

Abstract
Pooled genetic screens represent a powerful approach to identify vulnerabilities in cancer. Here we used pooled CRISPR/Cas9-based approaches to identify vulnerabilities associated with telomerase reverse transcriptase (TERT) promoter mutations (TPMs) found in >80% of glioblastomas. We first developed a platform to detect perturbations that cause long-term growth defects in a TPM-mutated glioblastoma cell line. However, we could not detect dependencies on either TERT itself or on an E-twenty six transcription (ETS) factor known to activate TPMs. To explore this finding, we cataloged TPM status for 441 cell lines and correlated this with genome-wide screening data. We found that TPM status was not associated with differential dependency on TERT, but that E-twenty six (ETS) transcription factors represent key dependencies in both TPM+ and TPM− lines. Further, we found that TPMs are associated with expression of gene programs regulated by a wide array of ETS-factors in both cell lines and primary glioblastoma tissues. This work contributes a unique TPM cell line reagent, establishes TPM status for many deeply-profiled cell lines, and catalogs TPM-associated vulnerabilities. The results highlight challenges in executing genetic screens to detect TPM-specific vulnerabilities, and suggest redundancy in the genetic network that regulates TPM function with therapeutic implications.
Introduction
Glioblastomas (GBM) are the most common primary malignant brain tumor, with a median overall survival of less than 2 years (1). Telomerase reverse transcriptase (TERT) gene promoter mutations are the most frequent somatic genetic alterations in GBMs (2,3). TERT promoter mutations (TPMs) are early somatic mutations (4,5) that occur in >80% of glioblastomas and in ~23% of all cancers (6–9). The two most frequent TPMs are heterozygous somatic point mutations that cause C>T transitions at 124 and 146 base pairs (bp), respectively, upstream of the TERT ATG start site (7). These are commonly referred to as C228T and C250T, respectively, based on their genomic coordinates.
TPMs function by reactivating TERT expression, which immortalizes GBM cells by facilitating indefinite telomere maintenance (10,11). Both C228T and C250T create a neomorphic 11-bp E-twenty six (ETS) transcription factor binding site which aberrantly recruits ETS-factors, including the transcription factor GA binding protein (GABP), to upregulate TERT expression (10).
Given their frequency and necessity for tumor cell immortalization, recent interest has focused on identifying methods to therapeutically target TPMs (2,3,12). Efforts to therapeutically target genetic alterations such as EGFR amplifications found in GBM have failed to extend patient survival (13–15), which may be due to intratumoral heterogeneity of those targets in GBM (16). However, TPMs are early clonal alterations found in GBM precursor cells that selectively sweep through the tumor cell population, suggesting that targeting TPMs could overcome the limitations seen with other targeted therapy approaches (4,5). Targeting telomerase itself is one attractive therapeutic concept for TPM+ GBM (17). Work in preclinical TPM+ GBM models suggests that targeting telomerase may be effective in low-volume disease states, but that resistance caused by tumor evolution may be a challenge (12). Further, clinical trials of anti-telomerase therapies such as imetelstat have so far shown limited efficacy in solid tumors (18), and considerable toxicities (19). This experience raises concerns that anti-telomerase therapies will be limited in their therapeutic window because normal stem cells rely heavily on telomerase function. Another approach under investigation is disruption of factors that regulate the mutant TERT promoter. For instance, disrupting the specific GABP isoform that regulates TPMs reverses the unlimited replication potential of TPM+ cell lines in the long-term (weeks to months) and sensitizes GBM cells to DNA damage in the short-term (days) (2,20). Unfortunately, therapeutically targeting transcription factors has historically presented a significant challenge (21).
The challenges with targeting telomerase itself or GABP inspired the current work to identify additional vulnerabilities associated with TPMs that could be therapeutically targeted. We generated a cell line to read out perturbations that specifically disrupt the ability for TPMs to drive TERT expression through a pooled genetic screen. We then used computational methods to conduct comparative analyses between the dependency and expression data of >400 cell lines and nearly 500 primary GBM tumors with known TPM status. Our results have important implications for therapeutic vulnerabilities in TPM+ GBM, and our experience provides guidance for future efforts to examine and target TPMs.
Results
Construction of a TPM+ cell line with inducible TERT
To effectively therapeutically target TPMs, a given perturbation must reverse the selective advantages conferred by TPMs to tumors (ie. reactivation of TERT expression). So, to hone in on perturbations specific to TPMs, we sought to develop a cell line with inducible exogenous TERT to facilitate a counter-screening methodology that could be carried out over long-term timelines (Figure 1a). For this, we selected the T98G GBM cell line since it contains a TPM and has previously been shown to be dependent on the TPMs for long-term growth (2,12,20,22). We found that T98G is homozygous for a C250T TPM (Figure 1b). TetON expression was detected by qPCR (Figure 1c). We next introduced an inducible cassette to express a codon-optimized TERT. Codon optimization was done to protect this exogenous TERT construct from CRISPR/Cas9 targeting directed at specific sequences in endogenous TERT (Figure 1d). To enable robust detection of exogenous TERT, N-terminal HA and Flag tags were engineered into the open reading frame (Supplementary Table 1). Notably, while N-terminal tagging may be expected to disrupt TERT function to some extent, N-terminal Flag tags of TERT have been shown to support near-wild-type telomerase catalytic activity (23). After selection, a stable cell pool was generated. qPCR revealed robust induction of TERT mRNA in the presence of doxycycline (dox), and a small amount of TERT mRNA in the absence of dox (Figure 1e). However, immunoblots for HA and Flag revealed apparent expression of TERT in the cell pool in both the absence and presence of dox (Figure 1f). This finding raises the possibility that in the cell pool, cells existed with leaky expression of TERT in the absence of dox.
Figure 1. Generation of TPM+ GBM cell line with inducible, exogenous TERT expression.
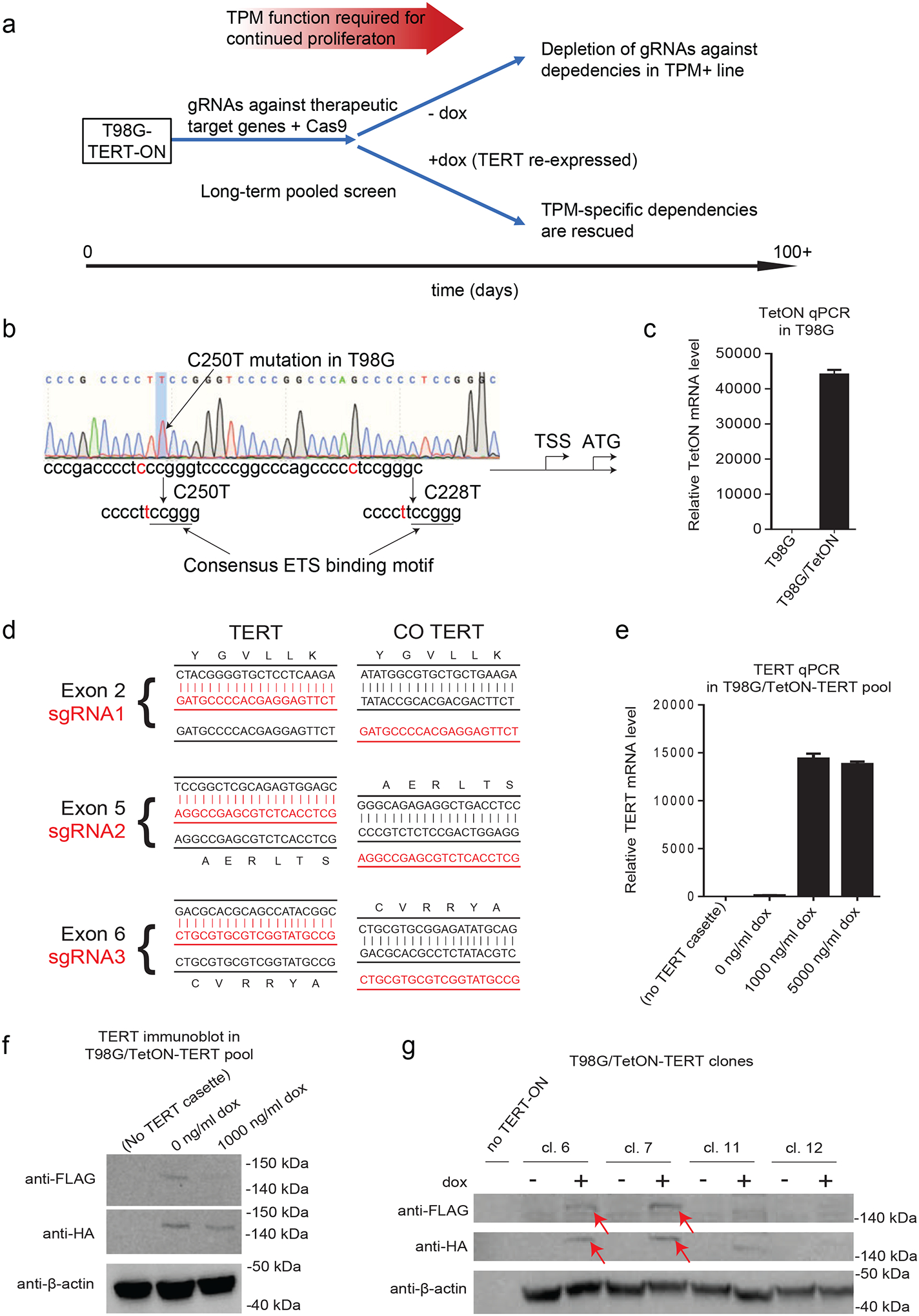
(A) Schema of genome-wide screen to identify dependencies associated with TPMs. gRNAs and Cas9 are introduced to cells at day 0 using lentivirus vectors which are then continuously passaged in the absence of dox. At a predefined timepoint, cells are divided into two separate pools. One pool is cultured in the absence of dox allowing for gRNAs to be depleted from culture. To distinguish TPM specific from non-specific hits, the other pool is induced with dox to re-express TERT. TERT re-expression will rescue cells containing gRNAs that disrupt the TPMs from replicative senescence, but gRNAs that nonspecifically disrupt cell growth will still be depleted.
(B) Sanger sequencing showing C250T mutation in T98G cell line. TSS, transcription start site; ATG, ATG start codon.
(C) TET-ON cassette mRNA expression in parental T98G cells and T98G with TetON cassette (T98G/TetON). Data are for mean +/− S.E.M. for n=3 technical qPCR replicates.
(D) Sequence alignment between the endogenous TERT sequences and the codon optimized sequence for each sgRNA. Codon optimized TERT was not targeted by sgRNAs. CO = codon optimized.
(E) TERT mRNA expression as assessed by qPCR for parental T98G cells (no TERT-ON cassette), and stable T98G/TetON-TERT pool treated with 0–5000 ng/ml dox. Data are for mean +/− S.E.M. for n=3 technical qPCR replicates.
(F) Immunoblot for HA and FLAG showing Tag-TERT expression in stable cell pool. Lane 1 is the parental T98G cell line. Lanes 2–3 are T98G/TetON-TERT cells with and without 1000 ng/ml dox added.
(G) Immunoblot for HA and FLAG showing Tag-TERT expression in subclones, either with (+) or without (−) addition of 1000 ng/ml dox.
Next, we isolated stable TERT-inducible clones. We sought to achieve a low expression of TERT to mimic physiologic low levels. Four clones were obtained and expanded. HA and FLAG detected induction of a band after dox treatment in clones C6 and C7 (Figure 1g). These results confirm the construction of a TPM+ GBM cell line with dox-inducible expression of an epitope-tagged, codon-optimized TERT at physiologic levels. Clone C7 (T98G-TERT-ON) was chosen and used for further experiments described below.
Mini pooled CRISPR/Cas9 screen for TPM-associated vulnerabilities.
We carried out a mini pooled CRISPR screen on T98G-TERT-ON as a preliminary experiment to gauge the feasibility of a genome-wide screen. We tested if TERT knockout decreased long-term growth of T98G as described previously (1) in a pooled experiment. We also included gRNAs against the GABPB1L subunit of GABP which has been shown to be required for GABP to drive TPM function in T98G (20). Since TPMs increase replicative immortality over a relatively long time-scale, we performed a long-term growth assay to assess the ability for cells to avoid senescence (Figure 2a). Cells were transduced with lentiviruses to deliver Cas9 and gRNAs against TERT (3 unique gRNAs) or GABPB1L (2 unique gRNAs), or control gRNAs (2 unique gRNAs targeting nonfunctional AAVS1). We analyzed cells transduced with each single gRNA in parallel. Additionally, we transduced a parallel pool of cells with equal amounts of each of these seven gRNAs (n=2 replicate pools). Since previous literature indicates that TERT disruption leads to decreased cell viability at late time points of >100 days (2), the experiment was carried out over a long time-scale of 134 days. The culture was split into cultures with and without dox at day 75.
Figure 2. Mini pooled screen to identify genetic dependencies that can be rescued by exogenous TERT expression.
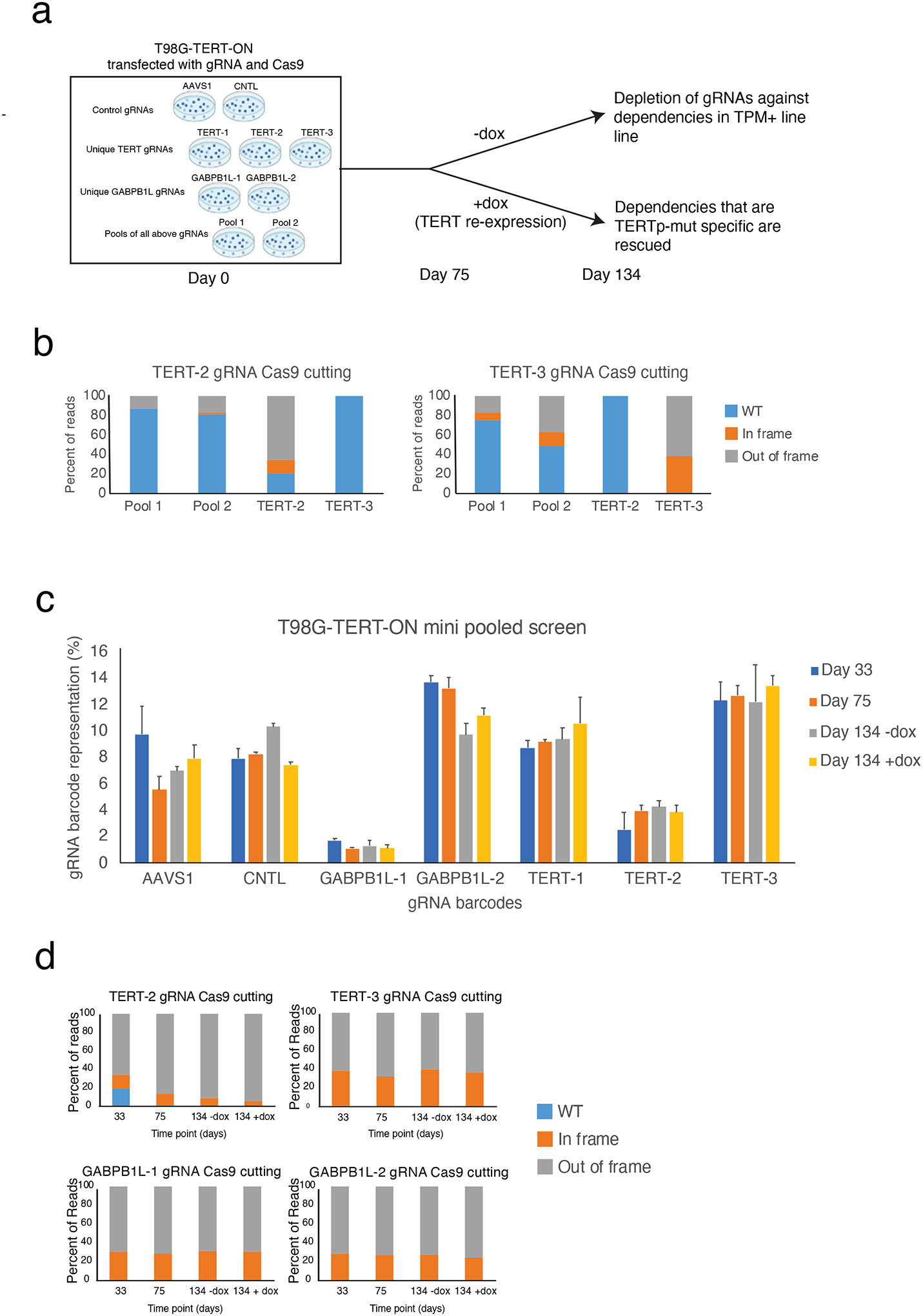
(A) Schematic for individual gRNA experiments and mini pooled screens. T98G-TERT-ON cells were transfected with either individual gRNAs or a pool of all gRNAs as shown. Antibiotic selection was added and cells were continuously cultured. At day 75, cells were divided into two parallel cultures to which dox or mock treatment was added. Cells were collected at multiple time points during the experiment for NGS analysis, including day 134.
(B) Percent Cas9 cutting induced by TERT-2 and TERT-3 gRNAs at 33 days post-transduction as assessed by targeted NGS of the gRNA target sites in Pool 1 and Pool 2, and in the cells transduced with the TERT-2 and TERT-3 gRNAs.
(C) Relative abundance of gRNA barcodes in pooled mini screen experiments. Percent representation determined by NGS sequencing of LentiCRISPRv2 barcodes, ~50,000 reads per sample. Data are for mean + S.E.M. for n=2 independent pooled screens.
(D) Percent Cas9 cutting induced by TERT-2, TERT-3, GABPB1L-1, and GABPB1L-2 gRNAs as assessed by targeted NGS of the target sites in cells transfected with each respective gRNA and Cas9.
We used next generation sequencing approaches to quantify Cas9 cutting and gRNA barcode representation. 80–100% of TERT genomic DNA contained expected TERT-directed cutting by day 33 post-transduction (Figure 2b). A proportionally lower amount of TERT genomic DNA was disrupted in the pooled screening experiment, in line with the fact that only 1/7th of the gRNAs were directed at each specific locus in the pooled experiment. We next quantified gRNA representation at three timepoints: after lentiviral transduction and initial selection, prior to dox induction, and after one month of culture with or without dox. We could not detect appreciable depletion of TERT or GABPB1L gRNAs over this timespan (Figure 2c). Similarly, we could not detect any evidence of negative selection against Cas9 cutting of TERT or GABPB1L over time, or differential selection for TERT or GABPB1L in the presence or absence of TERT re-expression. Indeed, the percentage of cells with damaging TERT or GABPB1L alterations induced by specific gRNAs remained stable and high over time, and was not affected by the addition of dox (Figure 2d). These results indicate that while TERT was appropriately disrupted by CRISPR/Cas9, there was no negative selection against TERT or GABPB1L-deficient cells in this system. We also found that TERT gRNAs were not preferentially targeting a certain allele of TERT, or that a specific allele of TERT underwent positive or negative selection over time after CRISPR/Cas9 disruption (Supplemental Note).
TERT is not a dependency in TPM+ GBM lines in genome-wide pooled screens
Since we could not detect a genetic dependency for TERT or GABPB1L in our TPM+ GBM cell line, we investigated if other TPM+ GBM cell lines were dependent on TERT or on GABP complex components. To do so, we compared CRISPR/Cas9 and RNAi screening data for TPM+ cell lines to TPM− cell lines in the Cancer Dependency Map database (DepMap, v22Q2, https://depmap.org/portal/) (24–29). Recent data indicates that TPM disruption causes near-term anti-growth effects in GBM within less than one month (20), which would be detected in the DepMap screening format. Dependency scores, which represent the degree of dependency based on positive or negative selection seen in CRISPR/Cas9 and RNAi screening experiments, have been calculated for nearly all genes in all lines (gene effect < −0.50 was considered dependent). Since TPM status is not available in the DepMap dataset, we established a consensus TPM status for >400 cell lines including n=36 glioblastoma lines by a combination of in-house genotyping and literature review (Figure 3a, Supplementary Table 2, see Methods).
Figure 3. TERT dependency scores in DepMap lines.
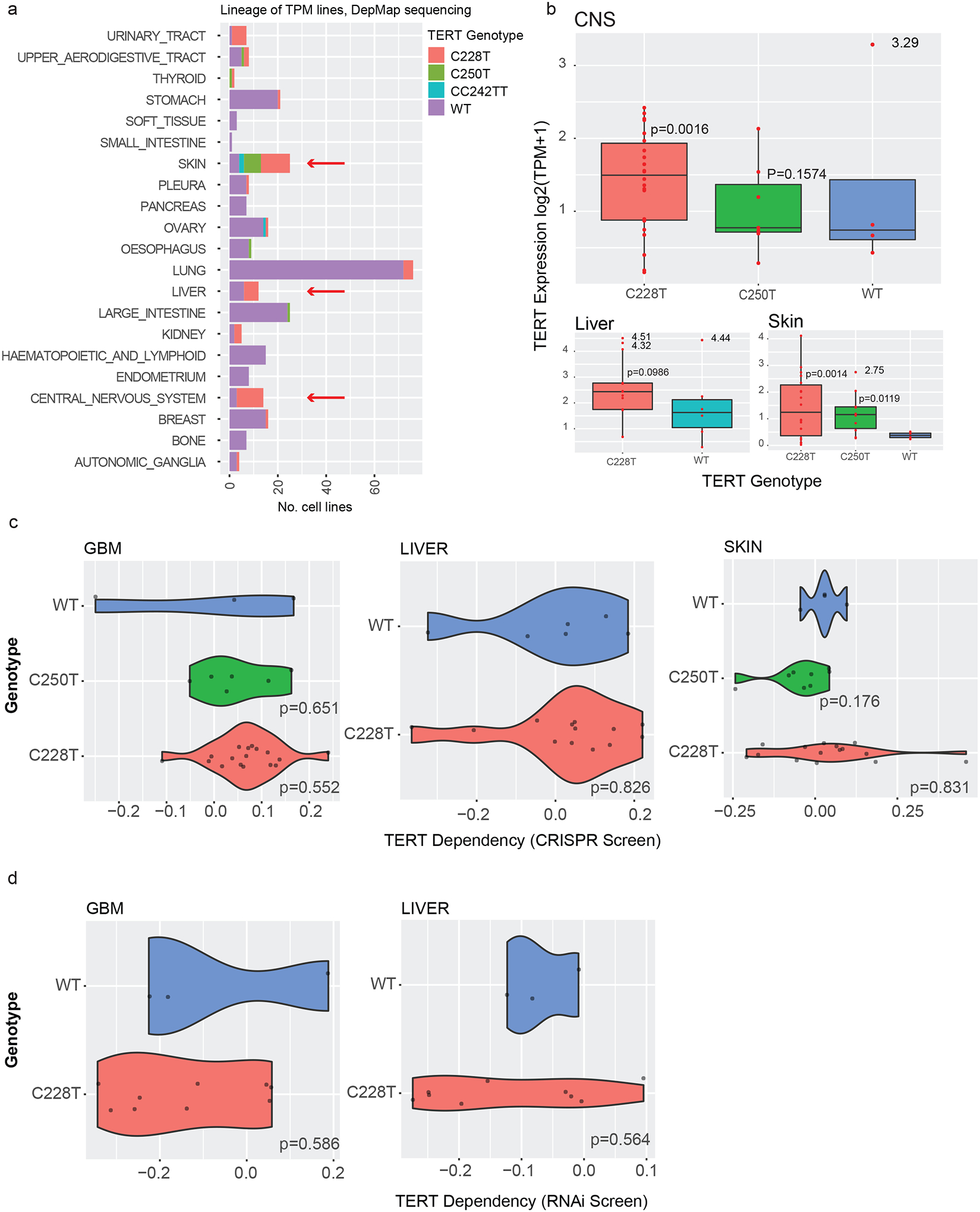
(A) Consensus TPM status for 441 cell lines. CNS, liver, and skin lineages (arrows) all contain at least three TPM+ and at least three TPM− lines, allowing comparative analyses within these lineages.
(B) Association between TPM status and TERT mRNA expression in DepMap cell lines in CNS, liver, and skin lineages. Welch’s t-test compared to the TPM-WT group, *p<0.05, **p<0.01, outliers excluded.
(C) Dependency scores in CRISPR/Cas9 screens in CNS, liver, and skin DepMap cell lines with and without TPMs. P-values were calculated with a linear hypothesis test, p<0.05 considered significant.
(D) Dependency scores in RNAi screens in CNS and liver DepMap cell lines with and without TPMs. Skin lines did not contain sufficient information for analysis in the RNAi dataset. P-values were calculated with a linear hypothesis test, p<0.05 considered significant.
We tested if TPM status was associated with dependency on TERT. We decided a priori to focus on GBMs, liver cancers (LC), and melanomas (MEL) since these histologies contained multiple TPM− and TPM+ cell lines necessary for statistical comparisons (Figure 3a). As expected, the presence of a TPM was generally associated with higher TERT mRNA expression (Figure 3b). However, TERT was not associated with a dependency in the CRISPR or RNAi DepMap database among GBM, LC, or MEL lines (Figure 3c, d). Very few cell lines scored as a CRISPR dependency on TERT (10/1086), none of which were TPM+ GBM, LC, or MEL lines. Thus, similar to our in vitro pooled genetic screening experiment on T98G, cell lines in the DepMap database did not appear to exhibit dependency on TERT regardless of TPM status.
GABP subunits demonstrate dependencies in both TPM+ and TPM− cell lines
We next sought to test whether TPMs were associated with dependencies on the GABP transcription factor subunits that are thought to regulate their function (2). First, we examined GABPB1. Though the DepMap does not contain information specifically on the previously studied GABPB1L isoform (2), gRNAs targeted at GABPB1 would be expected to deplete GABPB1L, in addition to other GABPB1 isoforms (Figure 4a). Significant differences (p<0.05) in GABPB1 dependency were detected between TPM+ and TPM− GBM lines (Figure 4b), though the gene still seemed to be somewhat dependent in WT cells. The GABP complex has previously been shown to be needed for TPM function in liver, urothelial, and melanoma cell lines (10), but we did not detect significant differential GABPB1 dependency scores between TPM+ and TPM− lines among any of these lineages (Figure 4c,d). Out of the 690 GABPB1-dependent lines, most are TERT promoter wild type and from lineages that infrequently harbor TPMs, such as breast and colon lines (Figure 4e). Considering orthogonal data, GBM and LC lines also did not demonstrate a differential dependency on GABPB1 in RNAi-based screens (Figure 4b,c). These data suggest that GABPB1 is especially important in promoting the replication capacity of C228T cells, and has weaker evidence for dependency in TPM− cells. We conducted similar analyses for GABPB2 and GABPA. We did not find CRISPR/Cas9 or RNAi dependency for GABPB2 in GBM lines (Figure 4b). GABPA scored as a strong dependency in C228T GBMs and within 856/1078 of all cancer cell lines; GABPA dependency was also observed in the limited number of available TPM− GBM lines (Figure 4b).
Figure 4. GABP subunit dependency scores in DepMap cell lines.
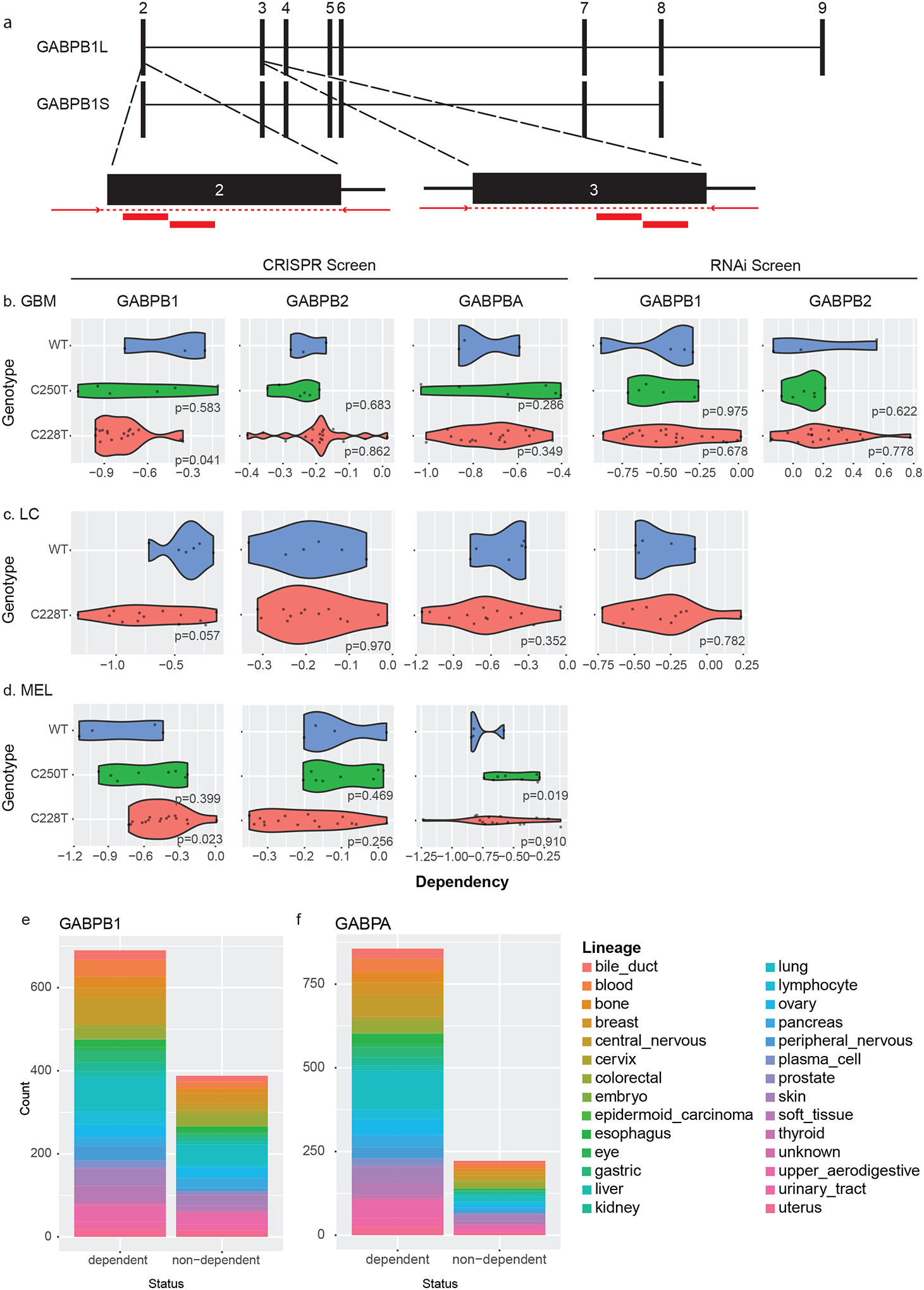
(A) Schematic showing locations of GABB1L gRNAs on B1S and B1L isoforms. Exons 2–9 are shown, with exon 1 located far upstream of exon 2.
(B) CRISPR-based dependency scores for GABPB1, GABPB2, and GABPA and RNAi-based dependency scores for GABPB1 and GABPB2 are shown for TPM+ and TPM− GBM cell lines (linear hypothesis test, p<0.05 considered significant).
(C) CRISPR-based dependency scores for GABPB1, GABPB2, and GABPA and RNAi-based dependency scores for GABPB1 are shown for TPM+ and TPM- liver cancer cell lines (linear hypothesis test, p<0.05 considered significant).
(D) CRISPR-based dependency scores for GABPB1, GABPB2, and GABPA are shown for TPM+ and TPM− skin cancer cell lines (linear hypothesis test, p<0.05 considered significant).
(E) Lineage of 742/1086 cell lines for which GABPB1 scored as a dependency based on CRISPR pooled screening experiments, compared to 344/1086 for which GABPB1 did not score as a dependency.
(F) Lineage of 930/1086 cell lines for which GABPA scored as a dependency based on CRISPR pooled screening experiments, compared to 156/1086 for which GABPA did not score as a dependency.
Comparing all genetic dependencies in DepMap between TPM+ and TPM− lines yielded relatively few differential dependencies after false discovery rate (FDR) correction (Supplementary Figure 2, Supplementary Table 3). Our ability to detect differential dependencies when correcting for multiple hypothesis testing for >10,000 genes was likely hampered by the small number of lines in the TPM− group.
TPM+ cell lines differentially express genes that are regulated by ETS-factors
To identify molecular features of the TPM+ and TPM− cell lines in the DepMap dataset that may explain our inability to detect TPM-specific dependencies, we examined RNA expression profiles between the two groups. We identified 147 differentially expressed genes between C228T vs WT cell lines, (DEGs, q<0.05, Figure 5a). Protein interaction network analysis (PPI) demonstrated a strong network enrichment (p=0.0003) suggesting that the proteins were biologically connected as a group (Figure 5b). Because transcription factors tend to regulate genes with similar sets of functions, we hypothesized that the biological interconnectedness might be explained by transcriptional reprogramming. If true, we would expect genes downstream of certain transcription factors to be heavily dysregulated.
Figure 5. Expression patterns associated with TPM status in GBM cell lines.
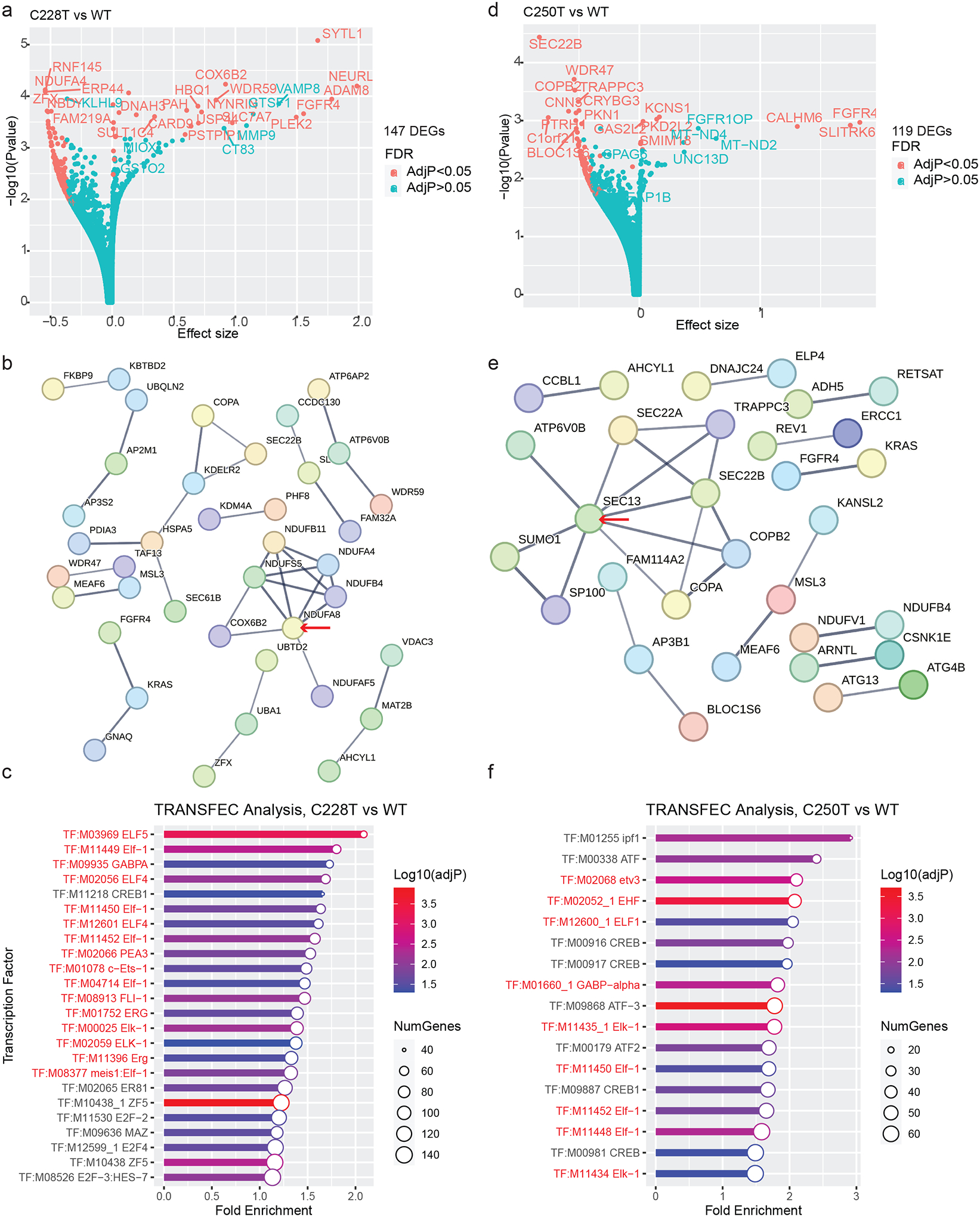
(A) Volcano plot showing DEGs between TPM-C228T vs. TPM-WT GBM cell lines. Top DEGs are labeled.
(B) Protein interaction network of DEGs identified from TPM C228T GBM cell lines. Red arrow denotes the gene with the highest number of interaction edges.
(C) TRANSFAC analysis of DEGs in TPM C228T GBM cell lines. Transcription factors whose regulated genes are significantly enriched among the DEGs are shown. ETS-factors are highlighted in red text.
(D) Volcano plot showing DEGs between TPM C250T vs. TPM-WT GBM cell lines. Top DEGs are labeled.
(E) Protein interaction network of DEGs in TERTp.C250TGBM cell lines. Red arrow denotes the gene with the highest number of interaction edges.
(F) TRANSFAC Analysis of DEGs in TERTp.C250T GBM cell lines. ETS-factors are highlighted in red text.
To test whether specific transcription factors were linked to the TPM-associated DEGs, we used gene set enrichment analysis for transcription factor targets as annotated by the TRANSFAC database (30–32) (Figure 5c). Sixteen out of the 24 significantly enriched transcription factors identified by TRANSFAC analysis were within the ETS family, indicating enrichment for ETS-factors, such as GABP, above expected levels (chi-square, p<8.52 × 10−30). Though the ETS transcription factors were not differentially expressed themselves, they are known to be regulated by post-translational modifications (33). Although there are fewer C250T cell lines in the DepMap dataset, analysis of C250T vs TPM− cells also demonstrated that ETS-factors are important mediators of DEG in C250T cell lines (PPI p<0.00007, chi-square, p=1.44 × 10−16, Figure 5d–f).
Expression of multiple ETS-factors correlate with TERT expression in TPM+ GBM tissues
To extend our findings from TPM+ cell lines to patient data, we examined GBM expression data from a dataset of nearly 500 primary GBM tissues. We identified 659 DEGs (adjP<0.05, |Log2(FC)| < 1) when comparing C228T vs WT (Figure 6a) and 892 DEGs when comparing C250T vs WT (Figure 6b). There were few DEGs when comparing C228T vs C250T, suggesting that the two groups were biologically more similar to each other than to the TPM− group (Figure 6c). Indeed, there were 505 DEGs that overlapped between the C228T vs TPM− and C250T vs TPM− comparisons (Figure 6d). K-means clustering (k=3, based on the three TPM alleles) of GBM samples according to the top 50 most variable genes (Figure 6e) differentiated between TPM− and TPM status. Specifically, cluster 2 (p=7.0E-04) and cluster 3 (p=1.7E-03) were significantly enriched with the TPM genotype, whereas cluster 1 is significantly enriched with the WT genotype (p=6.09E-05) (Supplementary Table 4). Thus, the TPMs delineate transcriptionally distinct subsets of primary GBMs in this dataset. However, clusters 2 and 3 did not differentiate between C228T and C250T, suggesting that the biologies underlying these two TPMs were comparable. The differences between clusters 2 and 3 were likely driven by differences in underlying transcriptional heterogeneity due to clinical, cellular composition, and other genetic factors. For instance, the two groups demonstrated differential expression of a set of seven genes including ATRX, PRRC2A, C4B, UTY, HLA-B, LTF, and RPS4Y1. Computation of overlaps between this 7-gene and the Molecular Signatures Database (MSigDB) (34) revealed significant overlap with an infection-related gene set (ATRX, HLA-B, C4B, HP_Unusual_infection_by_anatomical_site, q = 0.014). ATRX has been linked with immunomodulatory functions in IDH-mutated gliomas (35). The current data link ATRX expression to immunomodulatory functions among TERT promoter mutant, IDH WT gliomas. (36)
Figure 6. Expression patterns associated with TPM status in 500 primary GBM tumors.
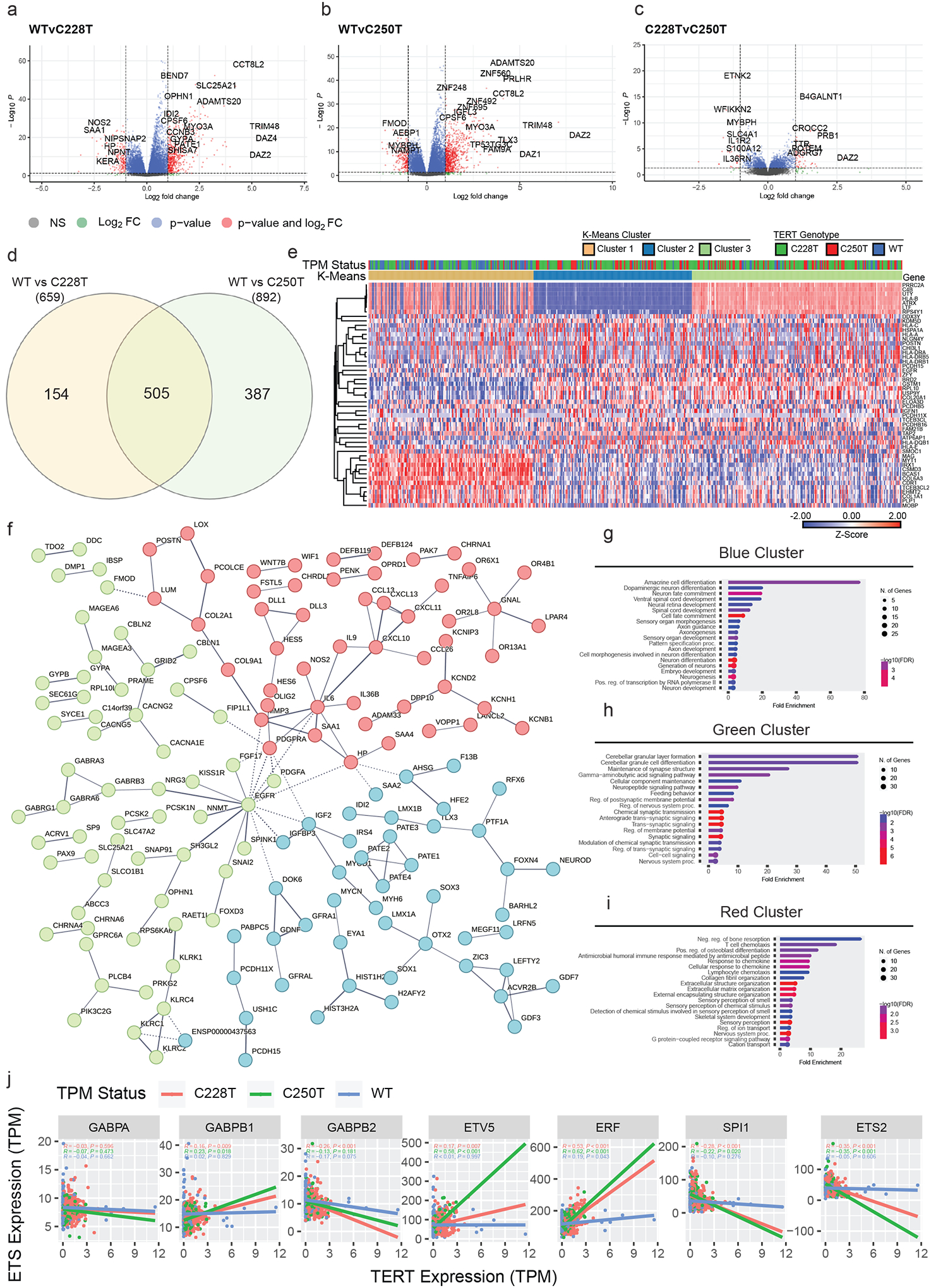
(A) Volcano plot showing DEGs between TPM-C228T and TPM-WT GBM primary tissues.
(B) Volcano plot showing DEGs between TPM-C250T and TPM-WT GBM primary tissues.
(C) Volcano plot showing DEGs between TPM-C228T and TPM-C250T GBM primary tissues.
(D) Shared DEGs between C228T and C250T GBM primary tissues.
(E) Heat map of the z-scores of expression for the top 50 most variable genes (rows) from GBM primary tissue expression profiles (columns). Rows are grouped by unsupervised hierarchical clustering and the columns are grouped by k-means clustering. TPM genotype and k-means cluster are indicated in the color strip above the heat map. One minus person correlation with averages linkage was used for all clustering.
(F) Protein interaction analysis of shared DEGs between C228T and C250T GBM primary tissues. Three clusters (blue, green, red) are developed by k-means clustering.
(G) Gene ontology analysis of genes from blue cluster derived from k-means clustering of consensus DEGs based on associated protein interaction networks.
(H) Gene ontology analysis of genes from green cluster derived from k-means clustering of consensus DEGs based on associated protein interaction networks.
(I) Gene ontology analysis of genes from red cluster derived from k-means clustering of consensus DEGs based on associated protein interaction networks.
(J) Selected pairwise correlations between TERT expression and ETS-factor expression. Pearson r statistic, associated p-values, and linear regression lines are shown for correlations between each respective ETS-factor expression and TERT expression. For each pairwise correlation, statistics were calculated among subgroups of TPM-C228T tumors, TPM-C250T tumors, and TPM-WT tumors.
We next explored the biological interactions that may underlie the expression patterns we found to be enriched in TPM+ GBM tissues. We used the 505 consensus DEGs shared by C228T and C250T tumors to construct a PPI (37). K-means clusters within the network (k=3) (Figure 6f) demonstrated enrichment in ontologies related to differentiation and development (Figure 6g), intercellular communication (Figure 6h), and immune response signaling (Figure 6i). Overall, these analyses suggest that TPM+ GBMs are associated with lineage-, stemness-, and immune-related transcriptional programs.
We then explored the relationship between ETS transcription factor expression and TERT expression in our primary GBM tissues. TERT expression is upregulated by a variety of ETS- factors, including GABP, in a dose-dependent fashion (10). We therefore hypothesized that the expression of specific ETS-factors responsible for driving TERT expression from the TPM+ promoter should correlate with TERT expression among TPM+ tumors, but not among TPM− tumors. We correlated TERT expression with the expression of every ETS transcription factor, respectively (Supplementary Figure 3). Several correlations are highlighted in Figure 6j. GABPB1 expression was correlated with TERT expression among TPM+ tumors (C228T p=0.009, C250T p=0.018), but not among TPM− tumors (TPM−, p=0.8). However, GABPA and GABPB2 expression were not significantly correlated with TERT expression among any groups. Interestingly, even stronger associations were observed in TPM+ tumors for ETV5 (C228T p=0.007, C250T p<0.001). We also found significant negative associations between TERT expression and SPI1 and ETS2. There were also very strong associations with ETV1, ETV2, and ETV4 (Supplementary Figure 3). The group of TPM− tumors exhibited very few significant correlations between TERT expression and ETS transcription factor expression. These results suggest that multiple ETS-factors may drive TERT expression via interactions with the mutant promoter in TPM+ GBM.
Discussion
Here, we leveraged pooled genetic screening approaches to identify vulnerabilities associated with TPMs. We developed T98G-TERT-ON, a GBM cell line platform harboring an endogenous TPM in which exogenous TERT can be induced by the addition of doxycycline to the culture media. We used this system to screen for dependencies that could be rescued by exogenous TERT expression. Surprisingly, we found that TERT itself and GABPB1L, a transcription factor previously shown to regulate TERT expression (2), were not dependencies in this platform. This discrepancy compared to prior studies (2,12) could potentially be due to differences in cellular contexts such as time scale of the experiments, clonality or pooled nature of the system, and/or in vivo vs. in vitro nature of the experiments. Similarly, we could not detect an association between TPMs and TERT dependency in a large number of cell lines within the DepMap dataset (Supplementary Table 2). GABPB1 and GABPA were strong dependencies in TPM+ GBMs, and also in TPM− GBMs as well as in the majority of cancer cell lines. Expression profiles between TPM+ and TPM− cell lines and GBM tumors support the deregulation of several ETS-factors. Together, these results support a model in which multiple ETS-factors may play an interchangeable and/or redundant role in regulating TERT expression in TPM+ GBM cells in a highly cellular context-dependent manner.
Our work provides important lessons to guide ongoing efforts to target TPMs. While our data supports previous results demonstrating a differential dependency to GABPB1 in TPM cells as well as increased ETS-like signaling (2), it also offers insights regarding the essentiality of GABPB1 in WT lines as well as the potential redundancy of ETS activity driving TERT expression, which has implications in therapeutic development. Such a finding has also been noted before in thyroid cancer (38). Our study extends upon previous studies by highlighting specific members within the ETS family that may play crucial roles in glioblastoma with TPM, offers valuable lessons for the use of genetic screens in cancer research, and provides valuable insights into the underlying regulators of TERT biology.
Another key insight is that pooled screens as executed here and by the DepMap do not find TERT as a dependency in TPM+ GBM lines. One possibility is that tumor cells have enough plasticity to activate other telomere-elongating processes in response to TERT knockout, such as activation of the ALT mechanism (39). Because the cell lines were not found to be dependent upon TERT, perhaps the cellular dependency upon pathways “upstream” of TERT formed as a result of its regulation of other tumorigenic processes. Thus, pooled knockout screens have proven a challenging approach for the identification of negative genetic interactions with TPMs.
Our study has several limitations. While we hypothesized that gRNAs directed at TERT or GABPB1L would be depleted in a mini pooled CRISPR/Cas9 screen, this was not observed. Several explanations could underlie this finding. Even though we confirmed efficient CRISPR/Cas9-mediated gene disruption activity, T98G has been reported to be a hyperdiploid line with substantial chromosomal instability (22). Therefore, even with a majority of TERT alleles disrupted, the possibility remains that there would still be at least one intact TERT allele in the cells. However, this is unable to explain the results in their entirety; for example, TERT-2 gRNA-guided cutting ultimately resulted in frameshifts in >90% of TERT by day 134. In addition, prior studies showed TERT dependency in subclones with TERT or GABPB1L disruption (2). Thus, it is possible that the pooled screening format allowed the emergence of cellular adaptations that were unable to develop in clonal populations. Yet another possibility is that the described clones themselves had cellular changes not present in the entire T98G population that caused TERT and/or GABPB1L to be non-essential. Another limitation in this study was the leakiness of TERT expression from the exogenous TERT promoter in the absence of doxycycline. Since even small amounts of TERT expression can fulfill its physiological functions in cells, this may explain why the cellular model did not demonstrate TERT dependency in the absence of doxycycline. This could be strengthened in future studies by an additional control group where the TERT overexpression cassette is absent.
A potential limitation of the computational analysis was that we did not consider telomeric content within our analysis. The time period the DepMap CRISPR screen was conducted over may be insufficient to observe telomere crisis in cell lines with high telomere content. Interestingly, a previous study used genome sequencing data within the DepMap to estimate telomere content within the cell lines to further annotate the C228T/C250T status of the cell lines (40). Such analysis of telomeric content found context-dependent dependencies on telomere maintenance and shelterin genes. Whereas our approach was more focused on targeting TPM status rather than telomeric abundance, this additional data may also be useful for better understanding in which contexts TPM vulnerabilities appear. For instance, examination of telomere content and dependency datasets for 344 cell lines (40) showed that dependencies on the ETS transcription factors ELF1 and ETV4 were significantly correlated with telomere content (P=6.0E-03 and P=8.9E-04, respectively), while this was not the case for GABPB1 (P>0.05). ELF1 and ETV4 expression were also strongly associated with TERT expression in our dataset (Supplementary Figure 3). These findings suggest that there may be complex interactions between various ETS transcription factors and telomere content maintenance, highlighting the need for comprehensive investigations into the regulation of telomerase expression in future studies. Further experiments focusing on the direct effects of the ETS factors nominated here on TERT expression may help define the TERT regulatory network as well as the contexts in which they are activated.
The time frames for the current experiments are critical considerations for interpreting the results. Since a primary mechanism of TPMs relates to cellular immortality, we initially hypothesized that a long-term screen involving >50 cell divisions over >100 days would be needed to detect dependencies related to the TPM disruption. However, recent data emerged indicating that TPM disruption can cause short-term growth defects (20), which would be expected to be detected in the approximately month-long DepMap CRISPR screens. Therefore, when difficulties arose in establishing a long-term senescence-based screening system, we turned our attention towards leveraging the DepMap’s publicly-available data. A drawback of this approach is the lack of an isogenic system to identify dependencies that are likely to be caused by the presence of a TPM rather than other confounding properties of the cell lines. Further, given the high frequency of TPMs in GBM, relatively few TPM− GBM cell lines exist limiting the statistical power for many analyses. In addition, this approach does not account for differences in baseline cell line proliferation rates and cell death rates that can alter the rates and degrees of depletion of gRNAs targeting essential genes. Along the same lines, baseline variability in copy numbers of essential genes between cell lines can result in apparent differences in gene essentiality. Additionally, the context of examining short- and long-term experiments needs to be taken into account for future analyses given the different mechanisms of action of TPM disruption on cell growth at different time scales (20).
While one initial goal of this work was to identify therapeutically-actionable upstream genetic pathways that are requisite for TPM oncogenic function (Figure 1a, Supplementary Figure 4a,b), our findings indicate that approaches to target other aspects of TPM biology also merit attention. For instance, our recent preclinical investigation of a 6-thio-deoxyguanosine drug was shown to specifically induce telomeric DNA damage in a telomerase-dependent manner in TPM+ GBMs (41). This study highlights that targeting TPM-associated biology downstream of the TPM may also be of value in therapeutic approach and development. Further studies are needed to fully understand the underlying mechanisms of TPM biology and to optimize the clinical application of this approach.
Methods
T98G-TERT-ON cell line development
T98G cells (ATCC, authenticated using Sanger sequencing) were cultured in EMEM (Gibco, 30–2003) 10% FBS (Gibco 10099–141-C) at 37C. All cells were tested routinely for mycoplasma and found to be negative. Inducible expression was accomplished using the Tet-On 3G system according to the manufacturer’s instructions (Takara, Cat. No. 631339). Briefly, transfection-grade Lenti-TetON 3G-Neo plasmid was used to generate lentiviruses to introduce TetOn expression. Cells were cultured in additional 400 ug/ml geneticin (Gibco) to select for a stable pool with Tet-On expression. Transfection-grade Lenti-TRE-hygro-TERT was generated using gene synthesis of an HA-Flag-TERT construct (Supplementary Table 1, Genscript). Lentiviruses were generated from Lenti-TRE-hygro-TERT. Stable pools containing the Lenti-TRE-hygro-TERT construct were selected using an additional 150 ug/ml hygromycin B (Invitrogen). Subcloning of inducible TERT cell lines was carried out according to the manufacturer’s instructions. Quantitative reverse transcriptase PCR (qPCR) was carried out using primers for TetON (forward 5’-GTATTGGAGGAACAGGAG-3’, reverse 5’-ATTGCTTGTTTCAGAAGTG-3’) and primers for TERT (forward 5’-GGCATGGAACCACTCTGTGA-3’, reverse 5’-GGTGTTCTCTCTGGCTCAGG-3’). Immunoblot and flow analysis was carried out with anti-FLAG (A00187–100, Genscript) and anti-HA antibodies (A01244–100) and a secondary goat-anti-mouse antibody (A00160, Genscript).
LentiCRISPRv2 constructs
Two published gRNAs for GABPB1L were used (5’-GCAGCTCCTAAAGAAAGAAC-3’, 5’-AGAGGCCTACAGACAGAAGT-3’) (2). A nontargeting control gRNA (5’-ATAATAATACGTACAGGCCC-3’) and a control targeting nonfunctional AAVS1 (5’-GGGGCCACtagggacaggat-3’) were used. TERT-1, TERT-2, and TERT-3 gRNAs were 5’-TcTTGAGGAgcaccCCGTAG-3’, 5’-AGGCCGAGCGTCtCACctcg-3’, and 5’-CTGCGtGCGTCGgtatgcCG-3’, respectively. Viruses were generated as described (42).
CRISPR/Cas9 mini pooled screen
Cells were seeded in EMEM containing hygromycin and blasticidin as above. Media was removed and LentiCRISPRv2 lentiviruses were added at multiplicity of infection of 0.2 in serum-free media. 3 volumes of serum-containing media were added 8 hours later. Puromycin selection (1 ug/ml, Gibco) was initiated by adding puromycin 2 days after lentivirus transduction. Cells were split 1:10 when they reached 90% confluency by removing media, washing 1x with PBS, adding 0.25% prewarmed trypsin to cover the cells, incubating at 37C for 5 minutes, and then placing 10% of the volume into a fresh vessel with fresh media. All experiments were done in replicates (n=2). Cells were passaged before reaching confluency as needed up to 3 times per week to maintain at least 50,000-fold pooled library coverage throughout the experiment. CRISPR/Cas9 gene disruption confirmation was completed as described in Supplemental Note 2.
Dependency and expression analysis of DepMap cell lines
Consensus TPM status for CCLE cell lines was determined by examining the status from (28). In-house Sanger sequencing as described previously (6) and TPM status from additional sources were used to supplement this list (6,7,41,43–46). Dependency scores from DepMap (v22Q2) CRISPR knockout screens and RNAi dependency scores from project Achilles (24–28) were downloaded from DepMap Public 22Q2 CRISPR_gene_effect.csv and Achilles_gene_effect.csv. Custom R scripts (v4.0.3) were used to generate differential dependency scores by carrying out pairwise t-tests on two groups (either TPM+ and TPM-, or C228T vs. C250T). Multiple hypothesis testing was accomplished using the Benjamini Hochberg method. Common essential genes as defined by DepMap in the CRISPR_common_essentials.csv file were disregarded as targetable vulnerabilities due to the likelihood for off-target effects. A gene effect of −0.50 was considered dependent. P<0.05 and q<0.05 was considered significant unless otherwise noted.
Expression data from DepMap from project DEMETER2 (v22Q2) (24–28) were downloaded from DepMap Public 22Q2 CCLE_expression.csv. R (v4.0.3) was used to identify differential expressed genes (DEG). Gene expression effect size, as defined by DepMap, was used to compare the expression of two groups (either C228T vs. WT, or C250T vs. WT).
Gene set enrichment analysis and protein interaction networks
Transcription factor enrichment analysis was performed with the TRANSFAC database through g:Profiler (30,31). ETS-factors taxonomy was considered as described in (32). Chi-squared tests were used to determine if ETS-factors were significantly dysregulated. P<0.05 was considered significant unless otherwise noted.
Protein interaction networks were constructed through the STRING database (37). We drew on all active interaction sources within the database and required a strict minimum interaction score of 0.700. In-house k-means clustering was used to cluster protein interactions. Gene ontology analysis was performed through ShinyGO (47). We calculated p-value with students-t-test and FDR with hypergeometric test. FDR<0.05 was defined as significant. We calculated fold enrichment by dividing the percentage of DEGs belonging to a GO term by the corresponding percentage in the genome (~20,000 genes).
Primary GBM dataset analysis
The primary glioblastoma tissue data used for this research was made available under a research use license from American Society of Clinical Oncology, Inc. (ASCO) and Tempus Labs, Inc. We obtained a dataset of expression and sequencing data from 500 primary GBM tumors. This project was approved by the Duke IRB (Protocol number 00108183), approved March 31, 2021. We grouped patients into groups carrying C228T, C250T, or WT TERT genotypes based on whole genome sequencing reads. Patients carrying TERT missense mutations (3 patients) or both TPMs (1 patient) were excluded to reduce confounding factors. Patients with low sequencing coverage (<50) were also excluded (25 patients). This resulted in 252 patients with C228T TPMs, 108 patients with C250T TPMs, and 111 WT patients.
We used DESeq2 to identify DEGs using raw RNA-seq data (48). We confirmed variance was similar between the TPM groups. We defined DEGs as adjP<0.05 and |Log2FC|>1. Dataset quality analyses are included in Supplementary Figure 5. ETS expression given in the transcripts per million format was linearly regressed separately with the same TERT expression.
Supplementary Material
Acknowledgements
We thank the Tempus/ASCO collaborative for access to their primary tumor databanks and their kind partnership. We thank David G. Kirsch for helpful critical feedback on the project.
We thank sources of funding including NCI K08256045 Mentored Clinician Scientist Development Award, U19CA264385 Glioblastoma Therapeutics Network and developmental funds from the Cancer Center Support Grant P30CA014236 to ZJR, and Fund to Retain Clinician Scientists at Duke from the Doris Duke Foundation to ZJR, and support for ZJR career development including the ChadTough Defeat DIPG Foundation, the SoSo Strong Foundation, the Pediatric Brain Tumor Foundation, the St. Baldrick’s Foundation, NCI P50CA190991 Duke SPORE in Brain Cancer developmental funds to ZJR. K.J.T. was supported by a scholarship from the Amgen Foundation, Banneker/Key Scholarship, and Churchill Scholarship.
Footnotes
Conflict of interest statement: ZJR is listed as an inventor for intellectual property related to genetic testing for TERT and other alterations in brain tumors that is managed by Duke Office of Licensing and Ventures and has been licensed to Genetron Health. The other authors have no conflicts of interest to report.
Data Availability
The genetic screen data datasets generated during and/or analyzed during the current study are available in the supplemental materials and DepMap database (https://depmap.org/portal/). The datasets generated during and/or analyzed during the current study are also available from the corresponding author on reasonable request. The primary glioblastoma data that support the findings of this study are available from TEMPUS and ASCO but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of TEMPUS and ASCO.
References
- 1.Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, et al. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol Biomarkers Prev. 2014. Oct 1;23(10):1985–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mancini A, Xavier-Magalhães A, Woods WS, Nguyen KT, Amen AM, Hayes JL, et al. Disruption of the β1L Isoform of GABP Reverses Glioblastoma Replicative Immortality in a TERT Promoter Mutation-Dependent Manner. Cancer Cell. 2018. Sep;34(3):513–528.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reitman ZJ, Pirozzi CJ, Yan H. Promoting a new brain tumor mutation: TERT promoter mutations in CNS tumors. Acta Neuropathol (Berl). 2013. Dec;126(6):789–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Körber V, Yang J, Barah P, Wu Y, Stichel D, Gu Z, et al. Evolutionary Trajectories of IDHWT Glioblastomas Reveal a Common Path of Early Tumorigenesis Instigated Years ahead of Initial Diagnosis. Cancer Cell. 2019. Apr;35(4):692–704.e12. [DOI] [PubMed] [Google Scholar]
- 5.Lee JH, Lee JE, Kahng JY, Kim SH, Park JS, Yoon SJ, et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature. 2018. Aug;560(7717):243–7. [DOI] [PubMed] [Google Scholar]
- 6.Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci. 2013. Apr 9;110(15):6021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science. 2013. Feb 22;339(6122):957–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science. 2013. Feb 22;339(6122):959–61. [DOI] [PubMed] [Google Scholar]
- 9.Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017. Mar;49(3):349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bell RJA, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, et al. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. 2015. May 29;348(6238):1036–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiba K, Lorbeer FK, Shain AH, McSwiggen DT, Schruf E, Oh A, et al. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science. 2017. Sep 29;357(6358):1416–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aquilanti E, Kageler L, Watson J, Baird DM, Jones RE, Hodges M, et al. Telomerase inhibition is an effective therapeutic strategy in TERT promoter-mutant glioblastoma models with low tumor volume. Neuro-Oncol. 2023. Jul 6;25(7):1275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wojton J, Meisen WH, Jacob NK, Thorne AH, Hardcastle J, Denton N, et al. SapC-DOPS-induced lysosomal cell death synergizes with TMZ in glioblastoma. Oncotarget. 2014. Oct 30;5(20):9703–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandes AA, Franceschi E, Tosoni A, Hegi ME, Stupp R. Epidermal Growth Factor Receptor Inhibitors in Neuro-oncology: Hopes and Disappointments. Clin Cancer Res. 2008. Feb 15;14(4):957–60. [DOI] [PubMed] [Google Scholar]
- 15.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017. Oct;18(10):1373–85. [DOI] [PubMed] [Google Scholar]
- 16.Ramkissoon SH, Bi WL, Schumacher SE, Ramkissoon LA, Haidar S, Knoff D, et al. Clinical implementation of integrated whole-genome copy number and mutation profiling for glioblastoma. Neuro-Oncol. 2015. Oct;17(10):1344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aquilanti E, Kageler L, Wen PY, Meyerson M. Telomerase as a therapeutic target in glioblastoma. Neuro-Oncol. 2021. Dec 1;23(12):2004–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiappori AA, Kolevska T, Spigel DR, Hager S, Rarick M, Gadgeel S, et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann Oncol. 2015. Feb;26(2):354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salloum R, Hummel TR, Kumar SS, Dorris K, Li S, Lin T, et al. A molecular biology and phase II study of imetelstat (GRN163L) in children with recurrent or refractory central nervous system malignancies: a pediatric brain tumor consortium study. J Neurooncol. 2016. Sep;129(3):443–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amen AM, Fellmann C, Soczek KM, Ren SM, Lew RJ, Knott GJ, et al. Cancer-specific loss of TERT activation sensitizes glioblastoma to DNA damage. Proc Natl Acad Sci. 2021. Mar 30;118(13):e2008772118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bushweller JH. Targeting transcription factors in cancer — from undruggable to reality. Nat Rev Cancer. 2019. Nov;19(11):611–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stein GH. T98G: An anchorage-independent human tumor cell line that exhibits stationary phase G1 arrest in vitro. J Cell Physiol. 1979. Apr;99(1):43–54. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt JC, Zaug AJ, Cech TR. Live Cell Imaging Reveals the Dynamics of Telomerase Recruitment to Telomeres. Cell. 2016. Aug;166(5):1188–1197.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat Genet. 2017. Dec;49(12):1779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dempster JM, Pacini C, Pantel S, Behan FM, Green T, Krill-Burger J, et al. Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat Commun. 2019. Dec 20;10(1):5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dempster JM, Boyle I, Vazquez F, Root DE, Boehm JS, Hahn WC, et al. Chronos: a cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021. Dec;22(1):343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pacini C, Dempster JM, Boyle I, Gonçalves E, Najgebauer H, Karakoc E, et al. Integrated cross-study datasets of genetic dependencies in cancer. Nat Commun. 2021. Mar 12;12(1):1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, et al. Next generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019. May;569(7757):503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell. 2017. Jul;170(3):564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019. Jul 2;47(W1):W191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matys V TRANSFAC(R): transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003. Jan 1;31(1):374–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gutierrez-Hartmann A, Duval DL, Bradford AP. ETS transcription factors in endocrine systems. Trends Endocrinol Metab. 2007. May;18(4):150–8. [DOI] [PubMed] [Google Scholar]
- 33.Tootle TL, Rebay I. Post-translational modifications influence transcription factor activity: A view from the ETS superfamily. BioEssays. 2005. Mar;27(3):285–98. [DOI] [PubMed] [Google Scholar]
- 34.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015. Dec;1(6):417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu C, Wang K, Damon C, Fu Y, Ma T, Kratz L, et al. ATRX loss promotes immunosuppressive mechanisms in IDH1 mutant glioma. Neuro-Oncol. 2022. Jun 1;24(6):888–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ebrahimi A, Skardelly M, Bonzheim I, Ott I, Mühleisen H, Eckert F, et al. ATRX immunostaining predicts IDH and H3F3A status in gliomas. Acta Neuropathol Commun. 2016. Dec;4(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021. Jan 8;49(D1):D605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thornton CEM, Hao J, Tamarapu PP, Landa I. Multiple ETS Factors Participate in the Transcriptional Control of TERT Mutant Promoter in Thyroid Cancers. Cancers. 2022. Jan 12;14(2):357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang JM, Zou L. Alternative lengthening of telomeres: from molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020. Dec;10(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu K, Ghandi M, Huang FW. Integrated evaluation of telomerase activation and telomere maintenance across cancer cell lines. eLife. 2021. Sep 6;10:e66198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu S, Wei S, Savani M, Lin X, Du K, Mender I, et al. A Modified Nucleoside 6-Thio-2′-Deoxyguanosine Exhibits Antitumor Activity in Gliomas. Clin Cancer Res. 2021. Dec 15;27(24):6800–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khadka P, Reitman ZJ, Lu S, Buchan G, Gionet G, Dubois F, et al. PPM1D mutations are oncogenic drivers of de novo diffuse midline glioma formation. Nat Commun. 2022. Feb 1;13(1):604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang FW, Bielski CM, Rinne ML, Hahn WC, Sellers WR, Stegmeier F, et al. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis. 2015. Dec;4(12):e176–e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johanns TM, Fu Y, Kobayashi DK, Mei Y, Dunn IF, Mao DD, et al. High incidence of TERT mutation in brain tumor cell lines. Brain Tumor Pathol. 2016. Jul;33(3):222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Zhou QL, Sun W, Chandrasekharan P, Cheng HS, Ying Z, et al. Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol. 2015. Oct;17(10):1327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bairoch A The Cellosaurus, a Cell-Line Knowledge Resource. J Biomol Tech JBT. 2018. Jul;29(2):25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ge SX, Jung D, Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Valencia A, editor. Bioinformatics. 2020. Apr 15;36(8):2628–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014. Dec;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genetic screen data datasets generated during and/or analyzed during the current study are available in the supplemental materials and DepMap database (https://depmap.org/portal/). The datasets generated during and/or analyzed during the current study are also available from the corresponding author on reasonable request. The primary glioblastoma data that support the findings of this study are available from TEMPUS and ASCO but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of TEMPUS and ASCO.