

Abstract
Aortic valve disease (AVD) is one of the leading causes of cardiovascular mortality. Abnormal expression of hyaluronan (HA) and its synthesizing/degrading enzymes have been observed during latent AVD however, the mechanism of impaired hyaluronan homeostasis prior to and after the onset of AVD remains unexplored. Transforming growth factor beta (TGFβ) pathway defects and biomechanical dysfunction are hallmarks of AVD, however their association with altered hyaluronan regulation is understudied. Expression of HA homeostatic markers was evaluated in diseased human aortic valves and TGFβ1-cultured porcine aortic valve tissues using histology, immunohistochemistry and western blotting. Further, porcine valve interstitial cell cultures were stretched (using Flexcell) and simultaneously treated with exogenous TGFβ1 ± inhibitors for activated Smad2/3 (SB431542) and ERK1/2 (U0126) pathways, and differential HA regulation was assessed using qRT-PCR. Pathological heavy chain HA together with abnormal regional expression of the enzymes HAS2, HYAL1, KIAA1199, TSG6 and IαI was demonstrated in calcified valve tissues identifying the collapse of HA homeostatic machinery during human AVD. Heightened TSG6 activity likely preceded the end-stage of disease, with the existence of a transitional, pre-calcific phase characterized by HA dysregulation. TGFβ1 elicited a fibrotic remodeling response in porcine aortic valves similar to human disease pathology, with increased collagen and HYAL to HAS ratio, and site-specific abnormalities in the expression of CD44 and RHAMM receptors. Further in these porcine valves, expression of HAS2 and HYAL1 was found to be differentially regulated by the Smad2/3 and ERK1/2 pathways, and CD44 expression was highly responsive to biomechanical strain. Leveraging the regulatory pathways that control both HA maintenance in normal valves and early postnatal dysregulation of HA homeostasis may identify new mechanistic insight into AVD pathogenesis.
Keywords: Whole leaflet organ culture, cell stretching, TGFβ pathways, KIAA1199, Inter-alpha-inhibitor, heavy chain hyaluronan
1. Introduction
Surgical intervention for aortic valve disease (AVD) is the second most common cardiothoracic procedure in the US. Over 100,000 valve surgical procedures are performed each year in the US, and AVD results in more than 25,000 deaths annually [1–4]. Aging is a significant clinical risk factor, and calcific AVD is the most common type of AVD occurring in 2% of the aged population [5–7]. Structurally, the aortic valve consists of three leaflets, each composed of three layers of organized extracellular matrix (ECM) with collagen in the fibrosa layer (outflow side), elastic fibers in the ventricularis layer (inflow side), and proteoglycans and glycosaminoglycans (GAGs) separating these in the intermediate spongiosa layer [8]. Valve ECM is primarily synthesized by valve interstitial cells (VICs), the predominant, mechanosensitive cells. The GAG hyaluronan (HA) is an abundant valve ECM component that maintains tissue hydration and flexibility, and impacts tissue viscoelasticity. Studies have shown that HA plays diverse biological roles in ECM assembly, cell proliferation and migration, and tissue development [9]. However, the molecular roles of HA in aortic valves are understudied.
HA is regulated through a homeostatic balance between enzymes that produce HA (HA synthases; HAS), enzymes that degrade HA (hyaluronidases; HYAL), and receptors that bind HA for retention and/or signaling, such as CD44 and RHAMM (the receptor for HA mediated motility) [10]. Any alterations in HA maintenance during disease is potentially indicative of coordinated pathogenesis involving these molecules. Most human valve HA studies have focused on mitral valve prolapse in which HA and proteoglycan accumulation leads to myxomatous change and valve dysfunction [11, 12]. However, calcific AVD results in a fibrotic aortic valve phenotype with increased collagen expression and associated GAG reduction at advanced age, and since aortic valves demonstrate tissue collagen deposition even with normal aging [13], analysis of collagen usually takes precedence over GAGs in AVD studies. As a result, GAG expression during AVD is largely underappreciated [14]. Previous studies from our group and others have shown abnormal expression of HA and its homeostatic markers in calcified aortic valve tissues [15–17], however the mechanistic regulation of HA homeostasis in aortic valves at early postnatal or diseased/end stages is poorly understood.
Development of novel therapies for AVD requires improved understanding of molecular pathways, such as transforming growth factor beta (TGFβ) signaling, in regulating valve ECM composition prior to disease. The TGFβ cytokine signaling pathways regulate valve cell phenotype and ECM remodeling through their ligands TGFβ1/2/3 via the activation of Smad2/3 by the TGFβ receptor complex (canonical pathway) [18]. The importance of other (non-canonical) pathways including the activation of mitogen-activated protein kinase (MAPK) cascades, notably the phosphorylation of ERK1/2 to pERK1/2 by TGFβ in valves, has only been recently established [19, 20]. Diseased aortic valves demonstrate increased VIC activation, as well as increased expression of TGFβ and its pathway activation factors such as pSmad2/3 and pERK1/2 [21–24]. Further, TGFβ promotes VIC proliferation and activation and is known to regulate GAG fine structure as well as HA synthesis in cultured VICs [23, 25–27]. Therefore these molecular pathways may play a significant role in both pre-calcific and late-stage calcified valves.
AVD manifests as stenosis and/or regurgitation and causes hemodynamic perturbations that often affect VIC phenotype initiating a maladaptive ECM remodeling response and resulting in a progressive disease process. Biomechanical studies on cultured VICs have shown that both calcific and myxomatous changes in valves are strain dependent. In particular, mechanical strain together with TGFβ enhances the activation, remodeling, and calcification potential of cultured VICs [28, 29]. HA secretion by VICs is mechanosensitive [30–32]; however, the mechanical regulation of its homeostatic factors is unclear. Along with biomechanics, aging provides an environmental factor that amplifies the early genetic predisposition associated with aortic valve malformation, resulting in AVD later in life [5–7]. Significant age-induced alterations in GAG content and biomechanical properties have been identified in aortic valves from humans and other animals [33, 34]. Therefore, simultaneous investigation of HA homeostasis, TGFβ, biomechanics, and aging is important to understand their mechanistic interactions during AVD and to elucidate structure-function relationships in native as well as diseased valves.
Overall, the goal of this study was to combine molecular and biomechanical approaches to determine the differential effects of Smad2/3 and ERK1/2 pathways on markers of HA homeostasis (regulatory enzymes / receptors) particularly in presence of biomechanical stretch, in the framework of postnatal AVD. We tested the hypothesis that Smad2/3 and ERK1/2 pathway inhibitors rescue the defective regulation of HA homeostatic regulators in VICs exposed to TGFβ1 and biomechanical strain, and that the rescue effect of these inhibitors is additive. We show that TGFβ caused fibrotic remodeling and regional abnormalities in the expression patterns of HA homeostatic regulators in porcine aortic valves, similar to diseased human valves. Further, the Smad2/3 and ERK1/2 pathways differentially regulated HA enzymes and there is an imbalance of HAS to HYAL ratio in valves exposed to TGFβ1 and biomechanical stretch. These results advance our understanding of HA homeostasis in AVD pathogenesis and may provide insight for future mechanistic studies.
2. Methods
2.1. Human Tissue Acquisition
Human hearts with normal (young and aged) and sclerotic (aged) aortic valves were obtained from donors through the National Disease Research Interchange program. The donors were under the age 20 (N=4) or over the age 50 (N=7; 4 normal, 3 sclerotic), for the young and aged stages respectively. They demonstrated normal cardiac structure and function, and died of non-cardiac causes with a warm ischemia time of less than 6 hrs. As revealed by patient autopsy information reports, healthy donors with normal aortic valves possessed none of the clinical risk factors for AVD such as diabetes, hypertension, hyperlipidemia, or heavy smoking, whereas the donors with sclerotic aortic valves exhibited at least two of these factors (Supplemental Table 1). Presence of sclerotic lesions and mild mineralization in these valves was visually confirmed by three independent investigators. Overtly calcified aortic valve tissues from patients over the age 50 (N=4) were procured during valve replacement surgeries at the Houston Methodist Hospital (Houston, TX) and decalcified [15, 16]. The valve leaflets from normal young, normal aged, sclerotic aged, and calcified aged patients were processed for histology. All tissue handling protocols have been approved by Rice University Institutional Review Board.
2.2. Porcine Whole Valve Leaflet Culture
Whole aortic valve leaflets were dissected from young (1–3 month old, N=6), adult (6–9 month old, N=24) and aged (>2 years old, N=6) pig hearts obtained from a local commercial abattoir (Animal Technologies, Tyler, TX). All the young and aged leaflets, and some of the adult leaflets (6/24) were directly processed for histology as fresh tissues. The remaining 18 adult leaflets were secured in 6-well plates for static leaflet cultures (Sapp et al., in Review). Briefly, 6-well plates were coated with polydimethylsiloxane (PDMS, to discourage cell attachment) and heated for 12 hrs at 80°C to cure the polymer. A 12 mm insect pin was inserted through the center of each valve leaflet and secured in the PDMS layer. Each well was filled with 8 mL of cell culture media (10% bovine growth serum (BGS) in Dulbecco’s modified Eagle medium (DMEM, Hyclone, Logan, UT) with 5 mmol/L glucose and 1% antibiotics) to ensure the leaflets were completely submerged. Leaflets were cultured for 2 weeks in a 37°C incubator with 5% CO2, and media was changed every 4 days. Cultures were split into 3 groups that included 2 ng/mL TGFβ1 (PeproTech, ON, Canada), 10 mM beta-glycerophosphate (βGP; Sigma-Aldrich, St. Louis, MO), or a no treatment culture control (6 per each treatment). After 2 weeks of culture, the leaflets were processed for histology. A heavily calcified aortic valve obtained randomly during one of the porcine valve harvests was also saved and processed for histology as a disease (positive) control.
2.3. Histology and Immunohistochemistry (IHC)
To evaluate regional ECM composition and organization, histochemistry was performed on 5 μm tissue sections using Movat’s modified pentachrome stain, which colors elastic fibers black, collagen fibers yellow, proteoglycans and GAGs blue, muscle red, and cell nuclei purple. Von Kossa histological stain was used to visualize valve tissue mineralization/calcification [35]. Antibodies directed against hyaluronan-binding protein (HABP, 1:100, Millipore), IαI (Inter-alpha-Inhibitor, 1:100, Dako), HAS2 (Abcam, 1:150), HYAL1 (Abcam, 1:200), KIAA1199 (Abcam, 1:200), CD44 (Calbiochem, 1:200), RHAMM (Receptor for HA Mediated Motility, Novus Biologicals, 1:100), TSG6 (Tumor Necrosis Factor-Inducible Gene 6 Protein, Santa Cruz Biotechnology, 1:200) and pERK1/2 (Cell Signaling Technology, 1:50) were used to determine protein expression and localization using standard or double immunofluorescence (HABP/IαI), or streptavidin/biotin colorimetry and diaminobenzidine (DAB) detection (all others) [36, 37]. Apoptosis in cell cultures was examined using the TACS® 2 TdT-DAB In Situ Apoptosis Detection Kit (Trevigen, Gaithersburg, MD) according to the manufacturer’s specifications. Antigen retrieval was performed using heat-mediated citrate buffer or Proteinase K. Imaging was done by confocal (Figs. 4 and 7) and bright-field microscopy (Figs. 1–3 and 7; Supplemental Figs. 1–3 and 5).
2.4. Analysis of Staining
ImageJ (NIH, Bethesda, MD) was used to quantitatively analyze IHC and Movat pentachrome stains, as recently described [38]. The ImageJ color thresholding function was used to analyze percent area coverage of markers that stained ECM alone or both cells and ECM in whole leaflet sections. All sections were thresholded to a brightness value that excluded slide background while including all of the tissue area. The Movat sections were thresholded according to a range of hues corresponding to the ECM stain colors. For DAB IHC of cells and ECM, tissue sections were thresholded to hues corresponding to the brown DAB color and the blue counterstain, hematoxylin. IHC that stained cells and ECM concurrently was quantified as percent area coverage of the stain over the total area of the tissue. For IHC that stained only cells, the ImageJ analyze particles function was used to quantify the percentage of cells stained with a cell-specific marker vs. the total number of cells in the whole leaflet section (Sapp et al., in Review).
2.5. Mechanical and TGFβ1/Inhibitor Stimulation of Cultured VICs
VICs were isolated from aortic valves from young and aged porcine hearts (n=6–9 per stage) using collagenase digests according to previous published methods [39], and cultured in Flexcell culture plates (with dimensions of a standard 6-well plate; Flexcell International, Hillsborough, NC) containing DMEM (5 mmol/L glucose), Ham’s F12 (Hyclone, Logan, UT), 10% BGS, and 1% antibiotic under sterile conditions. Media was changed every 2 days. Once confluent, the VIC cultures were serum-deprived by incubation in DMEM (5 mmol/L glucose) with 0.1% BGS for 24 hrs. Cells were then subjected to 8 different treatment conditions: no treatment (control), TGFβ1 only (2 ng/mL), SB431542 only (Smad2/3 pathway inhibitor, 3 μM), U0126 only (ERK1/2 pathway inhibitor, 5 μM), TGFβ1 + SB431542, TGFβ1 + U0126, SB431542 + U0126, and TGFβ1 + SB431542 + U0126. The optimal culture concentrations of TGFβ1 and the inhibitors were derived from previous studies from our group and others [20, 23]. These treatments were added to the low-serum media, and the culture plates were then loaded within base plates attached to a Flexcell system in a 37°C incubator. The cells were subjected to uniaxial cyclic stretch at 5% strain magnitude with 1 Hz frequency for 24 hrs [19, 28]. In total, 6 Flexcell runs were performed, each consisting of 4 plates (i.e., 24 wells) for 8 treatments (i.e., 3 wells per treatment per run). Static controls, i.e., VICs cultured on Flexcell plates with no applied stretch, were also included for all the treatments.
2.6. Molecular and Biochemical Analysis of Cultured VICs and the Conditioned Media
After cell culture, the conditioned media from different treatments was saved, and RNA from the VICs was extracted using Trizol (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. The mRNA was reverse transcribed into cDNA using Primescript 1st Strand cDNA Synthesis Kit (Takara Bio, Otsu, Japan). Quantitative RT-PCR (qRT-PCR) was performed on the cDNA using 2X QuantiTect SYBR Green PCR Master Mix (Clontech, Mountain View, CA) with a Realplex Mastercycler (Eppendorf, Hamburg, Germany) to measure differences in gene expression levels for HA homeostatic regulators (HAS1, HAS2, HAS3, HYAL1, HYAL2, CD44) and VIC activation (αSMA) between the different treatments (TGFβ1/inhibitors/strain) [38]. This was performed in triplicate for each treatment. The gene expression corresponding to each treatment was normalized to expression levels for “no treatment, static” following the mathematical model for relative qRT-PCR [40], with GAPDH being the housekeeping gene. All DNA primers were purchased from Integrated DNA Technologies (Coralville, IA).
Secreted GAGs and collagens in the conditioned media were quantified using Blyscan and Sircol assays (Biocolor, Carrickfergus, UK) respectively [41]. Colorimetric data was analyzed using a Spectramax M2 (Molecular Devices, Sunnyvale, CA) and compared to standard curves prepared using known quantities of rat-tail collagen or glucuronic acid for the Sircol or Blyscan assay respectively.
2.7. Analysis of HA Heavy Chain Modification and TSG6 Activity in Calcified Human Valves
Hyaluronidase extraction of heavy chains (HCs) from the pathological HC-HA complexes was performed using Western blot analysis as previously described [37]. Briefly, a 50 μL minced calcified valve tissue suspension was prepared in PBS and 10 μL of Streptomyces hyaluronidase (0.5 turbidity unit/ml; Seikagaku, East Falmouth, MA) was added. The mixture was centrifuged at 13,000 × g at 4°C for 5 min, and the supernatant was incubated for 30 min at 37°C. Subsequently, 25 μL of this digest was added per lane on 4–15% Mini-PROTEAN TGX gels (Bio-Rad, Hercules, CA) and blotted using the Bio-Rad nitrocellulose and Trans-Blot Turbo System. The blots were blocked with Li-Cor blocking buffer (Li-Cor, Lincoln, NE), and then probed with an antibody against IαI (1:8000, Dako North America, Carpinteria, CA). The final blots were imaged on an Odyssey Infrared Imaging System (Li-Cor).
Activation of the enzyme TSG6 is responsible for the formation of HC-HA complexes, and for the measurement of endogenous TSG6 activity in calcified valves, 2 μL of human serum was added to 50 μL of the tissue extract to provide a HC donor (i.e., IαI). 2 μL of an HA oligosaccharide (HA14, 1 mg/mL; Hyalose LLC, Oklahoma City, OK) and 3 μL of PBS were then added to 50 μL of the tissue extract and incubated at 37°C for 2 hrs [42]. For positive control, 2 μL of recombinant TSG6 (R&D Systems, Minneapolis, MN) was included. The mixture was centrifuged at 13,000 × g for 5 min at 4°C, and the supernatant was used for Western blot analysis.
2.8. Statistical Analysis
The statistical analyses for Movat Pentachrome and DAB immunostaining were performed using an ANOVA with a post-hoc Tukey’s test to analyze differences due to treatment (fresh, culture control, TGFβ1, βGP) and age (young, adult, aged) on cultured tissues. For qRT-PCR and Blyscan/Sircol data analysis, ANOVA and post hoc Bonferroni multiple comparison tests were used to determine the effect of treatment (TGFβ1 ± SB431542 ± U0126) and age (young, aged) on gene/protein expression in cultured cells. All analysis was performed using R studio software (version 0.98.953). All variables were expressed as mean ± standard deviation, and results were considered significant at p<0.05.
3. Results
3.1. Regional Misexpression of HA Homeostatic Markers in Diseased Human Aortic Valves
In young human aortic valves, HAS2 expression was primarily restricted to VICs (i.e., not expressed in the ECM) and was present in all three layers of the valve, whereas its expression significantly increased and expanded to the ECM in older valves where it was mostly present in the spongiosa and ventricularis layers (Fig 1A1, A2). HYAL1 was localized to the spongiosa layer of both normal young and aged valves, and was not significantly different between the two ages. While young valves showed HYAL1 confinement to a narrow, concentrated region, older valves demonstrated a wider and more diffuse HYAL1 expression pattern (Fig 1B1, B2). Sclerotic aged and calcified aged aortic valves demonstrated a reduction in HAS2 expression compared to normal aged valves, largely due to shrinkage of the GAG-rich spongiosa layer and associated enlargement of the collagen-rich fibrosa layer (Fig 1A3, A4; Supplemental Fig 1). HYAL1 presence was consistently observed in sclerotic as well as calcified valves (Fig 1B3, B4), however their expression was not significantly different from normal valves. Interestingly, there was HAS2 and HYAL1 overexpression in focal regions of the fibrosa layer in both sclerotic and calcified valves (arrowheads, Fig 1A3, A4, B3, B4).
Figure 1.
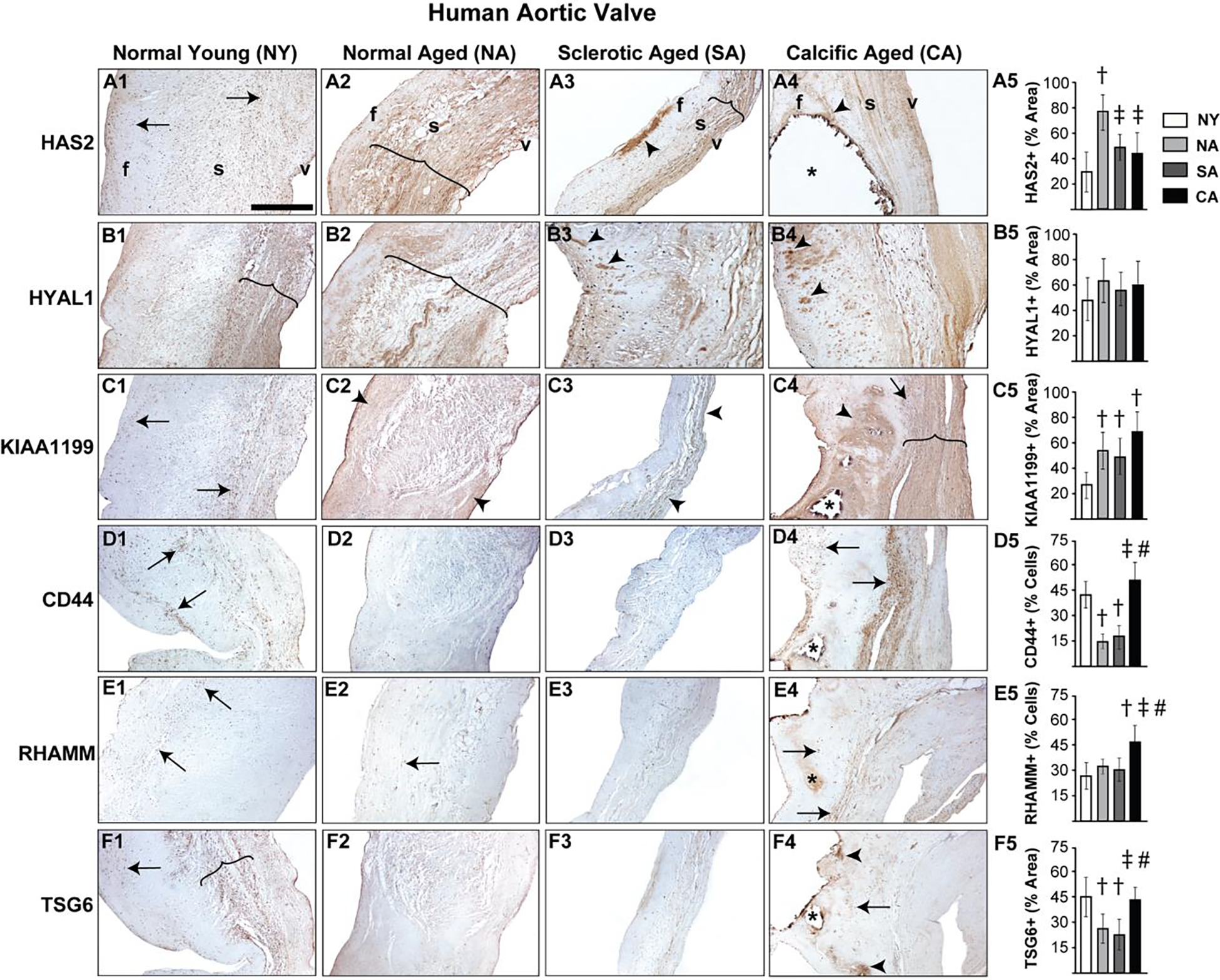
Diseased human aortic valves demonstrate abnormalities in regional expression of HA homeostatic markers. Expression patterns of HAS2 (A1-A5), HYAL1 (B1-B5), KIAA1199 (C1-C5), CD44 (D1-D5), RHAMM (E1-E5) and TSG6 (F1-F5) in valves classified as young (15–20 year old) or aged (>50 year old) and either normal or abnormal (sclerotic/calcific) are shown. Arrows and arrowheads denote cell and ECM expression respectively. The region of expression is marked by parenthesis. Calcific nodules are indicated by asterisk. †: vs normal young p<0.05; ‡: vs normal aged p<0.05; #: vs sclerotic aged p<0.05. f, fibrosa; s, spongiosa; v, ventricularis. The orientation provided by the use of f, s, v in panels A1-A4 apply to all the microscopic image panels in this figure. Scale bar: 200 μm.
KIAA1199 is a largely understudied HA depolymerization enzyme, and recent studies have shown its expression in cultured porcine VICs [43, 44]. In human aortic valve tissues, KIAA1199 increased with age, was mostly expressed in the cells at the young stage and in the ECM at advanced age (Fig 1C1, C2). Calcified aortic valves demonstrated heightened KIAA1199 expression as well, especially around the calcific nodules in the fibrosa layer (Fig 1C4).
CD44 and RHAMM receptors were ubiquitous in young aortic valves. Whereas CD44 expression diminished with age, RHAMM was unchanged (arrows, Fig 1D1, D2, E1, E2). TSG6, a HA-binding protein, was predominantly expressed in the cells and ECM of the valve spongiosa layer in young valves, and was downregulated in older valves (Fig 1F1, F2). However, calcified aortic valves exhibited marked upregulation of CD44 and RHAMM, and focal expression of TSG6, notably in the periphery of the calcific nodules (arrowheads, Fig 1D4, E4, F4). Taken together, these findings suggest dysregulation of enzymes that synthesize and degrade HA, combined with defective HA receptor expression, resulting in abnormal HA homeostasis during human AVD.
3.2. Aberrant Regulation of HA Homeostasis due to TGFβ in Porcine Aortic Valve Tissue
It is well established that porcine aortic valves are the most representative animals for human valve studies and exogenous TGFβ1-based in vitro porcine valve cultures are commonly used models for AVD research [19, 29, 45, 46], therefore we sought to examine the expression of HA homeostatic markers in normal and TGFβ1-treated porcine aortic valve tissues. Both HAS2 and HYAL1 expression in normal porcine valves was limited to spongiosa VICs at young stages, but similar to human valves, they showed overall expansion, and significantly enhanced expression in both VICs and the ECM in all layers of the valve at the aged stage (Fig 2A, B). When cultured in TGFβ1, porcine aortic valves exhibited fibrotic remodeling with significant reduction in GAG to collagen ratio compared to the fresh and culture controls (Fig 3A), consistent with previous findings [29]. Further, these TGFβ1-cultured valves demonstrated significantly increased HAS2 expression (~45% area coverage, p<0.05) and HYAL1 expression (>80% area coverage, p<0.001) when compared with controls (Fig 3C, D).
Figure 2.
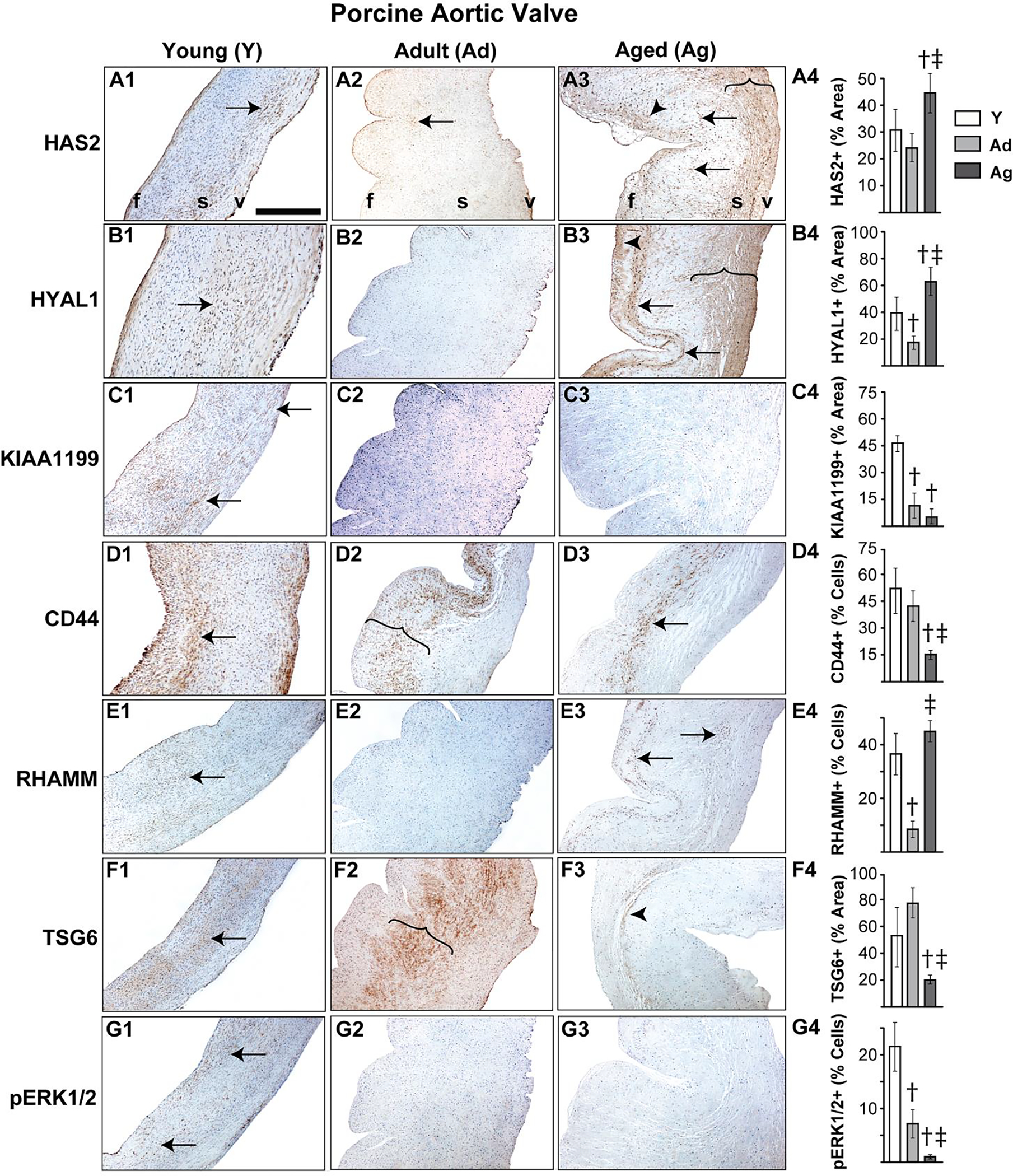
Porcine aortic valves demonstrate altered expression of HA homeostatic markers with age. Expression patterns of HAS2 (A1–4), HYAL1 (B1–4), KIAA1199 (C1–4), CD44 (D1–4), RHAMM (E1–4), TSG6 (F1–4) and pERK1/2 (G1–4) in valves classified as young (1–3 month old), adult (6–9 month old) or aged (>2 year old) are shown. Arrows and arrowheads denote cell and ECM expression respectively. Parentheses mark the regions of expression. †: vs young p<0.05; ‡: vs adult p<0.05. f, fibrosa; s, spongiosa; v, ventricularis. The orientation provided by the use of f, s, v in panels A-D apply to all the microscopic image panels in this figure. Scale bar: 200 μm.
Figure 3.
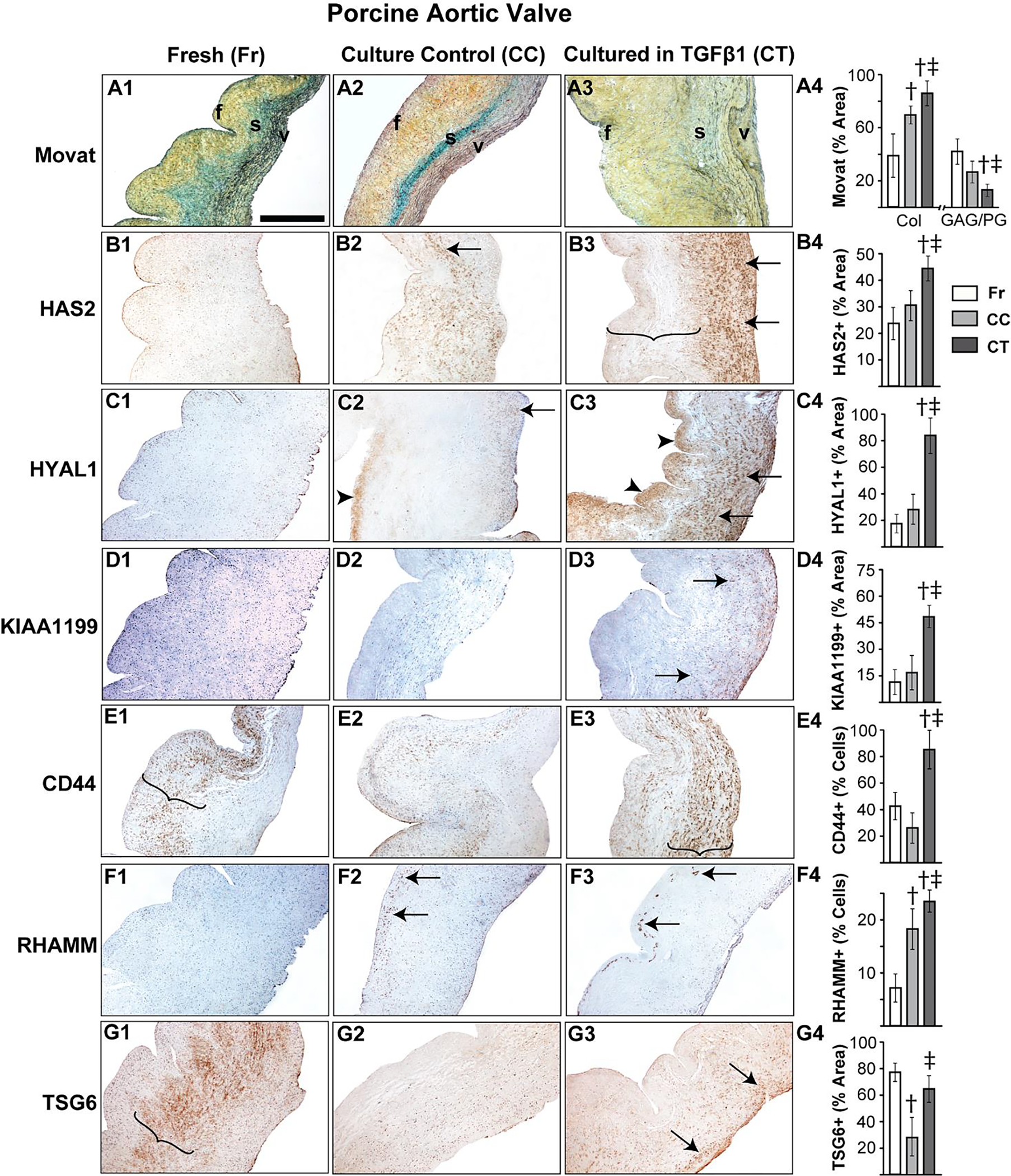
TGFβ1 causes aberrant regulation of HA homeostasis in porcine aortic valve tissue. Movat histochemical staining (A1–4) and expression patterns of HAS2 (B1–4), HYAL1 (C1–4), KIAA1199 (D1–4), CD44 (E1–4), RHAMM (F1–4) and TSG6 (G1–4) in valves classified as fresh, culture control or cultured in TGFβ1 are shown. Arrows and arrowheads denote cell and ECM expression respectively. Parenthesis mark the regions of expression. †: vs fresh p<0.05; ‡: vs culture control p<0.05. f, fibrosa; s, spongiosa; v, ventricularis. The orientation provided by the use of f, s, v in panels A-D apply to all the microscopic image panels in this figure. Scale bar: 200 μm.
Unlike human valves, normal porcine aortic valves showed only cellular expression, and hardly any ECM-located expression, of KIAA1199 at the young stage and, surprisingly, very low expression overall at the adult and aged stages (Fig 2C). However, aortic valves cultured in TGFβ1 showed significantly increased cell-specific KIAA1199 expression compared to fresh and culture controls (Fig 3D). CD44 and TSG6 expression was significantly reduced with age, as observed with human valves, and cellular RHAMM expression was widespread in young and old valves. All these proteins were largely present in the spongiosa layer (Fig 2D–F). TGFβ1 caused a significant upregulation of both CD44 and RHAMM receptors compared to fresh controls (Fig 3E, F); in particular, RHAMM was focally expressed in the subendothelial aspect of the fibrosa layer in TGFβ1-cultured valves (arrows, Fig 3F3). TSG6 was not significantly different between TGFβ1-cultured valves and the fresh controls (Fig 3G). Further, pERK1/2 showed a marked reduction in expression over time, suggesting the decline of non-canonical TGFβ activity following tissue maturation (Fig 2G). Overall, dysregulation of HA homeostasis is induced by TGFβ abnormalities during AVD.
In addition to TGFβ1, studies have shown that βGP is an established inducer of valve calcification in vitro [35], and therefore we sought to examine its exogenous regulation of HA homeostatic markers in porcine aortic valves. Valves cultured in βGP demonstrated fibrotic remodeling similar to TGFβ1-cultured valves, and its detailed findings and interpretations have been included in Supplemental results and a figure (Supplemental Fig 2). A porcine calcified aortic valve identified during one of the random harvests and included as a positive (disease) control showed extensive calcification as revealed by the Von Kossa stain (Supplemental Fig 3C). While there was modest HAS2 and HYAL1 distribution in this valve throughout, KIAA1199 showed heightened expression in the vicinity of calcific nodules. CD44, pERK1/2 and TSG6 were highly expressed in the perinodular regions, whereas RHAMM was mostly observed near outer edges of the valve away from the nodules (Supplemental Fig 3D–J).
3.3. Differential Regulation of Markers of HA and ECM Homeostasis by Canonical and Non-canonical TGFβ Pathways
Our Flexcell studies on aortic VICs cultured with TGFβ1 and/or its canonical and non-canonical inhibitors showed that HAS2 (in young VICs) and HYAL1 gene expression (in aged VICs) increased in the presence of TGFβ1 (Fig 4A, B). HYAL1 expression was significantly higher in older VICs compared to young VICs with or without TGFβ1. Interestingly at 5% strain, HYAL1 was upregulated even in young VICs exposed to TGFβ1. HAS2 expression was significantly reduced in older VICs in presence of both TGFβ1 and cyclic strain; however, HYAL1 expression was quite high under these conditions indicating an alteration in HAS to HYAL ratio. Even in the absence of any biomechanical stimulation, there was an aging-induced increase in expression of both HAS2 and HYAL1 and this confirmed our organ culture bioreactor findings (Fig 2A, B). In presence of TGFβ pathway inhibitors SB431542, U0126, or both, HAS2 (p<0.001 for SB431, p<0.05 for U0126 and p<0.0001 for both) was significantly downregulated at both young and old ages, and HYAL1 (p<0.05 for either, both) was downregulated at old age alone, compared to the no treatment control. However, HYAL1 expression was higher at the aged than at the young stage in VICs exposed to the inhibitors. When the inhibitors were added along with TGFβ1, SB431542 recovered HAS2 to control levels at both young and aged stages while U0126 recovered HAS2 levels only in young VICs. In contrast, SB431 had negligible effects on HYAL1 expression whereas U0126 completely recovered HYAL1 to control levels in both young and aged VICs, suggesting differential pathway mediation of HAS and HYAL turnover in VICs. HAS1 and HAS3 expression levels were modest in all treatment and strain conditions (data not shown), whereas HYAL2 did not show any significant difference between the groups (Fig 4C).
Figure 4.
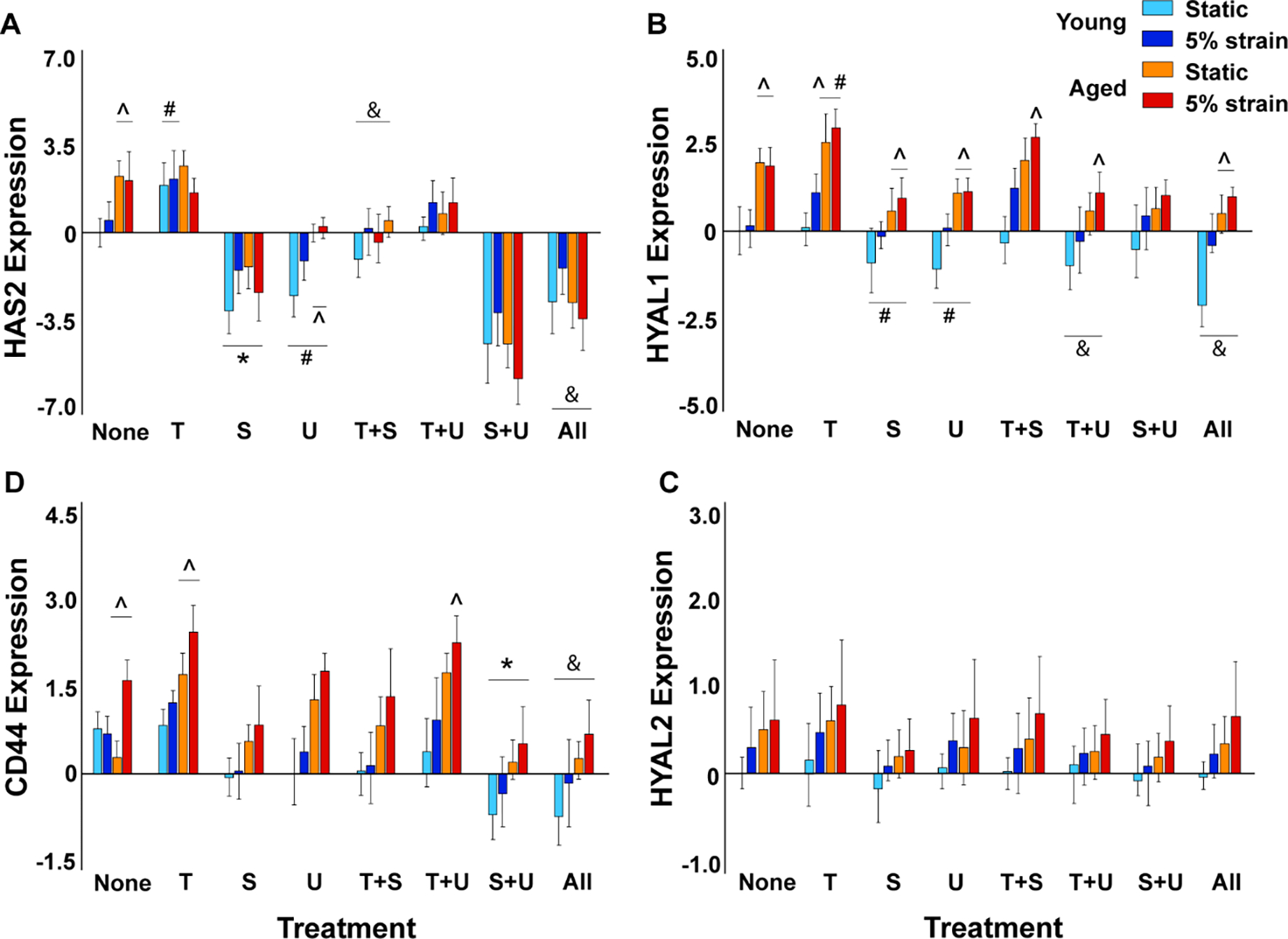
Regulation of HA homeostatic marker gene expression by exogenous TGFβ1, its inhibitors, biomechanical strain and aging in cultured porcine aortic VICs as measured by qRT-PCR. Gene expression (on y-axis) is presented as relative log2 expression, and all data for each gene is normalized to young, static with no treatment. “None”, no treatment; “T+S”, TGFβ1 + SB431542; “T+U”, TGFβ1 + U0126; “S+U”, SB431542 + U0126; “All”, TGFβ1 + SB431542 + U0126. #, vs. none, p<0.05; *, vs. none, p<0.001; &, vs. T, p<0.001; ^, vs. young, p<0.0001. Another version of the same figure with more elaborate statistics is provided as Supplemental Fig 4.
CD44 gene expression was downregulated with age (consistent with findings in Fig 2) under no strain conditions (Fig 4D). However, 5% strain significantly upregulated CD44 expression at advanced age and this effect was aggravated in TGFβ1-treated VICs. Neither SB431 nor U0126 had any effect on CD44 expression, however their combined treatment dramatically lowered CD44 expression. When the inhibitors were added along with TGFβ1, SB431 was more effective than U0126 in recovering CD44 levels, especially in older VICs. αSMA gene expression was consistently higher in older VICs compared to young VICs, and TGFβ1 worsened the activated phenotype overall [26] (Fig 5A). Neither inhibitor rescued the cell phenotype. Interestingly, young VICs cultured under static conditions demonstrated very low αSMA through all treatments, which may be attributed to lack of stiffness of the Flexcell membrane [47]. TUNEL, an apoptosis assay, confirmed no large scale cell death in VIC Flexcell cultures, both with and without the inhibitor treatments for up to 48 hrs (Fig 5B).
Figure 5.
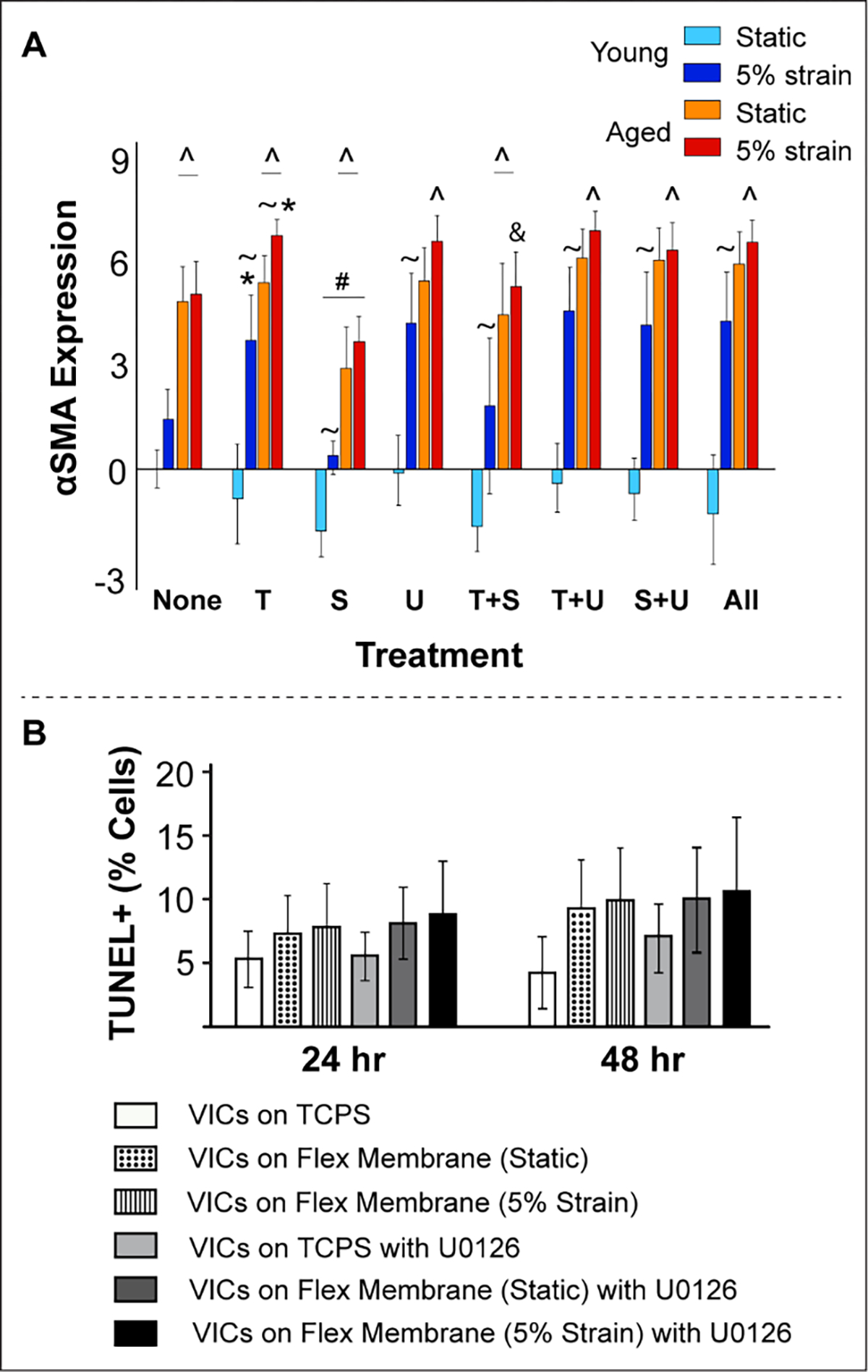
A) Regulation of αSMA (VIC activation) gene expression by exogenous TGFβ1, its inhibitors, biomechanical strain and aging in cultured porcine aortic VICs as measured by qRT-PCR. Gene expression (on y-axis) is presented as relative log2 expression, and all data for each gene is normalized to young, static with no treatment. “None”, no treatment; “T+S”, TGFβ1 + SB431542; “T+U”, TGFβ1 + U0126; “S+U”, SB431542 + U0126; “All”, TGFβ1 + SB431542 + U0126. #, vs. none, p<0.05; *, vs. none, p<0.001; &, vs. T, p<0.001; ^, vs. young, p<0.0001; ~, vs. age-matched static, p<0.05. B) TUNEL assay for apoptosis confirmed no significant cell death in VICs cultured for up to 48 hrs in either static or dynamic conditions and with or without the inhibitor. VICs cultured on tissue culture polystyrene dishes were used as controls.
ECM homeostasis was examined by estimating GAG and collagen levels in the conditioned media of these cultured porcine VICs. In young VICs, TGFβ1 elevated GAG synthesis in both static and 5% strain cultures, while reducing collagen synthesis in static and increasing its synthesis at 5% strain (Fig 6, Supplemental Fig 5). In aged VICs, both GAGs and collagen increased with TGFβ1 addition, in static as well as 5% strain conditions. Both the inhibitors independently rescued collagen as well as GAG levels in presence of TGFβ1 in young VICs, and their effects were additive. However in aged VICs in presence of TGFβ1, only SB431 and the combined inhibitor treatment recovered GAGs to control levels whereas collagen recovery was possible with either or both the inhibitors [46]. Overall, our gene expression studies revealed a disproportionate HAS to HYAL expression ratio, and other HA and ECM homeostatic abnormalities due to TGFβ1 and aging; these HA-specific findings were consistent with porcine and human tissue pathology (Sections 3.1, 3.2). Further, TGFβ pathway inhibitors differentially rescued HAS, HYAL, GAGs and collagen following TGFβ1 and biomechanical stimulation.
Figure 6.
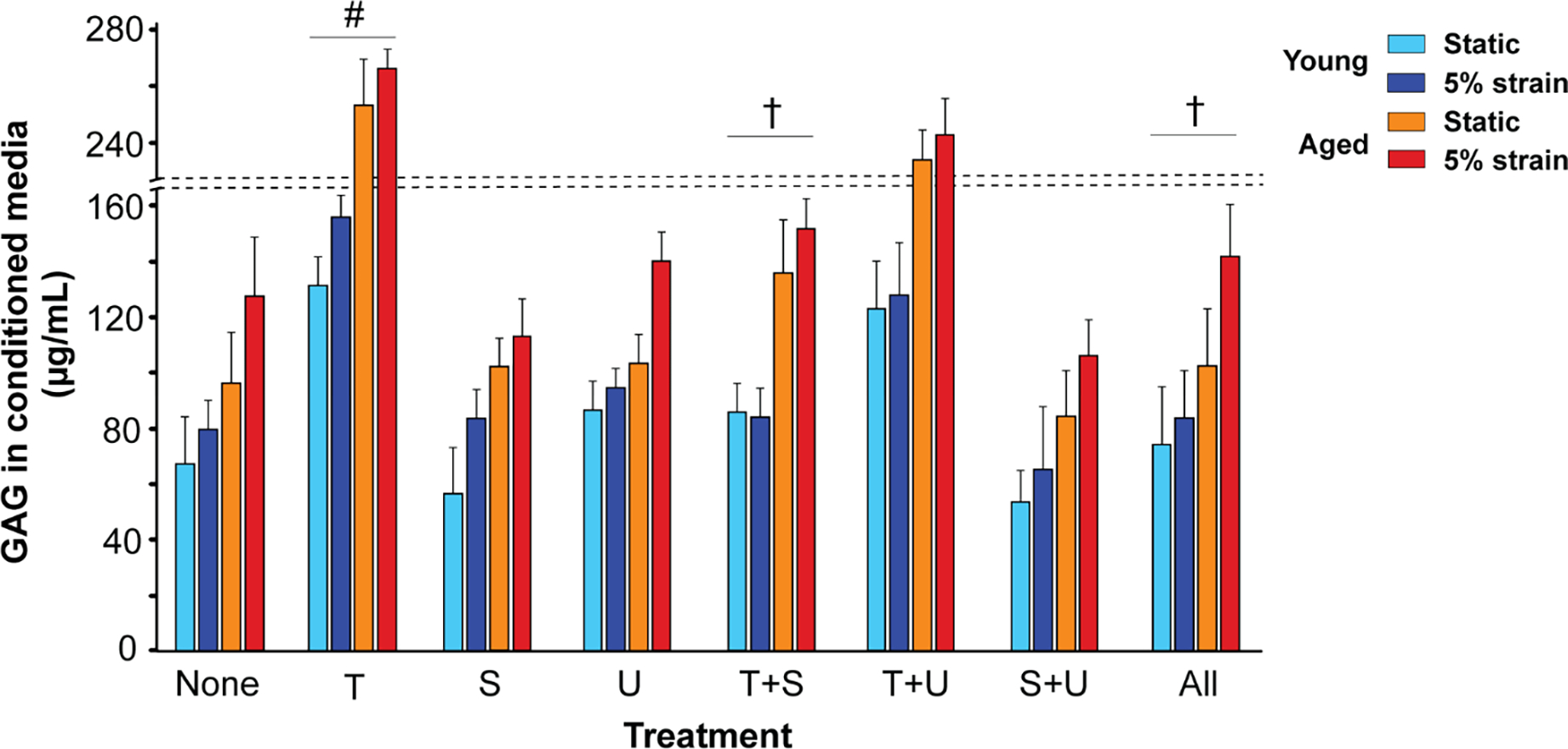
Regulation of GAG secretion in the conditioned media of cultured young and aged porcine VICs by exogenous TGFβ1, its inhibitors, biomechanical strain and aging. “None”, no treatment; “T+S”, TGFβ1 + SB431542; “T+U”, TGFβ1 + U0126; “S+U”, SB431542 + U0126; “All”, TGFβ1 + SB431542 + U0126. #, vs. none, p<0.001; †, vs. T, p<0.05.
3.4. Pathological Modification of HA in Calcified Human Aortic Valves
Although HA in normal tissues lacks any protein modifications, studies have shown that during disease, a unique protein modification occurs on HA where the activated form of the enzyme TSG6 covalently and irreversibly transfers heavy chains (HCs) from the serum-derived proteoglycan IαI to HA to form the pathological HC-HA complex [48]. Our IHC analysis showed the prevalence of HA and IαI in calcified human aortic valve tissues (Fig 7A–D). HA distribution in these valves was consistent with previous findings [16]. HA was co-localized with IαI (Fig 7E), suggesting HA was modified with HCs and the HCs of IαI may be covalently attached to the HA backbone in diseased valves [37]. As expected, there were significant amounts of pathological HC-HA complex in the calcified aortic valves demonstrated by western blotting (Fig 7F) and by hyaluronidase extraction of HC-HA from the tissues (Supplemental Fig 6). However, TSG6 activity was absent overall (Fig 7G), despite its focal expression in these valves (Fig 1F4) [15], and high levels of the enzymatic product of TSG6 (i.e. the HC-HA complex seen in Fig 7F) in these tissues. Therefore, it is possible that TSG6 activity was present earlier in AVD pathogenesis while its product (HC-HA) chronically remained in the calcified aortic valve.
Figure 7.
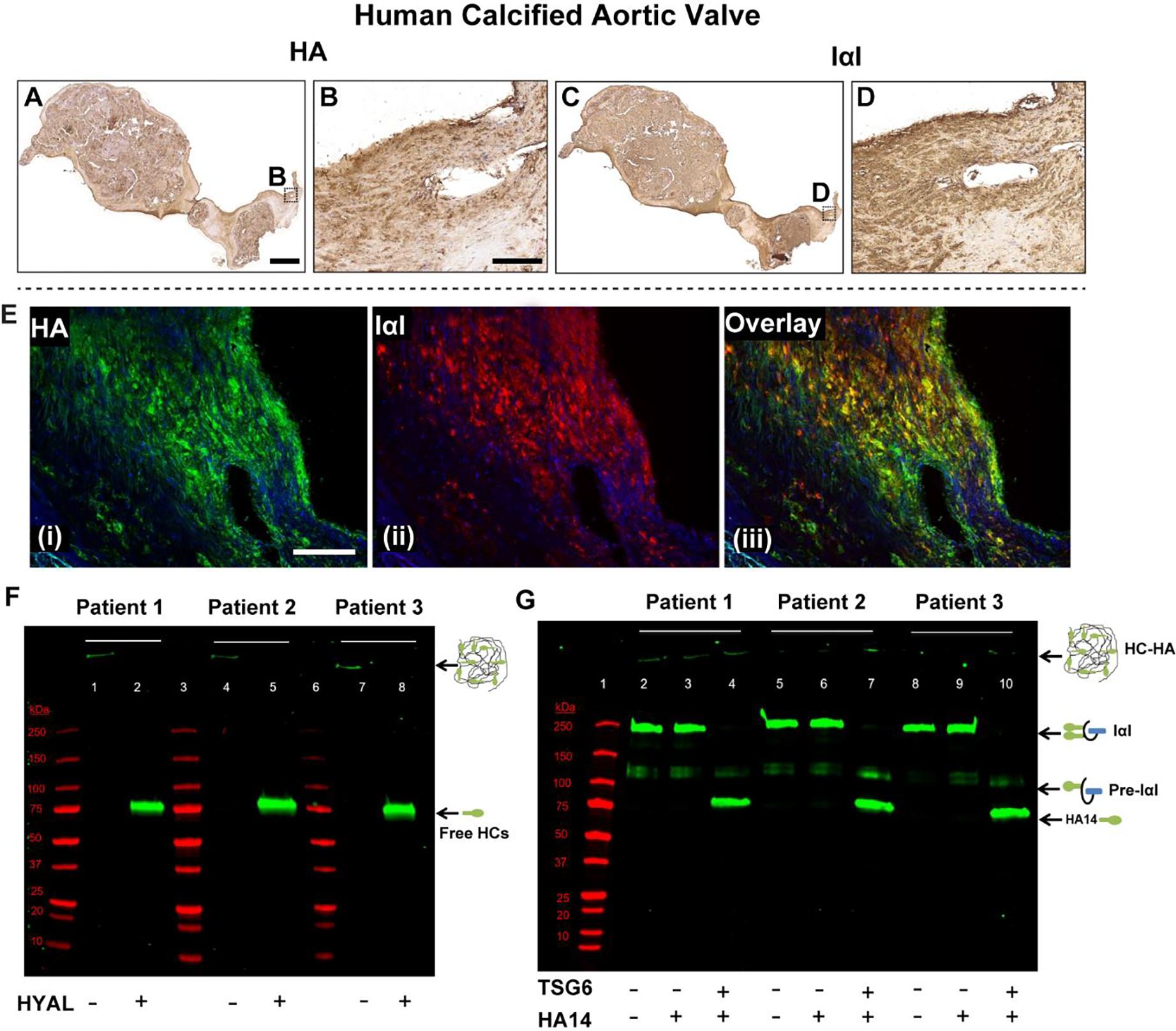
Human calcified aortic valves demonstrate extensive HA and IαI expression, and their co-localization (A-E). Their secondary-only controls are included in Supplemental Fig 5 for comparison. F) Western blot demonstrating presence of the pathological form of HA (i.e., heavy chain (HC)-HA complex) in these valves. When hyaluronidase (HYAL) is added to the minced tissue, HC-HA, which is too large to enter the gel, is digested, releasing free HCs that migrate and appear as a single band at ~85 kDa (green bands; lanes 2, 5, 8). G) The TSG6 activity assay works by supplying IαI (endogenous, from the tissue) and a HA oligo (exogenous) 14 monosaccharides in length (HA14) as an artificial heavy chain acceptor. If endogenous TSG6 activity was present, it would have transferred the HCs from IαI to HA14, and there would have been a gel shift from the 250 kDa IαI band to the ~85 kDa HC band in lanes 3, 6 and 9. This was demonstrated in the positive controls where recombinant TSG6 was supplied (green bands; lanes 4, 7, 10). TSG6 activity was largely absent in the calcified human aortic valves. Molecular weight standards are shown in red in both F and G. Scale bar: A, C: 0.5 mm; B, D, E: 200 μm.
4. Discussion
HA has long been perceived to play a fundamental role in valve function and disease, and its structural heterogeneity and multi-regulatory nature is known to contribute to the overall complexity of the valve ECM; yet its homeostatic regulation during normal and disease states have been underrepresented. Based on the findings of this study, we propose that the activation of Smad2/3 (canonical) and ERK1/2 (non-canonical) pathways and hemodynamic perturbations, causes disruption of HA homeostatic machinery with imbalance of enzymes that synthesize and breakdown HA, leading to maladaptive, fibrotic ECM remodeling and latent AVD (Fig 8A). Whereas SB431542 rescued HAS2 and CD44 but not HYAL1 expression in young and aged VICs, U0126 rescued HYAL1 at all ages but was ineffective on CD44 regulation and rescued HAS2 only in young VICs. Further, the rescue effect for these inhibitors was additive only for HAS2 and CD44. The age-related expression patterns for human vs. porcine aortic valve tissues were predominantly consistent, and the fibrotic remodeling with abnormalities in regional expression of HA regulators observed in TGFβ1-cultured porcine valves, was similar to diseased human valves. By evaluating the regulatory pathways that control HA turnover and postnatal dysregulation of its homeostasis in aortic valves, these studies identify mechanistic insights that are informative in the search for novel therapeutic targets for early AVD treatment e.g., pharmacologic intervention prior to the mineralization/end stage of valve calcification.
Figure 8.
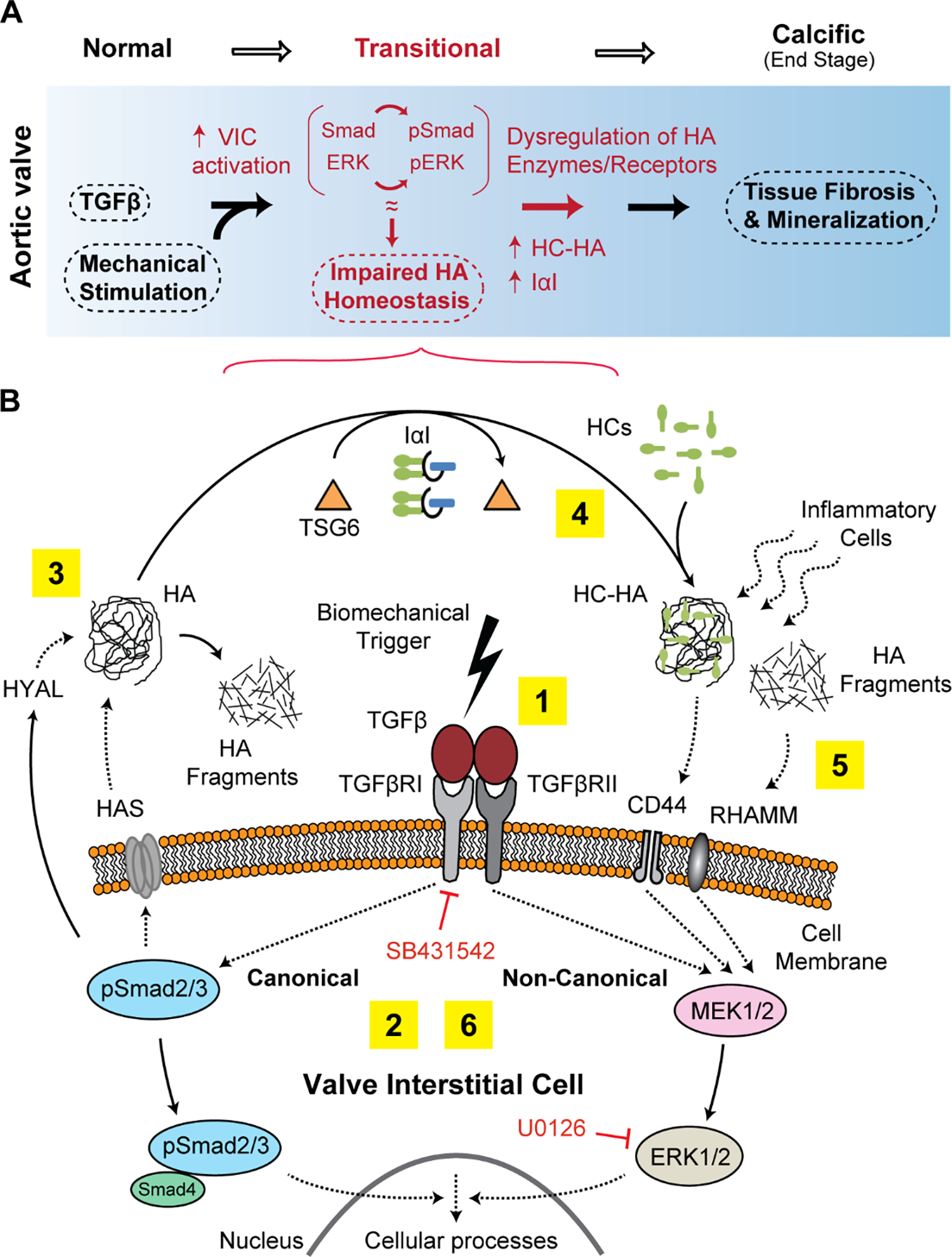
Schematic mechanism of the association between TGFβ signaling, biomechanics and HA homeostasis in calcific AVD progression. A) We propose that activation of intracellular TGFβ pathways together with biomechanical trigger cause a transitional, pre-calcific phase (red markings) with increased cell activation, ECM remodeling and dysregulation of enzymes that synthesize and breakdown HA. B) HA abnormalities manifesting as either accumulation of HA fragments (due to increased HYAL, denoted by 3) or HC-HA (due to increased TSG6 activity, denoted by 4) may impair normal HA receptor function and may attract inflammatory cells to the site of calcific lesions (denoted by 5). The numbers 1 through 6 denote the proposed sequence of events; dotted arrow between the moieties means “acts on”, and solid arrow means “converts to”.
The findings of the present study advance the idea that AVD pathogenesis originates in the valve fibrosa region. We observed that during human aortic valve aging and disease, the collagenous fibrosa layer expanded and the proportion of proteoglycans and GAGs decreased concomitantly, consistent with previous findings [13], and that the calcific nodules in diseased valves were mostly localized to the fibrosa layer (Supplemental Fig 1). Similarly, in porcine models of AVD Interestingly, we also noticed abnormal focal expression of HAS2 and HYAL1 in these calcified human valves and RHAMM in TGFβ-cultured porcine valves; all preferentially occurring on the fibrosa side. Fibrosa is known to be the most affected region during valve calcification, and our findings here confirm previously shown strong correlations in the expressions of HAS, HYAL and RHAMM with ossification markers in the vicinity of calcific nodules in diseased valves [15, 49, 50]. Therefore, this region seems to have the highest vulnerability to HA homeostatic alterations.
While TGFβ1-based in vitro organ culture of valve leaflets and cell culture of VICs particularly in presence of biomechanical stimulation have previously been employed to investigate VIC activation, ECM remodeling, and recapitulate AVD [19, 26, 29, 45, 46], our studies extend their application to assess HA homeostasis during disease pathogenesis. TGFβ is a ubiquitous regulatory cytokine that regulates VIC activation and ECM remodeling in valves [18], and studies have shown heightened expression of TGFβ and pSmad2/3 in the vicinity of calcific nodules and an overall increase in pERK1/2 expression in diseased valves [21–25]. The inhibitors of Smad2/3 and ERK1/2 pathways, i.e., SB431542 and U0126 respectively, have been well optimized for VIC studies in vitro. Whereas SB431542 reverses GAG chain elongation caused by TGFβ1 [23], U0126 has been used as a suppressor of calcification [20, 51]. In addition, the mechanistic association between TGFβ and HA in valves has been reported only in the context of early development (in endocardial cushions) or abnormal embryogenesis, not latent disease [52–54]. And to our knowledge, there has been only one valve study so far where the direct effect of exogenous TGFβ on HA production was analyzed [25]. Given HA’s major role in normal valve structure-function and disease, investigating the interplay between TGFβ pathways and regulation of HA homeostasis could help identify mechanistic targets for therapeutic intervention.
Our results demonstrate abnormal expression patterns of HAS, HYAL and KIAA1199 in diseased human valves and in TGFβ1-stimulated porcine valves suggesting an imbalance of the enzymes that synthesize and breakdown HA, during AVD pathology. We show HAS2 and HYAL1 expression increased with age in both human and porcine valves, and were primarily regulated through Smad2/3 and ERK1/2 pathways respectively. KIAA1199, a HA depolymerization enzyme, was previously shown to be expressed in porcine VICs in vitro [43]. Here we observed a gradual decrease in its expression in porcine aortic valve tissue over age. In contrast, KIAA1199 was present in normal human aortic valves at all postnatal stages, and particularly demonstrated heightened expression around the calcified nodules of diseased valves. Unlike HYALs, KIAA1199 is quite understudied in HA catabolism. It has previously been identified in the context of hearing loss and cancer [55, 56], and recent in vitro studies in human dermal fibroblasts have shown a marked reduction in KIAA1199 expression after TGFβ exposure [44, 57]; however, its mechanistic regulation is largely unknown otherwise. While HAS and HYAL are usually regulated through pSmad and/or MAPK pathways, KIAA1199 is likely controlled through the PI3K-Akt signaling pathway [57]. The specific roles of KIAA1199 during valve maintenance and disease, and its association with signaling pathways need to be investigated. Further, transgenic HAS and HYAL knock out mice that exhibit defective aortic valve morphogenesis with ECM disorganization and altered HA synthesis, may serve as indispensable animal models to investigate abnormal progression of HA homeostasis from early valve development through latent disease [58, 59].
CD44 expression in normal human and porcine valves was downregulated with age, with a marked upregulation observed in calcific valves and TGFβ1/βGP-cultured valves, suggesting HA degradation [60]. CD44 is a principal receptor for HA; HA-CD44 interaction has been implicated in cell proliferation and differentiation. Mechanistically, both high and low molecular weight HA are known to interact with CD44, and HA ligation to CD44 and/or RHAMM may mediate VIC activation via ERK1/2 signaling (Figure 8B) [61–64]. Our results further showed that biomechanical strain had a more dramatic effect on CD44 gene expression than HAS2 and HYAL1 in porcine VICs at both young and aged stages, particularly in presence of TGFβ1. Studies have shown that large in vivo strains on aortic valve during the cardiac cycles are usually transduced from the leaflet ECM to VICs via integrins, and VICs respond to these strains through development of focal adhesion complexes, to then synthesize structural proteins or remodeling enzymes and maintain valve structural integrity [65]. While studies focusing specific roles of CD44 in valve disease or biomechanics are limited [66, 67], the interaction among integrin receptors, non-integrin receptors such as CD44, and other mechano-accessory proteins in the context of abnormal hemodynamics in valves warrants further study [68].
Though HA has previously been shown to be differentially expressed in aged calcified human aortic valves [16], our findings here demonstrate co-localization of HA and IαI together with accumulation of HC-HA in calcified human aortic valves. It is well established that under normal conditions, HA is synthesized as a large GAG (>1500 kDa), lacking any protein modifications. Studies have shown that during inflammation, a pathological change occurs to HA in which the enzyme TSG6 transfers HCs (~85 kDa) from the serum-derived proteoglycan IαI to the 6-OH of N-acetylglucosamine residues in HA to form a covalent HC-HA complex (>3000 kDa) [69, 70]. HC-HA has recently been shown to accumulate abnormally during rheumatoid arthritis, idiopathic pulmonary arterial hypertension, asthma and ulcerative colitis [37, 42, 48]. Inflammation with leukocyte clustering in valves is a hallmark of calcific AVD [5, 71], and HC-HA is known to exacerbate inflammation via its leukocyte adhesion properties [72, 73]. Therefore HC-HA possibly initiates the inflammatory response during AVD (Fig 8B), however these mechanisms in valves warrant further investigation.
From our analysis, TSG6 activity was not detected in these aged calcified human aortic valves. This is not surprising since TSG6 activity is rarely detectable in end-stage diseased tissues, such as in the colon during ulcerative colitis and lungs during asthma, in which inflammation has reached a chronic and advanced state [42]. However, these tissues demonstrate HC-HA as well, indicating that endogenous TSG6 activity likely preceded the overt manifestation of disease. Additionally, HA and TSG6 share an interesting association - HA competitively binds TSG6 and promotes valve calcification, and this binding is dependent on HA size. Therefore, the degradation products of HA (i.e., smaller HA fragments generated by HYAL) may not effectively bind TSG6, leading to inhibition of calcification [15]. However, these HA fragments are known to bind RHAMM to promote activation of VICs, and the components of TGFβ pathway (Fig 8B) [15, 74, 75]. They also stimulate cytokine production for macrophage differentiation [76–78], and monocyte infiltration, macrophage differentiation along with heightened remodeling enzyme expression have been observed in calcified valves [5]. Taken together, both HC-HA and fragmented HA are pro-inflammatory and the ratio of different components of HA (intact vs. fragmented vs. HC) may be altered during AVD, which could set the stage for subsequent downstream inflammatory and ECM remodeling events. The pharmacologic potential of TSG6 neutralizing antibodies, or HA oligosaccharides to remove HCs from HC-HA and dissociate these pathologic complexes in diseased valves, may be explored in future [48].
Limitations of this study include the use of semiquantitative IHC analysis for human valves, and the need for a larger sample size for humans given the inherent variability between specimens. Second, the regulation of multiple isoforms of HAS and HYAL is complex [79, 80], and valve chondrogenic remodeling during development and disease is not regulated by TGFβ alone, but by other pathways such Wnt/β-catenin as well [81]. Hence there may be additional mechanisms of HA homeostasis, and changes due to TGFβ may be considered as just one of the factors that affect HA regulation in valves. Third, while the human and porcine valves mostly exhibit similar data trends, any difference between the findings, such as with KIAA1199 expression, may be due in part to interspecies differences in valve composition, and the wide distribution of sample ages of these valves [82]. The age classifications of the pigs in our study were based on our previous studies [38]; whereas 2 year old pigs are not considered very old, their aortic valves are quite mature with a prevalence of age-induced endothelial dysfunction and heightened VIC calcific potential similar to aged humans. In addition, considering the anatomy and structural similarity, 1–3 month old (young) and >2 year old (aged) pigs in our study closely correspond to “young” and “aged” humans respectively [83].
It may be noted that the porcine calcified aortic valve (used as a positive control) is a random one with unknown genetic background, and therefore future bioreactor culture studies could employ diseased valves from established in vivo porcine models of AVD [84, 85]. Although the TGFβ1 cultures in our study do not completely replicate the complexity and pathophysiology of AVD, these in vitro organ and cell culture models are widely used [86], and have been shown to adequately stimulate VIC activation and calcification, and recapitulate human disease [19, 26, 29, 45, 46]. Further, the advantages of short experiment time and better control over multiple variables, e.g., biomechanical stimulation or concentration-dependent inhibition of specific pathways, make in vitro cultures valuable tools for valve research. Lastly, given the non-specific nature of inhibitors such as U0126, future studies could use additional ERK1/2 inhibitors such as PD-98059 to confirm findings [20]. Short term (e.g., 2–4 hr as opposed to 24–48 hr) cell culture studies using Flexcell may also establish more direct links to ERK1/2 and TGFβ.
In summary, combining engineering and molecular approaches to understand differential VIC regulation of HA homeostasis due to TGFβ1 and its inhibitors highlight the multipotent nature of these development and disease pathways. Ultimately, these studies contribute to elucidating the predisposition of a diseased valve towards calcific vs. myxomatous phenotypes, and provide insight into AVD pathogenesis and identify therapeutic targets.
Supplementary Material
Acknowledgments
This paper is dedicated to the memory of Mark E. Lauer, our dear friend and collaborator and a great researcher. We thank Andy Zhang, Kristi Lim, Dr. Jennifer Connell (Rice University), Dr. Suneel Apte (Cleveland Clinic) and Dr. Robert Hinton (Cincinnati Children’s Hospital Medical Center) for their assistance. This study was supported by AHA 13POST16350003 (VKK), NIH PO1HL107147 and RO1HL113325 (MEL), and AHA 12EIA9300065 (JGA).
Abbreviations
- AVD
aortic valve disease
- βGP
beta-glycerophosphate
- HAS
hyaluronan synthase
- HYAL
hyaluronidase
- IαI
Inter-alpha-Inhibitor
- MAPK
mitogen-activated protein kinase
- PDMS
polydimethylsiloxane
- RHAMM
the receptor for hyaluronan mediated motility
- TGFβ
transforming growth factor beta
- TSG6
Tumor necrosis factor-inducible gene 6 protein
Footnotes
Disclosures: None
References:
- [1].Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M, Burden of valvular heart diseases: a population-based study, Lancet 368(9540) (2006) 1005–11. [DOI] [PubMed] [Google Scholar]
- [2].Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB, American C Heart Association Statistics, S. Stroke Statistics, Heart disease and stroke statistics––2015 update: a report from the American Heart Association, Circulation 131(4) (2015) e29–322. [DOI] [PubMed] [Google Scholar]
- [3].Roberts WC, Ko JM, Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation, Circulation 111(7) (2005) 920–5. [DOI] [PubMed] [Google Scholar]
- [4].Krishnamurthy VK, Godby RC, Liu GR, Smith JM, Hiratzka LF, Narmoneva DA, Hinton RB, Review of molecular and mechanical interactions in the aortic valve and aorta: implications for the shared pathogenesis of aortic valve disease and aortopathy, J Cardiovasc Transl Res 7(9) (2014) 823–46. [DOI] [PubMed] [Google Scholar]
- [5].Freeman RV, Otto CM, Spectrum of calcific aortic valve disease: pathogenesis, disease progression, and treatment strategies, Circulation 111(24) (2005) 3316–26. [DOI] [PubMed] [Google Scholar]
- [6].Otto CM, Valvular aortic stenosis: disease severity and timing of intervention, J Am Coll Cardiol 47(11) (2006) 2141–51. [DOI] [PubMed] [Google Scholar]
- [7].Tzemos N, Therrien J, Yip J, Thanassoulis G, Tremblay S, Jamorski MT, Webb GD, Siu SC, Outcomes in adults with bicuspid aortic valves, Jama 300(11) (2008) 1317–25. [DOI] [PubMed] [Google Scholar]
- [8].Hinton RB, Yutzey KE, Heart valve structure and function in development and disease, Annual review of physiology 73 (2011) 29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Toole BP, Hyaluronan: from extracellular glue to pericellular cue, Nat Rev Cancer 4(7) (2004) 528–39. [DOI] [PubMed] [Google Scholar]
- [10].Tammi MI, Day AJ, Turley EA, Hyaluronan and homeostasis: a balancing act, The Journal of biological chemistry 277(7) (2002) 4581–4. [DOI] [PubMed] [Google Scholar]
- [11].Gupta V, Barzilla JE, Mendez JS, Stephens EH, Lee EL, Collard CD, Laucirica R, Weigel PH, Grande-Allen KJ, Abundance and location of proteoglycans and hyaluronan within normal and myxomatous mitral valves, Cardiovascular pathology : the official journal of the Society for Cardiovascular Pathology 18(4) (2009) 191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Watanabe C, Sugiura M, Ohkawa S, Ito Y, Toku A, Maeda S, Kuboki K, Imai T, [Pathology and histochemistry of mitral valve prolapse], J Cardiol 23(1) (1993) 69–77. [PubMed] [Google Scholar]
- [13].Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD, Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies, Circulation 90(2) (1994) 844–53. [DOI] [PubMed] [Google Scholar]
- [14].Grande-Allen KJ, Osman N, Ballinger ML, Dadlani H, Marasco S, Little PJ, Glycosaminoglycan synthesis and structure as targets for the prevention of calcific aortic valve disease, Cardiovasc Res 76(1) (2007) 19–28. [DOI] [PubMed] [Google Scholar]
- [15].Stephens EH, Saltarrelli JG Jr., Balaoing LR, Baggett LS, Nandi I, Anderson KM, Morrisett JD, Reardon MJ, Simpson MA, Weigel PH, Olmsted-Davis EA, Davis AR, Grande-Allen KJ, Hyaluronan turnover and hypoxic brown adipocytic differentiation are co-localized with ossification in calcified human aortic valves, Pathol Res Pract 208(11) (2012) 642–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stephens EH, Saltarrelli JG, Baggett LS, Nandi I, Kuo JJ, Davis AR, Olmsted-Davis EA, Reardon MJ, Morrisett JD, Grande-Allen KJ, Differential proteoglycan and hyaluronan distribution in calcified aortic valves, Cardiovascular pathology : the official journal of the Society for Cardiovascular Pathology 20(6) (2011) 334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Johansson B, Holmgren A, Hedstrom M, Engstrom-Laurent A, Engstrom KG, Evaluation of hyaluronan and calcifications in stenotic and regurgitant aortic valves, Eur J Cardiothorac Surg 39(1) (2011) 27–32. [DOI] [PubMed] [Google Scholar]
- [18].Conway SJ, Doetschman T, Azhar M, The inter-relationship of periostin, TGF beta, and BMP in heart valve development and valvular heart diseases, ScientificWorldJournal 11 (2011) 1509–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hutcheson JD, Chen J, Sewell-Loftin MK, Ryzhova LM, Fisher CI, Su YR, Merryman WD, Cadherin-11 regulates cell-cell tension necessary for calcific nodule formation by valvular myofibroblasts, Arteriosclerosis, thrombosis, and vascular biology 33(1) (2013) 114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gu X, Masters KS, Role of the MAPK/ERK pathway in valvular interstitial cell calcification, Am J Physiol Heart Circ Physiol 296(6) (2009) H1748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Clark-Greuel JN, Connolly JM, Sorichillo E, Narula NR, Rapoport HS, Mohler ER 3rd, Gorman JH 3rd, Gorman RC, Levy RJ, Transforming growth factor-beta1 mechanisms in aortic valve calcification: increased alkaline phosphatase and related events, The Annals of thoracic surgery 83(3) (2007) 946–53. [DOI] [PubMed] [Google Scholar]
- [22].Miller JD, Weiss RM, Heistad DD, Calcific aortic valve stenosis: methods, models, and mechanisms, Circulation research 108(11) (2011) 1392–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Osman N, Grande-Allen KJ, Ballinger ML, Getachew R, Marasco S, O’Brien KD, Little PJ, Smad2-dependent glycosaminoglycan elongation in aortic valve interstitial cells enhances binding of LDL to proteoglycans, Cardiovasc Pathol 22(2) (2013) 146–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Anger T, El-Chafchak J, Habib A, Stumpf C, Weyand M, Daniel WG, Hombach V, Hoeher M, Garlichs CD, Statins stimulate RGS-regulated ERK 1/2 activation in human calcified and stenotic aortic valves, Experimental and molecular pathology 85(2) (2008) 101–11. [DOI] [PubMed] [Google Scholar]
- [25].Jian B, Narula N, Li QY, Mohler ER 3rd, Levy RJ, Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis, The Annals of thoracic surgery 75(2) (2003) 457–65; discussion 465–6. [DOI] [PubMed] [Google Scholar]
- [26].Liu AC, Gotlieb AI, Transforming growth factor-beta regulates in vitro heart valve repair by activated valve interstitial cells, Am J Pathol 173(5) (2008) 1275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li C, Gotlieb AI, Transforming growth factor-beta regulates the growth of valve interstitial cells in vitro, Am J Pathol 179(4) (2011) 1746–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fisher CI, Chen J, Merryman WD, Calcific nodule morphogenesis by heart valve interstitial cells is strain dependent, Biomechanics and modeling in mechanobiology 12(1) (2013) 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Merryman WD, Lukoff HD, Long RA, Engelmayr GC Jr., Hopkins RA, Sacks MS, Synergistic effects of cyclic tension and transforming growth factor-beta1 on the aortic valve myofibroblast, Cardiovasc Pathol 16(5) (2007) 268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Momberger TS, Levick JR, Mason RM, Hyaluronan secretion by synoviocytes is mechanosensitive, Matrix biology : journal of the International Society for Matrix Biology 24(8) (2005) 510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gupta V, Tseng H, Lawrence BD, Grande-Allen KJ, Effect of cyclic mechanical strain on glycosaminoglycan and proteoglycan synthesis by heart valve cells, Acta Biomater 5(2) (2009) 531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gupta V, Werdenberg JA, Lawrence BD, Mendez JS, Stephens EH, Grande-Allen KJ, Reversible secretion of glycosaminoglycans and proteoglycans by cyclically stretched valvular cells in 3D culture, Ann Biomed Eng 36(7) (2008) 1092–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Stephens EH, de Jonge N, McNeill MP, Durst CA, Grande-Allen KJ, Age-related changes in material behavior of porcine mitral and aortic valves and correlation to matrix composition, Tissue Eng Part A 16(3) (2010) 867–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Krishnamurthy VK, Guilak F, Narmoneva DA, Hinton RB, Regional structure-function relationships in mouse aortic valve tissue, J Biomech 44(1) (2011) 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kumar A, Wiltz DC, Grande-Allen KJ, Gentamicin Reduces Calcific Nodule Formation by Aortic Valve Interstitial Cells In Vitro, Cardiovasc Eng Technol 4(1) (2013) 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Krishnamurthy VK, Opoka AM, Kern CB, Guilak F, Narmoneva DA, Hinton RB, Maladaptive matrix remodeling and regional biomechanical dysfunction in a mouse model of aortic valve disease, Matrix biology : journal of the International Society for Matrix Biology 31(3) (2012) 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lauer ME, Aytekin M, Comhair SA, Loftis J, Tian L, Farver CF, Hascall VC, Dweik RA, Modification of hyaluronan by heavy chains of inter-alpha-inhibitor in idiopathic pulmonary arterial hypertension, J Biol Chem 289(10) (2014) 6791–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Balaoing LR, Post AD, Liu H, Minn KT, Grande-Allen KJ, Age-related changes in aortic valve hemostatic protein regulation, Arterioscler Thromb Vasc Biol 34(1) (2014) 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Stephens EH, Carroll JL, Grande-Allen KJ, The use of collagenase III for the isolation of porcine aortic valvular interstitial cells: rationale and optimization, The Journal of heart valve disease 16(2) (2007) 175–83. [PubMed] [Google Scholar]
- [40].Pfaffl MW, A new mathematical model for relative quantification in real-time RT-PCR, Nucleic Acids Res 29(9) (2001) e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Connell PS, Azimuddin AF, Kim SE, Ramirez F, Jackson MS, Little SH, Grande-Allen KJ, Regurgitation Hemodynamics Alone Cause Mitral Valve Remodeling Characteristic of Clinical Disease States In Vitro, Ann Biomed Eng (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lauer ME, Loftis J, de la Motte C, Hascall VC, Analysis of the heavy-chain modification and TSG-6 activity in pathological hyaluronan matrices, Methods Mol Biol 1229 (2015) 543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Puperi DS, O’Connell RW, Punske ZE, Wu Y, West JL, Grande-Allen KJ, Hyaluronan Hydrogels for a Biomimetic Spongiosa Layer of Tissue Engineered Heart Valve Scaffolds, Biomacromolecules 17(5) (2016) 1766–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yoshida H, Nagaoka A, Kusaka-Kikushima A, Tobiishi M, Kawabata K, Sayo T, Sakai S, Sugiyama Y, Enomoto H, Okada Y, Inoue S, KIAA1199, a deafness gene of unknown function, is a new hyaluronan binding protein involved in hyaluronan depolymerization, Proc Natl Acad Sci U S A 110(14) (2013) 5612–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].McCoy CM, Nicholas DQ, Masters KS, Sex-related differences in gene expression by porcine aortic valvular interstitial cells, PLoS One 7(7) (2012) e39980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Das D, Holmes A, Murphy GA, Mishra K, Rosenkranz AC, Horowitz JD, Kennedy JA, TGF-beta1-Induced MAPK activation promotes collagen synthesis, nodule formation, redox stress and cellular senescence in porcine aortic valve interstitial cells, J Heart Valve Dis 22(5) (2013) 621–30. [PubMed] [Google Scholar]
- [47].Quinlan AM, Billiar KL, Investigating the role of substrate stiffness in the persistence of valvular interstitial cell activation, J Biomed Mater Res A 100(9) (2012) 2474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lauer ME, Glant TT, Mikecz K, DeAngelis PL, Haller FM, Husni ME, Hascall VC, Calabro A, Irreversible heavy chain transfer to hyaluronan oligosaccharides by tumor necrosis factor-stimulated gene-6, J Biol Chem 288(1) (2013) 205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ankeny RF, Thourani VH, Weiss D, Vega JD, Taylor WR, Nerem RM, Jo H, Preferential activation of SMAD1/5/8 on the fibrosa endothelium in calcified human aortic valves--association with low BMP antagonists and SMAD6, PLoS One 6(6) (2011) e20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].O’Brien KD, Reichenbach DD, Marcovina SM, Kuusisto J, Alpers CE, Otto CM, Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis, Arterioscler Thromb Vasc Biol 16(4) (1996) 523–32. [DOI] [PubMed] [Google Scholar]
- [51].Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM, Identification of a novel inhibitor of mitogen-activated protein kinase kinase, J Biol Chem 273(29) (1998) 18623–32. [DOI] [PubMed] [Google Scholar]
- [52].Schroeder JA, Jackson LF, Lee DC, Camenisch TD, Form and function of developing heart valves: coordination by extracellular matrix and growth factor signaling, J Mol Med (Berl) 81(7) (2003) 392–403. [DOI] [PubMed] [Google Scholar]
- [53].Azhar M, Brown K, Gard C, Chen H, Rajan S, Elliott DA, Stevens MV, Camenisch TD, Conway SJ, Doetschman T, Transforming growth factor Beta2 is required for valve remodeling during heart development, Dev Dyn 240(9) (2011) 2127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lagendijk AK, Goumans MJ, Burkhard SB, Bakkers J, MicroRNA-23 restricts cardiac valve formation by inhibiting Has2 and extracellular hyaluronic acid production, Circ Res 109(6) (2011) 649–57. [DOI] [PubMed] [Google Scholar]
- [55].Abe S, Usami S, Nakamura Y, Mutations in the gene encoding KIAA1199 protein, an inner-ear protein expressed in Deiters’ cells and the fibrocytes, as the cause of nonsyndromic hearing loss, J Hum Genet 48(11) (2003) 564–70. [DOI] [PubMed] [Google Scholar]
- [56].Tiwari A, Schneider M, Fiorino A, Haider R, Okoniewski MJ, Roschitzki B, Uzozie A, Menigatti M, Jiricny J, Marra G, Early insights into the function of KIAA1199, a markedly overexpressed protein in human colorectal tumors, PLoS One 8(7) (2013) e69473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Nagaoka A, Yoshida H, Nakamura S, Morikawa T, Kawabata K, Kobayashi M, Sakai S, Takahashi Y, Okada Y, Inoue S, Regulation of Hyaluronan (HA) Metabolism Mediated by HYBID (Hyaluronan-binding Protein Involved in HA Depolymerization, KIAA1199) and HA Synthases in Growth Factor-stimulated Fibroblasts, J Biol Chem 290(52) (2015) 30910–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chowdhury B, Hemming R, Hombach-Klonisch S, Flamion B, Triggs-Raine B, Murine hyaluronidase 2 deficiency results in extracellular hyaluronan accumulation and severe cardiopulmonary dysfunction, J Biol Chem 288(1) (2013) 520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A Jr., Kubalak S, Klewer SE, McDonald JA, Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme, J Clin Invest 106(3) (2000) 349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Knudson W, Chow G, Knudson CB, CD44-mediated uptake and degradation of hyaluronan, Matrix Biol 21(1) (2002) 15–23. [DOI] [PubMed] [Google Scholar]
- [61].Fujita Y, Kitagawa M, Nakamura S, Azuma K, Ishii G, Higashi M, Kishi H, Hiwasa T, Koda K, Nakajima N, Harigaya K, CD44 signaling through focal adhesion kinase and its anti-apoptotic effect, FEBS Lett 528(1–3) (2002) 101–8. [DOI] [PubMed] [Google Scholar]
- [62].Zhang S, Chang MC, Zylka D, Turley S, Harrison R, Turley EA, The hyaluronan receptor RHAMM regulates extracellular-regulated kinase, The Journal of biological chemistry 273(18) (1998) 11342–8. [DOI] [PubMed] [Google Scholar]
- [63].Carthy JM, Boroomand S, McManus BM, Versican and CD44 in in vitro valvular interstitial cell injury and repair, Cardiovascular pathology : the official journal of the Society for Cardiovascular Pathology 21(2) (2012) 74–82. [DOI] [PubMed] [Google Scholar]
- [64].Orian-Rousseau V, Sleeman J, CD44 is a multidomain signaling platform that integrates extracellular matrix cues with growth factor and cytokine signals, Adv Cancer Res 123 (2014) 231–54. [DOI] [PubMed] [Google Scholar]
- [65].Chester AH, El-Hamamsy I, Butcher JT, Latif N, Bertazzo S, Yacoub MH, The living aortic valve: From molecules to function, Glob Cardiol Sci Pract 2014(1) (2014) 52–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Poggio P, Branchetti E, Grau JB, Lai EK, Gorman RC, Gorman JH 3rd, Sacks MS, Bavaria JE, Ferrari G, Osteopontin-CD44v6 interaction mediates calcium deposition via phospho-Akt in valve interstitial cells from patients with noncalcified aortic valve sclerosis, Arterioscler Thromb Vasc Biol 34(9) (2014) 2086–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wang JHC, Thampatty BP, Lin JS, Im HJ, Mechanoregulation of gene expression in fibroblasts, Gene 391(1–2) (2007) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Knudson W, Loeser RF, CD44 and integrin matrix receptors participate in cartilage homeostasis, Cell Mol Life Sci 59(1) (2002) 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Milner CM, Tongsoongnoen W, Rugg MS, Day AJ, The molecular basis of inter-alpha-inhibitor heavy chain transfer on to hyaluronan, Biochem Soc Trans 35(Pt 4) (2007) 672–6. [DOI] [PubMed] [Google Scholar]
- [70].Milner CM, Higman VA, Day AJ, TSG-6: a pluripotent inflammatory mediator?, Biochem Soc Trans 34(Pt 3) (2006) 446–50. [DOI] [PubMed] [Google Scholar]
- [71].Cote N, Mahmut A, Bosse Y, Couture C, Page S, Trahan S, Boulanger MC, Fournier D, Pibarot P, Mathieu P, Inflammation is associated with the remodeling of calcific aortic valve disease, Inflammation 36(3) (2013) 573–81. [DOI] [PubMed] [Google Scholar]
- [72].Lauer ME, Cheng G, Swaidani S, Aronica MA, Weigel PH, Hascall VC, Tumor necrosis factor-stimulated gene-6 (TSG-6) amplifies hyaluronan synthesis by airway smooth muscle cells, J Biol Chem 288(1) (2013) 423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhuo L, Kanamori A, Kannagi R, Itano N, Wu J, Hamaguchi M, Ishiguro N, Kimata K, SHAP potentiates the CD44-mediated leukocyte adhesion to the hyaluronan substratum, J Biol Chem 281(29) (2006) 20303–14. [DOI] [PubMed] [Google Scholar]
- [74].Krupinski J, Ethirajan P, Font MA, Turu MM, Gaffney J, Kumar P, Slevin M, Changes in Hyaluronan Metabolism and RHAMM Receptor Expression Accompany Formation of Complicated Carotid Lesions and May be Pro-Angiogenic Mediators of Intimal Neovessel Growth, Biomarker insights 2 (2008) 361–7. [PMC free article] [PubMed] [Google Scholar]
- [75].Gao F, Yang CX, Mo W, Liu YW, He YQ, Hyaluronan oligosaccharides are potential stimulators to angiogenesis via RHAMM mediated signal pathway in wound healing, Clin Invest Med 31(3) (2008) E106–16. [DOI] [PubMed] [Google Scholar]
- [76].McKee CM, Penno MB, Cowman M, Burdick MD, Strieter RM, Bao C, Noble PW, Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages. The role of HA size and CD44, J Clin Invest 98(10) (1996) 2403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].David-Raoudi M, Tranchepain F, Deschrevel B, Vincent JC, Bogdanowicz P, Boumediene K, Pujol JP, Differential effects of hyaluronan and its fragments on fibroblasts: relation to wound healing, Wound Repair Regen 16(2) (2008) 274–87. [DOI] [PubMed] [Google Scholar]
- [78].Masters KS, Shah DN, Leinwand LA, Anseth KS, Crosslinked hyaluronan scaffolds as a biologically active carrier for valvular interstitial cells, Biomaterials 26(15) (2005) 2517–25. [DOI] [PubMed] [Google Scholar]
- [79].Tammi RH, Passi AG, Rilla K, Karousou E, Vigetti D, Makkonen K, Tammi MI, Transcriptional and post-translational regulation of hyaluronan synthesis, FEBS J 278(9) (2011) 1419–28. [DOI] [PubMed] [Google Scholar]
- [80].Stern R, Jedrzejas MJ, Hyaluronidases: Their genomics, structures, and mechanisms of action, Chem Rev 106(3) (2006) 818–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Fang M, Alfieri CM, Hulin A, Conway SJ, Yutzey KE, Loss of beta-catenin promotes chondrogenic differentiation of aortic valve interstitial cells, Arterioscler Thromb Vasc Biol 34(12) (2014) 2601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Stephens EH, Chu CK, Grande-Allen KJ, Valve proteoglycan content and glycosaminoglycan fine structure are unique to microstructure, mechanical load and age: Relevance to an age-specific tissue-engineered heart valve, Acta biomaterialia 4(5) (2008) 1148–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Stephens EH, Grande-Allen KJ, Age-related changes in collagen synthesis and turnover in porcine heart valves, The Journal of heart valve disease 16(6) (2007) 672–82. [PubMed] [Google Scholar]
- [84].Porras AM, Shanmuganayagam D, Meudt JJ, Krueger CG, Hacker TA, Rahko PS, Reed JD, Masters KS, Development of Aortic Valve Disease in Familial Hypercholesterolemic Swine: Implications for Elucidating Disease Etiology, J Am Heart Assoc 4(10) (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Sider KL, Zhu C, Kwong AV, Mirzaei Z, de Lange CF, Simmons CA, Evaluation of a porcine model of early aortic valve sclerosis, Cardiovasc Pathol 23(5) (2014) 289–97. [DOI] [PubMed] [Google Scholar]
- [86].Bowler MA, Merryman WD, In vitro models of aortic valve calcification: solidifying a system, Cardiovasc Pathol 24(1) (2015) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.