

Abstract
Human immunodeficiency virus (HIV) causes complex metabolic changes in infected CD4+ T cells that lead to cell cycle arrest and cell death by necrosis. To study the viral functions responsible for deleterious effects on the host cell, we quantitated the course of HIV type 1 infection in tissue cultures by using flow cytometry for a virally encoded marker protein, heat-stable antigen (HSA). We found that HSA appeared on the surface of the target cells in two phases: passive acquisition due to association and fusion of virions with target cells, followed by active protein expression from transcription of the integrated provirus. The latter event was necessary for decreased target cell viability. We developed a general mathematical model of viral dynamics in vitro in terms of three effective time-dependent rates: those of cell proliferation, infection, and death. Using this model we show that the predominant contribution to the depletion of viable target cells results from direct cell death rather than cell cycle blockade. This allows us to derive accurate bounds on the time-dependent death rates of infected cells. We infer that the death rate of HIV-infected cells is 80 times greater than that of uninfected cells and that the elimination of the vpr protein reduces the death rate by half. Our approach provides a general method for estimating time-dependent death rates that can be applied to study the dynamics of other viruses.
AIDS results from infection with the human immunodeficiency virus (HIV), and nearly 40 million adults and children worldwide live with HIV/AIDS (32). The opportunistic infections characteristic of AIDS result from the progressive depletion of CD4+ T lymphocytes in infected individuals (9). During the acute phase, virus levels increase rapidly with a concomitant loss of CD4+ T cells. As cellular and humoral antiviral immune responses take effect, virus levels decrease and CD4+ T-cell numbers temporarily recover. During the subsequent phase of clinical latency, CD4+ T-cell numbers slowly, but inexorably, decline. Finally, as the immune system is exhausted by HIV, clinical immunodeficiency and lethal infections occur (29).
Models of the viral-cellular dynamics of HIV infection have provided different explanations for the CD4+ T-cell decline. CD4+ T cells are vulnerable to HIV, both in vitro and in vivo, because the virus uses the CD4 molecule as its principal receptor (6, 17). HIV selectively infects this class of lymphocytes and causes direct cell killing as well as cell cycle arrest in the G2 phase (4, 8, 13, 16, 28, 38). It was originally suggested that low numbers of circulating infected cells during clinical latency could not account for the overall decline in CD4+ T cells, implying that bystander CD4+ T-cell death was occurring (8). However, more sensitive methods of viral quantitation and mathematical modeling of infection kinetics in vivo revealed that the viral load in the peripheral blood and lymphoid organs was actually higher than previously believed (28, 29). HIV protease inhibitors, by powerfully suppressing virus infection in vivo, permitted a new dynamic view of HIV infection (13, 30, 31, 38). Protease inhibitors decreased circulating virus to low or undetectable levels and dramatically increased CD4+ T cells within 6 to 8 weeks (13, 23, 38). These effects were explained by a high-turnover model, in which the low level of circulating virus is maintained by continuous, high-level virus production and infection of new CD4+ T cells followed by rapid destruction of infected cells (7, 13, 21, 24). However, there may also be more complex effects, such as the redistribution of a fraction of CD4+ T cells into the circulating pool (21, 27). In the absence of protease inhibitors, CD4+ T-cell destruction is presumably partly masked by heightened cellular production, producing only a slow decline in CD4+ T cells. Ultimately, the cumulative direct killing of these cells is presumed to cause the collapse of the immune system (24).
We have studied how HIV causes the death of transformed CD4+ T-cell lines and peripheral blood CD4+ T cells in vitro. Some investigators have proposed that apoptosis is the cause for HIV-induced death of CD4+ T cells (2, 10). However, we previously showed that a novel form of necrosis, not apoptosis, is the primary mode of death in vitro (4, 16). In those studies, we also used the vesicular stomatitis virus glycoprotein (VSV-G) to pseudotype viral particles containing a mutated version of HIV env and found that viral cytopathicity does not require Env (1, 3, 19). However, assessing the role of other HIV type 1 (HIV-1) proteins in viral cytopathicity is more difficult. Certain HIV-1 proteins, such as Vpr, can cause cell cycle blockade and cell death (5, 6, 8, 11-13, 18, 25, 26, 28, 34, 36, 39). Therefore, we sought a means of analysis that would allow us to determine if the loss of viable cells is due to cell cycle blockade or death.
Another interesting facet of host cell-virus interaction is that Vpr has been shown to induce cell cycle arrest when passively delivered by virion fusion to target cells without proviral integration and expression (37). We therefore wanted to distinguish the phase of virion delivery of viral proteins from the phase of de novo viral protein synthesis from an integrated provirus. This demarcation would allow us to infer the direct cytopathicity of different HIV-1 mutants from the culture dynamics of single-round infections in vitro.
In this study, we have used an HIV-1 strain containing the mouse heat-stable antigen (HSA) cell surface marker (NL4-3HSA) to distinguish two stages of viral protein expression by the target cell: (i) cell surface association and fusion of virions and (ii) provirus integration and expression. We also introduce a new mathematical method for quantifying the cytopathicity of viral mutants, independently of their effects on cell cycle progression or other parameters of the in vitro cell culture system. Using these approaches, we show that Vpr decreases the number of viable cells infected by HIV-1 primarily by its cytopathic effect rather than by cell cycle blockade. We also describe a general means of determining time-dependent death rates of infected cells in tissue culture systems.
MATERIALS AND METHODS
Cell lines and cell cultures.
Hi-CD4 Jurkat T cells (Jurkat 1.9 cells) were maintained in complete RPMI medium (RPMI 1640; BioWhittaker), which included 10% heat-activated fetal calf serum, 100 U of penicillin and streptomycin/ml, 2 mM l-glutamine, and 50 μM β-mercaptoethanol (4).
Preparation of NL4-3HSA virus mutants.
Mutation of the NL4-3 vpr gene was performed by shuttling the PflMI-to-SalI fragment (488 bp) of pHXB2 (kindly provided by Andrew Leavitt, University of California, San Francisco) containing vpr into NL4-3HSA. The resulting strain of NL4-3, NL4-3HSA/HXB2 (P-S), hereupon referred to as P-S, encodes HXB2 vpr, which has a premature stop codon at amino acid 79 due to a thymidine insert at nucleotide 216. Truncation of the terminal 18 amino acids of Vpr results in an unstable protein that is poorly expressed and nonfunctional (data not shown).
HIV virus stocks and infections.
Mouse HSA is inserted in place of the nef-coding sequence in NL4-3HSA, a derivative of the HIV-1 laboratory strain NL4-3HSA. The original viral stock and virus plasmid were provided by Ned Landau (Salk Institute) (12). Viral stocks of NL4-3HSA and various mutants were generated by 293T cell transfection using FuGENE (Boehringer-Mannheim) according to the manufacturer's recommendations. The NL4-3HSA Env− strain was pseudotyped with VSV-G by cotransfection of 2 μg of HIV-1 plasmid and 1 μg of pVSV-G. Viral supernatants were harvested 48 h posttransfection, and stocks were frozen at −80°C. Virus titers were determined using the MAGI assay (15, 33). A total of 106 cells were infected per sample using 5 μg of Polybrene/ml. Complete RPMI medium was added to obtain a total volume of 3 ml in 12-well plates. Plates were centrifuged for 30 min at 800 × g, 25°C. After centrifugation, cultures were maintained at 37°C by refeeding as needed. Using HSA as a reporter gene, we determined the viability of infected and uninfected cells by cell surface staining and flow cytometry. For extended spin infections used in specified experiments, samples were centrifuged at 800 × g at 30°C for 30 h, with short interruptions to collect cells for HSA or intracellular p24 expression analysis. Lamivudine (3TC) and saquinavir (SQV) were obtained through the AIDS Research and Reference Reagent Program, National Institute of Allergy and Infectious Diseases, National Institutes of Health. Reverse transcription was blocked by preincubation of Jurkat cells with 10 μM 3TC 24 h prior to infection, with replenishment every 24 h. SQV was added to infection cultures at 1 μΜ and replenished every 48 h. Cells were washed in phosphate-buffered saline (PBS), treated with proteinase K at 0.5 mg/ml (Sigma) at 4°C for 30 min, and washed twice in PBS before immunostaining for flow cytometric analysis.
Quantification of cell viability and infection.
Flow cytometry was used to monitor cell viability and infection level on a FACSCalibur (Becton Dickinson and Co.). Culture viability was assessed using a live gate showing the percentage of cells exhibiting a forward scatter-side scatter profile determined by the analysis of live uninfected cells (mock control). The level of infection was quantified by the surface staining of HSA with anti-HSA phycoerythrin stain (CD24a PE; Pharmingen). Intracellular HIV-1 p24 production was measured by cytoplasmic immunostaining of cells permeabilized with the Cytofix/Cytoperm kit (BD Biosciences, Pharmingen) according to the manufacturer's instructions. Fixed cells were incubated in 1:1,000-diluted anti-p24 PE antibody, KC57-RD1 (Beckman Coulter, Fullerton, Calif.) at 4°C for 30 min, washed twice, and analyzed by flow cytometry. Cell cycle times were measured by dilution of 5,6-carboxyfluorescein diacetate succinimidyl ester (Molecular Probes) according to the manufacturer's instructions. Cell cycle stage analysis was carried out using flow cytometry detection of DNA content stained with propidium iodide as previously described (12). Flow cytometry data were analyzed using CellQuest (Becton Dickinson and Co.) or Flowjo (Treestar, Inc.) software.
Mathematical analysis.
The objective of this analysis was to obtain, from simple measurements of culture dynamics, accurate estimates of the rate at which different HIV-1 strains kill the T cells they infect. We distinguished the following cell populations: x represents live, uninfected cells; xd represents dead, uninfected cells; y represents live, infected (HSA+) cells; and yd represents dead, infected (HSA+) cells.
Though these populations were not directly determined experimentally, inferences about them were based on flow cytometric measurements for two derived variables: viability (v), the fraction of live cells in the culture, and infectivity (f), the fraction of live cells that is infected. Cells were considered infected if their level of HSA staining measured by flow cytometry fell within a predetermined gate of high-level expression relative to the mock sample (see Fig. 1 and 2, below). We denoted these samples as HSA+.
FIG. 1.

Expression of the HSA marker protein correlates with two phases of infection. (A) Jurkat 1.9 cells (106) were infected with serial dilutions of HIV-1 NL4-3HSA Env− virus. Each histogram shows the HSA expression of live cells, and the percentage of gated HSA+ cells (compared to mock) is given at 8 and 23 h p.i. Virus was serially diluted, and the MOIs were as follows: (a) mock infected, MOI = 0; (b) 1:243 serial dilution of virus, MOI = 0.03; (c) 1:81 serial dilution of virus, MOI = 0.09; (d) 1:27 serial dilution of virus, MOI = 0.28; (e) 1:9 serial dilution of virus, MOI = 0.83; (f) 1:3 serial dilution of virus, MOI = 2.5; (g) undiluted virus stock, MOI = 7.5. In the bottom right corner (h) of each group of panels, the viability profile of sample g as detected by forward light scatter (x axis) and side light scatter (y axis) is shown. Viable cells are shown within the polygon, and the fraction of total events is given. (B) Representative experiment in which the sample identity and viral titer were the same as those described for panel A, analyzed 23 and 46 h after the start of infection. The units on the x axis are arbitrary fluorescence units, and the units on the y axis are event counts. A total of 5,000 events were collected for each sample.
FIG. 2.

Relationship of HSA expression levels to cell viability. (A) Thresholds of HSA staining distinguish levels of infection associated with different fates of the infected cell. Data are derived from sample number 4 on day 4 shown in panel B. Low-negative expression indicates HSA-negative cells or transient HSA passively acquired from virion-cell interaction. Cells expressing HSA at the medium level (Med) are productively infected but have not yet reached high levels of expression. High-level expressers (Hi) are at the peak of infection, which leads to cell death. (B) Quantification of samples gated on HSA expression as for panel A. The various times and MOIs are shown. (C) Viability profiles for the infected samples represented in panel B. Serial dilutions of NL4-3HSA Env− virus were used for Jurkat 1.9 cell infection, which was monitored by HSA expression.
Thus, v and f are related to the various cell populations by the following expressions:
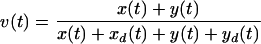 |
(1) |
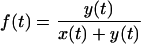 |
(2) |
A precise model of the dynamics of these cell populations would have to include several complications. For instance, the death rate of infected cells increases with the expression level of viral proteins in the cell, which tends to increase with time (see Fig. 1 and 2, below). Mathematically, this implies that the death rate of an infected cell depends on the length of time since infection. Consequently, the age structure of the infected cell population becomes important in accounting for this complication. A further complication is that infected cells may be infected more than once, and the propensity for death or cell cycle arrest appears to increase with the number of times the cell is infected or engaged by virus (16). The infection rate itself depends on the amount of free virus in the culture, which is depleted through both infection and decay. Finally, there is a variable delay between infection of a cell and expression of viral proteins within the cell (conversion time). A model that explicitly implements all these complexities would contain too many parameters to practically and meaningfully fit the data. We thus opted to develop an approach in which we describe the culture dynamics by introducing a number of effective rates which are time dependent and aim to subsume the aforementioned complications. Formally, we assume the following general culture dynamics, which are supported by measurements of uninfected cultures:
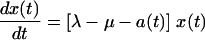 |
(3) |
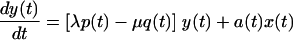 |
(4) |
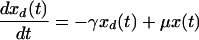 |
(5) |
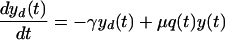 |
(6) |
Thus, we assume that uninfected cells (x) proliferate at a constant rate (λ) and die at a constant rate (μ) and that dead cells (xd and yd) decay at a constant rate (γ), estimated by measurements of uninfected cultures. For cultures that include both infected and uninfected cells, we make the general assumption that uninfected cells become infected at an effective rate a(t) which incorporates the effects of changing levels of infectious virus in the culture. Thus, in previous models such as that described in reference 32, in which virus, V(t), was an explicit variable, one would have a(t) = kV(t), where k is the infection rate constant. The infected cells proliferate at an effective rate λp(t), which incorporates the effects of cell cycle blockade, and die at an effective rate μq(t), which incorporates the effects on cytopathicity of changing expression levels of viral proteins. The function p(t) relates the proliferation rates of uninfected and infected cells. When p(t) = 0, the cell cycle of infected cells is completely blocked, whereas at p(t) = 1, infected cells proliferate at the same rate as uninfected cells. We assume that infected cells cannot proliferate faster than uninfected cells, i.e., that p(t) is ≤1. This assumption is consistent with our experimental observations. In particular, infected cells have never been observed to proliferate faster than uninfected cells (D. L. Bolton and M. J. Lenardo, unpublished data). Likewise, the function q(t) relates the death rates of uninfected and infected cells. When q(t) is >1, infected cells have an enhanced death rate compared to uninfected cells.
As shown in the supplemental material, equations 3 to 6can be rewritten in terms of the viability v(t) and infectivity f(t), yielding
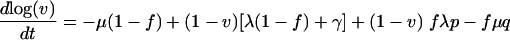 |
(7) |
We approximate the functions log(v), v(t), and f(t) by linear functions between consecutive measurement points. The validity of this approximation is supported by the smooth appearance of the curves that result from connecting measured values v or f at consecutive measurement points. Consider now a time point t that lies halfway between the time ti, at which the ith measurement was taken, and the time ti+1, at which the (i + 1)th measurement was taken. We denote the values of p and q at this time point as p and q. The values of v, f, and d[log(v)]/dt at this time are then given by the following equation:
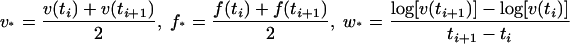 |
(8) |
Since v and f were measured in triplicate, the values of v(ti) and f(ti) in the equations were assumed to equal the averages of the three measurements. Using equations 7and 8, one then obtains q in terms of p:
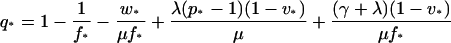 |
(9) |
We show in the text that q changes relatively little as p runs from 0 to 1, allowing us to estimate q(t) from the data.
Our analysis thus consists of three steps. First, we estimate the parameters λ, μ, and γ from measurements of ν(ti) on cultures of uninfected cells (see Results). Then, using these estimates, we use equation 9 to derive bounds on the function q(t) from a time course of viability, ν, and infectivity, f, measurements in cultures with both infected and uninfected cells. Finally, we use the inferred bounds on q(t) to compare the cytopathic effects of different virus strains in cell culture infections.
RESULTS
Target cells display viral proteins in two distinct phases. Our infection system consisted of a T-cell culture that had been exposed to HIV under conditions of a single-round infection, using an NL4-3HSA Env− virus (4). Because the virus lacks Env, no new infectious virions were produced in the cell culture postinfection (p.i.). In NL4-3HSA, HSA is encoded in the viral genome, so HSA expression is driven by the HIV-1 long terminal repeat. HSA is a mouse surface protein that is not normally expressed in human cells and, in this context, it becomes a specific viral marker. We therefore quantified the number of cells that had interacted with the virus by flow cytometric analysis for HSA.
A representative time course of infection revealed that low levels of HSA were evident at 8 h p.i. (Fig. 1A). Low-level HSA expression was followed by very-high-level HSA expression, which was clearly evident at 23 h (Fig. 1A). Transition between the two levels of HSA began at 8 h p.i. and was more advanced with higher multiplicities of infection (MOIs) and longer times (Fig. 1B). The effects of the MOI and time were clearly demonstrated when we assessed HSA expression in three flow cytometric gates: low-negative, medium, and high (Fig. 2A). HSA expression always proceeded from low to high as infection progressed, especially with greater MOIs (Fig. 2B). After 1 to 2 days following the expression of high HSA levels, cell viability decreased dramatically (Fig. 2C). These findings are consistent with our previous observations that high-level virus expression is essential for cell killing (4, 16) and support a model in which the persistent high-level expression of viral proteins ultimately leads to cellular demise.
Next, we examined the low-negative and medium ranges of HSA expression, which were both characteristically proportional to the amount of virus used and detectable as early as 2 to 4 h p.i. (data not shown). This is too soon to be due to provirus integration and expression (14). Therefore, for further analysis, we considered low-negative and medium HSA-expressing cells to be a single population (low HSA) with features distinct from cells expressing high HSA.
We hypothesized that low HSA was due to the passive “donation” of HSA from the virus envelope to the target cell during the virus-cell interaction, before viral integration. This has been previously described for other cell membrane proteins and internal virion proteins (35, 37). To test this hypothesis, we treated cells with 3TC, a reverse transcriptase inhibitor, in order to block productive infection and then exposed the cells to NL4-3HSA virus. We found early HSA was detectable on the target cells regardless of productive reverse transcription (Fig. 3A). Controls showed that early HSA was not blocked by addition of the protease inhibitor SQV, nor was it accompanied by expression of p24 (Fig. 3A, inset). These data suggested that the early low-level HSA expression was passively acquired and not due to transcription from an integrated provirus. Furthermore, we observed that the mixture of virus and cells at 3°C, a temperature known to block virus-cell interaction and fusion (22), or treatment with a blocking antibody against CD4 inhibited early HSA expression (Fig. 3B and data not shown). Taken together, these data indicate that the early low level of HSA is due to passive acquisition from viral particles. However, these data do not distinguish whether the low level of HSA was acquired by virus-cell fusion or binding of virus particles to the cell surface without fusion.
FIG. 3.

HSA detected on the cell surface during infection represents two mechanisms of acquisition. (A) Jurkat cells were cultured in plain medium or in the presence of either the nucleoside reverse transcriptase inhibitor 3TC or the protease inhibitor SQV. 3TC (10 μM) was added for 24 h prior to the infection and replenished every 24 h. Alternatively, the protease inhibitor SQV (1 μM) was added to Jurkat cells at the time of infection and replenished every 48 h. Surface expression of HSA and the levels of intracellular p24 (inset) were determined by flow cytometry. (B) Mixing virus and cells at 3°C significantly reduces detectable HSA on the cell surface. A total of 5 × 105 Jurkat 1.9 cells were spin infected with various dilutions of a concentrated stock of NL4-3HSA for 2.5 h, at 800 × g, at either 3 or 37°C. Immediately following centrifugation, samples were analyzed for surface expression of HSA (virion HSA) by flow cytometry. The samples and all reagents were maintained at 3°C following the spin infection to prevent subsequent viral fusion. The average percentage of HSA-positive cells from four independent samples is graphed, with error bars indicating one standard deviation. (C) Cell surface HSA staining in early stages of infection is partially sensitive to proteinase K cleavage. A total of 5 × 105 mock- or NL4-3HSA-infected Jurkat 1.9 CD4+ T cells were washed in PBS, incubated for 45 min in a rotary shaker at 4°C in either PBS (left panels) or 0.5 mg of proteinase K/ml (right panels), washed again in PBS, and stained for HSA. Control stains showed that CD4 was completely removed, but CXCR4 was unaffected by protease treatment (data not shown). HSA levels on HIV-1-infected cells were analyzed at day 1 (upper panels) and day 3 (lower panels) p.i. The fraction of HSA+ viable cells is shown. Ten thousand live events were acquired for each sample.
HSA is a glycosylphosphatidylinositol (GPI)-linked cell surface molecule that resists protease cleavage. We used this feature to distinguish viral particles that were bound to the CD4 receptor on the cell surface or those that had actually fused and donated HSA to the target cell membrane. We treated cells for 30 min with proteinase K, which removed surface-associated virus (20) and completely removed CD4 but not CXCR4 from the cell surface (data not shown). By treating cells under similar digestion conditions just before staining, we found that the low levels of HSA on day 1 were reduced from 79 to 47%, whereas the high HSA on day 3 was unaffected (Fig. 3C). Thus, early low-level HSA is due to two types of virion-cell interactions. The first is a protease-sensitive fraction, which we believe is comprised of unfused viral particles bound to the cell surface through protein-protein interactions. The second is a protease-resistant fraction that represents HSA tethered to the cell surface by a GPI linkage following fusion of the viral particle to the cell membrane. We will henceforth refer to both biochemical fractions of early HSA collectively as “virion HSA.”
As p.i. time increases, an increasing fraction of cells express a high level of HSA (Fig. 2B). This fraction has several distinct features compared to virion HSA. High-level HSA occurs at later times and is completely blocked by reverse transcriptase inhibition. Also, it is protease resistant and correlated with productive infection, as indicated by the generation of other viral proteins and, eventually, cell death. We will designate the high-level HSA as “proviral HSA,” since all of these characteristics are consistent with HSA newly produced from integrated proviruses.
Culture dynamics of uninfected cells.
To estimate the proliferation rate (λ) of uninfected cells, we used measurements of the flow cytometric distribution of the fluorescent marker carboxyfluorescein diacetate succinimidyl ester in a culture of uninfected cells as a function of time. Every time a cell divides the marker will be divided approximately equally to its two daughters. The concentration of marker in the daughters will therefore be roughly half of the concentration of marker in the mother cell. If cells divide at a rate λ, and 〈x〉 denotes the average of the logarithm of marker concentration (averaged over all cells in the cultures), then one can show (see the supplemental material) that 〈x〉 decreases linearly with time: d〈x〉/dt = −2log(2)λ. Using this equation we found that in uninfected Jurkat cells λ is 0.026 h−1, or 0.624 day−1.
To estimate the death rate μ of uninfected cells and the decay rate γ of dead cells, we examined data sets in which the viability of uninfected cells was measured as a function of time (mock data sets). Since no virus was added, we set y(t) = yd(t) = a(t) = 0 in the model's differential equations and denoted the ratio of dead to live cells at time zero as s0. Solving equations 3 and 5 and by using equation 1, the following expression for the viability is obtained (see also the supplemental material):
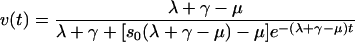 |
(10) |
We set the proliferation rate λ to 0.026 h−1 and then fit μ, γ, and s0 to the experimental data (Fig. 4) as follows. For each time point tj, we have three viability measurements: vj1, vj2, and vj3. We assume that there is Gaussian noise in these measurements of an unknown standard deviation, allowing for a different magnitude of noise at each time point. Under these assumptions, the likelihood L of the data given the theoretical viability curve v(t) is given by the following equation (see also the supplemental material):
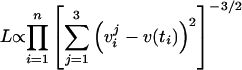 |
(11) |
The estimated values for the death rate and decay rate that we obtained are μ = 0.000764 h−1 and γ = 0.00785 h−1. These values correspond to a half-life of almost 38 days for live uninfected cells and a disintegration half-life of 3.7 days for dead cells.
FIG. 4.

Range of likely fits for uninfected cells. A time course of triplicate measurements of viability in cultures was made with uninfected cells (dots). The solid line is the maximum likelihood fit to the data. The dashed lines show fits with a likelihood that is a factor e lower than the maximum likelihood fit.
Estimating death rates of infected cells.
Next, we analyzed the dynamics of cultures with both uninfected and infected cells. As mentioned above, equations 3 to 6can be rewritten in terms of the viability v and infectivity f (defined in equations 1 and 2) to obtain the following equation:
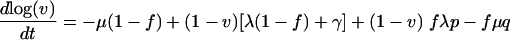 |
(12) |
Note that, for notational simplicity, we have suppressed the explicit time dependence of the variables v(t), f(t), p(t), and q(t). As discussed in Materials and Methods, we approximate log(v), v, and f by linear functions between consecutive measurement time points. We denote the approximation to a variable midway between two measurements with a subscript asterisk.
Using these approximations and equation 12, one obtains q in terms of p (see the supplemental material):
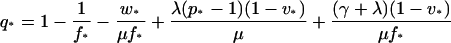 |
(13) |
where q and p are the approximations to q and p.
Fig. 5 shows the estimated values of q as a function of time for different viral strains and different viral concentrations as p runs from 0 (total cell cycle blockage) to 1 (no cell cycle blockage). The figure shows that the effect of infection on the proliferation rate, i.e., p, has only a relatively small effect on the estimate of q. We derive upper and lower bounds on q by assuming that P* = 0 or P* = 1. One then finds that q has to lie between the following:
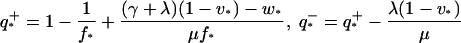 |
(14) |
FIG. 5.

Inferred effective death rates for infected cells as a function of time, viral strain, and viral concentration. The fold increase q(t) (z axis) of the death rate of infected cells compared to uninfected cells as a function of time (x axis) and the amount of cell cycle blockage p(t) of infected cells (y axis). Each plot shows the results for a different initial viral concentration: 0.5- (A), 1.0- (B), or 1.5-ml titer (C). Each surface corresponds to a particular viral strain. The top surface in each plot corresponds to wild-type virus. The middle surface corresponds to the vpr mutant strain, and the bottom surface corresponds to mock infections. Time runs from day 1 to day 6 after infection on the x axis. The proliferation rate p(t) runs from 0 (complete cell cycle blockage) to 1 (no cell cycle blockage) on the y axis. The fold change runs from 0 to 120 on the z axis, e.g., a value of 50 on the z axis indicates that we inferred from the measurements of infectivity and viability that, at that point of time in the infection and assuming that particular amount of cell cycle blockage, the infected cells were dying at a 50 times higher rate than uninfected cells.
The bounds that we obtained using equation 14 for different cultures and at different viral titers are shown in Fig. 6. For each data point, the top of the vertical error bar corresponds to the value of q that is obtained when p = 1, and the bottom of the bar corresponds to the value of q that is obtained when p = 0. As expected, there was hardly any increase in death rates for the mock data sets. Except for the third and fifth data points, where slight increases were apparent, all other data points were consistent with q(t) = 1, meaning that cells infected with mock virus died at the same rate as uninfected cells. In contrast, the wild-type virus had markedly higher cytopathicity, especially around day 4. Interestingly, the P-S virus with a mutation in the vpr gene exhibited cytopathicity that was reduced roughly twofold in comparison to wild type. We thus found that the effective death rate of cells infected by the wild-type virus was about 80 times higher than the death rate of uninfected cells and that of the P-S mutant virus was about 40 times higher (Fig. 6). Since uninfected cells have a half-life of almost 40 days, these death rates for the wild type and P-S virus correspond to infected cell half-lives of just under 1/2 day and just under 1 day, respectively. We also observed that the effective cytopathicity seemed to increase with time in both the wild-type and P-S mutant cultures, reaching a maximum value on day 4. Finally, although the results contained significant noise, it appears in general that the cytopathicity increases with the amount of virus. Finally, the last three time points for the mutant virus suggest that, at these late times, the cytopathicity of the virus increases significantly with the amount of virus. However, we stress these interpretations must be made with caution because of the noise in the measurements.
FIG. 6.

Inferred cytopathicity of different viral strains at different viral concentrations. The x-axis shows time in days and the y-axis shows the fold change in death rate of infected cells for the data in Fig. 5. Each bar indicates the inferred lower and upper bounds of the death rate that are obtained by setting p(t) = 0 and p(t) = 1, respectively. Green bars with triangles correspond to mock infection, blue bars with stars to the vpr mutant strain, and red bars with diamonds to wild-type virus. Each triplet of bars corresponds to cultures with viral-titers of 0.5 ml, 1 ml, and 1.5 ml, from left to right, respectively. Overlapping data sets were slightly shifted horizontally with respect with one another to make the figure easier to view. However, all samples for each set of viral titers were collected at the same time points.
DISCUSSION
To implicate specific HIV genes in cytopathicity, we required a method to compare death rates between viral stocks. Our ultimate goal is to determine the molecular mechanism by which HIV-1 kills infected CD4+ T cells. Our culture system, which gives high-efficiency infection, allows us to easily monitor infection and viability as a function of time by using flow cytometry for the surface marker HSA. Moreover, with Env− viruses, this can be analyzed in a one-step nearly synchronous infection (4, 16). We determined that there are two phases of infection that can be distinguished by HSA staining. Early on, low HSA levels correspond to the virion HSA that was passively transferred from virus particles to the target cells. At later data points, much higher levels of HSA were observed (generally 2 logs greater), corresponding to proviral HSA that resulted from the active transcription of integrated proviruses. This latter form of HSA is important in the quantification of virus infection that leads to cell death.
We have previously observed attenuated viral cytopathicity when the HIV-1 genome is devoid of its normal protein-coding sequences, indicating the presence of one or more cytopathic genes in the HIV-1 genome (4). The comparison of HIV derivatives with more selective mutations in various protein-coding regions, however, raised some analytical difficulties. First, certain HIV genes affect both cell cycle progression and viral cell killing. Therefore, changes in the fraction of viable cells could represent effects on either process. To address this issue, we developed a new mathematical approach that describes the culture dynamics in terms of effective time-dependent infection, proliferation, and death rates. Using this framework, we have rigorously shown that the death rates of infected cells can be estimated independently of the effect of infection on the cell cycle. Assuming only that the proliferation rate of infected cells lies between zero (total cell cycle blockade) and the proliferation rate of uninfected cells (no cell cycle blockade), we can derive accurate bounds on the death rates of different HIV variants. We found that the decrease in viable cells in infected cultures varies quantitatively with increased cell death but is relatively insensitive to cell cycle blockade. Hence, the measurements we make in our system will largely reflect viral cytopathicity effects.
To demonstrate the efficiency of our approach, we analyzed data from infections with wild-type NL4-3HSA virus and a mutant vpr virus NL4-3HSA that lacks the Vpr protein. Vpr has been implicated in both cell cycle blockade and cytopathic effects on infected cells (5, 11, 12, 18, 25, 26, 34, 36, 39). Compared to the wild-type virus, a vpr mutant showed a roughly twofold-attenuated cytopathicity. This is consistent with previous suggestions that Vpr could have a direct cytopathic effect (25, 26, 37, 39). Our model thus provides a framework in which to determine the alteration of death rates of infected cells by mutation of the viral genome, and it allows us to quantitatively demonstrate the decrease of the viral cytopathic potential.
The abilities to identify mutant HIV-1 strains with varied cytopathicity and to quantitate these differences are crucial for clearly identifying the cytopathic viral components. This may provide a deeper understanding of the role of HIV-1 in the immunodeficiency which results in AIDS. Finally, we note that our mathematical procedures are easily applicable to other model systems and may be used as a general tool for inferring differential cytopathicity or other effective rates in cell cultures.
Supplementary Material
Acknowledgments
We thank Andrew Leavitt, Ned Landau, John Coffin, Eric Freed, Malcolm Martin, and Steve Hughes for plasmids and advice offered in the course of this investigation. We also thank Francis Chan, Hyung Chun, Lixin Zheng, Viengngeun Bounkeua, Keiko Sakai, and Carol Trageser for their assistance in various aspects of this study.
Portions of this work were done under the auspices of the U.S. Department of Energy under contract W-7405-ENG-36 and supported by NIH grants AI28433 and RR06555 (A.S.P.). D.L.B. is a participant in the NIH-Johns Hopkins University Graduate Partnership Program in Biological Sciences.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Aiken, C. 1997. Pseudotyping human immunodeficiency virus type 1 (HIV-1) by the glycoprotein of vesicular stomatitis virus targets HIV-1 entry to an endocytic pathway and suppresses both the requirement for Nef and the sensitivity to cyclosporin A. J. Virol. 71:5871-5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ameisen, J. C., and A. Capron. 1991. Cell dysfunction and depletion in AIDS: the programmed cell death hypothesis. Immunol. Today 12:102-105. [DOI] [PubMed] [Google Scholar]
- 3.Bartz, S. R., and M. A. Vodicka. 1997. Production of high-titer human immunodeficiency virus type 1 pseudotyped with vesicular stomatitis virus glycoprotein. Methods 12:337-342. [DOI] [PubMed] [Google Scholar]
- 4.Bolton, D. L., B. I. Hahn, E. A. Park, L. L. Lehnhoff, F. Hornung, and M. J. Lenardo. 2002. Death of CD4+ T-cell lines caused by human immunodeficiency virus type 1 does not depend on caspases or apoptosis. J. Virol. 76:5094-5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brenner, C., and G. Kroemer. 2003. The mitochondriotoxic domain of Vpr determines HIV-1 virulence. J. Clin. Investig. 111:1455-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao, J., I. W. Park, A. Cooper, and J. Sodroski. 1996. Molecular determinants of acute single-cell lysis by human immunodeficiency virus type 1. J. Virol. 70:1340-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coffin, J. M. 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267:483-489. [DOI] [PubMed] [Google Scholar]
- 8.Fauci, A. S. 1988. The human immunodeficiency virus: infectivity and mechanisms of pathogenesis. Science 239:617-622. [DOI] [PubMed] [Google Scholar]
- 9.Fauci, A. S., and R. C. Desrosiers. 1997. Pathogenesis of HIV and SIV, p. 587-635. In H. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [PubMed]
- 10.Finkel, T. H., and N. K. Banda. 1994. Indirect mechanisms of HIV pathogenesis: how does HIV kill T cells? Curr. Opin. Immunol. 6:605-615. [DOI] [PubMed] [Google Scholar]
- 11.Gaynor, E. M., and I. S. Chen. 2001. Analysis of apoptosis induced by HIV-1 Vpr and examination of the possible role of the hHR23A protein. Exp. Cell Res. 267:243-257. [DOI] [PubMed] [Google Scholar]
- 12.He, J., S. Choe, R. Walker, P. Di Marzio, D. O. Morgan, and N. R. Landau. 1995. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69:6705-6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, and M. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123-126. [DOI] [PubMed] [Google Scholar]
- 14.Kim, S. Y., R. Byrn, J. Groopman, and D. Baltimore. 1989. Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infection: evidence for differential gene expression. J. Virol. 63:3708-3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimpton, J., and M. Emerman. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J. Virol. 66:2232-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenardo, M. J., S. B. Angleman, V. Bounkeua, J. Dimas, M. G. Duvall, M. B. Graubard, F. Hornung, M. C. Selkirk, C. K. Speirs, C. Trageser, J. O. Orenstein, and D. L. Bolton. 2002. Cytopathic killing of peripheral blood CD4+ T lymphocytes by human immunodeficiency virus type 1 appears necrotic rather than apoptotic and does not require env. J. Virol. 76:5082-5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levy, J. A. 1993. Pathogenesis of human immunodeficiency virus infection. Microbiol. Rev. 57:183-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lum, J. J., O. J. Cohen, Z. Nie, J. G. Weaver, T. S. Gomez, X. J. Yao, D. Lynch, A. A. Pilon, N. Hawley, J. E. Kim, Z. Chen, M. Montpetit, J. Sanchez-Dardon, E. A. Cohen, and A. D. Badley. 2003. Vpr R77Q is associated with long-term nonprogressive HIV infection and impaired induction of apoptosis. J. Clin. Investig. 111:1547-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manning, W. C., J. E. Murphy, D. J. Jolly, S. J. Mento, and R. O. Ralston. 1998. Use of a recombinant murine cytomegalovirus expressing vesicular stomatitis virus G protein to pseudotype retroviral vectors. J. Virol. Methods 73:31-39. [DOI] [PubMed] [Google Scholar]
- 20.Matlin, K. S., H. Reggio, A. Helenius, and K. Simons. 1982. Pathway of vesicular stomatitis virus entry leading to infection. J. Mol. Biol. 156:609-631. [DOI] [PubMed] [Google Scholar]
- 21.McCune, J. M. 2001. The dynamics of CD4+ T-cell depletion in HIV disease. Nature 410:974-979. [DOI] [PubMed] [Google Scholar]
- 22.Meng, G., X. Wei, X. Wu, M. T. Sellers, J. M. Decker, Z. Moldoveanu, J. M. Orenstein, M. F. Graham, J. C. Kappes, J. Mestecky, G. M. Shaw, and P. D. Smith. 2002. Primary intestinal epithelial cells selectively transfer R5 HIV-1 to CCR5+ cells. Nat. Med. 8:150-156. [DOI] [PubMed] [Google Scholar]
- 23.Mohri, H., A. S. Perelson, K. Tung, R. M. Ribeiro, B. Ramratnam, M. Markowitz, R. Kost, A. Hurley, L. Weinberger, D. Cesar, M. K. Hellerstein, and D. D. Ho. 2001. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J. Exp. Med. 194:1277-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mosier, D. E. 1995. HIV results in the frame: CD4+ cell turnover. Nature 375:193-194. [DOI] [PubMed] [Google Scholar]
- 25.Muthumani, K., A. Y. Choo, D. S. Hwang, M. A. Chattergoon, N. N. Dayes, D. Zhang, M. D. Lee, U. Duvvuri, and D. B. Weiner. 2003. Mechanism of HIV-1 viral protein R-induced apoptosis. Biochem. Biophys. Res. Commun. 304:583-592. [DOI] [PubMed] [Google Scholar]
- 26.Muthumani, K., D. S. Hwang, B. M. Desai, D. Zhang, N. Dayes, D. R. Green, and D. B. Weiner. 2002. HIV-1 Vpr induces apoptosis through caspase 9 in T cells and peripheral blood mononuclear cells. J. Biol. Chem. 277:37820-37831. [DOI] [PubMed] [Google Scholar]
- 27.Pakker, N. G., D. W. Notermans, R. J. de Boer, M. T. Roos, F. de Wolf, A. Hill, J. M. Leonard, S. A. Danner, F. Miedema, and P. T. Schellekens. 1998. Biphasic kinetics of peripheral blood T cells after triple combination therapy in HIV-1 infection: a composite of redistribution and proliferation. Nat. Med. 4:208-214. [DOI] [PubMed] [Google Scholar]
- 28.Pantaleo, G., C. Graziosi, J. F. Demarest, L. Butini, M. Montroni, C. H. Fox, J. M. Orenstein, D. P. Kotler, and A. S. Fauci. 1993. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature 362:355-358. [DOI] [PubMed] [Google Scholar]
- 29.Pantaleo, G., C. Graziosi, and A. S. Fauci. 1993. New concepts in the immunopathogenesis of human immunodeficiency virus infection. N. Engl. J. Med. 328:327-335. [DOI] [PubMed] [Google Scholar]
- 30.Perelson, A. S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D. D. Ho. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188-191. [DOI] [PubMed] [Google Scholar]
- 31.Perelson, A. S., A. U. Neumann, M. Markowitz, J. M. Leonard, and D. D. Ho. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582-1586. [DOI] [PubMed] [Google Scholar]
- 32.Piot, P., M. Bartos, P. D. Ghys, N. Walker, and B. Schwartlander. 2001. The global impact of HIV/AIDS. Nature 410:968-973. [DOI] [PubMed] [Google Scholar]
- 33.Rocancourt, D., C. Bonnerot, H. Jouin, M. Emerman, and J. F. Nicolas. 1990. Activation of a beta-galactosidase recombinant provirus: application to titration of human immunodeficiency virus (HIV) and HIV-infected cells. J. Virol. 64:2660-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roumier, T., H. L. Vieira, M. Castedo, K. F. Ferri, P. Boya, K. Andreau, S. Druillennec, N. Joza, J. M. Penninger, B. Roques, and G. Kroemer. 2002. The C-terminal moiety of HIV-1 Vpr induces cell death via a caspase-independent mitochondrial pathway. Cell. Death Differ. 9:1212-1219. [DOI] [PubMed] [Google Scholar]
- 35.Saifuddin, M., C. Crnich, T. Long, M. N. Saarloos, and G. T. Spear. 1998. Transfer of host T-cell membrane HLA-DR and CD25 to target cells by human retroviruses. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 17:196-202. [DOI] [PubMed] [Google Scholar]
- 36.Somasundaran, M., M. Sharkey, B. Brichacek, K. Luzuriaga, M. Emerman, J. L. Sullivan, and M. Stevenson. 2002. Evidence for a cytopathogenicity determinant in HIV-1 Vpr. Proc. Natl. Acad. Sci. USA 99:9503-9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stewart, S. A., B. Poon, J. B. Jowett, Y. Xie, and I. S. Chen. 1999. Lentiviral delivery of HIV-1 Vpr protein induces apoptosis in transformed cells. Proc. Natl. Acad. Sci. USA 96:12039-12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei, X., S. K. Ghosh, M. E. Taylor, V. A. Johnson, E. A. Emini, P. Deutsch, J. D. Lifson, S. Bonhoeffer, M. A. Nowak, B. H. Hahn, et al. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373:117-122. [DOI] [PubMed] [Google Scholar]
- 39.Yasuda, J., T. Miyao, M. Kamata, Y. Aida, and Y. Iwakura. 2001. T cell apoptosis causes peripheral T cell depletion in mice transgenic for the HIV-1 vpr gene. Virology 285:181-192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.