

SUMMARY
To maximize solute transport, epithelial cells build an apical “brush border”, where thousands of microvilli are linked to their neighbors by a protocadherin-containing intermicrovillar adhesion complexes (IMACs). Previous studies established that the IMAC is needed to build a mature brush border, but how this complex contributes to the accumulation of new microvilli during differentiation remains unclear. We found that, early in differentiation, mouse, human, and porcine epithelial cells exhibit a marginal accumulation of microvilli, which span junctions and interact with protrusions on neighboring cells using IMAC protocadherins. These transjunctional IMACs are highly stable and reinforced by tension across junctions. Finally, long-term live imaging showed that accumulation of microvilli at cell margins consistently leads accumulation in medial regions. Thus, nascent microvilli are stabilized by a marginal capture mechanism that depends on the formation of transjunctional IMACs. These results may offer insight into how apical specializations are assembled in diverse epithelial systems.
Keywords: transporting epithelia, actin, cadherins, differentiation, brush border, intermicrovillar adhesion
Graphical Abstract
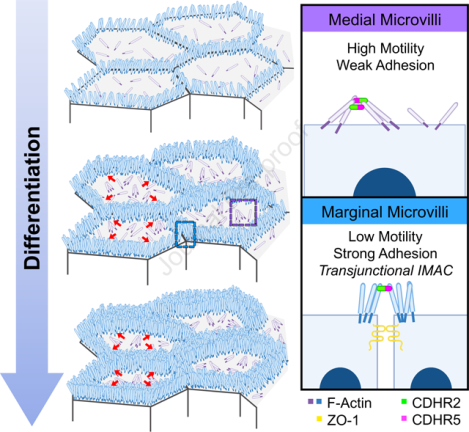
eTOC blurb:
Cencer et al. demonstrate that microvilli accumulation during the early stages of epithelial differentiation is driven by the formation of transjunctional adhesion complexes of CDHR2 and CDHR5, which link protrusions that extend from neighboring cells. Transjunctional adhesion stabilizes microvilli at these sites and ultimately drives their accumulation as differentiation progresses.
INTRODUCTION
Organ function depends on specialized cell types that have evolved morphologies to enable specific physiological tasks. Transporting epithelial cells like those found in the intestine and kidney proximal tubule, offer interesting examples of this phenomenon. As important sites of solute uptake, maximizing apical surface area is critical for these tissues. To meet this challenge, individual epithelial cells extend 1000s of bristle-like protrusions called microvilli, which collectively form the ‘brush border’1,2. A single microvillus is a cylinder-shaped, micron-scale membrane protrusion supported by a core actin bundle consisting of 20–40 actin filaments3,4. By scaffolding apical membrane in this way, microvilli amplify surface area available for solute transport and optimize solute uptake potential5–7. Microvilli first appear on the cell surface early in epithelial maturation; differentiating cells, like those found within intestinal stem cell-containing crypts, exhibit few, poorly organized microvilli8. However, differentiated, fully functional enterocytes, found on the villus or within the kidney tubule, present a well-organized and densely packed brush border3,9,10.
Previous studies established that tight microvillar packing is driven by a protocadherin-based intermicrovillar adhesion complex (IMAC), which physically links the distal tips of neighboring microvilli11–14. In the enterocyte, these adhesive interactions give rise to a hexagonal packing pattern when viewed en face, which represents maximum surface occupancy. Previous work also identified protocadherins CDHR2 and CDHR5 as the primary adhesive elements in these links, which form trans heterophilic adhesion complexes that are well suited for bridging the ~50 nm gap between neighboring microvilli11,15. CDHR2 and CDHR5 ectodomains contain multiple extracellular cadherin (EC) repeat motifs arranged in tandem, which are anchored to the membrane via a single spanning transmembrane domain16. Both protocadherins also contain cytoplasmic tails at their C-termini, which enable direct interactions with cytoplasmic IMAC binding partners including the actin-based motor, myosin-7B (MYO7B), and the scaffolding proteins, ankyrin repeat and sterile alpha motif domain containing 4B (ANKS4B) and usher syndrome 1C (USH1C)12,13,17,18. Recently, calmodulin-like protein 4 (CALML4) was also identified as a binding partner of MYO7B, making it another IMAC component19. KD studies of MYO7B indicate that this motor is required for the localization of CDHR2/CDHR5 adhesion complexes to the distal tips of microvilli12,13. In the CACO-2BBE intestinal epithelial cell culture model, disrupting the function of the IMAC via calcium chelators or KD of any single complex component leads to striking defects in microvillar growth and packing organization during differentiation11–13,19. Furthermore, CDHR2 KO from intestinal and kidney epithelia in a villin-Cre driven knockout (KO) mouse, causes shortening and loss of brush border microvilli, a consequential decrease in the apical enrichment of key solute transporters, and reduced animal growth rate14.
How new microvilli assemble and incorporate into a highly ordered brush border during differentiation remains unclear. Ultrastructural studies of native tissue and time-lapse imaging of epithelial cell culture models indicate that microvilli do not grow synchronously, but instead appear stochastically on the apical surface throughout differentiation8,11,20. One critical factor that promotes microvillar growth is the barbed end binder, epidermal growth factor receptor pathway substrate 8 (EPS8)20,21. Studies in multiple epithelial and non-epithelial systems have established that EPS8 is a highly specific marker of the distal ends of all forms of actin bundle supported protrusions21–24. Loss of this factor leads to shorter protrusions and increased length variability25,26. Strikingly, on the apical surface of differentiating epithelial cells, EPS8 arrives in diffraction-limited puncta at the membrane minutes before the subsequent growth of a core actin bundle and assembly of a microvillus at these sites20. Even once a core bundle begins to elongate, EPS8 puncta remain persistently associated with the distal end of the nascent structure. Following their initial growth, nascent microvilli are highly motile and translocate across the apical surface via a mechanism powered by treadmilling of the underlying core actin bundle27, an activity that is also regulated by EPS827. Remarkably, if the distal tip of a newly formed microvillus loses its EPS8 punctum, that structure rapidly collapses, suggesting that EPS8 serves as a microvillus survival factor20. These data point to a previously unrecognized dynamic microvillus lifecycle, consisting of distinct phases of structural stability and instability. For microvilli to eventually accumulate in large numbers on the apical surface, this cycle must ultimately tilt in favor of stability. How dynamic, nascent microvilli are stabilized on the apical surface so that they eventually accumulate in large numbers remains unknown.
Here we report our discovery of an adhesion-based mechanism that epithelial cells use to stabilize and in turn, drive the accumulation of microvilli during differentiation. Because microvillar growth takes place as differentiating enterocytes move through the crypt-villus transition8, we reasoned that we could gain insight on mechanisms of microvilli accumulation by careful inspection of apical morphology in this region. Using this approach, we discovered that crypt microvilli initially accumulate at cell margins, implying the existence of a mechanism for anchoring nascent protrusions at these sites. We observed similar marginal accumulation of microvilli on the surface of differentiating intestinal and kidney epithelial cell lines. In all models examined, microvilli extending from one cell span intercellular space to make physical contact with microvilli on a neighboring cell. Using super-resolution microscopy, mechanistic studies in epithelial cell culture models, and live imaging, we determined that these points of physical contact represent transjunctional IMACs containing both CDHR2 and CDHR5, which are highly stable complexes that capture nascent microvilli and constrain their motion. Consistent with this point, long-term live imaging revealed that microvilli accumulation at cell margins outpaces accumulation in medial regions of the surface early in differentiation. Thus, microvilli extending from neighboring epithelial cells participate in cell-cell contacts that promote apical surface maturation. The adhesion-based capture mechanism reported here might also extend our understanding of apical morphogenesis in other epithelial cell types that build elaborate surface specializations.
RESULTS
Differentiating epithelial cells exhibit a marginal enrichment of microvilli
To begin to understand how microvilli are stabilized and accumulate in large numbers during differentiation, we first examined the distribution of nascent protrusions early in the maturation process. To this end, we used scanning electron microscopy (SEM) to survey the apical surface of the crypt cells in fractured samples of mouse small intestine. Within the crypt, where immature enterocytes are actively assembling a brush border (Fig. 1A, zoom), we noted a striking enrichment of microvilli at cell margins (Fig. 1B, zoom 1 and 2 blue outlines). In contrast, medial regions of the apical surface presented only a few, sparse microvilli (Fig. 1B, zoom 1). Thus, in vivo, microvilli appear to accumulate at the edges of cells during the early stages of brush border assembly.
Figure 1: Microvilli of differentiating transporting epithelial cells concentrate at cell margins.
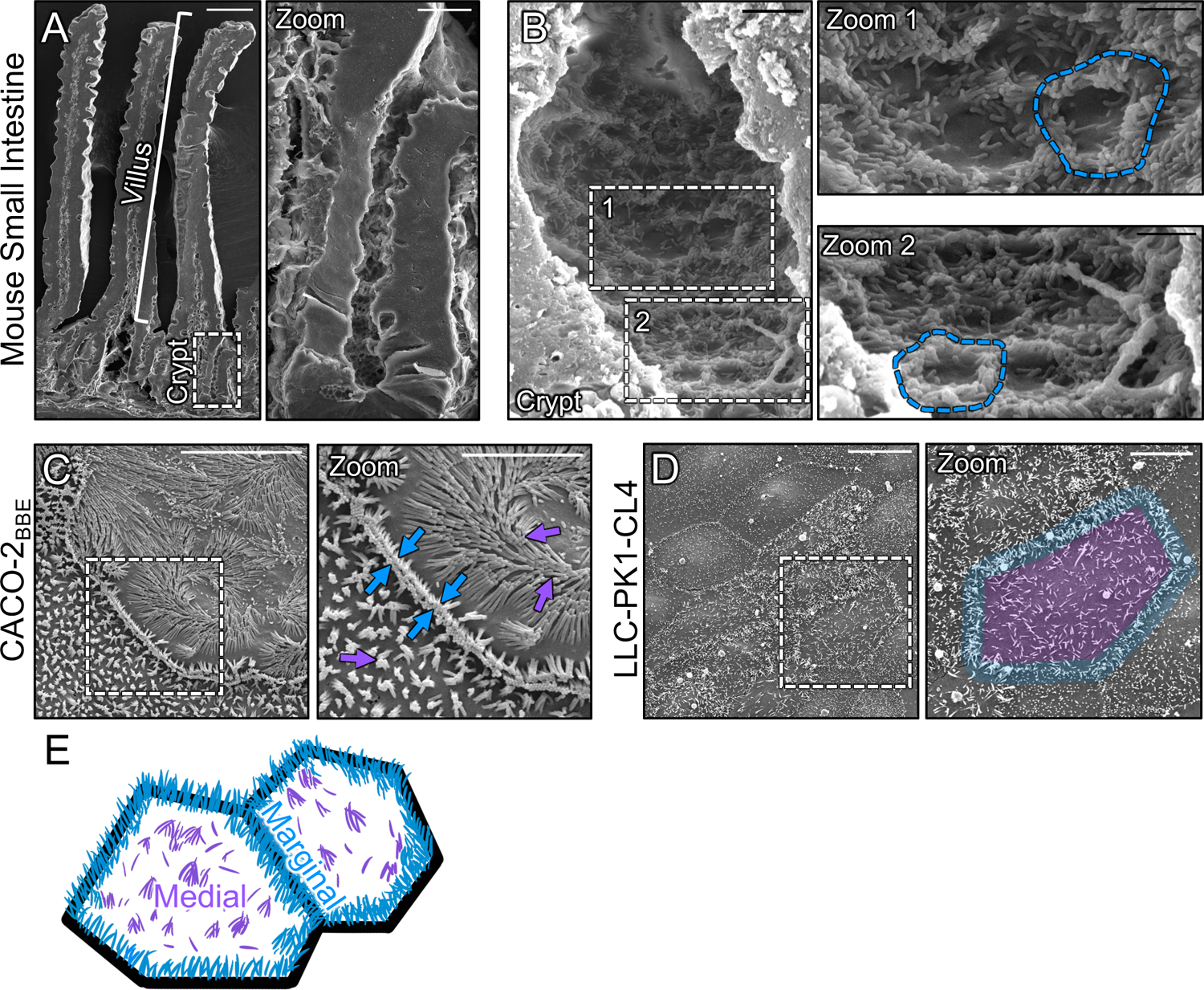
(A) Scanning electron micrograph (SEM) of native mouse small intestine crypt-villus axis. (A, zoom) Zoom of the dashed box in A showing the crypt and transit amplifying zone. (B) High-magnification view of the crypt base with (B, zooms 1 and 2) showing an enrichment of microvilli at the margins of crypt cells (dashed blue outline). (C) SEM of 8 days post-confluence polarized CACO-2BBE cells. Dashed box represents zoom area. Arrows denote medial (purple) and marginal microvilli (blue). (D) SEM of sub-confluent porcine kidney proximal tubule LLC-PK1-CL4 (CL4) cells. Dashed box represents zoom area. Pseudo coloring represents medial area (purple) and marginal microvillar area (blue). (E) Schematic of the two distinct organizations of microvilli found on differentiating transporting epithelial cells, medial (purple) and marginal (blue). Scale bars: 50 μm (A), 10 μm (A, zoom), 2 μm (B), 1 μm (B, zooms), 10 μm (C), 5 μm (C, zoom), 20 μm (D), 10 μm (D, zoom).
To determine if the marginal accumulation of microvilli observed on the surface of differentiating crypt cells in vivo could be recapitulated in vitro, we first turned to the CACO-2BBE line. CACO-2BBE cells are a human intestinal epithelial culture model that builds a well-organized brush border over the course of several weeks post-confluency28. SEM imaging of CACO-2BBE cells early in the differentiation time-course (8 days-post confluence) revealed a concentration of microvilli at cell margins similar to that observed in native crypts (Fig 1C). Moreover, protrusions in these regions appeared to span cell junctions and make physical contact with microvilli on neighboring cells (Fig. 1C, zoom blue arrows). As an additional point of comparison, we used SEM to examine sub-confluent, partially differentiated “islands”, of porcine kidney proximal tubule LLC-PK1 clone 4 (CL4) cells29,30, which also exhibited a marginal accumulation of microvilli (Fig. 1D, zoom blue outline), even at the earliest stages of cell surface organization (i.e. subconfluence). Based on these in vivo and in vitro observations, we conclude the differentiating apical surface is characterized by two distinct populations of microvilli, marginal and medial (Fig. 1E), with the marginal region demonstrating higher protrusion density at these early time points.
Microvilli adopt a vertical orientation upon arriving at cell margins
In the ultrastructural studies above, we noted that marginal microvilli appeared more vertically oriented relative to microvilli extending from medial parts of the cell surface. Here we use ‘vertical’ to describe an orientation that is parallel to the long (apicobasal) axis of the cell and perpendicular to the plane of the apical surface. To confirm this observation under hydrated conditions, we performed volume imaging of live sub-confluent CL4 cells expressing mCherry-Espin (ESPN), which serves as a highly specific marker of microvillar core actin bundles20,27,31,32 (Fig. 2A). Lateral views of reconstructed volumes enabled us to visualize individual microvilli and obtain measurements of their orientation relative to the plane of the apical surface. This analysis revealed that the marginal and medial microvilli demonstrate significant differences in their angle of protrusion, with marginal microvilli exhibiting a more vertical orientation (marginal, 77.3° ± 12.4° vs. medial, 46.5° ± 19.3, Fig. 2B).
Figure 2: Microvilli adopt a vertical orientation upon reaching cell margins.
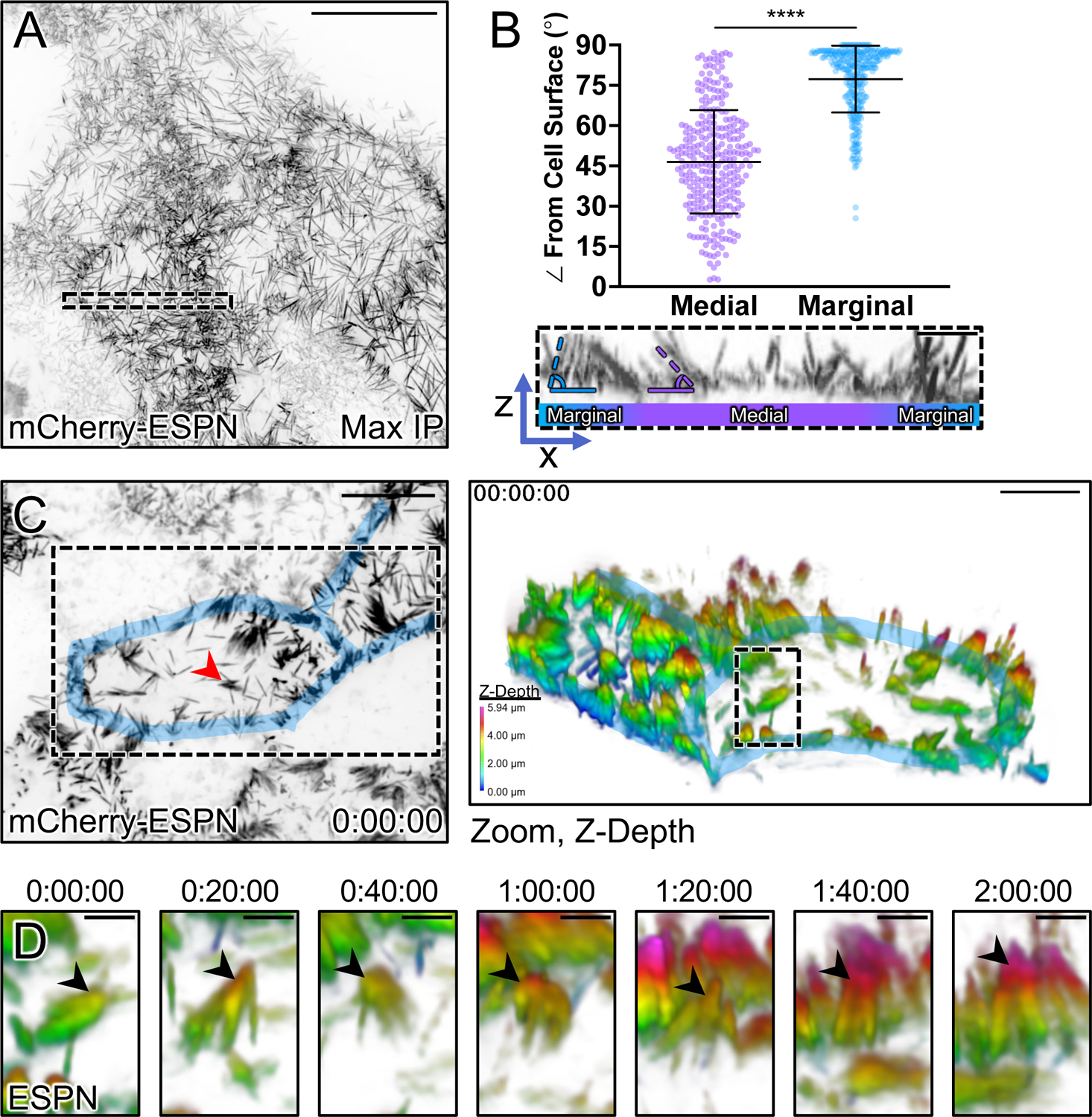
(A) Maximum intensity projection (MaxIP) of live CL4 cells expressing mCherry-ESPN. (B) Orientation measurements of the angle (dashed outlines) of microvilli to the cell surface of medial microvilli (purple) compared to marginal microvilli (blue). Sample ROI of Z-projection under plot is taken from the dashed box in (A). (C) t = 0 MaxIP image of live mCherry-ESPN CL4 cells. Two neighboring cell margins are highlighted in blue, while the red arrowhead points to the microvilli cluster followed in (D). Right panel shows a 3D tilted volume of the dashed box in (C), coded in Z for cell depth (bottom left key). Cell margins are highlighted in blue. (D) Montage over 2 hours following the cluster marked with the red arrowhead/dashed box in (C). Arrowheads mark the distal ends of microvilli that transition to a vertical orientation upon reaching the marginal cell area, as shown by a change in Z-depth coding. Each point on the graph represents one angle taken from 17 cells; total of n = 295 medial and n = 309 marginal angles. Error bars represent mean ± SD. **** p ≤ 0.0001 Welch’s unpaired t-test. Mean medial angle is 46.5° ± 19.3° and mean marginal angle is 77.3° ± 12.4°. Scale bars: 20 μm (A), 1 μm (B), 10 μm (C, left), 5 μm (C, right), 1.5 μm (D).
Previous studies established that nascent microvilli are highly dynamic, growing, collapsing, and adopting a range of angles while undergoing active movement across the medial cell surface20,27. With this in mind, we next sought to determine if microvilli grow in a vertical orientation at marginal sites or instead, grow medially and then adopt a vertical orientation upon arriving at the cell edge. To this end, we performed multi-hour time-lapse volume imaging to record microvillar motion and orientation in 3D. To help us interpret these complex datasets, we depth-coded volumes with a multi-color look-up table (LUT) so that image planes located further from the apical surface were rendered with warmer colors. While the dense accumulation of microvilli at cell margins impaired our ability to resolve individual growth events at these sites, we did observe individual protrusions and small adherent clusters of microvilli migrating while maintaining a small angle relative to the medial apical surface, as previously described20,27. Following these microvilli over 2 hrs revealed that upon reaching the cell margin, they become more vertically orientated as indicated by the distal tips acquiring a warmer color coding (Figs. 2C,D and Video S1). Although these data do not allow us to rule out the possibility that microvilli grow de novo in a vertical orientation in the marginal zone, they do indicate that medial microvilli can transition into the marginal zone and adopt a vertical orientation when doing so.
Marginal microvilli are less motile than medial microvilli
Vertically orientated microvilli are a defining feature of mature brush borders on the surface of villus enterocytes3. Based on this point, the vertically oriented microvilli found in the marginal zone may represent more mature, and potentially, more stable protrusions. To test this concept, we expressed EGFP-EPS8 to specifically mark the distal tips of microvilli20,33 in CL4 cells also expressing mCherry-ESPN (Fig. 3A, zooms). We then performed live volume imaging with the goal of using the punctate and stoichiometric EPS8 signal (one punctum per microvillus) as a marker for tracking microvillar dynamics over time. Temporal color coding of the ESPN channel over the course of 25 minutes revealed that medial microvilli are highly dynamic and demonstrate extensive movement as previously reported27 (Fig. 3B, zoom 1). In contrast, marginal microvilli dwelled for long periods near the edge of the cell, as indicated by the white band of color (merged colors of time points 0–25 min) in the projection (Fig. 3B, zoom 2). Next, we tracked individual EGFP-EPS8 puncta and generated rose plots of the resulting trajectories to examine the extent of motion demonstrated by individual microvilli. This analysis revealed that medial microvilli produce long trajectories consistent with directed motion, sampling an area of up to 6 μm2 during the time-lapse (Figs. 3C,D). By comparison, the trajectories of marginal microvilli were highly confined, with individual protrusions traveling less than 2 μm2 during the same period (Figs. 3F,G). Mean squared displacement (MSD) analysis of trajectory data also indicated that medial protrusions demonstrate active motion as previously reported27 (Fig. 3E), whereas marginal microvilli exhibit constrained diffusion with a sub-micron confinement radius (Fig. 3H). Together, these data suggest the existence of a mechanism for restricting the motion of microvilli at cell margins.
Figure 3: Tip tracking analysis reveals that marginal microvilli are constrained in their movement.
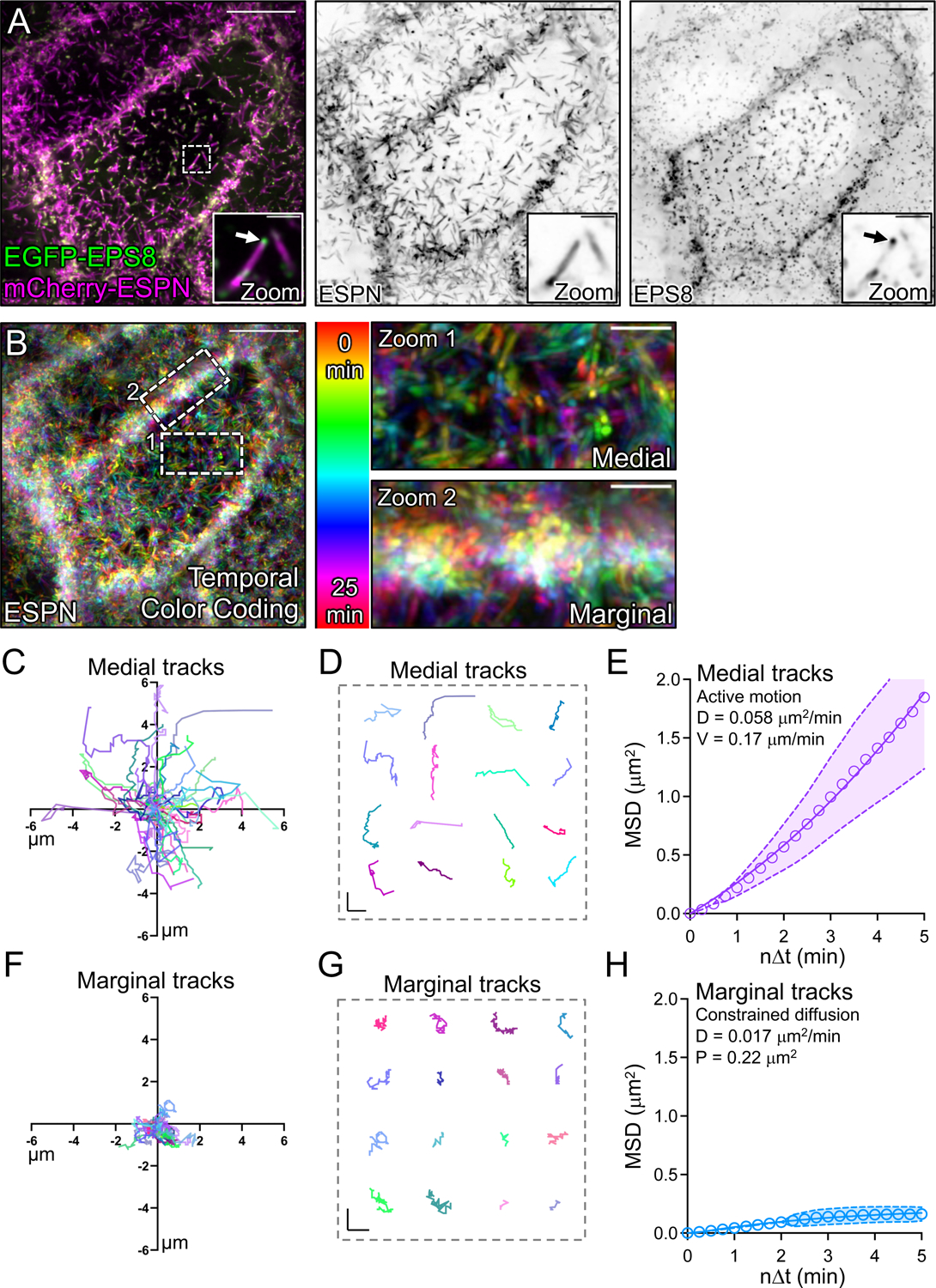
(A, left panel) Live CL4 cells co-expressing EGFP-EPS8 and mCherry-ESPN. Dashed box represents zoom area with arrow marking EPS8 at the tip of a single microvillus. (A, right panels) Single inverted channel MaxIP images showing mCherry-ESPN and EGFP-EPS8 alone. (B) Temporal color-coding over 25 minutes (see vertical color key). (B, zooms) of (1) medial and (2) marginal ROIs taken from the dashed boxes in (B). (C) Rose plot of n = 53 XY tracks (μm units) of medial microvilli over 25 minutes. (D) Representative medial microvilli tracks taken from (C). (E) Mean square displacement (MSD) of 50 medial microvilli imaged for 5 minutes over 15 second intervals; magenta open circles represent mean MSD values, magenta color band indicates the 95% CI, and the solid magenta line indicates a best fit of the data to an active motion model with D = 0.058 μm2/min and V = 0.17 μm/min. (F) Rose plot of n = 28 XY tracks of marginal microvilli taken from 3 independent live cell imaging experiments over 25 minutes. (G) Representative marginal microvilli tracks taken from (F). (H) MSD analysis of n = 88 marginal microvilli; blue open circles represent the mean MSD values, blue color band indicates the 95% CI, and the solid blue line indicates a best fit to a constrained diffusion model with D of 0.017 μm2/min and a plateau of confinement at 0.22 μm2. Scale bars: 10 μm (A), 1.5 μm (A, zooms), 10 μm (B), 2.5 μm (B, zooms), 1 μm (D, G).
Microvilli from neighboring cells are linked by transjunctional adhesion complexes containing CDHR2 and/or CDHR5
Our ultrastructural data suggested that marginal accumulations of microvilli might include protrusions from both cells of a neighboring pair (Fig. 1C, zoom). This led us to consider the possibility that microvilli extending from one cell may span the junctional space and physically contact microvilli from an adjacent cell; such interactions might in turn explain the upright orientation, reduced motility, and accumulation of microvilli at these sites. One potential mechanism for mediating such interactions involves the intermicrovillar adhesion complex (IMAC), which includes the protocadherins CDHR2 and CDHR5 as core components11. Previous studies established that CDHR2 and CDHR5 target to the distal tips of microvilli and interact with each other to form a Ca2+-dependent heterophilic extracellular adhesion complex that spans the ~50 nm between adjacent protrusions8–11. The resulting link promotes the tight packing of neighboring microvilli and contributes to minimizing length variability throughout the larger structure of the brush border14. Notably, these previous studies on IMAC function focused solely on medial microvilli, so the possibility that this complex might also link microvilli from neighboring cells remains unexplored.
To test this idea, we used an immunostaining approach and super-resolution structured illumination microscopy (SIM) to examine the localization of CDHR2, CDHR5, and F-actin relative to ZO-1, a critical component of tight junctions34. For these studies, we first examined native small intestinal tissues isolated from a new mouse model expressing CDHR2 tagged with EGFP at the endogenous locus (Fig. S1). SIM images revealed that both IMAC protocadherins are highly enriched at the tips of medial microvilli as previously reported (Fig. 4A)11. We also noted signal from CDHR2 and CDHR5 at the tips of microvilli at the margins of cells, with the adhesion protein signal spanning ZO-1 marked junctions (Fig. 4B). When viewing projected SIM volumes en face, we were unable to discern the position of the tight junctions based solely on the phalloidin, CDHR2, or CDHR5 signals, suggesting that the IMAC components form a continuous network that spans beyond the surface of a single cell (Fig. 4C). Similarly, on the surface of mature enterocytes viewed with electron microscopy, brush border microvilli are so tightly packed they appear form a continuous array that extends across the surface of multiple cells (Fig. S2A–B). However, the crowded nature of microvilli in these mature brush borders confounded our attempts to isolate and visualize interactions between the tips of individual protrusions at the margins of neighboring cells.
Figure 4: Marginal microvilli are linked via transjunctional CDHR2/CDHR5 adhesion complexes that extend across neighboring cell junctions.
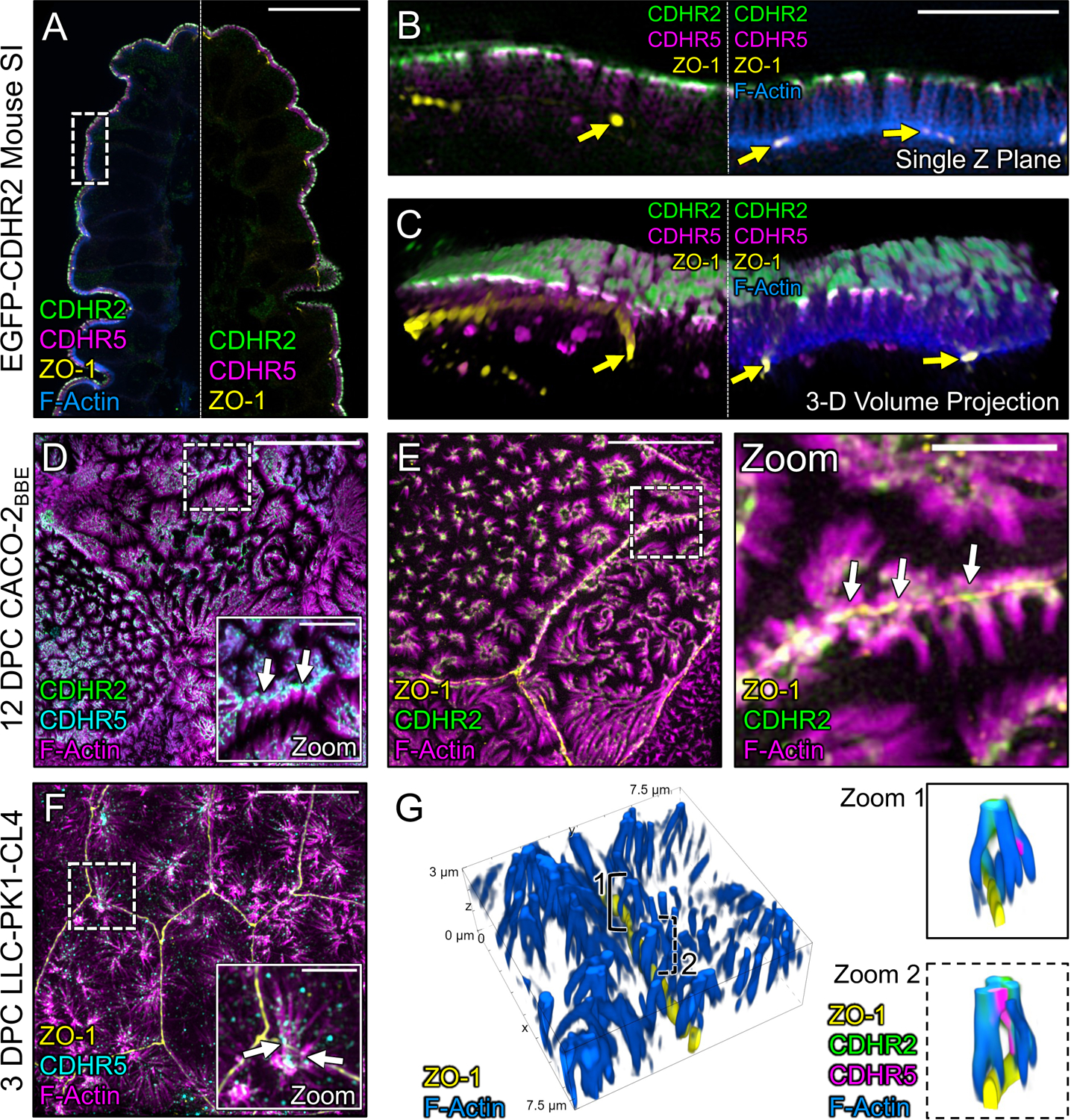
(A) Single Z-plane confocal image of CDHR2-EGFP mouse small intestine stained for ZO-1 (yellow), EGFP (green), CDHR5 (magenta), and F-actin (blue). (B) Single plane SIM image of the stained villus section; approximated area marked by the dashed box in (A). (C) 3D volume projection of the section in (B). Yellow arrows in both images mark ZO-1 labeled tight junctions. (D) MaxIP laser-scanning confocal image of 12 DPC CACO-2BBE cells stained for CDHR2 (green), CDHR5 (cyan), and F-actin (magenta). Dashed box represents zoom area. White arrows point to tip localized CDHR2/CDHR5 adhesion complexes at cell margins. (E) MaxIP SIM image of 12 DPC CACO-2BBE cells stained for ZO-1 (yellow), CDHR2 (green), and F-actin (magenta). Dashed box represents zoom area. White arrows point to CDHR2/CDHR5 marked complexes at the junction of neighboring cells. (F) MaxIP SIM image of 3 DPC CL4 cells stained for ZO-1 (yellow), CDHR5 (cyan), and F-actin (magenta). (G) 3D tilted volume projection of 3 DPC CL4 cells stained for ZO-1 (yellow), CDHR2 (green), CDHR5 (magenta), and F-actin (blue). Brackets highlight instances of marginal microvilli on adjacent cells linked via CDHR2/CDHR5 transjunctional adhesion complexes (zoom 1 and 2). Scale bars: 20 μm (A), 5 μm (B), 5 μm (D), 2.5 μm (D, zoom), 10 μm (E), 2.5 μm (E, zoom), 10 μm (F), 2.5 μm (F, zoom).
To work around the limitation imposed by microvillar crowding in native tissue, we used SIM to examine the apical surface of cultured CACO-2BBE cells at 12 days post-confluence (DPC), a time point before brush border assembly is complete, when microvillar packing density is low. Careful examination of phalloidin-stained CACO-2BBE monolayers revealed a striking enrichment and alignment of microvilli at the margins of cells (Fig. 4D,E), consistent with the SEM images described above. Immunofluorescence staining of these 12 DPC cultures revealed that marginally aligned microvilli do in fact span the cell junction marked by ZO-1 and exhibit enrichment of both protocadherins at their distal tips (Fig. 4D,E, white arrows). We observed similar structures and staining on the surface of CL4 monolayers at 3 DPC, a stage in differentiation when microvilli are still sparse but begin to form clusters and demonstrate marginal alignment (Fig. 4F). In this case, super-resolution lateral views clearly showed that individual microvilli from neighboring cells span the ZO-1-labeled tight junction and make contact via their distal tips, which are marked by both CDHR2 and CDHR5 (Fig. 4G, zooms). In combination, these results indicate that marginal microvilli on neighboring cells are physically linked via transjunctional IMACs that contain CDHR2 and CDHR5.
Heterophilic adhesion between CDHR2 and CDHR5 promotes robust association between microvilli from neighboring cells
Although IMAC protocadherin adhesion properties differ across species16, previous biochemical studies established that in humans, heterophilic complexes of CDHR2 and CDHR5 exhibit strong adhesion, CDHR2 demonstrates weak homophilic adhesion, and CDHR5 demonstrates no homophilic adhesion11. To further study the nature of transjunctional IMACs, we developed a cell mixing approach that enabled us to drive the formation of adhesion complexes consisting of different complements of CDHR2 and/or CDHR5 (Fig. 5A). For these experiments, we first transfected CL4 cells with either EGFP or mCherry-tagged constructs of H. sapiens CDHR2 and CDHR5. Stable selection and subsequent fluorescence-activated cell sorting (FACS) yielded robust populations of fluorescent protocadherin expressing cells (Fig. S3A). Strikingly, mixed monolayers composed of cells expressing CDHR2-EGFP or CDHR5-mCherry demonstrated robust alignment of protocadherin signals at mixed cell-cell contacts (Fig. 5B). Linescan analysis also revealed that CDHR2 and CDHR5 intensities were well correlated (mean r = 0.70) along these interfaces (Fig. 5C,D,K). These data are consistent with the formation of heterophilic adhesion complexes between microvilli of neighboring cells. In mixed monolayers composed of cells expressing CDHR2-EGFP or CDHR2-mCherry (Fig. 5E), mixed cell-cell contacts lacked the strong alignment of signals that we observed in the heterophilic case, and protocadherin intensities were poorly correlated (mean r = 0.07 (Fig. 5F,G,K). Mixed monolayers composed of cells expressing CDHR5-EGFP or CDHR5-mCherry also demonstrated a lack of signal alignment and poor intensity correlation along cell junctions (mean r = −0.19) (Fig. 5H–K). High-resolution imaging of the interfaces formed under each of these three conditions revealed that only heterophilic mixtures of cells expressing CDHR2-EGFP or CDHR5-mCherry aligned their microvilli at cell-cell contacts (Fig. S3B–D, white arrows). Based on these data, we conclude that heterophilic transjunctional IMACs containing CDHR2 and CDHR5 can drive robust interactions between microvilli extending from neighboring cell margins.
Figure 5: Cell mixing experiments reveal robust heterophilic adhesion complexes between marginal microvilli.
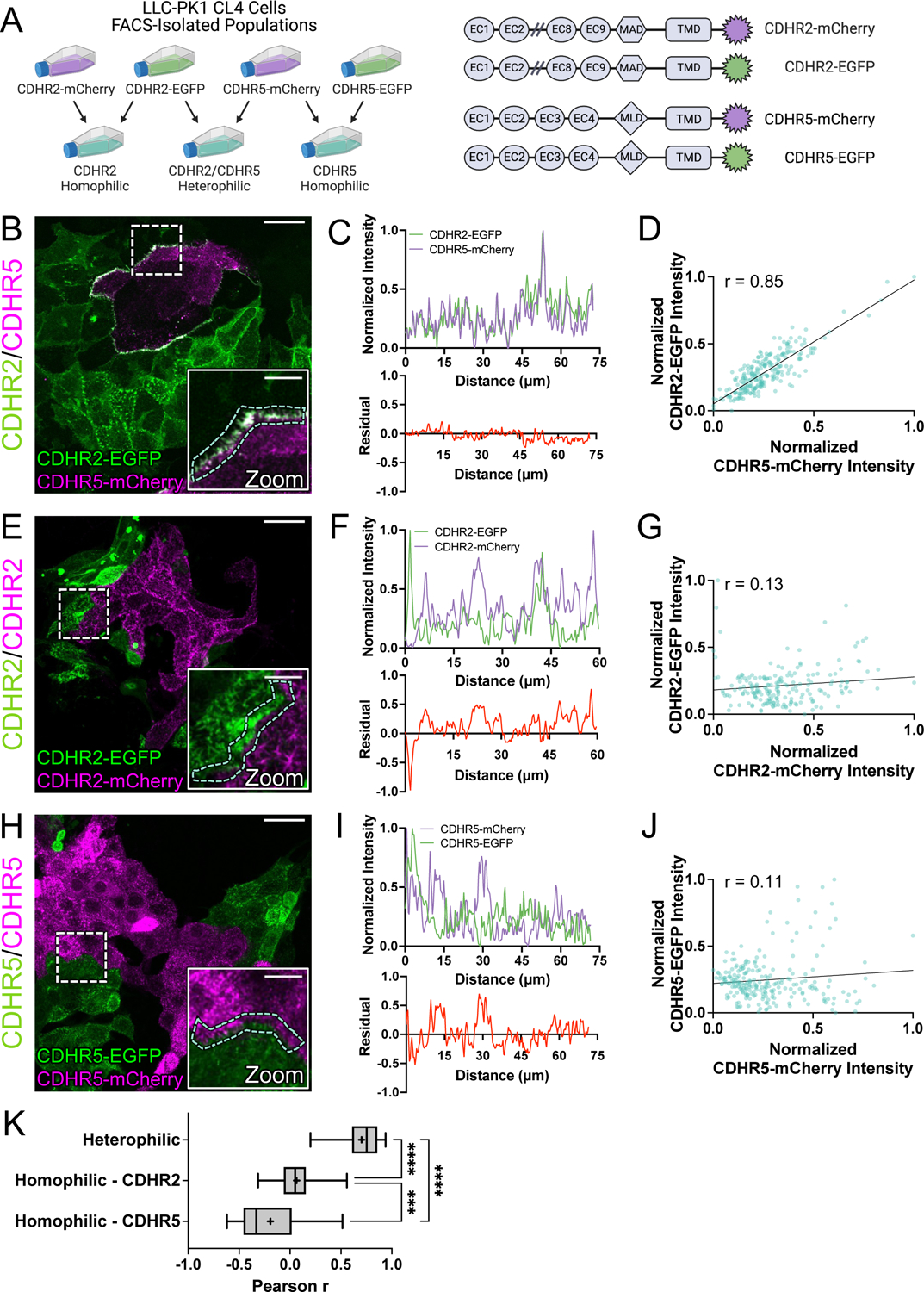
(A, left) Schematic depicting cell mixing method for the C-terminally tagged cadherin overexpression constructs (A, right). (B) MaxIP laser scanning confocal image of mixed heterophilic CDHR2-EGFP and CDHR5-mCherry CL4 cell populations. Dashed box represents zoom area and cyan dashed outline represents sample linescan. (C, top) Normalized fluorescence intensity plot taken from a representative linescan along the mixed cell interface. (C, bottom) Plotted difference (residual) of mCherry signal from EGFP signal from the top linescan plot. (D) Pearson’s r correlation plot from the linescan in (C); r = 0.85. (E) MaxIP of mixed homophilic CDHR2-EGFP and CDHR2-mCherry CL4 cells. (F-G) Representative linescan and respective Pearson’s r correlation; r = 0.13. (H) MaxIP of mixed homophilic CDHR5-EGFP and CDHR5-mCherry CL4 cells. (I-J) Representative linescan and respective Pearson’s r correlation; r = 0.11. (K) Combined Pearson’s r values from n = 30 individual linescans of each cell mixing scenario from 3 independent fixation and staining experiments (10 linescans per experiment). Mean Pearson’s r values are denoted by a “+” for each scenario where heterophilic r = 0.70, homophilic CDHR2 r = 0.07, and homophilic CDHR5 r = −0.19. Ordinary one-way ANOVA with multiple comparisons; **** p ≤ 0.0001 and *** p ≤ 0.001. Scale bars: 30 μm (B, E, H), 10 μm (zoom insets).
The cell mixing experiments described above indicate the heterophilic complexes of CDHR2 and CDHR5 are sufficient to drive microvilli alignment and accumulation at cell margins. We next asked if the endogenous IMAC protocadherins are required for transjunctional clustering of microvilli under normal conditions. To this end, we used CRISPR/Cas9 to generate CDHR2 KO in the CL4 line. Several KO lines were clonally selected and subsequently validated with genomic sequencing and antibody staining (Fig. S4). We then created mixed monolayers containing control and CDHR2 KO CL4 cells (Fig. S5A) and performed immunofluorescent staining to examine microvillar clustering; staining for endogenous CDHR2 enabled us to distinguish between control and CDHR2 KO cells in these samples. As expected, we noted robust accumulation of microvilli across control/control cell junctions (Fig. S5B, left panel). However, junctions formed between neighboring CDHR2 KO cells demonstrated little to no accumulation of microvilli; medial regions of the apical surface were also devoid of microvillar clusters (Fig. S5B, right panel). These data extend previous loss-of-function studies11,14 by revealing that CHDR2 is required for the formation of transjunctional IMACs, which are needed for the marginal accumulation of microvilli observed under normal conditions. Also consistent with previous studies13,14, CDHR2 KO cells demonstrated reduced CDHR5, most likely because this protocadherin becomes destabilized in the absence of its heterophilic binding partner. However, the remaining low level of CDHR5 was sufficient to support limited clustering of microvilli at junctions formed with control cells, presumably by complexing with CDHR2-presenting microvilli in those cells (Fig. S5B, middle panel).
Protocadherins in transjunctional IMACs exhibit limited turnover
Under normal conditions, epithelial cells express both CDHR2 and CDHR5, which target to the tips of all microvilli across the apical surface. Thus, heterophilic complexes are expected to form between the distal tips of microvilli in both the medial and marginal regions. However, the strong alignment of microvilli at cell-cell contacts in the heterophilic case outlined above led us to predict that transjunctional IMACs may be more stable relative to complexes that form medially. If true, this would offer a mechanistic explanation for the reduced motility of marginal microvilli, and in turn, the accumulation of microvilli at these sites. To determine if transjunctional IMACs are in fact longer lived than medial complexes, we performed fluorescence recovery after photobleaching (FRAP) analysis with CL4 monolayers formed using the cell mixing approach outlined above (Fig. 5A). Strikingly, photobleached ROIs positioned over junctional interfaces between heterophilic CDHR2-EGFP and CDHR5-mCherry expressing cells demonstrated extremely low signal recovery for both protocadherins (immobile fractions, 0.71 and 0.85, respectively; Figs. 6A,B and Video S2). In contrast, FRAP analysis of medially positioned ROIs on individual cells expressing both CDHR2-HALO and CDHR5-EGFP, revealed much lower immobile fractions for both protocadherins (0.47 and 0.56, respectively; Figs. 6C,D and Video S3). These results suggest that transjunctional IMACs formed between marginal microvilli are much longer lived relative to complexes formed between the tips of medial microvilli. We also examined recovery in photobleached ROIs positioned over junctional interfaces formed between homophilic CDHR2-EGFP and CDHR2-mCherry expressing cells (Figs. 6E,F and Video S4), as well as interfaces formed between homophilic CDHR5-EGFP and CDHR5-mCherry expressing cells (Figs. 6G,H and Video S5). Both homophilic scenarios exhibited higher levels of turnover and even lower immobile fractions. Together, these FRAP studies indicate that transjunctional IMACs composed of CDHR2 and CDHR5 are extremely stable, and the reduced turnover kinetics offer an explanation for the constrained motility and accumulation of microvilli observed at cell margins.
Figure 6: FRAP analysis suggests that heterophilic, transjunctional adhesion complexes are stable.
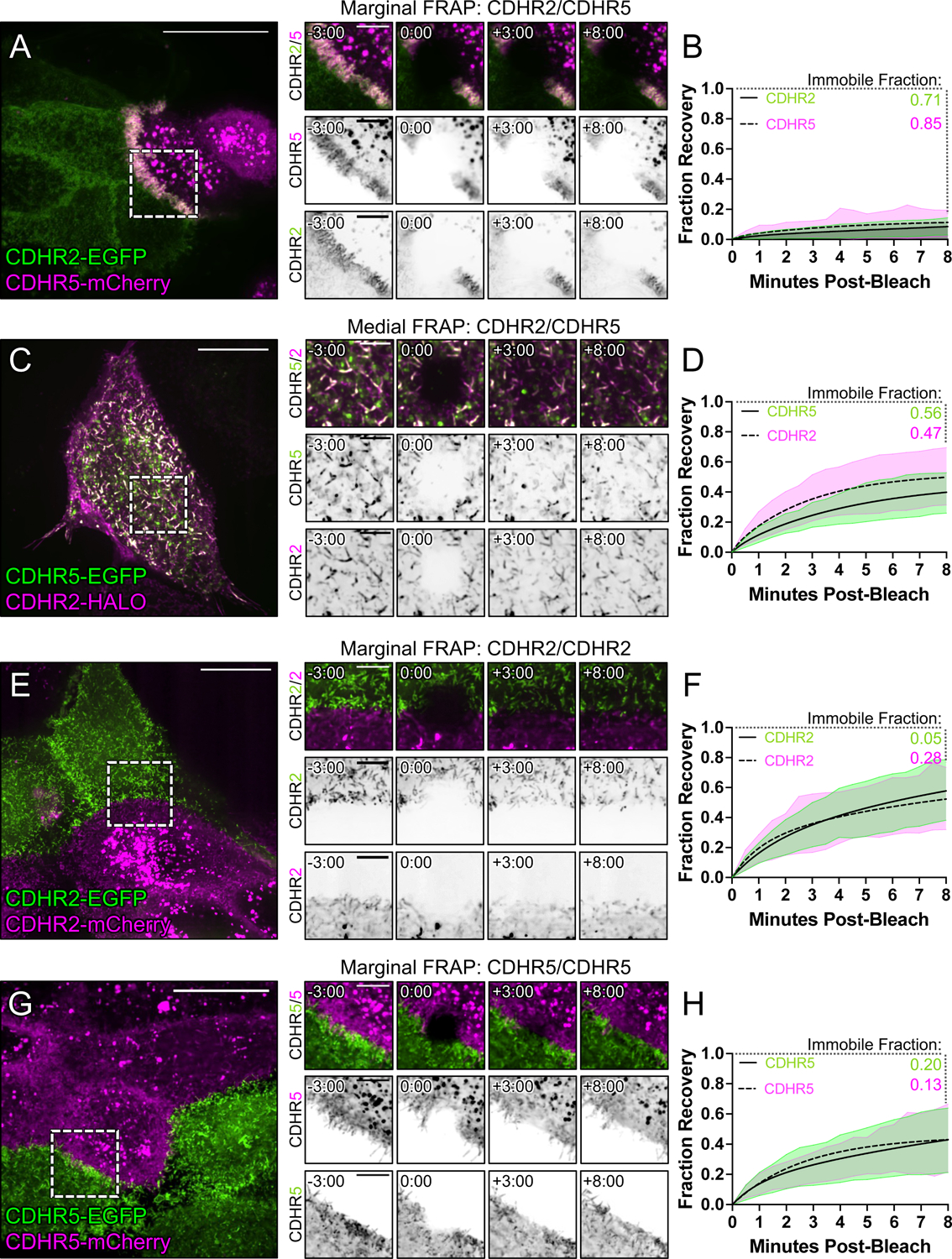
Mixed CL4 cells forming (A) marginal heterophilic, (C) medial heterophilic, (E) marginal homophilic CDHR2, and (G) marginal homophilic CDHR5 adhesion complex interfaces. Dashed boxes outline the photobleached ROI shown in the recovery montages on right. (B, D, F, H) Fluorescence recovery is plotted over the course of 8 minutes with the immobile fractions as written for each protein channel. Immobile fractions were calculated from a two-phase association curve by subtracting the predicted plateau from 1 (100% fluorescence recovery). All plots represent 3 independent FRAP experiments of n ≥ 20 ROIs from multiple cells. Scale bars: 20 μm (A, C, E, G), 5 μm (montages).
Myosin-2 dependent contractility stabilizes transjunctional clustering of microvilli
Why transjunctional IMACs exhibit longer lifetimes than medial IMACs remains unclear, but one possibility that is mechanical tension, which is high across cell junctions35, but low elsewhere on the apical surface, plays a role in specifically stabilizing these transjunctional complexes. Indeed, previous biophysical studies revealed catch bond-like behavior in cadherin family proteins, where the application of tensile force increases the lifetime of the adhesive complex36. Transjunctional tensile force has also been shown to promote the accumulation of junctional components, including E-cadherin, and driving the maturation of these contacts37,38. To determine if tension applied across cell junctions promotes the adhesive clustering and accumulation of microvilli at these sites, we inhibited non-muscle myosin-2, which accumulates in sub-apical networks and generates these large-scale forces39. For this experiment, we treated mCherry-ESPN expressing monolayers with blebbistatin and monitored marginal microvilli over 90 min using confocal microscopy. In alignment with previous studies that implicated myosin-2 in limiting microvillar core bundle length27,40, microvilli throughout the field elongated in response to blebbistatin compared to DMSO (Fig. S6A–B and C–D and Video S6). However, myosin-2 inhibition also markedly reduced the alignment and accumulation of microvilli at cell margins (Fig. S6C,F). These findings are consistent with the idea that mechanical tension applied across cell junctions plays a role, either directly or indirectly, in elongating the lifetime of transjunctional IMACs.
Microvilli accumulation at cell margins precedes accumulation in the medial zone during differentiation
Based on the stabilizing nature of transjunctional IMACs, we predicted that during differentiation, cells might assemble the brush border by packing microvilli inward from cell margins. To test this idea, we performed extended time-lapse imaging of CL4 cells expressing mCherry-ESPN to stoichiometrically label microvillar core actin bundles (Fig. 7A,B and Video S7)32. Comparing regional ESPN intensities on a per cell basis, we found that marginal ESPN intensity increased almost ~2-fold more than medial signal during 24 hrs of differentiation (Fig. 7C). These timelapse results are consistent with the idea that microvilli accumulate first at cell margins and then pack inwards from the edges of the cell over time as differentiation proceeds.
Figure 7: Long-term timelapse imaging reveals that microvilli accumulation at cell margins leads accumulation in medial regions during differentiation.
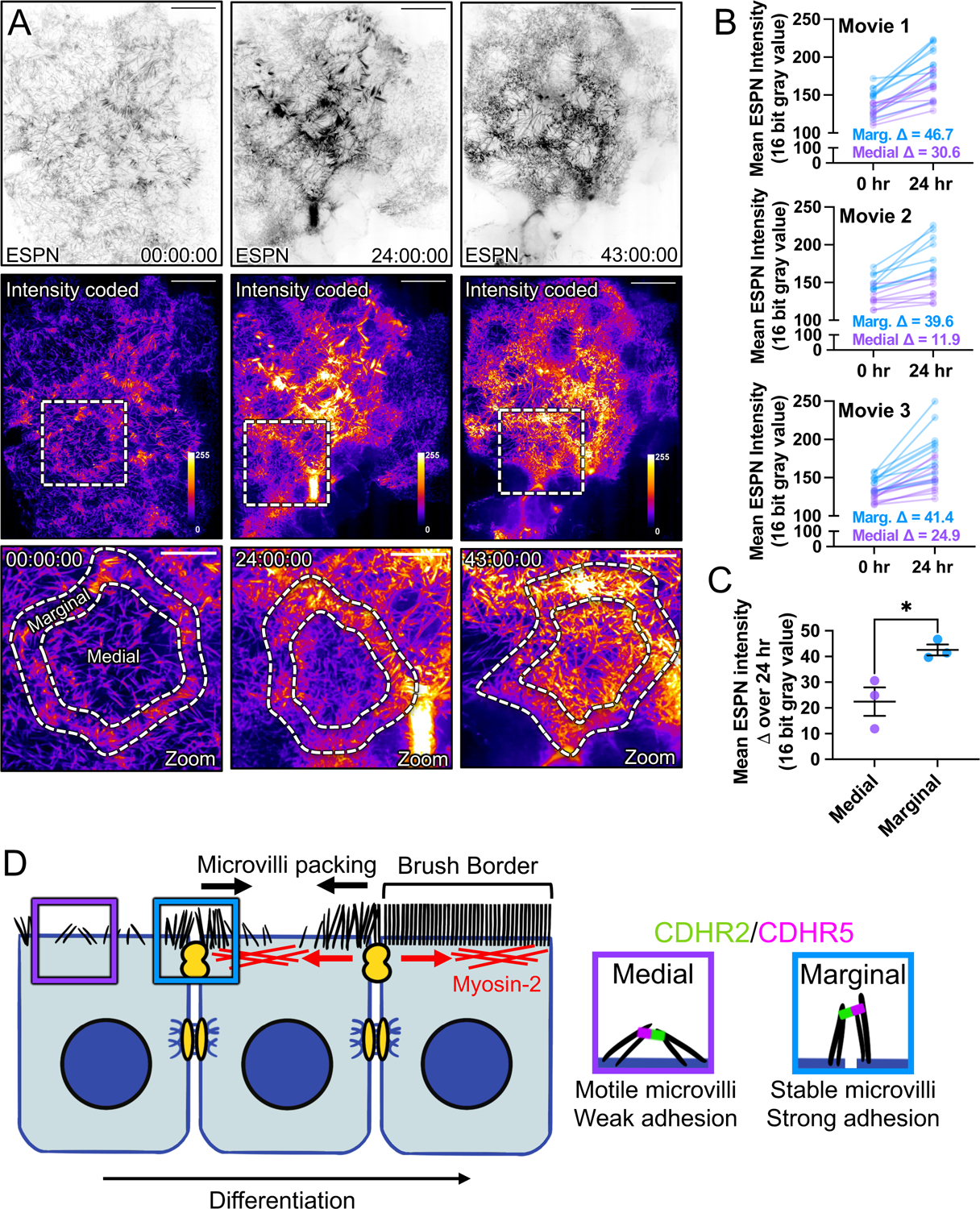
(A) MaxIP spinning disk confocal stills of live mCherry-ESPN expressing CL4 cells at t = 0, 24, and 43 hrs from a 43-hour acquisition. Intensity coded images show Fire LUT intensity profile of the mCherry-ESPN channel. Intensity scales from low (0; dark purple) to high (255; yellow/white) as denoted by LUT profile. Zooms at each time point are outlined by dashed boxes, with marginal and medial zones as marked. (B) Paired mean marginal and medial mCherry-ESPN intensities (16-bit gray value) at 0 and 24 hrs from three independent movies from n = 11 cells (movie 1), n = 8 cells (movie 2), and n = 11 cells (movie 3). The cut axes accounts for background mCherry signal. Mean Δ for the marginal and medial zones are denoted on each graph. (C) Mean ESPN intensity change over 24 hrs for medial (22.5 ± 5.7) and marginal (42.6 ± 3.5) zones. Error bars represent mean ± SEM. * p = 0.275 unpaired t-test. Scale bars: 20 μm (A-C), 10 μm (zooms). (D) An adhesion-based model for the marginal stabilization of microvilli during brush border assembly. Microvilli on nascent transporting epithelial cells initially organize into medial and marginal populations. Medial microvilli are highly motile while marginal microvilli are stable and stand at an orientation more vertical to the apical surface. Transjunctional CDHR2/CDHR5 heterophilic adhesion complexes span cell junctions and link marginal microvilli of neighboring cells. These complexes are long-lived, which leads to the accumulation of microvilli at the edges of cells that, in turn, may result in outside-in packing during differentiation.
DISCUSSION
Previous live imaging studies of epithelial cells at times points early in differentiation established that actively growing and newly formed microvilli are highly motile and unstable, undergoing rapid cycles of growth and collapse20,27. Those discoveries led us to question how dynamic, nascent microvilli are stabilized long-term to enable their timely accumulation in large numbers (i.e., thousands) by the end of differentiation. We approached this question by first examining the surface of the undifferentiated epithelial cells that line the interior of the intestinal crypt, where microvillar growth activity is high. Because the apical surface of cells in this region is not yet fully packed with protrusions, we were hoping to identify patterns in the distribution of nascent microvilli that might offer insight on underlying mechanisms of stability. Peering into the crypt is technically challenging given the tight confines of this invaginated compartment. Indeed, almost all previous ultrastructural studies of this region have been limited to conventional transmission EM of ultrathin sections41,42, which are difficult to interpret in the absence of 3D context. We worked around this obstacle using a combination of tissue fracturing and scanning EM, which allowed us to visualize the apical surface of immature intestinal epithelial cells within the crypt. Inspection of these images revealed that microvilli preferentially accumulate near the cell periphery at this point in differentiation. Cell culture models from the intestine (CACO-2BBE) and kidney (CL4) also demonstrated robust marginal accumulation of microvilli early in their maturation time course, suggesting that such patterning is not a function of the unique cellular packing geometry found in the crypt, nor is it tissue specific.
Accumulation at cell edges suggests that the marginal zone represents (i) a site of robust microvilli growth, (ii) a site of stabilization for nascent microvilli, or (iii) some combination of the two. Given the actin-rich junctional belt that surrounds the cell at the level of the terminal web43, it seems reasonable to expect that microvilli may grow more readily in this location. Although previous live imaging studies of CL4 cells characterized the properties of individual microvillar growth events20, those observations were limited to the medial regions of the cell where protrusion density is typically low; visualization of growth events in the marginal zone was confounded by the crowding of pre-existing microvilli in this region. While our data do not allow us to rule out the possibility that growth preferentially occurs at cell margins relative to medial regions, we were able to capture clear examples of clustered microvilli moving at a low angle relative to the cell surface, toward the edge of the cell and incorporating into the marginal population. Interestingly, these protrusions adopt the more vertical orientation of marginal microvilli upon reaching the cell edge. Because such upright orientation is a defining feature of microvilli in mature brush borders, the marginal population likely represents stabilized protrusions that persist into later stages of differentiation. Although we currently lack a method for tracking and measuring the lifetimes of individual microvilli over the course of days, our short-term tracking measurements using the tip marker, EPS8, confirm that marginal microvilli are less motile relative to medial microvilli, which is consistent with a physical capture mechanism near the cell edge.
Earlier work established that medial microvilli on the surface of mature villus enterocytes employ the protocadherins CDHR2 and CDHR5 to form intermicrovillar adhesion complexes (IMACs) that link the distal tips of neighboring microvilli11,15. Here we sought to test the possibility that IMACs form across cell junctions, between the protrusions that extend from neighboring cells. If so, this would offer a mechanistic explanation for the upright orientation and constrained motility that microvilli demonstrate at these sites, and potentially the long-term stabilization that enables microvillar accumulation on the apical surface in large numbers. Previous work in CACO-2BBE cells, native mouse intestinal tissue, and X-ray crystallography all indicate that the interacting ectodomains of CDHR2 and CDHR5 are structurally capable of spanning gaps up to 63 nm wide11,16, suggesting that they could easily reach across the ~15 nm tight junction between neighboring cells44. Indeed, in the current study, super-resolution imaging revealed that CDHR2 and CDHR5 span the intercellular space to form transjunctional IMACs that physically link marginal microvilli that extend from neighboring cells.
Does the formation of transjunctional IMACs explain the accumulation of microvilli at cell edges early in differentiation? Our mixed monolayer experiments with CL4 cells lacking CDHR2 or overexpressing tagged forms of CDHR2 and CDHR5, indicate that IMAC protocadherins are necessary and sufficient for the marginal accumulation of microvilli. Further, if transjunctional IMACs are more stable and exhibit longer lifetimes relative to IMACs that form medially, this would certainly offer a mechanistic underpinning for the increase in microvilli density at these sites. To test this hypothesis, we used the cell mixing approach to induce the formation of both homophilic and heterophilic transjunctional IMACs, to enable further characterization of their properties. FRAP analysis of the turnover dynamics of these complexes revealed that heterophilic (CDHR2/CDHR5) transjunctional IMACs are much longer lived relative to homophilic (CDHR2/CDHR2) complexes. These results from live epithelial cells echo previous in vitro data suggesting that homophilic (CDHR2/CDHR2) complexes are much weaker than heterophilic (CDHR2/CDHR5) complexes11. Interestingly, when we examined the dynamics of heterophilic complexes formed between microvilli in the medial population, we noted that these also turned over at a much higher rate relative to transjunctional heterophilic (CDHR2/CDHR5) complexes. Thus, the differential stability of transjunctional vs. medial IMACs indicated by our FRAP studies offers a mechanistic rationale for the accumulation of microvilli at cell margins.
Why are transjunctional IMACs more stable than those formed elsewhere on the apical surface? One possibility is that tensile forces, which are expected to be much higher across cell junctions relative to more medial regions of the cell surface35, exert a stabilizing effect on transjunctional CDHR2/CDHR5 adhesion. Indeed, transjunctional tension is known to drive the accumulation of junctional components including E-cadherin45 and F-actin46. Consistent with these ideas, we found that an inhibitor of myosin-2 contractility, blebbistatin, significantly reduced the accumulation of marginal microvilli in CL4 monolayers. A potential mechanism to explain such mechano-sensitivity in IMAC protocadherins is found in the biophysical literature on non-covalent bonds. When a tensile mechanical force is applied across a non-covalent bond formed between two proteins, the lifetime of that bond will be impacted in a way that depends on the structural nature of the bonding interface47,48. “Slip bonds” react to increased loading with a dramatic shortening of bond lifetime, whereas “catch bonds” respond by increasing bond lifetime; “ideal bonds” exhibit minimal response to mechanical loading36,47,49. Direct physical measurements provide strong evidence for catch bond behavior in structurally diverse proteins, ranging from myosin motor domains to cell surface molecules such as integrins50,51. Cadherins have been studied extensively in this regard and their bonding properties are complex. In the case of E-cadherin, adhesive interactions can exhibit slip or catch behavior depending on the conformation of the adhesive interface. In the canonical strand swapped conformation, E-cadherin exhibits slip bond behavior; while X-dimers of E-cadherin, which interact using a distinct extended structural interface demonstrate robust catch bond behavior36. In light of those findings, we speculate that IMACs might also exhibit catch bond properties. By bridging across cell junctions, transjunctional IMACs may be subject to higher tensile loads and exhibit increased adhesive lifetimes relative to IMACs that form elsewhere on the apical surface. Although rigorous testing of this concept awaits future biophysical studies, it is important to note that, based on the recently solved structures of mouse and human CDHR2 and CDHR5 ectodomains16, any catch bond behavior in the IMAC would emerge from a mechanism that is distinct from E-cadherin.
Given the adhesive capture of microvilli by stable transjunctional IMACs and our observations of marginal microvilli enrichment early in differentiation, we speculated that microvilli may pack from the margins of the apical surface inward during brush border assembly. To test this idea, we turned to multi-day time-lapse imaging of CL4 cells expressing mCherry-ESPN as a marker for microvilli. As expected, we noted that the marginal ESPN intensity was initially higher than in the medial region. After 24 hours of differentiation, the marginal region also demonstrated ~2-fold larger increase in signal relative to the medial zone, suggesting that microvillar packing density at the cell margin precedes packing of the interior apical surface (Fig. 7D). Moreover, intensity at the cell margin is consistently higher than medial signal, while both regions increase in intensity over the course of almost two days of imaging.
While previous work established that the IMAC is critical for maintaining brush border structure on mature enterocytes14, the current study highlights a role for this complex in apical surface maturation, by driving interactions between microvilli of neighboring cells. In the intestinal tract and other transporting epithelial tissue, cell-cell contacts are essential for barrier function and the maintenance of physical compartmentalization. Interestingly, Crohn’s disease patients exhibit a decrease in CDHR2 and CDHR5 mRNA expression52 while also experiencing increased intestinal permeability53. Transjunctional adhesion complexes may also form an additional layer of protection against colonizing pathogens. Infection by related pathogens Enteropathogenic and Enterohemorrhagic Escherichia coli (EPEC and EHEC) is characterized by effacement of brush border microvilli and F-actin pedestal formation54. CDHR2 has been identified as one of the initial EHEC targets during infection, which results in a significant decrease in CDHR2 expression55. Past reports on EPEC infection also show bacterial localization over cell junctions54,56. Exploring roles for transjunctional IMACs in maintaining epithelial barrier function in intestinal disease and infection should be a central goal of future studies.
LIMITATIONS OF THE STUDY
The main limitations to this study are technical in nature. Here we sought to investigate the dynamic apical surface remodeling that takes place during epithelial differentiation. One of the defining features of this process, and the focus of this study, is the accumulation of large numbers of microvilli, which serves to expand plasma membrane area and in turn, the functional capacity of the apical surface. Finding appropriate epithelial cell lines to model this complex process in the laboratory has been a challenge for many years. In this study, we leveraged two of the most highly characterized models for studying epithelial differentiation and apical morphogenesis – the LLC-PK-CL4 line from kidney and the CACO-2BBE line from the intestinal tract. Both models are grown as monolayers and progressively differentiate after reaching confluence. While these lines do exhibit features of apical maturation that are similar to native tissues (e.g. eventual accumulation of large numbers of microvilli, CDHR2/CDHR5 adhesion-dependent packing and organization of microvilli), they also demonstrate shortcomings that limit their utility. These include the prolonged time required for differentiation (several days in culture vs. hours in vivo), the artificial flattening of the monolayer by the glass coverslip substrate (required for high resolution microscopy), and the lack of other epithelial and non-epithelial cell types that may be relevant for promoting this process. Reducing the impact of these limitations will require the development of intravital imaging approaches in mice engineered to express key fluorescent reporters, or 2D or 3D organoid cultures derived from these animals.
STAR*METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Matthew J. Tyska (matthew.tyska@vanderbilt.edu).
Materials availability
Plasmids generated in this study will be made available from the lead contact on request.
Data code availability
No large-scale datasets or new code were generated in this study.
EXPERIMENTAL MODEL AND PARTICIPANT DETAILS
Cell culture models
LLC-PK1-CL4 (porcine kidney proximal tubule) cells were grown in 1X high glucose DMEM containing 2mM L-glutamine (Corning #10–013-CV) supplemented with 1% L-glutamine (Corning # 25–005-CI) and 10% fetal bovine serum (FBS) (R&D Systems). CACO-2BBE (human colonic adenocarcinoma) cells were grown in the same medium but supplemented with 20% FBS. Cells were maintained in culture incubated at 37°C and 5% CO2. Cells were tested for mycoplasma monthly using the MycoAlert PLUS Mycoplasma Detection Kit (Lonza #LT07–710) and/or MycoStrip Mycoplasma Dectection Kit (InvivoGen #rep-mys-50).
Animal models
CDHR2-EGFP mice were created in collaboration with the Vanderbilt Genome Editing Resource. A C57Bl/6N strain containing a CDHR2 C-terminal EGFP sequence insertion. [crRNA sequence: TGGACACCACAGATCTGTGA] Ribonucleoprotein complexes containing ctRNA and WT SpCas9 protein were targeted to the C-terminus of CDHR2 were assembled and injected with a single stranded 944 nt DNA donor into 1-cell C57Bl/6N embryos. crRNA, tracrRNA, and WT SpCas9 protein was sourced from Millipore Sigma. The single stranded DNA was produced by Genewiz. Pups were screened for CDHR2-EGFP sequence insertions by PCR and validated by Sanger sequencing (see Key Resources Table and Fig. S1). Animal experiments were carried out in accordance with Vanderbilt University Medical Center Institutional Animal Care and Use Committee guidelines under IACUC Protocol ID#: M1600206–02.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-PCLKC [CDHR2] (mouse) [1:25] | Abnova | Cat# H00054825M01; RRID: AB_490042 |
| anti-CDHR5 (rabbit) [1:250] | Sigma-Aldrich | Cat# HPA009173; RRID: AB_1079429 |
| anti-GFP (chicken) [1:200] | Aves Labs | Cat# GFP-1020; RRID: AB_10000240 |
| anti-ZO-1 (rat) [1:100] | Millipore | Cat# MABT11; RRID: AB_10616098 |
| anti-ZO-1 (rabbit) [1:50] | ThermoFisher | Cat# 61-7300; RRID: AB_2533938 |
| Goat anti-mouse Alexa Fluor 488 F(ab’)2 fragment [1:1000] | ThermoFisher | Cat# A-11017; RRID: AB_2534084 |
| Goat anti-rabbit Alexa Fluor 488 F(ab’)2 fragment [1:1000] | ThermoFisher | Cat# A-11070; RRID: AB_2534114 |
| Goat anti-rabbit Alexa Fluor 568 F(ab’)2 fragment [1:1000] | ThermoFisher | Cat# A-21069; RRID: AB_253573 |
| Goat anti-chicken Alexa Fluor 488 IgG (H+L) [1:1000] | ThermoFisher | Cat# A-11039; RRID: AB_142924 |
| Goat anti-rat Alexa Fluor 647 IgG (H+L) [1:200] | ThermoFisher | Cat# A-21247; RRID: AB_141778 |
| Bacterial and virus strains | ||
| E. coli DH5-Alpha competent cells | Molecular Cell Biology Resource Core, Vanderbilt Medical Center | Item# DH5 Alpha |
| Chemicals, peptides, and recombinant proteins | ||
| Alexa Fluor Plus 405 Phalloidin [1:200] | ThermoFisher | Cat# A30104 |
| Alexa Fluor 647 Phalloidin [1:200] | ThermoFisher | Cat# A22287 |
| Janelia Fluor 635 (JF635) | Janelia | N/A |
| Paraformaldehyde, 16% | Electron Microscopy Sciences | Cat# 15710 |
| Triton X-100 | Sigma | Cat# T8787 |
| ProLong Gold Antifade Reagent | Invitrogen | Cat# P36930 |
| FuGENE 6 | Promega | Cat# E2691 |
| Lipofectamine 2000 | ThermoFisher | Cat# 11668019 |
| Polybrene Infection Reagent | Sigma-Aldrich | Cat# TR-1003 |
| G418 Sulfate | Gold Biotechnology | Cat#G-418-1 |
| Puromycin | Gold Biotechnology | Cat# P-600-100 |
| Antibiotic-antimycotic (anti-anti) | Gibco | Cat# 15240062 |
| (-)-Blebbistatin | Selleck Chemicals | Cat# S7099 |
| DMSO | Sigma | Cat# D8418 |
| BsmBI-v2 enzyme | New England Biolabs | Cat# R0739 |
| Q5 High-Fidelity DNA Polymerase | New England Biolabs | Cat# M0491L |
| Q5 High GC Enhancer | New England Biolabs | Cat# B9028A |
| Q5 Reaction Buffer | New England Biolabs | Cat# B9027S |
| Deoxynucleotide Mix, 10mM | Sigma-Aldrich | Cat# D7295 |
| EDTA, 0.5M | Corning | Cat# 46-034-Cl |
| Nuclei Lysis Solution | Promega | Cat# A7941 |
| Protein Precipitation Solution | Promega | Cat# A795A |
| Proteinase K | Sigma | Cat# P4850 |
| Molecular Biology Grade Isopropanol | Fisher Scientific | Cat# BP2618500 |
| 70% Ethanol | Macherey-Nagel | N/A |
| TE-EF Redissolving Buffer | Macherey-Nagel | Cat# 740797.1 |
| Glutaraldehyde, 25% | Electron Microscopy Sciences | Cat# 16220 |
| Tannic Acid | Electron Microscopy Sciences | Cat# 21700 |
| Osmium Tetroxide | Electron Microscopy Sciences | Cat# 19112 |
| Experimental models: Cell lines | ||
| LLC-PK1-CL4 | Gift from Dr. Carolyn Slayman (Yale University) | N/A |
| CACO-2BBE | ATCC | Cat# CRL-2102 |
| Experimental models: Organisms/strains | ||
| CDHR2-EGFP mouse | Vanderbilt Genome Editing Resource | N/A |
| Oligonucleotides | ||
| CDHR2-Fwd: ATGGCCCAGCTATGGCTG | This paper | N/A |
| CDHR2-Rev: CAGGTCCGTGGTGTCCAGG | This paper | N/A |
| gRNA Exon 4(1) Sense: CACCGTAATGTCATCCGGAATACTG | This paper | N/A |
| gRNA Exon 4(1) Antisense: AAACCAGTATTCCGGATGACATTAC | This paper | N/A |
| gRNA Exon 4(2) Sense: CACCGGCCAACCTTCTGGACTACG | This paper | N/A |
| gRNA Exon 4(2) Antisense: AAACCGTAGTCCAGAAGGTTGGCC | This paper | N/A |
| CDHR2 KO Seq Fwd (cells): GTTTTCATGTCTTGGCCCTTCTAAC | This paper | N/A |
| CDHR2 KO Seq Rev (cells): CTGTGTGACCAAAAATGGACAAGTG | This paper | N/A |
| Cdhr2 Fwd1 (mouse): AACCCACCTGTACCACCCTTG | This paper | N/A |
| Cdhr2 eGFP Fwd (mouse): CCGACAACCACTACCTGAGCAC | This paper | N/A |
| Cdhr2 Rev1 (mouse): GTATTGGGAACAGGATGGAGGTC | This paper | N/A |
| Recombinant DNA | ||
| pmCherry-Espin (ESPN) | Gift from Dr. James Bartles, NWU | N/A |
| pLVX-mCherry-Espin (ESPN) | [20] | N/A |
| pEGFP-N3-CDHR2 (PCDH24-EGFP) | [11] | N/A |
| pmCherry-N3-CDHR2 | Tyska Laboratory | N/A |
| pEGFP-N3-CDHR5 | Tyska Laboratory | N/A |
| pmCherry-N3-CDHR5 | Tyska Laboratory | N/A |
| pHALO-N3-CDHR2 | This paper | N/A |
| pLentiCRISPRv2 | Addgene | Cat# 52961 |
| Software and algorithms | ||
| FIJI | https://fiji.sc | N/A |
| NIS AR Elements Analysis | Nikon | N/A |
| Prism 9 | GraphPad | N/A |
| Benchling CRISPR Guide design tool | Benchling | N/A |
| Inference of CRISPR Edits (ICE) | Synthego | N/A |
| BioRender | Biorender | N/A |
| Other | ||
| MycoAlert PLUS Mycoplasma Detection Kit | Lonza | Cat# LT07-710 |
| MycoStrip™ - Mycoplasma Detection Kit | InvivoGen | Cat# rep-mys-50 |
| pCR™8/GW/TOPO™ TA Cloning Kit | Invitrogen | Cat# K250020 |
| Gateway™ Vector Conversion System | Invitrogen | Cat# 11828029 |
| 35 mm #1.5 glass bottom dishes | CellVis | Cat# D35-20-1.5-N |
METHOD DETAILS
Frozen section tissue preparation
The proximal segment (duodenum to jejunum) of the mouse intestinal tube was excised and flushed with cold 1X phosphate-buffered saline (PBS). One end of the tube was clamped with a hemostat and the tube was filled with room temperature 2% paraformaldehyde (PFA) (Electron Microscopy Sciences) with a syringe and metal cannula. The other end of the tube was clamped with a hemostat and the tissue was laid in a petri dish containing excess 2% PFA and incubated for 15 minutes at room temperature. Hemostats were removed and the tissue was cut lengthwise into one flat piece. Tissue was then sub dissected into ~2mm2 pieces and fixed for an additional 30 minutes in a vial of 2% PFA at room temperature. After fixation, the tissue was washed 3 times with PBS and then placed, villi-side down, into a vial of cold 30% sucrose/1% sodium azide. The tissue was placed at 4°C, overnight until sections sank to the bottom of the tube. The next day, sections were passed through 3 separate blocks of optimal cutting temperature (OCT) compound (Electron Microscopy Sciences) to wash off the sucrose solution, oriented with villi parallel to the lab bench in a fresh block of OCT, and snap frozen in dry ice-cooled acetone. Samples were cut into 10 μm thin sections using a Leica CM1950 cryostat and mounted on plasma-cleaned #1.5H precision coverslips (Thorlabs). Coverslips were stored at −20°C until staining.
Frozen section immunofluorescence
Coverslips were thawed to room temperature and rinsed twice with 1X PBS to remove OCT. Sections were permeabilized with 0.2% Triton X-100 (diluted in PBS) for 10 minutes at room temperature. Sections were then rinsed once with PBS at room temperature and blocked in 10% bovine serum albumin (BSA) for 2 hours at 37°C in a humidified chamber.
After rinsing with PBS, primary antibody (diluted in 1% BSA) was applied overnight at 4°C in a humidified chamber. The next day, sections were rinsed with 1X PBS 4 times and secondary antibody (diluted in 1% BSA) was applied for 2 hours at room temperature in a dark, humidified chamber. Sections were rinsed 4 times with 1X PBS and coverslips were mounted onto glass slides with ProLong Gold. The following antibodies and dilutions were used for EGFP-CDHR2 mouse frozen section staining: anti-GFP (chicken Aves #GFP-1020), 1:200; anti-CDHR5 (rabbit, Sigma #HPA009173), 1:250; anti-ZO-1 clone R40.76 (rat, EMD Millipore Sigma #MABT11), 1:100; Alexa Fluor goat anti-chicken 488 (Invitrogen #A-11039), 1:1000; Alexa Fluor F(ab’)2 fragment goat anti-rabbit 568 (Invitrogen #A21069), 1:1000; Alexa Fluor goat anti-rat 647 (Invitrogen #A21247), 1:200; and Alexa Fluor Plus 405 Phalloidin (Invitrogen #A30104), 1:200 for actin staining. The secondary antibodies, not including phalloidin, were spun down for 10 minutes at 4°C and 21 × g prior to using.
Cloning and constructs
A C terminally tagged pHALO-N3-CDHR2 (CDHR2-HALO) construct was generated by taking full length CDHR2 via PCR from pEGFP-N3-PCDH24 (CDHR2-EGFP) with the primers CDHR2-Fwd: ATGGCCCAGCTATGGCTG and CDHR2-Rev: CAGGTCCGTGGTGTCCAGG. The product was then TOPO cloned into the pCR8/GW/TOPO vector (Invitrogen #K250020) and then placed into the pHALO-N3 backbone, adapted for Gateway cloning using the Gateway conversion kit (Invitrogen #11828029). All other overexpression constructs listed in this paper were previously created and/or reported as noted in the key resources table.
Cell line generation
Cells expressing one plasmid were transfected with FuGENE 6 (Promega #E2691) at a FuGENE:DNA (μL:μg) ratio of 3:1 following the reagent protocol in a T25 cell culture flask. The next day, all cells were split up to a T75 flask with the addition of 1mg/mL G418 sulfate for antibiotic selection. Cells were maintained in culture under constant G418 selection to create a stably expressing cell line. Cells co-expressing two plasmids were transiently transfected with Lipofectamine 2000 (Invitrogen #11668019) according to the manufacturer’s protocol. The next day, cells were split to plasma-cleaned 35mm glass bottom dishes (CellVis #D35-20-1.5-N) for subsequent imaging. The EGFP-EPS8/mCherry-ESPN CL4 stable cell line was previously created20 by transducing a G418-selected EGFP-EPS8 stable cell line with lentiviral mCherry-ESPN followed by 10 μg/mL puromycin selection. See citation for detailed protocol. The Halo-CDHR2/EGFP-CDHR5 co-expressing CL4 cells were a transient transfection, and not stably selected.
CRISPR CDHR2 knockout generation
Guide RNA’s targeting the porcine CDHR2 genetic locus were designed using the CRISPR Guide Benchling tool for single guides 20 bp in length with a NGG PAM within exon 4 of CDHR2. Two guides were selected based on the highest on-target scores provided by Benchling and assembled in silico into the lentiCRISPR v2 plasmid (Addgene #52961). Oligo sequences including overhangs for cloning (see Feng Zhang cloning protocol via Addgene) were exported and synthesized by Sigma-Aldrich via Vanderbilt’s DNA Core. Oligos were resuspended to 100 μM in molecular grade water. Cloning was performed following the lentiCRISPR v2 cloning protocol (Addgene, Feng Zhang). In brief, the plasmid was digested with BsmBI-v2 (NEB #R0739) and gel purified (Macherey-Nagel #740609.50). Phosphorylated and annealed gRNA oligos were ligated into the purified digested plasmid and the product was transformed into NEB Stable competent E. coli (NEB #C3040H). Bacterial clones were grown on carbenicillin agar plates, isolated, DNA purified, and sequenced using a U6 universal sequencing primer to validate correct gRNA insertion. CRISPR virus was harvested from HEK293FT cells as previously described30. Wildtype CL4 cells were transduced at 70% confluence in a 6-well plate with either gRNA 4(1), gRNA 4(2), or Control (no gRNA) lentiCRISPRv2 virus plus 10 μg/mL Polybrene and placed under 10 μg/mL puromycin selection 48 hours post-transduction. Cells were maintained for 3 weeks under puromycin selection alongside a non-transduced population of WT CL4 cells with puromycin as a marker for 100% cell death.
CRISPR clone screening
After puromycin selection, the mixed KO populations (gRNA exon 4.1 and gRNA exon 4.2) were seeded into a 24 well plate at a density of ≤ 1 cell per well. Once single cells had grown into small islands, one per well representing individual clonal populations, they were trypsinized and expanded for screening via antibody staining and DNA sequencing.
DNA Extraction:
Clonal “KO” populations were seeded in 6-well plates, 2 wells per clone, and grown to 100% confluence. Prior to DNA extraction, cells were rinsed in cold 1X DPBS (Corning Cat# 21–031-CV). A mixture of 484 μL Nuclei Lysis Solution (Promega Cat# A795A), 116 μL 0.5M EDTA (Corning Cat# 46–034-Cl), and 10 μL (20 mg/mL) Proteinase K (Sigma Cat# P4850) was added to the cells on ice, 300 μL per well. Cells were scraped and collected in 2 mL Eppendorf tubes and incubated for 1 hour in a 55°C water bath. 200 μL Protein Precipitation Solution (Promega Cat# A795A) was added, cells were vortexed for 20 s, and placed on ice for 5 min. After centrifugation for 10 min at 11,000Xg, the supernatant was collected, and 600 μL molecular grade isopropanol (Fisher Scientific Cat# BP2618500) was added and the tube was mixed by inverting. After pelleting by centrifugation for 5 min at 11,000Xg, the pellet was washed in 70% ethanol (Macherey Nagel) and resuspended in 25 μL TE Redissolving Buffer (Macherey Nagel Cat# 740797.1) and DNA concentration was measured on a NanoDrop.
Genomic PCR:
A region of 610bp encompassing exons 4 and 5 of the porcine CDHR2 genomic sequence (see Key Resources Table for primers) was generated by PCR following the Q5 High-Fidelity Polymerase (New England Biolabs Cat# M0491L) protocol with a genomic template of 200 ng from the clonal “KO” populations. An initial denaturation step of 3 min (98°C) was important for genomic PCR. The PCR products were run on a 1% agarose gel +EtBr (Bio-Rad Cat# 161–0433) and gel purified (Macherey-Nagel #740609.50). Samples were sent to Genewiz (Azenta Life Sciences) premixed with the same primers used for PCR for sequencing following Genewiz submission guidelines for a purified PCR product. The Synthego ICE CRISPR analysis tool was used to evaluate trace files from the KO clone sequencing results (Fig S4B)57.
Cell immunofluorescence
Prior to fixation and staining, CL4 and CACO-2BBE cells were grown to n days post-confluent (DPC) on acid-washed 22×22 mm #1.5H coverslips (Globe Scientific) in a 6-well plate to a time point with apical polarity representative of their native tissue, 3 DPC and 12 DPC, respectively. First, cells were rinsed in warm 1X PBS and fixed in 4% PFA for 15 minutes at 37°C. Cells were then washed three times, 5 minutes each, with 1X PBS and permeabilized with 0.1% Triton X-100 for 10 minutes at room temperature. 5% BSA was added for 1 hour at 37°C as blocking solution. After rinsing with 1X PBS, primary antibody (diluted in 1% BSA) was added for 1 hour at 37°C. Labeling with primary antibody was followed by washing 4 times, 5 minutes each, with 1X PBS. Secondary antibody (diluted in 1% BSA) was then applied for 1 hour at room temperature in the dark. After incubation in secondary antibody, cells were washed 4 times, 5 minutes each with 1X PBS and coverslips were mounted on glass slides with ProLong Gold. The following antibodies and dilutions were used for cell staining: anti-PCLKC (CDHR2) (mouse, Abnova #H00054825-M01), 1:25; anti-CDHR5 (Rabbit, Sigma #HPA009173), 1:250; anti-ZO-1 clone R40.76 in CL4 (rat, EMD Millipore Sigma #MABT11), 1:100; anti-ZO-1 in CACO-2BBE (rabbit, Invitrogen #61–7300), 1:50; Alexa Fluor F(ab’)2 fragment goat anti-mouse 488 (Invitrogen #A11017) and goat anti-rabbit 568 (Invitrogen #A21069), 1:1000; Alexa Fluor goat anti-rat 647 (Invitrogen #A21247), 1:200; and Alexa Fluor Plus 405 Phalloidin (Invitrogen # A30104) or Alexa Fluor 647 Phalloidin (Invitrogen # A22287), 1:200 for actin staining. The secondary antibodies, not including phalloidin, were spun down for 10 minutes at 4°C and 21 × g prior to using.
Fluorescence-activated cell sorting (FACS)
Cells were spun down into a pellet and resuspended in “pre-sort medium” containing Phenol free 1X DMEM (Gibco #21063–029) plus 5% FBS, and 1% L-glutamine. Cells were sorted by Vanderbilt University Medical Center’s Flow Cytometry Shared Resource on a 5-Laser FACS Aria III system with a 100 μm sized nozzle. All fluorescent positive cells (Fig. S3) were deposited into a single well of a 6-well plate containing “post-sort medium” of 1X DMEM (Corning #10–013-CV) with Phenol red, 10% FBS, 1% L-glutamine, and 10μL/mL anti-anti (Gibco #15240062). 24 hours post-sort, the media was changed to CL4 culture media (as detailed in cell culture methods) and 1 mg/mL G418 was added for maintaining stable plasmid overexpression. Sorted cell lines were maintained in this media and under antibiotic selection.
Cell mixing experiments
Fluorescently sorted CL4 cell populations were grown independently and under G418 antibiotic selective pressure to ~80% confluence, trypsinized, and resuspended in CL4 media to a density of ~850,000 cells/mL. 250 μL of each cell population were seeded in plasma-cleaned glass-bottom dishes or onto acid washed coverslips at a density of ~400,000 total cells at a mixing ratio of 1:1 (e.g. CDHR2-EGFP cells were mixed with CDHR5-mCherry cells). A similar mixing method was used for CRISPR Control and CDHR2 KO CL4 cells, at a mixing ration of 1:1. Immediately after seeding, cell populations were thoroughly mixed by pipette. Cells were grown to 3DPC for fixed cell staining or for 1 day for live cell imaging (FRAP).
Fixed sample microscopy
Laser scanning confocal microscopy was performed on a Nikon A1 microscope equipped with 488 nm, 568 nm, and 647 nm LASERs. Mixed CL4 cell populations for linescan analysis were imaged using a Plan Apo 40x/1.3 NA oil immersion objective. CACO-2BBE cells were imaged using an Apo TIRF 100x/1.49 NA TIRF oil immersion objective. Spinning disk confocal microscopy was used for CRISPR CDHR2 KO CL4 cell imaging and intensity analysis (Fig S4 C–E) using a Nikon Ti2 inverted light microscope with a Yokogawa CSU-W1 spinning disk head, a Photometrics Prime 95B sCMOS camera, and four excitation LASERs (488, 568, 647, and 405 nm) and a 60X/1.49 NA TIRF oil immersion objective. Structured illumination microscopy (SIM) was used for imaging frozen tissue sections and fixed cells with a Nikon N-SIM equipped with 405, 488, 468, and 647 nm LASERs, an Andor DU-897 EMCCD camera, and a TIRF 100X/1.49 NA TIRF oil immersion objective. All SIM images were reconstructed using Nikon Elements software.
Live imaging microscopy
Prior to live cell imaging, cells growing in 35mm glass bottom dishes were rinsed once with 1X DPBS (Corning #21–031-CV). FluoroBrite imaging media (Gibco #A18967–01) supplemented with 10% FBS and 1% L-glutamine was added to the dish. For CL4 cells expressing Halo-CDHR2, Janelia Fluor 635 dye (Janelia) was added to the FluoroBrite media at a concentration of 50 nM for 1 hour at 37°C immediately prior to imaging.
Spinning disk confocal microscopy was performed using a Nikon Ti2 inverted light microscope with a Yokogawa CSU-X1 spinning disk head, a Photometrics Prime 95B or Hamamatsu Fusion BT sCMOS camera, and three excitation LASERs (488, 568 and 647 nm). A 100X/1.49 NA TIRF oil immersion objective was used for all acquisitions. A stage incubator (Tokai Hit) maintained cells in a humidified environment at 37°C with 5% CO2.
Blebbistatin treatment
A 10 mM stock solution was created by dissolving Blebbistatin (Blebb) in DMSO (Sigma Cat# D8418) according to manufacturer’s guidelines (Selleck Chemicals Cat# S7099). Stable mCherry-ESPN CL4 cells were seeded on 35 mm glass bottom dishes and grown to 100% confluence. Immediately prior to live imaging, cells were rinsed once with 1X DPBS and 1mL of CL4 growth media was added to the dish. Cells were placed on the spinning disk confocal X1 microscope, described above, and live acquisition was started. After 5 min of baseline imaging, at 30 s intervals, 1mL of CL4 media containing 40 μM Blebb was added to the existing 1mL of media on the microscope, a final concentration of 20 μM. Control DMSO treatments were similarly performed, using the same volume of DMSO as Blebb in the 1mL of added media. Cells were imaged for a total of 1.5 hr, at 30 s intervals, with Blebb or DMSO.
Fluorescence recovery after photobleaching (FRAP)
A square ROI was drawn in Nikon Elements at marginal and/or medial microvilli regions. A stimulating 405 nm LASER controlled by a Bruker mini-scanner set at 70% power and a dwell time of 40 us was targeted to each ROI after the first 3 frames of the movie acquisition. Two ND time acquisitions were used for imaging fluorescence recovery at intervals of 15 s for 3 minutes, followed by intervals of 30 s for 10 minutes.
Electron microscopy – CACO-2BBE and LLC-PK1-CL4 cells and tissue
To prepare samples, cells were plated on glass coverslips washed once with warm SEM buffer (0.1M HEPES, pH 7.3) supplemented with 2 mM CaCl2, then fixed with 2.5% glutaraldehyde and then 4% paraformaldehyde in SEM buffer supplemented with 2mM CaCl2. Samples were washed in SEM buffer, then incubated in 1% tannic acid, washed with ddH2O, incubated with 1% OsO4, washed with ddH2O, incubated with 1% uranyl acetate, then washed with ddH2O. Samples were dehydrated in a graded ethanol series. Sampels were then dried using critical point drying and mounted on aluminum stubs and coated with gold/palladium using a sputter coater. SEM imaging was performed using Quanta 250 Environmental-SEM operated in high vacuum mode with an accelerating voltage of 5–10 kV, or imaged on a Zeiss Crossbeam 550 at 2keV. All reagents were purchased from Electron Microscopy Sciences. For more detailed methods, see27. TEM of mouse intestine (Fig. S2B) was performed as previously described14
Electron microscopy – crypt-villus axis
For SEM imaging of intestinal sections, immediately after euthanasia, ~5 mm murine duodenal sections were quickly fixed in a large volume (10 mL) of 2.5% glutaraldehyde and 4% paraformaldehyde in SEM buffer (described above). Sections were then washed in SEM buffer prior to embedding in Tissue-Tek OCT compound (Sakura Finetek #4583). To ensure stable support of the complex architecture within the explant lumens, samples were gently moved through 3 rounds of fresh OCT compound with gentle manipulation to ensure penetration of the OCT. Samples were then placed in cryomolds (with OCT) and frozen over a dry ice/ethanol slurry. Molds were stored at −80C once fully frozen. Frozen explants were subsequently sectioned on a Leica CM1950 crytostat at 50 μm/section and melted onto stainless steel AFM specimen discs (Electron Microscopy Sciences). Next, explant sections and disks were immersed in 1% OsO4, washed in ddH2O, then dehydrated through graded ethanol series. Of note, it was most common to experience detachment of the section from the AFM disk during the OsO4 and dehydration steps. Detached sections were recovered and gently adhered to an aluminum SEM specimen stub via conductive adhesive tab. SEM imaging was performed on a Quanta 250 environmental SEM, as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Microvilli orientation measurements
In Fiji, the first frames of three independent mCherry-ESPN CL4 cell movies were used for orientation measurements shown in Fig. 2B. A thin, rectangular ROI (height 12 pixels) was drawn across an entire cell to encompass both marginal and medial areas (sample ROI Fig 2A, dotted box). The ROI hyperstack was duplicated and 3D projected with rotation around the X axis. The marginal zone was defined as 10% of the ROI, at either end of the image (overlying cell junctions) and the medial zone was the remaining central 80% of the ROI. Using the Angle tool, a line was drawn down the length of each microvillus (dotted lines, Fig. 2B) with the angle base parallel to the cell surface (solid lines Fig. 2B). Angle measurements were plotted in Prism in a column chart and mean marginal and medial angles were compared using a Welch’s unpaired t-test.
Temporal color coding
Time frames for every 3 minutes were selected (18 total frames). Using the Temporal-Color Code function in Fiji, the ESPN channel was coded (start frame 1, end frame 18) using the Spectrum LUT (Fig. 3B).
Microvilli tracking using EGFP-EPS8 puncta
Denoised and deconvolved 3D movies were converted into max intensity projections in the Z plane. Next, a binary via the spot detection tool was applied to the FITC channel (EPS8 signal) with a diameter of 260 nm and a contrast value of 25 to threshold EPS8 puncta representing individual microvilli in the ROI (medial or marginal). Tracking parameters did not allow for the detection of new tracks after the first frame, allowed for a maximum of 3 gaps in a given track, and a standard deviation multiplier of 2. Using the tracking tool, binaries, representing EPS8 puncta, were tracked and any points lying outside of the ROI were deselected. Track data, time and X Y positions, were then exported to Excel and analyzed and plotted in Prism as radial X Y positions over time by subtracting each position in X or Y from the respective point position at time 0, making the first position (0,0) (Fig 3C and 3F). Three independent live cell imaging experiments were used for the analysis.
Mean squared displacement analysis
Marginal and medial EPS8 puncta trajectories representing microvillar motion were analyzed using mean squared displacement (MSD) analysis as previously described27.
MSD curves were subsequently fit with two models58,59: (1) an active motion model in the form MSD (nΔt) = 4Dt + V2* nΔt2, where D is the diffusion coefficient and V is the velocity of active motion, and (2) a constrained diffusion model in the form of MSD (nΔt) = P[1-e(−4D*nΔt/P)], where P is the plateau of confinement and D is the diffusion coefficient. Non-linear curve fitting was performed using PRISM v.9 and sum-of-squares F-tests were used to select the best model for fitting.
Cell mixing linescans
Using Fiji, a segmented line with a width of 6 was drawn across mixed cell interfaces to encapsulate signal at the mixed cell (marginal) interfaces. Lines with a minimum length of 20 μm and maximum length of 75 μm were used in analysis. Fig. 5 shows one representative linescan from the large dataset for each cell mixing scenario, with line length (μm) shown on the X axis and mCherry and EGFP construct intensities on the Y axis. Intensities were normalized from 0 to 1 in Prism with 0 being the lowest gray value in the linescan and 1 being the highest. The residual plots shown were calculated from the respective representative linescan by subtracting mCherry intensity from EGFP intensity at each length in X. 30 individual linescans from each cell mixing scenario were plotted on their own XY correlation plot in Prism. Combined Pearson’s r values from the 30 individual correlations were plotted in Fig 5K, and mean r values were compared in Prism using an Ordinary one-way ANOVA with multiple comparisons.
FRAP fraction recovery analysis
A background ROI and reference ROI were used to account for photobleaching and background fluorescence in both channels. Fraction recovery over time was calculated from . Recovery curves were fitted with a two-phase association equation in Prism and the immobile fraction was calculated from 1 minus the plateau. Images shown in Fig. 6 were denoised and deconvolved in Nikon Elements for presentation clarity, however all analyzed measurements presented in the FRAP plots were taken from raw, unprocessed movies.
CDHR2 KO CL4 cell intensity measurements
A total of 45 60X fields were used for each condition, control or KO, from stained coverslips. (For the KO, 15 60X fields were taken for each sequenced clone, for a compiled total of 45 KO fields.) Raw images were maximally projected in Z in FIJI and mean intensity of the entire field was measured for the CDHR2 and CDHR5 channels. The measured intensities were plotted in Prism and statistics were quantified with an unpaired t-test.
ESPN intensity measurements
Blebbistatin Treatment:
Movies from three independent control (DMSO) and blebbistatin treatments were maximally projected in Z in FIJI. Frames at time 0 hr (pre-Blebb or DMSO) and 1.5 hr (post-Blebb or DMSO) were isolated and used for intensity measurements. A segmented line with a width of 40 was drawn along the same cell-cell interfaces at both time points. Paired mean ESPN intensities (16-bit gray value) were plotted in Prism and the mean change in intensity (Δ) from 0 hr to 1.5 hr was quantified. A total of 20 pairs were used from each treatment analysis from 2 independent movies (4 movies total). A paired t-test was used for statistical testing of intensity differences.
24-hour ESPN movies:
Movies were maximum intensity projected through Z using FIJI. A segmented line with a width of 40 was drawn along the entire intact marginal perimeter of the same cells at 0 hr and 24 hr to measure mean marginal ESPN intensities. Medial intensities were measured by maximum intensity projected through Z using Nikon Elements. A Bezier ROI was drawn by hand to encompass the central, medial region of the same cells at 0 hr and 24 hr to measure mean medial ESPN intensities. Paired marginal and medial ESPN intensities (16-bit gray values) were plotted in Prism. Three separate live cell movies were used for the quantification as delineated by runs 1, 2, 3 (Fig 7D). The mean change in ESPN intensity (Δ) for each region from each movie at 0 hr and 24 hr were also potted (Fig. 7E) and subject to unpaired t-testing in Prism.
Supplementary Material
Video S1: Microvilli adopt a vertical orientation upon reaching cell margins, related to Figure 2. 3D volume projection depth-coded in Z of live mCherry-ESPN expressing CL4 cells. Spinning disk confocal movie taken over 2 hours with 2-minute intervals of the microvilli cluster marked in Fig 2C,D. Arrow follows a cluster of medial microvilli that transition to a vertical orientation upon reaching the marginal cell area (as marked by Z-depth color profile). Scale bar: 5 μm.
Video S2: Marginal heterophilic adhesion complexes have low signal recovery, suggesting high complex stability, related to Figure 6A. Mixed CL4 cells expressing CDHR2-EGFP (green) and CDHR5-mCherry (magenta) photobleached at two marginal ROIs; 30s intervals shown. Scale bar: 5 μm.
Video S3: Medial heterophilic adhesion complexes have higher signal recovery, suggesting lower complex stability, related to Figure 6C. CL4 cell co-expressing CDHR2-Halo (magenta) and CDHR5-EGFP (green) photobleached at a medial ROI; 15s and 30s intervals shown. Scale bar: 5 μm.
Video S4: Marginal homophilic CDHR2 adhesion complexes have higher signal recovery, suggesting lower complex stability, related to Figure 6E. Mixed CL4 cells expressing CDHR2-EGFP (green) and CDHR2-mCherry (magenta) photobleached at a marginal ROI; 15s intervals shown. Scale bar: 5 μm.
Video S5: Marginal homophilic CDHR5 adhesion complexes have higher signal recovery, suggesting lower complex stability, related to Figure 6G. Mixed CL4 cells expressing CDHR5-EGFP (green) and CDHR5-mCherry (magenta) photobleached at a marginal ROI; 30s intervals shown. Scale bar: 5 μm.
Video S6: Myosin-2 dependent contractility stabilizes transjunctional clustering of microvilli, related to Figure S6. Live mCherry-ESPN CL4 cell monolayers (100% confluence) treated with either DMSO (left) or 20 μM blebbistatin (right). Treatment was added at 5 min and cells were imaged for an additional 1.5 hrs at 30 s intervals. Scale bar: 20 μm.
Video S7: Long-term imaging reveals that microvilli accumulation at cell margins precedes medial accumulation over the course of differentiation, related to Figure 7. (Left) CL4 cells expressing mCherry-ESPN imaged at 3-minute intervals for 43 hours. (Right) Fire LUT intensity profile of the ESPN channel. Intensity scales from low (0; dark purple) to high (255; yellow/white) as denoted by LUT profile. StackReg ImageJ plugin was used to align all frames of the movie. Scale bar: 10 μm.
Highlights:
Nascent microvilli accumulate at cell margins early in epithelial differentiation
Marginal microvilli exhibit reduced motility and a more vertical orientation
Marginal microvilli from neighboring cells are linked by complexes of CDHR2/CDHR5
Transjunctional CDHR2/CDHR5 links are long-lived and drive microvilli accumulation
ACKNOWLEDGMENTS
The authors would like to thank all members of the M.J.T. laboratory for their constructive feedback. Special thanks go to laboratory manager Suli Mao, for her work with mouse husbandry and genotyping. We acknowledge the Translational Pathology Shared Resource (NCI Cancer Center Support Grant 5P30CA68485-19) and also thank the Vanderbilt Genome Editing Resource (RRID: SCR_018826). Microscopy was performed in part through the Vanderbilt Cell Imaging Shared Resource. Some SEM images were collected on a Zeiss Crossbeam funded by S10 OD028704. Flow Cytometry experiments were performed in the VUMC Flow Cytometry Shared Resource supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). This work was supported by Department of Veterans Affairs Career Development Award 1IK2-BX004885 (L.M.M.), the NIH grants DK095811, DK125546, DK111949 (M.J.T.) and the Training Program in Developmental Biology 2T32 HD007502-20 (C.S.C.).
INCLUSION AND DIVERSITY
The Tyska laboratory is committed to the inclusive, diverse, and equitable conduct of biomedical research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Coudrier E, Kerjaschki D, and Louvard D (1988). Cytoskeleton organization and submembranous interactions in intestinal and renal brush borders. Kidney Int 34, 309–320. 10.1038/ki.1988.183. [DOI] [PubMed] [Google Scholar]
- 2.Sauvanet C, Wayt J, Pelaseyed T, and Bretscher A (2015). Structure, regulation, and functional diversity of microvilli on the apical domain of epithelial cells. Annu Rev Cell Dev Biol 31, 593–621. 10.1146/annurev-cellbio-100814-125234. [DOI] [PubMed] [Google Scholar]
- 3.Mooseker MS, and Tilney LG (1975). Organization of an actin filament-membrane complex. Filament polarity and membrane attachment in the microvilli of intestinal epithelial cells. J Cell Biol 67, 725–743. 10.1083/jcb.67.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohta K, Higashi R, Sawaguchi A, and Nakamura K (2012). Helical arrangement of filaments in microvillar actin bundles. Journal of structural biology 177, 513–519. 10.1016/j.jsb.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 5.Helander HF, and Fandriks L (2014). Surface area of the digestive tract - revisited. Scand J Gastroenterol 49, 681–689. 10.3109/00365521.2014.898326. [DOI] [PubMed] [Google Scholar]
- 6.Wessely O, Cerqueira DM, Tran U, Kumar V, Hassey JM, and Romaker D (2014). The bigger the better: determining nephron size in kidney. Pediatr Nephrol 29, 525–530. 10.1007/s00467-013-2581-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delacour D, Salomon J, Robine S, and Louvard D (2016). Plasticity of the brush border - the yin and yang of intestinal homeostasis. Nat Rev Gastroenterol Hepatol 13, 161–174. 10.1038/nrgastro.2016.5. [DOI] [PubMed] [Google Scholar]
- 8.Fath KR, Obenauf SD, and Burgess DR (1990). Cytoskeletal protein and mRNA accumulation during brush border formation in adult chicken enterocytes. Development 109, 449–459. [DOI] [PubMed] [Google Scholar]
- 9.Specian RD, and Neutra MR (1981). The surface topography of the colonic crypt in rabbit and monkey. The American journal of anatomy 160, 461–472. 10.1002/aja.1001600409. [DOI] [PubMed] [Google Scholar]
- 10.Rice WL, Van Hoek AN, Paunescu TG, Huynh C, Goetze B, Singh B, Scipioni L, Stern LA, and Brown D (2013). High resolution helium ion scanning microscopy of the rat kidney. PLoS One 8, e57051. 10.1371/journal.pone.0057051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crawley SW, Shifrin DA Jr., Grega-Larson NE, McConnell RE, Benesh AE, Mao S, Zheng Y, Zheng QY, Nam KT, Millis BA, et al. (2014). Intestinal brush border assembly driven by protocadherin-based intermicrovillar adhesion. Cell 157, 433–446. 10.1016/j.cell.2014.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weck ML, Crawley SW, Stone CR, and Tyska MJ (2016). Myosin-7b Promotes Distal Tip Localization of the Intermicrovillar Adhesion Complex. Curr Biol 26, 2717–2728. 10.1016/j.cub.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawley SW, Weck ML, Grega-Larson NE, Shifrin DA Jr., and Tyska MJ (2016). ANKS4B Is Essential for Intermicrovillar Adhesion Complex Formation. Dev Cell 36, 190–200. 10.1016/j.devcel.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinette JA, Mao S, Millis BA, Krystofiak ES, Faust JJ, and Tyska MJ (2019). Brush border protocadherin CDHR2 promotes the elongation and maximized packing of microvilli in vivo. Mol Biol Cell 30, 108–118. 10.1091/mbc.E18-09-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McConnell RE, Benesh AE, Mao S, Tabb DL, and Tyska MJ (2011). Proteomic analysis of the enterocyte brush border. American journal of physiology. Gastrointestinal and liver physiology 300, G914–926. 10.1152/ajpgi.00005.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gray ME, Johnson ZR, Modak D, Tamilselvan E, Tyska MJ, and Sotomayor M (2021). Heterophilic and homophilic cadherin interactions in intestinal intermicrovillar links are species dependent. PLoS Biol 19, e3001463. 10.1371/journal.pbio.3001463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, He Y, Lu Q, and Zhang M (2016). Mechanistic Basis of Organization of the Harmonin/USH1C-Mediated Brush Border Microvilli Tip-Link Complex. Dev Cell 36, 179–189. 10.1016/j.devcel.2015.12.020. [DOI] [PubMed] [Google Scholar]
- 18.Li J, He Y, Weck ML, Lu Q, Tyska MJ, and Zhang M (2017). Structure of Myo7b/USH1C complex suggests a general PDZ domain binding mode by MyTH4-FERM myosins. Proc Natl Acad Sci U S A 114, E3776–E3785. 10.1073/pnas.1702251114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi MS, Graves MJ, Matoo S, Storad ZA, El Sheikh Idris RA, Weck ML, Smith ZB, Tyska MJ, and Crawley SW (2020). The small EF-hand protein CALML4 functions as a critical myosin light chain within the intermicrovillar adhesion complex. J Biol Chem 295, 9281–9296. 10.1074/jbc.RA120.012820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaeta IM, Meenderink LM, Postema MM, Cencer CS, and Tyska MJ (2021). Direct visualization of epithelial microvilli biogenesis. Curr Biol 31, 2561–2575 e2566. 10.1016/j.cub.2021.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Croce A, Cassata G, Disanza A, Gagliani MC, Tacchetti C, Malabarba MG, Carlier MF, Scita G, Baumeister R, and Di Fiore PP (2004). A novel actin barbed-end-capping activity in EPS-8 regulates apical morphogenesis in intestinal cells of Caenorhabditis elegans. Nat Cell Biol 6, 1173–1179. [DOI] [PubMed] [Google Scholar]
- 22.Manor U, Disanza A, Grati M, Andrade L, Lin H, Di Fiore PP, Scita G, and Kachar B (2011). Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr Biol 21, 167–172. 10.1016/j.cub.2010.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Disanza A, Mantoani S, Hertzog M, Gerboth S, Frittoli E, Steffen A, Berhoerster K, Kreienkamp HJ, Milanesi F, Di Fiore PP, et al. (2006). Regulation of cell shape by Cdc42 is mediated by the synergic actin-bundling activity of the Eps8-IRSp53 complex. Nat Cell Biol 8, 1337–1347. 10.1038/ncb1502. [DOI] [PubMed] [Google Scholar]
- 24.Postema MM, Grega-Larson NE, Neininger AC, and Tyska MJ (2018). IRTKS (BAIAP2L1) Elongates Epithelial Microvilli Using EPS8-Dependent and Independent Mechanisms. Curr Biol 28, 2876–2888 e2874. 10.1016/j.cub.2018.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tocchetti A, Soppo CB, Zani F, Bianchi F, Gagliani MC, Pozzi B, Rozman J, Elvert R, Ehrhardt N, Rathkolb B, et al. (2010). Loss of the actin remodeler Eps8 causes intestinal defects and improved metabolic status in mice. PLoS One 5, e9468. 10.1371/journal.pone.0009468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zampini V, Ruttiger L, Johnson SL, Franz C, Furness DN, Waldhaus J, Xiong H, Hackney CM, Holley MC, Offenhauser N, et al. (2011). Eps8 regulates hair bundle length and functional maturation of mammalian auditory hair cells. PLoS Biol 9, e1001048. 10.1371/journal.pbio.1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meenderink LM, Gaeta IM, Postema MM, Cencer CS, Chinowsky CR, Krystofiak ES, Millis BA, and Tyska MJ (2019). Actin Dynamics Drive Microvillar Motility and Clustering during Brush Border Assembly. Dev Cell 50, 545–556 e544. 10.1016/j.devcel.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peterson MD, and Mooseker MS (1992). Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci 102 ( Pt 3), 581–600. 10.1242/jcs.102.3.581. [DOI] [PubMed] [Google Scholar]
- 29.Nielsen R, Birn H, Moestrup SK, Nielsen M, Verroust P, and Christensen EI (1998). Characterization of a kidney proximal tubule cell line, LLC-PK1, expressing endocytotic active megalin. J Am Soc Nephrol 9, 1767–1776. 10.1681/ASN.V9101767. [DOI] [PubMed] [Google Scholar]
- 30.Gaeta IM, Meenderink LM, and Tyska MJ (2021). A protocol for imaging microvilli biogenesis on the surface of cultured porcine kidney epithelial cell monolayers. STAR Protoc 2, 100998. 10.1016/j.xpro.2021.100998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartles JR, Zheng L, Li A, Wierda A, and Chen B (1998). Small espin: a third actin-bundling protein and potential forked protein ortholog in brush border microvilli. J Cell Biol 143, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loomis PA, Zheng L, Sekerkova G, Changyaleket B, Mugnaini E, and Bartles JR (2003). Espin cross-links cause the elongation of microvillus-type parallel actin bundles in vivo. J Cell Biol 163, 1045–1055. 10.1083/jcb.200309093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higgs HN (2004). There goes the neighbourhood: Eps8 joins the barbed-end crowd. Nat Cell Biol 6, 1147–1149. 10.1038/ncb1204-1147. [DOI] [PubMed] [Google Scholar]
- 34.Stevenson BR, Siliciano JD, Mooseker MS, and Goodenough DA (1986). Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol 103, 755–766. 10.1083/jcb.103.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charras G, and Yap AS (2018). Tensile Forces and Mechanotransduction at Cell-Cell Junctions. Curr Biol 28, R445–R457. 10.1016/j.cub.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 36.Rakshit S, Zhang Y, Manibog K, Shafraz O, and Sivasankar S (2012). Ideal, catch, and slip bonds in cadherin adhesion. Proc Natl Acad Sci U S A 109, 18815–18820. 10.1073/pnas.1208349109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffman BD, and Yap AS (2015). Towards a Dynamic Understanding of Cadherin-Based Mechanobiology. Trends Cell Biol 25, 803–814. 10.1016/j.tcb.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Pinheiro D, and Bellaïche Y (2018). Mechanical Force-Driven Adherens Junction Remodeling and Epithelial Dynamics. Dev Cell 47, 3–19. 10.1016/j.devcel.2018.09.014. [DOI] [PubMed] [Google Scholar]
- 39.Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, and Madara JL (1997). Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol 273, C1378–1385. 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- 40.Chinowsky CR, Pinette JA, Meenderink LM, Lau KS, and Tyska MJ (2020). Nonmuscle myosin-2 contractility-dependent actin turnover limits the length of epithelial microvilli. Mol Biol Cell 31, 2803–2815. 10.1091/mbc.E20-09-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trier JS (1963). Studies on Small Intestinal Crypt Epithelium. I. The Fine Structure of the Crypt Epithelium of the Proximal Small Intestine of Fasting Humans. J Cell Biol 18, 599–620. 10.1083/jcb.18.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Li J Jr., Zhang SY, Li RX, Lin X, Mi YL, and Zhang CQ (2018). Culture and characterization of chicken small intestinal crypts. Poult Sci 97, 1536–1543. 10.3382/ps/pey010. [DOI] [PubMed] [Google Scholar]
- 43.Wu SK, Gomez GA, Michael M, Verma S, Cox HL, Lefevre JG, Parton RG, Hamilton NA, Neufeld Z, and Yap AS (2014). Cortical F-actin stabilization generates apical-lateral patterns of junctional contractility that integrate cells into epithelia. Nat Cell Biol 16, 167–178. 10.1038/ncb2900. [DOI] [PubMed] [Google Scholar]
- 44.Vanslembrouck B, Chen JH, Larabell C, and van Hengel J (2022). Microscopic Visualization of Cell-Cell Adhesion Complexes at Micro and Nanoscale. Front Cell Dev Biol 10, 819534. 10.3389/fcell.2022.819534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kale GR, Yang X, Philippe JM, Mani M, Lenne PF, and Lecuit T (2018). Distinct contributions of tensile and shear stress on E-cadherin levels during morphogenesis. Nat Commun 9, 5021. 10.1038/s41467-018-07448-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leerberg JM, Gomez GA, Verma S, Moussa EJ, Wu SK, Priya R, Hoffman BD, Grashoff C, Schwartz MA, and Yap AS (2014). Tension-sensitive actin assembly supports contractility at the epithelial zonula adherens. Curr Biol 24, 1689–1699. 10.1016/j.cub.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sokurenko EV, Vogel V, and Thomas WE (2008). Catch-bond mechanism of force-enhanced adhesion: counterintuitive, elusive, but … widespread? Cell Host Microbe 4, 314–323. 10.1016/j.chom.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas W (2006). For catch bonds, it all hinges on the interdomain region. J Cell Biol 174, 911–913. 10.1083/jcb.200609029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas W (2008). Catch bonds in adhesion. Annu Rev Biomed Eng 10, 39–57. 10.1146/annurev.bioeng.10.061807.160427. [DOI] [PubMed] [Google Scholar]
- 50.Guo B, and Guilford WH (2006). Mechanics of actomyosin bonds in different nucleotide states are tuned to muscle contraction. Proc Natl Acad Sci U S A 103, 9844–9849. 10.1073/pnas.0601255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kong F, Garcia AJ, Mould AP, Humphries MJ, and Zhu C (2009). Demonstration of catch bonds between an integrin and its ligand. J Cell Biol 185, 1275–1284. 10.1083/jcb.200810002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.VanDussen KL, Stojmirovic A, Li K, Liu TC, Kimes PK, Muegge BD, Simpson KF, Ciorba MA, Perrigoue JG, Friedman JR, et al. (2018). Abnormal Small Intestinal Epithelial Microvilli in Patients With Crohn’s Disease. Gastroenterology 155, 815–828. 10.1053/j.gastro.2018.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGuckin MA, Eri R, Simms LA, Florin TH, and Radford-Smith G (2009). Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm Bowel Dis 15, 100–113. 10.1002/ibd.20539. [DOI] [PubMed] [Google Scholar]
- 54.Velle KB, and Campellone KG (2017). Extracellular motility and cell-to-cell transmission of enterohemorrhagic E. coli is driven by EspFU-mediated actin assembly. PLoS Pathog 13, e1006501. 10.1371/journal.ppat.1006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.In J, Foulke-Abel J, Zachos NC, Hansen AM, Kaper JB, Bernstein HD, Halushka M, Blutt S, Estes MK, Donowitz M, and Kovbasnjuk O (2016). Enterohemorrhagic Escherichia coli reduce mucus and intermicrovillar bridges in human stem cell-derived colonoids. Cell Mol Gastroenterol Hepatol 2, 48–62 e43. 10.1016/j.jcmgh.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pedersen GA, Jensen HH, Schelde AB, Toft C, Pedersen HN, Ulrichsen M, Login FH, Amieva MR, and Nejsum LN (2017). The basolateral vesicle sorting machinery and basolateral proteins are recruited to the site of enteropathogenic E. coli microcolony growth at the apical membrane. PLoS One 12, e0179122. 10.1371/journal.pone.0179122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conant D, Hsiau T, Rossi N, Oki J, Maures T, Waite K, Yang J, Joshi S, Kelso R, Holden K, et al. (2022). Inference of CRISPR Edits from Sanger Trace Data. CRISPR J 5, 123–130. 10.1089/crispr.2021.0113. [DOI] [PubMed] [Google Scholar]
- 58.Qian H, Sheetz MP, and Elson EL (1991). Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys J 60, 910–921. S0006–3495(91)82125–7 [pii] 10.1016/S0006-3495(91)82125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheetz MP, Turney S, Qian H, and Elson EL (1989). Nanometre-level analysis demonstrates that lipid flow does not drive membrane glycoprotein movements. Nature 340, 284–288. 10.1038/340284a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1: Microvilli adopt a vertical orientation upon reaching cell margins, related to Figure 2. 3D volume projection depth-coded in Z of live mCherry-ESPN expressing CL4 cells. Spinning disk confocal movie taken over 2 hours with 2-minute intervals of the microvilli cluster marked in Fig 2C,D. Arrow follows a cluster of medial microvilli that transition to a vertical orientation upon reaching the marginal cell area (as marked by Z-depth color profile). Scale bar: 5 μm.
Video S2: Marginal heterophilic adhesion complexes have low signal recovery, suggesting high complex stability, related to Figure 6A. Mixed CL4 cells expressing CDHR2-EGFP (green) and CDHR5-mCherry (magenta) photobleached at two marginal ROIs; 30s intervals shown. Scale bar: 5 μm.
Video S3: Medial heterophilic adhesion complexes have higher signal recovery, suggesting lower complex stability, related to Figure 6C. CL4 cell co-expressing CDHR2-Halo (magenta) and CDHR5-EGFP (green) photobleached at a medial ROI; 15s and 30s intervals shown. Scale bar: 5 μm.
Video S4: Marginal homophilic CDHR2 adhesion complexes have higher signal recovery, suggesting lower complex stability, related to Figure 6E. Mixed CL4 cells expressing CDHR2-EGFP (green) and CDHR2-mCherry (magenta) photobleached at a marginal ROI; 15s intervals shown. Scale bar: 5 μm.
Video S5: Marginal homophilic CDHR5 adhesion complexes have higher signal recovery, suggesting lower complex stability, related to Figure 6G. Mixed CL4 cells expressing CDHR5-EGFP (green) and CDHR5-mCherry (magenta) photobleached at a marginal ROI; 30s intervals shown. Scale bar: 5 μm.
Video S6: Myosin-2 dependent contractility stabilizes transjunctional clustering of microvilli, related to Figure S6. Live mCherry-ESPN CL4 cell monolayers (100% confluence) treated with either DMSO (left) or 20 μM blebbistatin (right). Treatment was added at 5 min and cells were imaged for an additional 1.5 hrs at 30 s intervals. Scale bar: 20 μm.
Video S7: Long-term imaging reveals that microvilli accumulation at cell margins precedes medial accumulation over the course of differentiation, related to Figure 7. (Left) CL4 cells expressing mCherry-ESPN imaged at 3-minute intervals for 43 hours. (Right) Fire LUT intensity profile of the ESPN channel. Intensity scales from low (0; dark purple) to high (255; yellow/white) as denoted by LUT profile. StackReg ImageJ plugin was used to align all frames of the movie. Scale bar: 10 μm.