

Abstract
Aeroallergen sensitization, mainly mediated by lung epithelium and dendritic cells (DCs), is integral to allergic asthma pathogenesis and progression. IL-10 has a dual role in immune responses, as it inhibits myeloid cell activation but promotes B-cell responses and epithelial cell proliferation. Here, we report a proinflammatory function of B-cell-derived IL-10 modulated by Bcl-3 in allergic asthma. Specifically, Bcl-3−/− mice showed elevated IL-10 levels and were found to be highly vulnerable to allergic asthma induced by house dust mites (HDMs). IL-10 had a positive correlation with the levels of the DC chemoattractant CCL-20 in HDM-sensitized mice and in patients with asthma and induced a selective increase in CCL-20 production by mouse lung epithelial cells. Blockade of IL-10 or IL-10 receptors during sensitization dampened both HDM-induced sensitization and asthma development. IL-10 levels peaked 4 h post sensitization with HDM and IL-10 was primarily produced by B cells under Bcl-3–Blimp-1–Bcl-6 regulation. Mice lacking B-cell-derived IL-10 displayed decreased lung epithelial CCL-20 production and diminished DC recruitment to the lungs upon HDM sensitization, thereby demonstrating resistance to HDM-induced asthma. Moreover, responses to HDM stimulation in Bcl-3−/− mice lacking B-cell-derived IL-10 were comparable to those in Bcl-3+/+ mice. The results revealed an unexpected role of B-cell-derived IL-10 in promoting allergic sensitization and demonstrated that Bcl-3 prevents HDM-induced asthma by inhibiting B-cell-derived IL-10 production. Thus, targeting the Bcl-3/IL-10 axis to inhibit allergic sensitization is a promising approach for treating allergic asthma.
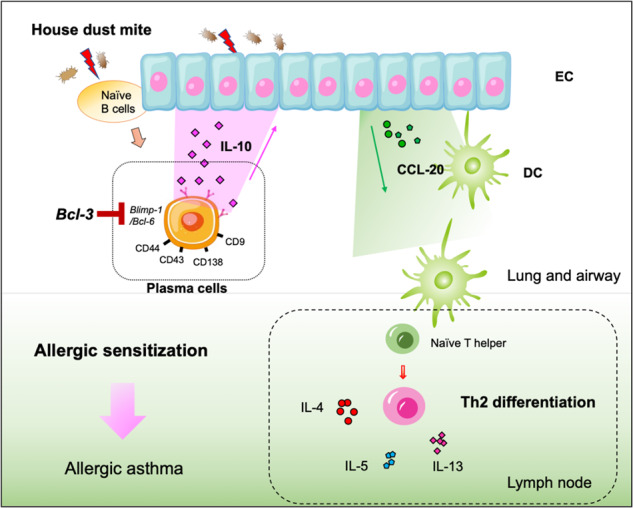
IL-10 is released rapidly from lung plasma cells under Bcl-3–Blimp-1–Bcl-6 regulation upon house dust mite exposure and amplifies lung epithelial cell (EC)‐derived CCL-20 production and subsequent dendritic cell (DC) recruitment to promote allergic sensitization in asthma.
Keywords: Bcl-3, Plasma cell, IL-10, Allergic sensitization, Airway inflammation, Asthma
Subject terms: Chronic inflammation, Interleukins, Mechanisms of disease
Introduction
Asthma is an allergic airway inflammatory disease caused by excessive Th2 responses to environmental aeroallergens, such as house dust mites (HDMs), fungal spores, and pollen, leading to eosinophilia, mucus overproduction, and airway obstruction [1, 2]. Allergen-specific Th2 cells produce cytokines, such as IL-4, IL-5, and IL-13, which cause many of the critical characteristics of asthma, including eosinophilic lung inflammation, goblet cell metaplasia, and airway hyperresponsiveness [1, 3].
Allergic sensitization is integral to asthma pathogenesis and progression [4, 5]. Once sensitized to aeroallergens, individuals will progress to allergic asthma when rechallenged by the sensitizing allergen [5]. Targeting allergen sensitization can be a promising therapeutic approach for controlling asthma [5, 6]. Sensitization to aeroallergens is primarily mediated by lung epithelial cells (ECs) and dendritic cells (DCs) [5, 7]. As the initial barrier of defense against allergens and pathogens, lung ECs initiate immune responses to stimuli and function in coordination with DCs to induce and amplify adaptive Th2 immunity [2, 8, 9]. Several epithelial-derived cytokines, such as CCL-20 (a DC chemoattractant), GM-CSF (a DC maturation promoter), IL-25, IL-33, CSF-1, and TSLP, can condition DCs to induce Th2 polarization and airway inflammation [2, 10–12]. Lung resident CD11b+ DCs can take up allergens and migrate to the mediastinal lymph node (MLN) to trigger proper Th2 immunity [10, 13].
B cells are essential in humoral immunity to inhaled allergens [14, 15]. Previous studies on B-cell functions in pathological situations have yielded controversial results, revealing the multifaceted roles of B cells in asthma [14, 16, 17]. Recently, a novel B-cell subset, IL-10-producing B cells, was reported to suppress established airway allergic inflammation via IL-10 production [18]. However, IL-10-producing B cells are also crucial for modulating the immune response in parasitic diseases by contributing to Th2 cell development [19, 20]. Moreover, IL-10 induces Th2 immunity and eosinophil infiltration in allergic dermatitis [21] and aggravates allergen-induced smooth muscle hyperresponsiveness [22, 23]. These findings indicated that B cells can secrete IL-10 upon stimulation by inhaled allergens; however, the precise role of IL-10 and IL-10-producing B cells in allergic disease remains controversial.
B-cell lymphoma-3 (Bcl-3) is an atypical member of the IκB family that regulates NF-κB transcription [24, 25]. Our group, as well as some others, have shown that Bcl-3 can modulate IL-10 production, which might be essential for the pathogenesis of several inflammatory diseases [24–27]. In addition, IL-10 production induced by LPS (a common contaminant varying across HDM batches) is regulated by Bcl-3 [1, 28]. However, whether Bcl-3 is involved in asthma pathogenesis via IL-10 remains unclear. This study demonstrated a novel function of IL-10, which is mainly secreted by B cells during the sensitization phase, in promoting allergic asthma. Bcl-3 limits the secretion of B-cell-derived IL-10 during allergic sensitization, thereby preventing HDM-induced asthma.
Materials and methods
Mice
Bcl-3−/−, Il10fl/fl, and Mb1-Cre mice have been described previously [25, 29]. All mice were backcrossed with C57BL/6 J mice for 10 or more generations. Bcl-3−/−IL-10B-KO mice were produced by crossing Bcl-3−/− and Il10fl/fl Mb1Cre mice. IL-10-IRES-EGFP mice, referred to as Tiger mice, were kindly gifted by Dr. Zhinan Yin and have been described previously [30]. Bcl-3−/− Tiger mice were generated by crossing Bcl-3−/− and Tiger mice. B-cell-deficient μMT mice (C57BL/6 J background; Jackson Laboratory) were kindly provided by Dr. Nan Shen (Renji Hospital, Shanghai Jiao Tong University).
Female littermates between 6 and 10 weeks of age, generated from heterozygous parents, were utilized in this study. These mice were cohoused throughout the experimental procedures.
Murine models for allergic asthma
To induce allergic asthma, Bcl-3+/+, Bcl-3−/−, IL-10B-WT, IL-10B-KO, μMT, and Bcl-3−/− IL-10B-KO mice were first sensitized intranasally with 5 µg of HDM (Greer laboratories, Lenoir, NC, USA) in 40 µl of PBS on days 0, 1, and 2 and subsequently challenged with 5 µg of HDM intranasally on days 8–12. In IL-10 supply experiments, 5 µg of HDM mixed with 10 ng of IL-10 was instilled intranasally during sensitization.
For the asthma model induced by ovalbumin (OVA) (Sigma, Billerica, MA, USA), mice were sensitized via intraperitoneal injection with a mixture of OVA (50 µg) and alum (50 µg, Sigma) on days 0 and 7 and then challenged with OVA (50 µg) intranasally daily on days 14–16. The model mice were analyzed on day 17.
In the IL-10 blocking experiments, 10 µg of IL-10 (PeproTech, Rocky Hill, NJ, USA) monoclonal antibody or rat/IgG2b isotype (eB149/10H5, eBioscience, Waltham, MA, USA) was mixed with 5 µg of HDM in 40 µl of PBS and administered intranasally during the sensitization phases. In the IL-10R blockade experiments, anti-IL-10R (50 µg; BioLegend, San Diego, CA, USA) or rat IgG1 isotype (Ebrg1, Invitrogen, Waltham, MA, USA) was mixed with 5 µg of HDM in 40 µl of PBS and administered intranasally in the sensitization phase.
Bronchoalveolar lavage fluid (BALF) analysis
BALF was collected from model mice by flushing their airways with PBS, and cell counting was performed using a Cellometer (Nexcelom, Boston, MA, USA). The differentiation and counting of eosinophils, neutrophils, macrophages, and lymphocytes were assessed after cytospin and Wright–Giemsa staining; eosinophils were identified by a blue bilobed nucleus and reddish granules, neutrophils by a dark blue multilobed nucleus and pale pink cytoplasm, macrophages by a purple nucleus with sky blue cytoplasm, larger than other leukocytes, and lymphocytes by a large round nucleus with a high nuclear/cytoplasm ratio [31, 32].
Airway hyperresponsiveness (AHR) measurement
Lung resistance was assessed by a Buxco Electronics device (Wilmington, NC, USA), as described elsewhere [10]. Briefly, on day 14, a tube was inserted in the trachea of each model mouse, and the lung was mechanically ventilated. After administering escalating aerosolized methacholine (5–40 mg/ml; Sigma), lung resistance (RL) was evaluated.
ELISA
ELISA kits for IL-4, IL-5, IL-10, IL-13, IL-17, CCL-20, GM-CSF, and CXCL1 (KC) were purchased from BD (San Jose, CA, USA) or R&D Systems (Minneapolis, MN, USA). All procedures were performed in accordance with the manufacturer’s instructions. To assess serum HDM-specific immunoglobulins, HDM allergens (5 µg/ml) dissolved in coating buffer were coated onto high-affinity 96-well plates (Costar, New York, NY, USA) overnight at 4 °C. The antibodies employed for detection were goat anti-mouse IgG2c (Novus Biologicals, Inc.), IgE (Southern Biotechnology), and IgG1 (Santa Cruz Biotechnology), all of which were conjugated with horseradish peroxidase.
Flow cytometry and cell sorting
Mouse lung tissue samples were cut into small pieces and enzymatically digested in RPMI-1640 medium containing 1 mg/ml collagenase IV (Sigma) and 5 U/ml DNase I (Sigma) for 1 h at 37 °C and vortexed occasionally. Single lung cells were extracted after the digested lungs were filtered and red blood cells were removed. Subsequently, the obtained single-cell suspensions were incubated with an Fc-blocking anti-mouse CD16/32 (BioLegend) antibody for 40 min on ice and then stained with fluorochrome-conjugated antibodies. Single lung cells were treated with anti-mouse CD45 (BioLegend) and CD326 (BD) for ECs or with anti-mouse CD45 and CD19 (BD) for B cells and then sorted on a Beckman Coulter Moflo Astrios to isolate primary lung ECs or B cells.
Histological analysis
Lungs fixed in 2% paraformaldehyde were paraffin-embedded, sectioned, and subjected to H&E staining. The pathological changes were visualized using light microscopy (Nikon, Tokyo, Japan).
Immunofluorescence analysis
IL-10 GFP mice were sensitized to HDM (5 µg), and lung tissue was harvested 4 h later for cryosection preparation. The resulting sections were stained with anti-CD19 antibody and analyzed using immunofluorescence on a Zeiss microscope (Zeiss, Tubingen, Germany).
scRNA-seq
Live lung B cells (CD45+ CD19+) were isolated by cell sorting from Bcl-3+/+ and Bcl-3−/− mice 4 h post HDM sensitization. Subsequently, viable B-cell suspensions were loaded onto a Chromium Controller Instrument (10× Genomics, Pleasanton, CA, USA) to generate single-cell gel bead-in-emulsions (GEMs) using the Single Cell 3’ Library and Gel Bead Kit V3.1 (10× Genomics, 1000121) according to the manufacturer’s instructions. All libraries were sequenced on an Illumina NovaSeq 6000 with a custom paired-end sequencing mode of 150 bp (read 1) × 150 bp (read 2). Raw data were preprocessed using Cell Ranger Single-Cell Software Suite (v 5.0.0) with default parameters. Reads were aligned to the genomic reference (Ensemble_ release 106 Mus musculus). For quality control, the cells that had either gene counts below 250 or over 4500, UMI counts <27,000 per cell or mitochondrial genes >25% were removed. The top variable genes across single cells were identified, and their log-transformed gene-barcode matrices were subjected to principal component analysis to minimize their dimensionality. Using Seurat (v 3.1.1), a t-SNE plot was constructed to identify the deviations across all kinds of B cells.
Statistical analysis
Data are presented as the mean ± standard error of the mean. If the data were normally distributed, an unpaired two-tailed Student’s t test was used to compare the differences between two groups. Otherwise, the Mann–Whitney U test was used. One-way or two-way analysis of variance revealed group differences. PRISM (GraphPad, San Diego, CA, USA) was used to conduct all statistical analyses. The statistical significance threshold was established at P < 0.05 (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Study approval
All animal procedures were approved by the Institutional Biomedical Research Ethics Committee of Shanghai Institutes for Biological Sciences (project number SIBS-2019-ZXR-1) and Guangzhou Medical University (project number 2019-273, G2023-270). All methods were conducted in accordance with the Guide for the Care and Use of Laboratory Animals.
Results
Bcl-3 inhibited IL-10 production and hindered HDM-induced allergic asthma
To investigate whether Bcl-3 and IL-10 contribute to asthma development, we retrieved BCL-3 and IL-10 mRNA expression data from the bronchial epithelial samples of 20 healthy controls and 38 patients with asthma from the public Gene Expression Omnibus (GEO) database (GSE43696) [33]. We found the mRNA levels of BCL-3 to be lower and those of IL-10 to be higher in patients with asthma than in healthy controls (Fig. S1A). Correlation analysis demonstrated an inverse correlation between the mRNA abundance of IL-10 and BCL-3 in patients with asthma (Fig. S1B).
Next, we explored whether Bcl-3 is essential in modulating allergic asthma through IL-10. Bcl-3+/+ and Bcl-3−/− mice were subjected to HDM-induced asthma (Fig. 1A). We observed that HDM-treated Bcl-3−/− mice had considerably increased infiltration of eosinophils, neutrophils, and lymphocytes in BALF in contrast to HDM-treated Bcl-3+/+ mice (Fig. 1B). Similarly, H&E staining revealed that HDM aggravated peribronchial inflammatory infiltrates in the Bcl-3−/− lung tissue and airways (Fig. 1C). In addition, Bcl-3−/− mice exhibited considerably higher production of IL-4, IL-5, IL-13 (Th2 cytokines) and IFN-γ (Th1 cytokine) in the lungs after HDM exposure than Bcl-3+/+ mice (Figs. 1D, S1C). Notably, IL-10 production was also elevated in Bcl-3−/− lungs (Figs. 1D, S1C). No difference was observed in IL-17 levels between the Bcl-3+/+ and Bcl-3−/− mice. Furthermore, compared to those in Bcl-3+/+ mice, Bcl-3−/− mice showed elevated serum IgE and IgG1 levels, which are Th2-specific antibodies, indicating increased Th2 responses in Bcl-3−/− mice (Fig. 1E).
Fig. 1.

Bcl-3 inhibited IL-10 production and HDM-induced allergic asthma. A Experimental scheme of HDM sensitization and challenge. Mice were sensitized with HDM (1.36 µg protein) or PBS on days 0, 1, and 2 and challenged daily from days 8–12 with 5 µg of HDM or PBS. Mice were euthanized on day 14 for analysis. B Differential cell counts in bronchoalveolar lavage fluid of PBS- or HDM-treated Bcl-3+/+ and Bcl-3−/− mice. C Representative lung sections stained with H&E (scale bar, 100 μm). D Cytokine levels in mouse lungs. E Levels of HDM-specific immunoglobulins in mouse sera. F Lung resistance in response to increasing doses of methacholine. Data in (B–F) were combined from four experiments; n = 6–15. B, E Student’s t test. (D) Mann‒Whitney test. F Two-way ANOVA
To further determine HDM-specific Th2 responses in our model, MLN cells were restimulated in vitro with HDM allergen. We found that MLN cell cultures from Bcl-3−/− mice had remarkably higher IL-4, IL-5, and IL-13 levels than those from Bcl-3+/+ mice (Fig. S1D). IL-17 production was similar between HDM-treated Bcl-3+/+ and Bcl-3−/− MLN cells (Fig. S1D). In addition, HDM-treated Bcl-3−/− mice had a higher number of Th2 cells (IL-4+/IL-5+ Th cells) and a similar number of Th17 cells (IL17+ Th cells) in the lung and MLN (Fig. S1E, F), consistent with the results of cytokine expression in the lung homogenates and MLN cell cultures. However, Th1 cells (IFN-γ+ Th cells) were also increased in HDM-treated Bcl-3−/− lungs and MLNs (Fig. S1E, F), suggesting that Bcl-3 does not limit Th2 development by modulating the Th1/Th2 balance [34] in our model. Furthermore, HDM-treated Bcl-3−/− mice had significantly increased AHR compared to Bcl-3+/+ mice (Fig. 1F).
Therefore, upon HDM stimulation, the absence of Bcl-3 increased the production of IL-10 in the lung and promoted significant eosinophilic lung inflammation, higher Th2 responses, and AHR, providing strong evidence that Bcl-3 protects against allergic asthma caused by HDM.
Bcl-3 limited pulmonary DC responses upon HDM exposure
Sensitization to HDMs depends on several lung DC subsets that migrate to the MLNs to initiate Th2 responses [9, 13]. As deficiency of Bcl-3 leads to augmented Th1/Th2 responses to HDM in the lung and MLN, we investigated whether it could be due to increased DC recruitment and activation during the sensitization phase. For this purpose, once Bcl-3+/+ or Bcl-3−/− mice were sensitized to HDM, the recruitment and activation of DCs in their lungs and MLNs were determined (Fig. S2A). DCs were identified as CD11chi MHCIIhi autofluorescentlo cells, and they were further segregated as CD103+ DCs and CD11b+ DCs (including monocyte-derived CD11b+ CD64+ DCs and conventional CD11b+ CD64− DCs; Fig. 2A), as described previously [10, 13]. After sensitization to HDM, the lungs of Bcl-3+/+ mice had significantly more DCs, especially inflammatory CD11b+ DCs and monocyte-derived DCs, than those of PBS control mice (Figs. 2B, S2B). In contrast to the Bcl-3+/+ lungs, Bcl-3−/− lungs had more CD11b+ DCs after HDM sensitization (Figs. 2A, B, S2B); CD11b+ DCs are the primary initiators of allergic Th2 responses [13, 35], and these results are consistent with the robust Th2 response induced in the HDM-challenged Bcl-3−/− lungs (Fig. 1). Similar results were observed in MLNs (Fig. S2C, D). The proportion and number of CD103+ DCs did not differ significantly between HDM-sensitized Bcl-3+/+ and Bcl-3−/− mice (Figs. 2B, S2B), supporting the notion that CD11b+ DCs play more critical roles than CD103+ DCs in initiating Th2 responses upon HDM stimulation [10, 13].
Fig. 2.

Bcl-3 mitigated DC/epithelial responses and IL-10 secretion during HDM sensitization. A–D Gating strategy, cell number, and activation marker CD86 in DCs in Bcl-3+/+ and Bcl-3−/− lungs. Mice were sensitized intranasally with HDM on days 0, 1, and 2 and analyzed on day 4. E Chemokine/cytokine concentrations in Bcl-3+/+ and Bcl-3−/− bronchoalveolar lavage fluid. F, G Cytokine/chemokine and Toll-like receptor signaling inhibitor mRNA levels in Bcl-3+/+ and Bcl-3−/− lung homogenates. E–G Mice were sensitized intranasally with HDM for 4 h. The data in (B–G) were combined from three experiments; n = 7–12. B Mann–Whitney test. C–G Student’s t test
Next, we measured the levels of the costimulatory molecule CD86 in different DC subsets and found that the CD11b+ DCs from HDM-sensitized Bcl-3−/− mice had increased levels of CD86 compared with those from HDM-treated Bcl-3+/+ mice (Fig. 2C), indicating that CD11b+ DCs from Bcl-3−/− mice were more activated upon HDM stimulation than those from Bcl-3+/+ mice. CD86 expression levels were similar between CD103+ DCs in the lungs in HDM-sensitized Bcl-3+/+ and Bcl-3−/− mice (Fig. 2D). Collectively, our data demonstrated that Bcl-3 limits DC responses upon HDM stimulation during the sensitization phase, thereby hindering the subsequent Th2 responses.
Bcl-3 blunted IL-10 production and pulmonary epithelial responses during HDM sensitization
Inhaled HDM allergens stimulate the production of proinflammatory chemokines/cytokines by barrier ECs, thereby promoting DC responses to initiate Th2 immunity [1, 2]. Considering the essential role of lung ECs in initiating allergic sensitization, we assumed that deficiency of Bcl-3 would augment lung epithelial responses to HDMs. To test this hypothesis, we sensitized Bcl-3+/+ and Bcl-3−/− mice intranasally using 5 µg of HDM (Fig. S2E) and measured the levels of CCL-20 (a DC chemoattractant), GM-CSF (a DC maturation promoter), KC, IL-33, IL-25, and CSF-1, which are critical for initiating lung DC responses and are primarily secreted by lung ECs [1, 2]. We found that HDM-sensitized Bcl-3−/− mice had higher BALF CCL-20, GM-CSF, and KC levels than did Bcl-3+/+ mice (Fig. 2E). Furthermore, Bcl-3−/− mice had increased levels of lung CCL-20 and GM-CSF in response to HDM (Fig. S2F). Remarkably, we discovered that IL-10 levels were augmented in HDM-sensitized Bcl-3−/− BALF and lung homogenates compared to those in their counterparts from Bcl-3+/+ mice (Figs. 2E, S2F). Other cytokines/chemokines, including IL-33, IL-25, and CSF-1, were present at comparable levels in the lungs of HDM-sensitized Bcl-3+/+ and Bcl-3−/− mice (Fig. 2F). As TLR4 stimulation of lung ECs is essential for Th2 response induction following HDM stimulation [9, 10, 36], we investigated whether the levels of negative TLR signaling regulators, including Tnfaip3 (encoding A20), Cish (encoding SOCS), Inpp5d (encoding SHIP), and Irak3 (encoding IRAK-M), were decreased in Bcl-3−/− lungs after HDM exposure. However, we found that Bcl-3+/+ and Bcl-3−/− lungs had similar levels of negative regulators of TLR signaling in response to HDM (Fig. 2G). These findings demonstrated that Bcl-3 inhibits IL-10 production in the lung and limits the production of the proallergic factors CCL-20 and GM-CSF by lung ECs upon HDM sensitization, which is consistent with the robust DC responses seen in Bcl-3−/− lungs upon HDM sensitization.
IL-10 promoted HDM-induced Th2 sensitization
Although IL-10 is commonly regarded as an anti-inflammatory cytokine, it exerts certain immunostimulatory and proinflammatory functions depending on the context [23, 28, 37, 38]. Given the elevated lung IL-10 levels in Bcl-3−/− mice upon HDM stimulation, along with the robust allergic sensitization and Th2 responses observed, we investigated whether IL-10 could serve as an activator/enhancer of aeroallergen sensitization, thereby promoting the subsequent development of allergic asthma. First, we observed a positive association between IL-10 and CCL-20 in HDM-sensitized mice and in patients with asthma (GEO: GSE103166) [39] (Fig. 3A, B). Next, we instilled anti-IL-10 with HDM to block endogenous IL-10 upon HDM sensitization and explored whether the elevated IL-10 levels could account for the enhanced epithelial and DC responses in HDM-sensitized Bcl-3−/− mice (Fig. S3A). HDM-sensitized Bcl-3−/− mice showed a substantial reduction in the secretion of BALF CCL-20 and in the recruitment of DCs (especially inflammatory CD11b+ DCs) to the lung when IL-10 was neutralized (Fig. 3C, D). In addition, no difference was observed between HDM-sensitized Bcl-3+/+ and Bcl-3−/− mice in initiating epithelial and DC responses when IL-10 was neutralized (Fig. 3C, D). The results demonstrated that IL-10 is crucial for amplifying lung epithelial and DC responses under the regulation of Bcl-3.
Fig. 3.

IL-10 promoted HDM sensitization for asthma development. A Correlations between lung Il10 mRNA levels and Ccl20 mRNA levels in HDM-sensitized mice. B Correlations of bronchial IL-10 mRNA levels versus CCL-20 mRNA levels in patients with asthma (n = 56). A and B were determined by Spearman’s rank correlation. C Mice were sensitized intranasally to HDM with or without anti-IL-10; 4 h later, CCL-20 levels in bronchoalveolar lavage fluid (BALF) were measured. D Mice were sensitized intranasally to HDM with or without anti-IL-10 on days 0, 1, and 2. Lung DCs were analyzed on day 4. E Mice were sensitized with HDM or HDM plus IL-10; 4 h later, lung epithelial cells were sorted using flow cytometry, and Ccl20 mRNA was measured. F Mice were sensitized with HDM or HDM plus IL-10 on days 0, 1, and 2. Lung DCs were analyzed on day 4. G Cells in BALF were analyzed on day 14 when supplying IL-10 during the sensitization phase in the HDM-induced asthma model. H Mice were intranasally instilled with IL-10; 4 h later, lung epithelial cells were sorted, and the Ccl20 mRNA level was measured. I Mice were instilled intranasally with IL-10 on days 0, 1, and 2. Lung DCs were analyzed on day 4. Data in (A, C–I) were combined from two or three experiments; n = 5–15. C, D, H, and I Mann‒Whitney test. E, F, and G Student’s t test
Next, we administered a small amount of IL-10 (10 ng) with HDM during the sensitization phase (Fig. S3B) to ascertain whether an exogenous supply of IL-10 could promote allergic sensitization to inhaled HDM allergens. Compared to the HDM-sensitized group, mice showed increased levels of the DC chemoattractant CCL20 in the lung, which did not influence other cytokines (GM-CSF and KC), when HDM was administered with IL-10 (Fig. S3C). Next, to directly confirm whether IL-10 promotes epithelial production of CCL-20, we sorted CD45− CD326+ lung ECs and found that those from mice exposed to HDM plus IL-10 had increased Ccl20 mRNA levels in contrast to those from HDM-exposed mice (Fig. 3E). Moreover, we discovered that mice had elevated DC responses (Figs. S3B, 3F) and increased eosinophilic inflammation when both HDM and IL-10 were instilled during sensitization (Fig. S3D-above; i.n. HDM + IL-10 during sensitization while i.n. HDM during challenge, Fig. 3G). However, when IL-10 was administered with HDM in the challenge phase (Fig. S3D-below; i.n. HDM during sensitization while i.n. HDM + IL-10 during challenge), we found decreased airway inflammation, manifested by diminished lung eosinophilia and lymphocytosis (Fig. S3E). Furthermore, we observed that the addition of IL-10 to HDM during sensitization promoted the production of Th2 cytokines in the lung, whereas the addition of IL-10 to HDM during challenge inhibited Th2 responses (Fig. S3F). These findings suggest that IL-10 contributes to the induction of the Th2 response while also inhibiting the established Th2 response.
To directly confirm the contribution of IL-10 in increasing epithelial chemokine production to promote DC responses, we instilled mice with IL-10 intranasally and isolated primary lung ECs to assess Ccl20 mRNA levels. We found that IL-10 could induce CCL-20 expression by lung ECs (Fig. 3H) and instigate the infiltration of DCs (especially inflammatory CD11b+ DCs) into the lung (Fig. 3I). Collectively, the findings demonstrated that IL-10 can amplify the release of the DC-attracting chemokine CCL-20 by lung ECs, thereby enhancing lung DC recruitment to increase Th2 sensitization upon HDM stimulation.
IL-10 was primarily secreted by B cells during HDM sensitization
Although Th2 cells were the first identified cellular source of IL-10, other cells, including macrophages, monocytes, DCs, B cells, and ECs, also produce this cytokine [28]. Considering the crucial role of IL-10 in allergic sensitization, we next determined the primary cellular sources of IL-10 following lung sensitization with HDM. To this end, analyses were performed 4 h after intranasal administration of HDM, when IL-10 levels in sensitized mice were the highest (Fig. S4A). Lung cells capable of IL-10 production were identified in our model via Tiger (IL-10-internal ribosome entry site-GFP-enhanced reporter) mice [30]. To validate whether GFP expression reflected the IL-10 mRNA levels in our model, we sorted the GFPhigh and GFPlow cells from HDM-sensitized lungs and found a 20-fold difference in IL-10 levels between GFPhigh and GFPlow cells (Fig. S4B). Using a gating strategy modified from that reported in a previously described study (Fig. S4C, D) [40, 41], we found that B cells (FSClo, CD11c−, CD19+, and MHCII+) produced significantly more IL-10 than other reported IL-10-producing cells, such as macrophages/monocytes (CD11c+ and MHC-IImid–low), DCs (CD11c+ and MHC-IIhi), T cells (FSClo, CD11c−, CD3+, and MHCII−), and ECs (CD45− and CD326+), following stimulation with HDM (Figs. 4A, S4E). HDM-sensitized mice had significantly more IL-10-producing B cells in the lung than the PBS control mice (Fig. 4B). Additionally, lung cells were stimulated with ionomycin and PMA in vitro and stained for intracellular IL-10 expression to confirm whether B cells were the predominant source of IL-10 following HDM stimulation. The results demonstrated that B cells are a major population of IL-10+ cells in HDM-treated WT mice (Fig. S4F). Immunofluorescence further confirmed these results (Fig. 4C). Additionally, we sorted lung B cells after mice were exposed to HDM and found that B cells had increased Il10 mRNA levels upon HDM stimulation (Fig. S4G). Thus, inhalation of HDMs resulted in IL-10 secretion primarily by B cells.
Fig. 4.

IL-10 secreted by B cells was crucial for initiating HDM-induced asthma. A Number of different GFP (IL-10)-producing cell subsets in the lungs of PBS- or HDM-treated Tiger mice. B Flow cytometry (left) and cell number (right) of GFP(IL-10)+ B cells in the lungs of PBS- or HDM-treated Tiger mice. C Representative immunofluorescence of the lungs of mice after 4 h of HDM exposure. Scale bar, 20 μm. A–C Mice were sensitized to HDM for 4 h. D Concentrations of IL-10 and CCL-20 in IL-10 B-WT and IL-10 B-KO BALF. Mice were sensitized intranasally to HDM with or without IL-10 for 4 h. E Number of lung DCs. Mice were sensitized to HDM with or without IL-10 on days 0, 1, and 2 and analyzed on day 4. F Mice were sensitized and challenged with HDM. On day 14, BALF cells were analyzed. G Representative lung sections stained with H&E (scale bars, 100 μm). H Lung levels of IL-10, IL-5, and IL-13. I Levels of HDM-specific IgE and IgG1 in mouse sera. J Lung resistance in response to increasing doses of methacholine. Data in (A–I) were combined from two or three experiments; n = 6–15. A, J Two-way ANOVA. B, I Mann‒Whitney test. C–H Student’s t test
IL-10 in B cells was responsible for HDM-induced asthma
To investigate whether IL-10-producing B cells are required for asthma development, we established our HDM model in μMT (B-cell-deficient) mice. Our data showed that HDM-treated μMT mice exhibited decreased allergic airway inflammation, including diminished BALF influx of eosinophils and lymphocytes and lower lung IL-10 and Th2 cytokine production, in contrast to HDM-treated WT mice (Fig. S4H, I). DC responses in the sensitization phase were also decreased in the absence of B cells (Fig. S4J).
Next, we specifically deleted IL-10 in B cells (IL-10B-KO) by crossing Il10fl/fl mice with Mb1-Cre mice and sensitized them with HDM to measure lung EC and DC responses in order to investigate whether IL-10 from B cells is critical for initiating or amplifying allergic responses. HDM-sensitized IL-10B-KO mice showed significantly lower IL-10 and CCL-20 levels in BALF and less lung DC recruitment than IL-10B-WT mice (Fig. 4D, E). Notably, when IL-10 was administered to HDM-treated IL-10B-KO mice, CCL-20 production in the BALF and lung DC recruitment in IL-10B-KO mice were restored (Fig. 4D, E), strongly suggesting that IL-10 produced by B cells can promote HDM sensitization by increasing the production of the DC chemoattractant CCL-20, thereby promoting inflammatory DC recruitment to the lung. However, the MLN size of HDM-sensitized IL-10B-KO mice was so small that we could not measure the DC response in MLNs in this experiment.
We next sought to determine whether the decreased allergic sensitization to HDM in IL-10B-KO mice alleviated asthma development. As expected, HDM-treated IL-10B-KO mice showed significantly decreased alveolar cellular infiltration and eosinophilia compared to IL-10B-WT mice (Fig. 4F). HDM-treated IL-10B-KO mice also developed mild lung pathology but with decreased inflammation (Fig. 4G). Moreover, lung IL-10 and Th2 cytokine levels were markedly reduced in HDM-treated IL-10B-KO mice compared to those in IL-10B-WT mice (Fig. 4H). Additionally, HDM-specific IgE and IgG1 levels were decreased in the sera of HDM-treated IL-10B-KO mice (Fig. 4I). HDM-treated IL-10B-KO mice also had significantly reduced AHR in comparison to IL-10B-WT mice (Fig. 4J). Therefore, these findings demonstrated that B-cell-derived IL-10 promoted allergic HDM sensitization and is therefore required for the development of HDM-induced asthma.
B-cell-derived IL-10 was not required for asthma development when sensitization bypassed the lung epithelium
To investigate whether the decreased allergic responses to HDM in IL-10B-KO mice are caused by the absence of B-cell-derived IL-10 (which might facilitate lung sensitization to aeroallergens, thereby promoting allergic asthma) and not due to unknown effects related to specific IL-10 loss in B cells, we used a traditional allergic asthma model, the OVA/alum-induced model, in which sensitization to the OVA allergen bypasses the lung epithelium (Fig. S5A) [10, 42]. IL-10B-WT and IL-10B-KO mice showed similar infiltration of BALF inflammatory cells (Fig. S5B) and comparable pulmonary levels of Th2 and Th17 cytokines (Fig. S5C), indicating that IL-10B-KO mice are not intrinsically incapable of mounting Th2 responses and that IL-10 derived from B cells promotes allergic asthma in a context-dependent manner, mainly for local airway sensitization but not for systemic sensitization.
Bcl-3 regulated allergic HDM-induced asthma through B-cell-derived IL-10
Bcl-3 is a critical IL-10 regulator during the innate immune response [24, 27]. Consistent with the findings of previous studies, we found that Bcl-3−/− mice had more IL-10 than Bcl-3+/+ mice in response to natural allergens (Figs. 1, 2) [24]. Moreover, as IL-10 derived from B cells was necessary for the promotion of Th2 sensitization upon HDM stimulation in the lung (Figs. 3, 4), we investigated whether Bcl-3 regulated allergic asthma through B-cell-derived IL-10. First, we measured Bcl-3 levels in the lung using qRT‒PCR and found Bcl-3 to be highly expressed in B cells (Fig. S6A), hence confirming the potential role of Bcl-3 in regulating B-cell-derived IL-10. Consistent with the results from WT Tiger mice (Fig. 4), B cells were found to be the primary lung IL-10-producing populations in Bcl-3-deficient mice upon HDM stimulation (Fig. 5A, S6B). Notably, Bcl-3-deficient mice had a higher number of IL-10-producing B cells upon HDM stimulation than WT mice (Fig. 5B, S6C), suggesting that Bcl-3 is an inhibitor of B-cell-derived IL-10 upon HDM stimulation. Furthermore, we confirmed the results by sorting B cells and T cells (the secondary IL-10-secreting cells in Bcl-3−/− mice) from HDM-treated lungs and found that Bcl-3 deficiency led to increased IL-10 levels in B cells upon HDM stimulation (Fig. S6D). In contrast, IL-10 expression in T cells did not differ between the Bcl-3+/+ PBS and Bcl-3+/+ HDM groups or between the Bcl-3+/+ HDM and Bcl-3−/− HDM groups, indicating that T-cell-derived IL-10 may be redundant for allergic sensitization in this model (Fig. S6D).
Fig. 5.

Bcl-3 regulated HDM-induced asthma through B-cell-derived IL-10. A Number of different GFP(IL-10)-producing cell subsets in the lungs of PBS- or HDM-treated Bcl-3−/−Tiger mice. B Flow cytometry (left) and cell number (right) of GFP(IL-10)+ B cells in Bcl-3+/+ Tiger and Bcl-3−/− Tiger lungs. A, B Mice were sensitized to HDM for 4 h. C Bcl-3+/+, Bcl-3−/−, and Bcl-3−/−IL-10 B-KO mice were sensitized and challenged with HDM. On day 14, bronchoalveolar lavage fluid cells were analyzed. D Lung levels of Th2 cytokines. E Bcl-3+/+, Bcl-3−/−, and Bcl-3−/−IL-10 B-KO mice were instilled intranasally with HDM on days 0, 1, and 2. Lung DCs were analyzed on day 4. F Concentrations of CCL-20, GM-CSF, and KC in Bcl-3+/+, Bcl-3−/−, and Bcl-3−/−IL-10 B-KO in bronchoalveolar lavage fluid. Mice were sensitized with HDM intranasally for 4 h. Data in (A–F) were combined from two or three experiments; n = 6–12. A Two-way ANOVA. B–F Student’s t test
Next, we crossed Bcl-3−/− mice with IL-10fl/fl Mb1Cre mice to deplete both Bcl-3 and B-cell-derived IL-10 (Bcl-3−/−IL10B-KO) and stimulated the animals with HDM. The increase in recruited eosinophils and lymphocytes in the BALF observed in Bcl-3−/− mice compared to Bcl-3+/+ mice following HDM exposure was abolished in Bcl-3−/−IL10B-KO mice (Fig. 5C). Additionally, the levels of the Th2 cytokines IL-4 and IL-5 were decreased in the lung homogenates of HDM-treated Bcl-3−/−IL-10B-KO mice compared to those in Bcl-3−/− mice (Fig. 5D). Loss of B-cell-derived IL-10 in Bcl-3−/− mice also led to reduced lung DC and epithelial responses during HDM sensitization (Fig. 5E, F). No difference was observed in eosinophilic airway inflammation, Th2 immune response, lung DC recruitment, or lung epithelial responses between HDM-treated Bcl-3+/+ and HDM-treated Bcl-3−/−IL-10B-KO mice (Fig. 5C–F), indicating that increased IL-10 production by B cells was necessary for elevated HDM-induced Th2 immunity in Bcl-3−/− mice. Thus, the results demonstrated that Bcl-3 limits allergic asthma by regulating the secretion of pro-allergic IL-10 from B cells.
Characterization of IL-10-secreting B cells during HDM sensitization
Given the predominant role of IL-10-secreting B cells in initiating aeroallergen sensitization, we sought to characterize the cells during HDM sensitization by single-cell RNA sequencing (scRNA-seq). Live lung B cells (CD45+ CD19+) from HDM-sensitized Bcl-3+/+ and Bcl-3−/− mice were isolated by cell sorting. A total of 5052 B cells from Bcl-3+/+ lungs and 5984 B cells from Bcl-3−/− lungs met the quality control criteria and were used in subsequent analyses. Cells were visualized using t-distributed stochastic neighbor embedding (t-SNE), and eight clusters of lung B cells were identified for both groups (Fig. 6A, S7A). Clusters 4, 6, 7, and 8 showed significantly different proportions between the Bcl-3+/+ and Bcl-3−/− groups, demonstrating that Bcl-3 modified the heterogeneity of lung B cells in response to HDM stimulation (Fig. 6A). Notably, we found IL-10 to be mainly expressed in cluster 7 (Fig. 6B, S7A). Next, we classified the eight clusters into four distinct subsets, including naïve B cells (clusters 1, 2, 3, and 8), transitional B cells (clusters 5 and 6), plasmablasts (cluster 4), and plasma cells (cluster 7) (Fig. 6C, S7B), by the hierarchical tree algorithm using Seurat’s Louvain algorithm and established marker genes of B-cell populations [43, 44]. In line with the results from IL-10 GFP mice (Fig. 5B), the IL-10+ plasma cell (cluster 7) proportion was increased in Bcl-3−/− mice (Fig. 6A, S7B). Furthermore, we found that the expression of several marker genes of murine IL-10-secreting B cells, characterized by other research groups [45], including CD44, CD138, CD9, and Blimp-1 (Prdm1), were upregulated in plasma cells (the main IL-10-secreting B cells in our model) compared to other subsets of B cells (Fig. S7C). Additionally, CD43, a previously identified marker gene for human IL-10-secreting B cells in alleviating inflammatory bowel disease [46], was also highly expressed in our IL-10-secreting plasma cells.
Fig. 6.

Characterization of lung IL-10-producing B cells upon HDM sensitization. A t-distributed stochastic neighbor embedding (t-SNE) plots of scRNA-seq data of lung B cells from the indicated mice clustered (left). Relative cell frequencies of each cluster are shown on the right. B IL-10 expression in lung B cells at the single-cell level. C Merged t-SNE plot of Bcl-3+/+ and Bcl-3−/− mice displaying the different B-cell subsets at the single-cell level. D Developmental trajectory of HDM-sensitized B cells over pseudotime in two-dimensional space. E Bubble plots showing the abundance of reported IL-10-producing B-cell markers in different subsets of HDM-sensitized lung B cells. F Verification of identified marker expression on GFP(IL-10)+ B cells by flow cytometry (above) and qRT‒PCR (below). Tiger mice were sensitized to HDM; 4 h later, lung GFP(IL-10)+ B cells were analyzed or sorted using flow cytometry. G Heatmap showing the expression of reported IL-10 regulators in IL-10+ plasma cells and IL-10– plasma cells. H Violin plots showing IL-10 regulator gene expression in Bcl-3+/+ and Bcl-3−/− plasma cells. I Verification of Bcl-6 expression in GFP(IL-10)+ B cells by flow cytometry (left) and qRT‒PCR (right). J Mice were sensitized to HDM; 4 h later, lung B cells were sorted using flow cytometry, and Blimp-1 and Bcl-6 mRNA levels were measured by qRT‒PCR. Data in (F, I, and J) were combined from two experiments; n = 6–11. F, I, and J) Mann‒Whitney test
Upon HDM sensitization, the plasma cluster subpopulation was the predominant fraction of IL-10-secreting B cells (Fig. S7D–F); however, the precise differentiation trajectory of this subpopulation remains elusive. Therefore, we used Monocle [47] to investigate the single-cell transition process of the IL-10-secreting plasma cell cluster and observed naïve B-cell (pre-branch) differentiation into cells in the IL-10-secreting plasma cluster, which was divided into two major tree-like structures (including cell fate 1 and cell fate 2) (Figs. 6D, Fig. S7G). The pre-branch consisted mainly of naïve B cells, whereas cell fate 1 consisted of plasma cells, plasmablasts, and transitional B cells, and the cell fate 2 consisted of naïve B cells and other subsets of plasmablasts and plasma cells (Fig. 6D, Fig. S7G). Notably, we found IL-10 to be mainly expressed in the fate 1 plasma cell subset. Moreover, we identified CD44, CD43, CD138, CD9, and Blimp-1 as specific markers of IL-10-secreting plasma cells in contrast to other B-cell subsets, including IL-10− plasma cells (Fig. 6E). Similar results were obtained from in vivo experiments by flow cytometry and qRT‒PCR (Figs. 6F, S7H). In addition, we observed that the expression of Blimp-1, a well-known IL-10 promoter [48, 49], was significantly upregulated in IL-10+ plasma cells compared to IL-10− plasma cells, and that of its antagonistic transcription factor Bcl-6 was downregulated in IL-10+ plasma cells (Fig. 6G). Furthermore, a lack of Bcl-3 in plasma cells caused increased expression of Blimp-1 and decreased expression of Bcl-6, indicating that Bcl-3 modulates IL-10 in plasma cells possibly by downregulating Blimp-1 expression, thereby upregulating Bcl-6 expression to suppress the expression of IL-10 (Fig. 6H); these results were in line with previous findings that demonstrated that Blimp-1 was a critical activator of IL-10 in T cells [48–50]. The results were further confirmed by flow cytometry and qRT‒PCR (Fig. 6I, J).
In summary, we concluded that CD44+CD43+CD138+ CD9+Blimp-1+ plasma cells are the main population of IL-10-producing B cells in HDM-sensitized mice and that Bcl-3 may regulate the expression of IL-10 in B cells through the Blimp-1–Bcl-6 axis.
Blockade of IL-10 during sensitization showed therapeutic potential in asthma
Previous results have demonstrated the proinflammatory potential of IL-10 in HDM-induced allergic asthma. Hence, we investigated whether blocking IL-10 signaling could have any therapeutic potential in allergic asthma. We intranasally instilled anti-IL-10/anti-IL-10R antibodies with HDMs to block IL-10 signaling during HDM sensitization (Fig. 7A) and examined airway eosinophilia and lymphocytosis, which are hallmarks of HDM-driven asthma. Mice showed a substantial reduction in lung eosinophilia and lymphocytosis when IL-10 or IL-10R was neutralized or blocked during sensitization (Fig. 7B, C). In addition, the levels of the Th2 cytokines IL-4, IL-5, and IL-13 were significantly decreased in lung homogenates when IL-10 signaling was blocked during sensitization (Fig. 7D). Isotype antibodies had no effect on the cellular composition of BALF or Th2 responses (Fig. 7B–D). The results suggested that targeting the IL-10 signaling pathway to avoid allergen sensitization could be employed as a promising therapeutic strategy for asthma, as allergic sensitization is not only necessary for developing new allergic asthma but also for aggravating the existing pulmonary inflammation in patients with asthma [5, 6].
Fig. 7.

IL-10 blockade during sensitization ameliorated allergic inflammation in asthma. A Experimental scheme. Mice were sensitized to HDM intranasally, with or without anti-IL-10/anti-IL-10R, and challenged with HDM intranasally. Mice were euthanized on day 14 for analysis. B Different cell counts in BALF when IL-10 was neutralized (isotype control, IgG2b). C Different cell counts in the BALF when IL-10R was blocked (isotype control, IgG1). D Levels of Th2 cytokines in the lungs when neutralizing IL-10 or blocking IL-10R during the sensitization phase. Combined data from three or four experiments shown in (B–D); n = 6–17. B–D Student’s t test
Discussion
Th2 immune responses induced by aeroallergens are responsible for most allergic asthma cases [2]. Bcl-3 and IL-10 (a broadly recognized anti-inflammatory cytokine) are essential regulators of T-cell immune responses [24, 27, 51]. Moreover, Bcl-3 is an inhibitor of IL-10 [52, 53]. However, whether Bcl-3 modulates Th2 cell-mediated allergic asthma via IL-10 remains unclear. We observed reduced Bcl-3 levels and increased IL-10 levels in patients with asthma, indicating that Bcl-3 and IL-10 are essential for asthma pathogenesis. Given the inverse correlation between Bcl-3 and IL-10 observed in patients with asthma, we hypothesized that Bcl-3−/− mice would show increased production of IL-10 and, consequently, decreased susceptibility to inflammation processes, such as allergic airway inflammation. Indeed, we found that Bcl-3−/− mice exhibited increased IL-10 production in the lung after exposure to HDM during the sensitization and challenge phases compared to Bcl-3+/+ mice. Unexpectedly, Bcl-3−/− mice also showed increased eosinophilic airway inflammation, augmented airway goblet cell hyperplasia, and elevated airway hyperreactivity upon HDM stimulation compared to HDM-treated Bcl-3+/+ control mice. Furthermore, Bcl-3−/− lungs had increased levels of epithelial chemokines in response to HDM, leading to robust DC responses and elevated Th2 responses. Thus, our results demonstrated that Bcl-3 limits lung IL-10 production and inhibits lung inflammation and asthma pathogenesis in an HDM-induced mouse model, suggesting that Bcl-3 is a critical inhibitor of IL-10 and allergic asthma.
Several previous studies have suggested that IL-10 can play either a proinflammatory [23, 35, 49] or anti-inflammatory role [18, 54–56] in the development of allergic asthma. In the OVA-induced asthma model, researchers found that IL-10 can inhibit airway inflammation by using IL-10 knockout mice [56]. However, while OVA can induce allergies in real life, it is less relevant than other environmental antigens, such as HDMs, which are a common trigger for asthma [1, 2]. Therefore, the HDM-induced asthma model is considered more appropriate for studying asthma.
In the HDM asthma model, researchers obtained contradictory results for IL-10 by using IL-10Rα-specific knockout in T-cell mice (IL10RαCD4Cre) [49, 54]. If the mice were sensitized with HDM via the peritoneum and challenged with HDM via the airway, IL10RαCD4Cre mice exhibited aggravated asthma features [54]. However, if the mice were sensitized and challenged with HDM via the airway, IL10RαCD4Cre mice exhibited relieved asthma features [49]. Thus, IL-10 may play different roles in modulating asthma depending on the allergen, the route of sensitization, and the time of disease development.
Our study found that IL-10 produced by B cells can promote asthma sensitization by enhancing the immune response of lung epithelial cells upon HDM stimulation. This explains the seemingly contradictory results mentioned above since the activation of lung epithelial cells is necessary for HDM asthma models (i.n. HDM in the sensitization and challenge phase) but is dispensable for the systemic sensitization in the OVA model (i.p. OVA/alum in sensitization phase and i.n. OVA in the challenge phase). Meanwhile, it also explains the divergent findings regarding IL10RαCD4Cre mice sensitized with HDM via the peritoneum or the airway.
Additionally, we propose that IL-10 has a dual role in regulating Th responses—promoting Th responses during the induction phase but having an anti-inflammatory effect on established Th responses. Specifically, we observed that adding IL-10 during the sensitization phase promoted asthma, while adding IL-10 during the challenge phase inhibited asthma. This observation is consistent with other studies that demonstrate that IL-10 promotes epicutaneous sensitization for allergic dermatitis [21], while transferring Tregs [55] or IL-10-producing B cells [18] to HDM-sensitized mice inhibits the subsequent development of asthma.
Although IL-10 from DCs may promote Th2 responses in asthma by inhibiting Th1 responses [35, 57], our study reveals that B cells are the primary source of IL-10, with DCs playing a minor role during sensitization to HDM. Using Tiger mice and intracellular IL-10 FACS staining, we examined the source of IL-10 in the allergic lung. Although several different cell types can secrete IL-10 in the respiratory tract [18, 28, 58, 59], we found that IL-10 is mainly secreted by B cells upon HDM exposure. This was in line with a previous study by Habener and colleagues, who showed that lungs from HDM-sensitized mice exhibited elevated B-cell numbers and B-cell-specific IL-10 expression, with the highest IL-10 level produced by follicular mature B cells [60]. Therefore, we propose that IL-10 from both B cells and DCs collaboratively induces a Th2 response during the sensitization phase of asthma, which is essential in protecting the lungs from allergen invasion.
Unexpectedly, resident lung plasma cells rather than reactive plasmablasts or follicular mature B cells formed the main subset of IL-10-secreting B cells upon HDM stimulation in our model, which is consistent with the results of another previous study [61]. IL-10-secreting plasma cells in HDM-sensitized lungs differ from other B-cell populations in their higher expression of CD44, CD43, CD9, and Blimp-1, which can serve as markers. Notably, these markers distinguished our IL-10-secreting B cells from the so-called regulatory B cells (B10 cells, CD5+ CD1dhi) [62, 63]. Regarding the rapid response observed in our study, we hypothesize that these specific plasma cells arise from B cells during steady-state conditions, and upon HDM sensitization, antigens or other signals, such as LPS, could rapidly stimulate these cells to produce IL-10 through TLR signaling and/or BCR signaling. Previous studies support this hypothesis, showing fast IL-10 production by natural plasma cells that are controlled by TLR signaling in infection models [61], and the necessity of low-level TLR ligand stimulation for Th2 immunity development in response to HDM allergens [64, 65].
Although T-cell-derived IL-10 could possibly play a concomitant role with B-cell-derived IL-10, as Mb1Cre mice show low-level recombinase activity in T cells in the lymph nodes, spleen, and thymus [66], we demonstrated that HDM-sensitized mice from Bcl-3+/+ HDM and Bcl-3−/− HDM groups have similar Il10 expression in T cells compared to PBS-control mice, which is indicative of a redundant role of T-cell-derived IL-10 in the allergic sensitization phase. Additionally, given that allergen sensitization is essential for the subsequent development of allergic asthma and that IL-10 is principally sourced from B cells during HDM sensitization, which facilitates this progression, the resistance to HDM-induced asthma in mice lacking B-cell-derived IL-10 seemed quite reasonable.
Bcl-3 is highly expressed in lung B cells and plays a critical role in controlling the production of B-cell-derived IL-10 under HDM stimulation. This was evidenced by the fact that HDM-treated Bcl-3−/− mice had a significantly higher number of IL-10+ B cells than HDM-treated Bcl-3+/+ mice. When IL-10 was neutralized during HDM sensitization, both Bcl-3+/+ and Bcl-3−/− mice had similar lung epithelial responses and DC recruitment. Moreover, Bcl-3−/−IL-10B-KO mice exhibited similar lung epithelial responses, DC recruitment patterns, and Th2 inflammation in response to HDM compared to HDM-treated Bcl-3+/+ mice. Thus, despite the lack of direct evidence from the selective depletion of Bcl-3 in B-cell mice, our study clearly demonstrated that Bcl-3 inhibits the production of pro-inflammatory IL-10 from B cells during allergic sensitization, thereby preventing HDM-driven asthma.
Blimp-1 is essential for IL-10 production in effector CD4 T cells and T regulatory cells [48, 49]. Additionally, IL-10 can upregulate Blimp-1, which in turn acts on Bcl-6 and increases GATA3 expression in lung Th2 cells [49]. We demonstrated that Bcl-3 possibly modulates plasma-derived IL-10 via the Blimp-1/Bcl-6 axis, as manifested by increased Blimp-1 but decreased Bcl-6 expression in Bcl-3-deficient plasma cells during HDM sensitization. However, further studies are required to carefully investigate the role of the Bcl-3–Blimp-1–Bcl-6 axis in regulating IL-10 production in B cells to better understand the complex interactions in allergic asthma.
Although we reported that B-cell-derived IL-10 amplifies lung epithelium‐derived CCL-20 production and subsequent DC recruitment, the underlying molecular mechanism remains relatively unexplored. Moreover, additional studies are required to fully understand the effect of B-cell-derived IL-10 in patients with asthma. As the heterogeneity of human patients with asthma is as wide-ranging as that of the mouse models mentioned above, we assume that B-cell-derived IL-10 may contribute to asthmatic features in some patient groups, although probably not in all. Indeed, through the analysis of microarray data published in a previous article [39], we found that the level of IL-10 was elevated in patients with asthma, while others found less IL-10 present in the lungs of patients with asthma than in healthy people [28, 67]. In addition, the adoptive transfer of IL-10-sufficient B cells has different impacts on murine HDM-induced allergic lung inflammation depending on the transfer timing [18, 60]. Based on these findings, we predicted that asthma therapies targeting IL-10 and Bcl-3 would work best when considering the course of disease progression.
In conclusion, we demonstrated that IL-10 derived from B cells promotes lung epithelial sensitization and the DC response to HDMs. Bcl-3 is a negative regulator of B-cell-derived IL-10 and inhibits HDM-induced asthma by limiting the production of proinflammatory B-cell-derived IL-10 during the allergic sensitization phase. The findings supported the notion that blocking allergic sensitization can efficiently prevent/limit the subsequent allergic Th2-related inflammation that causes allergic asthma and suggested that targeting the IL-10 and Bcl-3 signaling pathways to avoid allergen sensitization could be a promising therapeutic strategy for allergic asthma.
Supplementary information
Acknowledgements
We thank Dr. Mingfang Lu, Dr. Wei Jiang, Dr. Guohong Hu, Dr. Jin Li, and Dr. Xubo Huang for providing advice as well as some reagents used in this study. We appreciate Dr. Michael Reth and Dr. Axel Roers for gifting Il10fl/fl and Mb1-Cre mice. This investigation was funded by the National Natural Science Foundation of China (grants 81901633 to GQ and 91949102 to XZ), the National Program on Key Research (2021YFA0804703 to XZ), and the Discipline Construction Projects of Guangzhou Medical University (02-445-2301246XM to XZ).
Author contributions
XZ, GQ, and HF conceptualized the study. GQ, XZ, and HF designed the experiments. GQ, WJ, DS, ZS conducted the experiments. AC and HWF helped to perform the experiments; JW completed the acquisition and processing of microarray datasets. GQ, WJ, DS and ZS analyzed the data. GQ wrote the manuscript. XZ and HF revised the manuscript. YL, ZY, and HW provided advice and some reagents.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Guojun Qian, Wenxia Jiang, Donglin Sun, Zhun Sun.
Contributor Information
Guojun Qian, Email: qianguojun@gzhmu.edu.cn.
Hao Fang, Email: drfanghao@163.com.
Xiaoren Zhang, Email: xrzhang@gzhmu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41423-023-01079-w.
References
- 1.Lambrecht BN, Hammad H, Fahy JV. The cytokines of asthma. Immunity. 2019;50:975–91. [DOI] [PubMed] [Google Scholar]
- 2.Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16:45–56. [DOI] [PubMed] [Google Scholar]
- 3.Kool M, Willart MA, van Nimwegen M, Bergen I, Pouliot P, Virchow JC, et al. An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity. 2011;34:527–40. [DOI] [PubMed] [Google Scholar]
- 4.Stoltz DJ, Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Gern JE, et al. Specific patterns of allergic sensitization in early childhood and asthma & rhinitis risk. Clin Exp Allergy. 2013;43:233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moon HG, Kim SJ, Lee MK, Kang H, Choi HS, Harijith A, et al. Colony-stimulating factor 1 and its receptor are new potential therapeutic targets for allergic asthma. Allergy. 2020;75:357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teach SJ, Gill MA, Togias A, Sorkness CA, Arbes SJ Jr, Calatroni A, et al. Preseasonal treatment with either omalizumab or an inhaled corticosteroid boost to prevent fall asthma exacerbations. J Allergy Clin Immunol. 2015;136:1476–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammad H, Lambrecht BN. The basic immunology of asthma. Cell. 2021;184:2521–2. [DOI] [PubMed] [Google Scholar]
- 8.Klassen C, Karabinskaya A, Dejager L, Vettorazzi S, Van Moorleghem J, Luhder F, et al. Airway epithelial cells are crucial targets of glucocorticoids in a mouse model of allergic asthma. J Immunol. 2017;199:48–61. [DOI] [PubMed] [Google Scholar]
- 9.Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science. 2015;349:1106–10. [DOI] [PubMed] [Google Scholar]
- 10.Qian G, Jiang W, Zou B, Feng J, Cheng X, Gu J, et al. LPS inactivation by a host lipase allows lung epithelial cell sensitization for allergic asthma. J Exp Med. 2018;215:2397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holgate ST. Innate and adaptive immune responses in asthma. Nat Med. 2012;18:673–83. [DOI] [PubMed] [Google Scholar]
- 12.Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MA, Kool M, et al. Inflammatory dendritic cells–not basophils–are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med. 2010;207:2097–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity. 2013;38:322–35. [DOI] [PubMed] [Google Scholar]
- 14.Ballesteros-Tato A, Randall TD, Lund FE, Spolski R, Leonard WJ, Leon BT. Follicular helper cell plasticity shapes pathogenic T helper 2 cell-mediated immunity to inhaled house dust mite. Immunity. 2016;44:259–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wypych TP, Marzi R, Wu GF, Lanzavecchia A, Sallusto F. Role of B cells in TH cell responses in a mouse model of asthma. J Allergy Clin Immunol. 2018;141:1395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korsgren M, Erjefalt JS, Korsgren O, Sundler F, Persson CG. Allergic eosinophil-rich inflammation develops in lungs and airways of B cell-deficient mice. J Exp Med. 1997;185:885–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dullaers M, Schuijs MJ, Willart M, Fierens K, Van Moorleghem J, Hammad H, et al. House dust mite-driven asthma and allergen-specific T cells depend on B cells when the amount of inhaled allergen is limiting. J Allergy Clin Immunol. 2017;140:76–88.e7. [DOI] [PubMed] [Google Scholar]
- 18.Braza F, Chesne J, Durand M, Dirou S, Brosseau C, Mahay G, et al. A regulatory CD9(+) B-cell subset inhibits HDM-induced allergic airway inflammation. Allergy. 2015;70:1421–31. [DOI] [PubMed] [Google Scholar]
- 19.Ronet C, Hauyon-La Torre Y, Revaz-Breton M, Mastelic B, Tacchini-Cottier F, Louis J, et al. Regulatory B cells shape the development of Th2 immune responses in BALB/c mice infected with Leishmania major through IL-10 production. J Immunol. 2010;184:886–94. [DOI] [PubMed] [Google Scholar]
- 20.Hernandez HJ, Wang Y, Stadecker MJ. In infection with Schistosoma mansoni, B cells are required for T helper type 2 cell responses but not for granuloma formation. J Immunol. 1997;158:4832–7. [PubMed] [Google Scholar]
- 21.Laouini D, Alenius H, Bryce P, Oettgen H, Tsitsikov E, Geha RS. IL-10 is critical for Th2 responses in a murine model of allergic dermatitis. J Clin Investig. 2003;112:1058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Justice JP, Shibata Y, Sur S, Mustafa J, Fan M, Van Scott MR. IL-10 gene knockout attenuates allergen-induced airway hyperresponsiveness in C57BL/6 mice. Am J Physiol Lung Cell Mol Physiol. 2001;280:L363–8. [DOI] [PubMed] [Google Scholar]
- 23.Mäkelä MJ, Kanehiro A, Borish L, Dakhama A, Loader J, Joetham A, et al. IL-10 is necessary for the expression of airway hyperresponsiveness but not pulmonary inflammation after allergic sensitization. Proc Natl Acad Sci. 2000;97:6007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative regulation of toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science. 2007;317:675–8. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Wang H, Claudio E, Brown K, Siebenlist U. A role for the IkappaB family member Bcl-3 in the control of central immunologic tolerance. Immunity. 2007;27:438–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang W, Wang H, Claudio E, Tassi I, Ha HL, Saret S, et al. The oncoprotein and transcriptional regulator Bcl-3 governs plasticity and pathogenicity of autoimmune T cells. Immunity. 2014;41:555–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reissig S, Tang Y, Nikolaev A, Gerlach K, Wolf C, Davari K, et al. Elevated levels of Bcl-3 inhibits Treg development and function resulting in spontaneous colitis. Nat Commun. 2017;8:15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saraiva M, Vieira P, O’Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med. 2020;217:e20190418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A, et al. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J Immunol. 2009;183:2312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galan JE, et al. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity. 2006;25:941–52. [DOI] [PubMed] [Google Scholar]
- 31.Kalidhindi RSR, Ambhore NS, Sathish V. Cellular and biochemical analysis of bronchoalveolar lavage fluid from murine lungs. Methods Mol Biol. 2021;2223:201–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Rijt LS, Kuipers H, Vos N, Hijdra D, Hoogsteden HC, Lambrecht BN. A rapid flow cytometric method for determining the cellular composition of bronchoalveolar lavage fluid cells in mouse models of asthma. J Immunol Methods. 2004;288:111–21. [DOI] [PubMed] [Google Scholar]
- 33.Voraphani N, Gladwin MT, Contreras AU, Kaminski N, Tedrow JR, Milosevic J, et al. An airway epithelial iNOS-DUOX2-thyroid peroxidase metabolome drives Th1/Th2 nitrative stress in human severe asthma. Mucosal Immunol. 2014;7:1175–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asayama K, Kobayashi T, D’Alessandro-Gabazza CN, Toda M, Yasuma T, Fujimoto H, et al. Protein S protects against allergic bronchial asthma by modulating Th1/Th2 balance. Allergy. 2020;75:2267–78. [DOI] [PubMed] [Google Scholar]
- 35.Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL, et al. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun. 2013;4:2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15:410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laidlaw BJ, Cui W, Amezquita RA, Gray SM, Guan T, Lu Y, et al. Production of IL-10 by CD4(+) regulatory T cells during the resolution of infection promotes the maturation of memory CD8(+) T cells. Nat Immunol. 2015;16:871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xin G, Zander R, Schauder DM, Chen Y, Weinstein JS, Drobyski WR, et al. Single-cell RNA sequencing unveils an IL-10-producing helper subset that sustains humoral immunity during persistent infection. Nat Commun. 2018;9:5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khoo SK, Read J, Franks K, Zhang G, Bizzintino J, Coleman L, et al. Upper airway cell transcriptomics identify a major new immunological phenotype with strong clinical correlates in young children with acute wheezing. J Immunol. 2019;202:1845–58. [DOI] [PubMed] [Google Scholar]
- 40.Haspeslagh E, Debeuf N, Hammad H, Lambrecht BN. Murine models of allergic asthma. Methods Mol Biol. 2017;1559:121–36. [DOI] [PubMed] [Google Scholar]
- 41.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. 2013;49:503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Debeuf N, Haspeslagh E, van Helden M, Hammad H, Lambrecht BN. Mouse models of asthma. Curr Protoc Mouse Biol. 2016;6:169–84. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Xu Y, Chen Z, Liang J, Lin Z, Liang H, et al. Liver immune profiling reveals pathogenesis and therapeutics for biliary atresia. Cell. 2020;183:1867–83.e26. [DOI] [PubMed] [Google Scholar]
- 44.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jansen K, Cevhertas L, Ma S, Satitsuksanoa P, Akdis M, van de Veen W. Regulatory B cells, A to Z. Allergy. 2021;76:2699–715. [DOI] [PubMed] [Google Scholar]
- 46.Chen X, Cai C, Xu D, Liu Q, Zheng S, Liu L, et al. Human mesenchymal stem cell-treated regulatory CD23(+)CD43(+) B cells alleviate intestinal inflammation. Theranostics. 2019;9:4633–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14:979–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parish IA, Marshall HD, Staron MM, Lang PA, Brustle A, Chen JH, et al. Chronic viral infection promotes sustained Th1-derived immunoregulatory IL-10 via BLIMP-1. J Clin Invest. 2014;124:3455–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He K, Hettinga A, Kale SL, Hu S, Xie MM, Dent AL, et al. Blimp-1 is essential for allergen-induced asthma and Th2 cell development in the lung. J Exp Med. 2020;217:e20190742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neumann C, Heinrich F, Neumann K, Junghans V, Mashreghi MF, Ahlers J, et al. Role of Blimp-1 in programing Th effector cells into IL-10 producers. J Exp Med. 2014;211:1807–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaiswal H, Ciucci T, Wang H, Tang W, Claudio E, Murphy PM, et al. The NF-kappaB regulator Bcl-3 restricts terminal differentiation and promotes memory cell formation of CD8+ T cells during viral infection. PLoS Pathog. 2021;17:e1009249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuwata H, Watanabe Y, Miyoshi H, Yamamoto M, Kaisho T, Takeda K, et al. IL-10-inducible Bcl-3 negatively regulates LPS-induced TNF-alpha production in macrophages. Blood. 2003;102:4123–9. [DOI] [PubMed] [Google Scholar]
- 53.Riemann M, Endres R, Liptay S, Pfeffer K, Schmid RM. The IkappaB protein Bcl-3 negatively regulates transcription of the IL-10 gene in macrophages. J Immunol. 2005;175:3560–8. [DOI] [PubMed] [Google Scholar]
- 54.Coomes SM, Kannan Y, Pelly VS, Entwistle LJ, Guidi R, Perez-Lloret J, et al. CD4(+) Th2 cells are directly regulated by IL-10 during allergic airway inflammation. Mucosal Immunol. 2017;10:150–61. [DOI] [PubMed] [Google Scholar]
- 55.Kearley J, Barker JE, Robinson DS, Lloyd CM. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J Exp Med. 2005;202:1539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tournoy KG, Kips JC, Pauwels RA. Endogenous interleukin-10 suppresses allergen-induced airway inflammation and nonspecific airway responsiveness. Clin Exp Allergy. 2000;30:775–83. [DOI] [PubMed] [Google Scholar]
- 57.Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol. 2010;11:647–55. [DOI] [PubMed] [Google Scholar]
- 58.Golebski K, Layhadi JA, Sahiner U, Steveling-Klein EH, Shamji MH. Induction of IL-10-producing type 2 innate lymphoid cells by allergen immunotherapy is associated with clinical response. Immunity 2021;54:291–307.e7. [DOI] [PubMed] [Google Scholar]
- 59.Branchett WJ, Stolting H, Oliver RA, Walker SA, Puttur F, Gregory LG, et al. A T cell-myeloid IL-10 axis regulates pathogenic IFN-gamma-dependent immunity in a mouse model of type 2-low asthma. J Allergy Clin Immunol. 2020;145:666–78.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Habener A, Happle C, Grychtol R, Skuljec J, Busse M, Daluge K, et al. Regulatory B cells control airway hyperreactivity and lung remodeling in a murine asthma model. J Allergy Clin Immunol. 2021;147:2281–2294.e7. [DOI] [PubMed]
- 61.Lino AC, Dang VD, Lampropoulou V, Welle A, Joedicke J, Pohar J, et al. LAG-3 inhibitory receptor expression identifies immunosuppressive natural regulatory plasma cells. Immunity. 2018;49:120–33.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Horikawa M, Weimer ET, DiLillo DJ, Venturi GM, Spolski R, Leonard WJ, et al. Regulatory B cell (B10 Cell) expansion during Listeria infection governs innate and cellular immune responses in mice. J Immunol. 2013;190:1158–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. 2015;42:607–12. [DOI] [PubMed] [Google Scholar]
- 64.Wang J, Zhou Y, Zhang H, Hu L, Liu J, Wang L, et al. Pathogenesis of allergic diseases and implications for therapeutic interventions. Signal Transduct Target Ther. 2023;8:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196:1645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci USA. 2006;103:13789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Howes A, Gabryšová L, O’Garra A. Role of IL-10 and the IL-10 Receptor in Immune Responses. Reference Module Biomed Sci. 2014;9780128012383:1–11.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.