

Abstract
The third open reading frame (ORF3) located at the 3′ end of the genomic RNA of feline calicivirus (FCV) encodes a small (12.2-kDa) minor structural protein of 106 amino acids designated VP2. Point mutations and deletions were introduced into an infectious FCV cDNA clone in order to evaluate the functional importance of ORF3 and its encoded protein, VP2. Deletion of the entire ORF3 sequence was lethal for the virus, and evidence was found for strong selective pressure to produce the VP2 protein. Extended deletions in the 5′ end and small deletions in the 3′ end of ORF3, as well as the introduction of stop codons into the ORF3 sequence, were tolerated by the viral replication machinery, but infectious virus could not be recovered. Infectious virus particles could be rescued from a full-length FCV cDNA clone encoding a nonfunctional VP2 when VP2 was provided in trans from a eukaryotic expression plasmid. Our data indicate that VP2, a protein apparently unique to the caliciviruses, is essential for productive replication that results in the synthesis and maturation of infectious virions and that the ORF3 nucleotide sequence itself overlaps a cis-acting RNA signal at the genomic 3′ end.
Caliciviruses are small, nonenveloped, positive-strand RNA viruses that infect a broad range of hosts. Their virions are 27 to 40 nm in diameter and have icosahedral symmetry. The virion capsid is comprised of 180 copies of the major structural protein, VP1 (29, 31), and a minor structural protein designated VP2 (7, 27, 38, 43). The capsid surrounds the VPg-linked polyadenylated RNA genome (2, 6, 14, 23), and in at least two caliciviruses, rabbit hemorrhagic disease virus (RHDV) and feline calicivirus (FCV), a VPg-linked ∼2.2- to 2.6-kb subgenomic-size RNA is also packaged into virions (23, 25). The RNA genomes of caliciviruses range in size from 7.3 to 8.3 kb and are organized into either two or three major open reading frames (ORFs) (9). In the genera Lagovirus and Sapovirus of the family Caliciviridae, the nonstructural proteins and the structural protein VP1 are encoded in the same ORF. In the genera Vesivirus and Norovirus, VP1 is encoded in a separate ORF. In all genera, a genomic-length RNA serves as a template for synthesis of the nonstructural polyprotein while the 2.2- to 2.6-kb bicistronic subgenomic RNA is used for translation of the VP1 and VP2 proteins (3, 26, 28).
All caliciviruses encode the VP2 protein in a separate ORF near the 3′ end of the genome (ORF3 for noroviruses and vesiviruses and ORF2 for sapoviruses and lagoviruses). Comparative sequence analyses of this terminal ORF and the deduced VP2 amino acid sequence indicate that this region of the genome is highly variable in sequence between genera, with as little as 10% amino acid similarity. In addition, these proteins show significant variation in length, ranging from 106 (FCV) to 268 (norovirus MD-145) amino acids (aa). The expression of VP2 in calicivirus-infected cells has been reported for FCV and RHDV (13, 17, 42). For FCV, the level of VP2 expression has been estimated to be ∼10% of that of VP1 (13). The mechanism controlling VP2 synthesis in infected cells is not understood, but it may be regulated at the translational level (13, 26). Evidence for the expression of VP2 from the subgenomic RNA was observed in translational analysis of subgenomic RNAs synthesized in vitro (13, 22, 38). One proposed mechanism for translation of VP2 from this RNA involved a ribosomal termination-initiation event at the site of overlap between the ORFs encoding VP1 and VP2 (13). There has also been evidence for the presence of possible RNA structural elements involved in the regulation of VP2 expression. The sequence of the corresponding ORF (ORF3) in the FCV genome was predicted to have a hairpin structure at the beginning of the VP2-encoding sequence that might slow the translating ribosome (26). Furthermore, the 3′-terminal 84 nucleotides (nt) of the coding sequence of the RHDV capsid protein were found to be crucial for expression of the viral VP2 in a transient system (22).
The function of the calicivirus VP2 protein is unknown. It was proposed that due to its basic characteristics, VP2 might interact with RNA and the internal acidic domains of the calicivirus virion, providing a structural basis for encapsidation of the viral RNA into particles (26, 30, 43). The sequence between aa 108 and 152 of Norwalk virus (NV) VP2 has been mapped as a domain responsible for protein-protein interactions between the NV VP2 and VP1 proteins (8), but no evidence for direct interactions between VP2 and RNA has been reported. Recently, it was demonstrated that the presence of VP2 could protect VP1 from protease degradation and increase the stability of NV recombinant virus-like particles (1). However, VP2 was found to be dispensable for self-assembly of empty viral capsids (19, 20).
In this study, we demonstrate that ORF3 of the FCV genome and the VP2 protein encoded in this ORF are essential for virus replication and the assembly of infectious particles. In addition, we provide the first evidence that a replication-competent calicivirus genome defective in expression of the full-length VP2 can be complemented by provision of VP2 in trans. Virus particles synthesized in these cells could infect a new cell monolayer, resulting in virus replication without the release of infectious viral progeny.
MATERIALS AND METHODS
Cells and viruses.
Crandell-Rees feline kidney (CRFK) cells were grown in Eagle's minimum essential medium containing amphotericin B (2.5 μg/ml), chlortetracycline (25 μg/ml), penicillin (250 U/ml), and streptomycin (250 μg/ml) supplemented with 10% heat-inactivated fetal bovine serum. The Urbana strain of FCV (GenBank accession no. L40021) was described previously (35).
Plasmid construction.
Standard recombinant DNA methods were used for plasmid construction (33). To introduce an AvrII cleavage site into the 5′ end of the FCV ORF3 sequence (next to the junction of the ORF2 and ORF3 sequences) in the FL clone pQ14 (35), ORF2 and ORF3 sequences were amplified from pQ14 using the primer pairs 5′-TAATACGACTCACTATAGTGTTCGAAGTTTGAGCATGTGC-3′ (designated A1) plus 5′-GTAACAGTATCAATCAAGCCtAggATTGAATTCATAGTTTAGTC-3′ (designated A2) and 5′-GACTAAACTATGAATTCAATccTaGGCTTGATTGATACTGTTAC-3′ (designated A3) plus 5′-AATTTAGGTGACACTATAG-3′, respectively. The sequence of A1 contained 23 nt corresponding to the beginning of the subgenomic RNA (nt 5297 to 5319) and included an NspV site (underlined). The A2 oligonucleotide was complementary to nt 7308 to 7351 of the FCV genome and included an engineered AvrII site (underlined). The A3 oligonucleotide was complementary to the A2 oligonucleotide, and both primers were engineered to convert the wild-type sequence into a unique AvrII site (nucleotide changes are shown in lowercase). The fourth oligonucleotide corresponded to nt 163 to 181 of the vector sequence (pSPORT1; Invitrogen Inc.) The purified PCR-generated ORF2 and ORF3 DNA fragments were treated with NspV and AvrII or AvrII and NotI, respectively, and ligated into NspV-NotI-linearized pQ14. Clones with the desired mutation were screened by restriction enzyme and sequence analysis, and the resulting plasmid, designated pR6, was selected for the construction of FCV genomes with a modified ORF3.
To introduce a terminator codon in the beginning of the ORF3 sequence, the ORF3 sequence was amplified from plasmid pR6 using the sense primer 5′-ATGAATTCAATCCTAGGCTAGATTGATACTGTTAC-3′ or 5′-ATGAATTCAATCCTAGGCTAGTAATGAACTGTTACAAATACAATTGGCAAAGCAC-3′ and an antisense primer, 5′-ATATAAGCGGCCGC(T20)CCCTGGGGTTAG-3′ (designated polyA-FCV). The sequences of the sense oligonucleotides corresponded to the beginning of ORF3 and contained engineered terminator codons (boldface). The sequence of the antisense oligonucleotide was complementary to the 3′ end of the FCV genome and contained a poly(T20) sequence and a NotI site (underlined). The resulting plasmids, pF3Nterm and pF3Nterm3, carried the FCV genome with the ORF3 sequence interrupted, beginning at position 7, with one or three terminator codons, respectively.
To introduce amino acid changes into the VP2 sequence at position 7, DNA fragments were amplified from plasmid pR6 by PCR using a sense primer, 5′-ATGAATTCAATCCTAGGCNNNATTGATACTGTTAC-3′, corresponding to nt 7317 to 7351 of the genome and an antisense polyA-FCV primer (described above). The sense primers contained the following nucleotides in positions 19 to 21 (NNN): GCG for Ala, AAT for Asn, GAT for Asp, ACG for Thr, TCT for Ser, AAG for Lys, GGT for Gly, GAG for Glu, TGT for Cys, and CAG for Gln. The PCR fragments were purified, treated with AvrII and NotI, and ligated into the compatible sites of pR6. The clones were screened by sequence analysis, and plasmids containing the desired mutations in the VP2 sequence were selected and designated pF3X7, where X corresponds to the introduced amino acid mutation.
To construct FL clones with consecutive in-frame deletions extending from the 5′ end of ORF3, sense primers (Table 1) and an antisense primer, polyA-FCV, were used to amplify DNA fragments from plasmid pR6. The sense primers were designed as follows: the 5′ end contained sequence corresponding to the first 18 nt of ORF3, while the rest of the primer sequence corresponded to the various regions of ORF3 toward its 3′ end. Purified DNA fragments were digested with AvrII and NotI and ligated into the AvrII-NotI-linearized pR6 vector. Selected clones were designated pF3N, with the position of the nucleotide deletion in the ORF3 sequence given in parentheses, and the clone with a deletion from nt 19 to 318 (to the end of ORF3) was designated pR6delF3 (Table 1).
TABLE 1.
Constructs and PCR primers used in generation of full-length clones with N-terminal deletions in the sequence of VP2 protein
| FL clones | Sequencea | Nucleotide and amino acid positions of deleted sequence (ORF3/VP2) |
|---|---|---|
| pF3N(19-48) | atgaattcaatcctaggcAAAGCACAACAAATTGAATTAGACAAGGC | 19-48/7-16 |
| pF3N(19-78) | atgaattcaatcctaggcGCGCTTGGACAACAACGCGAGCTGGC | 19-78/7-26 |
| pF3N(19-108) | atgaattcaatcctaggcCAGCGTATGAATCTGGATCGCC | 19-108/7-36 |
| pF3N(19-138) | atgaattcaatcctaggcAATAACCAAGTAGAGCAGTTTAAC | 19-138/7-46 |
| pF3N(19-168) | atgaattcaatcctaggcCTTGAGCAAAGGGTACAAGGC | 19-168/7-56 |
| pF3N(19-198) | atgaattcaatcctaggcTCTGTGCGATTAGCACGTGCTGC | 19-198/7-66 |
| pF3N(19-228) | atgaattcaatcctaggcAGGGTTGACCCTTACTCATAC | 19-228/7-76 |
| pF3N(19-258) | atgaattcaatcctaggcAATTTTTATGATGATCAATTG | 19-258/7-86 |
| pF3N(19-288) | atgaattcaatcctaggcCGGCTATCATATAGGAATTTGTTTAAG | 19-288/7-96 |
| pR6delF3 | atgaattcaatcctaggcTGACCACAAGTATCCCTTTGG | 19-318/7-106 |
The sequences of the oligonucleotides corresponding to the first 18 nt of the ORF3 sequence are shown in lowercase. The sequences of the oligonucleotides located immediately downstream of the engineered deletions are in uppercase.
To introduce an AflII cleavage site into the 3′ end of the FCV ORF3 sequence in the pR6 vector, the ORF3 sequence was amplified from plasmid pR6 using a sense primer, A3 (described above), and an antisense primer, 5′-GGGATACTTGTGGTCAATTCTTAAACAAATTCCTATAgctTAagCGAATTGCATTC-3′. The sequence of the antisense primer was complementary to nt 7595 to 7650 of the FCV genome and included an engineered AflII site (underlined). The nucleotide changes converting the wild-type sequence into an AflII site are shown in lowercase. Two additional PCR amplification steps were utilized to extend an amplified ORF3 sequence through the poly(A) sequence. The sense primer, A3, and the antisense primer, 5′-CCCTGGGGTTAGGCGCAAATGCGGCAGCCCAAAGGGATACTTGTGGTC-3′, which contained sequence complementary to nt 7636 to 7683 of the FCV genome, were employed in the first additional round of amplification. In the second round, the PCR fragment sequence was extended using polyA-FCV as an antisense primer. The purified DNA fragment was treated with AvrII and NotI and ligated into the AvrII-NotI-linearized pR6 vector. The clones were screened by restriction analysis and selected for further sequence analysis. The resulting plasmid was designated pRAfl.
For construction of FL clones with 3′-end ORF3 deletions, the ORF3 sequence was amplified using the sense primer A3 (described above) and antisense primers (Table 2). The antisense primers were designed as follows: the 5′ end contained a sequence complementary to nt 289 to 303 of ORF3, while the rest of the primer sequence corresponded to the various regions of ORF3 toward its 5′ end. The purified DNA fragments were digested with AvrII and AflII and ligated into the AvrII-AflII-linearized pRAfl vector. Selected clones were designated pF3C, with the position of the nucleotide deletion in the ORF3 sequence given in parentheses (Table 2).
TABLE 2.
Constructs and PCR primers used in construction of full-length clones with C-terminal deletions in the sequence of VP2 protein
| FL clones | Sequencea | Nucleotide and amino acid borders of deleted sequence (ORF3/VP2) |
|---|---|---|
| pF3C(259-288) | cctatagcttaagcgTTGATTTGTGTATGAGTAAGG | 259-288/87-96 |
| pF3C(229-288) | cctatagcttaagcgAAAACCAGCAGCACGTGC | 229-288/77-96 |
| pF3C(199-288) | cctatagcttaagcgCTGGAGGGGGCCTTGTACCCTTTG | 199-288/67-96 |
| pF3C(169-288) | cctatagcttaagcgAATTTTGTTAAACTGCTCTAC | 169-288/57-96 |
The sequences of the oligonucleotides complementary to nt 7605 to 7619 of the FCV genome sequence (in pRAfI) are shown in lowercase. The sequences of the oligonucleotides located immediately upstream of the engineered deletion are in uppercase.
To truncate the ORF3 sequence by engineering a terminator codon into position 7578, a DNA fragment was PCR amplified from plasmid pRAfl using a sense primer, A3 (described above), and an antisense primer, 5′-ATATATTAAATACTTAAGCGAATTGCATTCAATTGATCATCCTATTAATTTTGATTTGTGTATGAG-3′. The sequence of the second oligonucleotide was complementary to nt 7559 to 7612 of the FCV genome (in pRAfl) and included two engineered stop codons (boldface). An AvrII-AflII-treated DNA fragment was ligated into the AvrII-AflII-linearized pRAfl vector. The resulting plasmid was designated pF3Cterm2.
To construct plasmid pCiF3, a DNA fragment was amplified from plasmid pQ14 using a sense primer, 5′-ATATAACGCGTACTATGAATTCAATTTTG-3′, and an antisense primer, 5′-TTATATAGCGGCCGCTTATCAATTCTTAAACAAATTCC-3′. The sequence of the first oligonucleotide contained 15 nt corresponding to the beginning of ORF3 and included an MluI site (underlined). The antisense oligonucleotide corresponded to the sequence complementary to the 3′ end of ORF3 and contained a terminator codon (boldface) and a NotI site (underlined). The purified fragment was treated with MluI and NotI and ligated into a MluI-NotI-digested pCi vector (Promega Inc.). The resulting plasmid, pCiF3, was verified by sequence analysis.
Transfection experiments and virus recovery.
Capped FL genomic RNAs were synthesized in an in vitro system (Ribomax; Promega Inc.) using NotI-linearized FCV FL cDNA clones. The RNA transfection experiments were conducted using a protocol similar to that described previously (35).
Transfection of MVA-T7-infected CRFK cells with FCV FL cDNA clones and virus recovery of FCV were performed as described previously (36, 37). Virus replication was detected by immunofluorescent (IF) staining of transfected cells with serum raised in guinea pigs against FCV virions (35), followed by detection of bound antibodies with a fluorescein isothiocyanate-conjugated goat anti-guinea pig immunoglobulin G antibody (ICN). Recovery of infectious virus was monitored by detection of characteristic FCV cytopathic effect (CPE) in the monolayer of CRFK cells and further verified by plaque formation assay.
Radiolabeling and immunoprecipitation of virus-specific proteins.
For radiolabeling of virus-specific proteins in MVA-T7-infected CRFK cells transfected with the FCV FL cDNA clones, the monolayers (106 cells) were washed at 5 h posttransfection with methionine-free growth medium and incubated in the same medium for 30 min. [35S]methionine (>1,000 Ci/mmol; Amersham) was added to the cells at a concentration of 100 μCi/ml, and the cells were incubated for 16 h. Following incubation, the cells were lysed in 1 ml of radioimmunoprecipitation assay (RIPA) buffer (33). Radiolabeling and immunoprecipitation (IP) of viral proteins from infected cell lysates and in vitro translation mixtures were performed using capsid-specific serum as described previously (39).
Northern blotting.
Total RNA extraction and its analysis by Northern blotting were carried out similarly to a previously described protocol (10). Briefly, total RNA was extracted from MVA-T7-infected CRFK cells 18 h after their transfection with the FCV FL cDNA clones using Trizol reagent (Invitrogen Inc.). Ethanol-precipitated samples of RNA were resolved by electrophoresis in 1% agarose gel containing formaldehyde (33). The RNA was transferred to Nytran SuPerCharge membranes (Schleicher & Schuell BioScience, Inc.) by capillary blotting. Following transfer, the RNA was cross-linked to the membrane with UV light. After the membrane was prehybridized in HYB buffer (Quality Biological, Inc.) at 65°C for 30 min, the biotinylated probe specific to the virus sense RNA (10) was added, and incubation continued overnight at 65°C. Following washing, the detection of bound biotinylated probe was accomplished using the Bright Star BioDetect System (Ambion).
Nucleotide sequence analysis.
To verify the presence of engineered mutations in constructed plasmid sequences and to confirm that no mutations were introduced during the PCR amplification step, the plasmids were sequenced using the Big Dye Terminator version 3.1 Cycle Sequencing Ready Reaction kit, and the sequencing products were resolved on an ABI 3100 automated sequencer (Applied Biosystems).
The presence of the desired nucleotide substitutions in the genomes of recovered viruses was confirmed by direct sequence analysis of reverse transcriptase (RT) PCR products derived from viral RNA as described previously (39). The sequences of oligonucleotides used to amplify and sequence virus-specific cDNA fragments are available upon request.
RESULTS
Construction of FL VP2 cassette vectors and ORF3 knockouts.
To facilitate manipulation of the VP2 sequence in the FCV FL cDNA clone pQ14, we introduced three silent mutations at positions 7328, 7329, and 7331 of the virus genome (ATT7328→ATC7328 and TTG7331→CTA7331) to generate a new AvrII restriction site at the beginning of the ORF3 sequence (Fig. 1A). In the newly derived vector, designated pR6, the ORF3 sequence, 3′-end nontranslated region (NTR), and poly(A) tract were bordered by unique AvrII and NotI restriction sites that could be used to replace this entire area with a corresponding modified fragment. Plaque-purified and amplified virus recovered from pR6 was indistinguishable from virus recovered from the parental plasmid pQ14 in its growth characteristics (data not shown).
FIG. 1.

Cloning cassette vectors and plasmid constructions. (A) Schematic diagram showing the FCV genome organization and locations of silent nucleotide changes introduced into the FCV FL cDNA clone (pQ14) to generate a unique AvrII restriction site (underlined) in the beginning of the ORF3 sequence or a unique AflII restriction site (underlined) in the end of the ORF3 sequence to generate cloning cassette vectors pR6 and pRAFL, respectively. (B) Plasmid constructions containing engineered stop codons in the ORF3 sequence of FL clones: pF3Nterm (introduction of terminator codon, TAG, at nucleotide positions 7335 to 7337 [nt 19 to 21 of ORF3], pF3Nterm3 (introduction of three terminatorcodons, TAGTAATGA, at the nucleotide positions 7335 to 7343 [nt 19 to 27 of ORF3], and pF3Cterm2 (introduction of two terminator codons, TAATAG, at the nucleotide positions 7578 to 7583 [nt 261 to 267 of ORF3]). (C) Plasmid constructions containing engineered deletions in the ORF3 sequence: pR6delF3 (with nearly all of ORF3 deleted), the pF3N series (with sequentially larger N-terminal deletions), and the pF3C series (with sequentially larger C-terminal deletions). The plasmids were engineered as described in the text. The borders (nucleotide positions) of introduced deletions in ORF3 are given on the scale above the plasmid diagrams, and the corresponding amino acid deletions (Δ) in VP2 are indicated on the right.
To examine the effect of VP2 inactivation on FCV growth properties, we used two approaches to knock out the corresponding gene. First, by PCR mutagenesis and subsequent subcloning of mutagenized fragments into the pR6 vector, a stop codon (TAG) was introduced into the beginning of the ORF3 sequence by the T7336→A7336 replacement in the TTG codon of Leu7 (pF3Nterm) (Fig. 1B). Second, a construct was engineered in which the entire ORF3 was deleted, with the exception of the first six codons (pR6delF3) (Fig. 1C).
CRFK cells transfected with capped genomic-size RNA transcripts derived from pF3Nterm (in which synthesis of the VP2 protein was prevented by the introduction of a terminator codon in the beginning of the coding sequence) showed no signs of the development of virus-specific CPE (Fig. 2A). How ever, a few isolated IF-positive cells with a rounded shape comparable to that observed for individual CRFK cells infected with the wild-type virus were sometimes observed in these experiments (Fig. 2A). In addition, the cell morphology and IF staining (intracellular distribution of expressed capsid antigen) were reminiscent of those observed in experiments where cells were transfected with FL cDNA clones carrying capsid precursor cleavage site mutations in which replication occurred but the maturation and release of infectious particles were blocked at the level of proteolytic processing of the capsid precursor protein (39).
FIG. 2.


Analysis of replication of the ORF3 knockout mutants. (A to D) Immunofluorescent detection of expression of the FCV capsid protein in CRFK cells transfected with capped genomic RNA synthesized in vitro from pF3Nterm (A) and pF3Nterm3 (C) clones and the MVA-T7-infected CRFK cells transfected with their plasmid DNAs (B and D). CRFK cells were stained 20 h posttransfection with hyperimmune serum raised against purified FCV virions, and the binding of antibody was detected with a fluorescein isothiocyanate-conjugated anti-guinea pig serum. Photographs were taken at a magnification of ×400. (E) Immunoprecipitation analysis of FCV capsid protein expression in MVA-T7-infected CRFK cells transfected with ORF3 knockout clones. The S35 labeling and immunoprecipitation of the capsid protein were performed as described in the text. Lanes 2, 3, 4, and 5 are radiolabeled proteins immunoprecipitated from MVA-T7-infected CRFK cells transfected with pR6, pR6delF3, pF3Nterm, and pF3Nterm3, respectively. Lane 1 contains the radiolabeled capsid protein immunoprecipitated from FCV-infected CRFK cells as a control.
In order to increase the efficiency of expression from pF3Nterm, the MVA-T7-based recovery system for FCV (36) was utilized because of its higher virus yields compared to the RNA-based recovery system. Transfection of MVA-T7-infected cells with pF3Nterm plasmid DNA resulted in the appearance of isolated capsid antigen-positive cells that were morphologically similar to those in the RNA transfection (Fig. 2B), but the numbers (40 to 60 IF-positive cells on average per monolayer of ∼106 cells) were higher. The higher expression levels allowed an immunoprecipitation analysis that confirmed the synthesis of capsid protein (Fig. 2E, lane 4).
Passage of the medium collected from cells transfected with pF3Nterm in the MVA-T7 system characteristically did not result in the recovery of infectious virus; however, in one experiment, IF staining of the inoculated CRFK cells showed evidence of the presence of a slowly replicating virus. Replication of this virus led to the appearance of single infected cells spread throughout the monolayer, with their numbers increasing with each passage (data not shown). After four passages of the mutant virus, the virus RNA was purified and used as a template for RT-PCRs to amplify cDNA fragments overlapping the whole genome of the virus. Subsequent sequence analysis showed the existence of a single mutation in nucleotide position 7335 of the virus genome that corresponded to the conversion of the stop codon of pF3Nterm ORF3 to the glutamine codon: TAG→CAG (Table 3). Attempts to further propagate and amplify the mutant virus led to the selection of fast-growing lytic revertants. The presence of growing foci was readily detectable by IF at passage 5 and higher (data not shown). Sequencing of the virus RNA samples collected at passage 5 revealed an emerging heterogeneity of the virus ORF3 sequence (Table 3). Interestingly, it showed reversion at the amino acid level of the mutant ORF3 sequence to that of the wild type: the point mutation in position 7336 resulted in the replacement of a CAG (Gln) codon with a CTG (Leu) codon (Table 3). Conversion of this CAG codon to a CTG codon occurred consistently in a series of independent experiments involving passaging of the F3(CAG) mutant virus (data not shown).
TABLE 3.
Replication and reversion of the vF3Nterm mutant
| FCV FL cDNA clones and recovered viruses | ORF3 nucleotide sequence (codons 6-8)a | Encoded amino acid (codon 7) |
|---|---|---|
| pR6 | GGCTTGATT | Leu |
| pF3Nterm | GGCTAGATT | AMB |
| vF3Nterm (passage 4) | GGCCAGATT | Gln |
| vF3Nterm (passage 4, sample 1) | GGCCA/TGATT | Gln/Leu |
| vF3Nterm (passage 4, sample 2) | GGCCTGATT | Leu |
Comparison of the 5′-end sequences of ORF3 determined for the vF3Nterm mutant at passages 4 and 5. Passage 5 is represented by two virus samples collected independently from different wells of a multiwell plate at 24 h postinfection. The ORF3 codon corresponding to position 7 of the VP2 amino acid sequence is underlined.
To confirm that the virus genome could replicate in transfected CRFK cells in the absence of the VP2 protein, we introduced two more stop codons next to the stop codon in the ORF3 sequence of pF3Nterm (pF3Nterm3) (Fig. 1B), thus significantly reducing the probability of reversion of the mutant sequence to that of wild-type virus. IF analysis of CRFK cells transfected with RNA transcripts derived from the resulting plasmid, pF3Nterm3, or CRFK cells infected with MVA-T7 and transfected with the plasmid itself demonstrated the presence of cells expressing virus capsid protein (Fig. 2C and D, respectively). Furthermore, an IP analysis showed the synthesis of capsid protein in the transfected cells (Fig. 2E, lane 5). However, attempts to pass the virus to the next monolayer were unsuccessful, and revertants were not detected. These data again confirmed that interruption of VP2 expression allowed replication and synthesis of the subgenomic RNA, but infectious viral progeny could not be produced.
Transfection of CRFK cells with capped genomic-size RNA transcripts derived from pR6delF3 (in which nearly the entire ORF3 was deleted) or transfection of the MVA-T7-infected CRFK cells with the plasmid pR6delF3 did not result in virus recovery or detectable replication of the virus, as determined by IF staining or Northern blotting (data not shown). Furthermore, no evidence for synthesis of the capsid protein was observed in cells transfected with pR6delF3 (Fig. 2E, lane 3). The failure to detect virus recovery and capsid protein synthesis with the deleted ORF3 suggested that ORF3 itself contained nucleotide sequences essential for replication.
Effects of amino acid substitutions at position 7 of VP2 on virus viability.
The emergence of a revertant virus following the introduction of a single stop codon at amino acid position 7 suggested the presence of selective pressure to produce a wild-type VP2 protein with Leu in this position. To evaluate the effects of other amino acid substitutions in position 7 of VP2 on virus growth, we constructed a series of FL clones, pF3X7, where X represents Ala, Gly, Cys, Asp, Asn, Glu, Gln, Thr, Ser, or Lys. The engineered codons that were selected would require at least two nucleotide mutations to revert to the Leu codon (Table 4). Transfection of these plasmids into MVA-T7-infected cells resulted in the detection of FCV capsid antigen-positive cells (illustrated by IF analysis of cells transfected with pR6, pF3Ala7, pF3Asn7, pF3Asp7, and pF3Lys7 in Fig. 3A), indicating that synthesis of the capsid protein was not affected by the examined changes in the ORF3 and VP2 sequences. The synthesis of capsid protein was confirmed by immunoprecipitation with capsid-specific serum from cells transfected with each of the F3 position 7 mutant cDNA clones (Fig. 3B).
TABLE 4.
Effects of various amino acid substitutions at residue 7 of VP2 on virus recovery
| Amino acid | Introduced nucleotide changes (codon 7) | Virus recoverya | Observed nucleotide changes (codon 7) | Additional nucleotide changes |
|---|---|---|---|---|
| Ala | GCG | + | None | None |
| Asn | AAT | + | None | C7351→T (Thr12→Ile12) |
| Asp | GAT | − | NAb | NA |
| Thr | ACG | + | None | None |
| Ser | TCT | + | None | None |
| Lys | AAG | + | None | A7356→G (Thr14→Ala14) |
| Gly | GGT | (+) | TGT | None |
| Glu | GAG | − | NA | NA |
| Cys | TGT | + | None | None |
| Gln | CAG | (+) | CTG | None |
+, clones that yielded viable virus progeny; −, clones that did not yield viable virus progeny; (+), clones that produced virus with nucleotide changes in ORF3.
NA, not applicable.
FIG. 3.

Analysis of FCV capsid protein expression in MVA-T7-infected CRFK cells transfected with FL cDNA clones containing amino acid changes in position 7 of the VP2 amino acid sequence. (A) Fluorescence microscopy observation of the MVA-T7-infected CRFK cells transfected with pF3X7 mutants was conducted to detect replication of the virus. Recovery of infectious virus particles was confirmed by transfer of the transfected cell media to a new cell monolayer and by immunofluorescent staining of these cells 24 h postinoculation. Immunofluorescent staining was performed as described in the text, and photographs were taken at a magnification of ×100. The insets show photographs of single positive cells taken at a magnification of ×400. (B) Replication of the virus in the MVA-T7-infected CRFK cells transfected with pF3X7 mutants was verified by immunoprecipitation of the radiolabeled virus capsid protein from lysates of the transfected cells. Lanes 2 to 7 and 9 to 13 are radiolabeled proteins immunoprecipitated with the FCV virion-specific serum from the cells transfected with pF3Ser7, pF3Thr7, pF3Ala7, pF3Gly7, pF3Cys7, pF3Lys7, pF3Asn7, pF3Asp7, pF3Gln7, pF3Glu7, and pR6, respectively. The radiolabeled capsid protein immunoprecipitated from CRFK cells infected with wild-type FCV was included as a positive control in lane 1. Radiolabeled proteins from mock-transfected MVA-T7-infected CRFK cells were analyzed by immunoprecipitation with the capsid-specific serum as a control in lane 8.
The cell culture fluids from these transfections were transferred to fresh CRFK cell monolayers in order to determine whether infectious viral progeny were generated. Although all constructs had shown evidence of capsid expression in the original transfection, the substitution of the amino acid Asp or Glu at position 7 was lethal for virus recovery, as illustrated by the absence of CPE or IF-positive cells in passage 1 of the Asp7 clone (Fig. 3A). In contrast, viruses were readily recovered with the Ala7, Gly7, Cys7, Ser7, and Thr7 mutations, and they replicated similarly to the parental-type virus, infecting nearly the entire monolayer after 24 h (illustrated by Ala7 in Fig. 3A). Viruses recovered with the Gln and Asn mutations showed single isolated cells at 24 h, with a gradual emergence of discrete foci over time, as illustrated by the analysis of Asn7 in Fig. 3A. Only a few positive cells were observed in the CRFK cell monolayer inoculated with the VP2 Lys7 mutant (Fig. 3A); however, further passage of the culture fluids resulted in selection of a fast-growing virus (data not shown).
The genetic stability of the recovered viruses was examined at passage 3 (Table 4). RT-PCR sequence analysis of the purified viral RNA confirmed the presence of the introduced mutations in the recovered Ala, Cys, Ser, and Thr mutants. Sequence analysis of the initially slow-growing Gln mutant showed reversion to the wild-type sequence of VP2 at residue 7 (Leu); however, the CAG codon introduced into the FL cDNA clone was replaced with a CTG codon (TTG in the wild-type sequence). This change was identical to the codon conversion observed for virus derived from pF3term, and recovery of this mutant from pF3Gln7 was observed in two independent experiments. Three independent attempts to recover virus with a Gly7 substitution resulted in selection of mutants in which a GGT codon was converted into a TGT codon (Cys residue). Sequence analysis of the pF3Asn7- and pF3Lys7-derived viruses demonstrated the presence of the original mutations in the ORF3 sequences of their genomes. However, in addition to the introduced changes, their ORF3 sequences acquired single-nucleotide substitutions in other positions, resulting in amino acid changes: C7351→T7351 (Thr12→Ile12) for the Asn7 mutant and A7356→G7356 (Thr14→Ala14) for the Lys7 mutant (Table 4).
The growth characteristics of mutant viruses selected after three passages in CRFK cells were similar; infected at a multiplicity of infection of 0.01, they produced titers in a range of 1.4 × 107 to 1 × 108 in 34 h (data not shown).
N- and C-terminal deletions in VP2.
Capsid expression in the cells transfected with virus genomes carrying VP2 knockout genes (pF3Nterm and pF3Nterm3) suggested that VP2 might not be required for virus RNA replication and, in particular, for the synthesis of the subgenomic RNA that serves as the mRNA template for expression of the capsid precursor and VP2. Nevertheless, the absence of virus replication in R6delF3-transfected cells indicated that the ORF3 nucleotide sequence itself might contain signals essential for replication. To examine this possibility, two series of FL clone constructs, designated pF3N and pF3C, were engineered with 5′- and 3′-end sequence deletions, respectively, in ORF3 (Fig. 1C). The introduced deletions had a 30-nt step increase in size and did not interfere with translation of the remaining VP2. Analysis of cells transfected with plasmids pF3N(19-48), pF3N(19-78), pF3N(19-108), pF3N(19-138), and pF3N(19-168) by IF showed the presence of 10 to 50 capsid antigen-positive cells per well (106 cells) (Fig. 4A), and IP confirmed the expression of the VP1 protein in cells transfected with pF3N(19-48), pF3N(19-78), pF3N(19-108), pF3N(19-138), and pF3N(19-168) (Fig. 4B), thus providing indirect evidence of subgenomic RNA synthesis. Further deletions in the ORF3 sequence from the 5′ end, pF3N(19-198), pF3N(19-228), pF3N(19-258), and pF3N(19-288), resulted in no detectable capsid protein synthesis in IF and IP analyses (Fig. 4). Our attempts to detect RNA replication directly by Northern blot analysis failed (data not shown), likely due to the small number of positive cells in the monolayer.
FIG. 4.


5′-end deletion mutagenesis of the ORF3 sequence in the FCV FL cDNA clone and analysis of the virus capsid protein expression in the MVA-T7-infected CRFK cells transfected with the corresponding clones. (A) Fluorescence microscopy observation of the MVA-T7-infected CRFK cells transfected with pF3N(del) mutants 20 h posttransfection. Immunofluorescent staining was performed as described in the text, and photographs were taken at a magnification of ×100. The insets show photographs of single positive cells taken at a magnification of ×400. (B) Replication of the virus in the MVA-T7-infected CRFK cells transfected with pF3N(del) mutants was verified by immunoprecipitation of the radiolabeled virus capsid protein from the lysates of the transfected cells. Lanes 2 to 11 are radiolabeled proteins immunoprecipitated with the FCV virion-specific serum from the cells transfected with pF3N(19-48), pF3N(19-78), pF3N(19-108), pF3N(19-138), pF3N(19-168), pF3N(19-198), pF3N(19-228), pF3N(19-258), pF3N(19-288), and pR6, respectively. The radiolabeled capsid protein immunoprecipitated from CRFK cells infected with wild-type FCV was included as a positive control in lane 12. Radiolabeled proteins precipitated from mock-transfected MVA-T7-infected CRFK cells were included as a control in lane 1.
Replication of the virus in cells transfected with pF3N(19-48), pF3N(19-78), pF3N(19-108), pF3N(19-138), and pF3N(19-168) did not result in the production of infectious virus particles. Consequently, we did not observe virus CPE development or virus-specific IF staining of monolayers of cells inoculated with the medium collected from wells with the transfected cells (data not shown).
Transfection of the plasmids with 3′-end deletions of the ORF3 sequence, pF3C(169-288), pF3C(199-288), and pF3C(229-288), into MVA-T7-infected CRFK cells did not result in virus replication and expression of the capsid antigen (data not shown). However, ∼3 to 10 positively stained cells were observed in experiments with transfection of pF3C(259-288) (data not shown).
Transfection of MVA-T7-infected CRFK cells with pF3Cterm2, in which two stop codons were introduced at nucleotide positions 7578 to 7583 (nt 261 to 267 of ORF3), resulted in detection of antigen-positive cells. Similar to the finding with the pF3Nterm3 plasmid, the observed replication did not result in production of infectious virus (data not shown). Synthesis of the VP1 protein in cells transfected with pF3Cterm2 and pF3C(259-288) was confirmed by IP analysis using anticapsid serum (Fig. 5).
FIG. 5.

3′-end deletion mutagenesis of the ORF3 sequence in the FCV FL cDNA clone and analysis of virus capsid protein expression in the MVA-T7-infected CRFK cells transfected with the indicated clones. Replication of the virus in the MVA-T7-infected CRFK cells transfected with pF3Cterm2 and pF3C(del) mutants was verified by IP of the radiolabeled virus capsid protein from the lysates of the transfected cells. Lanes 2, 3, and 5 to 8 are radiolabeled proteins immunoprecipitated with the FCV virion-specific serum from the cells transfected with pR6, pF3Cterm2, pF3C(169-288), pF3C(199-288), pF3C(229-288), and pF3C(259-288), respectively. The radiolabeled capsid protein immunoprecipitated from CRFK cells infected with wild-type FCV was included as a positive control in lane 1. Radiolabeled proteins precipitated from mock-transfected MVA-T7-infected CRFK cells were included as a control in lane 4.
VP2 trans-complementation.
The failure to recover virus from viral genomes encoding modified VP2 proteins suggested a possible role for VP2 in the formation of infectious particles. To examine whether this function of the VP2 protein could be restored by provision of the protein in trans, we subcloned ORF3 sequence into the pCI vector (Promega). In the resulting plasmid, pCiF3, the expression of the VP2 protein could be driven by either cytomegalovirus or T7 RNA polymerase promoters located upstream of the ORF3 sequence. The highest level of VP2 expression was achieved when pCiF3 was transfected into MVA-T7-infected CRFK cells (Fig. 6).
FIG. 6.
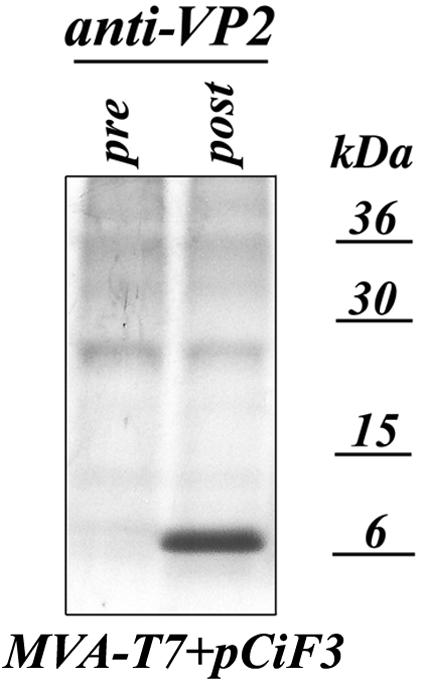
Expression of the VP2 protein from pCiF3 vector in MVA-T7-transfected CRFK cells. The CRFK cells were infected with MVA-T7 at a multiplicity of infection of 3 and transfected with pCiF3 2 h later. Proteins were metabolically labeled with [S35]methionine as described in the text, and radiolabeled VP2 was immunoprecipitated from cell lysates with VP2-specific postimmunization serum (38), subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and visualized with autoradiography. As a negative control, the radiolabeled proteins were immunoprecipitated from the same cell lysates using preimmunization VP2 serum.
When pCiF3 was cotransfected into MVA-T7-infected CRFK cells with FL cDNA clones encoding N-terminally truncated VP2, synthesis of the capsid protein was detected only for plasmids pF3N(19-48), pF3N(19-78), pF3N(19-108), pF3N(19-138), and pF3N(19-168) and not for the plasmids with N-terminal deletions extending beyond nt 168 of ORF3 (data not shown). This observation was consistent with those described above when the plasmids were transfected without pCiF3. Unexpectedly, the number of cells expressing capsid antigen was higher than that for cells transfected only with FL clones (data not shown). For example, the numbers were doubled in the case of pF3N(19-48) and pF3N(19-108). Northern blot analysis of the total RNAs isolated from transfected cells showed a correlation between synthesis of the virus subgenomic RNA and detection of the virus capsid protein by IF staining (Fig. 7). The 2.3- to 2.5-kb subgenomic RNA bands were detected only in cells transfected with pF3N(19-48), pF3N(19-78), pF3N(19-108), pF3N(19-138), and pF3N(19-168) (Fig. 7).
FIG. 7.

Subgenomic RNA synthesis in cells cotransfected with pCiF3 and FL cDNA clones containing sequential 5′-end deletions of the ORF3 sequence. Lanes 2 to 12, samples of total RNA purified from CRFK cells infected with MVA-T7 and cotransfected with pCiF3 and pF3N(19-48), pF3N(19-78), pF3N(19-108), pF3N(19-138), pF3N(19-168), pF3N(19-198), pF3N(19-228), pF3N(19-258), pF3N(19-288), pR6delF3, and pR6, respectively, were subjected to Northern blot analysis using an antisense RNA probe specific for FCV ORF2 (10). Lane 1, total RNA isolated from MVA-T7-infected cells transfected with pCiF3 only. Lane 13, polyadenylated RNA purified from FCV-infected CRFK cells using PolyAT tract system 1000 (Promega).
To test whether virus replication in transfected cells led to the production of infectious particles, cells were subjected three times to freeze-thawing and the culture fluid was transferred to a new CRFK monolayer. The cells were incubated for 1 to 2 h at 37°C and then thoroughly washed and incubated for an additional 12 h. Virus replication was examined by IF staining to detect capsid protein expression. Inoculation of cells with culture fluids from wells with cells transfected with pF3N(19-48), pF3N(19-78), pF3N(19-108), pF3N(19-138), or pF3N(19-168) resulted in the detection of capsid antigen-positive cells in the new monolayer; however, CPE did not develop and no plaques were observed (Fig. 8). This infectious material could be efficiently neutralized by the addition of 100-fold-diluted capsid-specific serum (data not shown). Attempts to pass the infectious material one more time to the next monolayer were unsuccessful, and only provision of VP2 in trans using an MVA-T7/pCiF3 system allowed the serial passage of the replication-competent particles. The number of fluorescence-positive cells (expressing virus capsid antigen) did not increase with serial passage, remaining between 1,500 and 2,000 cells per well. These data suggested that the provision of VP2 in trans rescued the generation of infectious particles.
FIG. 8.

trans-complementation of the FCV ORF3 5′-end deletion mutants. Limited virus replication was observed in CRFK cells inoculated with culture fluids from MVA-T7-infected cells cotransfected with pF3N(19-48), pF3N(19-78), pF3N(19-108), pF3N(19-138), pF3N(19-168), and pCiF3. Expression of the virus capsid antigen was observed using capsid-specific immunofluorescent staining 13 to 14 h postinoculation.
DISCUSSION
Calicivirus virions contain a minor structural VP2 protein that has no apparent homology with known viral or cellular proteins. The goal of this study was to address its function using a reverse-genetics approach. We demonstrate here that the VP2 protein is essential for the production of infectious FCV virions. Our data are consistent with previous suggestions (7, 27, 38, 43) that VP2 is involved in the maturation and assembly of calicivirus particles.
Our deletional analysis of the VP2 domains responsible for the production of infectious FCV particles mapped essential domains to both the N- and C-terminal parts of the VP2 protein. However, a single-amino-acid substitution in residue 7 of the VP2 sequence could also affect its function. Replacement of the wild-type leucine at position 7 of the VP2 sequence with negatively charged glutamic and aspartic acid residues blocked the production of infectious particles, while a glutamine mutation resulted in recovery of a slow-growing mutant virus. This mutant virus reverted with serial passage to a fast-growing virus bearing the wild-type leucine at position 7, providing additional evidence for the importance of the N-terminal sequence of VP2 and suggesting a strong selective pressure to produce intact VP2. Early passages of certain other mutants resulted in a rapid selection of VP2 sequences that acquired additional (probably compensatory) changes in their N-terminal parts, which also showed the strong selective pressure for a functional VP2. Taken together, the mutational analysis showed that an intact VP2 is critical for the production of infectious viral progeny. It is also likely that the VP2 amino acid sequence cannot tolerate substitutions that disrupt the overall conformation of the protein.
The mechanism by which the VP2 protein mediates virion production, as well as the reason for its incorporation into calicivirus particles, is unclear. The domain of the NV VP2 involved in the protein-protein interactions with VP1 was mapped to an internal region located between aa 108 and 152 (8) that has been described as relatively conserved among the VP2 proteins of different caliciviruses (8). Interestingly, this region corresponds to aa 60 to 85 of FCV VP2 and shows 41% amino acid similarity to the VP2 protein of NV. Deletion of this region in the FCV VP2 protein [clones pF3N(19-198), pF3N(19-228), pF3N(19-258), and pF3N(19-288)] dramatically affects the production of infectious virus, which is consistent with the proposal by Glass et al. (8) that this region might play a critical role in the interaction between VP1 and VP2. However, the presence of this sequence alone is not sufficient for the formation of infectious FCV particles, because our data indicate that VP2 domains responsible for the assembly of the infectious FCV particles might be located in the N- and C-terminal parts of the FCV VP2 protein as well. Supporting that idea, truncation of the VP2 sequence at the N terminus [clones pF3N(19-48), pF3N(19-78), pF3N(19-108), pF3N(19-138), and pF3N(19-168)], as well as at the C terminus [clone pF3Cterm2], prevented production of infectious virus particles, suggesting that the N- and C-terminal modifications of the VP2 sequence were likely to affect the folding of this protein and its function in particle assembly. In addition to interactions with VP1, protein-protein interactions of the highly basic VP2 with the second minor protein found inside calicivirus virions, VPg, cannot be excluded. The primary sequences of calicivirus VPgs have common structural features, such as the presence of clusters of positively and negatively charged amino acids (6, 40), and the polar organization of these amino acid clusters might promote protein-protein interactions inside the virion. The interactions of any or all of the three virion proteins (VP1, VP2, and VPg) with genomic RNA might also influence the role of VP2 in the production of infectious virions. The elucidation of the coordinated events involved in the maturation of calicivirus particles and analysis of the role of VP2 may provide insight into a possibly unique pathway used by the caliciviruses to package (or release) their VPg-linked RNA genomes.
Our mutagenesis studies showed that the ORF3 nucleotide sequence itself may contain structural elements involved in replication of the viral RNA. The location of the ORF3 sequence at the extreme 3′ end of the genome indicates that it could overlap sequences involved in the formation of a 3′-end RNA replication signal. The 3′-end sequence motifs regulating virus replication have been identified for a number of animal and plant viruses with positive-strand RNA genomes (4, 5, 12, 15, 16, 21, 24, 32, 44). These motifs have been described as parts of conserved RNA secondary and tertiary structural elements, such as stem-loops and pseudoknots, that participate directly or through interactions with viral or cellular proteins in the promotion of viral RNA synthesis. Recently, it was suggested that the predicted 47-nt stem-loop structure of the NV 3′-end NTR was involved in the formation of stable RNA-protein complexes (11). In in vitro UV cross-linking assays, the corresponding RNA was specifically recognized by at least 10 proteins from HeLa cells, including La and polypyrimidine tract-binding proteins, which are known to be involved in the replication of several viruses with positive-strand RNA genomes (11, 18). The presence of the stem-loop structures in the 3′-end NTR of the calicivirus genome was also predicted for the genomes of rabbit hemorrhagic disease virus, San Miguel sea lion virus, and FCV (34). According to this prediction, the 3′-end NTR of the FCV genome could have up to three possible duplexes incorporated in the single stem-loop structure (34). Our preliminary analysis of the secondary structure of the 3′-end part of the FCV RNA genome showed the presence of additional stem-loop elements located within the 3′ end of the ORF3 sequence (data not shown). Studies to elucidate the functional importance of these elements in virus replication are in progress.
It is likely that the ORF3 RNA sequence functions in cis, because the negative effect of its entire or partial removal was not compensated for in trans-complementation experiments. As evidenced by Northern blot analysis of subgenomic RNA synthesis in cells transfected with FL cDNA clones with extended ORF3 sequence lesions, expression of this ORF in trans did not result in restoration of virus replication. In contrast, it is likely that the protein encoded by ORF3, VP2, can function in trans because production of infectious particles was restored when intact VP2 protein was synthesized separately in the same cells.
The replication of the FCV genome in the absence of productive virus amplification creates a new replicon system that is different from that previously reported (41). Furthermore, this is the first demonstration of trans-complementation of a calicivirus genome defective in the synthesis of one of its proteins. These new tools for the study of calicivirus RNA replication and protein function should help elucidate the molecular basis for productive virus infection, which could lead to the identification of unique targets for antiviral drug development for this important group of pathogens.
Acknowledgments
We thank Tanaji Mitra for his dedicated technical support. We extend our appreciation to Albert Z. Kapikian and Robert H. Purcell, LID, NIAID, NIH, for continuing support.
REFERENCES
- 1.Bertolotti-Ciarlet, A., S. E. Crawford, A. M. Hutson, and M. K. Estes. 2003. The 3′ end of Norwalk virus mRNA contains determinants that regulate the expression and stability of the viral capsid protein VP1: a novel function for the VP2 protein. J. Virol. 77:11603-11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burroughs, J. N., and F. Brown. 1978. Presence of a covalently linked protein on calicivirus RNA. J. Gen. Virol. 41:443-446. [DOI] [PubMed] [Google Scholar]
- 3.Carter, M. J., I. D. Milton, P. C. Turner, J. Meanger, M. Bennett, and R. M. Gaskell. 1992. Identification and sequence determination of the capsid protein gene of feline calicivirus. Arch. Virol. 122:223-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, C. J., M. D. Kuo, L. J. Chien, S. L. Hsu, Y. M. Wang, and J. H. Lin. 1997. RNA-protein interactions: involvement of NS3, NS5, and 3′ noncoding regions of Japanese encephalitis virus genomic RNA. J. Virol. 71:3466-3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deiman, B. A., R. M. Kortlever, and C. W. Pleij. 1997. The role of the pseudoknot at the 3′ end of turnip yellow mosaic virus RNA in minus-strand synthesis by the viral RNA-dependent RNA polymerase. J. Virol. 71:5990-5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunham, D. M., X. Jiang, T. Berke, A. W. Smith, and D. O. Matson. 1998. Genomic mapping of a calicivirus VPg. Arch. Virol. 143:2421-2430. [DOI] [PubMed] [Google Scholar]
- 7.Glass, P. J., L. J. White, J. M. Ball, I. Leparc-Goffart, M. E. Hardy, and M. K. Estes. 2000. Norwalk virus open reading frame 3 encodes a minor structural protein. J. Virol. 74:6581-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glass, P. J., C. Q. Zeng, and M. K. Estes. 2003. Two nonoverlapping domains on the Norwalk virus open reading frame 3 (ORF3) protein are involved in the formation of the phosphorylated 35K protein and in ORF3-capsid protein interactions. J. Virol. 77:3569-3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green, K. Y., T. Ando, M. S. Balayan, I. N. Clarke, M. K. Estes, D. O. Matson, S. Nakata, J. D. Neill, M. J. Studdert, and H.-J. Thiel. 2000. Caliciviridae, p. 725-735. In M. H. V. Regenmortel, C. M. Fauquet, D. H. L. Bishop, E. B. Carsten, M. K. Estes, S. M. Lemon, J. Maniloff, M. A. Mayo, D. J. McGeoch, C. R. Pringle, and R. B. Wickner (ed.), Virus taxonomy: the classification and nomenclature of viruses. The seventh report of the International Committee on Taxonomy of Viruses. Springer-Verlag, Vienna, Austria.
- 10.Green, K. Y., A. Mory, M. H. Fogg, A. Weisberg, G. Belliot, M. Wagner, T. Mitra, E. Ehrenfeld, C. E. Cameron, and S. V. Sosnovtsev. 2002. Isolation of enzymatically active replication complexes from feline calicivirus-infected cells. J. Virol. 76:8582-8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutierrez-Escolano, A. L., M. Vazquez-Ochoa, J. Escobar-Herrera, and J. Hernandez-Acosta. 2003. La, PTB, and PAB proteins bind to the 3′ untranslated region of Norwalk virus genomic RNA. Biochem. Biophys. Res. Commun. 311:759-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris, K. S., W. Xiang, L. Alexander, W. S. Lane, A. V. Paul, and E. Wimmer. 1994. Interaction of poliovirus polypeptide 3CDpro with the 5′ and 3′ termini of the poliovirus genome. Identification of viral and cellular cofactors needed for efficient binding. J. Biol. Chem. 269:27004-27014. [PubMed] [Google Scholar]
- 13.Herbert, T. P., I. Brierley, and T. D. Brown. 1996. Detection of the ORF3 polypeptide of feline calicivirus in infected cells and evidence for its expression from a single, functionally bicistronic, subgenomic mRNA. J. Gen. Virol. 77:123-127. [DOI] [PubMed] [Google Scholar]
- 14.Herbert, T. P., I. Brierley, and T. D. Brown. 1997. Identification of a protein linked to the genomic and subgenomic mRNAs of feline calicivirus and its role in translation. J. Gen. Virol. 78:1033-1040. [DOI] [PubMed] [Google Scholar]
- 15.Isken, O., C. W. Grassmann, R. T. Sarisky, M. Kann, S. Zhang, F. Grosse, P. N. Kao, and S. E. Behrens. 2003. Members of the NF90/NFAR protein group are involved in the life cycle of a positive-strand RNA virus. EMBO J. 22:5655-5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khromykh, A. A., N. Kondratieva, J. Y. Sgro, A. Palmenberg, and E. G. Westaway. 2003. Significance in replication of the terminal nucleotides of the flavivirus genome. J. Virol. 77:10623-10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Konig, M., H. J. Thiel, and G. Meyers. 1998. Detection of viral proteins after infection of cultured hepatocytes with rabbit hemorrhagic disease virus. J. Virol. 72:4492-4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai, M. M. 1998. Cellular factors in the transcription and replication of viral RNA genomes: a parallel to DNA-dependent RNA transcription. Virology 244:1-12. [DOI] [PubMed] [Google Scholar]
- 19.Laurent, S., J. F. Vautherot, M. F. Madelaine, G. Le Gall, and D. Rasschaert. 1994. Recombinant rabbit hemorrhagic disease virus capsid protein expressed in baculovirus self-assembles into virus-like particles and induces protection. J. Virol. 68:6794-6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leite, J. P., T. Ando, J. S. Noel, B. Jiang, C. D. Humphrey, J. F. Lew, K. Y. Green, R. I. Glass, and S. S. Monroe. 1996. Characterization of Toronto virus capsid protein expressed in baculovirus. Arch. Virol. 141:865-875. [DOI] [PubMed] [Google Scholar]
- 21.Mellits, K. H., J. M. Meredith, J. B. Rohll, D. J. Evans, and J. W. Almond. 1998. Binding of a cellular factor to the 3′ untranslated region of the RNA genomes of entero- and rhinoviruses plays a role in virus replication. J. Gen. Virol. 79:1715-1723. [DOI] [PubMed] [Google Scholar]
- 22.Meyers, G. 2003. Translation of the minor capsid protein of a calicivirus is initiated by a novel termination-dependent reinitiation mechanism. J. Biol. Chem. 278:34051-34060. [DOI] [PubMed] [Google Scholar]
- 23.Meyers, G., C. Wirblich, and H. J. Thiel. 1991. Genomic and subgenomic RNAs of rabbit hemorrhagic disease virus are both protein-linked and packaged into particles. Virology 184:677-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mirmomeni, M. H., P. J. Hughes, and G. Stanway. 1997. An RNA tertiary structure in the 3′ untranslated region of enteroviruses is necessary for efficient replication. J. Virol. 71:2363-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neill, J. D. 2002. The subgenomic RNA of feline calicivirus is packaged into viral particles during infection. Virus Res. 87:89-93. [DOI] [PubMed] [Google Scholar]
- 26.Neill, J. D., I. M. Reardon, and R. L. Heinrikson. 1991. Nucleotide sequence and expression of the capsid protein gene of feline calicivirus. J. Virol. 65:5440-5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oehmig, A., M. Buttner, F. Weiland, W. Werz, K. Bergemann, and E. Pfaff. 2003. Identification of a calicivirus isolate of unknown origin. J. Gen. Virol. 84:2837-2845. [DOI] [PubMed] [Google Scholar]
- 28.Parra, F., J. A. Boga, M. S. Marin, and R. Casais. 1993. The amino terminal sequence of VP60 from rabbit hemorrhagic disease virus supports its putative subgenomic origin. Virus Res. 27:219-228. [DOI] [PubMed] [Google Scholar]
- 29.Peterson, J. E., and M. J. Studdert. 1970. Feline picornavirus. Structure of the virus and electron microscopic observations on infected cell cultures. Arch. Gesamte Virusforsch. 32:249-260. [PubMed] [Google Scholar]
- 30.Prasad, B. V., M. E. Hardy, T. Dokland, J. Bella, M. G. Rossmann, and M. K. Estes. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287-290. [DOI] [PubMed] [Google Scholar]
- 31.Prasad, B. V., D. O. Matson, and A. W. Smith. 1994. Three-dimensional structure of calicivirus. J. Mol. Biol. 240:256-264. [DOI] [PubMed] [Google Scholar]
- 32.Rohll, J. B., N. Percy, R. Ley, D. J. Evans, J. W. Almond, and W. S. Barclay. 1994. The 5′-untranslated regions of picornavirus RNAs contain independent functional domains essential for RNA replication and translation. J. Virol. 68:4384-4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 34.Seal, B. S., J. D. Neill, and J. F. Ridpath. 1994. Predicted stem-loop structures and variation in nucleotide sequence of 3′ noncoding regions among animal calicivirus genomes. Virus Genes 8:243-247. [DOI] [PubMed] [Google Scholar]
- 35.Sosnovtsev, S., and K. Y. Green. 1995. RNA transcripts derived from a cloned full-length copy of the feline calicivirus genome do not require VPg for infectivity. Virology 210:383-390. [DOI] [PubMed] [Google Scholar]
- 36.Sosnovtsev, S., S. Sosnovtseva, and K. Y. Green. 1996. Recovery of feline calicivirus from plasmid DNA containing a full-length copy of the genome, p. 125-130. In D. Chasey, R. M. Gaskell, and I. N. Clarke (ed.), The 1st International Symposium on Caliciviruses. European Society for Veterinary Virology and Central Veterinary Laboratory, Reading, United Kingdom.
- 37.Sosnovtsev, S. V., M. Garfield, and K. Y. Green. 2002. Processing map and essential cleavage sites of the nonstructural polyprotein encoded by ORF1 of the feline calicivirus genome. J. Virol. 76:7060-7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sosnovtsev, S. V., and K. Y. Green. 2000. Identification and genomic mapping of the ORF3 and VPg proteins in feline calicivirus virions. Virology 277:193-203. [DOI] [PubMed] [Google Scholar]
- 39.Sosnovtsev, S. V., S. A. Sosnovtseva, and K. Y. Green. 1998. Cleavage of the feline calicivirus capsid precursor is mediated by a virus-encoded proteinase. J. Virol. 72:3051-3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sosnovtseva, S. A., S. V. Sosnovtsev, and K. Y. Green. 1999. Mapping of the feline calicivirus proteinase responsible for autocatalytic processing of the nonstructural polyprotein and identification of a stable proteinase-polymerase precursor protein. J. Virol. 73:6626-6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thumfart, J. O., and G. Meyers. 2002. Feline calicivirus: recovery of wild-type and recombinant viruses after transfection of cRNA or cDNA constructs. J. Virol. 76:6398-6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tohya, Y., H. Shinchi, Y. Matsuura, K. Maeda, S. Ishiguro, M. Mochizuki, and T. Sugimura. 1999. Analysis of the N-terminal polypeptide of the capsid precursor protein and the ORF3 product of feline calicivirus. J. Vet. Med. Sci. 61:1043-1047. [DOI] [PubMed] [Google Scholar]
- 43.Wirblich, C., H. J. Thiel, and G. Meyers. 1996. Genetic map of the calicivirus rabbit hemorrhagic disease virus as deduced from in vitro translation studies. J. Virol. 70:7974-7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu, H., C. W. Grassmann, and S. E. Behrens. 1999. Sequence and structural elements at the 3′ terminus of bovine viral diarrhea virus genomic RNA: functional role during RNA replication. J. Virol. 73:3638-3648. [DOI] [PMC free article] [PubMed] [Google Scholar]