Google Scholar]
- Schmitz N, Beksac M, Bacigalupo A, Ruutu T, Nagler A, Gluckman E, et al. Filgrastim‐mobilized peripheral blood progenitor cells versus bone marrow transplantation for treating leukemia: 3‐year results from the EBMT randomized trial. Haematologica 2005;90(5):643‐8. [PubMed] [Google Scholar]
- Schmitz N, Beksac M, Hasenclever D, Bacigalupo A, Ruutu T, Nagler A, et al. Transplantation of mobilized peripheral blood cells to HLA‐identical siblings with standard‐risk leukemia. Blood 2002;100(3):761‐7. [DOI] [PubMed] [Google Scholar]
Heldal 2003 {published data only}
- Abrahamson IW, Somme S, Heldal D, Egeland T, Kvale D, Tjonnfjord GE. Immune reconstitution after allogeneic stem cell transplantation: the impact of stem cell source and graft‐versus‐host disease. Haematologica 2005;90(1):86‐93. [PubMed] [Google Scholar]
- Heldal D, Brinch L, Tjonnfjord G, Solheim BG, Egeland T, Albrechtsen D, et al. Fewer relapses and increased chronic GVHD in patients transplanted with blood stem cells: a 5‐year follow‐up in a single centre study. Bone Marrow Transplantation 2003;32(3):257‐64. [DOI] [PubMed] [Google Scholar]
Mahmoud 1999 {published data only}
- Mahmoud HK, Fahmy OA. Peripheral blood vs bone marrow as a source for allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplantation 1999;24:355‐58. [DOI] [PubMed] [Google Scholar]
Mielcarek 2011 {published data only}
- Bensinger WI, Martin PJ, Storer B, Clift R, Forman SJ, Negrin R, et al. Transplantation of bone marrow as compared with peripheral‐blood cells from HLA‐identical relatives in patients with hematologic cancers. The New England Journal of Medicine 2001;344(3):175‐81. [DOI] [PubMed] [Google Scholar]
- Flowers ME, Parker PM, Johnston LJ, Matos AV, Storer B, Bensinger WI, et al. Comparison of chronic graft‐versus‐host disease after transplantation of peripheral blood stem cells versus bone marrow in allogeneic recipients: long‐term follow‐up of a randomized trial. Blood 2002;100(2):415‐9. [DOI] [PubMed] [Google Scholar]
- Mielcarek M, Storer B, Martin PJ, Forman SJ, Negrin RS, Flowers ME, et al. Long‐term outcomes after transplantation of HLA‐identical related G‐CSF‐mobilized peripheral blood mononuclear versus bone marrow. Blood 2012;119:2675‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehler VG, Radich JP, Storer B, Blume KG, Chauncey T, Clift R, et al. Randomized trial of allogeneic related bone marrow transplantation versus peripheral blood stem cell transplantation for chronic myeloid leukemia. Biology of Blood and Marrow Transplantation 2005;11(2):85‐92. [DOI] [PubMed] [Google Scholar]
Mohty 2002 {published data only}
- Blaise D, Keuntz M, Fortanier C, Bourhis JH, Milpied N, Sutton L, et al. Randomized trial of bone marrow versus lenograstim‐primed blood cell allogeneic transplantation in patients with early‐stage leukemia: a report from the Société Française de Greffe de Moelle. Journal of Clinical Oncology 2000;18(3):537‐46. [DOI] [PubMed] [Google Scholar]
- Mohty M, Kuentz M, Michallet M, Bourhis JH, Milpied N, Sutton L, et al. Chronic graft‐versus‐host disease after allogeneic blood stem cell transplantation: long‐term results of a randomized study. Blood 2002;100(9):3128‐34. [DOI] [PubMed] [Google Scholar]
Powles 2002 {published data only}
- Powles R, Mehta J, Kulkarni S, Treleaven J, Millar B, Marsden J, et al. Allogeneic blood and bone‐marrow stem‐cell transplantation in haematological malignant diseases: a randomised trial. Lancet 2000;355(9211):1231‐7. [DOI] [PubMed] [Google Scholar]
Vigorito 2001 {published data only}
- Souza CA, Durães MI, Vigorito AC, Penteado Aranha FJ, Oliveira GB, De Brito, et al. Quality of life in patients randomized to receive a bone marrow or a peripheral blood allograft. Haematologica 2002;87:1281‐85. [PubMed] [Google Scholar]
- Nucci M, Andrade F, Vigorito A, Trabasso P, Aranha JF, Maiolino A, et al. Infectious complications in patients randomized to receive allogeneic bone marrow or peripheral blood transplantation. Transplant Infectious Disease 2003;5(4):167‐73. [DOI] [PubMed] [Google Scholar]
- Vigorito AC, Azevedo WM, Marques JF, Azevedo AM, Eid KA, Aranha FJ, et al. A randomised, prospective comparison of allogeneic bone marrow and peripheral blood progenitor cell transplantation in the treatment of haematological malignancies. Bone Marrow Transplantation 1998;22(12):1145‐51. [DOI] [PubMed] [Google Scholar]
- Vigorito AC, Marques Junior JF, Aranha FJ, Oliveira GB, Miranda EC, Souza CA. A randomized, prospective comparison of allogeneic bone marrow and peripheral blood progenitor cell transplantation in the treatment of hematologic malignancies: an update. Haematologica 2001;86(6):665‐6. [PubMed] [Google Scholar]
References to studies excluded from this review
Bacigalupo 2012 {published data only}
- Bacigalupo A, Socié G, Schrezenmeier H, Tichelli A, Locasciulli A, Fuehrer M, et al. Bone marrow versus peripheral blood as the stem cell source for sibling transplants in acquired aplastic anemia: survival advantage for bone marrow in all age groups. Haematologica 2012;97(8):1142‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Champlin 2000 {published data only}
- Champlin RE, Schmitz N, Horowitz MM, Chapuis B, Chopra R, Cornelissen JJ, et al. Blood stem cells compared with bone marrow as a source of hematopoietic cells for allogeneic transplantation. IBMTR Histocompatibility and Stem Cell Sources Working Committee and the European Group for Blood and Marrow Transplantation (EBMT). Blood 2000;95(12):3702‐9. [PubMed] [Google Scholar]
Cornelissen 2003 {published data only}
- Cornelissen JJ, van der Holt B, Petersen EJ, Vindelov L, Russel CA, Höglund M et al. A randomized multicenter comparison of CD34(+)‐selected progenitor cells from blood vs from bone marrow in recipients of HLA‐identical allogeneic transplants for hematological malignancies. Experimental Hematology 2003;31(10):855‐64. [DOI] [PubMed] [Google Scholar]
Favre 2003 {published data only}
- Favre G, Beksaç M, Bacigalupo A, Ruutu T, Nagler A, Gluckman E et al. Differences between graft product and donor side effects following bone marrow or stem cell donation. Bone Marrow Transplantation 2003;32(9):873‐80. [DOI] [PubMed] [Google Scholar]
Kirschbaum 2012 {published data only}
- Kirschbaum M, O’Donnell M, Spielberger R, Bhatia R, Pullarkat V, Stein A, et al. Peripheral blood stem cells vs bone marrow for matched sibling transplant in AML and ALL remission [abstract]. Blood 2012;104(11):3321. [Google Scholar]
Morton 2001 {published data only}
- Morton J, Hutchins C, Durrant S. Granulocyte‐colony‐stimulating factor (G‐CSF)‐primed allogeneic bone marrow: significantly less graft‐versus‐host disease and comparable engraftment to G‐CSF‐mobilized peripheral blood stem cells. Blood 2001;98(12):3186‐91. [DOI] [PubMed] [Google Scholar]
Pidala 2009 {published data only}
- Pidala J, Anasetti C, Kharfan‐Dabaja MA, Cutler C, Sheldon A, Djulbegovic B. Decision analysis of peripheral blood versus bone marrow hematopoietic stem cells for allogeneic hematopoietic cell transplantation. Biology of Blood and Marrow Transplantation 2009;15(11):1415‐21. [DOI] [PubMed] [Google Scholar]
Robinet 2003 {published data only}
- Robinet E, Lapierre V, Tayebi H, Kuentz M, Blaise D, Tiberghien P. Blood versus marrow hematopoietic allogeneic graft. Transfusion and Apheresis Science 2003;29(1):53‐9. [DOI] [PubMed] [Google Scholar]
Storek 2001 {published data only}
- Storek J, Dawson MA, Storer B, Stevens‐Ayers T, Maloney DG, Marr KA et al. Immune reconstitution after allogeneic marrow transplantation compared with blood stem cell transplantation. Blood 2001;97(11):3380‐9. [DOI] [PubMed] [Google Scholar]
Additional references
Appelbaum 2007
- Appelbaum FR. Hematopoietic‐cell transplantation at 50. The New England Journal of Medicine 2007;357(15):1472‐5. [DOI] [PubMed] [Google Scholar]
Baldomero 2011
- Baldomero H, Gratwohl M, Gratwohl A, Tichelli A, Niederwieser D, Madrigal A, et al. The EBMT activity survey 2009: trends over the past 5 years. Bone Marrow Transplantation 2011;46(4):485‐501. [DOI] [PubMed] [Google Scholar]
Bensinger 2001
- Bensinger WI, Martin PJ, Storer B, Clift R, Forman SJ, Negrin R, et al. Transplantation of bone marrow as compared with peripheral‐blood cells from HLA‐identical relatives in patients with hematologic cancers. The New England Journal of Medicine 2001;344(3):175‐81. [DOI] [PubMed] [Google Scholar]
Bensinger 2012
- Bensinger WI. Allogeneic transplantation: peripheral blood vs. bone marrow. Current Opinion in Oncology 2012;24(2):191‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Blaise 2000
- Blaise D, Kuentz M, Fortanier C, Bourhis JH, Milpied N, Sutton L et al. Randomized trial of bone marrow versus lenograstim‐primed blood cell allogeneic transplantation in patients with early‐stage leukemia: a report from the Société Française de Greffe de Moelle. Journal of Clinical Oncology 2000;18(3):537‐46. [DOI] [PubMed] [Google Scholar]
Brunner 2013
- Brunner AM, Kim HT, Coughlin E, Alyea EP, Armand P, Ballen KK, et al. Outcomes of patients age 70 or older undergoing allogeneic hematopoietic stem cell transplantation for hematologic malignancies. Biology of Blood and Marrow Transplantation 2013;19(9):1374‐80. [DOI] [PubMed] [Google Scholar]
Chang 2012
- Chang YJ, Weng CL, Sun LX, Zhao YT. Allogeneic bone marrow transplantation compared to peripheral blood stem cell transplantation for the treatment of hematologic malignancies: a meta‐analysis based on time‐to‐event data from randomized controlled trials. Annals of Hematology 2012;91(3):427‐37. [PUBMED: 21789620] [DOI] [PubMed] [Google Scholar]
Confer 2007
- Confer DL, Miller JP. Long‐term safety of filgrastim (rhG‐CSF) administration. British Journal of Haematology 2007;137:77‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Confer 2009
- Confer DL, Miller JP, Chell JW. Bone marrow and peripheral blood cell donors and donor registries. In: Frederick R. Appelbaum, Stephen J. Forman, Robert S. Negrin, Karl G. Blume editor(s). Thomas' Hematopietic Cell Transplantation. 4th Edition. Oxford, UK: Wiley‐Blackwell, 2009. doi: 10.1002/9781444303537.ch38:544‐58. [Google Scholar]
de Fabritiis 2001
- de Fabritiis P, Iori AP, Mengarelli A, Gozzer M, Ferrazza G, De Propris MS, et al. CD34+ cell mobilization for allogeneic progenitor cell transplantation: efficacy of a short course of G‐CSF. Transfusion 2001;41(2):190‐5. [DOI] [PubMed] [Google Scholar]
De Souza 2002
- Souza CA, Durães MI, Vigorito AC, Penteado Aranha FJ, Oliveira GB, De Brito, et al. Quality of life in patients randomized to receive a bone marrow or a peripheral blood allograft. Haematologica 2002;87(12):1281‐5. [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Eapen 2007
- Eapen M, Logan BR, Confer DL, Haagenson M, Wagner JE, Weisdorf DJ, et al. Peripheral blood grafts from unrelated donors are associated with increased acute and chronic graft‐versus‐host disease without improved survival. Biology of Blood and Marrow Transplantation 2007;13(12):1461‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ferrera 2009
- Ferrara JL, Levine JE, Reddy P, Holler E. Graft‐versus‐host disease. Lancet 2009;373(9674):1550‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Filipovich 2005
- Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft‐versus‐host disease: I. Diagnosis and staging working group report. Biology of Blood and Marrow Transplantation 2005;11(12):945‐56. [DOI] [PubMed] [Google Scholar]
Gluckman 2006
- Gluckman E, Rocha V. Donor selection for unrelated cord blood transplants. Current Opinion in Immunology 2006;18(5):565‐70. [DOI] [PubMed] [Google Scholar]
Glucksberg 1974
- Glucksberg H, Storb R, Fefer A, Buckner C, Neiman P, Clift R. Clinical manifestations of graft‐versus‐host disease in human recipients of marrow from HLA‐matched sibling donors. Transplantation 1974;18(4):295‐304. [DOI] [PubMed] [Google Scholar]
Goldberg 2013
- Goldberg JD, Giralt S. Assessing response of therapy for acute and chronic graft‐versus‐host disease. Expert Reviews in Hematology 2013;6(1):103‐7. [DOI] [PubMed] [Google Scholar]
Gratwohl 2007
- Gratwohl A, Baldomero H, Frauendorfer K, Urbano‐Ispizua A, Niederwieser D. Results of EBMT activity survey 2005 on hematopoietic stem cell transplantation: Focus on increasing use of unrelated donors. Bone Marrow Transplantation 2007;39:71‐87. [DOI] [PubMed] [Google Scholar]
Gratwohl 2009
- Gratwohl A, Baldomero H, Schwendener A, Rocha V, Apperley J, Frauendorfer K, et al. The EBMT activity survey 2007 with focus on allogeneic HSCT for AML and novel cellular therapies. Bone Marrow Transplantation 2009;43(4):275‐91. [DOI] [PubMed] [Google Scholar]
Gratwohl 2013
- Gratwohl A, Baldomero H, Gratwohl M, Aljurf MD, Bouzas LF, Horowitz M, et al. Quantitative and qualitative differences in use and trends of hematopoietic stem cell transplantation: a Global Observational Study. Haematologica 2013;98(8):1282‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Deeks JJ (editors). Chapter 7: Selecting studies and collecting data. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011c
- Higgins JPT, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Horwitz 2006
- Horwitz ME, Sullivan KM. Chronic graft‐versus‐host disease. Blood Reviews 2006;20(1):15‐27. [DOI] [PubMed] [Google Scholar]
Jenq 2010
- Jenq RR, Brink MRM. Allogeneic hematopoietic stem cell transplantation: individualized stem cell and immunotherapy of cancer. Nature Reviews of Cancer 2010;10(3):213‐21. [DOI] [PubMed] [Google Scholar]
Kalbfleisch 1980
- Kalbfleisch JD, Prentice RL. The Statistical Analysis of Failure Time Data . New York, NY, USA: John Wiley, 1980. [Google Scholar]
Kuzmina 2012
- Kuzmina Z, Eder S, Bohm A, Pernicka E, Vormittag L, Kalhs P, et al. Significantly worse survival of patients with NIH‐defined chronic graft‐versus‐host disease and thrombocytopenia or progressive onset type: results of a prospective study. Leukemia 2012;26(4):746‐56. [DOI] [PubMed] [Google Scholar]
Labopin 2003
- Labopin M, Iacobelli S. Statistical guidelines for EBMT. http://www.ebmt.org/1WhatisEBMT/Op_Manual/OPMAN_StatGuidelines_oct2003.pdf (accessed 01 March 2012).
Labopin 2009
- Labopin M, Latouche A, Suciu S, Santucci A, Canals C, Iacobelli S, et al. Definitions and evaluation of endpoints following stem cells transplantation ‐ recommendation from the European group for Blood and Marrow transplantation (EBMT). CLINT, Work Package 5 2009. http://portal.ebmt.org/sites/clint2/clint/Documents/Statistical%20Endpoints_CLINT%20Project_final%20version.pdf (accessed 25 March 2014).
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The Prisma Group. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. Journal of Clinical Epidemiology 2009;62(10):1006‐12. [DOI] [PubMed] [Google Scholar]
Nagler 2012
- Nagler A, Labopin M, Shimoni A, Niederwieser D, Mufti GJ, Zander AR, et al. Mobilized peripheral blood stem cells compared with bone marrow as the stem cell source for unrelated donor allogeneic transplantation with reduced‐intensity conditioning in patients with acute myeloid leukemia in complete remission: an analysis from the Acute Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Biology of Blood and Marrow Transplantation 2012;18(9):1422‐9. [DOI] [PubMed] [Google Scholar]
Norkin 2012
- Norkin M, Hsu JW, Wingard JR. Quality of life, social challenges, and psychosocial support for long‐term survivors after allogeneic hematopoietic stem‐cell transplantation. Seminars in Hematology 2012;49(1):104‐9. [DOI] [PubMed] [Google Scholar]
Paczesny 2009
- Paczesny S, Hanauer D, Sun Y, Reddy P. New perspectives on the biology of acute GVHD. Bone Marrow Transplantation 2009;45(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
Pasquini 2013
- Pasquini MC, Wang Z. Current use and outcome of hematopoietic stem cell transplantation: CIBMTR Summary Slides, 2013. Available at: http://www.cibmtr.org.
Petersdorf 2001
- Petersdorf E, Hansen J, Martin P, Woolfrey A, Malkki M, Gooley T, et al. Major‐histocompatibility‐complex class I alleles and antigens in hematopoietic‐cell transplantation. The New England Journal of Medicine 2001;345(25):1794‐800. [DOI] [PubMed] [Google Scholar]
Pidala 2009a
- Pidala J, Anasetti C, Kharfan‐Dabaja MA, Cutler C, Sheldon A, Djulbegovic B. Decision analysis of peripheral blood versus bone marrow hematopoietic stem cells for allogeneic hematopoietic cell transplantation. Biology of Blood and Marrow Transplantation 2009;15(11):1415‐21. [DOI] [PubMed] [Google Scholar]
Pidala 2009b
- Pidala J, Anasetti C, Jim H. Quality of life after allogeneic hematopoietic cell transplantation. Blood 2009;114(1):7‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pollack 2009
- Pollack SM, O’Connor TP, Hashash J, Tabbara IA. Nonmyeloablative and reduced‐intensity conditioning for allogeneic hematopoietic stem cell transplantation. American Journal of Clinical Oncology 2009;32(6):618‐28. [DOI] [PubMed] [Google Scholar]
Review Manager 2011 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Ringden 2009
- Ringden O, Karlsson H, Olsson R, Omazic B, Uhlin M. The allogeneic graft‐versus‐cancer effect. British Journal of Haematology 2009;147(5):614‐33. [DOI] [PubMed] [Google Scholar]
Ringden 2012
- Ringden O, Labopin M, Beelen DW, Volin L, Ehninger G, Finke J, et al. Bone marrow or peripheral blood stem cell transplantation from unrelated donors in adult patients with acute myeloid leukaemia, an Acute Leukaemia Working Party analysis in 2262 patients. Journal of Internal Medicine 2012;272(5):472‐83. [DOI] [PubMed] [Google Scholar]
Schmitz 2002
- Schmitz N, Beksac M, Hasenclever D, Bacigalupo A, Ruutu T, Nagler A et al. Transplantation of mobilized peripheral blood cells to HLA‐identical siblings with standard‐risk leukemia. Blood 2002;100(3):761‐7. [DOI] [PubMed] [Google Scholar]
Schmitz 2005
- Schmitz N, Beksac M, Bacigalupo A, Ruutu T, Nagler A, Gluckman E et al. Filgrastim‐mobilized peripheral blood progenitor cells versus bone marrow transplantation for treating leukemia: 3‐year results from the EBMT randomized trial. Haematologica 2005;90(5):643‐8. [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and 'Summary of findings' tables. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Siddiq 2009
- Siddiq S, Pamphilon D, Brunskill S, Doree C, Hyde C, Stanworth S. Bone marrow harvest versus peripheral stem cell collection for haemopoietic stem cell donation in healthy donors. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD006406.pub2] [DOI] [PubMed] [Google Scholar]
Sorror 2011
- Sorror ML, Sandmaier BM, Storer BE, Franke GN, Laport GG, Chauncey TR, et al. Long‐term outcomes among older patients following nonmyeloablative conditioning and allogeneic hematopoietic cell transplantation for advanced hematologic malignancies. Journal of the American Medical Association 2011;306(17):1874‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stem Cell Trialists' Collaborative Group 2005
- Stem Cell Trialists' Collaborative Croup. Allogeneic peripheral blood stem‐cell compared with bone marrow transplantation in the management of hematological malignancies: an individual patient data meta‐analysis of nine randomised trials. Journal of Clinical Oncology 2005;23(22):5074‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stem Cell Trialists' Collaborative Group 2006
- Stem Cell Trialists' Collaborative Group. Individual patient data meta‐analysis of allogeneic peripheral blood stem cell transplant vs. bone marrow transplant in the management of hematological malignancies: indirect assessment of the effect of the day 11 methotrexate administration. Bone Marrow Transplantation 2006;38(6):539‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D (editors). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Trials 2007;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vigorito 1998
- Vigorito AC, Azevedo WM, Marques JF, Azevedo AM, Eid KA, Aranha FJ et al. A randomised, prospective comparison of allogeneic bone marrow and peripheral blood progenitor cell transplantation in the treatment of haematological malignancies. Bone Marrow Transplantation 1998;22(12):1145‐51. [DOI] [PubMed] [Google Scholar]
Weisdorf 2012
- Weisdorf D, Zhang MJ, Arora M, Horowitz MM, Rizzo JD, Eapen M. Graft‐versus‐host disease induced graft‐versus‐leukemia effect: greater impact on relapse and disease‐free survival after reduced intensity conditioning. Biology of Blood and Marrow Transplantation 2012;18(11):1727‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wood 2013
- Wood WA, Chai X, Weisdorf D, Martin PJ, Cutler C, Inamoto Y, et al. Comorbidity burden in patients with chronic GVHD. Bone Marrow Transplantation 2013;48(11):1429‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]