

Abstract
Introduction
Uveal melanoma (UM) responds poorly to targeted therapies or immune checkpoint inhibitors. Adenosine monophosphate-activated protein kinase (AMPK) is a pivotal serine/threonine protein kinase that coordinates vital processes such as cell growth. Targeting AMPK pathway, which represents a critical mechanism mediating the survival of UM cells, may prove to be a novel treatment strategy for UM. We aimed to demonstrate the effects of AMPK modulation on UM cells.
Methods
In silico analyses were performed to compare UM and normal melanocyte cells via Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Set Enrichment Analysis (GSEA). The effects of AMPK modulation on cell viability and proliferation in UM cell lines with different molecular profiles (i.e., 92-1, MP46, OMM2.5, and Mel270) were investigated via XTT cell viability and proliferation assays after treating the cells with varying concentrations of A-769662 (AMPK activator) or dorsomorphin (AMPK inhibitor).
Results
KEGG/GSEA studies demonstrated that genes implicated in the AMPK signaling pathway were differentially regulated in UM. Gene sets comprising genes involved in AMPK signaling and genes involved in energy-dependent regulation of mammalian target of rapamycin by liver kinase B1-AMPK were downregulated in UM. We observed gradual decreases in the numbers of viable UM cells as the concentration of A-769662 treatment increased. All UM cells demonstrated statistically significant decreases in cell viability when treated with 200 µm A-769662. Moreover, the effects of AMPK inhibition on UM cells were potent, since low doses of dorsomorphin treatment resulted in significant decreases in viabilities of UM cells. The half maximal inhibitory concentration (IC50) values confirmed the potency of dorsomorphin treatment against UM in vitro.
Conclusion
AMPK may act like a friend or a foe in cancer depending on the context. As such, the current study contributes to the literature in determining the effects of therapeutic strategies targeting AMPK in several UM cells. We propose a new perspective in the treatment of UM. Targeting AMPK pathway may open up new avenues in developing novel therapeutic approaches to improve overall survival in UM.
Keywords: Uveal melanoma, AMPK, Tumor metabolism, A-769662, Dorsomorphin
Introduction
Uveal melanoma (UM) is the most common intraocular primary malignant tumor in adults [1]. It originates from melanocytes located in the iris, ciliary body, and choroid [1]. Ocular melanomas constitute 3% of all melanomas [2]. The mean age of diagnosis is 60 years and UM is rarely seen under 30 years of age [3–5]. The yearly incidence has been reported as 5–8 per million [3–5]. Based on tumor size and location, the treatment options of UM include photodynamic therapy, transpupillary thermotherapy, plaque brachytherapy, loaded particle radiotherapy, fractionated stereotactic radiotherapy, local resection, enucleation, and exenteration [6–14]. Metastatic UM responds poorly to targeted therapies and immune checkpoint inhibitors [10, 15]. It is estimated that metastasis is responsible for about 50% of UM death, despite the appropriate treatment [16]. Unfortunately, survival and metastasis rates do not differ in conservative and radical treatments [16, 17]. The most common sites of hematogenous metastasis include the liver (89%), lung (29%), and bones (17%) [18]. The life expectancy after metastasis is 6–12 months [18]. Only 20% of patients who develop metastases reach 1-year lifetime [18, 19]. Indeed, there has been no significant improvement in the life expectancy of UM patients, especially those with metastatic UM, in the last 30 years [20, 21].
Mechanisms regulating the carcinogenic processes in UM are still mostly unclear. Recently, Chua et al. reported that AMP (adenosine monophosphate)-activated protein kinase (AMPK) pathway might be upregulated in UM. AMPK is a pivotal serine/threonine protein kinase that provides energy homeostasis by detecting the AMP/ATP ratio in the cells [22–25]. Once AMPK is activated, it phosphorylates key enzymes and transcription factors in order to regulate metabolism and expression of various genes [26]. As a result, catabolic reactions such as fatty acid oxidation and glycolysis are upregulated under stress conditions such as nutrient deficiency and hypoxia via AMPK activation. On the other hand, anabolic reactions such as fatty acid, cholesterol, and protein synthesis are suppressed [27]. The AMPK signaling pathway coordinates vital processes such as cell growth, autophagy, and apoptosis [28]. Cell cycle control by AMPK is mediated by the upregulation of the p53-p21 axis as well as the regulation of TSC2-mammalian target of rapamycin (mTOR) pathway [28–31]. AMPK promotes maintenance of a resting cell state in mature tissues that do not require proliferative responses to maintain their functional states. In addition, AMPK was also implicated in the protection of cells from transformation by oncogenic stimulation [30]. Such findings demonstrate that the AMPK pathway plays a crucial role in the regulation of cell proliferation [30].
AMPK signaling has been shown to be dysregulated in various types of cancer as well as in diabetes, heart muscle disorders, inflammatory diseases, and viral infections [22]. In the process of oncogenic transformation, tumor cells have to maintain their energy homeostasis in order to grow and survive despite stresses such as hypoxia, nutrient deficiency, oncogene activation as well as radiotherapy and chemotherapy [22, 26, 27, 32–39]. As such, AMPK, the main regulator of energy and oxidoreduction homeostasis, plays a significant role in the cell’s response to such stresses [22]. Indeed, AMPK assists tumor cells to survive under conditions of hypoxia and nutrient deficiency [22, 33], as well as promoting metastasis [33, 40]. In an animal study conducted with astrocytic tumors, phosphorylation of retinoblastoma protein with AMPK activation increased tumor cell proliferation [41]. In another study that utilized real-time cell analysis, activation of AMPK was shown to increase cancer cell proliferation and aggressiveness in breast and hepatocellular cancers [42].
Given these findings, the notion that “AMPK keeps tumor cells from starving to death” seems to be gaining importance [43–45]. Moreover, recent findings suggest that AMPK pathway represents a critical mechanism mediating the survival of UM cells [15]. Thus, targeting the AMPK pathway may prove to be a novel treatment strategy for UM. In this proof-of-concept study, we aimed to demonstrate the effects of AMPK modulation on UM cell lines in order to present a new perspective in the treatment of UM. We first performed in silico analyses to compare UM (92-1) and normal melanocyte (PIG1) cells via the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Set Enrichment Analysis (GSEA). We then compared the AMPK expression levels in UM cells with those in noncancerous retinal epithelial and noncancerous fibroblast cells. We also analyzed the dependency of UM cells on AMPK through CRISPR-cas9 screening data at the Cancer Dependency Map (DepMap) database. Then, we investigated the effects of AMPK modulation on cell viability and proliferation in several UM cell lines with different molecular profiles (i.e., 92-1, MP46, OMM2.5, and Mel270).
Materials and Methods
In silico Analyses
In silico analyses were performed using the GSE176345 and GSE181125 expression profiling by high-throughput sequencing datasets as well as GSE22138 expression profiling by array dataset in the Gene Expression Omnibus (GEO) database [46, 47]. FASTQ files were downloaded through TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA resources) servers via NCBI SRA-toolkit (http://ncbi.github.io/sra-tools/). Numerical computations of the RNA sequence dataset were performed on Galaxy servers [48]. Data files were aligned with the GRCh38 (hg38) human reference genome via Minimap2 sequencing package [49]. BAM files were counted via featureCounts package [50]. Estimating variance-mean dependence in count data from high-throughput sequencing assays and testing for differential expression were based on a model using the negative binomial distribution with DESeq2 [51]. Finally, the obtained results were compared using the KEGG [52] and GSEA [53, 54] analysis tools. Pathview package was used in KEGG analyses [55]. The entire pipeline has been implemented from Reference-based RNA-Seq data analysis (Galaxy Training materials) [56]. The “24-AMPK-gene set” score was calculated based on the mean expressions of the relevant genes as previously reported (SLC2A4, FOXO3, PPP2CB, PIK3CD, CAB39L, CCNA1, FBP1, FBP2, FOXO1, HMGCR, IRS2, PIK3R1, SIRT1, TBC1D1, PPARGC1A, PPP2R2C, MLYCD, PFKFB3, PPP2R2B, PRKAA2, LEPR, CAB39, IRS1, and PFKFB1) [57].
Determination of the Dependency of UM Cells on Protein Kinase AMP-Activated Alpha 1 Catalytic Subunit
The Cancer Cell Line Encyclopedia (CCLE) allows for predictive modeling of anticancer drug sensitivity [58]. We compared AMPK (protein kinase AMP-activated alpha 1 [PRKAA1]) expression levels in UM cells with those of noncancerous retinal epithelial cells (RPE) and noncancerous fibroblast cells by utilizing CCLE. In addition, we utilized the DepMap, which serves as a valuable database for systematically identifying genetic and pharmacologic dependencies and the biomarkers that predict them. It employs RNA interference and/or CRISPR-Cas9 technology to screen and assess these targets [59]. By accessing the DepMap database (https://depmap.org/portal/), gene dependency information pertaining to different cancer cell lines can be acquired. AMPK (PRKAA1) gene effect was displayed with Chronos, an algorithm for inferring gene knockout fitness effects based on an explicit model of the dynamics of cell proliferation after CRISPR gene knockout [60]. A negative score serves as an indication that the gene disruption impedes the proliferation and survival of the cell lines. A lower score implies a greater degree of importance attributed to the gene. In this particular study, we analyzed the essentiality of AMPK for the survival of UM cells and/or noncancerous RPE by determining their Chronos scores [61].
Materials
AMPK activator A-769662 and AMPK inhibitor dorsomorphin (compound C) were purchased from Abcam, Cambridge, UK. A-769662 is an effective and reversible AMPK activator. A-769662 activates AMPK both allosterically and by inhibiting AMPK dephosphorylation via mimicking AMP. Dorsomorphin is an effective and reversible AMPK inhibitor.
100 mm A-769662 stock solution and 10 mm dorsomorphin stock solution were prepared in dimethyl sulfoxide (SERVA, Heidelberg, Germany). UM cell lines (i.e., 92-1, MP46, OMM2.5, and Mel270) were treated with varying (freshly prepared) concentrations of either AMPK activator or inhibitor (A-769662: 200 µm, 143 µm, 102 µm, 72.9 µm, 52.1 µm, 37.2 µm, 26.6 µm, 19 µm, and 13.6 µm) (dorsomorphin: 20 µm, 14.3 µm, 10.2 µm, 7.29 µm, 5.21 µm, 3.72 µm, 2.66 µm, 1.9 µm, and 1.36 µm), similar to previously reported [62–65].
Cell Culture Studies
92-1, OMM2.5, and Mel270 UM cell lines [66–68] (kind gift of Prof. Martine Jager, MD, PhD, Leiden University) were grown in RPMI-1640 Dutch Modified medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Biological Industries, Kibbutz Beit-Haemek, Israel), 2 mml-glutamine (Biological Industries, Kibbutz Beit-Haemek, Israel), and 2% penicillin/streptomycin (Biological Industries, Kibbutz Beit-Haemek, Israel). MP46 UM cell line [69] (also a kind gift of Prof. Martine Jager, MD, PhD, Leiden University) was grown in IMDM (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) medium supplemented with 20% FBS (Biological Industries, Kibbutz Beit-Haemek, Israel), 3 mml-glutamine (Biological Industries, Kibbutz Beit-Haemek, Israel), and 2% penicillin/streptomycin (Biological Industries, Kibbutz Beit-Haemek, Israel). The cells were incubated in a cell culture incubator at 37°C, 5% CO2, and 60% humidity. The cell cultures were maintained by the replacement of media 2–3 times per week. Cells growing as a monolayer were subcultured by trypsin-EDTA once a week.
XTT Cell Viability and Proliferation Assay
The XTT cell proliferation assay determines the cell proliferation rate and, conversely, the reduction in cell viability, when metabolic events lead to apoptosis or necrosis. XTT is a highly sensitive and nonradioactive assay.
A-769662 and dorsomorphin were diluted in cell culture media and assay concentrations were freshly prepared. Briefly, 50 µL UM cell suspensions in culture medium containing 3 × 103 92-1 cells, 4 × 103 MP46 cells, 4 × 103 OMM2.5 cells, or 5 × 103 Mel270 cells were plated in 96-well flat-bottom culture plates (Corning, MA, USA) and incubated for 12 h to recover from handling. Varying concentrations of A-769662 and/or dorsomorphin in cell culture media were added into each well in triplicate. Cell-free wells were also prepared in order to determine the background absorbance values. UM cells were treated with varying concentrations of A-769662 or dorsomorphin for 48 h at 37°C, 5% CO2, and 60% humidity. According to the XTT assay (Biological Industries, Kibbutz Beit-Haemek, Israel) protocol, XTT reagent solution and activation solution were defrosted immediately prior to use in a 37°C bath. 0.1 mL activation solution was added to 5 mL XTT reagent to prepare the reaction solution. Then, 50 µL of the XTT reaction solution was added to each well in order to assess cell viability at the end of the 48 h of incubation period with A-769662 or dorsomorphin. Following 4 h of incubation of the cells with the XTT reaction solution, the absorbance values (of each well) were measured at 465 nm in a microtiter plate reader (SpectraMax Plus, Molecular Devices, CA, USA) at 25°C. Cells incubated in culture medium only (without A-769662 or dorsomorphin) served as the control for cell viability. The half maximal inhibitory concentration (IC50) values of A-769662 and dorsomorphin for each UM cell line were estimated by fitting a model with nonlinear regression.
Statistical Analyses
Data are presented as mean ± SD. Student’s t test was used for pairwise comparisons. A 5% type 1 error level was used to infer statistical significance. All statistical analyses were carried out using IBM SPSS Statistics for Windows Software, version 23.
Results
AMPK Demonstrates a Plethora of Metabolic Effects and Genes Associated with AMPK Signaling Pathway Are Differentially Expressed in UM Cells Compared to Normal Melanocytes
When AMPK signaling pathway was investigated with KEGG analysis, AMPK has been shown to bear pivotal effects on glucose, protein, and fatty acid metabolisms as well as on various adipokines, insulin signaling pathway, inflammation, cell cycle regulation, cell growth, and autophagy (Fig. 1). Moreover, expressions of various genes that are implicated in AMPK signaling pathway are differentially regulated in UM cells (92-1) in contrast to their normal counterparts (PIG1), as shown by the gene expression heatmap in Figure 1. We also investigated AMPK expression in UM cells compared with noncancerous cells. UM cells appear to express lower levels of AMPK than noncancerous RPE and noncancerous fibroblast cells (Fig. 2a–c). Moreover, our DepMap analyses clearly demonstrated that a greater degree of essentiality was attributed to AMPK in UM cells than in noncancerous RPE (Fig. 2d–e). Moreover, comparison of PRKAA1 (AMPK) expression in UM cells and immortalized human melanocytes at a publicly available dataset (GSE181125) revealed that AMPK was expressed at lower levels in UM cells (log2FC: −1.545287, p < 0.05). Thus, pharmacological inhibition or activation of AMPK has a great potential for regulating key downstream processes related with cell cycle as well as glucose metabolism and biosynthesis in UM.
Fig. 1.

AMPK expression in UM cells. a KEGG graph of AMPK signaling pathway is shown. Gene expression data on KEGG graph were rendered by KEGG Pathway Painter (KPP) according to the imported list of genes that are differentially expressed in UM cells (92-1) compared to normal melanocyte cells (PIG1). Heatmap displays upregulated (red) and downregulated (green) genes associated with AMPK signaling pathway. b AMPK (PRKAA1) gene expression across nine UM cell lines with available DepMap data are shown (Expression 22Q2 Public). Each dot is annotated by the cell line. All genomic data from the CCLE are available at https://portals.broadinstitute.org/ccle/data. DepMap 22Q2 was used for the analyses.
Fig. 2.

Comparison of AMPK expression in UM cells with noncancerous cells. a AMPK (PRKAA1) gene expression in 92-1, MP46, OMM2.5, and Mel270 UM cells. b AMPK (PRKAA1) gene expression in noncancerous RPE. c AMPK (PRKAA1) gene expression in noncancerous fibroblast cells. Each dot is annotated by the cell line. All genomic data from the CCLE are available at https://portals.broadinstitute.org/ccle/data. DepMap 23Q2 was used for the analyses. d PRKAA1 (AMPK) gene effect in UM cells (DepMap). e PRKAA1 (AMPK) gene effect in noncancerous RPE (DepMap).
Gene Sets Associated with AMPK Signaling Are Differentially Expressed in UM Cells
In order to further investigate the distinction of UM cells from normal melanocytes, we analyzed a publicly available dataset to compare gene expression in 92-1 UM and PIG1 normal melanocyte cells (GSE176345). The experiment was analyzed by GSEA, as described previously [53, 70]. WP_AMPACTIVATED_PROTEIN_KINASE_AMPK_SIGNALING gene set [54, 71], which comprises genes involved in AMPK signaling, was found to be differentially expressed. Furthermore, this gene set appeared to be downregulated in UM cells compared with normal melanocyte cells (normalized enrichment score: 1.05, FDR: 0.32). Figure 3a and b show the enrichment plot and blue-pink O’gram of the genes that contribute to the enrichment result, respectively.
Fig. 3.

GSEA analysis results. Enrichment plots of normal melanocytes versus UM cells for the WP_AMPACTIVATED_PROTEIN_KINASE_AMPK_SIGNALING (NES: 1.05) (a) and REACTOME_ENERGY_DEPENDENT_REGULATION_OF_MTOR_BY_LKB1_AMPK (NES: 1.06) (c) gene sets are shown. Genes involved in AMPK signaling and energy-dependent regulation of mTOR by LKB1-AMPK were downregulated in UM cells compared with normal melanocyte cells. Gene expression data were acquired from NCBI Gene Expression Omnibus (GSE176345) and GSEA was performed as described previously [53]. Genes that contribute to the enrichment results within each gene set are shown in blue-pink O’grams of altered AMPK signaling and energy-dependent regulation of mTOR by LKB1-AMPK in (b) and (d), respectively. These GSEA results are consistent with our KEGG findings concerning AMPK signaling pathway gene expressions of UM and normal melanocyte cells. b, d Normal melanocyte cell population was enriched for expression of PRKAA1, which is a gene that encodes AMPK (i.e., third and first rows in blue-pink O’grams, respectively). d On the other hand, expressions of late endosomal/lysosomal adaptor and MAPK and mTOR activator (LAMTOR) 1, 2, 4, and 3 genes were upregulated in UM cells. LAMTOR is a scaffold protein complex that senses nutrients such as amino acids and lipids while integrating growth factor signaling [72]. LAMTOR1 functions as an anchor for the remaining LAMTOR subunits [73–77]. LAMTOR was also reported to activate mTORC1 signaling [78]. e The graph shows that patients with low AMPK level had significantly higher tumor mutational burden (TMB). f The “24-AMPK-gene set” scores of the patient samples. g The “24-AMPK-gene set” scores of the UM patients with or without metastasis. GSEA, Gene Set Enrichment Analysis; NES, normalized enrichment score; PRKAA1, protein kinase AMP-activated alpha 1 catalytic subunit; UM, uveal melanoma; LKB1, liver kinase B1; mTORC1, mTOR complex 1.
In addition, we also found that REACTOME_ENERGY_DEPENDENT_REGULATION_OF_MTOR_BY_LKB1_AMPK gene set [54, 71], which comprises genes involved in energy-dependent regulation of mTOR by liver kinase B1-AMPK, was differentially expressed. Similar to our previous findings, this gene set seem to be downregulated in UM cells in comparison to normal melanocyte cells (normalized enrichment score: 1.06, FDR:0.4). Figure 3c and d demonstrate the enrichment plot and blue-pink O’gram of the genes that contribute to the enrichment result, respectively. Even though not statistically significant, both of these GSEA results are close to statistical significance and are consistent with our findings concerning KEGG analysis and differential gene expression (Fig. 1), suggesting that UM represents a tumor with differential regulation of AMPK signaling compared with normal melanocytes. Moreover, we analyzed UM patient samples at The Cancer Genome Atlas (TCGA) and found that patients with low AMPK level had significantly higher tumor mutational burden, which was reported to be an independent and reliable indicator of poor patient outcomes (Fig. 3e) [79, 80].
Chang et al. [57] recently reported that a “24-AMPK-gene set” could successfully stratify patients into high- and low-risk groups in several types of tumors based on Cox regression and log-rank tests. As such, we investigated the significance of this gene set in UM patient samples at TCGA. Indeed, samples with high AMPK levels had higher “24-AMPK-gene set” scores, as expected (Fig. 3f). We then applied this scoring approach to the expression data from UM primary tumors (GSE22138) [81]. Our results demonstrated that UM patients with metastasis had significantly lower levels of the “24-AMPK-gene set” score (Fig. 3g). Accordingly, our in silico results (e.g., KEGG, GSEA, differential gene expression) provided a basis for investigating the activity of potential therapeutic approaches against AMPK, as well as helping us select the strategy with the highest potential activity for further in vitro experiments.
Modulation of AMPK Significantly Alters UM Cell Viability
Given that UM demonstrates differential activation of the AMPK signaling pathway, we set out to explore whether pharmacological inhibition or activation of AMPK has therapeutic value for the treatment of UM, since AMPK is known to regulate crucial downstream processes. Four different UM cell lines (i.e., 92-1, MP46, OMM2.5, and Mel270) were treated with varying concentrations of either A-769662 (AMPK activator) or dorsomorphin (AMPK inhibitor) for 48 h in order to determine cell viability and proliferation.
Treatment of UM cells with lower doses of A-769662 (e.g., 13.6 µm) did not appear to cause significant killing of the tumor cells (Fig. 4, points and error bars designate means and standard deviations, respectively). Indeed, we observed gradual decreases in the numbers of viable cells as the concentration of A-769662 increased, when compared to their corresponding control groups (i.e., the cells that were not treated with any chemicals). When the concentration of A-769662 increased to 200 µm, all UM cells demonstrated statistically significant decreases in cell viability. The most prominent cytotoxic effect of A-769662 was observed on Mel270 cells, as the normalized cell number decreased nearly 50% compared to the control group. It should be noted that 92-1 UM cells seemed to display a type of resistance to A-769662 up to 142.9 µm, since we observed a significant decrease in 92-1 cell number at 200 µm of A-769662.
Fig. 4.

Cell viabilities of UM cells treated with varying concentrations of AMPK activator (A-769662). 92-1 (a), MP46 (b), OMM2.5 (c), and Mel270 (d) UM cells were treated with A-769662 for 48 h and the effects of A-769662 on the cells were determined via XTT assay. Cells that were not treated with any chemicals were used as the control group. Normalized cell numbers are shown. 200 µm A-769662 resulted in statistically significant decreases in cell viabilities of all types of UM cells, when compared to their corresponding control groups. *p < 0.05 (n = 3). UM, uveal melanoma.
On the other hand, the effects of AMPK inhibition on UM cells seemed to be more prominent since lower doses of dorsomorphin treatment resulted in significant decreases in cell viabilities of UM cells (Fig. 5, points and error bars designate means and standard deviations, respectively). Indeed, we observed significant sharp decreases in the numbers of viable cells as the concentrations of dorsomorphin increased, when compared to their corresponding control groups (i.e., the cells that were not treated with any chemicals). Especially dorsomorphin concentrations over 5 µm resulted in significant cytotoxic effects on UM cells. Only OMM2.5 UM cells seemed to display a type of resistance to dorsomorphin over 5 µm. However, we could still achieve a statistically significant decrease in OMM2.5 cell number at 20 µm of dorsomorphin. Indeed, 20 µm dorsomorphin resulted in statistically significant decreases in cell viabilities of all types of UM cells that were studied. When 92-1 and Mel270 cells were treated with 20 µm of dorsomorphin, the normalized cell numbers decreased more than 90% compared to the corresponding control groups, whereas the same concentration of dorsomorphin could decrease the normalized cell number of MP46 cells nearly 80% (Fig. 5).
Fig. 5.

Cell viabilities of UM cells treated with varying concentrations of AMPK inhibitor (dorsomorphin). 92-1 (a), MP46 (b), OMM2.5 (c), and Mel270 (d) UM cells were treated with dorsomorphin for 48 h and the effects of dorsomorphin on the cells were determined via XTT assay. Cells that were not treated with any chemicals were used as the control group. Normalized cell numbers are shown. Cytotoxic effects of AMPK inhibition on UM cells appear to be more potent than those of AMPK activation (Fig. 4). 20 µm dorsomorphin achieved statistically significant cytotoxic effects on all types of UM cells, when compared to their corresponding control groups. *p < 0.05 (n = 3). UM, uveal melanoma.
The IC50 of AMPK inhibitor was determined for each UM cell line in order to measure the potency of dorsomorphin in killing different types of UM cells. The IC50 values of dorsomorphin (estimated by fitting models with nonlinear regression) for 92-1, MP46, OMM2.5, and Mel270 cells were found to be 6.526 µm, 10.13 µm, 31.45 µm, 8.39 µm, respectively (Fig. 6). Such IC50 values proved the potency of dorsomorphin treatment against these UM cells in vitro.
Fig. 6.
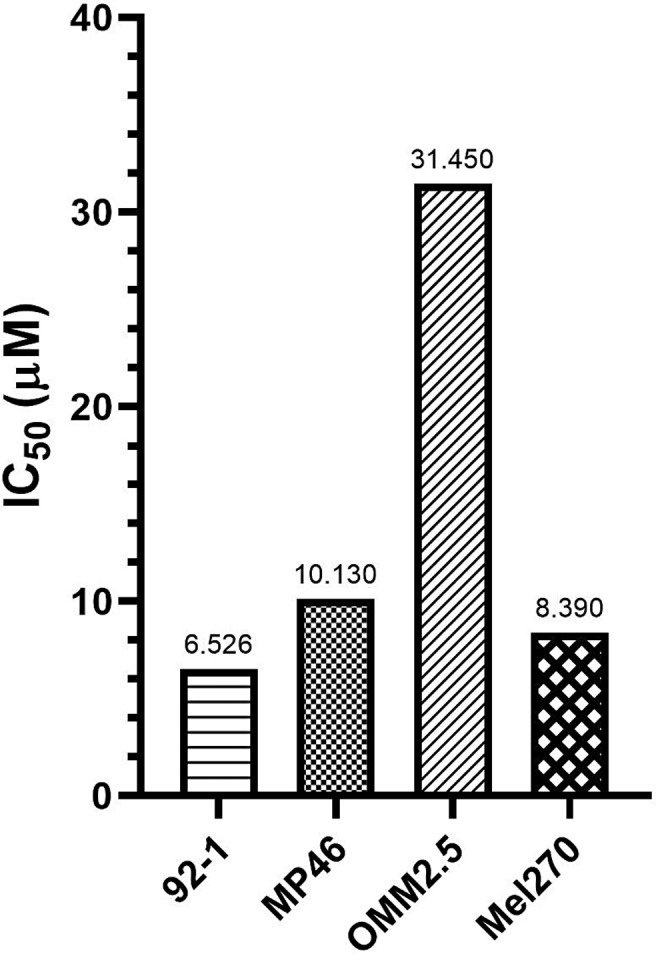
Graph of the IC50 values of dorsomorphin. Bar graph shows the IC50 values of dorsomorphin; hence, its cytotoxic effects, in a variety of UM cell lines after 48 h treatment. IC50 values were estimated by fitting models with nonlinear regression. UM, uveal melanoma.
Discussion
AMPK pathway has been recently implicated in key mechanisms that mediate the survival of UM cells [15]. As such, we performed in silico analyses to compare UM cells with normal melanocytes in terms of AMPK signaling. We demonstrated that genes associated with AMPK signaling pathway are differentially expressed in UM cells compared to normal melanocytes via KEGG analysis. Moreover, gene sets associated with AMPK signaling were shown via GSEA to be differentially expressed in UM cells. Such results confirmed that UM represents a tumor with differential regulation of AMPK signaling. Therefore, we thought that pharmacological targeting of AMPK in UM might hold therapeutic value.
As such, we then determined the effects of AMPK modulation on UM cells by XTT cell viability and proliferation assays after 48 h treatment with varying concentrations of either A-769662 or dorsomorphin. We demonstrated the cytotoxic effects of AMPK inhibition and/or activation in UM cell lines (i.e., 92-1, MP46, OMM2.5, and Mel270), which have different molecular profiles, in vitro. We observed incremental cytotoxic effects on UM cells as the concentration of A-769662 increased. The decrease in UM cell viability may be due to the cessation of cell division caused by the restraining influences of AMPK on growth [82].
Although we observed decreases in cell viabilities as a result of increasing the concentration of the AMPK activator in general, it should also be underlined that there exist significant differences in terms of the responses of different UM cell lines to A-769662 treatment. Such variations may stem from metabolic differences of those UM cell lines. Indeed, in a seminal article by Jager et al. [68], it was reported that UM cell lines could display significant differences. Such variations may result in differences in terms of the cells’ responses to AMPK modulation via other pathways that potentially increase the resistance of tumor cells to AMPK activation. We think that investigating such resistance mechanisms in various UM cell lines in the future via examining the intracellular pathways that are activated as a result of AMPK activation is critical.
Al-Moujahed et al. [83] reported decreases in cell proliferations when Mel270, Mel202, and 92-1 UM cell lines were treated with 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR, an AMPK activator). Moreover, AICAR treatment resulted in inhibition of eukaryotic translation initiation factor 4E-BP1 phosphorylation (a marker for mTOR pathway activity) as well as downregulating cyclins A and D, which is associated with cell cycle arrest at the G1-S phase. AICAR, which is an adenosine analog, provides nonselective AMPK activation [84]. As an AMP analog, AICAR can activate various AMP-dependent enzymes (e.g., fructose-1,6-bisphosphatase) [85]. Thus, inhibition of UM cell growth by AICAR treatment reported by Al-Moujahed et al. [83] may indeed be partially via the activation of AMPK in addition to various other mechanisms. Our current findings support such previous reports since genes involved in AMPK signaling as well as in energy-dependent regulation of mTOR by liver kinase B1-AMPK were found to be downregulated in UM cells compared with normal melanocyte cells, which were also enriched for the expression of PRKAA1. When AICAR is taken up into cells, it is phosphorylated by adenosine kinase, which generates AICAR monophosphate (an AMP-mimetic) [85]. AICAR monophosphate neither alters the ADP:ATP ratio nor changes oxygen uptake; it rather binds to site 3 on the AMPKγ subunit, similar to cellular AMP. However, the effects of AICAR are known to be more apparent in quiescent cells than in rapidly proliferating cells, since AICAR monophosphate is a natural intermediate in the purine nucleotide synthesis [85]. Given those drawbacks associated with AICAR, we decided to use A-769662 in order to directly activate AMPK in our study. Unlike AICAR, A-769662 demonstrates high specificity toward AMPK. In addition, A-769662 allosterically activates the AMPK complex and inhibits dephosphorylation of Thr-172 in AMPKα subunit, similar to AMP [85–87]. In another study, Sevim and Kiratli reported that patients with UM had significantly lower levels of serum adiponectin compared with controls. Moreover, metastatic patients had significantly lower serum levels of adiponectin compared with nonmetastatic patients [88]. Adiponectin is a cytokine that is produced by adipocytes [89]. It activates protein kinase A, which results in increased AMPK activation [89–92]. Consistent with such findings, adiponectin-null mice were found to have diminished AMPK signaling [22, 93]. Given the association of low serum levels of adiponectin with more aggressive clinical course, low serum adiponectin levels may play a role in promoting the growth of UM tumors indirectly via downregulating the AMPK signaling. In another study, inhibition of MEK/ATK pathways was reported to induce activation of AMPK in GNAQ-mutant UM cells [94]. Oncogenic mutations in GNAQ and GNA11 genes are present in 80% of UM, which result in the activation of RAF/MEK signaling pathway [94]. Thus, Ambrosini et al. [94] proposed that AMPK might be a pivotal regulator of mutant GNAQ signaling and a switch between autophagy and apoptosis. Given our current findings and those from previous studies, AMPK activators can be effective in the treatment of UM [83].
On the other hand, AMPK inhibition resulted in more significant cytotoxicity in UM cells, as lower doses of dorsomorphin achieved substantial decreases in cell viabilities of UM cells. We observed significant UM cytotoxicity at dorsomorphin concentrations over 2.5 µm/5 µm. Moreover, dorsomorphin treatment was shown to be capable of achieving potencies that may reach up to 90% cytotoxicity. Dorsomorphin currently represents one of the most commonly used methods as a means to inhibit AMPK; however, it is important to keep in mind some potential off-target effects that may arise when using this compound.
It may seem perplexing to observe that AMPK inhibition also results in UM cell cytotoxicity similar to (or even more potent than) AMPK activation. It is well known that tumor cells establish mechanisms to downregulate AMPK in order for them to escape its restraining influences on growth. Therefore, activation of AMPK as a means of therapeutic strategy sounds reasonable. However, the vital roles of AMPK, which is an evolutionarily conserved serine/threonine kinase, in maintaining cellular energy homeostasis should also be kept in mind [26, 95, 96]. Considering the role and significance of AMPK in cancer cell metabolism, its function is crucial for tumor cell survival as well as proliferation. Therefore, it would be “incompatible with life” for a tumor cell to completely knockout AMPK and it would remain indispensable for the tumor cells despite being downregulated. As such, a critical threshold level of AMPK may be implicated in determining the fate of a given tumor cell. Sub-physiological levels of AMPK expression may help tumor cells to grow; however, decreasing AMPK levels lower than that is essential for life can be deleterious for the tumor cell. This perspective brings forth the idea of a hypothetical therapeutic window for AMPK inhibition, which mandates overcoming a threshold to achieve therapeutic effects. Interestingly, our findings were indeed in agreement with this notion, as we observed sharp decreases in the numbers of viable 92-1, MP46, and Mel270 cells as the concentrations of dorsomorphin exceeded 5 µm, which might represent an arbitrary threshold effect. Our findings demonstrated that only OMM2.5 UM cells demonstrated a type of resistance to dorsomorphin over 5 µm, although 20 µm of dorsomorphin managed to achieve a statistically significant decrease in OMM2.5 cell viability. Indeed, among UM cells, OMM2.5 was found to be the least dependent on AMPK based on the DepMap analyses (Fig. 2d). Given the fact that OMM2.5 cells were not derived from primary tumors (unlike other UM cell lines) and were rather derived from metastases [68], such a predilection by OMM2.5 cells to resist lower levels of AMPK activity seems reasonable. Moreover, the hypothetical arbitrary AMPK threshold may indeed be unique for each and every UM type, which simply does not disprove the concept.
The microphthalmia-associated transcription factor (MITF) is a critical oncogene in melanoma. It is an important regulator of melanogenesis and melanocyte development [97]. MITF was suggested to have a dual role in benign and malignant melanocytic cells since it was proposed to promote local proliferation, while loss of MITF expression could increase invasion [97, 98]. Gelmi et al. [99] recently demonstrated that low MITF expression was associated with inflammatory markers and epithelial-mesenchymal transition. They proposed that MITF loss in UM could be related to de-differentiation to a less favorable epithelial-mesenchymal transition profile [99]. Borgdorff et al. [100] found that AMPK was an important regulator for the maintenance of MITF protein levels in melanocytic cells. Moreover, downregulation of MITF protein levels by AMPK inhibition was reported to be associated with decreased cell viability [100].
AMPK has long been known to display “multifaceted activities” in tumor progression and it can act like a friend or a foe in cancer depending on the context as well as the tumor cell type [101, 102]. As such, it is of utmost importance to investigate such metabolic mechanisms in different types of cells. For this reason, we investigated UM cell lines with different molecular profiles and tissue origins (i.e., 92-1, MP46, OMM2.5, and Mel270). In addition, the role of AMPK cannot be merely identified as either anti- or pro-tumorigenic; it rather appears to have two faces similar to a double-edged sword [40]. In this respect, we aimed to compare the correct strategy for AMPK modulation in terms of UM treatment in four different UM cell lines. To the best of our knowledge, this is the first study that investigates the effects of dual AMPK modulation in UM cells. In line with previous reports, our findings also indicate that the cells with different molecular expression profiles (even of the same tissue origin) may show distinct metabolic activities and varying responses to metabolic manipulations. In this context, the current study contributes to the literature in determining the effects of therapeutic strategies targeting AMPK in UM tumor cells.
Since a limitation of the current study was the absence of investigations concerning the mechanisms that are responsible from the decrease in cell viability, we plan to investigate the molecular mechanisms underlying the effects of AMPK modulation in UM carcinogenesis in future studies via experiments that utilize proteomics or AMPK-knock-down animals [42]. Nevertheless, we propose a new perspective in the treatment of UM. Indeed, targeting the AMPK pathway may open up new avenues in developing novel therapeutic approaches to improve overall survival in UM.
Statement of Ethics
92-1, OMM2.5, Mel270, and MP46 uveal melanoma cell lines used in this study were obtained from Prof. Martine Jager, MD, PhD, at Leiden University. Written informed consent was not required for the use of these cells in accordance with local/national guidelines. The study was conducted ethically in accordance with the World Medical Association Declaration of Helsinki. This study protocol was reviewed and approved by the Non-Interventional Clinical Research Ethics Committee of Hacettepe University (Approval # 2019/23-31).
Conflict of Interest Statement
The authors declare no competing interests.
Funding Sources
This study was supported by the Hacettepe University Scientific Research Projects Coordination Unit (Project # 18446) and the Turkish Ophthalmological Association.
Author Contributions
O.D: performed the experiments and wrote the paper; M.E.G: performed the experiments and analysis of data and wrote the paper; I.K: conception; G.G: conception and design, conduction of the study, analysis and interpretation of data, wrote the paper, and reviewed and edited the manuscript; and H.K: conception and conduction of the study. All authors read and approved the final manuscript.
Funding Statement
This study was supported by the Hacettepe University Scientific Research Projects Coordination Unit (Project # 18446) and the Turkish Ophthalmological Association.
Data Availability Statement
All data generated during this study are included in this article. Further inquiries can be directed to the corresponding authors.
References
- 1. Singh N, Bergman L, Seregard S, Singh AD. Uveal melanoma: epidemiologic aspects. Clinical ophthalmic oncology. Springer; 2014. p. 75–87. [Google Scholar]
- 2. Chang AE, Karnell LH, Menck HR. The national cancer data base report on cutaneous and noncutaneous melanoma: a summary of 84,836 cases from the past decade. The American college of surgeons commission on cancer and the American cancer society. Am Coll Surgeons Comm Cancer Am Cancer Soc Cancer. 1998 Oct 15;83(8):1664–78. . [DOI] [PubMed] [Google Scholar]
- 3. Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973-1997. Ophthalmology. 2003 May;110(5):956–61. 10.1016/S0161-6420(03)00078-2. [DOI] [PubMed] [Google Scholar]
- 4. Virgili G, Gatta G, Ciccolallo L, Capocaccia R, Biggeri A, Crocetti E, et al. Incidence of uveal melanoma in Europe. Ophthalmology. 2007 Dec;114(12):2309–15. 10.1016/j.ophtha.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 5. Mahendraraj K, Lau CS, Lee I, Chamberlain RS. Trends in incidence, survival, and management of uveal melanoma: a population-based study of 7,516 patients from the Surveillance, Epidemiology, and End Results database (1973–2012). Clin Ophthalmol. 2016;10:2113–9. 10.2147/OPTH.S113623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kiratli H, Yilmaz PT, Sargon M. Ultrastructural alterations in extraocular muscles following iodine-125 brachytherapy for uveal melanoma. Strabismus. 2007 Apr–Jun;15(2):103–9. 10.1080/09273970701431368. [DOI] [PubMed] [Google Scholar]
- 7. Yazici G, Kiratli H, Ozyigit G, Sari SY, Cengiz M, Tarlan B, et al. Stereotactic radiosurgery and fractionated stereotactic radiation therapy for the treatment of uveal melanoma. Int J Radiat Oncol Biol Phys. 2017 May 1;98(1):152–8. 10.1016/j.ijrobp.2017.02.017. [DOI] [PubMed] [Google Scholar]
- 8. Kiratli H, Koç İ. Orbital exenteration: institutional review of evolving trends in indications and rehabilitation techniques. Orbit. 2018 Jun;37(3):179–86. 10.1080/01676830.2017.1383466. [DOI] [PubMed] [Google Scholar]
- 9. Koç İ, Kıratlı H. Current management of conjunctival melanoma Part 2: treatment and future directions. Turk J Ophthalmol. 2020 Dec 29;50(6):362–70. 10.4274/tjo.galenos.2020.22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chua V, Mattei J, Han A, Johnston L, LiPira K, Selig SM, et al. The latest on uveal melanoma Research and clinical trials: updates from the cure ocular melanoma (CURE OM) science meeting. Clin Cancer Res. 2021 Jan 1;27(1):28–33. 10.1158/1078-0432.CCR-20-2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gunaydin G, Gedik ME, Ayan S. Photodynamic therapy for the treatment and diagnosis of cancer-A review of the current clinical status. Front Chem. 2021;9:686303. 10.3389/fchem.2021.686303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang JZ, Lin V, Toumi E, Wang K, Zhu H, Conway RM, et al. Development of new therapeutic options for the treatment of uveal melanoma. FEBS J. 2021 Nov;288(21):6226–49. 10.1111/febs.15869. [DOI] [PubMed] [Google Scholar]
- 13. Wei AZ, Maniar AB, Carvajal RD. New targeted and epigenetic therapeutic strategies for the treatment of uveal melanoma. Cancer Gene Ther. 2022 Dec;29(12):1819–26. 10.1038/s41417-022-00443-8. [DOI] [PubMed] [Google Scholar]
- 14. Yazici G, Kiratli H, Ozyigit G, Sari SY, Elmali A, Yilmaz MT, et al. Every other day stereotactic radiation therapy for the treatment of uveal melanoma decreases toxicity. Radiother Oncol. 2022 Nov;176:39–45. 10.1016/j.radonc.2022.09.010. [DOI] [PubMed] [Google Scholar]
- 15. Chua V, Han A, Bechtel N, Purwin TJ, Hunter E, Liao C, et al. The AMP-dependent kinase pathway is upregulated in BAP1 mutant uveal melanoma. Pigment Cell Melanoma Res. 2022 Jan;35(1):78–87. 10.1111/pcmr.13007. [DOI] [PubMed] [Google Scholar]
- 16. Seddon JM, Gragoudas ES, Egan KM, Glynn RJ, Howard S, Fante RG, et al. Relative survival rates after alternative therapies for uveal melanoma. Ophthalmology. 1990 Jun;97(6):769–77. 10.1016/s0161-6420(90)32512-5. [DOI] [PubMed] [Google Scholar]
- 17. Hawkins BS; Collaborative Ocular Melanoma Study Group . The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma: IV. Ten-year mortality findings and prognostic factors. COMS report number 24. Am J Ophthalmol. 2004 Dec;138(6):936–51. 10.1016/j.ajo.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 18. Diener-West M, Reynolds SM, Agugliaro DJ, Caldwell R, Cumming K, Earle JD, et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: collaborative Ocular Melanoma Study Group Report No. 26. Arch Ophthalmol. 2005 Dec;123(12):1639–43. 10.1001/archopht.123.12.1639. [DOI] [PubMed] [Google Scholar]
- 19. Augsburger JJ, Correa ZM, Shaikh AH. Effectiveness of treatments for metastatic uveal melanoma. Am J Ophthalmol. 2009 Jul;148(1):119–27. 10.1016/j.ajo.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 20. Koopmans AE, Klein A, Naus NC, Kilic E. Diagnosis and management of uveal melanoma. Journal-Diagnosis Management Uveal Melanoma. 2013;07(01):56. 10.17925/eor.2013.07.01.56. [DOI] [Google Scholar]
- 21. Violanti SS, Bononi I, Gallenga CE, Martini F, Tognon M, Perri P. New insights into molecular oncogenesis and therapy of uveal melanoma. Cancers. 2019;11(5):694. 10.3390/cancers11050694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009 Jul;89(3):1025–78. 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 23. Rajamohan F, Reyes AR, Frisbie RK, Hoth LR, Sahasrabudhe P, Magyar R, et al. Probing the enzyme kinetics, allosteric modulation and activation of α1- and α2-subunit-containing AMP-activated protein kinase (AMPK) heterotrimeric complexes by pharmacological and physiological activators. Biochem J. 2016 Mar 1;473(5):581–92. 10.1042/BJ20151051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yahsi B, Gunaydin G. Immunometabolism: the role of branched-chain amino acids. Front Immunol. 2022;13:886822. 10.3389/fimmu.2022.886822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yazici SE, Gedik ME, Leblebici CB, Kosemehmetoglu K, Gunaydin G, Dogrul AB. Can endocan serve as a molecular “hepatostat” in liver regeneration? Mol Med. 2023 Feb 27;29(1):29. 10.1186/s10020-023-00622-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016 Jul 15;48(7):e245. 10.1038/emm.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Z, Wang N, Liu P, Xie X. AMPK and cancer. AMP-activated protein kinase. Springer; 2016. p. 203–26. [Google Scholar]
- 28. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011 Sep 2;13(9):1016–23. 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005 Apr 29;18(3):283–93. 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 30. Motoshima H, Goldstein BJ, Igata M, Araki E. AMPK and cell proliferation–AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol. 2006;574(Pt 1):63–71. 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cerezo M, Tichet M, Abbe P, Ohanna M, Lehraiki A, Rouaud F, et al. Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol Cancer Ther. 2013 Aug;12(8):1605–15. 10.1158/1535-7163.MCT-12-1226-T. [DOI] [PubMed] [Google Scholar]
- 32. Gunaydin G, Altundag K. Ductal carcinoma in situ and bilateral atypical ductal hyperplasia in a 23-year-old man with gynecomastia. Am Surg. 2011 Sep;77(9):1272–3. 10.1177/000313481107700945. [DOI] [PubMed] [Google Scholar]
- 33. Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 2015 Jan 28;356(2 Pt A):165–70. 10.1016/j.canlet.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 34. Gok Yavuz B, Gunaydin G, Kosemehmetoglu K, Karakoc D, Ozgur F, Guc D. The effects of cancer-associated fibroblasts obtained from atypical ductal hyperplasia on anti-tumor immune responses. Breast J. 2018 Nov;24(6):1099–101. 10.1111/tbj.13139. [DOI] [PubMed] [Google Scholar]
- 35. Esim O, Gedik ME, Dogan AL, Gunaydin G, Hascicek C. Development of carboplatin loaded bovine serum albumin nanoparticles and evaluation of its effect on an ovarian cancer cell line. J Drug Deliv Sci Technol. 2021;64:102655. 10.1016/j.jddst.2021.102655. [DOI] [Google Scholar]
- 36. Esim O, Hascicek C, Gedik ME, Gunaydin G, Dogan AL. Carboplatin and decitabine loaded lipid-coated albumin nanoparticles for an efficient treatment of platinum-resistant ovarian cancer. J Drug Deliv Sci Technol. 2022;76:103801. 10.1016/j.jddst.2022.103801. [DOI] [Google Scholar]
- 37. Ozturk Gunduz E, Atajanov R, Gedik ME, Tanrıverdi Eçik E, Günaydın G, Okutan E. BODIPY-GO nanocomposites decorated with a biocompatible branched ethylene glycol moiety for targeted PDT. Dalton Trans. 2023 May 2;52(17):5466–77. 10.1039/d2dt04013a. [DOI] [PubMed] [Google Scholar]
- 38. Ozturk Gunduz E, Tasasız B, Gedik ME, Günaydın G, Okutan E. NI-BODIPY-GO nanocomposites for targeted PDT. ACS Omega. 2023 Mar 7;8(9):8320–31. 10.1021/acsomega.2c06900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pamuk AE, Gedik ME, Sutay Suslu N, Gunaydin G. Candidate angiogenesis-related biomarkers in patients with laryngeal carcinoma (AngLaC): a prospective cohort study. Otolaryngol Head Neck Surg. 2023 Jun;168(6):1433–42. 10.1002/ohn.219. [DOI] [PubMed] [Google Scholar]
- 40. Jeon SM, Hay N. The double-edged sword of AMPK signaling in cancer and its therapeutic implications. Arch Pharm Res (Seoul). 2015 Mar;38(3):346–57. 10.1007/s12272-015-0549-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ríos M, Foretz M, Viollet B, Prieto A, Fraga M, Costoya JA, et al. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res. 2013;73(8):2628–38. 10.1158/0008-5472.CAN-12-0861. [DOI] [PubMed] [Google Scholar]
- 42. Günaydın G, Gedik ME. Meme kanseri ve hepatosellüler kanser hücre dizilerinde AMPK modülasyonunun kanser hücre proliferasyonu üzerine etkisinin gerçek-zamanlı hücre analiz sistemi (xCelligence) aracılığıyla i̇ncelenmesi. Bozok Tıp Dergisi. 10.16919/bozoktip.434074. [DOI] [Google Scholar]
- 43. Shaw RJ. AMPK keeps tumor cells from starving to death. Cell Stem Cell. 2015 Nov 5;17(5):503–4. 10.1016/j.stem.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 44. Gunaydin G, Gedik ME. Effects of cellular energy homeostasis modulation through AMPK on regulation of protein translation and response to hypoxia. Turk J Biochem. 2019;44(5):611–20. 10.1515/tjb-2018-0338. [DOI] [Google Scholar]
- 45. Beduk Esen CS, Gedik ME, Canpinar H, Yedekci FY, Yildiz F, Gunaydin G, et al. Radiosensitising effects of metformin added to concomitant chemoradiotherapy with cisplatin in cervical cancer. Clin Oncol. 2023. 10.1016/j.clon.2023.08.007. [DOI] [PubMed] [Google Scholar]
- 46. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002 Jan 1;30(1):207–10. 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013 Jan;41(Database issue):D991–5. 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018 Jul 2;46(W1):W537–44. 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010 Mar 1;26(5):589–95. 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014 Apr 1;30(7):923–30. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 51. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000 Jan 1;28(1):27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005 Oct 25;102(43):15545–50. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011 Jun 15;27(12):1739–40. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013 Jul 15;29(14):1830–1. 10.1093/bioinformatics/btt285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Batut B, Hiltemann S, Bagnacani A, Baker D, Bhardwaj V, Blank C, et al. Community-driven data analysis training for biology. Cell Syst. 2018 Jun 27;6(6):752–8.e1. 10.1016/j.cels.2018.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chang WH, Lai AG. An integrative pan-cancer investigation reveals common genetic and transcriptional alterations of AMPK pathway genes as important predictors of clinical outcomes across major cancer types. BMC Cancer. 2020 Aug 17;20(1):773. 10.1186/s12885-020-07286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012 Mar 28;483(7391):603–7. 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017 Dec;49(12):1779–84. 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dempster JM, Boyle I, Vazquez F, Root DE, Boehm JS, Hahn WC, et al. Chronos: a cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021 Dec 20;22(1):343. 10.1186/s13059-021-02540-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li F, Lai L, You Z, Cheng H, Guo G, Tang C, et al. Identification of UBE2I as a novel biomarker in ccRCC based on a large-scale CRISPR-cas9 screening database and immunohistochemistry. Front Mol Biosci. 2022;9:813428. 10.3389/fmolb.2022.813428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, et al. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem. 2007 Nov 9;282(45):32549–60. 10.1074/jbc.M706536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Qin Y, Sekine I, Hanazono M, Morinaga T, Fan M, Takiguchi Y, et al. AMPK activation induced in pemetrexed-treated cells is associated with development of drug resistance independently of target enzyme expression. Mol Oncol. 2019 Jun;13(6):1419–32. 10.1002/1878-0261.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gao Y, Yan Y, Tripathi S, Pentinmikko N, Amaral A, Paivinen P, et al. LKB1 represses ATOH1 via PDK4 and energy metabolism and regulates intestinal stem cell fate. Gastroenterology. 2020 Apr;158(5):1389–401.e10. 10.1053/j.gastro.2019.12.033. [DOI] [PubMed] [Google Scholar]
- 65. Hanada Y, Ishihara N, Wang L, Otera H, Ishihara T, Koshiba T, et al. MAVS is energized by Mff which senses mitochondrial metabolism via AMPK for acute antiviral immunity. Nat Commun. 2020 Nov 11;11(1):5711. 10.1038/s41467-020-19287-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. De Waard-Siebinga I, Blom DJ, Griffioen M, Schrier PI, Hoogendoorn E, Beverstock G, et al. Establishment and characterization of an uveal-melanoma cell line. Int J Cancer. 1995 Jul 17;62(2):155–61. 10.1002/ijc.2910620208. [DOI] [PubMed] [Google Scholar]
- 67. Chen PW, Murray TG, Uno T, Salgaller ML, Reddy R, Ksander BR. Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. Clin Exp Metastasis. 1997 Sep;15(5):509–18. 10.1023/a:1018479011340. [DOI] [PubMed] [Google Scholar]
- 68. Jager MJ, Magner JA, Ksander BR, Dubovy SR. Uveal melanoma cell lines: where do they come from? (An American ophthalmological society thesis). Trans Am Ophthalmol Soc. 2016 Aug;114:T5. [PMC free article] [PubMed] [Google Scholar]
- 69. Amirouchene-Angelozzi N, Nemati F, Gentien D, Nicolas A, Dumont A, Carita G, et al. Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target. Mol Oncol. 2014 Dec;8(8):1508–20. 10.1016/j.molonc.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gunaydin G, Kesikli SA, Guc D. Cancer associated fibroblasts have phenotypic and functional characteristics similar to the fibrocytes that represent a novel MDSC subset. Oncoimmunology. 2015 Sep;4(9):e1034918. 10.1080/2162402X.2015.1034918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015 Dec 23;1(6):417–25. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017 Apr 6;169(2):361–71. 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 73. Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A, Weber MJ. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998 Sep 11;281(5383):1668–71. 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- 74. Wunderlich W, Fialka I, Teis D, Alpi A, Pfeifer A, Parton RG, et al. A novel 14-kilodalton protein interacts with the mitogen-activated protein kinase scaffold mp1 on a late endosomal/lysosomal compartment. J Cell Biol. 2001 Feb 19;152(4):765–76. 10.1083/jcb.152.4.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nada S, Hondo A, Kasai A, Koike M, Saito K, Uchiyama Y, et al. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J. 2009 Mar 4;28(5):477–89. 10.1038/emboj.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012 Sep 14;150(6):1196–208. 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lamberti G, De Smet CH, Angelova M, Kremser L, Taub N, Herrmann C, et al. LAMTOR/Ragulator regulates lipid metabolism in macrophages and foam cell differentiation. FEBS Lett. 2020 Jan;594(1):31–42. 10.1002/1873-3468.13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Filipek PA, de Araujo MEG, Vogel GF, De Smet CH, Eberharter D, Rebsamen M, et al. LAMTOR/Ragulator is a negative regulator of Arl8b- and BORC-dependent late endosomal positioning. J Cell Biol. 2017 Dec 4;216(12):4199–215. 10.1083/jcb.201703061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Riviere P, Goodman AM, Okamura R, Barkauskas DA, Whitchurch TJ, Lee S, et al. High tumor mutational burden correlates with longer survival in immunotherapy-naive patients with diverse cancers. Mol Cancer Ther. 2020 Oct;19(10):2139–45. 10.1158/1535-7163.MCT-20-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cakan E, Gunaydin G. Activation induced cytidine deaminase: an old friend with new faces. Front Immunol. 2022;13:965312. 10.3389/fimmu.2022.965312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Laurent C, Valet F, Planque N, Silveri L, Maacha S, Anezo O, et al. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Cancer Res. 2011 Feb 1;71(3):666–74. 10.1158/0008-5472.CAN-10-0605. [DOI] [PubMed] [Google Scholar]
- 82. Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011 Sep 15;25(18):1895–908. 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Al-Moujahed A, Nicolaou F, Brodowska K, Papakostas TD, Marmalidou A, Ksander BR, et al. Uveal melanoma cell growth is inhibited by aminoimidazole carboxamide ribonucleotide (AICAR) partially through activation of AMP-dependent kinase. Invest Ophthalmol Vis Sci. 2014;55(7):4175–85. 10.1167/iovs.13-12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LM, et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci U S A. 2014 Jan 28;111(4):E435–44. 10.1073/pnas.1311121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kim J, Yang G, Kim Y, Kim J, Ha J. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med. 2016 Apr 1;48(4):e224. 10.1038/emm.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Moreno D, Knecht E, Viollet B, Sanz P. A769662, a novel activator of AMP-activated protein kinase, inhibits non-proteolytic components of the 26S proteasome by an AMPK-independent mechanism. FEBS Lett. 2008 Jul 23;582(17):2650–4. 10.1016/j.febslet.2008.06.044. [DOI] [PubMed] [Google Scholar]
- 87. Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol. 2008 Nov 24;15(11):1220–30. 10.1016/j.chembiol.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 88. Sevim DG, Kiratli H. Serum adiponectin, insulin resistance, and uveal melanoma: clinicopathological correlations. Melanoma Res. 2016 Apr;26(2):164–72. 10.1097/CMR.0000000000000226. [DOI] [PubMed] [Google Scholar]
- 89. Medina EA, Oberheu K, Polusani SR, Ortega V, Velagaleti GV, Oyajobi BO. PKA/AMPK signaling in relation to adiponectin’s antiproliferative effect on multiple myeloma cells. Leukemia. 2014 Oct;28(10):2080–9. 10.1038/leu.2014.112. [DOI] [PubMed] [Google Scholar]
- 90. Tomas E, Tsao TS, Saha AK, Murrey HE, Zhang C, Itani SI, et al. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc Natl Acad Sci U S A. 2002 Dec 10;99(25):16309–13. 10.1073/pnas.222657499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002 Nov;8(11):1288–95. 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 92. Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007 Mar;13(3):332–9. 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- 93. Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med. 2004 Dec;10(12):1384–9. 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ambrosini G, Musi E, Ho AL, de Stanchina E, Schwartz GK. Inhibition of mutant GNAQ signaling in uveal melanoma induces AMPK-dependent autophagic cell death. Mol Cancer Ther. 2013 May;12(5):768–76. 10.1158/1535-7163.MCT-12-1020. [DOI] [PubMed] [Google Scholar]
- 95. Roustan V, Jain A, Teige M, Ebersberger I, Weckwerth W. An evolutionary perspective of AMPK-TOR signaling in the three domains of life. J Exp Bot. 2016 Jun;67(13):3897–907. 10.1093/jxb/erw211. [DOI] [PubMed] [Google Scholar]
- 96. Jain A, Roustan V, Weckwerth W, Ebersberger I. Studying AMPK in an evolutionary context. Methods Mol Biol. 2018;1732:111–42. 10.1007/978-1-4939-7598-3_8. [DOI] [PubMed] [Google Scholar]
- 97. Gelmi MC, Houtzagers LE, Strub T, Krossa I, Jager MJ. MITF in normal melanocytes, cutaneous and uveal melanoma: a delicate balance. Int J Mol Sci. 2022 May 26(11):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gelmi M, Marinkovic M, Vu THK, van der Velden PA, Luyten GPM, Jager MJ. MITF: a progression marker in uveal melanoma. Invest Ophthalmol Vis Sci. 2022;63(7):2353–A0022. [Google Scholar]
- 99. Gelmi MC, Verdijk RM, Houtzagers LE, van der Velden PA, Kroes WGM, Luyten GPM, et al. Microphthalmia-associated transcription factor: a differentiation marker in uveal melanoma. Int J Mol Sci. 2023 May 16;24(10):8861. 10.3390/ijms24108861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Borgdorff V, Rix U, Winter G, Gridling M, Müller A, Breitwieser F, et al. A chemical biology approach identifies AMPK as a modulator of melanoma oncogene MITF. Oncogene. 2014;33(19):2531–9. 10.1038/onc.2013.185. [DOI] [PubMed] [Google Scholar]
- 101. Bonini MG, Gantner BN. The multifaceted activities of AMPK in tumor progression--why the “one size fits all” definition does not fit at all? IUBMB Life. 2013 Nov;65(11):889–96. 10.1002/iub.1213. [DOI] [PubMed] [Google Scholar]
- 102. Hardie DG. Molecular pathways: is AMPK a friend or a foe in cancer? Clin Cancer Res. 2015 Sep 1;21(17):3836–40. 10.1158/1078-0432.CCR-14-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated during this study are included in this article. Further inquiries can be directed to the corresponding authors.