

Abstract
Neurodegenerative diseases are characterized by a dysregulated neuro-glial microenvironment, culminating in functional deficits resulting from neuronal cell death. Inflammation is a hallmark of the neurodegenerative microenvironment and despite a critical role in tissue homeostasis, increasing evidence suggests that chronic inflammatory insult can contribute to progressive neuronal loss. Inflammation has been studied in the context of neurodegenerative disorders for decades but few anti-inflammatory treatments have advanced to clinical use. This is likely due to the related challenges of predicting and mitigating off-target effects impacting the normal immune response while detecting inflammatory signatures that are specific to the progression of neurological disorders. Inflammasomes are pro-inflammatory cytosolic pattern recognition receptors functioning in the innate immune system. Compelling pre-clinical data has prompted an intense interest in the role of the NLR family pyrin domain containing 3 (NLRP3) inflammasome in neurodegenerative disease. NLRP3 is typically inactive but can respond to sterile triggers commonly associated with neurodegenerative disorders including protein misfolding and aggregation, mitochondrial and oxidative stress, and exposure to disease-associated environmental toxicants. Clear evidence of enhanced NLRP3 inflammasome activity in common neurodegenerative diseases has coincided with rapid advancement of novel small molecule therapeutics making the NLRP3 inflammasome an attractive target for near-term interventional studies. In this review, we highlight evidence from model systems and patients indicating inflammasome activity in neurodegenerative disease associated with the NLRP3 inflammasome’s ability to recognize pathologic forms of amyloidβ, tau, and α-synuclein. We discuss inflammasome-driven pyroptotic processes highlighting the potential utility of evaluating extracellular inflammasome-related proteins in the context of biomarker discovery. We complete the report by pointing out gaps in our understanding of intracellular modifiers of inflammasome activity and mechanisms regulating the resolution of inflammasome activation. The literature review and perspectives provide a conceptual platform for continued analysis of inflammation in neurodegenerative diseases through the study of inflammasomes and pyroptosis, mechanisms of inflammation and cell death now recognized to function in multiple highly prevalent neurological disorders.
Background and Canonical Mechanisms
NLR Family Pyrin Domain Containing 3 (NLRP3) was first identified as a 514 base-pair human gene similar to the rat angiotensin/vasopressin receptor in CD34+ hematopoietic stem-like cells.1 Subsequently, genetic studies identified a shared linkage at 1q44 in 2 independently hereditable autoinflammatory disorders, Muckle-Wells Syndrome,2 and familial cold autoinflammatory syndrome.3 Based on these linkage studies, Hoffman and others used positional cloning to identify mutations in NLRP3 (originally referred to as CIAS1).4 These seminal discoveries expanded our understanding of the Cryopyrin-associated periodic syndromes (CAPS) and provided the initial characterization of NLRP3 as a mediator of inflammation and cell death. Over 2 decades, thousands of reports have detailed mechanisms of NLRP3 inflammasome signaling in health and disease. Among them are many outstanding reviews.5–10 Here we will provide a brief overview of canonical inflammasome signaling to contextualize mechanisms of translational interest in neurodegenerative disease which we discuss in more detail throughout the review.
The NLRP3 inflammasome exists among multiple families of inflammasomes that include the nucleotide-binding domain-like receptors (NLRs), absent in melanoma 2-like receptors (ALRs), and the pyrin family, each with distinct properties and abilities to respond to unique molecular patterns to initiate formation of a multi-protein inflammasome complex and a pro-inflammatory phenotype. The NLRP3 inflammasome is capable of responding to pathogen-associated molecular patterns (PAMPs). Microbial triggers range from bacterial pathogens11 to the hyperinflammatory SARS-CoV-2 N protein.12 Interest in the NLRP3 inflammasome in the context of neurodegenerative disorders however likely stems from its ability to respond to endogenous damage-associated molecular patterns (DAMPs). As discussed throughout this review, significant evidence has been generated that the NLRP3 inflammasome is activated by multiple danger signals associated with neurodegeneration. These include normal aging, debris from dying cells, protein aggregates, lysosomal dysfunction, and metabolic stress.
Similar to many inflammasomes, the NLRP3 inflammasome is activated by a “two-hit” mechanism involving priming and stimulus.13 Canonical priming is thought to occur after an initial exposure to certain exogenous stimuli, for example lipopolysaccharide (LPS) or other bacterial components, resulting in toll-like receptor (TLR) ligand-receptor interaction. This triggers NF-KB-mediated transcriptional upregulation of inflammasome components and targets, including NLRP3 itself, and the downstream target IL1B.14 Subsequent exposure to an inflammatory stimulus activates the NLRP3 inflammasome and the formation of the multi-protein inflammasome complex. The NLRP3 inflammasome complex includes the sensor protein NLRP3 , the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD), the inactive zymogen pro-caspase-1, and often the NIMA-related kinase NEK7.15 Formation of the NLRP3 complex drives the auto-catalysis of pro-caspase 1 generating the components of mature caspase-1. Active caspase-1 then processes targets including the pro-inflammatory cytokines IL-1B and IL-18 (Fig 1). Active caspase-1 can also cleave the protein Gasdermin D (GSDMD), the effector of pyroptotic cell death. Once cleaved, the N-terminal fragments of GSDMD are inserted into the plasma membrane whereby oligomeric pores are formed. These pores are important for the secretion of mature IL-1B and IL-18, ionic/osmotic flux, and cellular swelling.16,17 Pyroptosis is also characterized by vesicular shedding, hypothesized as a means of inter-cellular communication and as a mechanism to remove the harmful pores from the plasma membrane. However, if unresolved, the cell will undergo a lytic, pro-inflammatory demise, releasing all intracellular components into the extracellular environment (Fig 1). The potential to use proteins released during the pyroptotic process as biomarkers of disease is discussed is greater detail later in this review.
Fig 1.
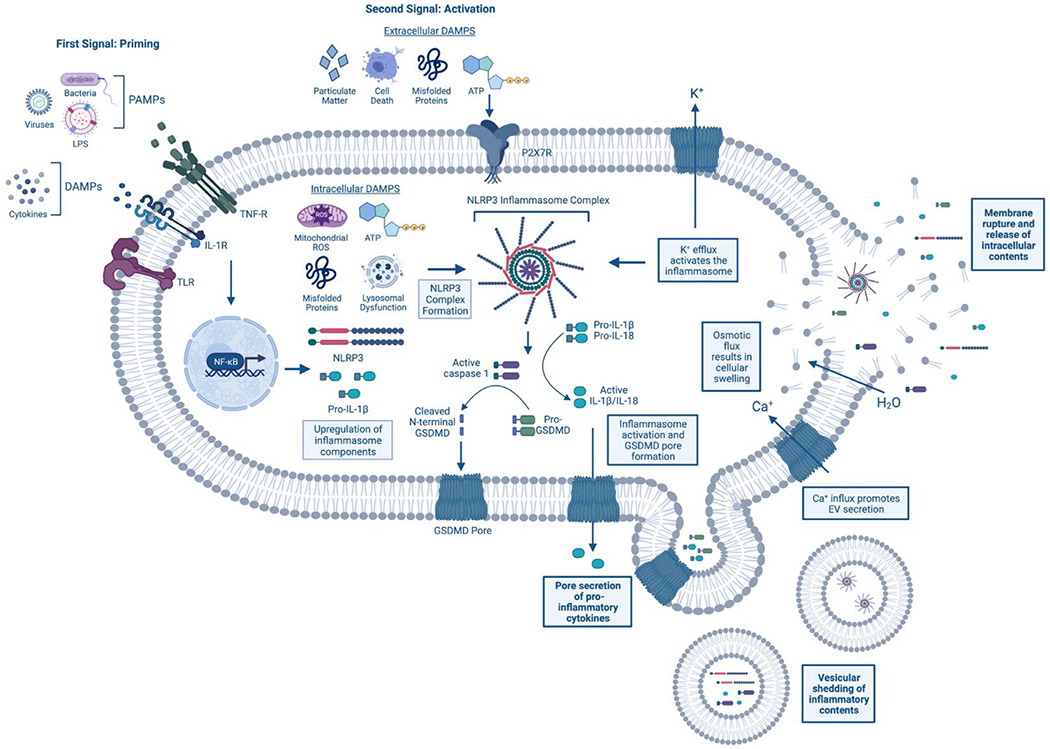
Mechanism of inflammasome activation and pyroptosis. The NLRP3 inflammasome is activated in a 2-step fashion. First, priming occurs at surface membrane receptors. This can include recognition of pathogen-associated molecular patterns (PAMPS) (eg, viruses and bacteria) at Toll-like receptors (TLRs) and/or danger-associated molecular patterns (DAMPs) (eg, cytokines) at the IL-1 receptor (IL-1R) or TNF-a receptor (TNF-R) for example. Following receptor activation, the transcription factor NF-KB translocates to the nucleus where it causes upregulation of inflammasome components, such as NLRP3 and pro-IL-1B. A secondary stimulus is necessary to cause inflammasome complex formation and activation. This second trigger can be either intra- and extra-cellular. Some common stimuli include both intra- and extra-cellular ATP and misfolded proteins, extracellular particulate matter (eg, silica, uric acid crystals) and components of cellular demise, and intracellular mitochondrial reactive oxygen species (ROS) and lysosomal dysfunction. After NLRP3 inflammasome complex formation, the pro-caspase-1 enzyme is cleaved into its active, mature form and can cleave both pro-inflammatory cytokines, IL-1B and IL-18, and the effector protein of pyroptosis, Gasdermin-D (GSDMD). Once cleaved, the N-terminal fragments of GSDMD form pores in the plasma membrane allowing for the extracellular release of the mature cytokines IL-1B and IL-18. Further, these pores promote osmotic and ionic flux, resulting in cellular swelling and the efflux of potassium (K+) and the influx of calcium (Ca2+). In a feed-forward mechanism, K+ efflux causes further inflammasome activation, while Ca2+ influx promotes the release of extracellular vesicles (EVs). Pyroptotic vesicular shedding results in the release of membrane-bound inflammatory proteins, including cytokines, inflammasome components, and even fully formed inflammasome complexes. It is thought that the release of these EVs is a means to resolve the GSDMD pore formation through their budding off the membrane. If unresolved, the osmotic flux and cellular swelling can cause the cell to burst, resulting in the pro-inflammatory release of the cell’s intracellular contents into the extracellular environment and cellular death. Created with BioRender.com. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Mechanistic questions remain related to the adaptability of the priming and stimulus model to explain NLRP3 inflammasome activation in non-microbial neurodegenerative disorders. A significant body of evidence delineates non-microbial or “sterile” induction of NLRP3 inflammasome activity, demonstrating that non-pathogenic danger signals are sufficient to prime the inflammasome.18 Reactive oxygen species (ROS) are by-products of mitochondrial oxidative metabolism, which in excess, can lead to oxidative stress and damage to structural molecules of the cell.19 Although classically thought of as an activator of the inflammasome,20 it has been shown that ROS may play a role in its priming as well. NLRP3 expression was decreased in mouse macrophages treated with a ROS scavenger, diphenyliodonium (DPI), and downstream targets, IL-1B and IL-18 were decreased only when macrophages were treated with DPI during the priming step, but not the activation step.21 Other sterile inflammatory triggers, including hypoxia,22,23 tricarboxylic acid (TCA) cycle metabolite itaconate,24 regulators of fatty acid synthesis25 cholesterol metabolism,26 and components of the complement system,27 have been shown to prime the NLRP3 inflammasome for activation. These data have major implications for neurodegenerative diseases where NLRP3 activation and inflammation occur seemingly without a pathogenic trigger. Interestingly, even man-made toxicants have been shown sufficient to drive NLRP3 priming, with rotenone, a mitochondrial respiratory chain inhibitor, exposure eliciting NLRP3 priming in bone-marrow derived macrophages (BMDMs).28 The NLRP3 inflammasome may provide an interesting mechanism by which environmental toxicant exposure may elicit neuroinflammation in neurodegenerative diseases.29 In addition, NLRP3 enters a hypersensitive state reminiscent of priming as a result of normal aging. This hypersensitive state has implicated NLRP3 as a key mediator of “inflammaging” (for review30,31). Once primed, the NLRP3 inflammasome requires a “second-hit” to be activated and the protein complex formed. Many triggers commonly seen in neurodegenerative-diseased brain, including misfolded proteins,32,33 extracellular ATP typically caused by nearby cell death,34,35 and lysosomal dysfunction36 are sufficient to activate the NLRP3 inflammasome. Although most work surrounding ATP and NLRP3 are in the context of extracellular ATP activating the inflammasome via the P2×7R, intracellular ATP has also been shown to mediate inflammasome activation.37,38 NLRP3 activity is now recognized in a host of sterile inflammatory disorders, including metabolic disorders, monogenic autoinflammatory disorders, and neuroinflammatory diseases that identify non-pathogenic mechanisms sufficient to drive NLRP3 inflammasome activation.18 Here, we highlight mechanisms of NLRP3 inflammasome activation identified in neurodegenerative disease.
Evidence of Inflammasome Activity in Neurodegenerative Disease
NLRP3 in Alzheimer’s disease
Alzheimer’s disease (AD) is the most common neurodegenerative disorder, affecting 5.8 million Americans 65 years or older today and is ranked the 6th leading cause of death in the US.39 Although age is the number 1 risk factor for the disease, other possibly modifiable lifestyle factors have been linked to the risk of developing the disorder including general vascular health, educational attainment, mental and physical activity, and depression.40 Symptomatically, AD is characterized by memory loss, executive dysfunction, and depression, with more advanced disease presenting with impaired communication, confusion, behavior changes, and difficulty performing everyday tasks.39 Loss of neurons within the hippocampus and neocortical areas resulting in brain atrophy are believed to underlie the clinical manifestations of AD.41 AD is characterized post-mortem by 2 histopathologic hallmarks: 1) extracellular amyloid beta (Aβ) plaques and 2) intracellular neurofibrillary tau tangles containing hyperphosphorylated tau.42 Questions related to toxicity, causality, and therapeutic relevance of the abnormal accumulations of protein observed in AD continue to fuel research efforts. Identification of mutations in amyloid precursor protein (APP), the gene encoding for Aβ itself, and in presenilin 1 (PSEN1/PS1) and 2 (PSEN2/PS2), 2 genes that encode proteins involved in the processing of Aβ, are the only genetic mutations that lead to the autosomal dominant form of AD, called familial AD (fAD). This genetic linkage has been used as evidence that aberrant protein aggregation is indeed a key driver of AD incidence and progression.43–45
The notion that NLRP3 was related to neurodegenerative disease was accelerated by Halle et al. who identified NLRP3 as a sensor of the fibrillar form of Aβ in microglia.36 Aβ oligomers and protofibrils were later shown to activate the inflammasome suggesting that the NLRP3 inflammasome may be a driver of early-stage disease progression.32 Heneka et al. reported caspase 1 activity in post-mortem brain tissue obtained from patients with mild cognitive impairment (MCI) and AD highlighting the importance of inflammasome signaling in both the early and late stages of AD pathology.46 The same group demonstrated that ipsilateral injection of brain homogenates from transgenic mice expressing chimeric mouse/human APP and mutant human PS1 (APP/PS1) increased the number of Aβ immunoreactive deposits compared to the contralateral injection of homogenates derived from wild-type mice in an Nlrp3-dependent manner.47 ASC specks were also found to bind Aβ and enhance Aβ aggregation48 and a synergistic effect of Aβ and ASC fibril formation exacerbated NLRP3 inflammasome activation and pyroptotic cell death in primary microglia.49 ASC cross seeding of amyloids such as Aβ, tau, alpha synuclein, may be a common mechanism by which NLRP3 activation may accelerate disease progression and neurotoxicity and presents a candidate common target for therapeutic intervention possibly via targeting of aggregated ASC with antibody-based therapy. The NLRP3 inflammasome has also been implicated in tau pathology both in AD and primary tauopathies where tau aggregation, but not Aβ aggregation, is the primary pathological hallmark.50 Injection of APP/PS1 brain homogenate induced tau hyperphosphorylation in the Tau22 mouse model, but not in Tau22/Asc−/− or Tau22/Nlrp3−/− mice indicating a requirement for NLRP3 activity in the Aβ-Tau cascade.47 K18 tau fibrils, a fragment containing the central, amyloidogenic, region of tau activates the inflammasome and inhibition of NLRP3 is protective in an in vivo tau seeding model utilizing these aggregated tau fragments.51 Hyperphosphorylated tau derived from neuronal cultures and mouse or human brains can prime and activate the inflammasome in an ASC-dependent manner and the suppression of ASC in microglia improves cognition in the hTau mouse model of primary tauopathy.52 The exact mechanism by which Aβ activates the inflammasome is unclear and there are likely several potential pathways. TLR4 ligation by Aβ has been shown to be a mechanism by which the inflammasome is activated, although this study pretreated cells with LPS which also binds to TL4 as its primary receptor making precise conclusions difficult.53 Conflicting reports suggest P2×7R receptor either mediates54 or has no effect55 on Aβ mediated activation of the NLRP3 inflammasome. Tau most likely primes the inflammasome through TLR interaction based on the key role of MyD88, a TLR adaptor protein, in mediating NF-κβ induced expression of inflammasome related genes,52 although the exact TLR has not been elucidated. The production of ROS and lysosomal rupture induced by aggregates have also been proposed as mechanisms of inflammasome activation in response to tau and Aβ.56–58
NLRP3 activation has also been shown in neurons within the context of AD. Aβ1-42 has been found to induce NLRP3-caspase-1 dependent pyroptosis in primary cortical neurons.59 Further, injection of Aβ1-42 was found to induce pyroptosis in the hippocampus of a rat model of AD that was ameliorated with administration of the biflavonoid antioxidant amentoflavone.60 There are conflicting reports as to whether astrocytes express functional NLRP3. Using primary murine cell cultures, Gustin et al. (2015) showed Aβ peptides activated the NLRP3 inflammasome in microglia but not astrocytes.61 Later work by Couturier et al. (2016) found that astrocyte cultures treated with Aβ showed IL-1B production in an ASC-dependent manner.62 Discrepancies in these two reports could stem from the treatment paradigms and Aβ peptide species used. Gustin et al. (2015) stimulated cells for 6 hours with 10 ng/mL LPS followed by 20 uM Aβ25-35 peptide exposure for 5 hours, while Couturier et al. (2016) treated cells for 3 hours with 1 ug/mL LPS prior to 10 uM Aβ1-42 peptide exposure for 3 hours. Supportive of astrocytic expression of NLRP3, Freeman et al. (2017) showed NLRP3 activation in both microglia and astrocytes following the sterile trigger lysophosphatidylcholine63 and Ebrahimi et al. 201864 found Aβ1-42 to upregulate NLRP3 mRNA/protein and caspase-1 activity in primary murine cortical astrocytes. Infiltrative immune cells provide an important source of inflammation in the diseased brain.65 Not only do these cells respond to immune changes within the periphery, but they traverse into the CNS, bringing with them vital information to influence the inflammatory microenvironment within this highly regulated system.66 Saresella et al. (2016) investigated NLRP3 pathway activation in peripheral monocytes derived from healthy control, MCI, mild AD, and severe AD patients. Following LPS-priming and stimulation with AB1-42, they saw increased inflammasome transcript and protein expression in mild and severe AD monocytes, suggesting global changes in NLRP3 activation in AD.67
NLRP3 in Parkinson’s disease
Parkinson’s disease (PD) is the most common neurodegenerative motor disorder, with an estimated 1.2 million Americans living with the disease by 2030.68 Pathologically, PD is characterized by the loss of dopaminergic (DA) neurons of the substantia nigra pars compacta (SNpC). These DA neurons project to the striatum, releasing DA from their terminals which allows for proper motor function. When these neurons are lost, less DA is produced, resulting in severe motor impairment.69 PD is defined clinically by 4 cardinal motor symptoms: bradykinesia, rest tremor, rigidity, and postural instability.70 However, non-motor symptoms including olfactory dysfunction, constipation, sleep disturbances, and depression are common in PD and can manifest decades before the onset of motor symptoms.71 Definitive diagnosis of PD can be made post-mortem and is largely based on the observation of dopaminergic neuronal cell loss in the substantia nigra and the presence of intracellular Lewy bodies and Lewy neurites.72 The observation of early-stage alterations and the estimation that 30%–70% of DA neurons are already lost when a patient clinically presents with the disease underly considerations of a “prodromal” period of PD during which neuroprotective measures may be able to modify the disease course.73,74 Lewy histopathology is highly immunoreactive for misfolded and post-translationally modified alpha-synuclein (α-syn), encoded by SNCA. SNCA mutations,75 duplications,76 and triplications77 observed in familial PD suggest a causal role for SNCA in PD etiology. In animal models, pre-formed exogenous α-syn fibrils can seed endogenous α-syn, promoting widespread deposition of α-syn aggregates in mice, associated with the development of PD-like pathologies.78 Neuroinflammation co-occurs with proteinopathy in PD.79 Reactive microglia were first described within the SNpC of PD patients post-mortem by McGeer et al. in 1988.80 The use of certain non-steroidal anti-inflammatory drugs has been linked to a decreased risk of PD,81,82 and genetic mutations in various immune genes, including in the human leukocyte antigen (HLA) family83–85 have been shown to alter PD risk. The presence of cell death, self-proteins that may take on pathologic conformations, normal aging, and the observation of inflammasome activity under similar condition in AD has prompted the study of the NLRP3 inflammasome in PD.
Evidence of inflammasome signaling in PD patient tissue was first described by Wang et al. who reported co-localization of caspase-1 and α-syn in Lewy bodies derived from PD tissues.86 These findings were supported by our laboratory and others demonstrating enhanced NLRP3 immunoreactivity in mesencephalic tissues obtained from PD patients,87 an increased abundance of the activated caspase-1 p20 fragment and ASC adapter within the PD SNpC,88 and evidence of enhanced NLRP3 expression in dopaminergic neurons from PD patients compared to healthy controls.89,90 The designer drug MPTP caused dopaminergic neuron death and PD symptomatology in humans and animal models.91,92 In the MPTP-based mouse model of PD, loss of Nlrp3 results in decreased microglial activation, sparing of DA neurons, and rescue of motor deficits.93 Loss of caspase-1 also has been shown to prevent DA loss and motor dysfunction in the MPTP-model of PD.94 Attenuation of hepatic NLRP3 alone can be sufficient to reduce MPTP-induced microglial activation and DA neuron loss in vivo.95 Further, a recent study shows that MPTP causes DA cell loss through pyroptotic mechanisms, and inhibition of this pathway ameliorates MPTP-induced DA death.96 Conversely, DA-neuron specific expression of a hyperactivating NLRP3-allele results in neuroinflammation and motor deficits in aged mice.97 Although controversial, some have reported astrocytic NLRP3 activation (see above), with 1 study finding the dopamine D2 receptor (Drd2) inhibited NLRP3 activation in cultured astrocytes treated with LPS and ATP. In the same study, they showed a Drd2 agonist inhibited inflammasome activation in the midbrain of mice treated with MPTP.98 Further work is needed to delineate the role of neurons and astrocytes in NLRP3-mediated pathogenesis in PD.
Significant attention has been devoted to the determination of whether pathologic α-syn is a direct inflammasome activator. α-syn fibrils or monomers can prime and activate the inflammasome in human monocytes via interactions with TLR2 in association with α-syn fibril phagocytosis, ROS production, and cathepsin B release from the lysosome into the cytosol.99 The differential ability of various forms of α-syn to prime and activate the NLRP3 inflammasome was further elucidated in a recent paper from Scheiblich et al. where they showed that α-syn monomer and non-fibrillar oligomers are able to the prime and activate the inflammasome in a 1-step process in primary mouse microglia without the addition of the microbial priming agent LPS whereas sonicated α-syn fibrils require priming using LPS. Findings indicted that activation was primarily mediated by TLR2, and to a lesser extent TLR5 interactions. In this model, inhibition of the inflammasome increased the degradation rate of α-syn oligomers whereas no change in degradation efficiency is seen for monomers or sonicated fibrils.33 A second study provides evidence that sonicated α-syn fibrils benefit from an LPS priming step to achieve robust NLRP3 activation, but the authors found, in contradiction to the previous study, that sonicated α-syn alone could induce NLRP3 activation to a lesser extent than if a priming step was used.88 Finally, another study showed that α-syn fibrils can prime and activate the inflammasome in a 1 step process,100 although these fibrils were not sonicated to fragment them into smaller fibrils as compared to the study by Scheiblich et al.33 and Gordon et al.88. α-syn induction of NLRP3 activation was also shown in primary human microglia derived from post-mortem brains where fibrils, but not monomers, induced inflammasome priming and activation in a 1-step process.101 Similar findings were reported in microglia derived from human induced pluripotent stem cells (hIPSCs) where activation was mediated by TLR2. The study demonstrated that oligomers contaminated with proto-fibrils prime and activate the inflammasome via TLR2 ligation and mitochondrial ROS production whereas monomers have no effect. The authors then depleted α-syn in a hiPSC-derived microglia/hiPSC-derived dopaminergic neuron co-culture system in a paradigm that models anti-α-syn antibody-based therapies for PD currently in clinical trials. Unexpectedly, the authors found that the α-syn antibody complex exacerbated inflammasome activation instead of attenuating it which has significant implications in the context of α-syn antibodies being developed for clinical use.102
The heterogeneity observed across these studies regarding the specific proteoform of α-syn used to prime and activate the inflammasome highlights a distinct difficulty across studies addressing α-syn, Aβ, and tau induced NLRP3 priming and activation. That is the fact that none of these studies are directly comparable due to differences in oligomer and fibril preparation, specifically relating to if the α-syn fibril is or is not sonicated, how the oligomeric species are prepared, and the specific assembly conditions used to aggregate protein into oligomers or fibrils. It is difficult to define the oligomeric state of amyloidogenic proteins, including α-syn and Aβ, in general due to their disordered nature and tendency to sample thousands of conformational states along a continuum of aggregation from monomer to mature fibril103–105 thus making both the identification of a distinct neurotoxic oligomeric species and the reliable preparation of similar oligomers virtually impossible. Whether the mature fibrils are sonicated or not is also suggested to play a key role in NLRP3 activation dynamics where sonicated fibrils require priming the microglia with LPS33 and unsonicated fibrils appear to not require priming with LPS.100 This distinction in sonication state is important because one of the primary in vivo mouse models of PD and synucleinopathies is the injection of WT mice with sonicated, fibrillar α-syn fibrils which directly templates the aggregation of endogenous mouse α-syn, neuroinflammation, and neuronal loss.78,106,107 Thus, it seems that in vitro models of microglia reactivity to aberrant α-syn species and accompanying NLRP3 activation should follow the widely used in vivo model even if that requires priming with LPS, which is not required in the in vivo model but appears to be required in vitro. Finally, the conditions in which protein is aggregated to form oligomers or fibrils also plays a large role in the final ultrastructure of the aggregates and the level of toxicity that these aggregates have. A recent report used 2 preparations of α-syn fibrils using different buffers to create distinct fibrillar polymorphs that had differential abilities to activate the inflammasome in primary microglia33 highlighting the strain specific differences between different protein assemblies. Other parameters of aggregation assembly including ionic composition and strength, pH, protein concentration, agitation, and temperature also play a role in determining the ultimate fibril structure and resultant toxicity.108–110 Taken together, these 3 qualities of describing the specific protein species used to activate the inflammasome are important to keep in mind when performing in vitro studies and parity in the preparations used across the field will increase the reproducibility and rigor when probing NLRP3 dynamics in response to amyloids.
It should also be noted that inflammasome activation itself may directly modulate α-syn pathology. Caspase-1 can directly cleave α-syn producing a species more prone to aggregation.86 Gordon et al., also showed the converse is true, demonstrating that blocking NLRP3 inflammasome activation with the small molecular inhibitor MCC950 reduces α-syn aggregation and motor deficits in several in vivo mouse models including the α-syn PFF model.78,88 The mice used in this study were treated with the NLRP3 inhibitor MCC950 for the entire duration of their exposure to α-syn fibrils and were sacrificed at a single time point at 8- months post injection of α-syn. This time point is somewhat late in the progression of the model and the report does not determine whether changes are observable at earlier time points after α-syn administration.111 It would be of interest to measure NLRP3 associated changes at different stages of disease progression in the model both with and without treatment with MCC950 as distinct neuroimmune progression has been observed in this model over time.107 It is possible that early NLRP3 activation may be protective and as the burden of aberrant α-syn grows, the NLRP3 inflammasome becomes over-activated and eventually neurotoxic. This highlights the importance of temporal profiling of NLRP3 dynamics in this model and others. Although pathogenic, viral infections have been suggested as triggers of neurodegenerative disease.112 A recent report found that the SARS-CoV-2 spike glycoprotein can act as both a priming and an activation signal of the NLRP3 inflammasome in microglia and that the presence of α-syn amplified its activation.113 This data is intriguing in the context of case reports describing individuals who suddenly developed PD following infection with SARS-CoV-2.114–116
Peripheral immune involvement in PD and other neurodegenerative disorders is a topic of increasing interest. Notably, NLRP3 activation is reported in peripheral blood mononuclear cells (PBMCs) of PD patients. Fan et al. (2020) collected PBMCs from PD patients and healthy controls for NLPR3 pathway transcript and protein expression profiling. They discovered elevated levels of NLRP3 pathway components at both the RNA and protein level in in PD PBMCS. Further, they found elevated IL-18 in the plasma of PD patients which correlated to cognitive and motor clinical scores. Circulating plasma-borne α-syn also correlated to plasma IL-1B levels, suggestive of a link between PD pathology and inflammasome activation.117 Data from our lab found elevated levels of circulating plasma-borne NLPR3 in PD patients compared to healthy controls87 prompting our discussion of pyroptosis as a platform for biomarker discovery in later sections.
NLRP3 in amyotrophic lateral sclerosis
Amyotrophic Lateral Sclerosis (ALS) is a progressive neurodegenerative disorder that primarily affects upper- and lower-motor neurons and is the third most prevalent progressive neurodegenerative disease after AD and PD.118 Like other neurodegenerative diseases, there is considerable genetic heterogeneity, with the most common genetic causes of ALS attributable to mutations in the proteins: chromosome 9 open reading frame 72 (C9orf72), superoxide dismutase 1 (SOD1), TAR DNA binding protein 43 (TDP-43), fused in sarcoma (FUS), and TANK-binding kinase 1 (TBK1).119 Together, mutations to these 5 proteins account for 15% of all ALS cases with the majority of ALS patients displaying no clear familial link to disease.119 The primary pathological feature of ALS is the presence TDP-43 positive inclusions bodies in the brain and spinal cord.120 Neuroinflammation is readily observed in ALS patients. Using a positron emission tomography (PET) tracer specific for reactive microglia, Turner et al. showed widespread microglia activation in human ALS patients where the magnitude of microglial activation correlates with the severity of motor neuron degeneration.121 A recent study shed light on the role of the NLRP3 inflammasome specially in ALS. Van Schoor et al. showed that expression of NLRP3 was increased in microglia located in the white matter of the motor cortex of ALS patients and this was accompanied by GSDMD antibody reactivity, indicating pyroptotic NLRP3 activation. Interestingly, this was isolated to the cortex and was not observed in microglia located in the spinal cord of ALS patients.122 Another recent study suggests that dysregulation of NLRP3 assembly, causing an increased propensity to assemble, results in reduced cognitive resiliency to non-motor symptoms in ALS patients.123
The primary rodent model of ALS expresses a familial mutation in SOD1 where glycine is changed to alanine at position 93 (SOD1-G93A) which results in decreased stability of SOD1 and an increased propensity to aggregate.124 Inflammasome components including IL-1β, NLRP3, ASC, and Caspase-1 have all been shown to be upregulated in the SOD1-G93A mouse model,125 in addition to TLR4 and NF-κβ.126 Blockade of IL-1β signaling in SOD1-G93A mice using an IL-1 receptor antagonist prolonged the lifespan of the mice and reduced inflammation,127 suggesting that IL-1β is the primary driver of neuroinflammation in ALS. ALS-associated proteins including aggregated TDP-43 and SOD1 readily activate the NLRP3 inflammasome in primary microglia.125,128 SOD1-G93A may also cause increased oxidative stress through increased peroxynitrite formation that directly leads to increased inflammasome activation.129 Astrocytic NLRP3 activation has also been suggested as a driver of neuroinflammation in ALS mice,130 although these results have since been called into question.125
NLRP3 in multiple sclerosis
Multiple sclerosis (MS) is the most common progressive neurodegenerative disorder affecting younger adults.131 A recent study estimated approximately 1 million Americans were living with the disorder in 2017.132 This disease disproportionally affects women, whose risk of developing the disorder is increased 3-fold compared with men.132 Genetic mutations in HLA genes account for approximately 20-30% of all MS genetic susceptibly.133 Environmental exposures and lifestyle factors influence disease development, with vitamin D deficiency,134 cigarette smoking,135 obesity,136 and exposure to the Epstein-Barr virus137,138 identified as risk modifiers. MS is an auto-inflammatory disorder, driven by an immune response targeting myelin sheaths in the central nervous system (CNS), resulting in axonal demyelination and eventual neuronal cell death. This neuronal loss results in slowly progressive brain atrophy.139 The adaptive and innate arms of the immune system play a role in MS pathology (for review140). Pathologic demyelination manifests as periods of acute neurologic deficit, with the symptomology dependent upon the location of the inflammatory lesion.141 Microglial activation and subsequent release of pro-inflammatory cytokines is believed to play a role in lesion formation.142 The clear association between MS and inflammation alongside the production of sterile triggers resulting from demyelination and cell death provides a platform for evaluation of DAMP sensitive pattern recognition receptors such as the NLRP3 inflammasome.
Multiple lines of evidence support a role for the NLRP3 inflammasome in MS. Polymorphisms in NLRP3,143,144 IL1B,145,146 and IL18147 genes confer an increased risk of MS. Increased levels of IL-1B have been detected in the blood148,149 and cerebrospinal fluid (CSF)149,150 of MS patients. CSF IL-1B levels correlate with the severity of demyelination within the brain151 and disease progression.152 NLRP3,153 caspase-1,154 IL-1B,153,155 have all been detected in MS lesions, while elevated mRNA levels of caspase-1 and IL-18 were seen in peripheral blood mononuclear cells (PBMCs) isolated from MS patients.156 A 2020 study suggests an inflammasome signature may be present in MS patients’ PBMCs that differs from healthy controls. Immunophenotyping and RNASeq showed an IL-1B signature in monocytes of patients with primary progressive MS. Monocytes from these individuals had higher baseline levels of ASC specks and upon LPS and ATP treatment had increased IL-1B production. Further, individuals with high IL-1B transcript levels had faster disease progression.153
Pronounced NLRP3 inflammasome activity is observed in the autoimmune encephalomyelitis (EAE) model of MS.157 The immune response in the EAE system is dependent on ASC,158 caspase-1,158,159 NLRP3,159,160 IL-18,159,160 and Gasdermin D (GSDMD), a key effector of pyroptosis.161 Enhancing the anti-inflammatory cytokine interferon-β (IFN-β) has been suggested as a treatment for MS. IFN-β inhibits NLRP3 inflammasome function162 and reduces IL-1B production163 in the EAE model while in variations of the model that negate the reliance on the NLRP3 inflammasome, there is no response to IFN-β.164 Direct inhibition of the NLRP3 inflammasome with small molecule MCC950 decreases IL-1B production, disease severity,165 relapse,166 astrocyte activation, and cognitive deficits167 in the EAE model. MCC950 in combination with rapamycin, an autophagy inducer, reduced the clinical symptomology and cytokine burden in this model as well.168 Other newly developed drugs JC-171169 and OLT1177 (Dapansutrile)170 which directly target the NLRP3 inflammasome, delayed the progression, reduced the severity, and ameliorated clinical symptomology of disease in the EAE model.
Oligodendrocytes (OLs) are the major cell type at risk in MS. There is some evidence to suggest that OLs express inflammasome components.171,172 In the context of AD, OLs from AD patients and the 5xFAD mouse model of AD showed immunoreactivity for GSDMD, the pyroptotic effector protein, alongside axonal damage and demyelination. In vitro, the treatment of OLs with AB1-42 caused an increase in NLRP3, cleaved GSDMD, and IL-1B expression, suggestive of inflammasome activation.172 Studies exploring NLRP3 activation in OLs in the context of MS are lacking, but we can infer from these data that OLs may be capable of mounting their own NLRP3-based response.
The influence of peripheral immune involvement is well-described in MS.173 Inflammasomes have been well-characterized within circulating cells of the peripheral immune system and they are most likely contributing to the inflammatory signature within not only the periphery, but also within the CNS. Interestingly, pyroptotic signaling within the periphery has proven vital for neuroinflammation within the EAE model of MS. Loss of GSDMD within peripheral myeloid cells in the EAE mouse model led to reduced inflammation and demyelination in the CNS. Further, it impaired the infiltration of peripheral T cells into the CNS.161 In a similar study, Inoue et al. (2012) found that in the EAE model NLRP3 played a role in T-cell migration to the CNS whereby inflammasome-activated cells primed CD4+ T-cells to upregulate chemotaxis-related proteins.174
Pyroptosis as a platform for biomarker discovery
Inflammasome activation drives pyroptosis, an inflammatory form of programmed cell death characterized by chromatin condensation, osmotic flux/cellular swelling, vesicular shedding, and potentially membrane rupture.175 Pyroptosis shares similarities and distinctions with apoptosis and necrosis, including the release of pro-inflammatory cytosolic proteins (Fig 2).16 This is of interest because it broadens the pool of potential extracellular biological indicators of inflammasome activation to include virtually the entire intracellular proteome. Pyroptosis is mediated by a cascade of molecular events which ultimately result in the cleavage of the protein effector of pyroptosis, GSDMD. For this reason, pyroptosis has been referred to as “GSDMD-mediated programmed necrosis.”176 GSDMD is a member of a family of related proteins that include GSDMA, GSDMB, GSDMC, GSDMD, GSDME (aka DFNA5) and PJVK (aka DFNB59), all of which play a role in inflammation and cell death.17 Three labs simultaneously discovered GSDMD as the pyroptosis effector protein.177–179 In canonical inflammasome signaling (Fig 1), DAMPs or PAMPs drive inflammasome complex formation leading to caspase 1 activation and resultant GSDMD cleavage and activation.180 Non-canonical activation of caspase 4/5 (caspase-11 in mice) through pathogenic stimuli (e.g., gram-negative bacteria) can also cause cleavage of GSDMD and pore formation.181,182 Regardless of its initial upstream modulator, GSDMD is cleaved within its central linker domain to produce a 31-kDa N-terminal fragment and a 22- kDa C-terminal fragment.177–179 The function of the 22 kDa fragment is to halt cell death, as it acts in an autoinhibitory fashion binding to the N-terminal fragment to prevent insertion into the plasma membrane. Once uncoupled from its inhibitory partner, 31-kDa N-terminal fragments of GSDMD associate with the inner leaflet of the plasma membrane, where they assemble into oligomeric pores.183–186 The N-terminal of GSDMD can directly interact with the plasma membrane, as it has a high affinity for acidic phospholipids, such as phosphoinositides,184 which are only found in the cytoplasmic leaflet of the plasma membrane. GSDMD pores range in size from 10 to 20 nm,187,188 however these pores are large enough for mature IL-1B (~5 nm in diameter) to be released upon canonical NLRP3 inflammasome activation.189,190 Although the entire mechanism of GSDMD membrane pore formation has not been fully delineated, recent studies suggests that monomeric N-terminal GSDMD fragments insert into the plasma membrane where they begin to form intermediate structures before eventually forming ring-shaped membrane-spanning pores.187 A recent study sought to determine the timeline of events which ultimately lead to pyroptosis. They report that GSDMD-mediated ion channels are the first to open, with a much smaller diameter than previously reported for GSDMD-pores. These channels allow the flux of ions, with Ca2+ influx being cited as occurring before complete membrane permeabilization. Osmotic swelling, mitochondrial depolarization, and lysosome leakage occur before the plasma membrane integrity of the cells is lost, which coincides late-stage nuclear condensation and release of intracellular components.191 Morphologically, end-stage pyroptosis is characterized by membrane blebbing, loss of plasma membrane integrity, and release of cytosolic material (Fig 1).
Fig 2.
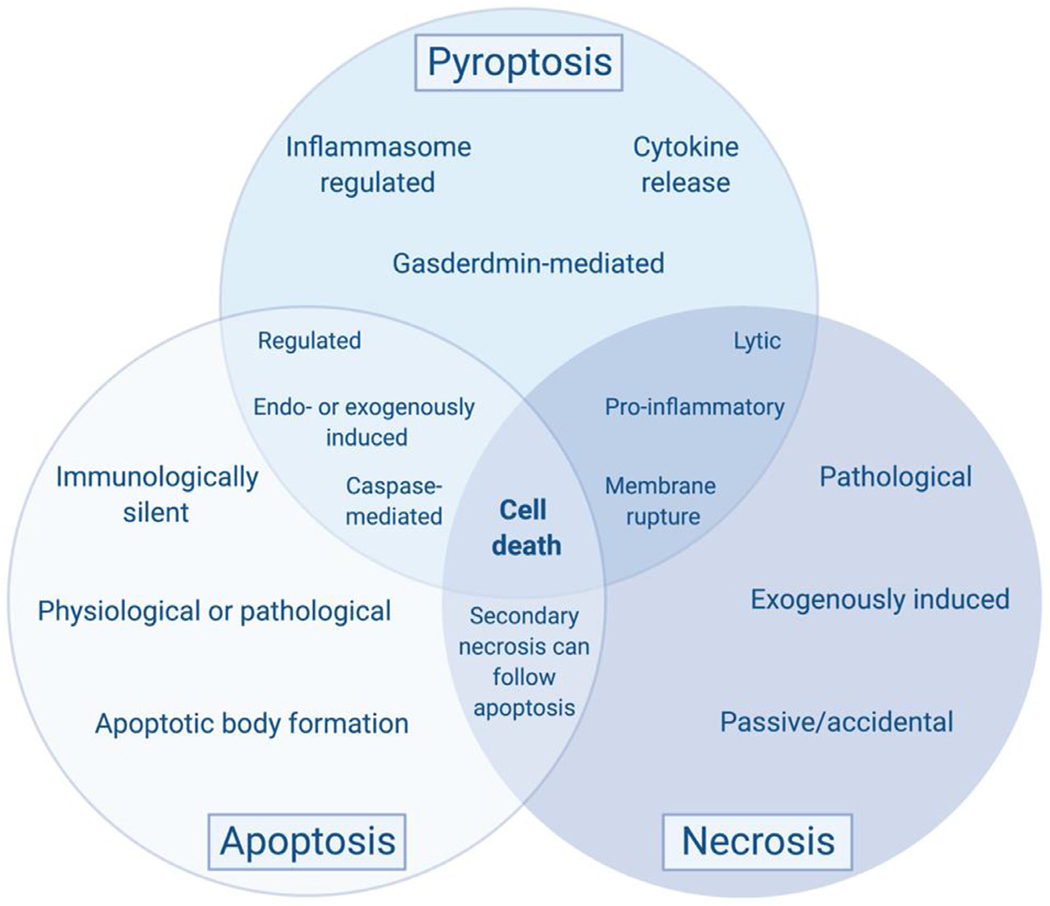
Comparison of necrosis, apoptosis, and pyroptosis. Apoptosis and pyroptosis, 2 forms of programmed cell death, share many commonalties with each other and with an accidental form of cell death, necrosis. These are not meant as absolutes, as there are many examples where these statements might not necessarily hold true, dependent on cell type, deadly trigger, and molecular platforms available. However, this framework is helpful to break these forms of cell death into distinct categories with general characteristics. Pyroptosis is typically inflammasome- and gasdermin-mediated, resulting in the release of inflammatory cytokines. Similar to apoptosis, it is highly regulated by caspase enzymes and can be triggered by both endogenous and exogenous triggers. However, apoptosis is immunologically silent resulting in apoptotic body formation, unlike the pro-inflammatory pyroptotic phenotype. Further, apoptosis can occur as a consequence of normal physiology, such as during development and normal cellular turnover, or in response to a harmful trigger or pathologic insult. Necrosis shares its greatest commonalties to pyroptosis as it is lytic, resulting in membrane rupture, and results in the pro-inflammatory release of intracellular contents into the extracellular environment. However, unlike either pyroptosis or apoptosis, it is accidental, not resulting from mechanistic regulation. Necrosis is exogenously induced from physical, chemical, or mechanical stimuli causing immediate cellular demise from pathologic injury. Interestingly, a process known as secondary necrosis can follow apoptosis if the apoptotic bodies cannot be phagocytosed, which is characterized by lytic release of intracellular material. Created with BioRender.com. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
The formation of GSDMD pores, although classically linked to membrane rupture and cellular lysis, is part of a protracted process that does not always translate into cellular demise. Lysis-independent GSDMD pore formation has been found to occur following inflammasome activation and is critical for the release of mature pro-inflammatory cytokines without causing cell death.189,190,192,193 During the sublytic phase of pyroptosis, GSDMD pore formation results in the secretion of pro-inflammatory cytokines, IL-1B and IL-18. There is some evidence to suggest that GSDMD pore formation, during non-canonical pyroptosis, can cause K+ efflux, resulting in NLRP3 inflammasome formation as well.178,179 Further, there has been some work to show that damage to the plasma membrane can be repaired and therefore is not an inherently fatal event.194 One of the major pyroptotic events – vesicular shedding – has in fact been shown to repair GSDMD-pore perforation of the plasma membrane. In a recent publication, endosomal sorting complexes required for transport (ESCRT) machinery were found to transport to the plasma membrane in response to GSDMD-pore induced Ca2+ influx and cause vesicular shedding of the perforated membrane.195 More recently, the release of such inflammasome-related vesicles was discovered to be dependent on the activation of GSDMD.196 Vesicular release from inflammasome-activated and pyroptotic cells provides an interesting avenue for potential biomarker investigation. There have been several studies showing release of IL-1B-containing vesicles following NLRP3 inflammasome activation from monocytes.197–199 Secretion of inflammasome-component containing extracellular vesicles (EVs) following NLRP3 inflammasome activation has been shown in a variety of cell types including macrophages200–202 and microglial cells.203 Although pyroptosis can be resolved, in most cases, the lytic phase eventually occurs resulting in plasma membrane rupture and release of cytosolic material, which includes the individual components of the inflammasome as well as some evidence to suggest, fully formed inflammasome complexes. Overall, pyroptosis provides 3 potential avenues by which intracellular proteins can be released into the extracellular environment: (1) through GSDMD pores (eg, IL-1B, IL-18), (2) EV shedding, and (3) bursting of the cell (Fig 1).
The conceptual platform of pyroptosis has fueled a significant effort to identify evidence of plasma-borne inflammasome indicators. Elevated expression of inflammasome-related transcripts in PBMCs and whole blood has been seen in multiple diseases including AD,204 PD,117 and ALS.205 Elevated levels of circulating IL-1B protein have been identified in a host of inflammatory disorders206 including PD.117,207,208 More recently, typically cytosolic inflammasome- or pyroptosis-related proteins and structures have been observed in the biofluids of patients in multiple disorders. Baroja-Mazo et al. identified oligomeric ASC particles in the serum of patients with active CAPS.209 Elevated circulating inflammasome proteins have been seen in the biofluids of patients with diabetes,210 human immunodeficiency virus (HIV-1),211 chronic obstructive pulmonary disease (COPD) and pneumonia,212 myelodysplastic syndromes,213 psoriasis/ psoriatic arthritis,214 and macular degeneration.215 Many disorders of the CNS also exhibit elevations in these circulating pyroptotic proteins. These include traumatic brain injury (TBI),216 stroke,217 MS,218 AD,219 and PD.87,117 Specifically, higher levels of ASC were found in the serum of MCI and AD patients compared to healthy controls, suggesting ASC as a novel biomarker of dementia.219 Elevated caspase-1 and NLRP3 have been reported in PD plasma.87,88 It is noteworthy that EVs are avidly released following inflammasome activation. Refinement of sampling to include isolation of the EV fraction may improve the precision of bioassays for inflammasome-related proteins. Elevated EV-bound ASC was observed in individuals who had suffered a stroke.217 Our laboratory reported higher levels of plasma-borne EV-contained NLRP3 in PD patients compared to healthy controls. Deeper characterization determined that ASC, the cleaved forms of CASP1 and GSDMD, and pro-IL-1B were also enriched in the EV fraction.87 These data suggest that inflammasome activation and pyroptosis result in the release of a subset of typically cytosolic proteins into the extracellular environment, that may be contained within EVs. Such evidence suggests that inflammatory, pathologic, neurodegenerative disease processes could be monitored through evaluation of pyroptotic EVs in peripheral biofluids.
Additional Topics
Resolution
Inflammation is an inherently beneficial process despite the prevailing focus on pathologic outcomes. Induction of the inflammatory response is critical for elimination of microbial pathogens, restoration of homeostasis in stressed or damaged tissues, and the elaboration of the adaptive response and immune memory. Resolution is defined by a series of cellular and molecular transitions that reduce the initial pro-inflammatory response and promote clearance of debris in damages tissues (for review,220–222). Mechanisms resolving the NLRP3-driven inflammatory response are poorly characterized, however, several lines of evidence have provided a framework for understanding this process.
The IκB kinase (IKK) complex subunit IKKa is a negative regulator of inflammation that contributes to resolution by inhibiting NF-kB interactions with pro-inflammatory gene promoters.223 IKKa also interacts with the inflammasome scaffold ASC, limiting NLRP3 inflammasome activation by sequestering ASC in the nucleus. Loss of IKKα kinase activity in murine bone marrow-derived macrophages (BMDMs) results in inflammasome hyperactivation.224 Ubiquitination and degradation of NLRP3 is a reproducibly observed mechanism of inflammasome inactivation. Bile acid driven signaling has been implicated in the control of inflammation and reported to inhibit NLRP3 inflammasome activation via TGR5-cAMP-PKA axis driven NLRP3 ubiquitination.225 Acetate inhibits NLRP3 inflammasome activation in BMDMs by activating soluble adenylyl cyclase, inducing PKA-mediated ubiquitination of NLRP3 and subsequent degradation through autophagic pathways.226 Resolvins are a well characterized family of pro-resolving lipid mediators with protective functions in neurodegenerative diseases (for review227). Resolvin D2 (RvD2) promotes the degradation of NLRP3 through autophagic pathways, and the inhibition of autophagy reversed RvD2-mediated suppression of NLRP3 inflammasome in murine peritoneal macrophages.228 Indirect mechanisms of inflammasome resolution are suggested in studies of downstream inflammasome effectors. RGS10 is a GTPase activating protein best characterized as an inhibitor of platelet activation, providing a “brake” critical for regulation of clotting and hemostasis.229 Anti-inflammatory and neuroprotective functions for RGS10 have been delineated in myeloid cells and PD models.230 Lee et al. demonstrated that Rgs10−/− macrophages produced higher levels of pro-inflammatory cytokines, including IL-1B, in response to inflammatory stimulus suggesting the potential of an antagonistic relationship between RGS10 and NLRP3.229 Taken together evidence indicates that there are active cellular mechanisms functioning to regulate and resolve the NLRP3 inflammasome-mediated inflammatory response. Further characterization of mechanisms of resolution specific to NLRP3 in the context of neurological disorders and aging has a high likelihood if identifying pathways of interest for novel treatments and preventative strategies.
Drug development
Pre-clinical studies have defined a key role for the NLRP3 inflammasome in neurodegenerative diseases, including PD, AD, and MS. Evidence also suggests that the inflammasome may function during the earliest stages of disease pathogenesis making the NLRP3 inflammasome an attractive target for therapeutic development. A rapid expansion of drug discovery efforts followed reports by Coll and others. These studies reinvigorated a molecule initially described as CRID3, a diarylsulfonylurea-containing compound among the Cytokine Release Inhibitory Drug (CRID) class.231 Collectively, these groundbreaking reports determined that CRID3 (more recently described as MCC950) directly targeted NLRP3 itself, interacting with the Walker B motif within the NLRP3 NACHT domain, blocking ATP hydrolysis, and subsequent NLRP3 activation (165,232 for additional review233).
Preclinical models have advanced the development of known NLRP3 targeting drugs specifically in neurodegenerative disease paradigms. Pharmacological inhibition of NLRP3 with MCC950 ameliorated inflammation, reduced Aß plaque burden, and improved behavioral outcomes in the APP/PS1 mouse model of AD.234 Dapansutrile (OLT1177) is well-tolerated in humans and effective in the treatment of gout (EU Clinical Trials Register, EudraCT 2016-000943-14).235 Oral administration of Dapansutrile (OLT1177) was tested in the APP/PS1 model of AD and it was found that disease phenotype was attenuated, although CNS pharmacokinetics (PK) were not performed so it is still unknown how much of the drug enters the CNS.236 Another group used a small molecule inhibitor, NIC7, which specifically targets the NLRP3 protein. The authors found that NIC7, administered via oral gavage, successfully ameliorated disease phenotype in an AD mouse model.237 Glibenclamide (glyburide), an ATP-sensitive K+ channel inhibitor used clinically to treat type 2 diabetes,238 has been shown to inhibit NLRP3 in vitro.239 The drug was used to treat a pesticide-induced PD mouse model by intraperitoneal injection and was shown to attenuate neuronal loss and inhibit the NLPR3 inflammasome and overall microglial reactivity.240 None of these animal studies explicitly tested the PK of their respective drug candidate so it remains unclear whether the compounds were able to enter the CNS.
Clinical studies of novel inflammasome inhibitors have advanced in the context of CAPS family of monogenic inflammatory disorders. Although rare, these disorders are a meaningful indication in early-stage testing of novel NLRP3 inhibitors due to a clear genetic link between CAPS and NLRP3 hyperactivation. Canakinumab, an antibody therapy targeting IL-1β was shown to significantly attenuate CAPS symptoms (NCT01302860)241 and there are several other ongoing clinical trials investigating the utility of therapies designed to inhibit ASC oligomerization (ZYIL1, NCT05186051) or target the NLRP3 protein itself (DFV890, NCT04868968; Inzomelid/IZD174, NCT04015076) in CAPS patients. Recent efforts are underway to target NLRP3 components specifically in diseases of the CNS. Treatment of CNS specific diseases is challenging due to lack of CNS bioavailability of many candidate compounds stemming from a failure to cross the blood brain barrier (BBB).242 Progress in overcoming this field-wide hurdle includes Inzomelid/IZD174 (NCT04015076), now known as Emlenoflast, a drug candidate that received promising results in phase I trials but has yet to enter stage II trials.
Concluding Remarks
Neurodegenerative disorders such as AD, PD, MS, and ALS represent major public health burdens. Despite years of study, the establishment of clear causes for these disorders remains elusive. In many cases, the initial degenerative processes are selective, however, as diseases progress, neuronal degeneration can become widespread,243 further complicating the identification of key pathologic mechanisms. Co-occurring pathobiologies offer inroads to understanding the degenerative process that may be leverages to prevent or slow disease progression. Chronic inflammation, both within the CNS244 and the peripheral immune system245 are common to many age-related neurodegenerative disorders. Glial cell activation,246 enhanced pro-inflammatory cytokine profiles,247 and infiltration of peripheral immune cells into the CNS66 are now recognized as key pathologies within many neurodegenerative diseases but an incomplete understanding of the molecular mechanisms underlying the inflammatory responses has impeded the development of anti-inflammatory therapies designed to treat neurodegenerative disorders. NLRP3 is clearly active in neurodegeneriative diseases yet the degree to which the inflammatory response causes, contributes to, or is a bystander to neurodegeneration remains unclear. This is especially important in the conundrum of proteinopathy. More work is needed to clarify whether the inflammatory milieu drives protein aggregation, responds to already misfolded proteins as it might to other pathogenic molecular patterns, or both. Understanding these mechanisms will have a transformative impact on our understanding of disease progression and the development of diagnostic and neuroprotective strategies to improve the care of aging and at-risk populations.
The rationale for consideration of pyroptosis as a key mechanism in neurodegeneration emerges from the recent realization that inflammasomes are active in neurodegeneration-associated inflammation. Inflammasome activation has been demonstrated in a variety of neurodegenerative diseases including AD,46,67 PD,87–89 and MS,157 and ALS.122,123 Inflammasome activity has been detected in multiple cell types in the CNS including microglia,36,248, neurons,89,97 and peripheral immune cells known to infiltrate the brain including monocytes.249 These findings are exciting because they provide a platform for exploring the prediction that pyroptosis links cell death and concomitant inflammation observed in neurodegenerative diseases. Studies of pyroptotic processes including the processing and release of vesicles, release of normally intracellular proteins into the extracellular environment, and pro-inflammatory cellular rupture, provides an interesting area of study towards understanding the complex multi-cellular interactions and stress processes observed in during neurodegeneration. In this review, we highlighted evidence of NLRP3 inflammasome activity and pyroptosis in the CNS and neurodegenerative disease, and discussed pyroptotic-extracellular vesicles (EVs) as a tractable means of detecting inflammation in neurological disorders.
NLRP3 has been largely characterized within peripheral immune cells, such as monocytes and macrophages, and resident immune cells of the CNS, namely microglia. Only recently has data emerged to suggest NLRP3 activation within neurons.87,89,90 Despite the NLRP3 inflammasome being the most widely studied inflammasome, other inflammasome complexes play important roles within the CNS, especially within non-immune cells. In particular, the NLRP1 inflammasome has largely been implicated in neuronal inflammasome activation. For example, the knockdown of NLRP1 in a mouse model of AD resulted in reduced neuronal pyroptosis and reversed cognitive impairments.250 Further, elevated levels of NLRP1 and caspase-1 have been seen in resected hippocampal sections from patients with intractable mesial temporal lobe epilepsy (mTLE) and knock-down of NLRP1 and caspase-1 in a rat model of epilepsy reduced neuronal pyroptosis.251 Also, while not explicitly linked to neuronal cell death, following ischemia-like conditions, such as glucose and oxygen-glucose deprivation, NLRP1 and NLRP3 proteins and cleaved caspase-1 levels were increased, and IL-1B and IL-18 precursor proteins matured in cultured cortical neurons, suggesting evidence of pyroptosis.252 The AIM2 inflammasome has also been widely associated with neuronal damage. Embryonic cortical neurons induced with aberrant synthetic DNA were found to undergo pyroptosis after AIM2 inflammasome activation of caspase-1.253 AIM2 and caspase-1 up-regulation resulting in inflammasome-induced pyroptosis has also been determined as a cell death mechanism in neurons after Enterovirus-A71 infection.254 Knockdown of the AIM2 inflammasome attenuated inflammasome activation and pyroptosis in a mouse model of ischemia-reperfusion injury.255 The AIM2 inflammasome has also been implicated in astrocyte256 and microglial257 activation in the EAE model of MS. For comprehensive reviews on inflammasomes and neurodegenerative disease, please refer to these resources.258,259
In conclusion, the NLRP3 inflammasome remains a focus of foundational and translational research while pointing towards other inflammasome classes as new frontiers for discovery in the context of neurological disorders. Novel bioassays and drugs provide an opportunity to conduct informed interventional trials aimed at modifying the progression of age-related disorders associated with chronic sterile inflammation. Based on the current knowledgebase, it is feasible that subsets of patients suffering from neurodegenerative disorders with detectably enhanced NLRP3 inflammasome activity could be treated with NLRP3 inhibitors (Fig 3). It stands to reason that these treatments could then be monitored over time to determine whether management of sterile inflammation could modify the disease course in debilitating conditions such as AD, PD, MS, and ALS, among others. More work will need to be done before this aspiration can be safely realized. This includes the optimization of universal biomarker assays that can reliably and efficiently detect inflammasome activation in living patients. Drug development hinges on the understanding of the strengths and weaknesses of peripheral versus central acting agents administered in oftentimes complex chronic disorders impacted by systemic factors such as aging and environmental exposure. Perhaps most importantly, foundational studies aimed at understanding secretory pathways, mechanisms of resolution, and cell and tissue specific mechanisms and modifiers of inflammasome activity merit prioritization to increase the scope of efforts to understand disease progression and target this critical pathway.
Fig 3.
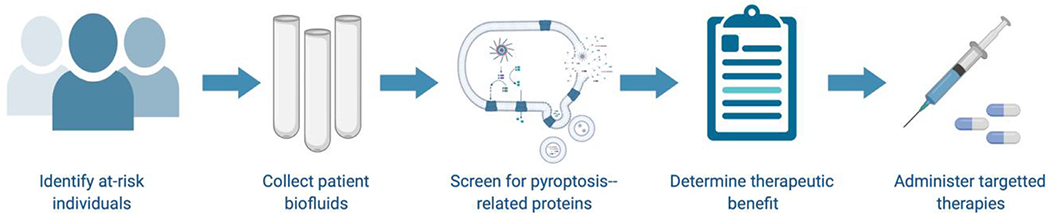
Using pyroptotic proteins as biomarkers for disease. We suggest identification of at-risk individuals and populations based on environmental exposures, familial history, and genetic testing. Collection and screening of patient biofluids for inflammasome-related proteins will allow for the generation of unique pyroptotic signatures. This information can be used by physicians to inform treatment strategies. With the development of new inflammasome pathway modulators, targeted therapies might one day exist for the direct treatment of pyroptosis-related disorders. Created with BioRender.com. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Acknowledgments
This project was supported by NIH/NIEHS 2R01ES024745-06 (MCH), 1R01ES033462-0 (MCH), F31ES030982-01(FLA), and the Michael J. Fox Foundation (MCH).
Abbreviations:
- ALRs
absent in melanoma 2-like receptors
- α-syn
alpha-synuclein
- AD
Alzheimer’s disease
- Aβ
amyloid beta
- APP
amyloid precursor protein
- ALS
amyotrophic lateral sclerosis
- ASC
apoptosis-associated speck-like protein containing a CARD
- BBB
blood brain barrier
- BMDMs
bone-marrow derived macrophages
- CARD
caspase activation and recruitment domain
- CNS
Central nervous system
- CSF
cerebrospinal fluid
- COPD
chronic obstructive pulmonary disease
- CRID
cytokine release inhibitory drug
- DAMPs
danger associated molecular patterns
- DPI
diphenyliodonium
- DA
dopaminergic
- Drd2
dopamine D2 receptor
- ESCRT
endosomal sorting complexes required for transport
- EAE
experimental autoimmune encephalomyelitis
- EVs
extracellular vesicles
- GSDMD
Gasdermin D
- hIPSCs
human induced pluripotent stem cells
- HIV-1
human immunodeficiency virus
- HLA
human leukocyte antigen
- IKK
IκB kinase
- IFN-β
interferon-β
- LPS
lipopolysaccharide
- MCI
mild cognitive impairment
- MS
Multiple Sclerosis
- MVBs
multivesicular bodies
- NLRP3
NLR family pyrin domain containing 3
- NLRs
nucleotide-binding domain-like receptors
- PD
Parkinson’s disease
- PAMPs
pathogen associated molecular patterns
- PET
positron emission tomography
- PK
pharmacokinetics
- PTMs
post-translational modifications
- PSEN1/PS1
presenilin 1
- PSEN2/PS2
presenilin 2
- ROS
Reactive oxygen species
- RvD2
Resolvin D2
- SOD1
superoxide dismutase 1
- SNpC
substantia nigra pars compacta
- TDP-43
TAR DNA binding protein 43
- TBI
traumatic brain injury
- TLR
toll-like receptor
- TCA
tricarboxylic acid
Footnotes
Conflicts of interest: The authors declare that they have no competing interests.
References
- 1.Mao M, et al. Identification of genes expressed in human CD34(+) hematopoietic stem/progenitor cells by expressed sequence tags and efficient full-length cDNA cloning. Proc Natl Acad Sci U S A, 1998;95:8175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cuisset L, et al. Genetic linkage of the Muckle-Wells syndrome to chromosome 1q44. Am J Hum Genet 1999;65:1054–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffman HM, et al. Identification of a locus on chromosome 1q44 for familial cold urticaria. Am J Hum Genet 2000;66:1693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffman HM, et al. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001;29:301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol 2013;13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 2016;41:1012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016;16:407–20. [DOI] [PubMed] [Google Scholar]
- 8.Zheng M, Kanneganti TD. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol Rev 2020;297:26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strowig T, et al. Inflammasomes in health and disease. Nature 2012;481:278–86. [DOI] [PubMed] [Google Scholar]
- 10.Schroder K, Tschopp J. The inflammasomes. Cell 2010;140:821–32. [DOI] [PubMed] [Google Scholar]
- 11.Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006;440:228–32. [DOI] [PubMed] [Google Scholar]
- 12.Pan P, et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun 2021;12:4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 2010;10:210–5. [DOI] [PubMed] [Google Scholar]
- 14.Bauernfeind FG, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009;183:787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He Y, et al. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016;530:354–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 2005;73:1907–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol 2020;20:143–57. [DOI] [PubMed] [Google Scholar]
- 18.Patel MN, et al. Inflammasome Priming in Sterile Inflammatory Disease. Trends Mol Med 2017;23:165–80. [DOI] [PubMed] [Google Scholar]
- 19.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou R, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011;469:221–6. [DOI] [PubMed] [Google Scholar]
- 21.Bauernfeind F, et al. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 2011;187:613–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Folco EJ, et al. Moderate hypoxia potentiates interleukin-1beta production in activated human macrophages. Circ Res 2014;115:875–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panchanathan R, Liu H, Choubey D. Hypoxia primes human normal prostate epithelial cells and cancer cell lines for the NLRP3 and AIM2 inflammasome activation. Oncotarget 2016;7:28183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lampropoulou V, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab 2016;24:158–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moon JS, et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest 2015;125:665–80. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Xiao H, et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation 2013;128:632–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suresh R, et al. Complement-mediated ‘bystander’ damage initiates host NLRP3 inflammasome activation. J Cell Sci 2016;129:1928–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Won JH, et al. Rotenone-induced impairment of mitochondrial electron transport chain confers a selective priming signal for NLRP3 inflammasome activation. J Biol Chem 2015;290:27425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson FL, et al. Inflammasomes: An Emerging Mechanism Translating Environmental Toxicant Exposure Into Neuroinflammation in Parkinson’s Disease. Toxicol Sci 2018;166:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Latz E, Duewell P. NLRP3 inflammasome activation in inflammaging. Semin Immunol 2018;40:61–73. [DOI] [PubMed] [Google Scholar]
- 31.Cicolari S, Catapano AL, Magni P. Inflammaging and neurodegenerative diseases: role of NLRP3 inflammasome activation in brain atherosclerotic vascular disease. Mech Ageing Dev 2021;195:111467. [DOI] [PubMed] [Google Scholar]
- 32.Luciunaite A, et al. Soluble Abeta oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J Neurochem 2019: e14945. [DOI] [PubMed] [Google Scholar]
- 33.Scheiblich H, et al. Microglial NLRP3 Inflammasome Activation upon TLR2 and TLR5 ligation by distinct α-synuclein assemblies. The J Immunol 2021;207:2143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amores-Iniesta J, et al. Extracellular ATP activates the NLRP3 inflammasome and is an early danger signal of skin allograft rejection. Cell Rep 2017;21:3414–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sadatomi D, et al. Mitochondrial function is required for extracellular ATP-induced NLRP3 inflammasome activation. J Biochem 2017;161:503–12. [DOI] [PubMed] [Google Scholar]
- 36.Halle A, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 2008;9:857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nomura J, et al. Intracellular ATP Decrease Mediates NLRP3 Inflammasome Activation upon Nigericin and Crystal Stimulation. J Immunol 2015;195:5718–24. [DOI] [PubMed] [Google Scholar]
- 38.Billingham LK, et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat Immunol 2022;23:692–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.2020 Alzheimer’s disease facts and figures. Alzheimers Dement, 2020. [DOI] [PubMed] [Google Scholar]
- 40.Norton S, et al. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol 2014;13:788–94. [DOI] [PubMed] [Google Scholar]
- 41.Donev R, et al. Neuronal death in Alzheimer’s disease and therapeutic opportunities. J Cell Mol Med 2009;13:4329–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 2014;71:505–8. [DOI] [PubMed] [Google Scholar]
- 43.Goate A, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991;349:704–6. [DOI] [PubMed] [Google Scholar]
- 44.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 2016;8:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Strooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell 2016;164:603–15. [DOI] [PubMed] [Google Scholar]
- 46.Heneka MT, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013;493:674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ising C, et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019;575:669–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venegas C, et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017;552:355–61. [DOI] [PubMed] [Google Scholar]
- 49.Friker LL, et al. beta-amyloid clustering around ASC fibrils boosts its toxicity in microglia. Cell Rep 2020;30:3743–54. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Götz J, Halliday G, Nisbet RM. Molecular pathogenesis of the tauopathies. Ann Rev Pathol: Mechan Dis 2019;14:239–61. [DOI] [PubMed] [Google Scholar]
- 51.Stancu IC, et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol 2019;137:599–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang S, et al. Proteopathic tau primes and activates interleukin-1β via myeloid-cell-specific MyD88- and NLRP3-ASC-inflammasome pathway. Cell Rep 2021;36:109720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Y, et al. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci Lett 2020;736:135279. [DOI] [PubMed] [Google Scholar]
- 54.Islam J, et al. GPCR19 regulates P2×7R-mediated NLRP3 inflammasomal activation of microglia by amyloid β in a mouse model of alzheimer’s disease. Front Immunol 2022;13:766919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bibič L, Stokes L. Revisiting the Idea That Amyloid-β Peptide Acts as an Agonist for P2×7. Front Mol Neurosci 2020;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sbai O, et al. AGE-TXNIP axis drives inflammation in Alzheimer’s by targeting Aβ to mitochondria in microglia. Cell Death Dis 2022;13:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okada M, et al. The Lysosome Rupture-activated TAK1-JNK Pathway Regulates NLRP3 Inflammasome Activation*. J Biol Chem 2014;289:32926–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flavin WP, et al. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol 2017;134:629–53. [DOI] [PubMed] [Google Scholar]
- 59.Han C, et al. New mechanism of nerve injury in Alzheimer’s disease: beta-amyloid-induced neuronal pyroptosis. J Cell Mol Med 2020;24:8078–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao N, et al. Amentoflavone suppresses amyloid beta1-42 neurotoxicity in Alzheimer’s disease through the inhibition of pyroptosis. Life Sci 2019;239:117043. [DOI] [PubMed] [Google Scholar]
- 61.Gustin A, et al. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS One 2015;10:e0130624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Couturier J, et al. Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J Neuroinflammation 2016;13:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Freeman L, et al. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med 2017;214:1351–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ebrahimi T, et al. alpha1-antitrypsin mitigates NLRP3-inflammasome activation in amyloid beta1-42-stimulated murine astrocytes. J Neuroinflammation 2018;15:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest 2010;120:1368–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rezai-Zadeh K, Gate D, Town T. CNS infiltration of peripheral immune cells: D-Day for neurodegenerative disease? J Neuroimmune Pharmacol 2009;4:462–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saresella M, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol neurodegeneration 2016;11. 23–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marras C, et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis 2018;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eriksen JL, Wszolek Z, Petrucelli L. Molecular pathogenesis of Parkinson disease. Arch Neurol 2005;62:353–7. [DOI] [PubMed] [Google Scholar]
- 70.Rodriguez-Oroz MC, et al. Initial clinical manifestations of Parkinson’s disease: features and pathophysiological mechanisms. Lancet Neurol 2009;8:1128–39. [DOI] [PubMed] [Google Scholar]
- 71.Postuma RB, et al. Identifying prodromal Parkinson’s disease: pre-motor disorders in Parkinson’s disease. Mov Disord 2012;27:617–26. [DOI] [PubMed] [Google Scholar]
- 72.Spillantini MG, et al. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A, 1998;95:6469–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 2010;67:715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 1991;114(Pt 5):2283–301. [DOI] [PubMed] [Google Scholar]
- 75.Polymeropoulos MH, et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 1996;274:1197–9. [DOI] [PubMed] [Google Scholar]
- 76.Chartier-Harlin MC, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004;364:1167–9. [DOI] [PubMed] [Google Scholar]
- 77.Singleton AB, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003;302:841. [DOI] [PubMed] [Google Scholar]
- 78.Luk KC, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012;338:949–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener 2015;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McGeer PL, et al. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988;38:1285–91. [DOI] [PubMed] [Google Scholar]
- 81.Chen H, et al. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol 2003;60:1059–64. [DOI] [PubMed] [Google Scholar]
- 82.Gao X, et al. Use of ibuprofen and risk of Parkinson disease. Neurology 2011;76:863–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hamza TH, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 2010;42:781–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.International Parkinson Disease Genomics C, et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 2011;377:641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hollenbach JA, et al. A specific amino acid motif of HLA-DRB1 mediates risk and interacts with smoking history in Parkinson’s disease. Proc Natl Acad Sci U S A, 2019;116:7419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang W, et al. Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein alpha-synuclein. Proc Natl Acad Sci U S A, 2016;113:9587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anderson FL, et al. Plasma-borne indicators of inflammasome activity in Parkinson’s disease patients. NPJ Parkinsons Dis 2021;7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gordon R, et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med 2018;10:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.von Herrmann KM, et al. NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Parkinsons Dis 2018;4:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Panicker N, et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson’s disease. Neuron 2022: 2422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Langston JW. The MPTP Story. J Parkinsons Dis 2017;7(s1):S11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Langston JW, et al. Selective nigral toxicity after systemic administration of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyrine (MPTP) in the squirrel monkey. Brain Res 1984;292:390–4. [DOI] [PubMed] [Google Scholar]
- 93.Lee E, et al. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ 2019;26:213–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Qiao C, et al. Caspase-1 deficiency alleviates dopaminergic neuronal death via inhibiting caspase-7/AIF Pathway in MPTP/p mouse model of Parkinson’s disease. Mol Neurobiol 2017;54:4292–302. [DOI] [PubMed] [Google Scholar]
- 95.Qiao C, et al. Inhibition of the hepatic Nlrp3 protects dopaminergic neurons via attenuating systemic inflammation in a MPTP/p mouse model of Parkinson’s disease. J Neuroinflammation 2018;15:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang X, et al. Salidroside ameliorates Parkinson’s disease by inhibiting NLRP3-dependent pyroptosis. Aging (Albany NY) 2020;12:9405–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.von Herrmann KM, et al. Slc6a3-dependent expression of a CAPS-associated Nlrp3 allele results in progressive behavioral abnormalities and neuroinflammation in aging mice. J Neuroinflammation 2020;17:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhu J, et al. Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of beta-arrestin2 and NLRP3. Cell Death Differ 2018;25:2037–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Codolo G, et al. , Triggering of inflammasome by aggregated a – synuclein, an inflammatory response in synucleinopathies. 2013; 8(1): e55375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Panicker N, et al. Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med 2019;216:1411–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pike AF, et al. α-synuclein evokes NLRP3 inflammasome-mediated IL-1β secretion from primary human microglia. Glia 2021;69:1413–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Trudler D,et al. Soluble α-synuclein–antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc Nationl Acad Sci 2021;118:e2025847118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fink AL. The Aggregation and Fibrillation of α-synuclein. Acc Chem Res 2006;39:628–34. [DOI] [PubMed] [Google Scholar]
- 104.Mehra S, Sahay S, Maji SK. α-synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim Biophys Acta Proteins Proteom 2019;1867:890–908. [DOI] [PubMed] [Google Scholar]
- 105.Teplow DB. On the subject of rigor in the study of amyloid β-protein assembly. Alzheimer’s Res Ther 2013;5:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Earls RH, et al. Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. Journal of Neuroinflammation 2019;16:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Duffy MF, et al. Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflam 2018;15:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Buell AK, et al. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc Nationl Acad Sci 2014;111:7671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lövestam S, et al. Assembly of recombinant tau into filaments identical to those of Alzheimer’s disease and chronic traumatic encephalopathy. Elife 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fujiwara S, et al. Dynamic properties of human α-synuclein related to propensity to amyloid fibril formation. J Mol Biol 2019;431:3229–45. [DOI] [PubMed] [Google Scholar]
- 111.Stoll AC, Sortwell CE. Leveraging the preformed fibril model to distinguish between alpha-synuclein inclusion- and nigrostriatal degeneration-associated immunogenicity. Neurobiol Dis 2022;171:105804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Deleidi M, Isacson O. Viral and inflammatory triggers of neurodegenerative diseases. Sci Transl Med 2012;4:121. ps3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Albornoz E SARS-CoV-2 drives NLRP3 inflammasome activation in human microglia through spike-ACE2 receptor interaction. bioRxiv 2022: 138–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cohen ME, et al. A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol 2020;19:804–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mendez-Guerrero A, et al. Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 2020;95:e2109–18. [DOI] [PubMed] [Google Scholar]
- 116.Faber I, et al. Coronavirus disease 2019 and Parkinsonism: a non-post-encephalitic case. Mov Disord 2020;35:1721–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fan Z, et al. Systemic activation of NLRP3 inflammasome and plasma alpha-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J Neuroinflammation 2020;17:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hardiman O, et al. Amyotrophic lateral sclerosis. Nature Rev Dis Primers 2017;3:17071. [DOI] [PubMed] [Google Scholar]
- 119.Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol 2020;27:1918–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–3. [DOI] [PubMed] [Google Scholar]
- 121.Turner MR, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis 2004;15:601–9. [DOI] [PubMed] [Google Scholar]
- 122.Van Schoor E, et al. Increased pyroptosis activation in white matter microglia is associated with neuronal loss in ALS motor cortex. Acta Neuropathol 2022: 393–411. [DOI] [PubMed] [Google Scholar]
- 123.Banerjee P, et al. NLRP3 inflammasome as a key molecular target underlying cognitive resilience in amyotrophic lateral sclerosis. The J Pathol 2022;256:262–8. [DOI] [PubMed] [Google Scholar]
- 124.Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994;264:1772–5. [DOI] [PubMed] [Google Scholar]
- 125.Deora V, et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 2020;68:407–21. [DOI] [PubMed] [Google Scholar]
- 126.Gugliandolo A, et al. NLRP3 inflammasome activation in a transgenic amyotrophic lateral sclerosis model. Inflammation 2018;41:93–103. [DOI] [PubMed] [Google Scholar]
- 127.Meissner F, Molawi K, Zychlinsky A. Mutant superoxide dismutase 1-induced IL-1beta accelerates ALS pathogenesis. Proc Natl Acad Sci U S A, 2010;107:13046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhao W, et al. TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp Neurol 2015;273:24–35. [DOI] [PubMed] [Google Scholar]
- 129.Bellezza I, et al. Peroxynitrite activates the NLRP3 inflammasome cascade in SOD1 (G93A) mouse model of amyotrophic lateral sclerosis. Mol Neurobiol 2018;55:2350–61. [DOI] [PubMed] [Google Scholar]
- 130.Johann S, et al. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 2015;63:2260–73. [DOI] [PubMed] [Google Scholar]
- 131.Evans C, et al. Incidence and prevalence of multiple sclerosis in the Americas: a systematic review. Neuroepidemiology 2013;40:195–210. [DOI] [PubMed] [Google Scholar]
- 132.Wallin MT, et al. The prevalence of MS in the United States: a population-based estimate using health claims data. Neurology 2019;92:e1029–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Haines JL, et al. Linkage of the MHC to familial multiple sclerosis suggests genetic heterogeneity. The multiple sclerosis genetics group. Hum Mol Genet 1998;7:1229–34. [DOI] [PubMed] [Google Scholar]
- 134.Sintzel MB, Rametta M, Reder AT. Vitamin D and multiple sclerosis. A Comprehensive Rev. Neurol Ther 2018;7:59–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wingerchuk DM. Smoking: effects on multiple sclerosis susceptibility and disease progression. Ther Adv Neurol Disord 2012;5:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gianfrancesco MA, Barcellos LF. Obesity and multiple sclerosis susceptibility: a review. J Neurol Neuromed 2016;1:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Levin LI, et al. Temporal relationship between elevation of epstein-barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA 2005;293:2496–500. [DOI] [PubMed] [Google Scholar]
- 138.Bjornevik K, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022;375:296–301. [DOI] [PubMed] [Google Scholar]
- 139.Frohman EM, Racke MK, Raine CS. Multiple sclerosis–the plaque and its pathogenesis. N Engl J Med 2006;354:942–55. [DOI] [PubMed] [Google Scholar]
- 140.Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol 2015;14:406–19. [DOI] [PubMed] [Google Scholar]
- 141.van der Valk P, De Groot CJ. Staging of multiple sclerosis (MS) lesions: pathology of the time frame of MS. Neuropathol Appl Neurobiol 2000;26:2–10. [DOI] [PubMed] [Google Scholar]
- 142.Palle P, et al. Cytokine signaling in multiple sclerosis and its therapeutic applications. Med Sci (Basel) 2017;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Compeyrot-Lacassagne S, et al. Brain multiple sclerosis-like lesions in a patient with Muckle-Wells syndrome. Rheumatology (Oxford) 2009;48:1618–9. [DOI] [PubMed] [Google Scholar]
- 144.Schuh E, et al. Expanding spectrum of neurologic manifestations in patients with NLRP3 low-penetrance mutations. Neurol Neuroimmunol Neuroinflamm 2015;2:e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mann CL, et al. Interleukin 1 genotypes in multiple sclerosis and relationship to disease severity. J Neuroimmunol 2002;129:197–204. [DOI] [PubMed] [Google Scholar]
- 146.Isik N, et al. Multiple sclerosis: association with the interleukin-1 gene family polymorphisms in the Turkish population. Int J Neurosci 2013;123:711–8. [DOI] [PubMed] [Google Scholar]
- 147.Karakas Celik S, et al. Interleukin 18 gene polymorphism is a risk factor for multiple sclerosis. Mol Biol Rep 2014;41:1653–8. [DOI] [PubMed] [Google Scholar]
- 148.de Jong BA, et al. Production of IL-1beta and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. J Neuroimmunol 2002;126:172–9. [DOI] [PubMed] [Google Scholar]
- 149.Dujmovic I, et al. The analysis of IL-1 beta and its naturally occurring inhibitors in multiple sclerosis: The elevation of IL-1 receptor antagonist and IL-1 receptor type II after steroid therapy. J Neuroimmunol 2009;207:101–6. [DOI] [PubMed] [Google Scholar]
- 150.Hauser SL, et al. Cytokine accumulations in CSF of multiple sclerosis patients: frequent detection of interleukin-1 and tumor necrosis factor but not interleukin-6. Neurology 1990;40:1735–9. [DOI] [PubMed] [Google Scholar]
- 151.Seppi D, et al. Cerebrospinal fluid IL-1beta correlates with cortical pathology load in multiple sclerosis at clinical onset. J Neuroimmunol 2014;270:56–60. [DOI] [PubMed] [Google Scholar]
- 152.Rossi S, et al. Cerebrospinal fluid detection of interleukin-1beta in phase of remission predicts disease progression in multiple sclerosis. J Neuroinflammation 2014;11:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Malhotra S, et al. NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain 2020;143:1414–30. [DOI] [PubMed] [Google Scholar]
- 154.Ming X, et al. Caspase-1 expression in multiple sclerosis plaques and cultured glial cells. J Neurol Sci 2002;197:9–18. [DOI] [PubMed] [Google Scholar]
- 155.Wucherpfennig KW, et al. T cell receptor V alpha-V beta repertoire and cytokine gene expression in active multiple sclerosis lesions. J Exp Med 1992;175:993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Huang WX, Huang P, Hillert J. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult Scler 2004;10:482–7. [DOI] [PubMed] [Google Scholar]
- 157.Barclay W, Shinohara ML. Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathol 2017;27:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shaw PJ, et al. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol 2010;184:4610–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jha S, et al. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci 2010;30:15811–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gris D, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol 2010;185:974–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Li S, et al. Gasdermin D in peripheral myeloid cells drives neuroinflammation in experimental autoimmune encephalomyelitis. J Exp Med 2019;216:2562–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Inoue M, et al. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal 2012;5:ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Guarda G, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011;34:213–23. [DOI] [PubMed] [Google Scholar]
- 164.Inoue M, et al. An interferon-beta-resistant and NLRP3 inflammasome-independent subtype of EAE with neuronal damage. Nat Neurosci 2016;19:1599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Coll RC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015;21:248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Khan N, et al. Pharmacological inhibition of the NLRP3 inflammasome as a potential target for multiple sclerosis induced central neuropathic pain. Inflammopharmacology 2018;26:77–86. [DOI] [PubMed] [Google Scholar]
- 167.Hou B, et al. Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis 2020;11:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Xu L, et al. Rapamycin combined with MCC950 to treat multiple sclerosis in experimental autoimmune encephalomyelitis. J Cell Biochem 2019;120:5160–8. [DOI] [PubMed] [Google Scholar]
- 169.Guo C, et al. Development and Characterization of a Hydroxyl-Sulfonamide Analogue, 5-Chloro-N-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a Novel NLRP3 Inflammasome Inhibitor for Potential Treatment of Multiple Sclerosis. ACS Chem Neurosci 2017;8:2194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sanchez-Fernandez A, et al. OLT1177 (Dapansutrile), a selective NLRP3 inflammasome inhibitor, ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front Immunol 2019;10:2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Maturana CJ, Aguirre A, Saez JC. High glucocorticoid levels during gestation activate the inflammasome in hippocampal oligodendrocytes of the offspring. Dev Neurobiol 2017;77:625–42. [DOI] [PubMed] [Google Scholar]
- 172.Zhang X, et al. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci Adv 2020;6(49):eabb8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hoglund RA, Maghazachi AA. Multiple sclerosis and the role of immune cells. World J Exp Med 2014;4:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Inoue M, et al. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A, 2012;109:10480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Galluzzi L, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci 2017;42:245–54. [DOI] [PubMed] [Google Scholar]
- 177.He WT, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 2015;25:1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kayagaki N, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015;526:666–71. [DOI] [PubMed] [Google Scholar]
- 179.Shi J, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015;526:660–5. [DOI] [PubMed] [Google Scholar]
- 180.Sharma D, Kanneganti TD. The cell biology of inflammasomes :. Mech inflammasome activ regul 2016;213:617–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kayagaki N, et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011;479:117–21. [DOI] [PubMed] [Google Scholar]
- 182.Huang X, et al. Caspase-11, a specific sensor for intracellular lipopolysaccharide recognition, mediates the non-canonical inflammatory pathway of pyroptosis. Cell Biosci 2019;9:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Aglietti RA, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A, 2016;113:7858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ding J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016;535:111–6. [DOI] [PubMed] [Google Scholar]
- 185.Liu X, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016;535:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sborgi L, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 2016;35:1766–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mulvihill E, et al. Mechanism of membrane pore formation by human gasdermin-D. EMBO J 2022;10. 10.1038/s41467-021-27692-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ruan J, et al. Cryo-EM structure of the gasdermin A3 membrane pore. Nature 2018;557:62–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Evavold CL, et al. The pore-forming protein Gasdermin D regulates Interleukin-1 secretion from living macrophages. Immunity 2018;48:35–44. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Heilig R, et al. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol 2018;48:584–92. [DOI] [PubMed] [Google Scholar]
- 191.de Vasconcelos NM, et al. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ 2019;26:146–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mangan DF, Welch GR, Wahl SM. Lipopolysaccharide, tumor necrosis factor-alpha, and IL-1 beta prevent programmed cell death (apoptosis) in human peripheral blood monocytes. J Immunol 1991;146:1541–6. [PubMed] [Google Scholar]
- 193.Ozawa T, et al. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 2014;26:288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cooper ST, McNeil PL. Membrane repair: mechanisms and pathophysiology. Physiol Rev 2015;95:1205–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ruhl S, et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018;362:956–60. [DOI] [PubMed] [Google Scholar]
- 196.Mitra S, Sarkar A. Microparticulate P2×7 and GSDM-D mediated regulation of functional IL-1beta release. Purinergic Signal 2019;15:119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rubartelli A, et al. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J 1990;9:1503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Andrei C, et al. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol Biol Cell 1999;10:1463–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.MacKenzie A, et al. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 2001;15:825–35. [DOI] [PubMed] [Google Scholar]
- 200.Baroja-Mazo A, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nature Immunol 2014;15:738–48. VN - r. [DOI] [PubMed] [Google Scholar]
- 201.Valimaki E, et al. Calpain activity is essential for ATP-driven unconventional vesicle-mediated protein secretion and inflammasome activation in human macrophages. J Immunol 2016;197:3315–25. [DOI] [PubMed] [Google Scholar]
- 202.Zhang Y, et al. Inflammasome-derived exosomes activate NF-kappaB signaling in macrophages. J Proteome Res 2017;16:170–8. [DOI] [PubMed] [Google Scholar]
- 203.Sarkar S, et al. Manganese activates NLRP3 inflammasome signaling and propagates exosomal release of ASC in microglial cells. Sci Signal 2019: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rui W, et al. Systemic inflammasome activation and pyroptosis associate with the progression of amnestic mild cognitive impairment and Alzheimer’s disease. J Neuroinflammation 2021;18:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Moreno-Garcia L, et al. Inflammasome in ALS skeletal muscle: NLRP3 as a potential biomarker. Int J Mol Sci 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011;117:3720–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Blum-Degen D, et al. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett 1995;202:17–20. [DOI] [PubMed] [Google Scholar]
- 208.Mogi M, et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett 1994;180:147–50. [DOI] [PubMed] [Google Scholar]
- 209.Baroja-Mazo A, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 2014;15:738–48. [DOI] [PubMed] [Google Scholar]
- 210.Wan Z, et al. NLRP3 inflammasome promotes diabetes-induced endothelial inflammation and atherosclerosis. Diabetes Metab Syndr Obes 2019;12:1931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ahmad F, et al. Evidence of inflammasome activation and formation of monocyte-derived ASC specks in HIV-1 positive patients. AIDS 2018;32:299–307. [DOI] [PubMed] [Google Scholar]
- 212.Franklin BS, et al. The adaptor ASC has extracellular and ’prionoid’ activities that propagate inflammation. Nat Immunol 2014;15:727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Basiorka AA, et al. Assessment of ASC specks as a putative biomarker of pyroptosis in myelodysplastic syndromes: an observational cohort study. Lancet Haematol 2018;5:e393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Forouzandeh M, et al. The inflammasome signaling proteins ASC and IL-18 as biomarkers of psoriasis. Front Pharmacol 2020;11:1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Weaver C, et al. Inflammasome proteins as inflammatory biomarkers of age-related macular degeneration. Transl Vis Sci Technol 2020;9:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kerr N, et al. Inflammasome proteins as biomarkers of traumatic brain injury. PLoS One 2018;13:e0210128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kerr N, et al. Inflammasome proteins in serum and serum-derived extracellular vesicles as biomarkers of stroke. Front Mol Neurosci 2018;11:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Keane RW, Dietrich WD, de Rivero Vaccari JP. Inflammasome proteins as biomarkers of multiple sclerosis. Front Neurol 2018;9:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Scott XO, et al. The inflammasome adaptor protein ASC in mild cognitive impairment and Alzheimer’s disease. Int J Mol Sci 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Feehan KT, Gilroy DW. Is resolution the end of inflammation? Trends Mol Med 2019;25:198–214. [DOI] [PubMed] [Google Scholar]
- 221.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol 2005;6:1191–7. [DOI] [PubMed] [Google Scholar]
- 222.Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov 2016;15:551–67. [DOI] [PubMed] [Google Scholar]
- 223.Lawrence T, et al. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature 2005;434:1138–43. [DOI] [PubMed] [Google Scholar]
- 224.Martin BN, et al. IKKalpha negatively regulates ASC-dependent inflammasome activation. Nat Commun 2014;5:4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Guo C, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 2016;45:802–16. [DOI] [PubMed] [Google Scholar]
- 226.Xu M, et al. Acetate attenuates inflammasome activation through GPR43-mediated Ca(2+)-dependent NLRP3 ubiquitination. Exp Mol Med 2019;51:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Li C, et al. Role of resolvins in the inflammatory resolution of neurological diseases. Front Pharmacol 2020;11:612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Cao L, et al. Resolvin D2 suppresses NLRP3 inflammasome by promoting autophagy in macrophages. Exp Ther Med 2021;22:1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Lee JK, et al. Critical role of regulator G-protein signaling 10 (RGS10) in modulating macrophage M1/M2 activation. PLoS One 2013;8:e81785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Lee JK, et al. Regulator of G-protein signaling-10 negatively regulates NF-kappaB in microglia and neuroprotects dopaminergic neurons in hemiparkinsonian rats. J Neurosci 2011;31:11879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Perregaux DG, et al. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J Pharmacol Exp Ther 2001;299:187–97. [PubMed] [Google Scholar]
- 232.Coll RC, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol 2019;15:556–9. [DOI] [PubMed] [Google Scholar]
- 233.Coll RC, Schroder K, Pelegrín P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends in Pharmacol Sci 2022: 653–68. [DOI] [PubMed] [Google Scholar]
- 234.Dempsey C, et al. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav Immun 2017;61:306–16. [DOI] [PubMed] [Google Scholar]
- 235.Klück V, et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol 2020;2:e270–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Lonnemann N, et al. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. In: Proc Nationl Acad Sci, 117; 2020. p. 202032145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Haseeb M, et al. Novel small-molecule inhibitor of NLRP3 inflammasome reverses cognitive impairment in an Alzheimer’s disease model. ACS Chemical Neuroscience 2022;13:818–33. [DOI] [PubMed] [Google Scholar]
- 238.Hardin MD, Glyburide JT. Treasure Island. StatPearls FL: StatPearls Publishing; 2022. [Internet]. [Google Scholar]
- 239.Lamkanfi M, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 2009;187:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Qiu X, et al. Inhibition of NLRP3 inflammasome by glibenclamide attenuated dopaminergic neurodegeneration and motor deficits in paraquat and maneb-induced mouse Parkinson’s disease model. Toxicol Letters 2021;349:1–11. [DOI] [PubMed] [Google Scholar]
- 241.Brogan PA, et al. Rapid and sustained long-term efficacy and safety of canakinumab in patients with cryopyrin-associated periodic syndrome ages five years and younger. Arthritis Rheumatol 2019;71:1955–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kadry H, Noorani B, Cucullo L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids and Barriers of the CNS 2020;17:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Braak H, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- 244.Amor S, et al. Inflammation in neurodegenerative diseases. Immunology 2010;129:154–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Trager U, Tabrizi SJ. Peripheral inflammation in neurodegeneration. J Mol Med (Berl) 2013;91:673–81. [DOI] [PubMed] [Google Scholar]
- 246.von Bernhardi R, Eugenin-von Bernhardi L, Eugenin J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci 2015;7:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci 2001;2:734–44. [DOI] [PubMed] [Google Scholar]
- 248.Martinez EM, et al. Editor’s highlight: Nlrp3 is required for inflammatory changes and nigral cell loss resulting from chronic intragastric rotenone exposure in mice. Toxicol Sci 2017;159:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Singer II, et al. The interleukin-1 beta-converting enzyme (ICE) is localized on the external cell surface membranes and in the cytoplasmic ground substance of human monocytes by immuno-electron microscopy. J Exp Med 1995;182:1447–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Tan MS, et al. Amyloid-beta induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis 2014;5:e1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Tan CC, et al. NLRP1 inflammasome is activated in patients with medial temporal lobe epilepsy and contributes to neuronal pyroptosis in amygdala kindling-induced rat model. J Neuroinflammation 2015;12:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Fann DY, et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis 2013;4:e790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Adamczak SE, et al. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J Cereb Blood Flow Metab 2014;34:621–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Yogarajah T, et al. AIM2 Inflammasome-Mediated Pyroptosis in Enterovirus A71-Infected Neuronal Cells Restricts Viral Replication. Sci Rep 2017;7:5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Li XQ, et al. Knockdown of the AIM2 molecule attenuates ischemia-reperfusion-induced spinal neuronal pyroptosis by inhibiting AIM2 inflammasome activation and subsequent release of cleaved caspase-1 and IL-1beta. Neuropharmacology 2019;160:107661. [DOI] [PubMed] [Google Scholar]
- 256.Barclay WE, et al. The AIM2 inflammasome is activated in astrocytes during the late phase of EAE. JCI Insight 2022;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ma C, et al. AIM2 controls microglial inflammation to prevent experimental autoimmune encephalomyelitis. J Exp Med 2021;218(5):e20201796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 2018;19:610–21. [DOI] [PubMed] [Google Scholar]
- 259.Voet S, et al. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med 2022;7(8):e155563. [DOI] [PMC free article] [PubMed] [Google Scholar]