

Abstract
Sendai virus (SeV) infection causes the transcriptional induction of many cellular genes that are also induced by interferon (IFN) or double-stranded RNA (dsRNA). We took advantage of various mutant cell lines to investigate the putative roles of the components of the IFN and dsRNA signaling pathways in the induction of those genes by SeV. Profiling the patterns of gene expression in SeV-infected cells demonstrated that Toll-like receptor 3, although essential for gene induction by dsRNA, was dispensable for gene induction by SeV. In contrast, Jak1, which mediates IFN signaling, was required for the induction of a small subset of genes by SeV. NF-κB and interferon regulatory factor 3 (IRF-3), the two major transcription factors activated by virus infection, were essential for the induction of two sets of genes by SeV. As expected, some of the IRF-3-dependent genes, such as ISG56, were more strongly induced by SeV in IRF-3-overexpressing cells. Surprisingly, in those cells, a number of NF-κB-dependent genes, such as the A20 gene, were induced poorly. Using a series of cell lines expressing increasing levels of IRF-3, we demonstrated that the degree of induction of A20 mRNA, upon SeV infection, was inversely proportional to the cellular level of IRF-3, whereas that of ISG56 mRNA was directly proportional. Thus, IRF-3 can suppress the expression of NF-κB-dependent genes in SeV-infected cells.
Many cellular genes, encoding proteins of diverse functions, are transcriptionally induced during viral infections (6, 15, 32, 40, 47). Because host-virus coevolution has tolerated this host response to virus infection, it is thought to be conducive to maintaining proper homeostasis between viral proliferation and host survival. A large number of virally induced proteins have direct or indirect antiviral effects. They may inhibit protein synthesis in the infected cells (17), impair viral assembly (7), initiate the innate immune response, or prime the adaptive immune response (29). Diverse components of the infecting viruses or viral intermediates, produced during its replication, can be the responsible agents that trigger the signaling pathways leading to cellular gene induction. Depending on the specific virus, viral envelope proteins (5), viral ribonucleoproteins (41), or viral single-stranded RNAs (ssRNAs) (28) or double-stranded RNAs (dsRNAs) (1, 11, 46) have been shown to be the critical agent. Among these, dsRNA has been viewed as the most important agent because it is produced in many virus-infected cells; moreover, synthetic dsRNA, when added to cells, can induce transcription of some of the same cellular genes that are induced by virus infection (14). A major transducer of signaling, generated by exogenously added dsRNA or intracellular viral dsRNA, is a member of the Toll-like receptor (TLR) family, TLR3 (1).
One set of common genes induced by many viruses and dsRNA encodes type I interferons (IFNs) (29). These secreted cytokines have strong antiviral effects. Surprisingly, many dsRNA-induced and virally induced cellular genes are also induced by IFNs (37), thus creating a positive feedback loop that reinforces induction of the same genes in infected cells.
To better understand the repertoires of genes induced by Sendai virus (SeV) infection of cells in culture, we have classified them into groups that are induced by IFNs, dsRNA, or other viral products. For identifying SeV-induced genes, we used cDNA microarrays customized for this purpose and based on genes that we previously identified whose transcription is induced by IFNs (9, 10) or dsRNA (14). In addition to many virally induced genes, dsRNA- and IFN-inducible genes were represented in the microarray used here. We had previously used mutant cell lines, which could not respond to IFN or dsRNA to delineate the signaling pathways activated by the different inducers (3, 24, 43). To assess the relative contributions of the IFN and dsRNA signaling pathways in gene induction by SeV, we again took advantage of some of those mutant cell lines. Other mutant cell lines were used to investigate the relative contributions of NF-κB and interferon regulatory factor 3 (IRF-3), the major transcription factors activated by SeV infection, to cellular gene induction (21, 36).
Our study revealed that among the genes that are induced immediately after SeV infection, only a few were dependent on IFN signaling and none were dependent on dsRNA signaling through TLR3. As expected, one class of genes required NF-κB, whereas another class required IRF-3 for induction in SeV-infected cells. Surprisingly, viral induction of a subset of NF-κB-dependent genes was negatively regulated by IRF-3, thus revealing a new aspect of cross talk between the two transcription factors.
MATERIALS AND METHODS
Cell culture.
The cell lines used in this study and their principal characteristics are listed in Table 1. All cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum, 2 mM l-glutamine, 50 U of penicillin/ml, and 50 μg of streptomycin/ml. 293/TLR3 cells were derived from 293 cells transfected with a TLR3 expression vector as described previously and cultured in 400 μg of G418/ml (38). Wild-type (wt) 2fTGH cells, Jak1-deficient U4C cells, and IRF-3-deficient P2.1 cells have been discussed previously (24). The generation of U4C.2 cells, a U4C-derived line overexpressing IRF-3, along with 2F-SR cells, which express the dominant negative IκBα super-repressor, were described by Peters et al. (36). These cells were cultured in the presence of puromycin and G418, respectively. 1080.10 cells are a clonal line derived from HT1080 cells cotransfected with the IRF-3 expression vector pCDNA3/hIRF-3 and the selection vector pBABE/Puro. Following selection with puromycin, 1080.10 and other clones were picked from the stably transfected cells and screened by Western blotting for increased IRF-3 expression.
TABLE 1.
Mutant cell lines used
| Cell line | Lineage | Relevant altered protein | Affected pathway(s) |
|---|---|---|---|
| HEK293 | None | TLR3 nonresponsive | |
| 293/TLR3 | HEK293 | TLR3 | TLR3 responsive |
| 2fTGH | HT1080 | None | None |
| 2F-SR | 2fTGH | IκB | NF-κB signaling |
| 1080.10 | HT1080 | Increased IRF-3 | IRF-3 signaling |
| U4C | 2fTGH | Jak1 | IFN signaling |
| U4C.2 | U4C | Increased IRF-3 | IRF-3 and IFN signaling |
| P2.1 | U4C | Decreased IRF-3 | IRF-3 and IFN signaling |
| P2.1.6 | P2.1 | Increased IRF-3 | IRF-3 and IFN signaling |
| P2.1.17 | P2.1 | Increased IRF-3 | IRF-3 and IFN signaling |
Treatment methods.
Treatment of cells with dsRNA was carried out as described previously (36). Briefly, poly(I)· ·poly(C) (Amersham) was added directly to the culture medium to a final concentration of 100 μg/ml for 6 h. Sendai virus infection was carried out as previously described by Heylbroeck et al. (20). Culture medium was removed from cells and replaced with serum-free DMEM. Sendai virus (Cantell strain; Charles River, SPAFAS) was added directly to the medium to a final concentration of 1 hemagglutinating unit/4.0 × 103 cells (∼10 to 15 PFU/cell) and incubated for 1 h with gentle shaking every 10 min. Following viral adsorption, the medium was removed and replaced with DMEM containing 10% fetal bovine serum for the remainder of the experiment.
RNA isolation.
For both microarray and RNase protection assay (RPA) experiments, total cellular RNA was isolated from cells by use of the RNA-Bee kit (Tel-Test, Friendswood, Tex.). Following isolation, RNA was washed twice in 70% ethyl alcohol and dissolved in water.
Array construction.
The array used in this study comprised a subset of sequence-verified cDNA clones from the Research Genetics 40K set. The clones included 950 genes containing adenylate-uridylate-rich elements and 18 genes potentially involved in AU-directed mRNA decay as previously described (13); 855 interferon-stimulated genes representing an expansion of a previously described clone set (10); 288 genes responsive to the viral analog poly(I)· ·poly(C), representing an expansion of the clone set described by Geiss et. al (14); and 85 housekeeping genes. DNA preparation and slide printing were done as previously described (13) except for the use of 50% dimethyl sulfoxide as printing solution.
RNA labeling and array hybridization.
Cy3- or Cy5-labeled cDNA was prepared by direct incorporation. One hundred micrograms of total RNA, 2 μg of dT12-18 primer (Invitrogen), and 1 μl of anti-RNase (Ambion, Austin, Tex.) were combined in a reaction volume of 30 μl and incubated for 10 min at 70°C. Reverse transcription was for 2 h at 42°C in a 50-μl reaction containing annealed RNA template; 1× first strand buffer (Invitrogen); 500 μM concentrations of dATP, dCTP, and dGTP; 300 μM dTTP; 250 μM dUTP (Sigma); 3 nmol Cy3-dUTP or Cy5-dUTP; 10 mM dithiothreitol; and 6 U of Superscript II/μl. For template hydrolysis, 10 μl of 0.1 M NaOH was added to the reverse transcription reaction and the mixture was incubated for 10 min at 70°C, allowed to cool at room temperature for 5 min, and neutralized by addition of 10 μl of 0.1 M HCl. The cDNA was purified with GFX columns following the manufacturer's instructions (Amersham Pharmacia Biotech), dried down, and resuspended in hybridization buffer containing 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 0.1% sodium dodecyl sulfate, 4 μg of poly(dA)40-60, and 4 μg of yeast tRNA. The Cy3- and Cy5-labeled cDNAs were pooled, and after 2 min of denaturation at 90°C, the hybridization mixture was applied to the microarray slide under a coverslip. Hybridization proceeded overnight in a sealed moist chamber in a 55°C water bath. Posthybridization, slides were washed successively for 5 min each in 2× SSC plus 0.1% sodium dodecyl sulfate at 55°C, 2× SSC at 55°C, and 0.2× SSC at room temperature; spun dry; and scanned on a GenePix 4000A scanner (Axon).
Data acquisition and normalization.
Data were acquired with a GenePix 4000A laser scanner and GenePix Pro 5.0 software as previously described (13). Raw data were imported into Genespring 6.0 software (Silicon Genetics) and normalized based on the distribution of all values with locally weighted linear regression (LOWESS) before further analysis.
To assure reproducibility, three microarray experiments, using different biological repeats and one dye swap, were performed for every cell line infected with SeV. For assessing the effects of dsRNA, two microarray experiments, using different biological samples, were conducted in both the 293 and 293/TLR3 cell lines. (For primary data from the microarray experiments, see the supplemental material.) Spots representing genes with fluorescent intensities of less than 200 U in the SeV- or dsRNA-treated samples were excluded when surveying for changes in expression. A gene was considered to be induced by SeV or dsRNA in a particular cell type if its expression in treated cells was at least twofold greater than its expression in uninfected cells in at least two microarray experiments.
RNase protection assay and Northern blotting.
RPAs were performed by using the RPA III kit (Ambion). The actin and 561 RPA probes were described previously by Peters et al. (36). The A20, follistatin, and NOXA RPA probes were made from their respective cDNA microarray constructs and corresponded to the final 220, 164, or 158 nucleotides of each mRNA. Northern blotting was performed as described previously (14). The full-length cDNA was used as a probe for 2′,5′-oligoadenylate synthetase (OAS) p69. Gene induction was measured with a Storm Imager and quantified with Image Quant 5.2 (Molecular Dynamics).
RESULTS
Role of TLR3 in gene induction by SeV.
To determine the importance of the TLR3-mediated dsRNA signaling pathway for gene induction by SeV, we used HEK293-derived cell lines. The parental cell line does not express any TLR3, whereas a derived line, 293/TLR3, does. In the absence of TLR3, 293 cells do not respond to exogenously added dsRNA; however, upon the expression of TLR3, the cells are able to respond robustly (1). Thus, a comparison of the genes induced by SeV in this pair of cell lines enabled us to assess the importance of TLR3-mediated viral dsRNA signaling. To focus on the primary cellular response to virus in the absence of paracrine or autocrine signaling, cells were infected with SeV at a high multiplicity of infection, and gene induction was measured 6 h after infection. From our previous experience, 6 h was the optimum time to observe the strong direct induction of genes by SeV, with minimal secondary induction (18).
For transcript profiling, a cDNA microarray of 2,196 genes, including 288 previously identified dsRNA-regulated genes (14) and 855 interferon-induced genes (10), was used. Because dsRNA and IFN induce many of the same genes that viral infection does, this array was well suited to study differential gene induction. Total RNA from infected cells and their uninfected counterparts was used to make fluorescently labeled cDNA, and RNA expression levels were compared to each other by microarray analysis. Individual gene induction or repression by SeV infection was determined from the ratio of relative fluorescent intensities between the infected and uninfected samples.
SeV infection was able to alter the expression of multiple genes in 293 cells; in virus-infected cells, the expression of 36 genes was elevated, whereas that of many others was reduced (Fig. 1A). In 293/TLR3 cells, the patterns were not markedly different (Fig. 1B), indicating that TLR3 plays an insignificant role in gene induction by SeV. In contrast, TLR3 was required for the responsiveness of the cells to dsRNA treatment (Fig. 1C and D).
FIG. 1.

Effect of TLR3 on the regulation of cellular genes by SeV and dsRNA. Shown are global changes in mRNA levels in 293 cells (A and C) or 293 cells expressing TLR3 (B and D) 6 h after SeV infection at 1 hemagglutinating unit/4.0 × 103 cells (A and B) or dsRNA treatment at 100 μg/ml (C and D) as determined by cDNA microarray experiment. Each point on the scatter plots represents the expression of an individual mRNA message, as determined by units of fluorescent intensity, in untreated cells (x axis) plotted against its expression 6 h after SeV infection or dsRNA treatment (y axis). The central diagonal line (black) represents equal expression in treated and untreated samples, while twofold differences in expression are indicated by the two flanking blue lines. (E) Regulation of specific genes by dsRNA and SeV in 293 (−) and 293/TLR3 (+) cells. The tiles show the increase (n-fold) in mRNA expression for specific genes in SeV- or dsRNA-treated cells relative to untreated cells as a function of color. Green, expression was unchanged; yellow to red, expression was induced to increasing degrees. (F) RPA of ISG56 induction in untreated (lanes 1 and 4), dsRNA-treated (lanes 2 and 5), or SeV-infected (lanes 3 and 6) 293 cells (lanes 1 to 3) or 293/TLR3 cells (lanes 4 to 6).
We next examined the induction of specific genes to verify the apparent lack of importance of TLR3 in global gene regulation by SeV. All of the genes that were induced over twofold by SeV in 293/TLR3 cells in at least two of three microarray experiments were also induced in 293 cells. Interferon-stimulated gene 15 (ISG15), ISG54, and ISG56, along with the A20 (TNFAIP3) and interleukin 8 (IL-8) genes, were all strongly induced by SeV infection and dsRNA treatment; the induction of all five genes by dsRNA required TLR3, but SeV was able to induce them all without TLR3 (Fig. 1E). To confirm the microarray data, we used RPAs to quantitate the levels of ISG56 mRNA (Fig. 1F). As expected, SeV induction of ISG56 was TLR3 independent, while its induction by dsRNA was TLR3 dependent. Moreover, the levels of ISG56 mRNA in SeV-infected 293 and 293/TLR3 cells were similar (Fig. 1F, lanes 3 and 6), indicating that even for the maximal induction of this mRNA, the viral signaling pathway did not require TLR3.
Although there was a differential need for TLR3, because both agents induced many common genes, we wondered whether the profiles of genes induced by dsRNA and SeV were completely overlapping. This was not the case. Of the 36 genes that were induced by SeV in 293 cells and the 35 genes induced by dsRNA in 293/TLR3 cells, only 9 genes were common. In addition to the five genes shown in Fig. 1E, FUT2, syndecan4, IκBα, and NOXA also belonged to this class. A few examples of differentially induced genes are given in Fig. 2. These results suggest that the signaling pathways used by the two agents are different, although some of them may be shared.
FIG. 2.

Differential induction of genes by SeV and dsRNA. Select genes were differentially regulated by SeV and dsRNA in 293 cells expressing (+) or not expressing (−) TLR3. Colors represent induction (n-fold) by SeV (left column) or dsRNA (right column) as described in the legend to Fig. 1.
Involvement of IFN signaling in gene induction by SeV.
SeV infection is known to induce IFN synthesis, and many of the genes induced by SeV are also induced by IFNs. Thus, it is possible that some genes are induced in SeV-infected cells by the autocrine action of newly synthesized IFN. To test this possibility, we compared gene induction in SeV-infected U4C and 2fTGH cells (Fig. 3). U4C cells, unlike their parental line 2fTGH, lack functional Jak1, which is required for both type I and type II IFN signaling. Consequently, IFNs cannot induce any genes in U4C cells (31). As in 293 cells, many genes were strongly induced in 2fTGH cells after 6 h of SeV infection (Fig. 3A). Using the criterion of twofold induction as the cutoff, we observed that 49 genes were induced in 2fTGH cells. Only a small subset of genes, 14 of the 49, was not induced more than twofold by SeV in U4C cells as well. Hence, the majority of genes induced in 2fTGH cells were also induced in U4C cells, indicating that Jak1, and hence IFN signaling, were not involved in their induction. The A20 and OAS p69 genes were selected as representatives of IFN-independent and -dependent genes, and induction of the corresponding mRNAs was monitored by RPA and Northern blotting, respectively. Those quantitative assays supported the conclusions drawn from the microarray analyses: A20 mRNA was induced by SeV infection of either cell line, but OAS mRNA was not induced in U4C cells (Fig. 3B). Surprisingly, another small subset of six genes was induced in U4C cells but not in 2fTGH cells, suggesting a suppressive function of Jak1 (Fig. 3A).
FIG. 3.

IFN-dependent regulation of SeV-induced genes. Select SeV-regulated genes display differential induction patterns in wt (2fTGH) and Jak1−/− (U4C) cells. (A) Microarray data showing average increase (n-fold) in mRNA expression 6 h after SeV infection in 2fTGH and U4C cells. Genes are grouped based on their independence of, dependence on, or impairment by Jak1 for induction. (B) RPA (A20) and Northern (OAS, p69) analysis of mRNA expression in 2fTGH and U4C cells, before or 6 h after SeV infection.
Role of NF-κB in SeV signaling.
One of the major transcription factors activated by viral infections is NF-κB. To assess its role in gene induction by SeV, we took advantage of another mutant cell line, 2F-SR. This line was derived from the 2fTGH line by expressing a nonphosphorylatable mutant of IκB, which acts as a super-repressor of NF-κB. Cytokines, dsRNA, or SeV cannot activate NF-κB in the 2F-SR cells (2, 36, 45). Of the 49 genes induced by SeV infection in 2fTGH cells, 28 of these were not induced in 2F-SR cells. Thus, almost 60% of the induced genes were NF-κB dependent, while just over 40% were not. Examples of both classes of genes are given in Fig. 4A. Quantitative assays again confirmed our conclusions: NOXA and ISG56 mRNAs were induced equally well in both cell lines, whereas A20 mRNA was not induced in 2F-SR cells (Fig. 4B). These results confirm the notion that NF-κB is a major transcription factor used by SeV to induce cellular genes.
FIG. 4.

Requirement for NF-κB for gene induction by SeV. Genes were regulated by SeV in wt (2fTGH) and NF-κB null (2F-SR) cells. (A) Average induction (n-fold) of SeV-regulated genes, grouped by dependence on NF-κB, as determined by microarray. (B) Quantitative analysis of NOXA, ISG56, and A20 mRNA induction by RPA.
Role of IRF-3 in gene induction by SeV.
In addition to NF-κB, another major transcription factor activated by SeV is IRF-3. To identify genes requiring IRF-3 for induction by SeV, we used P2.1 cells (24). These cells were derived from U4C cells and hence lack Jak1. In addition, their IRF-3 level is very low, and consequently, the IRF-3 signaling pathway is defective in these cells (36). As expected, we observed that one set genes, 30 of the 42 genes induced by SeV in U4C cells, was induced well in P2.1 cells, and another set, the remaining 12 genes, was not. Examples of the two sets of genes are given in Fig. 5. These results demonstrated that the induction of some genes by SeV infection required IRF-3.
FIG. 5.

IRF-3-dependent gene induction. Shown are examples of genes regulated by SeV in U4C cells and P2.1 cells in which IRF-3 expression is impaired. Genes are grouped by their dependence on IRF-3 for induction.
To further characterize this requirement, we used U4C.2 cells in which IRF-3 was overexpressed. The only difference between U4C cells and U4C.2 cells was the level of IRF-3 expressed in the two cell lines. Forty-five genes were induced in U4C.2 cells, including a group of 19 genes that was induced much better than in U4C cells (Fig. 6A). An extreme example of this group is follistatin; quantitative assays confirmed that its mRNA was barely induced in U4C cells but strongly induced in U4C.2 cells (Fig. 6C). The induction of another group of mRNAs was hardly affected by the IRF-3 level (Fig. 6B and D). These results indicate that among the IRF-3-dependent genes, some, but not all, can be induced efficiently only when the cellular IRF-3 level is high.
FIG. 6.

Regulation of SeV-induced genes by IRF-3 and altered expression of SeV-regulated genes in U4C cells and U4C.2 cells, which express high levels of IRF-3. (A and B) Microarray data for the induction (n-fold) of select SeV-regulated genes in U4C and U4C.2 cells. Genes are grouped as follows: IRF-3 enhanced, genes with augmented expression in IRF-3-overexpressing cells (i.e., induced more strongly in U4C.2 cells); IRF-3 neutral, genes unaffected by cellular IRF-3 levels (induced at equivalent levels in both cell lines); IRF-3 repressed, SeV-induced genes negatively regulated by IRF-3 (induced in U4C but not U4C.2 cells). Also shown are quantitative RPA analyses of follistatin (C), NOXA (D), and A20 (E) mRNA induction in U4C and U4C.2 cells before or after SeV infection.
Role of IRF-3 in suppressing gene induction by SeV infection.
In addition to the results discussed above, the array analysis shown in Fig. 6 produced an unexpected result. We observed that at least eight genes, or about 20% of those that were strongly induced in U4C, were poorly induced in U4C.2 cells, indicating that IRF-3 was negatively affecting their expression (Fig. 6B). RPA for A20 mRNA confirmed the conclusions from the microarray analysis (Fig. 6E).
To test further the possibility that IRF-3 was acting as a negative regulator of SeV-induced gene expression, five cell lines, all derived from the U4C line and expressing increasing levels of IRF-3 protein, were used (Fig. 7A). The cell lines were infected with SeV, and the levels of the ISG56 and A20 mRNAs were measured by RPA before and after virus infection. Actin mRNA levels were used for normalization of the data. Neither mRNA was detectable in any uninfected cell line. ISG56 mRNA was barely induced in P2.1 cells, which expressed the lowest level of IRF-3, and its induction levels increased with increasing levels of IRF-3 expression. Conversely, A20 mRNA was most strongly induced in P2.1 cells and very poorly induced in cells expressing high levels of IRF-3 (Fig. 7B).
FIG. 7.
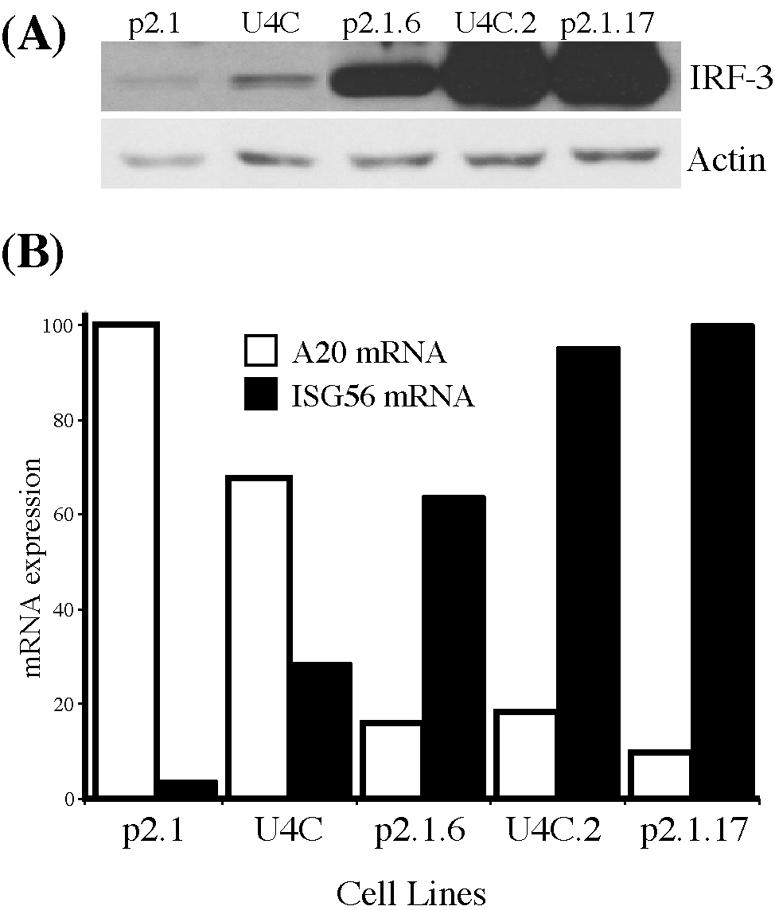
Modulation of A20 mRNA and ISG56 mRNA expression by cellular levels of IRF-3. Cell lines derived from U4C and P2.1 cells and expressing different levels of IRF-3 protein were infected with SeV and analyzed for the expression of A20 mRNA and ISG56 mRNA. (A) Western blot showing the relative levels of IRF-3 expression in the different cell lines relative to actin. (B) Percent maximum induction (n-fold) of A20 mRNA (white bars) and ISG56 mRNA (black bars) in cells 6 h after infection, normalized to actin mRNA expression as determined by RPA.
Although the above analysis validated our original observation, we were concerned that the phenomenon might be restricted to the U4C cells from which all of the above lines were derived. Because the U4C cells lacked Jak1 and were obtained by extensive mutagenesis of 2fTGH cells, it remained possible that the observed suppressing function of IRF-3 required either the absence of Jak1 or the presence of an unknown mutation in U4C cells. If that were the case, our observation would have restricted significance. To test the generality of the observation, a new cell line was derived from the wt HT1080 cells. This line, 1080.10, expressed a much higher level of IRF-3 than the parental cells (Fig. 8A). Confirming the results seen with the U4C derivative lines, A20 mRNA was induced strongly upon SeV infection in HT1080 cells but only weakly in 1080.10 cells (Fig. 8B). The above series of experiments allowed us to conclude that the cellular abundance of IRF-3 can both positively and negatively influence the extent of induction of specific cellular genes in response to infection by SeV.
FIG. 8.
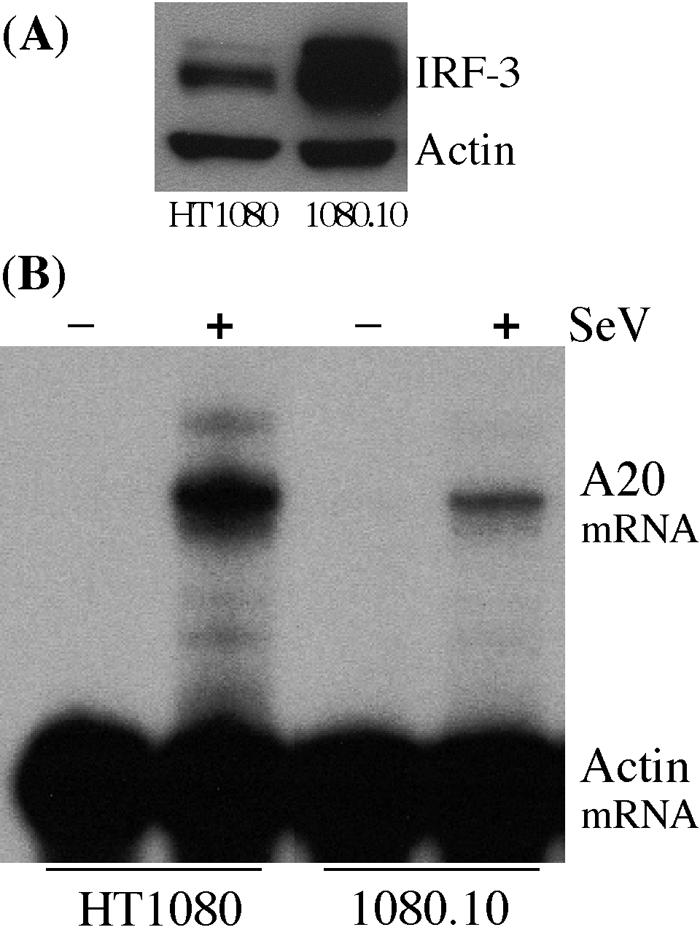
Negative regulation of SeV-induced A20 mRNA expression by IRF-3 in wt cells. (A) Western analysis of IRF-3 expression in HT1080 and 1080.10 cells. (B) RPA comparing A20 mRNA induction in untreated (−) and SeV-infected (+) HT1080 and 1080.10 cells.
DISCUSSION
In this study, we focused our attention on identifying the signaling pathways responsible for inducing specific sets of cellular genes early after SeV infection. The cDNA microarray screening used was convenient for profiling the expression of a large number of genes simultaneously, but to ensure reliability we had to perform multiple repeats of the same screen and accept an arbitrary cutoff of at least twofold change as significant. Consequently, our calculations of the number of genes induced in different cell lines are likely underestimates. The overall gene induction patterns in different SeV-infected cell lines are summarized in Table 2. Table 2 includes only the genes whose expression was induced by at least twofold in wt 2fTGH cells. Following classification of different induced genes into distinct groups, representative members were selected for further analysis by Northern blotting or RNase protection assays. The choice of these sentinel genes was dictated by their degree of inducibility and their abundance, because we could make much more reliable conclusions for genes that were highly induced and for which the corresponding mRNA levels were easily measurable.
TABLE 2.
Genes induced or repressed by SeV infection in various cell linesa
| Encoded protein | Unigene ID | Symbol | Effect of SeV infection in:
|
||||
|---|---|---|---|---|---|---|---|
| 2fTGH | U4C | 2F-SR | P2.1 | U4C.2 | |||
| 2′-5′-oligoadenylate synthetase 2 (69-71 kDa) | Hs.414332 | OAS2 | I | U | U | R | U |
| Activating transcription factor 3 | Hs.460 | ATF3 | I | I | U | I | I |
| Apoptosis inhibitor | Hs.75263 | cIAP1 | I | I | U | I | U |
| Apoptosis inhibitor 2 | Hs.127799 | cIAP2 | I | I | U | U | U |
| Bcl-2 binding component 3 | Hs.87246 | BBC3 (PUMA) | I | I | U | U | I |
| Chemokine (C-X-C motif) ligand 10 | Hs.2248 | IP-10 (CXCL10) | I | I | U | I | I |
| Colony-stimulating factor 1 (macrophage) | Hs.173894 | CSF1 | I | I | U | I | I |
| Fibroblast growth factor 2 (basic) | Hs.284244 | FGF2 | I | U | I | U | U |
| Fucosyltransferase 2 | Hs.292999 | FUT2 | I | I | I | I | I |
| General transcription factor IIF, polypeptide 1 | Hs.68257 | GTF2F1 | I | U | U | U | U |
| GTP cyclohydrolase 1 (dopa-responsive dystonia) | Hs.86724 | GCH1 | I | U | U | U | U |
| IFN-induced protein with tetratricopeptide repeats 1 | Hs.20315 | ISG56 | I | I | I | U | I |
| IFN-induced protein with tetratricopeptide repeats 2 | Hs.293797 | ISG54 | I | I | I | U | I |
| IFN-induced protein with tetratricopeptide repeats 4 | Hs.181874 | ISG60 | I | U | U | U | U |
| Inhibin, beta A (activin A, activin AB alpha polypeptide) | Hs.727 | INHBA | I | I | U | U | U |
| Inhibitor of kappa B alpha | Hs.81328 | IκBα | I | I | U | I | I |
| Interferon regulatory factor 1 | Hs.80645 | IRF-1 | I | I | U | U | U |
| Interferon regulatory factor 2 | Hs.83795 | IRF-2 | I | U | U | U | U |
| Interferon, alpha-inducible protein (clone IFI-15K) | Hs.833 | ISG15 | I | I | I | U | I |
| Interleukin 6 | Hs.93913 | IL-6 | I | I | U | U | I |
| Interleukin 8 | Hs.624 | IL-8 | I | I | U | I | I |
| Jun B proto-oncogene | Hs.515157 | JUNB | I | I | U | I | U |
| Macrophage inflammatory protein I alpha | Hs.73817 | MIP1α (CCL3) | I | I | U | I | I |
| Mannose-binding lectin (protein C) 2 | Hs.2314 | MBL2 | I | I | R | I | I |
| MHC class I, A | Hs.181244 | HLA-A | I | U | I | U | I |
| MHC class I, C | Hs.277477 | HLA-C | I | U | U | I | I |
| MHC class I, F | Hs.110309 | HLA-F | I | U | U | U | I |
| MHC class I, G | Hs.73885 | HLA-G | I | U | U | U | I |
| Monocyte chemotactic protein 1 | Hs.303649 | MCP1 (CCL2) | I | U | U | U | U |
| Natural killer cell transcript 4 | Hs.943 | NK4 | I | U | U | U | I |
| Nexin, plasminogen activator inhibitor type 1 | Hs.82085 | PAI1 (SERPINE1) | I | I | I | I | I |
| NF-κB p105 | Hs.83428 | NF-κB1 | I | I | U | I | I |
| Nuclear factor (erythroid-derived 2)-like 3 | Hs.22900 | NFE21.3 (NRF3) | I | I | U | I | U |
| Nucleoside phosphorylase | Hs.75514 | NP | I | U | I | U | I |
| Phorbol-12-myristate-13-acetate-induced protein 1 | Hs.96 | NOXA (PMAIP1) | I | I | I | I | I |
| Protein tyrosine phosphatase, nonreceptor type 11 | Hs.83572 | PTPN11 (SHP2) | I | I | U | U | I |
| Protein tyrosine phosphatase, receptor type kappa | Hs.79005 | PTPRK | I | U | U | U | U |
| Short stature homeobox | Hs.105932 | SHOX | I | U | U | U | I |
| Similar to S. cerevisiae SSM4 | Hs.380875 | TEB4 | I | I | I | I | U |
| Stanniocalcin | Hs.25590 | STC1 | I | I | I | U | U |
| Syndecan 4 (amphiglycan, ryudocan) | Hs.252189 | SDC4 | I | I | U | I | I |
| Tight junction protein 2 (zona occludens 2) | Hs.75608 | TJP2 | I | I | I | I | I |
| TNF receptor-associated factor 1 | Hs.2134 | TRAF1 | I | I | R | U | U |
| Transcription factor AP-2 gamma | Hs.61796 | ERF1 (TFAP2C) | I | U | U | I | U |
| Tumor necrosis factor alpha-induced protein 3 | Hs.211600 | TNFAIP3 (A20) | I | I | U | I | I |
| UDP-N-acetyl-alpha-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase 2 | Hs.130181 | GALNT2 | I | U | I | U | U |
| Vascular endothelial growth factor C | Hs.79141 | VEGFC | I | I | I | U | U |
| Viperin | Hs.17518 | CIG5 | I | U | I | U | I |
| Zinc finger CCCH type, antiviral 1 | Hs.35254 | ZC3HAV1 | I | I | I | U | U |
I, induced; U, unchanged; R, repressed.
Other groups have previously sought to profile some of the genes induced by SeV. Strahle et al. employed SeV mutants to profile the effects of viral proteins on the induction of cellular genes in virus-infected cells (40), and Matikainen et al. looked at chemokine induction by SeV (30). In the present study, we see the induction by SeV of many of the same genes that were previously identified, including IL-8, IP-10, and class I major histocompatibility complex (MHC) genes. Further, we have been able to characterize the signaling pathways involved in the induction of not only these genes but also numerous other genes not formerly known to be induced by SeV.
Given the parameters of our experiment, there did not appear to be a role for TLR3 in gene induction by SeV. The paramyxovirus replication cycle takes >10 h, so it remains possible that at later times after virus infection, TLR3 signaling is used by viral dsRNA for induction of the dsRNA-inducible genes that were not induced by SeV at 6 h (Fig. 2) (23). Since some genes that were induced by dsRNA and SeV were common, it is also possible that the virus used intracellular viral dsRNA to induce them and that TLR3 was required for exogenous dsRNA, but not for SeV, because it mediates efficient uptake of dsRNA. Although our data showed that TLR3 was not required for gene induction early after SeV infection, viral dsRNA could have used other cellular sensors of dsRNA. PKR and RIG-I are two such intracellular dsRNA receptors that have recently been shown to mediate dsRNA signaling (11, 46). The apparent irrelevance of TLR3 in gene induction by SeV is supported by the fact that TLR3−/− mice are not deficient in clearing infections with other ssRNA viruses in vivo (12). Further, vesicular stomatitis virus, another ssRNA virus, has also been shown to activate downstream signaling factors independently of TLR3 in TLR3-deficent mouse embryo fibroblasts (42). It is highly unlikely that viral ssRNA was the gene inducer in our experiments, because 293 cells do not respond to ssRNA without the expression of exogenous TLR7 or TLR8, the human TLRs that sense ssRNA (19, 26).
Our investigation of the role of IFN and the corresponding signaling pathway in gene induction by SeV produced both anticipated and unanticipated results. In U4C cells lacking functional Jak1, we observed that some genes were not induced compared to wt cells. This result was expected because some genes might be induced by the autocrine action of IFN induced by SeV infection. Many genes of this class, such as OAS2 and MHC class I genes, are known IFN-stimulated genes (Fig. 3). Further, our findings with SeV correspond with those observed in Newcastle disease virus-infected cells, where type I IFN signaling was required for OAS induction (33). However, other genes of the same family, such as ISG56 and ISG54, were induced even in U4C cells, confirming previous observations by us and others that these genes are directly induced upon infection with many viruses (18, 34). The fact that these genes were induced in infected cells independently of autocrine IFN signaling is not unexpected, because IRF-3, which is activated in the infected cells, can induce these genes directly. The surprising observation was that a cohort of genes was induced only in U4C cells, indicating that Jak1 or IFN signaling somehow negatively regulates the expression of these genes in wt cells. The implications and the mechanism of this regulation need to be further explored in the future. To distinguish between the needs for IFN signaling and other putative functions of Jak1, we also analyzed the gene induction profiles in SeV-infected U2A cells and U3A cells (data not shown), which are also defective in IFN signaling because they lack functional IRF-9 and STAT1, respectively. The MHC class I, which were not induced by SeV in U4C cells, were not induced in U2A and U3A cells either, indicating that IFN signaling was needed for their induction. In contrast, the Jak1-repressed genes (Fig. 3), although induced in U4C cells, were not induced in U2A and U3A cells (data not shown), suggesting that it is the absence of Jak1 or another mutation specific to the cell line and not the loss of IFN signaling that led to SeV-mediated induction of this class of genes in U4C cells. These results indicate that autocrine IFN signaling and Jak1 per se can influence the pattern of cellular gene induction by SeV infection.
For assessing the relative contributions of NF-κB and IRF-3, two major transcription factors activated by SeV infection, we took advantage of other mutant lines generated by us and our colleagues. The 2F-SR line was derived from 2fTGH cells by overexpressing a dominant negative inactivatable mutant of IκB. NF-κB cannot be activated by IL-1, tumor necrosis factor alpha (TNF-α), dsRNA, or SeV infection in these cells, as judged by electrophoretic mobility shift assays (36). Consequently, these cells are functionally NF-κB null cells. As expected, we observed that many genes could not be induced by SeV in the absence of NF-κB signaling (Fig. 4). Some of these genes, such as the IKKα, NF-κB p105, and A20 genes, encode proteins that are involved in the NF-κB signaling pathway itself. Similarly, another set of genes was completely dependent on IRF-3 (Fig. 5). For identifying them, we used P2.1 cells, derived from U4C cells, which express little IRF-3 (36). Many of these genes fall into the category of viral stress-inducible genes (37) because they can be independently induced by IFN, dsRNA, or virus infection. Although the same cis element, ISRE, that is present in their promoter is responsible for their induction by the three agents, different trans-acting factors mediate the process. Induction of these genes by IFNs is mediated by ISGF3, a trimeric complex of IRF-9, STAT1, and STAT2. Induction by dsRNA and viruses, on the other hand, is mediated by activated IRF-3, which also binds to ISRE. However, the signaling pathways, triggered by dsRNA and viruses, that lead to IRF-3 activation overlap only partially (39). Thus, it appears that viral stress-inducible genes can be induced by multiple inducers using multiple signaling pathways. There were also genes that were induced when either the NF-κB pathway or the IRF-3 pathway were inoperative (Fig. 4 and 5). These genes probably get induced through other transcription factors activated by virus infection.
One such NF-κB- and IRF-3-independent gene is the NOXA gene. NOXA, also known as PMAIP1 and originally discovered as a mediator of p53-induced apoptosis (35), has been shown to be induced by a constitutively active IRF-3 mutant (IRF-3 5D) (16, 21). However, in the context of induction by SeV, IRF-3 does not appear to play an important role in NOXA induction (Fig. 5 and 6B and D). In the absence of STAT1, NOXA induction through the p53 response element in its promoter is impaired (44); likewise, SeV fails to induce NOXA in STAT1−/− U3A cells (data not shown). This finding suggests that SeV may induce a subset of genes through the IRF-3-, NF-κB-, and IFN-independent activation of STAT1 or p53. It also raises the possibility that the constitutively active IRF-3 5D mutant and virally activated IRF-3 do not induce equivalent sets of genes. Active SeV infection causes additional phosphorylation of IRF-3 beyond that required for the IRF-3-mediated induction of ISG56 (8). These additional modifications to IRF-3 might very well impair its ability to mediate the induction of other genes, such as the NOXA gene.
Further explorations of the role of IRF-3 in gene induction by SeV produced interesting results. Using two related cell lines that expressed different levels of IRF-3, we could demonstrate that although some genes were induced equally well in both lines, other genes were induced better in U4C.2 cells, which expressed a higher level of IRF-3 (Fig. 6A). Thus, it seems that at least in the 2fTGH cell lineage, IRF-3 expression is the limiting factor and the degree of gene induction can be modulated easily by changing the cellular concentration of IRF-3. The above conclusion was confirmed by a more quantitative assay using a series of cell lines expressing increasing amounts of IRF-3 (Fig. 7). Similar results were obtained with the same cell lines when the inducer was dsRNA, not SeV (36). Among the genes induced by SeV in U4C.2 cells were the MHC genes which were not induced in U4C cells but induced in 2fTGH cells (Fig. 3 and 6). Thus, a high level of IRF-3 could mediate SeV induction of genes which otherwise required IFN signaling. Other genes, such as the follistatin gene, were induced by SeV very strongly in U4C.2 cells, whereas the induction was minimal in U4C or 2fTGH cells (Fig. 6A and C).
The most surprising observation was that a higher level of IRF-3 expression negatively affected the degree of induction of some genes (Fig. 6B and E). Using the A20 gene as the sentinel for this group of genes, we could demonstrate that unlike induction of ISG56, induction of A20 mRNA was inversely related to the level of IRF-3 (Fig. 7). This phenomenon was not restricted to the U4C cells and its derivatives; the same trend was also observed in wt HT1080 cells by overexpressing IRF-3 (Fig. 8). It is not clear how IRF-3 affects A20 gene induction by SeV. The promoter of this gene contains κB sites (22) but no consensus ISRE, and as expected, its induction required NF-κB (Fig. 4) but not IRF-3 (Fig. 5). Thus, the observed negative effect of IRF-3 is probably not exerted through its direct interaction with the A20 promoter. Further investigation should reveal the nature of the cross talk between the two major virus-activated signaling pathways.
What are the possible physiological implications of the above phenomenon? SeV and other viruses cause active and rapid degradation of IRF-3 in infected cells (27). Consequently, the degree of induction of genes, such as the A20 gene, can be temporally regulated in the infected cells. Moreover, the level of IRF-3 expression varies greatly among different cell types and different tissues (data not shown). From the observations reported here, one can predict that the profiles of gene induction, and hence the outcome of virus infection, will also vary greatly among different host cells. A20 is required for attenuation of NF-κB activity induced both through TNF-α (25) and through Toll-like receptors (4). Consequently, cells expressing high levels of IRF-3 would have to have sustained NF-κB and IRF-3 responses due to their inability to make A20. To further speculate about the possible cellular functions of the genes that are negatively regulated by IRF-3, it is interesting that several, such as A20, CIAP1, and CIAP2, have antiapoptotic functions. The encoded proteins may protect the infected cells from the actions of the proapoptotic proteins that are also induced with virus infection. It has been reported that apoptosis of SeV-infected cells is mediated by IRF-3 (20). Our results suggest that this function of IRF-3 may be mediated not only by inducing proapoptotic genes but also by suppressing the induction of antiapoptotic genes.
Supplementary Material
Acknowledgments
Our appreciation is extended to George Stark and Kristi Peters for use of various cell lines. We especially thank Roger Slee and Mat Frevel for their help and advice in conducting the microarray experiments. We are also grateful to Mat Frevel as well as Ge Guo for their contribution to the development of the customized cDNA microarray.
This work was supported in part by National Institutes of Health grants CA62220 (G.C.S.), CA68782 (G.C.S.), and AI34039 (B.R.G.W.) and Medical Scientist Training Grant GM07250 (C.P.E.) and by U.S. Army grant DAMD17-01-C-065 (B.R.G.W.), and a UNCF/Merck Research Fellowship (C.P.E.).
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Alexopoulou, L., A. C. Holt, R. Medzhitov, and R. A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732-738. [DOI] [PubMed] [Google Scholar]
- 2.Algarte, M., H. Nguyen, C. Heylbroeck, R. Lin, and J. Hiscott. 1999. IκB-mediated inhibition of virus-induced beta interferon transcription. J. Virol. 73:2694-2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bandyopadhyay, S. K., G. T. Leonard, Jr., T. Bandyopadhyay, G. R. Stark, and G. C. Sen. 1995. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J. Biol. Chem. 270:19624-19629. [DOI] [PubMed] [Google Scholar]
- 4.Boone, D. L., E. E. Turer, E. G. Lee, R. C. Ahmad, M. T. Wheeler, C. Tsui, P. Hurley, M. Chien, S. Chai, O. Hitotsumatsu, E. McNally, C. Pickart, and A. Ma. 2004. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 5:1052-1060. [DOI] [PubMed] [Google Scholar]
- 5.Boyle, K. A., R. L. Pietropaolo, and T. Compton. 1999. Engagement of the cellular receptor for glycoprotein B of human cytomegalovirus activates the interferon-responsive pathway. Mol. Cell. Biol. 19:3607-3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang, Y. E., and L. A. Laimins. 2000. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol. 74:4174-4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chin, K. C., and P. Cresswell. 2001. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc. Natl. Acad. Sci. USA 98:15125-15130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins, S. E., R. S. Noyce, and K. L. Mossman. 2004. Innate cellular response to virus particle entry requires IRF3 but not virus replication. J. Virol. 78:1706-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Der, S. D., A. Zhou, B. R. Williams, and R. H. Silverman. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 95:15623-15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Veer, M. J., M. Holko, M. Frevel, E. Walker, S. Der, J. M. Paranjape, R. H. Silverman, and B. R. Williams. 2001. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 69:912-920. [PubMed] [Google Scholar]
- 11.Diebold, S. S., M. Montoya, H. Unger, L. Alexopoulou, P. Roy, L. E. Haswell, A. Al-Shamkhani, R. Flavell, P. Borrow, and C. Reis e Sousa. 2003. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 424:324-328. [DOI] [PubMed] [Google Scholar]
- 12.Edelmann, K. H., S. Richardson-Burns, L. Alexopoulou, K. L. Tyler, R. A. Flavell, and M. B. Oldstone. 2004. Does Toll-like receptor 3 play a biological role in virus infections? Virology 322:231-238. [DOI] [PubMed] [Google Scholar]
- 13.Frevel, M. A., T. Bakheet, A. M. Silva, J. G. Hissong, K. S. Khabar, and B. R. Williams. 2003. p38 mitogen-activated protein kinase-dependent and -independent signaling of mRNA stability of AU-rich element-containing transcripts. Mol. Cell. Biol. 23:425-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geiss, G., G. Jin, J. Guo, R. Bumgarner, M. G. Katze, and G. C. Sen. 2001. A comprehensive view of regulation of gene expression by double-stranded RNA-mediated cell signaling. J. Biol. Chem. 276:30178-30182. [DOI] [PubMed] [Google Scholar]
- 15.Geiss, G. K., M. Salvatore, T. M. Tumpey, V. S. Carter, X. Wang, C. F. Basler, J. K. Taubenberger, R. E. Bumgarner, P. Palese, M. G. Katze, and A. Garcia-Sastre. 2002. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc. Natl. Acad. Sci. USA 99:10736-10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grandvaux, N., M. J. Servant, B. tenOever, G. C. Sen, S. Balachandran, G. N. Barber, R. Lin, and J. Hiscott. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol. 76:5532-5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo, J., D. J. Hui, W. C. Merrick, and G. C. Sen. 2000. A new pathway of translational regulation mediated by eukaryotic initiation factor 3. EMBO J. 19:6891-6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo, J., K. L. Peters, and G. C. Sen. 2000. Induction of the human protein P56 by interferon, double-stranded RNA, or virus infection. Virology 267:209-219. [DOI] [PubMed] [Google Scholar]
- 19.Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science 303:1526-1529. [DOI] [PubMed] [Google Scholar]
- 20.Heylbroeck, C., S. Balachandran, M. J. Servant, C. DeLuca, G. N. Barber, R. Lin, and J. Hiscott. 2000. The IRF-3 transcription factor mediates Sendai virus-induced apoptosis. J. Virol. 74:3781-3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiscott, J., N. Grandvaux, S. Sharma, B. R. Tenoever, M. J. Servant, and R. Lin. 2003. Convergence of the NF-kappaB and interferon signaling pathways in the regulation of antiviral defense and apoptosis. Ann. N. Y. Acad. Sci. 1010:237-248. [DOI] [PubMed] [Google Scholar]
- 22.Laherty, C. D., N. D. Perkins, and V. M. Dixit. 1993. Human T cell leukemia virus type I Tax and phorbol 12-myristate 13-acetate induce expression of the A20 zinc finger protein by distinct mechanisms involving nuclear factor kappa B. J. Biol. Chem. 268:5032-5039. [PubMed] [Google Scholar]
- 23.Lamb, R. A., and D. Kolakofsky. 2001. Paramyxoviridae: the viruses and their replication, p. 1305-1339. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 24.Leaman, D. W., A. Salvekar, R. Patel, G. C. Sen, and G. R. Stark. 1998. A mutant cell line defective in response to double-stranded RNA and in regulating basal expression of interferon-stimulated genes. Proc. Natl. Acad. Sci. USA 95:9442-9447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee, E. G., D. L. Boone, S. Chai, S. L. Libby, M. Chien, J. P. Lodolce, and A. Ma. 2000. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 289:2350-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, J., T. H. Chuang, V. Redecke, L. She, P. M. Pitha, D. A. Carson, E. Raz, and H. B. Cottam. 2003. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 100:6646-6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin, R., C. Heylbroeck, P. M. Pitha, and J. Hiscott. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986-2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lund, J. M., L. Alexopoulou, A. Sato, M. Karow, N. C. Adams, N. W. Gale, A. Iwasaki, and R. A. Flavell. 2004. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 101:5598-5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malmgaard, L. 2004. Induction and regulation of IFNs during viral infections. J. Interferon Cytokine Res. 24:439-454. [DOI] [PubMed] [Google Scholar]
- 30.Matikainen, S., J. Pirhonen, M. Miettinen, A. Lehtonen, C. Govenius-Vintola, T. Sareneva, and I. Julkunen. 2000. Influenza A and Sendai viruses induce differential chemokine gene expression and transcription factor activation in human macrophages. Virology 276:138-147. [DOI] [PubMed] [Google Scholar]
- 31.McKendry, R., J. John, D. Flavell, M. Muller, I. M. Kerr, and G. R. Stark. 1991. High-frequency mutagenesis of human cells and characterization of a mutant unresponsive to both alpha and gamma interferons. Proc. Natl. Acad. Sci. USA 88:11455-11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mossman, K. L., P. F. Macgregor, J. J. Rozmus, A. B. Goryachev, A. M. Edwards, and J. R. Smiley. 2001. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 75:750-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakaya, T., M. Sato, N. Hata, M. Asagiri, H. Suemori, S. Noguchi, N. Tanaka, and T. Taniguchi. 2001. Gene induction pathways mediated by distinct IRFs during viral infection. Biochem. Biophys. Res. Commun. 283:1150-1156. [DOI] [PubMed] [Google Scholar]
- 34.Navarro, L., K. Mowen, S. Rodems, B. Weaver, N. Reich, D. Spector, and M. David. 1998. Cytomegalovirus activates interferon immediate-early response gene expression and an interferon regulatory factor 3-containing interferon-stimulated response element-binding complex. Mol. Cell. Biol. 18:3796-3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oda, E., R. Ohki, H. Murasawa, J. Nemoto, T. Shibue, T. Yamashita, T. Tokino, T. Taniguchi, and N. Tanaka. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053-1058. [DOI] [PubMed] [Google Scholar]
- 36.Peters, K. L., H. L. Smith, G. R. Stark, and G. C. Sen. 2002. IRF-3-dependent, NFκB- and JNK-independent activation of the 561 and IFN-β genes in response to double-stranded RNA. Proc. Natl. Acad. Sci. USA 99:6322-6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarkar, S. N., and G. C. Sen. 2004. Novel functions of proteins encoded by viral stress-inducible genes. Pharmacol. Ther. 103:245-259. [DOI] [PubMed] [Google Scholar]
- 38.Sarkar, S. N., H. L. Smith, T. M. Rowe, and G. C. Sen. 2003. Double-stranded RNA signaling by Toll-like receptor 3 requires specific tyrosine residues in its cytoplasmic domain. J. Biol. Chem. 278:4393-4396. [DOI] [PubMed] [Google Scholar]
- 39.Servant, M. J., N. Grandvaux, and J. Hiscott. 2002. Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem. Pharmacol. 64:985-992. [DOI] [PubMed] [Google Scholar]
- 40.Strahle, L., D. Garcin, P. Le Mercier, J. F. Schlaak, and D. Kolakofsky. 2003. Sendai virus targets inflammatory responses, as well as the interferon-induced antiviral state, in a multifaceted manner. J. Virol. 77:7903-7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.tenOever, B. R., M. J. Servant, N. Grandvaux, R. Lin, and J. Hiscott. 2002. Recognition of the measles virus nucleocapsid as a mechanism of IRF-3 activation. J. Virol. 76:3659-3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.tenOever, B. R., S. Sharma, W. Zou, Q. Sun, N. Grandvaux, I. Julkunen, H. Hemmi, M. Yamamoto, S. Akira, W.-C. Yeh, R. Lin, and J. Hiscott. 2004. Activation of TBK1 and IKKε kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J. Virol. 78:10636-10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tiwari, R. K., J. Kusari, and G. C. Sen. 1987. Functional equivalents of interferon-mediated signals needed for induction of an mRNA can be generated by double-stranded RNA and growth factors. EMBO J. 6:3373-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Townsend, P. A., T. M. Scarabelli, S. M. Davidson, R. A. Knight, D. S. Latchman, and A. Stephanou. 2004. STAT-1 interacts with p53 to enhance DNA damage-induced apoptosis. J. Biol. Chem. 279:5811-5820. [DOI] [PubMed] [Google Scholar]
- 45.Van Antwerp, D. J., S. J. Martin, T. Kafri, D. R. Green, and I. M. Verma. 1996. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 274:787-789. [DOI] [PubMed] [Google Scholar]
- 46.Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi, M. Miyagishi, K. Taira, S. Akira, and T. Fujita. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730-737. [DOI] [PubMed] [Google Scholar]
- 47.Zhu, H., J. P. Cong, G. Mamtora, T. Gingeras, and T. Shenk. 1998. Cellular gene expression altered by human cytomegalovirus: global monitoring with oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 95:14470-14475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.