


Keywords: peritoneal dialysis, transplantation
Abstract
Key Points
Staphylococcus, Corynebacterium, Streptococcus, and Anaerococcus are the most common genera in the anterior nares.
The nasal abundance of Staphylococcus is inversely correlated with the nasal abundance of Corynebacterium.
Peritoneal dialysis patients have a distinctly diverse representation of Staphylococcus and Streptococcus in their anterior nares.
Background
The nasal passages harbor both commensal and pathogenic bacteria that can be associated with infectious complications. The nasal microbiome in peritoneal dialysis (PD) patients, however, has not been well characterized. In this study, we sought to characterize the anterior nasal microbiota in PD patients and assess its association with PD peritonitis.
Methods
In this study, we recruited 32 PD patients, 37 kidney transplant (KTx) recipients, and 22 living donor/healthy control (HC) participants and collected their anterior nasal swabs at a single point in time. We followed the PD patients for future development of peritonitis. We performed 16S ribosomal RNA (rRNA) gene sequencing of the V4–V5 hypervariable region to determine the nasal microbiota. We compared nasal abundance of common genera among the three groups using Wilcoxon rank-sum test with Benjamini–Hochberg adjustment. DESeq2 was also used to compare the groups at the amplicon sequence variant levels.
Results
In the entire cohort, the most abundant genera in the nasal microbiota included Staphylococcus, Corynebacterium, Streptococcus, and Anaerococcus. Correlational analyses revealed a significant inverse relationship between the nasal abundance of Staphylococcus and that of Corynebacterium. PD patients have a higher nasal abundance of Streptococcus than KTx recipients and HC participants. PD patients have a more diverse representation of Staphylococcus and Streptococcus than KTx recipients and HC participants. PD patients who concurrently have or who developed future Staphylococcus peritonitis had a numerically higher nasal abundance of Staphylococcus than PD patients who did not develop Staphylococcus peritonitis.
Conclusions
We find a distinct nasal microbiota signature in PD patients compared with KTx recipients and HC participants. Given the potential relationship between the nasal pathogenic bacteria and infectious complications, further studies are needed to define the nasal microbiota associated with these infectious complications and to conduct studies on the manipulation of the nasal microbiota to prevent such complications.
Introduction
The anterior nasal microbiota is at the interface between the external environment and the nasal passages and contains a combination of commensal and pathogenic bacteria. The most common genera defined in healthy individuals in the Human Microbiome Project are Staphylococcus, Corynebacterium, Propionibacterium, and Moraxella.1 Subsequent studies on the nasal microbiota have revealed microbiota dysbiosis in diseased states, such as chronic rhinosinusitis,2 and have linked the nasal microbiota to infectious complications after elective surgical procedures.3
Peritoneal dialysis (PD) patients undergo dialysis through PD catheter through their abdomen. Despite being taught sterile technique, PD patients experience both exit-site infections around the catheter and infectious peritonitis. Prior work has established that pathogenic bacteria in the nasal passages may be associated with infectious complications in PD patients. Luzar et al. reported that Staphylococcus aureus (S. aureus) nasal colonization was associated with exit-site infections in a cohort of 140 PD patients.4 Other studies have found that persistent nasal colonization with S. aureus was also associated with peritonitis.5,6 Decolonization with mupirocin has been suggested to prevent infections, and the MUPIROCIN Study Group found that nasal mupirocin prevented S. aureus exit-site infection.7 As such, the International Society of PD Catheter-Related Infection Recommendation 2017 Update recommends screening for S. aureus and treating with intranasal mupirocin if present.8 No study, to date, has comprehensively evaluated the anterior nasal microbiota in PD patients, which could be useful to understand how mupirocin could be beneficial.
In this pilot study, we evaluated the anterior nasal microbiota using 16S rRNA gene sequencing of the V4–V5 hypervariable region in PD patients, kidney transplant (KTx) recipients, and healthy controls (HCs).
Methods
Study Cohort Recruitment and Nasal Swab Specimen Collection
From August 2021 to January 2022, we recruited patients receiving PD, KTx recipients, and living donor/HC participants for anterior nasal swab specimen collection. All KTx recipients and living donor candidates were recruited from the clinic. Most PD patients were recruited in the PD clinic; several were recruited during hospitalization. The Weill Cornell Institutional Review Board approved this protocol (1604017181), and all participants provided written informed consent. This study adhered to the principles of the Declaration of Helsinki. The clinical and research activities being reported are consistent with the Principles of the Declaration of Istanbul as outlined in the “Declaration of Istanbul on Organ Trafficking and Transplant Tourism.”
Anterior nasal swab specimens were collected once from each participant using the Human Microbiome Project protocol. A Copan Eswab (Copan Diagnostics, Murietta, CA) was inserted into the anterior part of one nostril of the participant and turned twice and was then inserted into the anterior part of the other nostril and turned twice. The Copan Eswab was then placed into 1 ml of liquid Amies provided by the Copan Eswab technology and immediately stored on ice or 4°C. Aliquots of 300 µl were created in 2 ml cryovial and stored at −80°C within 12 hours.
16S rRNA Gene Sequencing of the V4–V5 Hypervariable Region
A single aliquot of approximately 285 μl was deposited into a Qiagen PowerBead glass 0.1-mm tube. Using a Promega Maxwell RSC PureFood GMO and Authentication Kit (AS1600), 1ml of CTAB buffer and 20 μl of RNAse A Solution were added to the PowerBead tube containing the sample. The sample/buffer was mixed for 10 seconds on a Vortex Genie2 and then incubated at 95°C for 5 minutes on an Eppendorf ThermoMixer F2.0, shaking at 1500 rpm. The tube was removed and clipped to a horizontal microtube attachment on a Vortex Genie2 (SI-H524) and vortexed at high speed for 20 minutes. The sample was removed from the Vortex and centrifuged on an Eppendorf Centrifuge 5430R at 40°C, 12,700 rpm for 10 minutes. On completion, the sample was centrifuged again for an additional 10 minutes to eliminate foam. The tube was then added to a Promega MaxPrep Liquid Handler tube rack. The Liquid Handler instrument was loaded with proteinase K tubes, lysis buffer, elution buffer, 1000 ml tips, 50 ml tips, 96-sample deep-well plate, and Promega Maxwell RSC 48 plunger tips. The Promega MaxPrep Liquid Handler instrument was programed to use 300 μl of sample and transfer all sample lysate into Promega Maxwell RSC 48 extraction cartridge for DNA extraction. On completion, the extraction cartridge was loaded into Promega Maxwell RSC 48 for DNA extraction and elution. DNA was eluted in 100 μl and transferred to a standard 96-well plate. DNA was quantified using Quant-iT double stranded DNA High-Sensitivity Assay Kit using Promega GloMax plate reader on a microplate (655087). 16S rRNA library generation followed the protocol from the Earth Microbiome Project.
Amplicon libraries were washed using Beckman Coulter AMPure XP magnetic beads. Library quality and size verification was performed using PerkinElmer LabChip GXII instrument with DNA 1K Reagent Kit (CLS760673). Library concentrations were quantified using Quant-iT double stranded DNA High-Sensitivity Assay Kit using Promega GloMax plate reader on a microplate (655087). Library molarity was calculated based on library peak size and concentration. Libraries were normalized to 2 nM using the PerkinElmer Zephyr G3 NGS Workstation (133750) and pooled together using the same volume across all normalized libraries into a 1.5 ml Eppendorf DNA tube (022431021). Sequencing was performed on an Illumina MiSeq instrument at loading concentration of 7 pM with 15% PhiX, paired-end 250 using MiSeq Reagent Kit v2, 500 cycles (MS-102-2003).
Bioinformatics Pipeline
Demultiplexed raw reads were processed using the Nextflow9 nf-core10 ampliseq pipeline,11 version 2.2.0, with the following parameters: profile singularity—input SampleSheet.tsv—FW_primer GTGYCAGCMGCCGCGGTAA—RV_primer CCGYCAATTYMTTTRAGTTT—metadata Metadata.tsv—outdir results—dada_ref_taxonomy silva—ignore_empty_input_files—ignore_failed_trimming—min_frequency 10—retain_untrimmed—trunclenf 240—trunclenr 160. Specifically, reads were trimmed with cutadapt,12 PhiX and quality filtering, and read pair merging, and amplicon sequence variant (ASV) resolution was performed with DADA2.13 Subsequent taxonomic assignment was also performed with DADA2, using the Silva reference database,14 version 138. Sequences that were assigned the families, Chloroplast and Mitochondria, were removed from downstream analyses.
Biostatistical Analyses
The distribution of categorical variables was compared using Fisher exact tests. The distribution of continuous variables was compared using Wilcoxon rank-sum tests, and to account for the comparison of multiple taxa, adjusted P values were calculated using Benjamini–Hochberg adjustment for multiple comparisons. DESeq2 was used to detect differences at the ASV between the groups using Benjamini–Hochberg adjustment. Comparison of correlations using a correlational matrix was adjusted for multiple comparisons using the Bonferroni method. All statistical tests were performed using R 4.1.3 in RStudio.
Data Availability
Sequencing data that support the findings of this study will be made available in the database of Genotype and Phenotype phs002251.v1.p1 after peer-reviewed acceptance. Local institutional review board approval will be needed to access the data.
Results
Characteristics of the Study Cohort and Nasal Microbial Sequencing
The microbiota in the anterior nares was performed using 16S rRNA gene sequencing of the V4–V5 hypervariable region in 32 PD patients, 37 KTx recipients, 22 HC participants, and three negative controls. A total of 1,116,291 reads with assigned taxonomy were obtained in the cohort of 91 participants with a median of 12,713 assigned reads with an interquartile range of 7132 and 16,018 assigned reads. The number of assigned reads in the three negative controls was 146, 308, and 529, below the number in the cohort of participants.
Table 1 presents the demographics of the participants. In general, the PD patients were older than the HC participants and similar in age to the KTx recipients. More than 50% of PD patients performed automated PD, and 15% had current Staphylococcus peritonitis or developed future Staphylococcus peritonitis within 10–12 months from the nasal specimen collection (last follow-up). Approximately one third of KTx recipients received deceased donor transplantation, and 32% were on trimethoprim/sulfamethoxazole (TMP-SMX) prophylaxis.
Table 1.
Demographics of the cohort
| Characteristic | PD Cohort (n=32) | KTx Cohort (n=37) | HC Cohort (n=22) |
|---|---|---|---|
| Age, yr | 63 (51–73) | 59 (52–67) | 51 (33–61) |
| Female sex | 20 (63%) | 17 (46%) | 13 (59%) |
| Ethnicity, n (%) | |||
| Hispanic | 3 (9) | 2 (5) | 3 (14) |
| Non-Hispanic | 27 (84) | 33 (89) | 16 (73) |
| Declined | 2 (6) | 2 (5) | 3 (14) |
| Race, n (%) | |||
| Asian | 4 (11) | 4 (11) | 1 (5) |
| Black | 13 (41) | 10 (27) | 5 (23) |
| White | 11 (34) | 20 (54) | 11 (50) |
| Other | 3 (9) | 2 (5) | 1 (5) |
| Declined | 1 (3) | 1 (3) | 4 (18) |
| History of hypertension, n (%) | 26 (81) | 36 (97) | 2 (9) |
| History of diabetes mellitus, n (%) | 9 (28) | 12 (32) | 0 (0) |
| Years on PD | 1.2 (0.6–2.4) | ||
| Automated PD, n (%) | 21 (66) | ||
| Concurrent or develops future, n (%) | |||
| Staphylococcus peritonitis | 6 (19) | ||
| Decreased donor transplantation | 14 (38) | ||
| Days post-transplantation | 364 (51–1637) | ||
| History of prior transplantation | 4 (11) | ||
| Maintenance immunosuppression, n (%) | |||
| Tacro/mycophenolic mofetil | 19 (51) | ||
| Tacro/mycophenolic mofetil/pred | 14 (38) | ||
| Tacro/mycophenolic acid | 1 (3) | ||
| Tacro/mycophenolic acid/pred | 3 (8) | ||
| TMP-SMX PPx | 12 (32) |
Categorical variables are represented by the number followed by the percentage in parentheses. Continuous variables are represented by the median followed by the interquartile range in parentheses. PD, peritoneal dialysis; KTx, kidney transplant; HC, healthy control; TMP-SMX, trimethoprim/sulfamethoxazole; ppx, prophylaxis; yr, year; Tacro, tacrolimus; pred, prednisone.
Anterior Nasal Microbial Diversity Differs across the Study Cohort
Microbial diversity among the study participants was measured at the ASV level using the Shannon diversity, an index that evaluates the richness and evenness in a community, and Chao1, an index that estimates the total number of ASVs in the specimens. Figure 1, A and B, shows box and whisker plots of these diversity indices and reveals that PD patients had a significantly higher Shannon diversity index and Chao1 diversity index than KTx recipients (P < 0.05, Wilcoxon rank-sum test) but similar to the HC participants (P > 0.05).
Figure 1.
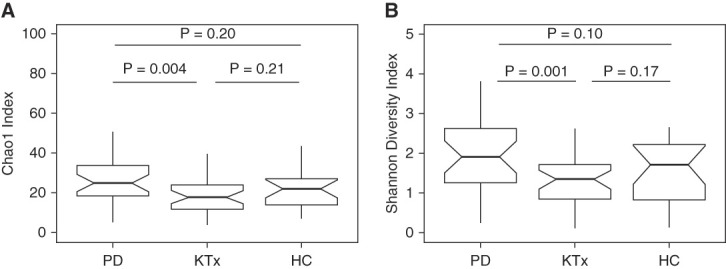
Distinct differences in Nasal Microbial Diversity among the study cohort. (A) Box and whisker plots of Chao1 index, the estimated number of ASVs, in the anterior nasal specimens from the PD cohort, the KTx cohort, and the living donor/HC cohort. The Chao1 index is on the y axis, and the study group is on the x axis. P value shown was calculated using the Wilcoxon rank-sum test. (B) Box and whisker plots of Shannon diversity index, a measure of evenness and richness, in the anterior nasal specimens from the three cohorts. The Shannon diversity index is on the y axis, and the study group is on the x axis. P values shown were calculated using the Wilcoxon rank-sum test. ASV, amplicon sequence variant; HC, healthy control; KTx, kidney transplant; PD, peritoneal dialysis.
Staphylococcus Abundance Negatively Correlates with Corynebacterium Abundance in Anterior Nasal Specimens
We further evaluated the anterior nasal microbiota among the study cohort at the genus level. At the genus level, the top abundant genera (>1% mean abundance across the cohort) included Staphylococcus, Corynebacterium, Anaerococcus, Streptococcus, unspecified Neisseriaceae, Moraxella, Cutibacterium, Peptoniphilus, and Finegoldia (Figure 2A). We performed a correlational matrix analysis among each of the genera (Figure 2B). The relative abundance of Staphylococcus was inversely correlated with that of Corynebacterium (Pearson r=−0.66, adjusted P value <0.10, Benjamini–Hochberg adjustment). The relative abundance of Peptoniphilus was positively associated with that of Anaerococcus (r=0.52, adjusted P value <0.10) and Finegoldia (r=0.27, adjusted P value <0.10). The relative abundance of Finegoldia was positively associated with that of Anaerococcus (r=0.60, adjusted P value <0.10). The relative abundance of unspecified Neisseriaceae was positively associated with that of Cutibacterium (r=0.31, adjusted P value <0.10).
Figure 2.
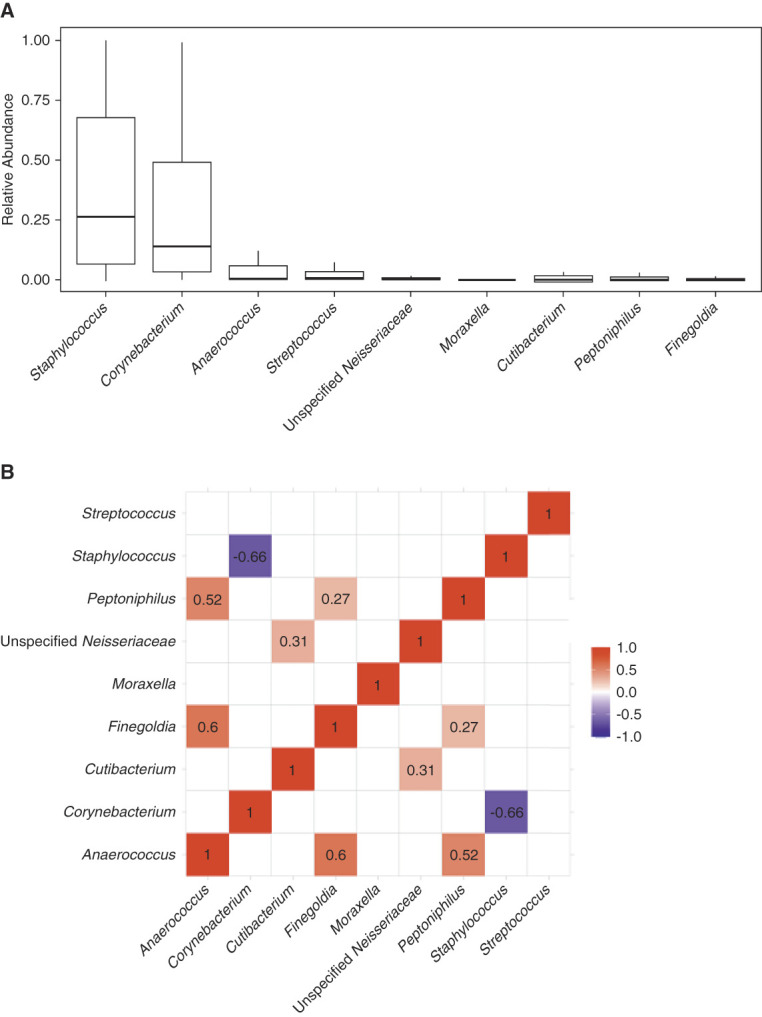
Significant correlations among the most common genera in the study cohort. (A) The most common genera in the anterior nasal microbiota (>1% mean relative abundance in the cohort). Box and whisker plots are represented to show the variation in the relative abundance of the genus (y axis) with the genus on the x axis. (B) A correlational matrix between the nasal abundance of the most common genera using Pearson r correlations with Benjamini–Hochberg adjustment for multiple hypotheses. The numbers shown are Pearson r correlations that had an adjusted P value <0.10. The color shows the strength of the correlation with red showing a positive correlation between two genera and blue showing a negative correlation between two genera.
Distinct Anterior Nasal Microbiota Define PD Patients and KTx Recipients
The individual profiles of the top genera in anterior nasal microbiota are shown in PD patients, KTx recipients, and HC participants (Figure 3). Figure 4 shows box and whisker plots of the top nine taxa among PD patients, KTx recipients, and HC participants, and Table 2 presents the comparisons among the groups using Wilcoxon rank-sum test with Benjamini–Hochberg adjustment. PD patients had a distinctly higher relative abundance of Streptococcus than KTx recipients or HC participants (adjusted P value <0.10, Wilcoxon rank-sum test, Benjamini–Hochberg adjustment) (Figure 4D). PD patients also had lower abundance of unspecified Neisseriaceae, Moraxella, Cutibacterium, and Peptoniphilus than HC participants (Figure 4G) (adjusted P value <0.10). KTx recipients had a lower abundance of Moraxella than HC participants (adjusted P value <0.10). Other than Streptococcus, KTx recipients had similar abundance of the top genera compared with PD patients (adjusted P value >0.10).
Figure 3.
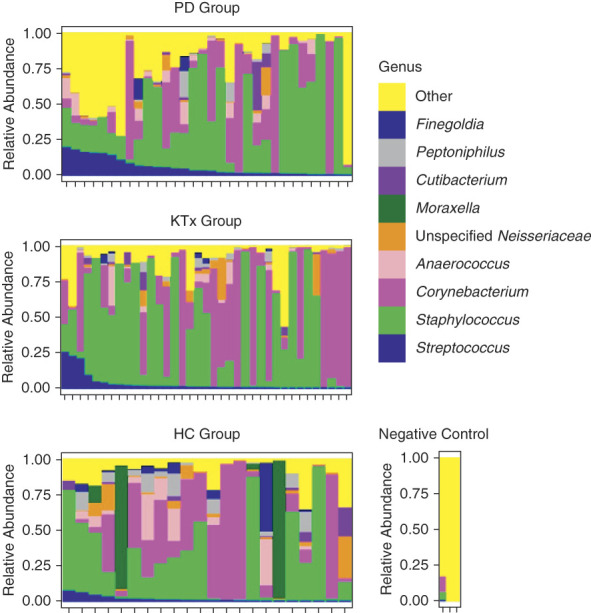
Individual microbiota profiles by the study cohort. The relative abundance of microbiota is on the y axis, and individual nasal specimens are on the x axis. The relative abundance of each genus is represented by color. The top panel represents anterior nasal microbiota profiles from the 32 PD patients, the middle panel represents the anterior nasal microbiota profiles from the 37 KTx patients, and the bottom panel represents the anterior nasal microbiota profiles from the 22 potential living donor/HC participants. The right panel represents the microbiota from three negative controls. HC, healthy control; KTx, kidney transplant; PD, peritoneal dialysis.
Figure 4.
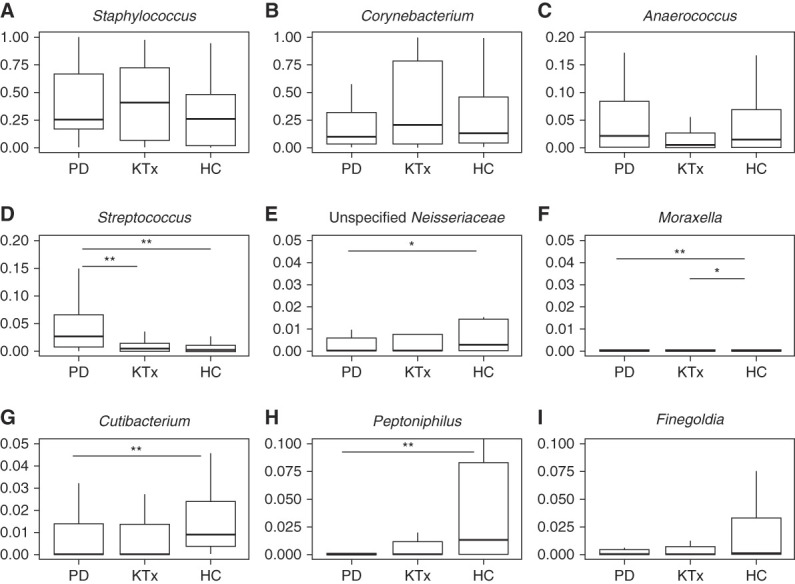
Distinct microbial differences among the cohort at the genus level. Box and whisker plots are represented with the relative abundance of individual genera on the y axis and the group on the x axis. PD cohort (n=32). KTx cohort (n=37). Living donor/HC cohort (n=22). P values were calculated using the Wilcoxon rank-sum test with Benjamini–Hochberg adjustment for multiple hypotheses. **Adjusted P value <0.05. *Adjusted P value <0.10. (A) Staphylococcus analysis. (B) Corynebacterium analysis. (C) Anaerococcus analysis. (D) Streptococcus analysis. (E) Unspecified Neisseriaceae analysis. (F) Moraxella analysis. (G) Cutibacterium analysis. (H) Peptoniphilus analysis. (I) Finegoldia analysis. HC, healthy control; KTx, kidney transplant; PD, peritoneal dialysis.
Table 2.
Comparison of the nasal abundance among the three cohorts at the genus level
| Genus | PD Cohort (n=32) | KTx Cohort (n=37) | HC Cohort (n=22) | PD versus HC | PD versus HC | PD versus KTx | PD versus KTx | KTx versus HC | KTx versus HC |
|---|---|---|---|---|---|---|---|---|---|
| Median Abundance | Median Abundance | Median Abundance | P Value | Adjusted P Value | P Value | Adjusted P Value | P Value | Adjusted P Value | |
| Anaerococcus | 0.020 | 0.004 | 0.014 | 0.999 | 0.999 | 0.238 | 0.722 | 0.303 | 0.389 |
| Corynebacterium | 0.098 | 0.200 | 0.127 | 0.509 | 0.573 | 0.271 | 0.722 | 0.701 | 0.701 |
| Cutibacterium | 0.000 | 0.000 | 0.009 | 0.016a | 0.049a | 0.942 | 0.942 | 0.029 | 0.133 |
| Finegoldia | 0.000 | 0.000 | 0.001 | 0.098 | 0.147 | 0.660 | 0.880 | 0.178 | 0.320 |
| Moraxella | 0.000 | 0.000 | 0.000 | 0.014a | 0.049a | NA | NA | 0.008a | 0.074a |
| Unspecified Neisseriaceae | 0.000 | 0.000 | 0.003 | 0.034a | 0.062a | 0.510 | 0.817 | 0.105 | 0.237 |
| Peptoniphilus | 0.000 | 0.000 | 0.013 | 0.022 | 0.049a | 0.382 | 0.763 | 0.046 | 0.139 |
| Staphylococcus | 0.249 | 0.403 | 0.257 | 0.406 | 0.522 | 0.891 | 0.942 | 0.285 | 0.389 |
| Streptococcus | 0.027 | 0.005 | 0.003 | 0.001a | 0.008a | 0.003a | 0.027a | 0.584 | 0.657 |
The median abundance of the most common genera is shown for the peritoneal dialysis cohort, the kidney transplant cohort, and the living donor/healthy control cohort. P values shown were calculated using the Wilcoxon rank-sum test between groups. Adjusted P values were calculated using Benjamini–Hochberg adjustment. P values with NA were unable to be calculated because the abundances were 0 in both groups. PD, peritoneal dialysis; KTx, kidney transplant; HC, healthy control; NA, not available.
P value or adjusted P value <0.10.
To gain further insight, we evaluated the taxa at the ASV level. We performed pairwise DESeq2 between the groups to identify ASVs that were consistently different among the groups. Figure 5 shows the significant log2-fold abundance changes between the groups, and Supplemental Tables 1–3 reveal the changes in the nasal abundances of the groups. Both PD patients and KTx recipients had significantly higher nasal abundances of Staphylococcus ASV 1 and Corynebacterium ASV 1 and lower abundance of Anaerococcus ASV 1 than HC participants (adjusted P value <0.10, Benjamini–Hochberg adjustment). PD patients had higher nasal abundance of Staphylococcus ASV 2, Abiotrophia ASV 1, and Porphyromonas ASV 1 than KTx recipients or HC participants (adjusted P value <0.10).
Figure 5.

Differential abundance analyses among the cohort at the ASV level. Differential abundance analyses were performed on the anterior nasal microbiota between the groups using DESeq2 with Benjamini–Hochberg adjustment for multiple hypothesis testing. On the y axis is the individual ASV with genus shown, and on the x axis is the fold difference in abundance. The fold difference directionality is represented above the graph. (A) Differential abundance analyses between the HC group and the PD group. (B) Differential abundance analyses between the KTx group and the PD group. (C) Differential abundance analyses between the HC group and the KTx group. ASV, amplicon sequence variant; HC, healthy control; KTx, kidney transplant; PD, peritoneal dialysis.
PD Patients Have a More Diverse Representation of Staphylococcus and Streptococcus than KTx Recipients and HC Participants
To further understand why PD patients have higher microbial diversity, we evaluated the diversity of ASVs in the most common genera: Staphylococcus, Corynebacterium, and Streptococcus (Figure 6). There were 61 different Staphylococcus ASVs identified in the whole cohort. PD patients had a significantly higher number of Staphylococcus ASVs per specimen than KTx patients (P = 0.03, Wilcoxon rank-sum test) and HC participants (P = 0.04) (Figure 6A). There were 95 different Corynebacterium ASVs identified in the whole cohort. PD patients, KTx patients, and HC participants had similar number of Corynebacterium ASVs per specimen (P > 0.10) (Figure 6B). There were 46 different Streptococcus ASVs identified in the whole cohort. PD patients had a significantly higher number of Streptococcus ASVs per specimen than KTx patients (P = 0.04) and HC participants (P = 0.05) (Figure 6C).
Figure 6.

Diverse representation of the most common genera in the study cohort. Each set of graphs represents the number of ASV from a particular genus by the group. PD group (n=32). KTx group (n=37). Living donor/HC group (n=22). The top graph presents box and whisker plots of the number of ASVs per specimen by the group. P value was calculated using the Wilcoxon rank-sum test: **P value <0.05, *P value <0.10. The bottom three graphs represent box and whisker plots of the relative abundance of individual ASVs from a particular genus. The relative abundance is on the y axis with the color representing individual ASVs, and anterior nasal specimens are on the x axis with the second top graph representing the PD cohort, the second bottom graph representing the KTx cohort, and the bottom graph representing the HC cohort. (A) The diversity of Staphylococcus ASVs in the study cohort. (B) The diversity of Corynebacterium ASVs in the study cohort. (C) The diversity of Streptococcus ASVs in the study cohort. ASV, amplicon sequence variant; HC, healthy control; KTx, kidney transplant; PD, peritoneal dialysis.
Clinical Factors, Outcomes, and the Nasal Microbiota
We next evaluated the relationship among the nasal microbiota, clinical factors, and outcomes in the cohort. There were no significant differences in the nasal abundances of the most common genera on the basis of age 65 years or older (Supplemental Table 4). The relative abundance of Peptoniphilus was significantly higher in male patients than in female patients (adjusted P value <0.10) (Supplemental Table 5). Within the entire cohort, 63 participants received antibiotics within 1 year of nasal swab collection and 28 participants did not. The relative abundance of Moraxella, Petoniphilus, and Cutibacterium was significantly lower in the participants who received antibiotics within 1 year than in the participants who did not (adjusted P value <0.10) (Supplemental Table 6). Among the KTx cohort, 12 KTx recipients were on TMP-SMX prophylaxis for Pneumocystic jirovecii prophylaxis and 35 were not. There were no significant differences in the nasal abundance of the most common genera between the KTx recipients on TMP-SMX and those who were not (Supplemental Table 7). In the PD cohort, six PD patients concurrently had Staphylococcus peritonitis or developed future Staphylococcus peritonitis within 10–12 months (last follow-up) (Staph Peritonitis Group) and 26 PD patients did not (No Staph Peritonitis Group). The nasal abundance of Staphylococcus was higher in the Staph Peritonitis Group than in the No Staph Peritonitis Group, but the difference was not statistically significant (median abundance 52% versus 24%, respectively, adjusted P value 0.73). There were no significant differences in the nasal abundance of the other most common genera between the Staph Peritonitis Group and the No Staph Peritonitis Group (Supplemental Table 8). In the PD cohort, ten PD patients had a history of peritonitis and 22 PD patients did not. The relative abundance of Finegoldia was significantly lower in the PD patients who had a history of peritonitis than in the PD patients who did not (adjusted P value <0.10) (Supplemental Table 9). PD patients who had a history of peritonitis had similar Shannon diversity index and Chao1 index compared with PD patients who did not have a history of peritonitis (Shannon diversity index, median 1.7 versus 2.1, P = 0.86; Chao1 index, median 29.5 versus 23.5, P = 0.67).
Discussion
This study aimed to describe the anterior nasal microbiota across different groups of patients with kidney disease. We detect a distinct microbial signature in the anterior nares of PD patients compared with KTx recipients and HC participants.
Many of the most common genera in the kidney cohort overlap with those reported in healthy individuals and include Staphylococccus, Corynebacterium, Finegoldia, and Cutibacterium.15,16 However, there were some distinct differences among the groups. PD patients had a higher nasal abundance of Streptococcus than HC participants or KTx recipients. Interestingly, having a higher nasal abundance of Streptococcus has been associated with respiratory infections, such as bronchiolitis, in infants.17 Although the most common type of infectious peritonitis is Staphylococcus in origin, Streptococcus peritonitis also occurs in PD patients. Our study was not able to directly address whether PD patients with nasal abundance of Streptococcus are associated with Streptococcus peritonitis and/or respiratory viral infections.
In our analysis, we noticed a higher nasal microbial diversity in the PD patients compared with KTx recipients and HC participants. Further analysis showed that part of this increased microbial diversity may be due to a more diverse representation of Staphylococcus and Streptococcus in PD patients (Figure 6). This may have interesting implications as prior data suggest that PD patients with S. aureus colonization had a higher incidence of exit-site infections.7 In our study, we did find an increased nasal abundance of Staphylococcus in PD patients who had a history of Staphylococcus peritonitis and/or developed Staphylococcus peritonitis. Although the association was not significant, it could be due to the low number of PD patients in the PD Staph peritonitis group as our study was not powered to detect the differences. At the 16S rRNA level, we were not able to determine Staphylococcus at the species level and this limitation prevented us to assess this association in more detail. It is possible that the relative abundance of Staphylococcus rather than the presence/absence of Staphylococcus in the anterior nares is associated with Staphylococcus peritonitis. Future studies using mupirocin to decrease the relative abundance of Staphylococcus in the anterior nares and possibly decrease the rates of peritonitis may be informative.
Our data also highlight a strong inverse association between the nasal abundance of Staphylococcus and that of Corynebacterium. Interesting mechanistic studies have shown a complicated relationship between these two taxa, which represent the most common taxa in the nasal microbiota. One study found that Corynebacterium species can secrete antimicrobial peptides against S. aureus18. Another study has shown that Corynebacterium species can decrease the virulence of S. aureus.19 Taken together, our data are consistent with the inverse relationship and suggest potential novel approaches to manipulate the nasal microbiota. For example, because Staphylococcus peritonitis is much more common than Corynebacterium peritonitis, establishing a Corynebacterium dominant nasal microbiota may be preventative of Staphylococcus in the nasal passages and possibly decrease the risk for Staphylococcus exit-site infection and/or peritonitis.
A surprising result is that we did not find an association between TMP/SMX and nasal microbiota differences. TMP/SMX has broad coverage against gram-positive cocci, including Staphylococcus species. There are few studies which have investigated the role of oral antibiotics on the nasal microbiota, and it is possible that intranasal antibiotics rather than oral antibiotics may more efficiently affect the nasal microbiota. Although our study is limited by the population size and the cross-sectional nature, our study raises this possibility.
There are several limitations to our study. As mentioned earlier, we are unable to assess species-level identification via 16S rRNA gene sequencing of the V4–V5 hypervariable region. Future studies using whole gene 16S rRNA gene sequencing or metagenomic sequencing may provide better resolution on the intricate intraspecies competition between the microbiota, particularly between Staphylococcus species and Corynebacterium species. Given the low biomass of the nasal microbiota, environmental contamination and/or contamination through the DNA processing steps could artificially introduce microbiota in our specimens. However, we did sequence negative controls (Figure 3), and the most abundant microbiota identified were not the most common nasal microbiota flora previously reported, suggesting that the nasal microbiota identified in our cohort was present in higher quantities and distinct. An important limitation is that demographics, cultural, and dietary practices could affect the nasal microbiota, and our study is not powered to detect the effect of these potential confounders, so a future larger study needs to be performed to validate our findings. Another important limitation is the cross-sectional design of the nasal microbiota component of the study. Although the cross-sectional nature of our study provides a snapshot of the microbiota across different groups of patients with kidney disease, it does not provide longitudinal changes. Such a longitudinal study may provide better insight into the relationship between the microbiota and outcomes in the populations.
In conclusion, we provide the first description of a distinct nasal microbiota signature in PD patients compared with KTx recipients and HC participants. We find a higher abundance of Streptococcus and a more diverse representation of Staphylococcus and Streptococcus in PD patients. Given the potential relationship between the nasal bacteria and infectious complications in PD patients, further studies are needed to define the nasal microbiota associated with these infectious complications and to conduct studies on the manipulation of the nasal microbiota to prevent such complications.
Supplementary Material
Acknowledgments
We thank Dr. Manikkam Suthanthiran for his overall guidance in this project. We thank the Microbiome Core Laboratory of Weill Cornell Medicine for processing the specimens.
Footnotes
V.S. and J.R.L. contributed equally to this work.
Disclosures
D.M. Dadhania reports the following: Employer: Weill Cornell Medical Center; Consultancy: Advisory Board—AlloVir Inc. and CareDx; Research Funding: AlloVir, CareDx, CSL Behring, and NIH; Patents or Royalties: Inventor on patent application W02018187521A2 entitled Methods of detecting cell-free DNA in biological samples; and Advisory or Leadership Role: Associate Editor, Transplantation; AST Committee Members; Member, LiveOnNY Medical Advisory Board; and Section Editor, Nephrology Dialysis Transplantation journal. I. De Vlaminck reports the following: Employer: Cornell University; Consultancy: Viracor Eurofins; Ownership Interest: Kanvas Biosciences and KariusDX; Patents or Royalties: Kanvas Biosciences, KariusDX, and Viracor Eurofins; and Advisory or Leadership Role: GenDX, Kanvas Biosciences, and KariusDX. C. Kraft reports the following: Employer: Emory University; Consultancy: Ferring/Rebiotix—consultant; and Advisory or Leadership Role: Advisory role, SERES Therapeutics; President, American Society for Microbiology. J.R. Lee reports the following: Employer: Weill Cornell Medicine; Research Funding: BioFire Diagnostics, LLC; Honoraria: Astellas; Patents or Royalties: Patent 2020-0048713-A1 “Methods of Detecting Cell-Free DNA in Biological Samples,” licensed to Eurofins Viracor; and Advisory or Leadership Role: Academic Editor, PLOS One; Chinese American Medical Society Board Member. M.J. Satlin reports the following: Employer: Weill Cornell Medicine; Consultancy: Shionogi: 2021: participation in a virtual advisory board; Research Funding: Biomérieux, Merck, and SNIPRBiome; Honoraria: AbbVie: 2023: participation in a data safety monitoring board; and Advisory or Leadership Role: Clinical and Laboratory Standards Institute, Co-Chair of Breakpoint Working Group and Subcommittee Member, and Subcommittee on Antimicrobial Susceptibility Testing. J. Silberzweig reports the following: Employer: The Rogosin Institute; Consultancy: I have done consultancy work for Alkahest, Bayer, Honeywell, Kaneka, and St. Gobain; Ownership Interest: My wife is an employee of Anthem and holds stock in the company. We hold stock in American Express, AT&T, IBM, and Wells Fargo; Research Funding: I am the national principal investigator for two clinical trials sponsored by Kaneka. One is using their Lixelle device for treatment of β-2-microglobulin amyloidosis in patients with ESKD; the other is using lipid apheresis for the treatment of focal glomerulosclerosis.; and Advisory or Leadership Role: American Society of Nephrology: Nephrologists Transforming Dialysis Safety COVID-19 and Other Current and Emerging Threats workgroup, COVID-19 Response team, Emergency Partnership Initiative. A.-C. Uhlemann reports the following: Employer: Columbia University; and Research Funding: Merck; unrelated to the current study. L.F. Westblade reports the following: Employer: Weill Cornell Medicine; and Research Funding: bioMerieux, Inc., Roche Molecular Systems, Inc., and Selux Diagnostics, Inc. S. Wu reports the following: Employer: CapitalRx. All remaining authors have nothing to disclose.
Funding
J.R. Lee: National Institute of Allergy and Infectious Diseases (R21AI164093).
Author Contributions
Conceptualization: Iman Khan, John Richard Lee, Vesh Srivatana, Anne-Catrin Uhlemann.
Data curation: Anika Hudson, Clayton Hughes, Iman Khan, John Richard Lee, Carol Li, Vesh Srivatana, Gabriel Stryjniak, Nicholas Tedrow, Sylvia Wu.
Formal analysis: Darshana M. Dadhania, Iwijn De Vlaminck, Iman Khan, Colleen Kraft, John Richard Lee, Michael J. Satlin, Jeffrey Silberzweig, Vesh Srivatana, Lars F. Westblade, Anne-Catrin Uhlemann, Sylvia Wu.
Supervision: John Richard Lee.
Writing – original draft: John Richard Lee.
Writing – review & editing: Darshana M. Dadhania, Iwijn De Vlaminck, Anika Hudson, Clayton Hughes, Iman Khan, Colleen Kraft, John Richard Lee, Carol Li, Michael J. Satlin, Jeffrey Silberzweig, Vesh Srivatana, Gabriel Stryjniak, Nicholas Tedrow, Anne-Catrin Uhlemann, Lars F. Westblade, Sylvia Wu.
Data Sharing Statement
Anonymized data created for the study are or will be available in a persistent repository on publication. Raw Data/Source Data; The Database of Genotypes and Phenotypes (dbGaP); Sequencing data that support the findings of this study will be made available in the database of Genotypes and Phenotypes (dbGaP) phs002251.v1.p1 after peer-reviewed acceptance. Partial restrictions to the data and/or materials apply. Local institutional review board approval will be needed to access the data.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/KN9/A388.
Supplemental Table 1. DESeq2 analysis of the nasal microbiota between the PD group and the HC group.
Supplemental Table 2. DESeq2 analysis of the nasal microbiota between the PD group and the KTx group.
Supplemental Table 3. DESeq2 analysis of the nasal microbiota between the KTx group and the HC group.
Supplemental Table 4. Comparison of nasal microbiota on the basis of patient's age at the genus level.
Supplemental Table 5. Comparison of nasal microbiota on the basis of patient's sex at the genus level.
Supplemental Table 6. Comparison of nasal microbiota on the basis of antibiotic usage within the past year at the genus level.
Supplemental Table 7. Comparison of nasal microbiota on the basis of trimethoprim/sulfamethoxazole usage in the kidney transplant recipients at the genus level.
Supplemental Table 8. Comparison of nasal microbiota on the basis of PD patients who developed Staphylococcus peritonitis status and PD patients who did not develop Staphylococcus peritonitis at the genus level.
Supplemental Table 9. Comparison of nasal microbiota on the basis of PD patients who had a history of peritonitis and PD patients who did not have a history of peritonitis at the genus level.
References
- 1.The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen F Gao W Yu C, et al. Age-associated changes of nasal bacterial microbiome in patients with chronic rhinosinusitis. Front Cell Infect Microbiol. 2022;12:786481. doi: 10.3389/fcimb.2022.786481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsiao CJ Paulson JN Singh S, et al. Nasal microbiota and infectious complications after elective surgical procedures. JAMA Netw Open. 2021;4(4):e218386. doi: 10.1001/jamanetworkopen.2021.8386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luzar MA Coles GA Faller B, et al. Staphylococcus aureus nasal carriage and infection in patients on continuous ambulatory peritoneal dialysis. N Engl J Med. 1990;322(8):505–509. doi: 10.1056/NEJM199002223220804 [DOI] [PubMed] [Google Scholar]
- 5.Nouwen JL, Fieren MW, Snijders S, Verbrugh HA, van Belkum A. Persistent (not intermittent) nasal carriage of Staphylococcus aureus is the determinant of CPD-related infections. Kidney Int. 2005;67(3):1084–1092. doi: 10.1111/j.1523-1755.2005.00174.x [DOI] [PubMed] [Google Scholar]
- 6.Ong LM Ch'ng CC Wee HC, et al. Risk of peritoneal dialysis-related peritonitis in a multi-racial Asian population. Perit Dial Int. 2017;37(1):35–43. doi: 10.3747/pdi.2015.00141 [DOI] [PubMed] [Google Scholar]
- 7.Mupirocin Study Group. Nasal mupirocin prevents Staphylococcus aureus exit-site infection during peritoneal dialysis. J Am Soc Nephrol. 1996;7(11):2403–2408. doi: 10.1681/ASN.V7112403 [DOI] [PubMed] [Google Scholar]
- 8.Szeto CC Li PK Johnson DW, et al. ISPD catheter-related infection recommendations: 2017 update. Perit Dial Int. 2017;37(2):141–154. doi: 10.3747/pdi.2016.00120 [DOI] [PubMed] [Google Scholar]
- 9.Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017;35(4):316–319. doi: 10.1038/nbt.3820 [DOI] [PubMed] [Google Scholar]
- 10.Ewels PA Peltzer A Fillinger S, et al. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020;38(3):276–278. doi: 10.1038/s41587-020-0439-x [DOI] [PubMed] [Google Scholar]
- 11.Straub D, Blackwell N, Langarica-Fuentes A, Peltzer A, Nahnsen S, Kleindienst S. Interpretations of environmental microbial community studies are biased by the selected 16S rRNA (gene) amplicon sequencing pipeline. Front Microbiol. 2020;11:550420. doi: 10.3389/fmicb.2020.550420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10. doi: 10.14806/ej.14817.14801.14200 [DOI] [Google Scholar]
- 13.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi: 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quast C Pruesse E Yilmaz P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41(D1):D590–D596. doi: 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oh J, Conlan S, Polley EC, Segre JA, Kong HH. Shifts in human skin and nares microbiota of healthy children and adults. Genome Med. 2012;4(10):77. doi: 10.1186/gm378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pereira PAB, Aho VTE, Paulin L, Pekkonen E, Auvinen P, Scheperjans F. Oral and nasal microbiota in Parkinson's disease. Parkinsonism Relat Disord. 2017;38:61–67. doi: 10.1016/j.parkreldis.2017.02.026 [DOI] [PubMed] [Google Scholar]
- 17.Hasegawa K Linnemann RW Mansbach JM, et al. Nasal airway microbiota profile and severe bronchiolitis in infants: a case-control study. Pediatr Infect Dis J. 2017;36(11):1044–1051. doi: 10.1097/INF.0000000000001500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Menberu MA Liu S Cooksley C, et al. Corynebacterium accolens has antimicrobial activity against Staphylococcus aureus and methicillin-resistant Staphylococcus aureus pathogens isolated from the sinonasal niche of chronic rhinosinusitis patients. Pathogens. 2021;10(2):207. doi: 10.3390/pathogens10020207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramsey MM, Freire MO, Gabrilska RA, Rumbaugh KP, Lemon KP. Staphylococcus aureus shifts toward commensalism in response to Corynebacterium species. Front Microbiol. 2016;7:1230. doi: 10.3389/fmicb.2016.01230 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data that support the findings of this study will be made available in the database of Genotype and Phenotype phs002251.v1.p1 after peer-reviewed acceptance. Local institutional review board approval will be needed to access the data.
Anonymized data created for the study are or will be available in a persistent repository on publication. Raw Data/Source Data; The Database of Genotypes and Phenotypes (dbGaP); Sequencing data that support the findings of this study will be made available in the database of Genotypes and Phenotypes (dbGaP) phs002251.v1.p1 after peer-reviewed acceptance. Partial restrictions to the data and/or materials apply. Local institutional review board approval will be needed to access the data.