

Abstract
The ability of the central nervous system (CNS) to generate innate immune responses was investigated in an in vitro model of CNS infection. Cultures containing CNS cells were infected with mouse hepatitis virus-JHM, which causes fatal encephalitis in mice. Immunostaining indicated that viral infection had a limited effect on culture characteristics, overall cell survival, or cell morphology at the early postinfection times studied. Results from Affymetrix gene array analysis, assessed on RNA isolated from virally and sham-infected cultures, were compared with parallel protein assays for cytokine, chemokine, and cell surface markers. Of the 126 transcripts found to be differentially expressed between viral and sham infections, the majority were related to immunological responses. Virally induced increases in interleukin-6 and tumor necrosis factor alpha mRNA and protein expression correlated with the genomic induction of acute-phase proteins. Genomic and protein analysis indicated that viral infection resulted in prominent expression of neutrophil and macrophage chemotactic proteins. In addition, mRNA expression of nonclassical class I molecules H2-T10, -T17, -M2, and -Q10, were enhanced three- to fivefold in virus-infected cells compared to sham-infected cells. Thus, upon infection, resident brain cells induced a breadth of innate immune responses that could be vital in directing the outcome of the infection and, in vivo, would provide signals which would summon the peripheral immune system to respond to the infection. Further understanding of how these innate responses participate in immune protection or immunopathology in the CNS will be critical in efforts to intervene in severe encephalitis.
Viral encephalitis is an emerging global health threat (60). Understanding the events that occur within the central nervous system (CNS) after viral exposure is necessary if effective therapeutic interventions against viral encephalitides are to be developed. The traditional concept that the brain is an immune-privileged site has given way to the current understanding that resident CNS cells, including microglia, astrocytes, and neurons, are capable of initiating innate immune responses (14, 54). Within the brain, these innate responses are critical in establishing protective immunity, and the defenses mounted by these cell types are the first to engage and counter viruses or other infectious agents. Innate immune responses also recruit leukocytes into the CNS and establish a microenvironment that can potentially direct the activity of infiltrating cells. However, innate immunity and its subsequent impact on adaptive responses can also contribute to virally induced immunopathology that can manifest as inflammatory neurologic disease (15, 38). Thus, the ability to effectively treat viral encephalitis requires an understanding of the role of innate immunity in disease resolution and pathogenesis.
Mouse hepatitis virus (MHV)-JHM is a coronavirus (genus Coronaviridae) that is particularly trophic for astrocytes but also infects oligodendrocytes and neurons (21, 48, 64). MHV-JHM is a highly virulent virus in the susceptible mouse. In the absence of prophylactic treatment, such as antiviral antibodies, adult C57BL/6 mice injected intracranially with 10 PFU of MHV-JHM succumb to fatal encephalitis within 5 to 7 days (8, 49). The extreme neurovirulence of MHV-JHM following intracranial or intranasal infection has been attributed to the extraordinarily rapid spread of the virus in the CNS relative to less-virulent variants (e.g., MHV-A59) and mutants (20). However, recent evidence suggests that the morbidity following MHV-JHM infection may arise from the lack of protective immunity capable of controlling viral replication and the generation of an immunopathogenic response (55, 56). The in vivo intracranial inoculation of MHV-JHM can induce high levels of macrophage inflammatory protein α (MIP-1α), MIP-1β, MIP-2, and interleukin-6 (IL-6) mRNA levels within the brain 3 days postinfection (55). Resident brain cells appeared to be the source of these immune mediators, as leukocyte infiltration did not reach a peak until days 5 and 8 after infection. The early chemokine response corresponded with a subsequent altered leukocyte infiltrate, consisting predominantly of macrophages. These results, supported by chimeric studies, indicated that the innate immune response (presumably induced by resident brain cells) contributes to disease pathology in the CNS (56).
However, immune events seen within the CNS are a net result of the combined actions of resident brain cells and the recruited leukocytes, making it difficult to identify the immune component that originates with resident cell populations. To address this issue, we used a well-defined in vitro model system, primary cultures of cerebellar cells derived from C57BL/6 mice. The in vitro model system is comprised of three types of resident CNS cells: neurons, astrocytes, and microglia. Employing this model, we investigated the CNS-derived innate immune responses generated following infection with MHV-JHM.
The response of resident brain cells to viral infection was evaluated at various time points following viral infection by Affymetrix gene profiling and measurement of intracellular or culture supernatant protein concentrations. The results reveal that the primary event upon viral infection in the brain is the production of a wide range of CNS immune responses, which have the capacity to influence disease outcome.
MATERIALS AND METHODS
Cerebellar cultures.
Neurons and glial cells were isolated from the cerebella of 7-day-old C57BL/6 mice according to standard enzyme dissociation methods as described previously (50), with the modification that antimitotics (e.g., 5-fluorodeoxyuridine) were not used. In brief, cerebella were dissociated in calcium-free saline with trypsin and DNase and then plated on MATRIGEL (Collaborative Products, Bedford, Mass.)-coated dishes. The cells, which are adherent, were grown in Dulbecco's modified Eagle medium--F-12 (Life Technologies, Gaithersburg, Md.) plus 10% horse serum supplemented with 30 mM glucose, 2 mM glutamine, 20 mM KCl, and penicillin (20 U/ml)-streptomycin (20 μg/ml).
Virus inoculation.
MHV-JHM was grown in DBT murine astrocytoma monolayers, and a lysate was prepared. The lysates were diluted over a 100-fold to 0.01 multiplicity of infection (MOI) in culture medium. The diluted lysate was added to the medium of the neuronal-glial cultures 5 days after establishment of the cultures, when the cells had achieved a mature culture phenotype. Control cultures were sham inoculated with equivalent amounts of culture medium. The medium was not exchanged after viral infection.
Plaque assays.
Viral production was measured in both the supernatant and cell fractions of the cultures by plaque assay on DBT murine astrocytoma monolayers grown at 80 to 90% confluence (43). Cell lysates and supernatants from control and infected CNS cultures were serially diluted, and titers were determined directly on monolayers. Cells from control and infected cultures were scraped and repeatedly pipetted through a narrow-bore pipette tip to disrupt cells. Cell lysates and supernatants were gently rocked for 90 min, and then the samples were aspirated. A 1% agarose (GIBCO BRL, Rockville, Md.) solution in Dulbecco's modified Eagle medium (GIBCO BRL) was overlaid for 3 days at 37°C. Cells were fixed with formaldehyde (Fisher Scientific) and stained with crystal violet before plaques were counted.
RNA isolation.
Total RNA was isolated from the control and virus-infected cultures by using the RNeasy mini kit (QIAGEN, Santa Clarita, Calif.). Briefly, the cell lysates were homogenized in the presence of guanidine isothiocyanate-containing buffer to inactivate RNases. The addition of ethanol promoted the binding of the RNA to the RNeasy membrane. DNase digestion was performed on a sample adsorbed to the membrane of an RNeasy spin column. Total RNA was eluted from the column with water. To confirm the integrity and size distribution of the collected RNA, the samples were run on a 1% agarose gel. Total RNA was estimated by measuring the A260.
Gene profiling.
Sham- and virus-infected cultures were set up in triplicate. One 24-h sample and two independent 72-h samples were evaluated. RNA was pooled from triplicate cultures for each sample. GeneChip analysis was performed by using Affymetrix reagents and protocols. Briefly, 10 μg of labeled cRNA was placed on a GeneChip (murine genome array U74Av2) and allowed to hybridize with probe pairs (37). Each probe pair consisted of one perfect match oligonucleotide of 25 bp and one mismatch oligonucleotide that contained a single mismatch at the 13th position. For each probe pair, a hybridization signal was calculated from the difference in hybridization between the perfect and mismatched probes. This signal is a quantitative measurement, representing the relative abundance of a transcript. The detection P value was based on a comparison of the probe pair intensities to a user-definable threshold. The distribution of probe pair intensities was analyzed by a one-sided Wilcoxon signed rank test which allows for the definition of a transcript as detected (present), not detected (absent), or marginal. All user-definable parameters necessary for these calculations were set at the Affymetrix default values. A detailed explanation of the analysis of the signal and detection algorithms can be obtained from Affymetrix. Analysis of GeneChip data was done with Affymetrix Microarray Suite, version 5.0, software. Genes that were defined as present in either the baseline or experiment sample and had a signal intensity of over 200 were selected for analysis. All gene changes at 24 and 72 h are shown as ratios of relative differences between virus and sham inoculations. The 72-h data represent means ± standard errors of the means (SEM).
ELISA.
Supernatants were collected from sham- and virus-inoculated cultures at 24, 48, 72, and 96 h after infection and analyzed for secretion of IL-6, tumor necrosis factor alpha (TNF-α), IL-1, MIP-1α, MIP-2, and GRO-α. Enzyme-linked immunosorbent assay (ELISA) antibodies, standards, and streptavidin-horseradish peroxidase were purchased as DuoSet kits from R&D Systems (Minneapolis, Minn.) and used according to the manufacturer's protocols. Ninety-six-well microplates (Fisher Scientific) were coated with capture antibody at concentrations given for each protocol and blocked in 1% bovine serum albumin (Sigma-Aldrich, St. Louis, Mo.) in phosphate-buffered saline (PBS, 100 mM, pH 7.3). Serially diluted culture supernatants and appropriate standards were bound by the corresponding detection antibody. The development reaction (H2O2 and tetramethyl benzidine; R&D Systems) was stopped with 2 N H2SO4 (Fisher Scientific), and plates were read at wavelengths of 595 to 450 nm.
Western blots.
Cultures were washed in ice-cold PBS, and lysates were prepared in lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.5% NP-40, a protease inhibitor cocktail tablet (Boehringer-Mannheim, Indianapolis, Ind.), and a cocktail of phosphatase inhibitors (4.5 mM sodium pyrophosphate, 10 mM β-glycerophosphate, 1 mM sodium fluoride, 1 mM sodium orthovanadate). After the addition of lysis buffer, cells were scraped and incubated on ice for 30 min and then centrifuged at 15,000 rpm in a Beckman J21 centrifuge for 30 min at 4°C. The supernatants were saved, and the protein concentrations were determined by using the Bio-Rad protein assay kit (Hercules, Calif.). Samples for each treatment were diluted with Laemmli sample buffer and then reduced for 10 min at 70°C prior to being size fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis with 10% Tris--glycine or 3 to 8% Tris--acetate gels (NuPage Novex; Invitrogen Life Technologies, Carlsbad, Calif.). Proteins were transferred to Immobilon P membranes (Millipore, Bedford, Mass.) overnight at 17 V. After blocking in 25% casein--PBS--0.1% Tween buffer, membranes were incubated in primary antibody solution at room temperature for 2 h and washed. The membranes were incubated for 1 h in secondary antibodies (anti-mouse or anti-rabbit; Pierce, Rockford, Ill.) coupled to horseradish peroxidase and then washed and visualized with a chemiluminescent system (Amersham, Encinitas, Calif.). Quantification was by densitometry measurements with NIH image software. The primary antibodies used for detection of cell-specific proteins were anti-glial fibrillary acidic protein (anti-GFAP, 1:5,000, clone MAB360; Chemicon International, Temecula, Calif.) for astrocytes, anti-F4/80 (1:100, clone CI:A3-1; Caltag, Burlingame, Calif.) and anti-microglial protein αM integrin (CD11b) (1:100, clone MAB 1458; Chemicon) for microglia, and anti-α-internexin (1:500, polyclonal AB5354; Chemicon International) and anti-synapsin 1 (1:1,000, polyclonal A-6442; Molecular Probes, Eugene, Oreg.) for neurons. Antibodies to procaspase 3 (1:000, polyclonal 06-735; Upstate, Lake Placid, N.Y.) and STAT-1α (1:000; polyclonal KAP-TF001; StressGen, Victoria, British Columbia, Canada) were also used. The antibodies used in this study have been well characterized by the companies that distribute them and by our laboratory.
Immunohistochemistry.
Cells were stained according to the Vectastain Elite kit protocol (Vector Laboratories, Burlingame, Calif.). Briefly, cells were fixed with 4% paraformaldehyde for 15 min. After rinsing with 5% sucrose three times for 10 min, cells were rinsed with PBS and incubated for 30 min in 0.05% Triton X-100 (Sigma) in PBS to permeablize the cells. Cells were rinsed in PBS and incubated with the endogenous biotin-blocking kit (Molecular Probes). Antibodies specific for the cell or molecule of interest were added overnight with appropriate normal serum and 0.5% bovine serum albumin to block nonspecific binding. As described above, anti-GFAP (1:800), anti-F4/80 (1:100), and α-internexin (1:5000) were used for the detection of astrocytes, microglia, and neurons, respectively. Oligodendrocytes were not detectable in the cultures as indicated by immunostaining with an antibody to myelin-oligodendrocyte-specific protein (1:750, clone MAB 328; Chemicon). Antibodies to IL-6 (5 μg/ml; R&D Systems, Inc.) and IL-1a (7 μg/ml; R&D Systems, Inc.) were also used. Following incubation with primary antibodies, cultures were washed with PBS and sequentially treated with biotinylated secondary antibody and the peroxidase-conjugated avidin-biotin complex provided in the Vectastain Elite ABC kit. Immunoreactivity was detected by the presence of a brown reaction product following incubation with 0.05% 3,3′-diaminobenzidine (Sigma).
RESULTS AND DISCUSSION
Viral infection has limited impact on culture characteristics at 72 h.
The general appearance of the mixed neuronal-glial cultures and the morphology of the cells in sham-infected cultures were comparable to that previously reported by this laboratory (50). Representative microscopic fields of the immunostained mixed neuronal-glial cultures are shown in Fig. 1A. The micrographs illustrate the relative contribution of each cell type to the cultures. The granule neurons were the most numerous cell type in the cultures (∼90% of cells) but were considerably smaller than the astrocytes (∼5% of cells) and microglia (∼5% of cells). The neurons were localized to neuronal clusters connected by fiber tracks. Astrocytes and microglia were primarily located in nonneuronal regions of the culture but could also be found within the neuronal clusters.
FIG. 1.

CNS culture characteristics. (A) Representative microscopic fields from control and virally infected cultures immunostained with cell-specific antibodies for neurons, astrocytes, and microglia. Immunostaining was carried out at 72 h postinfection. The unstained neuronal clusters (NC) are evident in the microscopic fields showing astrocyte (A) and microglial (M) immunostaining. (B) Western blot analysis demonstrates the relative level of the cell-specific proteins in the cultures. Results from virus-infected cultures were normalized (norm) to control values represented by the dotted line. Insets show representative Western blots. Means ± SEM are shown (two to five independent experiments; protein levels at 96 h reflect one experiment).
The ability of MHV-JHM to infect murine astrocytes, oligodendrocytes, and neurons in vitro and in vivo has been well documented (21, 34, 64). MHV-JHM was added to the neuronal-glial cultures at a dose of 0.01 MOI or 3.6 × 104 PFU/ml at 0 h. A low dose of MHV-JHM was used to simulate in vivo conditions after the inoculation of a low dose of MHV-JHM (10 PFU) was given to the whole brain. The initial low dose of inoculum allowed the in vitro infection to develop over a time span similar to that observed in an acute infection in vivo (20). Results indicated that the in vitro conditions were conducive to the development of infection and CNS response. By 72 h postinfection, viral titers in the cell lysates had increased over 10-fold and it is estimated that approximately 10% of cells were infected. The increase in viral titers indicated that active replication and secondary infection was occurring, simulating in vivo events (Fig. 2). Although correlating increases are observed in the supernatant, comparatively limited virus appeared to be shed into the supernatant (10-fold-larger increase in cell lysate at 72 h than in the supernatants). Cells and supernatants from sham cultures remained free of virus (data not shown).
FIG. 2.
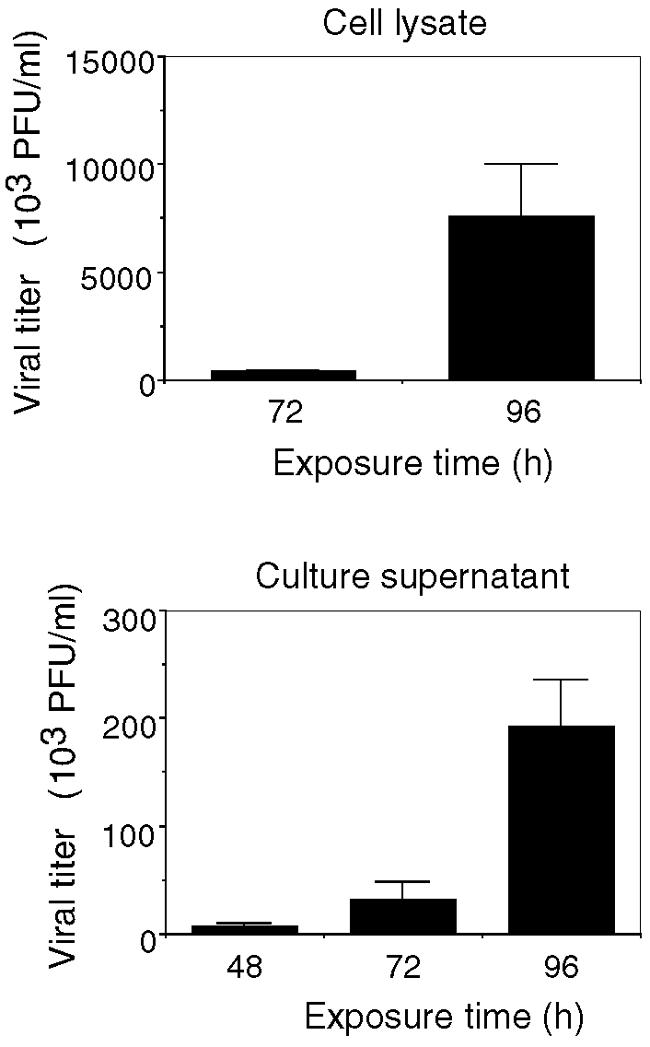
Viral titer kinetics. Cultures were either sham or MHV-JHM infected at 0 h. At the times indicated, cell lysates and culture supernatants were harvested and viral titers were determined by plaque assay. Virus was not detected in sham-infected cultures (data not shown). Means ± SEM are shown (two to seven independent experiments, duplicate cultures).
The impact of MHV-JHM infection on the culture characteristics and morphological features of the three cell types present in culture were evaluated with immunostaining and Western blot analysis of cell-specific proteins. Viral infection had limited cytopathic effects on the culture characteristics or cellular morphology at 72 h (Fig. 1). Although a slight decrease in the astrocyte marker GFAP production was apparent at 72 and 96 h, viral infection did not greatly alter the level of GFAP or the neuronal marker α-internexin, as determined by Western blotting (Fig. 1B). Similar results were obtained at the mRNA level by Affymetrix gene array analyses (data not shown). In contrast, in virally infected cultures, the levels of the microglial marker F4/80 (Fig. 1B) and αM integrin/CD11b (data not shown) were depressed at 24 h postinfection and increased with time, surpassing that of control cultures at 96 h (Fig. 1B). Changes in the microglial markers may have indicated an alteration in the activation stage, as numeric differences were not apparent by immunostaining.
The unchanged morphology at 72 h reaffirmed that the differences in cell biology (noted below) were the result of the natural development of the infection and not the consequence of a major toxic event due to overwhelming viral growth.
Cellular induction of innate immunity in response to viral infection.
To gain insight into the cellular events initiated by viral infection, genomic profiling and protein assays on the MHV-JHM- and sham-inoculated neuronal-glial cultures was performed. Limited changes in mRNA messages were observed at 24 h. In contrast, at 72 h, 126 mRNA messages were found by genomic profiling to be differentially expressed between virally and sham-infected cultures, reflecting the impact of the progression of infection. The overwhelming majority of these mRNA messages were immune-related genes (Fig. 3; Tables 1 to 5). Of the 32 genes upregulated over 10-fold in the presence of virus, all were directly involved in CNS immune responses (highlighted in Tables 1 to 5). Two ESTs, one thought to be immune related, were also enhanced 10-fold (data not shown). Virus-induced suppression of gene expression was observed for only 2 genes at 72 h (H19 and synapsin IIb) (Table 5). Together, these results clearly demonstrate that, upon viral infection, the primary response of cells within the CNS was to initiate an innate response, as opposed to modulation of other neuronal or glial cell functions. Note, a single gene may have one or more functions; thus, the tabular assignments, Tables 1 to 5, are attempts to categorize gene products for the purpose of discussion.
FIG. 3.

Gene expression categories by percentage. Gene array analysis was done as per Methods and Materials. Percentages represent a ≤2.5-fold increase in the expression genes from virus-infected cultures relative to sham-infected cultures at 72 h. ESTs, expressed sequence tags.
TABLE 1.
Cytokines and acute-phase reactants
| Gene product | Function or cellular expression and ligand | Affymetrix probe set identification | Gene symbol | GenBank accession no. | Change (n-fold) at time (h):
|
|
|---|---|---|---|---|---|---|
| 24 | 72 | |||||
| Cytokine | ||||||
| IL-6 | APRa neuropathogenic/protective | 102218_at | Il6 | X54542 | 1.3 | 37.3 ± 22 |
| TNF-α | APR, septic shock, proinflammatory | 102629_at | Tnf | D84196 | 0.9 | 100.0 ± 48 |
| IL-1α precursor | Fever, T-cell and M/Mo activation, neuropathogenic/protective | 94755_at | II1a | M14639 | 1.1 | 43.6 ± 11 |
| IL-1β | APR, T-cell and M/Mo activation, neuropathogenic/protective | 103486_at | II1b | M15131 | 1.1 | 41.4 ± 36 |
| IL-1 receptor antagonist | Inhibits IL-1 activity | 93871_at | II1rn | L32838 | 1.3 | 2.6 ± 0 |
| Macrophage colony-stimulating factor 1 | Macrophage growth factor | 101450_at | Csf1 | M21952 | 0.8 | 2.1 ± 0 |
| ISG-15 | Activation of NK cells (LPS), ubiquitin, homeostatic brain function | 161511_f_at | G1p2 | AV152244 | 0.9 | 15.5 ± 9 |
| ISG-15 | 98822_at | G1p2 | X56602 | 0.4 | 13.8 ± 2 | |
| Acute-phase response (IL-6 inducible) | ||||||
| Serum amyloid A-3 protein | 102712_at | Saa3 | X03505 | 1.6 | 73.7 ± 6 | |
| Preprocomplement component C3 | 93497_at | C3 | K02782 | 1.6 | 4.2 ± 1 | |
| Pentraxin-related protein 3 | 92731_at | Ptx3 | X83601 | 1.1 | 5.8 ± 0 | |
| C-type lectin, superfamily member 8 | 95951_at | Clecsf8 | AF061272 | 1.6 | 4.8 ± 1 | |
| Chemokine and cytokine receptors | ||||||
| P75 TNF receptor/TNFR2 | Endothelial cells, T cells, M/Mo; TNF-α, lymphotoxin-α | 94928_at | Tnfrsf1b | X87128 | 1.5 | 2.9 ± 0 |
| FPR-1 | Astrocytes, M/Mo, neutrophils; n-formyl- methionyl peptides (fMet-Leu-Phe) | 99387_at | Fpr1 | L22181 | 5.5 | 16.7 ± 0 |
| FPR-RS-2 | Microglia, M/Mo; amyloid beta 42 | 101800_at | Fpr-rs2 | AF071180 | 2.5 | 33.6 ± 7 |
APR, acute-phase response.
TABLE 5.
Growth, differentation, metabolism, and apoptosisa
| Gene product | Function | Affymetrix probe set identification | Gene symbol | GenBank accession no. | Change (n-fold) at time (h):
|
|
|---|---|---|---|---|---|---|
| 24 | 72 | |||||
| Glial cells | ||||||
| F4/80 gene, macrophages/microglia | Adhesion, signaling | 103507_at | Emr1 | X93328 | 0.7 | 3 |
| CD14 antigen, macrophages/microglia | Inflammatory responses | 98088_at | Cd14 | X13333 | 1.2 | 2 |
| Intracellular regulation | ||||||
| GADD/MYD | ||||||
| GADD45 gamma/MYD118 | MAP kinase (proliferation and apoptotic responses) activator | 101979_at | Gadd45g | AF055638 | 1.0 | 3.4 |
| GADD45 beta/MYD118 | MAP kinase activator | 102779_at | Gadd45b | X54149 | 1.0 | 4.7 |
| GADD45 beta/MYD118 | MAP kinase activator | 161666_f_at | Gadd45b | AV138783 | 1.6 | 8.0 |
| GADD34/MYD116 | Complexes with proliferating cell nuclear antigen | 160463_at | Myd116 | X51829 | 3.2 | 2.5 |
| JUNB | Transcriptional factor promoting cell proliferation | 102363_r_at | Junb | U20735 | 0.9 | 2.1 |
| Schlafen 2 | Inhibit cell proliferation, lymphocyte maturation | 92471_i_at | Slfn2 | AF099973 | 1.4 | 3.4 |
| B-cell leukemia/lymphoma 2A1d | Antiapoptosis | 93869_s_at | Bcl2A1d | U23781 | 1.1 | 2.9 |
| TNF-α-induced protein-3/A20 | Antiapoptosis | 99392_at | Tnfaip3 | U19463 | 0.8 | 29.5 |
| Zinc-finger protein 147 | Mediates estrogen actions | 100475_at | Trim25 | D63902 | 1.0 | 2.7 |
| H19 | Embryonic development | 93028_at | H19 | X58196 | 1.1 | 0.3 |
| Receptors | ||||||
| CD52 | Marker malignant T and B cells, maturing spermatozoa | 104606_a | Cd52 | M55561 | 1.2 | 3.8 |
| JTB | Proliferation, onogenesis | 102791_at | Jtb | AB016490 | 1.4 | 6.0 |
| Extracellular/secreted; proliferin/MRP embryonic growth factors | ||||||
| Proliferin 1/MRP-1 | 94838_r_at | Plf | K02245 | 0.9 | 4.0 | |
| Proliferin 2/MRP-2 | 93883_at | Plf2 | K03235 | 0.8 | 4.6 | |
| Proliferin 3/MRP-3 | 93929_s_at | plf3 | X16009 | 0.4 | 6.5 | |
| Neurons | ||||||
| Synapsin IIb | Neurotransmitter release | 160954_at | Syn2 | AF096867 | 0.8 | 0.6 |
| Purine nucleoside phosphorylase | Astrocyte regulation of adenosine guanosine | 93290_at | Pnp | U35374 | 1.1 | 4.3 |
| Adrenomedullin | Neuropeptide, hypotensive vasodilator | 102798_at | Adm | U77630 | 1.0 | 4.2 |
| Nerve injury-induced protein/ninjurin | Promotes axonal growth and nerve regeneration | 93318_at | Ninj1 | U91513 | 0.7 | 2.4 |
| Huntingtin-associated protein 1-A | Neuritic development and synaptic function | 102323_at | Hap1 | AJ002272 | 0.9 | 2.5 |
| Multiple cell types | ||||||
| *GTP cyclohydrolase 1 | Production of tetrahydrobiopterin and NO | 102313_at | Gch | L09737 | 0.6 | 5.4 |
| Ceruloplasmin | Multicopper oxidase | 92851_at | Cp | U49430 | 1.4 | 2.9 |
| Manganese superoxide dismutase | Oxidoreductase, manganese, mitochondrion | 96042_at | Sod2 | L35528 | 1.1 | 2.4 |
| Cytochrome P450 | Heme-thiolate monooxygenases, NADPH-dependent electron transport pathway; oxidizes steroids and fatty acids | 99979_at | Cyp1b1 | X78445 | 0.9 | 2.4 |
| Acyloxyacyl hydrolase | A phospholipase, lysophospholipase, diacylglycerollipase, and acyltransferase | 99838_at | Aoah | AF018172 | 0.3 | 8.8 |
| Cholesterol 25-hydroxylase | Cholesterol metabolism, oxidoreductase activity, steroid hydroxylase activity | 104509_at | Ch25h | AF059213 | 0.9 | 4.9 |
| Lipocalin 2 | Intracellular transport, neutrophil responses, sleep cycles | 160564_at | Lcn2 | X81627 | 0.8 | 3.4 |
| Spermidine synthase | Biosynthesis of spermidine from arginine | 92540_f_at | Srm | Z67748 | 1.1 | 2.7 |
GADD, growth and DNA damage inducible; MYD, myeloid differentiation response protein; JTB, jumping translocation breakpoint; MRP, mitogen-regulated protein; *GTP, guanidine triphosphate; MAP, mitogen-activated protein.
Induction of cytokine and acute-phase proteins.
A primary function of innate immunity is to provide an early defense against a pathogen within the tissue. This is accomplished, in part, by the induction of proinflammatory cytokines and acute-phase proteins. At 24 h postinfection, viral infection had negligible impact on cytokine mRNA levels at 24 h postinfection. In contrast, by 72 h, virally infected cultures demonstrated a greater than 10-fold upregulation of IL-6, TNF-α, pro-IL-1α, IL-1β, and interferon-stimulated protein 15 mRNA levels compared with sham-infected cultures (Table 1). Anti-inflammatory cytokines, IL-10 and transforming growth factor β (TGF-β) were not detected in either sham-infected or virally infected cultures, reinforcing the view that this response was proinflammatory nature (data not shown).
Supernatants from the virus- and sham-infected cultures were assayed for IL-6 and TNF-α by ELISA to determine whether the changes in mRNA expression resulted in parallel changes in protein levels. The culture supernatants showed limited increases in IL-6 and TNF-α protein concentrations at 24 and 48 h, reflecting mRNA levels (Fig. 4A). However, at 72 h, IL-6 and TNF-α levels in supernatants from virally infected cultures were elevated more than 300-fold compared to sham-infected culture supernatants, clearly indicating the ability of CNS cells to produce these cytokines in response to viral infection. IL-1α protein was not detected in the culture supernatants (data not shown) or upon immunohistochemical analysis (Fig. 4B), although elevated mRNA levels were observed in virally infected samples. This discrepancy may be a consequence of the inability to detect IL-1α-converting enzyme mRNA in the genomic analysis of our samples. Thus, the increased IL-1α mRNA expression seen upon infection with MHV-JHM and other encephalitic viruses may not correspond with simultaneous protein production (35, 47, 55). Alternatively, the IL-1 may be consumed rapidly at, or near, the site of synthesis and, thus, undetectable in the supernatant.
FIG. 4.

Virally induced cytokines and chemokines. (A) IL-6, TNF-α, MIP-1α, MIP-2, and GRO-α concentrations in the supernatants of control and virus-infected cultures at indicated times postinfection were determined by ELISA. A prominent increase in these proteins was observed 48 h postinfection. Means ± SEM are shown (2 to 14 cultures were measured for each time point). (B) IL-6 and IL-1β in cultures were evaluated by immunohistochemical staining. Strong immunostaining for IL-6 was observed in microglia in the virus-infected cultures, whereas no immunostaining was observed for IL-1β. The arrows indicate an unstained microglia. Panel 1 is a phase-contrast micrograph; panel 2 shows the bright-field image of the same field.
Previous studies have demonstrated that within days of either in vivo or in vitro viral infection of CNS cells, heightened IL-6 and TNF-α transcript levels result (35, 46, 55). Acutely expressed, IL-6 has been reported to contribute to neuroprotection (75), whereas chronic IL-6 synthesis has been implicated in aggravating CNS inflammatory diseases (10, 45). TNF-α production in the brain has been associated with neurotoxicity, although here there is limited evidence of toxicity under the conditions used in this study (28). Importantly, IL-6 and TNF-α are potent inducers of acute-phase responses and have the ability to act as mediators between the immune and endocrine systems (3, 6, 19). This activity of IL-6 and TNF-α is partially regulated through the transcription factor CCAAT/enhancer binding protein (C/EBP)-delta3, the mRNA level of which was enhanced (Table 4) (23, 27). In the CNS, acute-phase responses appear to be produced by the reactive astrocytes in response to proinflammatory cytokines (11). Following 72 h of MHV-JHM infection, serum amyloid A-3 transcripts were strongly upregulated (74-fold) along with procomplement C3 component and pentraxin-related protein 3 (Table 1). Procomplement C3 component, produced by microglia and astrocytes, is seen in the neuritic plaques of Alzheimer's disease and is thought to cause neuronal damage (7, 24). In contrast, pentraxin-related protein 3, made by astrocytes and some neurons in response to seizures and TNF-α, may be neuronal protective (52).
TABLE 4.
Intracellular regulation of innate immunity
| Gene product | Target response(s) | Affymetrix probe set identification | Gene symbol | GenBank accession no. | Change (n-fold) at time (h):
|
|
|---|---|---|---|---|---|---|
| 24 | 72 | |||||
| Transcription factor/DNA binding protein | ||||||
| IRF-7 | IFN responses | 104669_at | Irf7 | U73037 | 3.5 | 15.8 ± 3.0 |
| STAT-1 | IFN responses | 101465_a | Stat1 | U06924 | 2.1 | 12.3 ± 1.7 |
| ISGF-3 | IFN responses | 103634_at | Isgf3g | U51992 | 1.0 | 3.3 ± 1.2 |
| Interferon activated gene (IFI-205) | M/Mo responses | 94224_s_at | Ifi205 | M74123 | 0.9 | 17.6 ± 9.7 |
| C/EBP-3 | Immune and inflammatory responses | 160894_at | Cebpd | X61800 | 1.5 | 2.9 ± 0.4 |
| C/EBP-3 | Immune and inflammatory responses | 92925_at | Cebpd | M61007 | 1.4 | 2.5 ± 0.1 |
| Immediate early response | Immune and inflammatory responses | 94384_at | Ier3 | X67644 | 1.4 | 3.6 ± 0.4 |
| Nuclear factor-kappa B signaling | ||||||
| Iκ-B-zeta | Prevents NF-κB binding to DNA | 98988_at | Nfkbiz | AA614971 | 0.8 | 2.9 ± 0.4 |
| IκB-alpha | Traps NF-κB in the cytoplasm | 104149_at | Nfkbia | AI642048 | 1.4 | 2.8 ± 0.6 |
| IκB-alpha | Traps NF-κB in the cytoplasm | 101554_at | Nfkbia | U57524 | 0.9 | 2.7 ± 0.3 |
| 47-kDa GTPase family | ||||||
| IFN-γ-inducible protein 47 | B-cell responses | 104750_at | Irg47 | M63630 | 2.2 | 12.2 ± 2.7 |
| IFN-γ-induced GTPase | M/Mo cytokine, chemokine signaling | 96764_at | Iigp | AJ007971 | 1.0 | 29.1 ± 0.7 |
| IFN-γ-induced GTPase | M/Mo cytokine, chemokine signaling | 96764_at | Iigp | AJ007971 | 1.5 | 29.3 ± 12.3 |
| IFN-γ-induced GTPase | IFN responses | 98410_at | Gpti | AJ007972 | 1.2 | 5.4 ± 2.1 |
| IGTP | STAT1-dependent IFN responses | 160933_at | Igtp | U53219 | 0.9 | 9.5 ± 1.4 |
| T-cell-specific protein (TGTP) | T-cell or M/Mo responses | 102906_at | Tgtp | L38444 | 0.6 | 17.2 ± 8.2 |
| Other GTPases | ||||||
| Guanylate nucleotide binding protein 2 | M/Mo, fibroblast IFN responses | 104597_at | Gbp2 | AJ007970 | 2.5 | 31.7 ± 19.4 |
| Mx protein | Antiviral responses | 102699_a | Mx2 | J03368 | 1.1 | 6.9 ± 3.8 |
| Interferon inducible protein 1 | M/Mo IFN responses | 97409_at | Ifi1 | U19119 | 1.8 | 6.4 ± 2.4 |
| GARG/IFITa | ||||||
| GARG-16/IFIT-1 | Proinflammatory responses | 100981_at | Ifit1 | U43084 | 1.1 | 24.7 ± 7.9 |
| GARG-49/IFIT-3 | Proinflammatory responses | 93956_at | Ifit3 | U43086 | 1.9 | 16.9 ± 6.3 |
| GARG-39/IFIT-2 | Proinflammatory responses | 103639_at | Ifit2 | U43085 | 1.3 | 11.3 ± 5.1 |
| Translation factors/RNA binding protein, ELL-related RNA polymerase II | Elongation factor | 103891_i_at | Ell2 | AI197161 | 0.9 | 2.5 ± 0.1 |
| Miscellaneous | ||||||
| Ubiquitin specific protease 18 | Acts on ISG-15 | 95024_at | Usp18 | AW047653 | 1.8 | 33.3 ± 1.2 |
| Viperin/viral hemorrhagic septicemia virus induced gene 1 | Antiviral | 104177_at | Vip1 | AA204579 | 5.0 | 41.5 ± 6.9 |
IFIT, IFN-induced protein with tetratricopeptide repeats.
Expression of cytokine and chemokine receptors.
The potential for TNF-α to directly influence CNS cells is evident by the upregulation of TNF-α receptor mRNA in virus-infected cultures (Table 1). TNF-α receptors are expressed on microglia and astrocytes (2, 42). Virus infection also enhanced the expression of chemokine formyl peptide receptor (FPR) levels more than 16-fold (72 h) that of control cultures. Predominantly expressed in lymphoid tissues, FPR-1 is a high-affinity receptor for n-formyl-methionyl peptides (FMLP), which are chemotactic factors for neutrophils, eosinophils, and macrophages (29, 69). In macrophages, FMLP binding appears to activate intracellular calcium, priming the cells for antigen presentation and cytokine release (1). Whether similar events occur in microglia or astrocytes has not been investigated, although increases in both classical and nonclassical major histocompatibility complex (MHC) mRNA levels were observed in virally infected cells (see below) and an FMLP receptor has been previously detected in the CNS (33). FPR-RS-2 binds amyloid beta 42. Upregulated on microglia in response to inflammatory stimuli, such as TNF-α, FPR-RS-2 has been implicated in the pathogenesis of Alzheimer's disease (18).
Facilitating leukocyte recruitment in response to viral infection.
Another function of the CNS innate immune response is facilitating the recruitment of leukocytes by the coordinated upregulation of chemokines, adhesion molecules, and matrix metalloproteinases (MMPs). These molecules are critical in the transition from innate tissue responses to adaptive lymphocyte responses. Significant CNS chemokine activity upon infection was evident in the genomic data (Table 2). A marked increase in neutrophil-targeted chemokines was observed. MIP-2 mRNA was one of the few messages upregulated at 24 h postinfection (3.1-fold). At 72 h, MIP-2 transcript levels in virally infected cultures were increased 114-fold relative to levels in control cultures, consistent with our previous in vivo observations (55). Likewise, virus-induced GRO-α and LIX mRNA levels were 10-fold higher than in sham-infected cultures. These results were echoed in culture supernatant protein concentrations (Fig. 4A). MIP-2 and GRO-α supernatant protein levels were elevated 20- and 10-fold, respectively, at 72 h and further enhanced by 96 h. This increase may relate to the observation that neutrophils appear as the earliest infiltrates in the CNS after an attenuated MHV-JHM infection (76). In the milder infections, neutrophils appeared to be protective. However, neutrophils can also contribute substantially to immunopathology following more virulent infections (66), suggesting that their presence in severe encephalitis is harmful.
TABLE 2.
Leukocyte infiltration
| Gene product | Chemotactic target | Affymetrix probe set identification | Gene symbol | GenBank accession no. | Change (n-fold) at time (h):
|
|
|---|---|---|---|---|---|---|
| 24 | 72 | |||||
| Chemokines | ||||||
| KC/CXCL1 precursor | Neutrophils | 95348_at | Cxcl1 | J04596 | 1.3 | 11.8 ± 0.6 |
| KC/CXCL1 precursor | Neutrophils | 95349_g_at | Cxcl1 | J04596 | 1.4 | 16.6 ± 3.3 |
| LPS-inducible CXC chemokine/ ENA-78/CXCL5/GARG-39 | Neutrophils | 98772_at | Cxcl5 | U27267 | 1.0 | 11.3 ± 2.6 |
| RAS-related C3 botulinum substrate 2 | Neutrophils | 103579_a | Rac2 | X53247 | 1.1 | 3.2 ± 1.0 |
| MIP-2/CXCL2 | Neutrophils | 101160_at | Cxcl2 | X53798 | 3.1 | 113.7 ± 55.9 |
| MIP-1α/CCL3 | M/Mo, TH1 T cells, NK cells | 102424_at | Ccl3 | J04491 | 0.9 | 7.3 ± 0.7 |
| MIP-1β/CCL4 | M/Mo, TH1 T cells, NK cells | 94146_at | Ccl4 | X62502 | 0.4 | 8.4 ± 1.7 |
| MIP-1γ/CCL9 | Osteoclasts, leukocytes | 104388_at | Ccl9 | U49513 | 1.2 | 5.3 ± 2.6 |
| MCP-1/CCl2 (GARG) | T cells, M/Mo, basophils | 102736_at | Ccl2 | M19681 | 1.3 | 7.5 ± 4.4 |
| MCP-2/CCL8 | T cells, M/Mo, basophils, eosinophils | 92459_at | Ccl8 | AB023418 | 1.3 | 8.8 ± 0.6 |
| MCP-3/CCL7 (GARG) | T cells, M/Mo, basophils, eosinophils | 94761_at | Ccl7 | X70058 | 2.3 | 8.1 ± 4.8 |
| MCP-5/CCL12 | Monocytes | 93717_at | Ccl12 | U50712 | 1.0 | 43.1 ± 23.6 |
| IP-10/CXCL10 (GARG) | Activated TH1 T cells | 93858_at | Cxcl10 | M33266 | 0.6 | 72.9 ± 42.0 |
| RANTES/CCL5 | Memory TH1 T cells, NK cells, M/Mo, basophils, eosinophils | 98406_at | Ccl5 | AF065947 | 0.5 | 65.6 ± 53.2 |
| Cell adhesion | ||||||
| ICAM-1/CD54 | 96752_at | Icam1 | M90551 | 7.1 | 6.5 ± 1.2 | |
| VCAM-1/CD106 | 92558_at | Vcam1 | M84487 | 0.7 | 5.8 ± 1.4 | |
| VCAM-1/CD106 | 92559_at | Vcam1 | U12884 | 2.4 | 3.2 ± 0.8 | |
| Ly6e/thymic shared antigen 1 | 101487_f_a | Ly6e | U47737 | 1.1 | 3.0 ± 0.3 | |
| MMP | ||||||
| Stromelysin/MMP-3 | 98833_at | Mmp3 | X66402 | 0.5 | 13.5 ± 7.4 | |
| Procollagenase/MMP-13 | 100484_at | Mmp13 | X66473 | 0.7 | 18.7 ± 3.0 | |
In addition to the chemokines described above, there was a strong upregulation macrophage chemoattractant protein (MCP)- and MIP-1-related transcripts, along with MIP-1α protein expression. Although these chemokines are attractants for other types of leukocytes (Table 2), previous in vivo chimeric studies demonstrated that prior to MHV-JHM-induced mortality, high MIP-1α and MIP-1β mRNA levels were clearly associated with enhanced macrophage infiltration when compared with a less-virulent MHV strain (56). In the neuronal-glial cultures, astrocytes are a likely source of MIP-1β and -2, MCP-1, MCP-2, and interferon-inducible protein 10 (IP-10) (34). Our previously studies demonstrated that upon infection with a low dose of an MHV-JHM variant, expression of these chemokines was induced in astrocyte cultures and that the effect was abolished by UV inactivation of the viral infectivity (34).
Adhesion molecules and MMP are necessary to allow inflammatory lymphocytes to enter through the blood-brain barrier into the CNS. Intracellular adhesion molecule 1 (ICAM-1) was one of the few transcripts to be upregulated at 24 h (7.0-fold). However, unlike most transcripts that increased with time, its expression appeared stable between 24 and 72 h. Vascular cell adhesion molecule 1 (VCAM-1) and ICAM-1 expression along the blood-brain barrier allows leukocytes to attach and begin the process of infiltration into the CNS. This process is augmented by MMP digestion of the endothelial layer. Both MMP-3 and MMP-13 mRNA levels were enhanced in virus-infected cells (Table 2). Thus, these observations indicate that resident brain cells can initiate a vigorous recruitment of neutrophils and macrophages upon infection, which may subsequently result in either control of the infection, immunopathological injury, or death. In MHV-JHM infection and fatal encephalitis, this balance appears to lean towards immunopathological injury, as opposed to viral clearance.
Nonclassical antigen presentation upon viral infection.
Another critical function of CNS innate immunity supportive of the transition to adaptive immunity is resident cell interaction with infiltrating T cells in the context of MHC molecules (4, 14, 17, 53, 64). Small, but consistent, increases in classical MHC class I transcript expression were observed with virally infected cultures compared to sham-infected cultures (Table 3). These increases are likely to be microglial in origin, due to the differential propensity of microglia to express MHC class I compared to astrocytes following in vitro MHV-JHM infection and in other models (9, 17, 64). In an in vitro model of Theiler's murine encephalomyelitis virus infection, MHC class II, B7-1, B7-2, and CD40 protein expression were upregulated on the surface of microglia upon infection in a manner similar to the upregulation occurring with exposure to gamma interferon (IFN-γ) (46). Comparable expression of MHC class II, CD80, CD86, and CD40 was not observed in our studies. Thus, MHV-JHM-induced innate immunity in CNS cells does not appear to be supportive of antigen presentation, and an inadequate transition to adaptive immunity may result. This possibility is corroborated by studies of in vivo MHV-JHM infection where limited T-cell activity in the brain was observed (55).
TABLE 3.
Antigen presentation
| Gene product | Cell type presented to | Affymetrix probe set identification | Gene symbol | GenBank accession no. | Change (n-fold) at time (h):
|
|
|---|---|---|---|---|---|---|
| 24 | 72 | |||||
| MHC class I | ||||||
| β2 microglobulin | 93088_at | B2m | X01838 | 0.8 | 2.4 ± 0.0 | |
| Histocompatibility 2, K region | CD8 T | 93120_f_a | H2-K | V00746 | 1.3 | 2.7 ± 0.4 |
| Histocompatibility 2, D region locus 1 | CD8 T | 101886_f_at | H2-D1 | X52490 | 0.9 | 2.6 ± 0.5 |
| Histocompatibility 2, D region locus 1 | CD8 T | 97540_f_at | H2-D1 | M69069 | 1.3 | 3.2 ± 0.2 |
| Proteasome activator 28-alpha subunit | 101510_at | Psme1 | AB007136 | 1.1 | 2.8 ± 0.1 | |
| Proteasome activator 28-beta subunit | 100588_at | Psme2 | U60329 | 1.0 | 2.1 ± 0.1 | |
| Proteasome subunit type 10γ/MECI-1 | 101486_at | Psmb10 | Y10875 | 1.0 | 3.6 ± 0.5 | |
| TAP binding protein | 102689_at | Tapbp | AF110520 | 0.9 | 3.5 ± 0.1 | |
| TAP binding protein | 100154_at | Tapbp | AI836367 | 1.4 | 3.7 ± 0.8 | |
| Nonclassical MHC class 1 | ||||||
| Histocompatibility 2, M region locus 2 | Unknown | 101300_at | M26156 | 1.5 | 4.6 ± 0.8 | |
| Histocompatibility 2, Q region locus 1 | Unknown | 99378_f_a | H2-Q1 | M18837 | 1.0 | 3.5 ± 0.2 |
| Histocompatibility 2, Q region locus 2-k | Unknown | 102161_f_at | H2-Q2-k | X58609 | 1.2 | 2.4 ± 0.4 |
| Histocompatibility 2, Q region locus 10 | Alloreactive CD8 T, NK | 97173_f_at | H2-Q10 | M27134 | 0.6 | 4.9 ± 1.6 |
| Histocompatibility 2, T region locus 10 | γδ T | 93865_s_at | H2-T10 | M35244 | 1.1 | 4.2 ± 0.8 |
| Histocompatibility 2, T region locus 17 | γδ T | 101876_s_at | H2-T17 | M35247 | 1.2 | 3.7 ± 0.7 |
| Receptors to nonclassical MHC class I | ||||||
| Gp49 | Mast cell development, NK cell cytotoxicity | 100325_at | Gp49a | M65027 | 1.9 | 3.4 ± 0.2 |
| Gp49B | Mast cell development, NK cell cytotoxicity | 92217_s_at | Gp29b | U05265 | 1.7 | 3.8 ± 0.5 |
Viral infection enhanced nonclassical MHC class I molecule expression up to fivefold over sham-infected cultures. Nonclassical MHC class I may have less dependence on IFN-γ regulation than classical MHC (22), which could explain the enhanced nonclassical MHC expression in the absence of IFN-γ. Nonclassical class I molecules associate with β2-microglobulin in a manner similar to class I molecules but have restricted polymorphism (63). Little is known about the impact of nonclassical MHC presentation on disease outcome in the periphery, much less in the CNS. H2-T10 and H2-T17 present to γδ T cells, which along with NK T cells and NK cells can migrate into the CNS following parasitic helminth infection and the induction of experimental autoimmune encephalomyelitis (12, 62). Generally, the presence of γδ T cells, NK T cells, and NK cells had been associated with exacerbating disease outcome, although it has been suggested that nonclassical MHC class I expression on neurons might be protective (31, 36). Q10, thought to be liver specific, appears to bind a classical peptide repertoire and can interact with CD8 T cells and NK cells, potentially providing some protective immunity against viral infection (71). Q2-k and Q1 are also thought to be limited to the liver and to the thymus and intestinal epithelium, respectively (68). It is assumed that these molecules are presenting to, or interacting with, infiltrating lymphocytes (68, 71). However, the upregulation of Gp49 mRNA in virus-infected cells is curious. Gp49 is a membrane receptor that contains immunoreceptor tyrosine-based inhibitory motifs. Expressed on NK cells, it inhibits their activation and release of immune reactants in response to nonclassical MHC class I expression and other stimuli, suggesting that a resident brain cell may act in a similar manner (30, 67). The potential role of this molecule expressed in virally infected CNS cells is unknown.
Transcription factors and regulators.
The expression of the secreted and membrane-bound immune mediators is regulated by intracellular events (Table 4). The discovery of many of the gene products listed was accomplished by evaluating cellular activity in response to IFN-γ or lipopolysaccharide (LPS), potent initiators of immune responses. Here, the presence of IFN-α/β or IFN-γ mRNA in the gene profiling experiments was limited, and IFN-γ protein was not detected in supernatants from virally infected cultures (data not shown). In addition, the IFN-γ receptor message level was not significantly different between sham and infected cultures at 72 h (mean signal intensities, 571 ± 155 and 473 ± 125, respectively). With restricted amounts of interferons, interferon regulatory factor 7 (IRF-7) was the likely intermediary between viral replication and induction of interferon sensitive transcription factors. IRF-7 is a distinct and essential transcription factor that regulates interferon antiviral responses (58). IRF-7 was one of the few transcripts enhanced at 24 h, supporting its ability to activate subsequent events observed at 72 h (Table 4). First discovered as a factor in Epstein-Barr virus pathogenesis and latency, IRF-7 can be activated by Epstein-Barr virus latent membrane protein 1, other viral components, NF-κB, and signal transducer and activation of transcription 1 (STAT1) (39, 58). Latent membrane protein 1 directly induces the expression, phosphorylation, and nuclear translocation of IRF-7, which can regulate both viral and cellular genes, including GTPases (44, 73). MHV-JHM spike and hemagglutinin esterase have also been shown to induce specific CNS genes (56, 74). The potential of these proteins to interact with IRF-7 is a subject for future investigation.
STAT1 mRNA levels and protein concentrations were substantially increased following viral infection of neuronal-glial cultures (Table 4; Fig. 5). STAT1 is considered the primary mediator of IFN-γ responses; nonetheless, it can also be activated by IFN-α/β, IL-6, granulocyte colony stimulating factor, and other mediators (51). Activated STAT1 binds to interferon-dependent positive-acting transcription factor 3 (ISGF-3)/p48 (a member of the IRF of the transcription factor family), which is also upregulated (Table 4). Together, they form the ISGF-3 complex, which translocates into the nucleus and binds to interferon-stimulated response elements in the promoters of interferon-inducible genes, including many GTPases, cytokines, and chemokines.
FIG. 5.
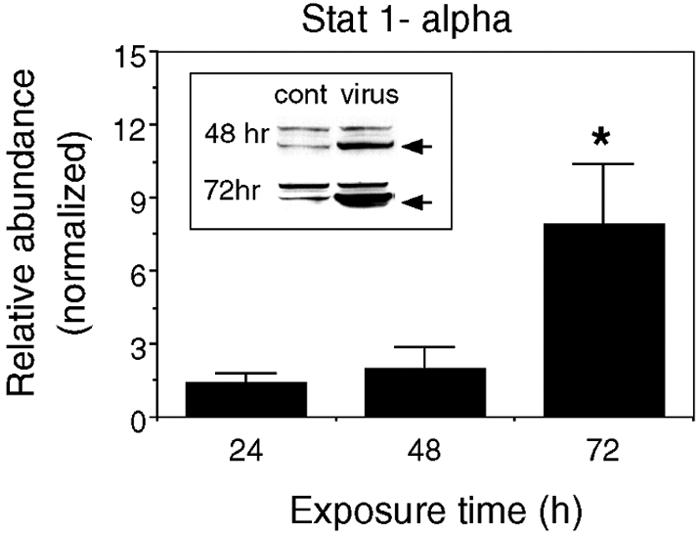
Viral infection induces expression of STAT1α. Mean values (± SEM) for the expression of STAT1α protein in virus-infected cultures relative to control (cont) cultures are shown (two to five independent experiments; *, P < 0.05). The inset shows representative Western blots (arrows indicate STAT1α bands).
Unlike many of the gene products listed in these tables, NF-κB and its pathways have been well characterized. Triggered by cytokines, bacterial products, and viruses, NF-κB is involved in the regulation of over 100 genes of the apoptotic, growth, immune, and other pathways (26). Little has been reported about the impact of MHV on NF-κB pathways. Increased expression in negative regulators of NF-κB may indicate an internal attempt to control its function (Table 4). The outcome can be surmised by the other events described here.
GTPases and glucocorticoid attenuated response gene (GARG).
MHV-JHM infection resulted in considerable elevation of members of the p47 GTPase superfamily. These proteins appear to localize to the endoplasmic reticulum, presenting the possibility that they function in processing or trafficking of proteins to the cell surface (5). Five of the six known members of this family, IIGP, GPTI, TGTP, IFN-inducible GTPase (IGTP), and IFN-γ-inducible protein 47 (with the exception of LRG-47), were expressed at higher levels (5- to 29-fold) in MHV infected cells. Reports indicate that these GTPases are involved in the generation of innate immune responses against specific pathogens (13, 57, 65, 72). IGTP knockout mice appeared highly susceptible to the protozoan parasite Toxoplasma gondii but not to Listeria monocytogenes and cytomegalovirus infections (65). Although IGTP upregulation upon T. gondii infection is IFN-γ independent, it appears to be STAT1 (Table 4) dependent (16). In contrast, IIGP seems necessary for protection against intracellular pathogens such as L. monocytogenes (72). IIGP had been reported to be upregulated in the CNS, along with Mx, upon transmissible spongiform encephalopathy infection (57). TGTP may, in a manner similar to Mx, contribute to protection against negative-strand RNA viruses (13). Whether TGTP and Mx (or any of the GTPases upregulated here) offer protection against positive-stranded RNA viruses, such as MHV, aggravate the proinflammatory response leading to fatal encephalitis, or both, is unknown. The ability of Mx to bind to viral proteins and interfere with assembly and transport of intact virion out of the cell is the best characterized of these GTPases (25). Mx and other larger GTPases can be directly activated by IRF-7 in the absence of interferons (59). However, with the exception of IGTP, studies investigating IRF-7 and/or STAT1 regulation of p47 GTPases have not been published.
MHV-JHM infection of CNS cultures resulted in an 11- to 25-fold change in GARG-16, -39, and -49 mRNAs relative to control cultures. GARGs are highly conserved proteins with multiple repeat domains that are inhibited by glucocorticoids (61). Although inducible by IFN-γ and LPS, this report suggests they are not IFN-γ dependent. GARG-16 production by microglia has been described and is thought to play a role in the regulation of inflammatory responses in the CNS (32).
Thus, despite the limited appearance of interferons, considerable upregulation of genes (and proteins) known to be interferon inducible was evident. Similar results have been found in models of spontaneous experimental autoimmune encephalomyelitis and spongiform encephalopathies (40, 57). Although these studies were performed with whole animal tissues, allowing for the infiltration of leukocytes from the periphery, they support our finding that interferons, particularly IFN-γ, may not be required for the activation of these responses within the CNS.
Viral effects on cell viability.
Consistent with the culture characteristics (Fig. 1) at 72 h, apoptosis was not evident in either virus-infected or control cultures. mRNA levels for caspases and death domain proteins were not upregulated in virus-infected cultures relative to sham-infected cultures (data not shown). Western blot analysis for procaspase 3, the precursor of active caspase 3, did not exhibit increased expression upon viral infection (Fig. 6A), and active caspase 3 was undetectable in both sham- and virus-infected cultures (data not shown). Instead, MHV-JHM infection resulted in elevated levels of genes associated with antiapoptotic activity (Bc1-2A1d and A20) and cell growth (GADD34, JUNB, JTB, and proliferin) (Table 5). Finally, total protein levels in sham- and virus-infected cultures were similar (Fig. 6B). Although some of these genes (e.g., GADD45) can also participate in apoptosis, together, these results indicate that virally induced apoptosis is limited at 24 and 72 h. Therefore, the observed alterations in mRNA and protein levels following viral infection appear to be a consequence of initial cellular responses to infection as opposed to a function of apoptotic events.
FIG. 6.
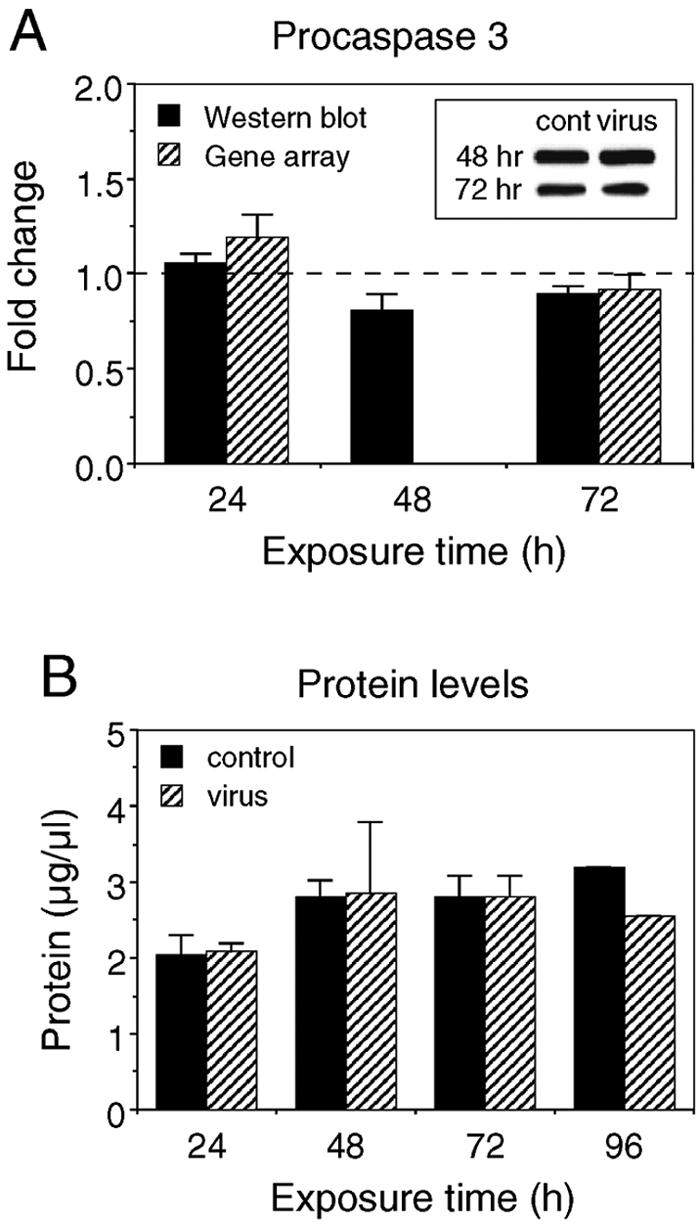
Cell viability was not altered by viral infection at 72 h. (A) Western blot analysis for procaspase 3 at indicated times postinfection. Results from virus-infected cultures are normalized to control (cont) values, represented by the dotted line. Insets show representative Western blots. (B) Protein levels in the cultures at indicated times postinfection. Means ± SEM are shown (two to five independent experiments; results for protein level at 96 h are from one experiment).
Neuronal effects of viral infection.
The predominant cell type in the neuronal-glial cultures is neurons. However, only a few of the mRNAs differentially regulated between sham- and virus-infected cultures in the genomic analysis are neuron specific (Table 5). For example, the gene array data indicated that viral infection did not alter the expression of mRNA for synapsin 1, a synaptic protein involved in neurotransmitter release that is found at high abundance in CNS neurons (data not shown). Western blot analysis corroborated this finding at the protein level (Fig. 7A). The majority of the genes related to neuronal growth or function that were altered upon viral infection are thought to favor neuronal survival, including ninjurin and Huntington-associated protein 1-A.
FIG. 7.

Viral infection slightly alters neuronal properties. (A) Western blot and gene array data showing the relative level of mRNA and protein for the alpha-1 subunit of the synaptic vesicle protein synapsin 1 at 72 h. (B) Representative recordings of intracellular Ca2+ in a neuron of a control (cont) and virus-infected culture at 48 h postinfection. The neurons were stimulated at the time indicated by the arrow with a brief pulse (1 s) of high-K+ saline applied from a micropipette placed near the neuron. (C) Digitized micrographs of fura-2-loaded granule neurons used for measurement of intracellular Ca2+ levels. (D) Mean (± SEM) values for the peak amplitude of the Ca2+ signals (with resting Ca2+ levels subtracted) in the population of neurons studied in control and virus-infected cultures at different times postinfection. Data are normalized to the mean value for control cultures at the same time point. (E) Mean (± SEM) resting Ca2+ levels at the three time periods studied postinfection. A significant increase in resting Ca2+ levels was observed at 72 h postinfection. The neuronal population is the same as shown in panel D (*, P = 0.05). (F) L-type Ca2+ channel Western blot and gene array analysis. Insets shows representative Western blots.
To further assess the impact of viral infection on the functional properties of the neurons, we examined their ability to respond to a depolarizing stimulus, the brief application of high-K+ saline. The high-K+ saline depolarizes the neuronal membrane potential, resulting in Ca2+ influx through voltage-gated Ca2+ channels, a process similar to that occurring during action potential generation. The Ca2+ signal was measured by using fura-2-based Ca2+ imaging. At 48 h postinfection, the Ca2+ signal produced by K+ application to neurons in the virus-infected cultures was smaller than the Ca2+ signal produced in control neurons. However, this effect was transient, and no difference in the Ca2+ signals was observed at the later times studied (Fig. 7B to D). Resting Ca2+ levels were comparable in control and virus-infected neurons until 72 h postinfection, when neurons in the virus-infected cultures demonstrated an elevation in resting Ca2+ levels relative to neurons in control cultures (Fig. 7E). Consistent with these results, Western blot analysis indicated that, prior to 72 h postinfection, the virus had limited effect on the level of protein for the L-type Ca2+ channel (Fig. 7F), the primary Ca2+ channel involved in the generation of the Ca2+ signal following K+ application. Together, these results suggest that at 72 h postinfection neuronal function was slowly beginning to be affected. However, the disruption of neuronal function does not appear to be a primary outcome at the early stages of infection investigated here.
Summary and implication of results.
The innate capacity of resident brain cells to respond to local insults within the brain is still poorly understood. To elucidate how early CNS responses may participate immunologically in the outcome of fatal encephalitis, the impact of MHV-JHM infection on mixed neural-glial cell cultures was evaluated by gene profiling and protein analysis. Viral infection of CNS cells resulted in the strong induction of proinflammatory responses by 72 h postinfection (Fig. 3; Tables 1 to 4). These results suggest that beyond cytokines, chemokines, and classical MHC class I products, a broader scope of innate immune molecules (including interferon-stimulated protein 15 [ISG-15], FPRs, nonclassical MHC class I, IRF-7, STAT1, small-size GTPases, GARGs, GADD45, and proliferin) may participate in fatal viral encephalitis. In contrast, limited differences in cell survival, metabolism, and neuronal function, aside from promoting neuronal growth between sham- and virus-infected cultures were observed (Fig. 2 and 7; Table 5).
Because a low MOI was used in our experiments, the observed changes in host gene expression probably represent both direct viral effects on the infected cells and bystander effects, as seen upon in vivo infection. Production of immune factors by a limited number of MHV-JHM-infected cells could also result in the altered gene expression of uninfected cells. Resident brain cells can express receptors that correspond to immune receptors, as demonstrated in this study by the expression of TNF-α receptor and FPR transcripts (Table 1), and produce immune factors when stimulated by appropriate signals.
The robust proinflammatory responses by CNS cells in vitro parallel those of previous in vivo reports (4, 10, 11, 34). However, in vitro CNS preparations allowed unequivocal discrimination of virally induced innate CNS responses from infiltrating leukocyte responses. This immunological activity can contribute to either protective or pathogenic mechanisms. Moreover, protective immune responses can also be immunopathological, as in the case of MHV-induced demyelination. Our previous studies of chimeric viruses demonstrated that the MHV-JHM spike glycoprotein was sufficient to trigger immunopathology leading to death. On a virus background (MHV-A59) that allowed for viral clearance, the presence of the MHV-JHM spike protein resulted in cytokine (IL-6) and chemokine (MIP-2 and MIP-1α) mRNA expression concomitant with high mortality (56). The discrepancy between virus clearance and corresponding neurological disease has been documented for other viral agents. Reports indicated that, following resolution of herpes simplex virus titers upon treatment, approximately one-third of encephalitis survivors subsequently die or become neurologically impaired (41). Moreover, cytokine levels or the severity of clinical outcomes did not correlate with cerebrospinal fluid herpes simplex virus loads (70). Therefore, the successful treatment of severe encephalitis may require viral elimination and down regulation of immunopathogenic responses. This study provides potential new targets for research and therapeutic intervention in severe viral encephalitis.
Acknowledgments
This work was supported by grants from the NIH, MH63712 (to D.L.G.) and AI 25913 and AI 43103 (to M.J.B.), core support from P30 MH62261-04, and fellowships from the National MS Society and the MS Society of Canada (to J.D.R.).
We thank The Scripps Research Institute DNA Core for assistance with the gene profiling experiments.
REFERENCES
- 1.Ayala, A., and I. H. Chaudry. 1994. Hemorrhage induces a reduction in the capacity of macrophages to mobilize intracellular calcium secondary to formyl-methionyl-leucyl-phenylalanine stimulation: association with alterations in cells surface Fc receptor expression and increased prostaglandin release. Shock 1:228-235. [DOI] [PubMed] [Google Scholar]
- 2.Becher, B., M. Blain, and J. P. Antel. 2000. CD40 engagement stimulates IL-12 p70 production by human microglial cells: basis for Th1 polarization in the CNS. J. Neuroimmunol. 102:44-50. [DOI] [PubMed] [Google Scholar]
- 3.Berczi, I., D. A. Chow, and E. R. Sabbadini. 1998. Neuroimmunoregulation and natural immunity. Domest. Anim. Endocrinol. 15:273-281. [DOI] [PubMed] [Google Scholar]
- 4.Bi, Z., M. Barna, T. Komatsu, and C. S. Reiss. 1995. Vesicular stomatitis virus infection of the central nervous system activates both innate and acquired immunity. J. Virol. 69:6466-6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boehm, U., L. Guethlein, T. Klamp, K. Ozbek, A. Schaub, A. Futterer, K. Pfeffer, and J. C. Howard. 1998. Two families of GTPases dominate the complex cellular response to IFN-gamma. J. Immunol. 161:6715-6723. [PubMed] [Google Scholar]
- 6.Bopst, M., C. Haas, B. Car, and H. P. Eugster. 1998. The combined inactivation of tumor necrosis factor and interleukin-6 prevents induction of the major acute phase proteins by endotoxin. Eur. J. Immunol. 28:4130-4137. [DOI] [PubMed] [Google Scholar]
- 7.Bradt, B. M., W. P. Kolb, and N. R. Cooper. 1998. Complement-dependent proinflammatory properties of the Alzheimer's disease beta-peptide. J. Exp. Med. 188:431-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchmeier, M. J., H. A. Lewicki, P. J. Talbot, and R. L. Knobler. 1984. Murine hepatitis virus-4 (strain JHM)-induced neurologic disease is modulated in vivo by monoclonal antibody. Virology 132:261-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cabarrocas, J., J. Bauer, E. Piaggio, R. Liblau, and H. Lassmann. 2003. Effective and selective immune surveillance of the brain by MHC class I-restricted cytotoxic T lymphocytes. Eur. J. Immunol. 33:1174-1182. [DOI] [PubMed] [Google Scholar]
- 10.Campbell, I. L., A. K. Stalder, C. S. Chiang, R. Bellinger, C. J. Heyser, S. Steffensen, E. Masliah, H. C. Powell, L. H. Gold, S. J. Henriksen, and G. R. Siggins. 1997. Transgenic models to assess the pathogenic actions of cytokines in the central nervous system. Mol. Psychiatry 2:125-129. [DOI] [PubMed] [Google Scholar]
- 11.Cardinaux, J. R., I. Allaman, and P. J. Magistretti. 2000. Pro-inflammatory cytokines induce the transcription factors C/EBPbeta and C/EBPdelta in astrocytes. Glia 29:91-97. [PubMed] [Google Scholar]
- 12.Cardona, A. E., P. A. Gonzalez, and J. M. Teale. 2003. CC chemokines mediate leukocyte trafficking into the central nervous system during murine neurocysticercosis: role of gamma delta T cells in amplification of the host immune response. Infect. Immun. 71:2634-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlow, D. A., S. J. Teh, and H. S. Teh. 1998. Specific antiviral activity demonstrated by TGTP, a member of a new family of interferon-induced GTPases. J. Immunol. 161:2348-2355. [PubMed] [Google Scholar]
- 14.Carson, M. J. 2002. Microglia as liaisons between the immune and central nervous systems: functional implications for multiple sclerosis. Glia 40:218-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charles, P. C., J. Trgovcich, N. L. Davis, and R. E. Johnston. 2001. Immunopathogenesis and immune modulation of Venezuelan equine encephalitis virus-induced disease in the mouse. Virology 284:190-202. [DOI] [PubMed] [Google Scholar]
- 16.Collazo, C. M., G. S. Yap, S. Hieny, P. Caspar, C. G. Feng, G. A. Taylor, and A. Sher. 2002. The function of gamma interferon-inducible GTP-binding protein IGTP in host resistance to Toxoplasma gondii is Stat1 dependent and requires expression in both hematopoietic and nonhematopoietic cellular compartments. Infect. Immun. 70:6933-6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Correale, J., S. Li, L. P. Weiner, and W. Gilmore. 1995. Effect of persistent mouse hepatitis virus infection on MHC class I expression in murine astrocytes. J. Neurosci. Res. 40:10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui, Y. H., Y. Le, X. Zhang, W. Gong, K. Abe, R. Sun, J. Van Damme, P. Proost, and J. M. Wang. 2002. Up-regulation of FPR2, a chemotactic receptor for amyloid beta 1-42 (A beta 42), in murine microglial cells by TNF alpha. Neurobiol. Dis. 10:366-377. [DOI] [PubMed] [Google Scholar]
- 19.Dunn, A. J. 2000. Cytokine activation of the HPA axis. Ann. N. Y. Acad. Sci. 917:608-617. [DOI] [PubMed] [Google Scholar]
- 20.Fazakerley, J. K., S. E. Parker, F. Bloom, and M. J. Buchmeier. 1992. The V5A13.1 envelope glycoprotein deletion mutant of mouse hepatitis virus type-4 is neuroattenuated by its reduced rate of spread in the central nervous system. Virology 187:178-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilmore, W., J. Correale, and L. P. Weiner. 1994. Coronavirus induction of class I major histocompatibility complex expression in murine astrocytes is virus strain specific. J. Exp. Med. 180:1013-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gobin, S. J., M. van Zutphen, A. M. Woltman, and P. J. van den Elsen. 1999. Transactivation of classical and nonclassical HLA class I genes through the IFN-stimulated response element. J. Immunol. 163:1428-1434. [PubMed] [Google Scholar]
- 23.Greenwel, P., S. Tanaka, D. Penkov, W. Zhang, M. Olive, J. Moll, C. Vinson, M. Di Liberto, and F. Ramirez. 2000. Tumor necrosis factor alpha inhibits type I collagen synthesis through repressive CCAAT/enhancer-binding proteins. Mol. Cell. Biol. 20:912-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haga, S., K. Ikeda, M. Sato, and T. Ishii. 1993. Synthetic Alzheimer amyloid beta/A4 peptides enhance production of complement C3 component by cultured microglial cells. Brain Res. 601:88-94. [DOI] [PubMed] [Google Scholar]
- 25.Haller, O., and G. Kochs. 2002. Interferon-induced mx proteins: dynamin-like GTPases with antiviral activity. Traffic 3:710-717. [DOI] [PubMed] [Google Scholar]
- 26.Hawiger, J. 2001. Innate immunity and inflammation: a transcriptional paradigm. Immunol. Res. 23:99-109. [DOI] [PubMed] [Google Scholar]
- 27.Huang, J. H., and W. S. Liao. 1994. Induction of the mouse serum amyloid A3 gene by cytokines requires both C/EBP family proteins and a novel constitutive nuclear factor. Mol. Cell. Biol. 14:4475-4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kakinuma, C., Y. Hamada, Y. Futamura, C. Kuwayama, A. Shimoi, and Y. Shibutani. 1999. Human natural tumor necrosis factor alpha induces multiple endocrine and hematologic disorders in rats. Toxicol. Pathol. 27:402-411. [DOI] [PubMed] [Google Scholar]
- 29.Karsten, V., S. Tritschler, K. Mandes, A. Belcourt, M. Pinget, and L. Kessler. 2000. Chemotaxis activation of peritoneal murine macrophages induced by the transplantation of free and encapsulated pancreatic rat islets. Cell Transplant. 9:39-43. [DOI] [PubMed] [Google Scholar]
- 30.Katz, H. R. 2002. Inhibition of anaphylactic inflammation by the gp49B1 receptor on mast cells. Mol. Immunol. 38:1301-1305. [DOI] [PubMed] [Google Scholar]
- 31.Kimura, T., and D. E. Griffin. 2000. The role of CD8+ T cells and major histocompatibility complex class I expression in the central nervous system of mice infected with neurovirulent Sindbis virus. J. Virol. 74:6117-6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitamura, Y., O. Spleiss, H. Li, T. Taniguchi, H. Kimura, Y. Nomura, and P. J. Gebicke-Haerter. 2001. Lipopolysaccharide-induced switch between retinoid receptor (RXR) alpha and glucocorticoid attenuated response gene (GARG)-16 messenger RNAs in cultured rat microglia. J. Neurosci. Res. 64:553-563. [DOI] [PubMed] [Google Scholar]
- 33.Lacy, M., J. Jones, S. R. Whittemore, D. L. Haviland, R. A. Wetsel, and S. R. Barnum. 1995. Expression of the receptors for the C5a anaphylatoxin, interleukin-8 and FMLP by human astrocytes and microglia. J. Neuroimmunol. 61:71-78. [DOI] [PubMed] [Google Scholar]
- 34.Lane, T. E., V. C. Asensio, N. Yu, A. D. Paoletti, I. L. Campbell, and M. J. Buchmeier. 1998. Dynamic regulation of alpha- and beta-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J. Immunol. 160:970-978. [PubMed] [Google Scholar]
- 35.Li, Y., L. Fu, D. M. Gonzales, and E. Lavi. 2004. Coronavirus neurovirulence correlates with the ability of the virus to induce proinflammatory cytokine signals from astrocytes and microglia. J. Virol. 78:3398-3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lidman, O., T. Olsson, and F. Piehl. 1999. Expression of nonclassical MHC class I (RT1-U) in certain neuronal populations of the central nervous system. Eur. J. Neurosci. 11:4468-4472. [DOI] [PubMed] [Google Scholar]
- 37.Lipshutz, R. J., S. P. Fodor, T. R. Gingeras, and D. J. Lockhart. 1999. High density synthetic oligonucleotide arrays. Nat. Genet. 21:20-24. [DOI] [PubMed] [Google Scholar]
- 38.Lokensgard, J. R., M. C. Cheeran, S. Hu, G. Gekker, and P. K. Peterson. 2002. Glial cell responses to herpesvirus infections: role in defense and immunopathogenesis. J. Infect. Dis. 186(Suppl. 2):S171-S179. [DOI] [PubMed] [Google Scholar]
- 39.Lu, R., P. A. Moore, and P. M. Pitha. 2002. Stimulation of IRF-7 gene expression by tumor necrosis factor alpha: requirement for NFkappa B transcription factor and gene accessibility. J. Biol. Chem. 277:16592-16598. [DOI] [PubMed] [Google Scholar]
- 40.Matejuk, A., C. Hopke, J. Dwyer, S. Subramanian, R. E. Jones, D. N. Bourdette, A. A. Vandenbark, and H. Offner. 2003. CNS gene expression pattern associated with spontaneous experimental autoimmune encephalomyelitis. J. Neurosci. Res. 73:667-678. [DOI] [PubMed] [Google Scholar]
- 41.McGrath, N., N. E. Anderson, M. C. Croxson, and K. F. Powell. 1997. Herpes simplex encephalitis treated with acyclovir: diagnosis and long term outcome. J. Neurol. Neurosurg. Psychiatry 63:321-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nadeau, S., and S. Rivest. 2000. Role of microglial-derived tumor necrosis factor in mediating CD14 transcription and nuclear factor kappa B activity in the brain during endotoxemia. J. Neurosci. 20:3456-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nash, T. C., and M. J. Buchmeier. 1996. Spike glycoprotein-mediated fusion in biliary glycoprotein-independent cell-associated spread of mouse hepatitis virus infection. Virology 223:68-78. [DOI] [PubMed] [Google Scholar]
- 44.Ning, S., A. M. Hahn, L. E. Huye, and J. S. Pagano. 2003. Interferon regulatory factor 7 regulates expression of Epstein-Barr virus latent membrane protein 1: a regulatory circuit. J. Virol. 77:9359-9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okuda, Y., S. Sakoda, H. Fujimura, Y. Saeki, T. Kishimoto, and T. Yanagihara. 1999. IL-6 plays a crucial role in the induction phase of myelin oligodendrocyte glucoprotein 35-55 induced experimental autoimmune encephalomyelitis. J. Neuroimmunol. 101:188-196. [DOI] [PubMed] [Google Scholar]
- 46.Olson, J. K., A. M. Girvin, and S. D. Miller. 2001. Direct activation of innate and antigen-presenting functions of microglia following infection with Theiler's virus. J. Virol. 75:9780-9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parra, B., D. R. Hinton, M. T. Lin, D. J. Cua, and S. A. Stohlman. 1997. Kinetics of cytokine mRNA expression in the central nervous system following lethal and nonlethal coronavirus-induced acute encephalomyelitis. Virology 233:260-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perlman, S., and D. Ries. 1987. The astrocyte is a target cell in mice persistently infected with mouse hepatitis virus, strain JHM. Microb. Pathog. 3:309-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perlman, S., R. Schelper, and D. Ries. 1987. Maternal antibody-modulated MHV-JHM infection in C57BL/6 and BALB/c mice. Adv. Exp. Med. Biol. 218:297-305. [DOI] [PubMed] [Google Scholar]
- 50.Qiu, Z., K. L. Parsons, and D. L. Gruol. 1995. Interleukin-6 selectively enhances the intracellular calcium response to NMDA in developing CNS neurons. J. Neurosci. 15:6688-6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramana, C. V., M. Chatterjee-Kishore, H. Nguyen, and G. R. Stark. 2000. Complex roles of Stat1 in regulating gene expression. Oncogene 19:2619-2627. [DOI] [PubMed] [Google Scholar]
- 52.Ravizza, T., D. Moneta, B. Bottazzi, G. Peri, C. Garlanda, E. Hirsch, G. J. Richards, A. Mantovani, and A. Vezzani. 2001. Dynamic induction of the long pentraxin PTX3 in the CNS after limbic seizures: evidence for a protective role in seizure-induced neurodegeneration. Neuroscience 105:43-53. [DOI] [PubMed] [Google Scholar]
- 53.Redwine, J. M., M. J. Buchmeier, and C. F. Evans. 2001. In vivo expression of major histocompatibility complex molecules on oligodendrocytes and neurons during viral infection. Am. J. Pathol. 159:1219-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reiss, C. S., D. A. Chesler, J. Hodges, D. D. Ireland, and N. Chen. 2002. Innate immune responses in viral encephalitis. Curr. Top. Microbiol. Immunol. 265:63-94. [DOI] [PubMed] [Google Scholar]
- 55.Rempel, J. D., S. Murray, J. Meisner, and M. J. Buchmeier. 2004. Differential regulation of innate and adaptive immune responses in viral encephalitis. Virology 318:381-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rempel, J. D., S. Murray, J. Meisner, and M. J. Buchmeier. 2004. Mouse hepatitis virus neurovirulence; evidence of a linkage between spike glycoprotein expression and immunopathology. Virology 318:45-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Riemer, C., I. Queck, D. Simon, R. Kurth, and M. Baier. 2000. Identification of upregulated genes in scrapie-infected brain tissue. J. Virol. 74:10245-10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sato, M., H. Suemori, N. Hata, M. Asagiri, K. Ogasawara, K. Nakao, T. Nakaya, M. Katsuki, S. Noguchi, N. Tanaka, and T. Taniguchi. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539-548. [DOI] [PubMed] [Google Scholar]
- 59.Servant, M. J., B. Tenoever, and R. Lin. 2002. Overlapping and distinct mechanisms regulating IRF-3 and IRF-7 function. J. Interferon Cytokine Res. 22:49-58. [DOI] [PubMed] [Google Scholar]
- 60.Shoji, H., K. Azuma, Y. Nishimura, H. Fujimoto, Y. Sugita, and Y. Eizuru. 2002. Acute viral encephalitis: the recent progress. Intern. Med. 41:420-428. [DOI] [PubMed] [Google Scholar]
- 61.Smith, J. B., and H. R. Herschman. 1996. The glucocorticoid attenuated response genes GARG-16, GARG-39, and GARG-49/IRG2 encode inducible proteins containing multiple tetratricopeptide repeat domains. Arch. Biochem. Biophys. 330:290-300. [DOI] [PubMed] [Google Scholar]
- 62.Storch, M. K., R. Weissert, A. Steffer, R. Birnbacher, E. Wallstrom, I. Dahlman, C. G. Ostensson, C. Linington, T. Olsson, and H. Lassmann. 2002. MHC gene related effects on microglia and macrophages in experimental autoimmune encephalomyelitis determine the extent of axonal injury. Brain Pathol. 12:287-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stroynowski, I. 1990. Molecules related to class-I major histocompatibility complex antigens. Annu. Rev. Immunol. 8:501-530. [DOI] [PubMed] [Google Scholar]
- 64.Sun, N., D. Grzybicki, R. F. Castro, S. Murphy, and S. Perlman. 1995. Activation of astrocytes in the spinal cord of mice chronically infected with a neurotropic coronavirus. Virology 213:482-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Taylor, G. A., C. M. Collazo, G. S. Yap, K. Nguyen, T. A. Gregorio, L. S. Taylor, B. Eagleson, L. Secrest, E. A. Southon, S. W. Reid, L. Tessarollo, M. Bray, D. W. McVicar, K. L. Komschlies, H. A. Young, C. A. Biron, A. Sher, and G. F. Vande Woude. 2000. Pathogen-specific loss of host resistance in mice lacking the IFN-gamma-inducible gene IGTP. Proc. Natl. Acad. Sci. USA 97:751-755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thomas, J., S. Gangappa, S. Kanangat, and B. T. Rouse. 1997. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J. Immunol. 158:1383-1391. [PubMed] [Google Scholar]
- 67.Wagtmann, N. 1999. gp49: an Ig-like receptor with inhibitory properties on mast cells and natural killer cells. Curr. Top. Microbiol. Immunol. 244:107-113. [DOI] [PubMed] [Google Scholar]
- 68.Wang, Q., and L. Flaherty. 1996. Developmental expression of the mouse MHC Q genes. Eur. J. Immunogenet. 23:121-127. [DOI] [PubMed] [Google Scholar]
- 69.Warringa, R. A., H. J. Mengelers, J. A. Raaijmakers, P. L. Bruijnzeel, and L. Koenderman. 1993. Upregulation of formyl-peptide and interleukin-8-induced eosinophil chemotaxis in patients with allergic asthma. J. Allergy Clin. Immunol. 91:1198-1205. [DOI] [PubMed] [Google Scholar]
- 70.Wildemann, B., K. Ehrhart, B. Storch-Hagenlocher, U. Meyding-Lamade, S. Steinvorth, W. Hacke, and J. Haas. 1997. Quantitation of herpes simplex virus type 1 DNA in cells of cerebrospinal fluid of patients with herpes simplex virus encephalitis. Neurology 48:1341-1346. [DOI] [PubMed] [Google Scholar]
- 71.Zappacosta, F., P. Tabaczewski, K. C. Parker, J. E. Coligan, and I. Stroynowski. 2000. The murine liver-specific nonclassical MHC class I molecule Q10 binds a classical peptide repertoire. J. Immunol. 164:1906-1915. [DOI] [PubMed] [Google Scholar]
- 72.Zerrahn, J., U. E. Schaible, V. Brinkmann, U. Guhlich, and S. H. Kaufmann. 2002. The IFN-inducible Golgi- and endoplasmic reticulum-associated 47-kDa GTPase IIGP is transiently expressed during listeriosis. J. Immunol. 168:3428-3436. [DOI] [PubMed] [Google Scholar]
- 73.Zhang, L., and J. S. Pagano. 2001. Interferon regulatory factor 7: a key cellular mediator of LMP-1 in EBV latency and transformation. Semin. Cancer Biol. 11:445-453. [DOI] [PubMed] [Google Scholar]
- 74.Zhang, X., D. Hinton, S. Park, C. L. Liao, M. M. Lai, and S. Stohlman. 1998. Using a defective-interfering RNA system to express the HE protein of mouse hepatitis virus for studying viral pathogenesis. Adv. Exp. Med. Biol. 440:521-528. [DOI] [PubMed] [Google Scholar]
- 75.Zhang, X., D. R. Hinton, S. Park, B. Parra, C. L. Liao, M. M. Lai, and S. A. Stohlman. 1998. Expression of hemagglutinin/esterase by a mouse hepatitis virus coronavirus defective-interfering RNA alters viral pathogenesis. Virology 242:170-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou, J., S. A. Stohlman, D. R. Hinton, and N. W. Marten. 2003. Neutrophils promote mononuclear cell infiltration during viral-induced encephalitis. J. Immunol. 170:3331-3336. [DOI] [PubMed] [Google Scholar]