

Abstract
Since testicular orphan nuclear receptor 4 (TR4) was cloned, its physiological function has remained largely unknown. Throughout postnatal development, TR4-knockout (TR4−/−) mice exhibited behavioral deficits in motor coordination, suggesting impaired cerebellar function. Histological examination of the postnatal TR4−/− cerebellum revealed gross abnormalities in foliation; specifically, lobule VII in the anterior vermis was missing. Further analyses demonstrated that the laminations of the TR4−/− cerebellar cortex were changed, including reductions in the thickness of the molecular layer and the internal granule layer, as well as delayed disappearance of the external granule cell layer (EGL). These lamination irregularities may result from interference with granule cell proliferation within the EGL, delayed inward migration of postmitotic granule cells, and a higher incidence of apoptotis. In addition, abnormal development of Purkinje cells was observed in the postnatal TR4−/− cerebellum, as evidenced by aberrant dendritic arborization and reduced calbindin staining intensity. Expression of Pax-6, Sonic Hedgehog (Shh), astrotactin (Astn), reelin, and Cdk-5, genes correlated with the morphological development of the cerebellum, is reduced in the developing TR4−/− cerebellum. Together, our findings suggest that TR4 is required for normal cerebellar development.
Testicular orphan nuclear receptor 4 (TR4) was cloned from human and rat testis and prostate, as well as from mouse brain cDNA libraries, and is a member of the nuclear receptor superfamily (5). No biological ligand has been identified; however, TR4 was shown to be essential for normal spermatogenesis in the testis (28). In the rodent, TR4 expression can be detected in the peripheral organs, such as the adrenal gland, spleen, testis, thyroid and pituitary glands, and prostate. Interestingly, its expression in the central nervous system (CNS) is even higher. Brain regions with particularly high TR4 expression include the hypothalamus, hippocampus, and cerebellum (5), suggesting that TR4 may have a physiological role in the nervous system as well.
Previous studies have demonstrated that TR4 promotes the binding of several nuclear receptors, including the thyroid hormone and chicken ovalbumin upstream protein transcription factor (COUP-TF) receptors, to the promoter regions of target genes (19). In addition, TR4 acts as a modulator for retinoic acid signaling by competing with the retinoic acid receptor (RAR) and the retinoid X receptor (RXR) for binding to the direct-repeat (AGGTCA) site (21, 23). Related studies of the COUP-TF receptor, as well as of RAR and RXR, further demonstrated that nuclear receptors that can be modulated by TR4 are important in neurogenesis. The role of COUP-TF1 in neurogenesis was demonstrated by using COUP-TF1 knockout mice. In the cerebral cortices of COUP-TF1-null animals, a failure of innervation of thalamocortical projections causes extensive cell death and results in the absence of cortical layer IV (42). Retinoid signaling in neurogenesis has been demonstrated to promote neuronal generation, as shown by the effects of direct administration of RAR/RXR mRNA to Xenopus embryos (34). Besides interacting with other nuclear receptors, TR4 directly regulates the expression of ciliary neurotrophic factor receptor alpha (CNTFRα) (41) and apolipoprotein E (ApoE) (22). The role of CNTFRα in the development of the nervous system has been revealed by work with CNTFRα-null mice, which die soon after birth due to severe motor neuron deficiency (7). ApoE has been suggested to be involved in cerebral amyloid angiopathy by facilitating the deposition of amyloid-β in the cerebral vessels and subsequently causing hemorrhage and infarction, which can be found in most cases of Alzheimer's disease (10). The regulation by TR4 of these nuclear receptors that are important in nervous system development and degeneration, as well as the abundant expression of TR4 in multiple brain regions, indicates an important role for the receptor in the CNS.
A recent study showed that the expression patterns of TR4 in the embryonic and neonatal cerebella are correlated with neurogenesis (37), suggesting a significant role of TR4 in regulating the development of the cerebellum. To study the putative physiological roles of TR4 in the CNS, TR4−/− mice were generated by homologous recombination in embryonic stem cells (6). Throughout postnatal development, behavioral deficits in motor coordination were observed in mice lacking a functional TR4 gene, suggesting abnormal development and/or function of the TR4−/− cerebellum.
The mouse cerebellum is a particularly useful organ for studying neurogenesis due to the profound morphological transformations that occur during development. Histological examination revealed that cerebellar development in both embryonic and postnatal TR4−/− mice is abnormal. Moreover, the expression of genes that were correlated with the morphological changes was observed in the TR4−/− cerebellum. The results of this study demonstrate the importance of TR4 in the process of cerebellar development.
MATERIALS AND METHODS
Animals.
TR4−/− mice were obtained from Lexicon Genetics Incorporated and were generated as described elsewhere (6). Briefly, mice heterozygous for disruption of TR4 were maintained on a C57BL/6 × 129SvEv hybrid background. TR4+/+ and age-matched TR4−/− embryos or pups for this study were produced from heterozygous breeding pairs. The day on which a vaginal plug was observed was considered embryonic day 0 (E0), and the day of birth was postnatal day 0 (P0). The genotype of offspring was determined by PCR analysis of genomic DNA prepared from tail samples as described previously (6). Mice were housed in the vivarium facility of the University of Rochester Medical Center. The animals were provided a standard diet with constant access to food and water and were exposed to a 12-h light-dark cycle.
Behavioral analysis.
The general sensorimotor capabilities of the mice were evaluated using the ledge test (32). The ledge was 0.75 by 30 cm2. The time to fall was calculated from the moment each mouse was placed on the ledge. A maximum of 60 s was established for the task. The wire-hanging test was used to evaluate the prehensile reflex (35). A wire, 40 cm long and 0.2 cm in diameter, was placed 30 cm above the floor. A maximum time of 60 s was set for this task. Three trials per animal, separated by 20-min intervals, were conducted.
Histological analysis and immunohistochemical assays.
Animals were sacrificed with a lethal dose of sodium pentobarbital (250 mg/kg of body weight) in accordance with the guidelines of the University Committee on Animal Resources. Embryos or cerebella of TR4+/+ and TR4 −/− mice at different developmental stages were fixed overnight or longer in fresh 10% buffered paraformaldehyde and were then dehydrated through a series of graded alcohols before being embedded in paraffin. Sagittal sections from TR4+/+ and TR4−/− cerebella were cut at a thickness of 4 μm and placed on slides for staining. For immunohistochemical staining, sections were first incubated with anti-calbindin (Chemicon, Temecula, Calif.), antibromodeoxyuridine (anti-BrdU) (Zymed, San Francisco, Calif.), anti-TR4 (Santa Cruz Biotechnology, Inc., Santa Cruz, Calif.), anti-NeuN (Chemicon), or anti-Reelin (Chemicon) antibodies. By using the ABC kit (Vector, Burlingame, Calif.), the sections were then incubated with biotin-conjugated secondary antibodies and visualized under bright-field microscopy with a diaminobenzidene substrate (DAB) kit (Vector). Sections were counterstained with hematoxylin or cresyl violet. For reelin staining, the binding of the antibody was detected by Alexa Fluor goat anti-mouse immunoglobulin G (Molecular Probes, Eugene, Oreg.). For the in vivo migration assay, P7 littermates were injected with BrdU (10 μg/g, intraperitoneally [i.p.]) and harvested 24, 48, or 72 h after administration. To assess cell proliferation, BrdU was injected either into pregnant mice (50 μg/g, i.p.) or into pups after birth (10 μg/g, i.p.). Following a 2- to 4-h incorporation period, anti-BrdU staining was conducted. To assess cell death, a terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay was conducted using the In Situ Cell Death Detection kit (Roche, Penzberg, Germany) as directed. For RNA detection, in situ reverse transcription-PCR (RT in situ PCR) was performed according to the procedure published by Nuovo (29). Five pups or embryos were used per age group for each procedure.
Calculations of cerebellar cortical area.
The sizes of the external granule cell layer (EGL) and internal granule layer (IGL) in the developing cerebellar cortices of TR4+/+ and TR4 −/− mice were determined by photographing Nissel-stained transverse sections by using a digital camera (Diagnostic Instruments, Inc.) mounted on a Nikon microscope. Randomly selected regions, 10 per sample, in the center of lobules at identical anteroposterior and mediolateral coordinates, were measured. SPOT software (Diagnostic Instruments, Inc) was used to examine the sizes of cerebellar layers. Quantitative data were obtained from 4 sections per mouse and 5 mice per age group.
Reverse transcription and real-time PCR.
Total RNA was isolated from the cerebella of age-matched TR4+/+ and TR4 −/− mice by using the TRIzol reagent (Invitrogen, Carlsbad, Calif.). For each sample, first-strand cDNA synthesis was performed with the Superscript II RNase H reverse transcriptase kit (Invitrogen) from 2 μg of total RNA. Real-time PCR was performed with the iCycler iQ PCR and detection system (Bio-Rad Laboratories, Hercules, Calif.). cDNA was amplified under the following conditions: 95°C for 5 min; 45 cycles of 15 s at 95°C, 8 s at 55°C, and 25 s at 72°C. Quantitative analysis was performed using iCycler analysis software (Bio-Rad Laboratories). Measurements were normalized to those of β-actin to correct for variability in the RNA amount in each sample. The quantification of each sample relative to the control was calculated using the 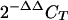 method (24).
method (24).
RESULTS
Impaired motor coordination in TR4−/− mice.
Although TR4−/− mice were viable, a higher incidence of death occurred among these animals than among TR4+/+ littermates during the first 2 months of postnatal life (2.04% for TR4+/+ versus 44.44% for TR4−/− mice). The body weight and overall brain size of TR4−/− mice were significantly reduced throughout the entire period of postnatal development. Even as adults, TR4−/− mice displayed 45% lower body weight (6). In addition to general differences in physical appearance, developing and mature TR4−/− mice exhibited a variety of behavioral abnormalities, such as mild trembling, unsteady gait, hyperreactivity upon manipulation, and a decreased tendency to explore their surroundings. When lifted by the tail, TR4−/− mice at P21 clasped their hind limbs, whereas TR4+/+ mice extended their legs (Fig. 1A). To characterize these behavioral observations, we performed a set of performance tests assessing motor coordination and balance for both TR4+/+ and TR4−/− mice (Fig. 1B). In ledge tests, which require animals to maintain their balance in order to avoid falling, the mean latency to fall for TR4−/− mice was significantly shorter than that for TR4+/+ controls (P < 0.005 by Student's t test). While all TR4+/+ mice remained on the ledge for at least 60 s, 6 out of 7 TR4−/− mice were unable to stay on the ledge for more than 30 s. Furthermore, TR4+/+ and TR4−/− mice were placed on a thin horizontal wire. Consistent with results from the ledge test, the mean latency to fall for TR4−/− mice was significantly shorter than that for TR4+/+ controls in wire-hanging tests (P < 0.005 by Student's t test). All TR4−/− mice fell off the wire within 15 s, whereas TR4+/+ mice had no difficulty in hanging on for 60 s. In adult TR4−/− mice, additional severe symptoms were present. As revealed by footprinting, mature TR4−/− mice dragged their hind limbs while walking and were more likely to stray from a straight path (Fig. 1C). These deficits in performance indicate that the motor coordination and balance of TR4−/− mice are impaired.
FIG.1.

Motor dysfunction and abnormal behavior in 21-day-old and adult TR4−/− mice. (A) Photographs of TR4+/+ and TR4−/− mice at P21, as they were suspended by the tail. TR4+/+ mice usually extended their hind and front limbs when in this position, whereas the limbs of TR4−/− mice were often crossed or clasped. (B) Results of the ledge (left) and wire hanging (right) tests. The latency to fall in both tests was significantly shorter for TR4−/− mice than for TR4+/+ controls. Solid bars, TR4+/+ mice (n = 7); open bars, TR4−/− mice (n = 7). Asterisks indicate significant differences between TR4+/+ and TR4−/− mice. Data shown are means ± standard errors of the means. *, P < 0.005 by Student's t test. (C) Images of inked footprints of TR4+/+ and TR4−/− mice. Normal adults place equal weight on all four limbs when walking, while adult TR4−/− mice appeared to drag their hind limbs. Arrow indicates the direction of movement. (D) Photographs of gross brain samples from P7 and P14 TR4+/+ and TR4−/− mice showing reduced cerebellar size in the TR4−/− mice. (E) The adult TR4−/− cerebellum in the midsagittal section is obviously smaller than that of the TR4+/+ control. Asterisk indicates correlated folium, which shows abnormal folium development in TR4−/− mice.
Disturbed cerebellar development in TR4−/− mice.
In a previous study, we found that the cerebella of TR4−/− mice were reduced by 25% in volume relative to those of TR4+/+ mice, while no obvious difference was observed in the cerebral cortices (data not shown). Midsagittal sections of adult TR4−/− cerebella revealed a reduced hemisphere size as well as stunted folium formation, particularly an absence of lobule VII, relative to those of controls (Fig. 1E). This phenotype can be observed even earlier, during postnatal stages. At P7 and P14, the size of the cerebellum and the formation of cerebellar fissures were reduced in TR4−/− mice relative to those of control littermates (Fig. 1D). Histologically, the formation of cerebellar folia in TR4−/− mice was stunted at all ages, from P0 to P21 (Fig. 2). At P0, a reduced depth of the cerebellar fissures was observed in TR4−/− mice compared to corresponding areas of TR4+/+ cerebella (Fig. 2A and B) In particular, lobule VII consistently failed to develop morphological distinction from lobule VI in TR4−/− mice at P7, P14, and P21 (Fig. 2D, F, and H). Under higher magnification, it was observed that the fine laminar structure was stunted in the cerebellar cortices of TR4−/− mice. At P14, the EGLs of TR4−/− mice were abnormally thicker, and the molecular layers (ML) were abnormally thinner, than those of TR4+/+ mice (Fig. 2E and F, insets, and Fig. 3C and D). By P21, whereas most of the EGL has disappeared in the TR4+/+ cerebellum, a significant EGL presence could still be observed in the TR4−/− cerebellum (Fig. 3F).
FIG. 2.

Cerebellar morphology in developing TR4+/+ and TR4−/− mice. Paraffin-embedded, Nissel-stained midsagittal sections reveal abnormal cerebellar structure at various postnatal stages in TR4−/− mice. Aberrant folium development in TR4−/− cerebellar sections is indicated by arrowheads. Insets in panels E and F show magnified views of the EGL, which is abnormally thick in the TR4−/− cerebellum. Mouse ages and genotypes are as indicated. Cerebellar lobules are indicated by Roman numerals. PCL, Purkinje cell layer. Bars in panels A and B, 250 μm; bars in panels C to H, 500 μm.
FIG. 3.

Differences in cortical layer thickness in TR4+/+ and TR4−/− cerebella. Nissel-stained sections from comparable lobules of TR4+/+ littermates and TR4−/− mice at P7, P14, and P21 reveal that both the lamination and the dispersion of the EGL are delayed in TR4−/− mice. Arrows designate migrating granule cells; arrowheads point to apoptotic bodies. PCL, Purkinje cell layer. Bars, 50 μm.
The lamination irregularities in the TR4−/− EGL and ML suggest that the development of cerebellar granule and Purkinje cells may be affected by the absence of TR4. To examine this possibility, immunohistochemistry was performed using an antibody recognizing a C-terminal region of TR4; it showed that TR4 was expressed in the cerebellar cortices of wild-type mice (Fig. 4A), a finding consistent with previous reports (5, 37). Interestingly, positive signals were observed not only in the granule cells of the IGL but also among postmitotic granule cells of the EGL (Fig. 4C), migrating granule cells of the ML (Fig. 4D), and Purkinje cells (Fig. 4E) in the postnatal control cerebellum. This finding suggests that the specific expression of TR4 in cerebellar granule and Purkinje cells during early postnatal stages may be related to the proper development of hindbrain regions, as supported by the abnormal lamination that occurs in the age-matched TR4−/− cerebellar cortex (Fig. 4B).
FIG. 4.

Expression of TR4 in the postnatal cerebellar cortex. Midsagittal cerebellar sections were immunostained with the anti-TR4 antibody at P12. Arrowheads point to the cells that show positive signals. (A and B) Cerebellar sections from TR4+/+ and TR4−/− mice, respectively. (C, D, E, and F) Higher-magnification views of the cells indicated by the arrowheads in panel A. Bars in panels A and B, 100 μm; bars in panels C to F, 10 μm.
Delayed postmitotic granule cell migration in the TR4−/− cortex.
It is known that the proper migration of postmitotic granule cells from the EGL toward the IGL plays an important role in cerebellar organogenesis. During postnatal development of the cerebellum, granule cells proliferate in the EGL, subsequently migrate inward along the radial glial fiber through the ML, and penetrate the single layer of Purkinje cells to reach their final destination, the IGL (11). Interestingly, at all three postnatal stages, the numbers of teardrop-shaped granule cells in the ML of TR4−/− cerebella were lower than those for age-matched control littermates (Fig. 3). In contrast, round granule cells in the ML were more abundant in TR4−/− than in TR4+/+ cerebella (Fig. 3). Because these changes in granule cell shape are linked to migratory behavior (1), our findings suggest that the abnormal thickness of the EGL in the TR4−/− cerebellum may result from impaired migration of granule cells. To test this hypothesis, long-term BrdU incorporation assays were performed 24, 48, and 72 h following injection at P7. At 24 h post-BrdU injection, cerebellar granule cells in wild-type mice began to migrate inward toward the IGL, as demonstrated by the presence of BrdU-labeled cells in the inner layer of the EGL. However, fewer BrdU-labeled cells were observed in the inner layers of the EGLs of TR4−/− cerebella (Fig. 5B) than in controls. After 48 h, fewer granule cells reached the IGL in TR4−/− cerebella than in controls. This was demonstrated by the findings that fewer BrdU-labeled cells were located in the IGLs of TR4−/− cerebella than in those of TR4+/+ cerebella (Fig. 5C and D) and that more BrdU-labeled granule cells were observed in the ML of TR4−/− cerebella than in those of TR4+/+ cerebella (Fig. 5C and D). Thus, these abnormalities suggest that the migration profile of differentiated granule cells in the TR4−/− cerebellum was altered, causing a delay in their migration into the IGL. This is further supported by real-time RT-PCR analysis, which revealed that the expression of genes related to granule cell migration was reduced in the TR4−/− cerebellum at P7. Specifically, Pax-6 and Astn expression in the TR4−/− cerebellum was down-regulated to 51.76 and 22.92% of the expression levels in the TR4+/+ cerebellum, respectively (Table 1). Even after 72 h of BrdU incorporation, the number of BrdU-labeled granule cells in the IGL was dramatically lower than that in age-matched controls (Fig. 5E and F). Interestingly, dark spots could be seen in the TR4−/− ML (Fig. 3), indicating that cells were undergoing apoptosis. Thus, in addition to the interruption of granule cell migration, a higher rate of cell turnover was occurring simultaneously in the TR4−/− cerebellum.
FIG. 5.

Disrupted migration of granule cells in the TR4−/− cerebellum. Midsagittal sections of mouse cerebella after BrdU incorporation for different lengths of time revealed an abnormal granule cell migration pattern in the postnatal TR4−/− cerebellum. Arrowheads point to granule cells in the inner EGL (A and B) or to migrating granule cells in the ML (C and D). Arrows indicate granule cells in the IGL. BrdU incorporation time and genotypes are as indicated. PCL, Purkinje cell layer. Bars, 100 μm.
TABLE 1.
Relative expression of genes in the developing TR4−/− cerebellum
| Time | Avg (range) relative gene expression in TR4−/− cerebellaa
|
|||||
|---|---|---|---|---|---|---|
| Pax-6 | Astn | Shh | Reelin | Cdk-5 | ROR-α | |
| E18.5 | 176.13 (6.23-489.10) | 27.66 (1.53-40.6) | 43.28 (11.63-98.45) | 156.91 (83.23-295.82) | 96.59 (26.25-355.47) | 132.46 (66.65-263.23) |
| P0 | 224.50 (109.42-460.59) | 43.28 (10.59-91.17) | 40.50 (18.28-89.71) | 37.89 (7.46-52.45) | 85.01 (37.61-92.41) | |
| P7 | 51.76 (35.19-76.14) | 22.92 (11.33-46.38) | 35.97 (15.91-81.33) | 50.58 (27.28-93.78) | 35.63 (20.14-63.01) | 72.92 (58.57-90.79) |
| P14 | 2.63 (1.47-4.71) | 127.40 (60.54-268.32) | 9.15 (4.37-19.18) | 41.56 (22.48-76.86) | 97.72 (26.10-365.80) | |
Calculated as a percentage of gene expression in TR4+/+ cerebella. Four mice were used for each genotype, and mean expression levels were used for comparison. The expression levels of each gene were normalized by using β-actin expressions and the final values were then calculated as percentages of expression in TR4+/+ cerebella. Error ranges were calculated by using the 2−ΔΔCT method.
Altered proliferation pattern of external granule cell layer neurons in TR4−/− mice.
The rate of progenitor cell proliferation in the EGL may also determine the thickness of the EGL in the early postnatal cerebellum. To examine this hypothesis, the immunocytochemical method of short-term BrdU incorporation (2 to 4 h) was performed with mice at different developmental stages (Fig. 6). The number of labeled cells, as well as the intensity of BrdU labeling in the proliferative zone of the EGL, was reduced in the TR4−/− cerebellum at P7 (Fig. 6D). Diminished neurogenesis in the proliferative zones occurred even earlier, as indicated by reduced BrdU labeling of P0 and E16.5 samples (Fig. 6D and J). In addition, sections of cerebella from TR4−/− mice stained with crystal violet demonstrated a thinner neuroepithelial layer and rhombic lip at E16.5 than sections from TR4+/+ controls (Fig. 6I and J). These observations indicate that the increased thickness of the EGL in the P7 TR4−/− cerebellum was due not to increased proliferation of progenitors but rather to a transient delay in the migration of these cells toward the IGL. This is supported by analysis of real-time RT-PCR of P7 cerebellum samples, which revealed that the expression of Shh, a mitogen for proliferation of granule cell progenitors, was decreased to 35.97% of TR4+/+ control levels (Table 1). To further characterize the expression of Shh in Purkinje cells specifically, RT in situ PCR was performed. In P7 TR4−/− cerebellar sections, the expression of Shh was reduced (Fig. 7), suggesting that the suppressed neurogenesis in the TR4−/− cerebellar EGL before P7 may result from decreased expression of Shh in Purkinje cells. In the P12 cerebellum, more BrdU-labeled cells were found in the proliferative zones of the EGLs of TR4−/− mice than in those of TR4+/+ mice (Fig. 6E and F). Cells in the EGL continued to be labeled by BrdU at later postnatal stages, as late as P18 (Fig. 6G and H). Thus, unlike that in earlier developmental stages, the accumulation of postmitotic granule cells in the EGL of the TR4−/− cerebellum at P12 and beyond may be accounted for by both increased proliferation and delayed migration of granule cells.
FIG. 6.

Short-term BrdU incorporation in the TR4+/+ and TR4−/− cerebella at different developmental stages. (A to H) Arrows point to proliferating granule cell precursors. Mouse ages and genotypes are as indicated. PCL, Purkinje cell layer. Bars, 50 μm. (I to L) The TR4−/− cerebellar primordium at a late embryonic stage exhibits germinal cell deficiency. (I and J) Nissel-stained parasagittal sections of E16.5 TR4+/+ (I) and TR4−/− (J) cerebella. The neuroepithelium (NE) and rhombic lip (RL) are designated in each micrograph. CP, choroid plexus. (K and L) BrdU staining in correlated germinal zones. Arrowheads point to proliferating granule cell precursors. Bars, 250 μm.
FIG. 7.

(A to D) Decreased Sonic Hedgehog RNA level in Purkinje cells in the TR4−/− cerebellum. (A and B) RT in situ PCR of Shh was performed on P7 midsagittal cerebellar sections from TR4+/+ and TR4−/− mice. Arrowheads indicate Purkinje cells with positive signals. Bars, 200 μm. (C and D) High-magnification views from panels A and B, respectively. Bars, 50 μm. (E to G) Increased apoptosis of granule cells in the IGL. (E and F) TUNEL staining of P7 cerebellar sections from TR4+/+ and TR4−/− mice. Arrows indicate cells with positive signals. Bars, 50 μm. (G) Relative numbers of TUNEL-positive cells in correlated areas of the IGL of TR4+/+ and TR4−/− cerebella at P7. Asterisks indicate significant differences between TR4+/+ and TR4−/− mice. Values are means ± standard errors of the means. *, P < 0.05.
Increased neuronal apoptosis in the postnatal TR4−/− cerebellum.
In contrast to the effects observed in the EGL of the TR4−/− cerebellum, the thickness and cell density of the IGL were decreased at P7, P14, and P21 (Fig. 3; see also S1A and S1B in the supplemental material). To determine whether the abnormalities in the IGL of the TR4−/− cerebellum resulted from an increase in cell death, the TUNEL assay was performed. In the TR4−/− cerebellum, we observed an increase in the number of TUNEL-positive cells in the IGL (Fig. 7F and G) and the ML at P7. In fact, a higher incidence of TUNEL labeling occurred in the IGL of the TR4−/− cerebellum at all postnatal stages. These cells were confirmed to be granule cells by their size and location in serial cut sections, as well as by staining with the NeuroN antibody (data not shown). These results suggest that a higher rate of apoptosis results in abnormal thinning and decreased cell density of the IGL in the TR4−/− cerebellum during postnatal development.
Abnormalities in the development of Purkinje cells of the postnatal TR4−/− cerebellum.
Previous studies of cerebellar development have suggested the importance of Purkinje cells in neurogenesis. During normal mouse cerebellar formation, Purkinje cell progenitors begin to differentiate, by approximately E10.5, at the neuroepithelium of the ventricular zone in the metencephalon. After mitosis, Purkinje cells migrate outward in a radial direction over the deep cerebellar nuclei and form a temporary plate-like structure beneath the secondary germinal zone, the EGL (36, 39). At the same time, the dendrites of Purkinje cells extend into the ML to establish synapses with granule cell axons. A deficit in Purkinje cell development in the TR4−/− cerebellum is indirectly suggested by the observation that the thickness of the ML in these animals was reduced to 36, 85, and 62% of that in the TR4+/+ cerebellum at P7, P14, and P21 (Fig. 3; see also S1D in the supplemental material). To examine this possibility, immunocytochemical analysis of calbindin, a specific marker for the perikaryonic region and dendrites of Purkinje cells, was conducted. At P0 and P7, calbindin intensities were weak in the TR4−/− cerebellum (Fig. 8B, E, and F) compared to the levels present in the TR4+/+ cerebellum. However, there was no obvious difference in the number of Purkinje cells. Under higher magnification, we also observed reduced dendritic arborization of Purkinje cells at P7 and P14 in the TR4−/− cerebellum compared to that occurring in the TR4+/+ cerebellum (Fig. 8C to J). In addition, a lower number of BrdU-labeled cells and fewer layers of the neuroepithelium were found in the E16.5 TR4−/− cerebellum (Fig. 6L) than in controls. These observations suggest that the developmental deficiency of Purkinje cells may occur at earlier embryonic stages. To determine whether the expression of genes related to Purkinje cell development was disrupted in TR4−/− mice, real-time RT-PCR analysis was performed. Two genes that have been suggested to be involved in the development of Purkinje cells, reelin and Cdk-5 (3), showed levels of expression in TR4−/− cerebellum samples that were only 50.58 and 35.63%, respectively, of control levels at P7 (Table 1). Immunohistochemical analysis indicated that reelin and Cdk-5 signaling were impaired in Purkinje cells of the TR4−/− cerebellum (Fig. 9). At P14 and P21, although dendrites extended through the molecular layer in both TR4+/+ and TR4−/− mice, there was an obvious decrease in the length of dendritic branches and the thickness of primary bundles (Fig. 8J and L), as well as a reduction in ML thickness in the TR4−/− cerebellum. These data demonstrate that, during cerebellar development, the differentiation and maturation of Purkinje cells are disrupted in the cerebellum of the TR4−/− mouse.
FIG. 8.

Abnormal neonatal development of Purkinje cells in the TR4−/− cerebellum. Midsagittal cerebellar sections at different developmental stages were immunostained with an anticalbindin antibody. Mouse ages and genotypes are as indicated. Arrows point to Purkinje cells. Arrowheads indicate primary bundles of Purkinje cell dendritic trees. Bars, 200 μm (A and B), 50 μm (C, E, G, and I), and 30 μm (D, F, H, J, K, and L).
FIG. 9.

Decreased expression of Cdk-5 and reelin in the TR4−/− cerebellum. Midsagittal cerebellar sections at different developmental stages were immunostained with anti-Cdk-5 (A to D) or anti-Reelin (E to H) antibodies. Mouse ages and genotypes are as indicated. (A to D) Arrows indicate Purkinje cells; arrowheads point to migrating granule cells. (E, G, and H) Arrowheads indicate Purkinje cells. Bars, 50 μm.
DISCUSSION
In this study, histological results reveal that the overall size and folium structure of the cerebellum, as well as the relative portions of different cerebellar cortical layers, are altered in TR4−/− mice during postnatal development. In addition to histological differences between TR4−/− and TR4+/+ cerebella, impaired motor coordination is apparent in TR4−/− mice at the age of 2 weeks and persists into adulthood. These changes in cerebellar cytoarchitecture and this abnormal behavior in postnatal TR4−/− mice suggest that TR4 may play an important role in cerebellar development.
Further analysis of the developing TR4−/− cerebellum supports the idea that alterations in cerebellar cytoarchitecture, especially the reduction in IGL density and size, may result from a combination of effects including disturbances in the proliferation of granule cell progenitors, delayed inward migration of postmitotic granule cells, and higher apoptotic incidence. Intriguingly, the proliferation of granule cell progenitors in the TR4−/− EGL exhibits a pattern opposite that in the EGL of the control TR4+/+ cerebellum during postnatal development. In the TR4+/+ EGL, the proliferation of granule cell progenitors increases during the first postnatal week and then declines to undetectable levels during the second and third postnatal weeks. However, in the TR4−/− EGL, fewer proliferation signals can be detected at P0 and P7 than in the control EGL. Conversely, a prolonged period of granule cell proliferation occurs. In TR4−/− mice, proliferating granule cells can be found at P14 and even as late as P18, while no proliferation can be detected in the controls at these ages. This shift in the proliferation pattern suggests that TR4 may have different functions depending on developmental time. Before P7, the expression of TR4 may be essential for the proliferation of granule cells. However, after P7, TR4 may be involved in controlling exit from the cell cycle. Such a switch in the function of TR4 in granule cell proliferation could arise from changes in the composition of participating coregulators or other nuclear receptors. Previous in vitro studies have demonstrated that TR4 has the ability to cross talk or form dimer transcriptional complexes with numerous other nuclear receptors. In addition to the abnormal proliferation pattern of granule cells in the TR4−/− EGL, delayed migration of postmitotic granule cells and higher incidences of apoptosis in both the ML and IGL are observed in the TR4−/− cerebellum. Thus, granule cell proliferation is prolonged in the TR4−/− EGL, but the subsequently differentiated granule cells fail to migrate into the IGL at the appropriate time. This failure to reach the correct location at the proper time may induce programmed cell death in these postmitotic granule cells, resulting in reduced width and cellular density of the TR4−/− IGL. This interpretation is supported by the significant reduction in the number of BrdU-labeled cells in the TR4−/− IGL and ML 72 h after administration, during which time cells are normally evenly distributed in the control IGL. Taken together, prolonged proliferation and slower migration of granule cells in the TR4-deficient cerebellum can explain the delayed disappearance of the EGL during the late development stages (P14 to P21) of TR4−/− mice.
In addition to the abnormalities observed in granule cells, dendritic arborization of Purkinje cells in the TR4−/− cerebellum is considerably stunted, and the labeling intensity of calbindin is significantly reduced, during the first two postnatal weeks. These findings further suggest that Purkinje cell differentiation or function may be compromised in the cerebella of TR4-deficient mice early in postnatal development. It has been demonstrated that when postmitotic granule cells depart from the inner portion of the EGL and migrate radically into the IGL, their axons extend horizontally to establish synapses with the dendrites of Purkinje cells. During this process, the ML grows considerably in size to accommodate the extensions of neurites of both cell types. Thus, the stunted formation of dendritic trees of Purkinje cells and the delayed migration of granule cells indirectly suggest that the extension of neurites of both Purkinje and granule cells is affected and may account for the reduced width of the ML in the TR4−/− cerebellar cortex. During cerebellar development, the interaction between Purkinje and granule cells guides other aspects of their cell survival and behavior (11, 20). Although expression of TR4 can be detected in Purkinje and granule cells in the normal postnatal cerebellum, in the present model we are not able to determine whether the alterations in granule cell proliferation and migration, as well as Purkinje cell development, are cell autonomous. These effects may reflect abnormal interactions between Purkinje and granule cells, due to deficits in function of either or both cell types. However, examination of the number and distribution of basket and stellate cells in the ML, as well as of Golgi cells in the IGL, reveals no obvious differences in these inhibitory interneurons between TR4−/− and age-matched TR4+/+ mice. Thus, it is likely that TR4 is specifically involved in the development of granule and Purkinje cells, which in turn affects the cerebellar structures they populate.
Interestingly, morphological alterations occur in the CNS even earlier, at embryonic stages of the TR4−/− cerebellum. This finding is consistent with a previous study showing the predominant expression of TR4 in the mesencephalon at the embryonic stage, which is believed to be the origin of cerebellar progenitors (37). In the TR4−/− embryo, the size of the rhombic lip and the thickness of cell layers in the neuroepithelium are reduced, while no obvious difference appears in the structure of other organs relative to those of control littermates. As revealed by BrdU incorporation assays in TR4−/− embryos, the reduced number of cells in these two germinal zones results from interference with the proliferation of these progenitors. At E18.5, the expression of Zic1 and Pax-6, which have been suggested to play fundamental roles in the neurogenesis of cerebellar granule cells (2, 34), increased 1.6- and 1.8-fold, respectively, in the TR4−/− cerebellum over that in controls. This result suggests that during the commencement of cerebellar development, some compensatory processes may be triggered to overcome the effects of losing TR4. Thus, our results from the TR4−/− embryo further demonstrate that TR4 is important very early in neurogenesis.
Previous studies with staggerer (sg) and RORα−/− mice, which express deficits in the thyroid hormone/RORα signaling pathway, revealed a cell-autonomous defect of the Purkinje cells. Purkinje cells of sg and RORα−/− mice fail to establish synaptic contacts with granule cell parallel fibers, causing granule cell numbers to decrease. In addition, the proliferation and migration of granule cells are disrupted in sg and RORα−/− mice. Both types of mutant mice exhibit perturbed cerebellar development and present with tremor and impaired balance (8, 12, 13, 14, 15, 16). The abnormalities in sg and RORα−/− mice are similar to those we observed in TR4−/− mice. Because TR4 is able to modulate thyroid hormone and retinoic acid signaling (21, 23), it is possible that the defects observed in TR4−/− cerebellar structure may result, in part, from disturbed thyroid hormone signaling. However, the aberrant development of Purkinje cells in the TR4−/− cerebellum is distinct from that in sg and RORα−/− mice. Although the growth of dendritic branches of Purkinje cells is stunted in the cerebella of all three types of mice, only sg and RORα−/− mice show a reduction in the number of Purkinje cells at late developmental stages. In contrast, the Purkinje cell dendrites in the TR4−/− cerebellum can extend into the ML at late developmental stages, although the thickness of the primary bundle and the length of dendritic trees are obviously reduced relative to those of the TR4+/+ cerebellum. To determine whether thyroid hormone signaling genes are affected, we examined the expression of the RORα gene, which has been implicated as the major effector for thyroid hormone signaling in cerebellar development. RORα gene expression in the TR4−/− cerebellum is reduced to 72.92% of the level in TR4+/+ controls at P7, when the TR4−/− cerebellum shows a pattern of disorganization most similar to that of thyroid hormone-deficient mice. Interestingly, at E18.5, RORα expression is up-regulated 1.3-fold in the TR4−/− cerebellum. Thus, we surmise that during cerebellar development, TR4 may cross talk with RORα in regulating the expression of some genes that are essential for cerebellar development. The fact that TR4−/− mice survive beyond the age of 4 weeks, when most sg and RORα−/− mice die, suggests that other genes may compensate for the deficiency of TR4.
Besides the thyroid hormone deficiency model, several other null mice models have been studied in order to understand neurogenesis. TR4-deficient mice also share several similarities in abnormal cerebellar development with those mutant mice, including defects in the proliferation and migration of granule cells and in the development of Purkinje cells. Therefore, we further examined those well-documented genes which have been suggested to be involved in cerebellar development in order to gain insight into the physiological function of TR4 in neurogenesis. Previous studies with reeler and Cdk-5−/− mice have shown that disturbances in the arrangement of Purkinje cells cause a failure in cerebellar development (26, 30). Related studies further demonstrated that reelin, the mutant gene in reeler mice, is critical for neuronal migration and cell position in the CNS (25). In the reeler cerebellum, Purkinje cells fail to form an appropriate plate structure to support the proliferation of granule cells in the EGL, resulting in reductions in the size and foliation of the cerebellum (27). The disturbed arrangement of Purkinje cells in the neonatal TR4−/−cerebellum, and the ability of TR4 to regulate the expression of ApoE, whose receptor has been shown to be involved in the degradation of neuronal adaptor protein Disabled-1, the key molecule in reelin signaling (4), led us to hypothesize that reelin signaling may be affected. Consistent with this hypothesis, reelin expression is reduced in the TR4−/− cerebellum at different postnatal stages. Interestingly, in reeler mice, although the Purkinje cells are disoriented, no disturbance is found in granule cell migration. This result is distinct from our observation for the TR4−/− cerebellum. Moreover, at later developmental stages, the Purkinje cells in the TR4−/− cerebellar cortex seem to align normally, although severely stunted dendritic arborization occurs at P7. Cdk-5 has been demonstrated to be important in neurogenesis, specifically for neuronal migration and neurite outgrowth (17, 30). Based on the stunted Purkinje cell arborization and the deficit in granule cell migration in the postnatal TR4−/− cerebellum, we predicted that the expression of Cdk-5 may also be altered. As revealed by real-time RT-PCR and immunohistochemistry, the expression level of Cdk-5 is reduced, specifically in Purkinje and granule cells. Thus, the present data suggest that the abnormal development of Purkinje cells in the TR4−/− cerebellum may result in part from impaired reelin and Cdk-5 signaling. In the cerebellum, in addition to making direct synaptic contacts with granule cells, Purkinje cells also secrete mitogenic factors, such as Shh, to stimulate the proliferation of granule cell progenitors in the secondary proliferative zone, the EGL, and may be involved in guiding the migration of postmitotic granule cells inward (31, 38, 40). In the postnatal TR4−/− cerebellum, the development of Purkinje cells is stunted and the level of Shh is decreased at postnatal stages (P0 and P7) relative to those in the TR4+/+ cerebellum. These results further support the idea that the function of Purkinje cells is compromised when TR4 is absent, and they may explain the diminished proliferation of granule cell progenitors in the TR4−/− EGL at early developmental stages.
It is known that proper inward migration of postmitotic granule cells plays an important role in cerebellar organogenesis. In the developing TR4−/− cerebellum, granule cell proliferation and Purkinje cell differentiation are affected. Moreover, the profile of granule cell migration from the EGL to the IGL is interrupted. A previous study with Cdk-5-null mice indicated that Cdk-5 is not only essential for Purkinje cell orientation but also important for granule cell migration (30). The reduced Cdk-5 expression in TR4−/− granule and Purkinje cells implies that the delayed migration of neuronal cells may be cell autonomous and that TR4 may play a role in regulating Cdk-5 expression in neurons. On the other hand, expression of Pax-6, which has been demonstrated to be important for postmitotic granule cell migration and the formation of parallel fibers (9, 18), is first increased and then decreased in the TR4−/− cerebellum. The observation of a lower number of progenitors at the primary proliferative zone but with higher expression of Pax-6 and reelin at embryonic and neonatal stages in the TR4−/− cerebellum suggests that these mechanisms may be involved in compensating for the lack of TR4 in neurogenesis rather than retarding cerebellar development. Abnormal granule cell migration may also reflect disrupted establishment of neuronal-glial contact during neurogenesis. Astn, a neuronal-glial ligand required for normal migration of neuronal cells (1), is down-regulated in the TR4−/− cerebellum at early developmental stages. This raises the possibility that the microenvironment between the granule cells and glial fibers may also be altered in the TR4−/− cerebellum. Thus, TR4 may play a role in neuronal migration in two ways: regulating the expression of reelin, Cdk-5, and Pax-6 in cerebellar neurons and establishing the proper contact in neuronal-glial circuitry.
Interestingly, the defects in the TR4−/− cerebellum are similar, but not identical, to those found in several mouse models which concern cerebellar development, including stagger, RORα−/−, reeler, Cdk-5−/−, small eye, and astn−/− mice (1, 8, 12, 14, 25, 30, 33). Furthermore, the expression of some of these genes is not simply reduced in TR4−/− mice. An initial increase in expression is observed for Pax-6 and reelin at embryonic stages (Table 1), and expression subsequently declines to below normal levels during later postnatal development. These results implicate TR4 in differentially modulating the expression of genes required for the development of cerebellar neurons, either directly or indirectly. Abolishing TR4 function alters granule cell proliferation and migration, influences Purkinje cell development, and/or affects cytoskeletal organization between glia and migrating granule cells, which is required for proper cerebellar development. Although the expression of genes which are important for cerebellar development is altered in the TR4-deficient cerebellum, the specific cellular and molecular mechanisms underlying these changes still need to be determined. Immunocytochemically, the TR4 signal is observed ubiquitously in normally developing cerebella (Fig. 4). Specifically, TR4 is most highly expressed in postmitotic granule cells in the EGL, ML, and IGL, as well as in Purkinje cells. The presence of TR4 in these cells raises the possibility that the abnormalities in EGL and IGL thickness, increased numbers of rounded granule cells in the ML, and stunted growth of Purkinje cell dendritic trees in the TR4−/− cerebellum may be specifically due to the absence of functional TR4 in these cells. Therefore, it would be interesting to study conditional or cell-specific TR4-null models in order to further clarify the function of TR4 in neurogenesis.
In conclusion, our results show that multiple abnormalities observed in the developing TR4−/− cerebellum may be due to the loss of TR4 function in the CNS. However, whether the cerebellar defects are direct effects of deletion of the functional TR4 gene specifically in Purkinje and/or granule cells or indirect effects due to loss of TR4 in other cell types remains to be determined. Most likely, multiple mechanisms exist by which TR4 regulates the development of the cerebellum, as suggested by our findings. Our results provide important insights into the physiological role of TR4 during cerebellar development and may serve as the basis for further exploration of the function of TR4 in CNS development.
Supplementary Material
Acknowledgments
We thank Chiayu Chiu for comments on the manuscript.
This work was supported by grants DK 56984 and DK 63212 from the National Institutes of Health, as well as the George Whipple Professorship Endowment. The TR4 knockout mice were generated in collaboration with Lexicon Genetics Inc.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Adams, N. C., T. Tomoda, M. Cooper, G. Dietz, and M. E. Hatten. 2002. Mice that lack astrotactin have slowed neuronal migration. Development 129:965-972. [DOI] [PubMed] [Google Scholar]
- 2.Aruga, J., O. Minowa, H. Yaginuma, J. Kuno, T. Nagai, T. Noda, and K. Mikoshiba. 1998. Mouse Zic1 is involved in cerebellar development. J. Neurosci. 18:284-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beffert, U., E. J. Weeber, G. Morfini, J. Ko, S. T. Brady, L.-H. Tsai, J. D. Sweatt, and J. Herz. 2004. Reelin and cyclin-dependent kinase 5-dependent signals cooperate in regulating neuronal migration and synaptic transmission. J. Neurosci. 24:1897-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bock, H. H., Y. Jossin, P. May, O. Bergner, and J. Herz. 2004. Apolipoprotein E receptors are required for reelin-induced proteasomal degradation of the neuronal adaptor protein Disabled-1. J. Biol. Chem. 279:33471-33479. [DOI] [PubMed] [Google Scholar]
- 5.Chang, C., S. L. Da Silva, R. Ideta, Y. Lee, S. Yeh, and J. P. Burbach. 1994. Human and rat TR4 orphan receptors specify a subclass of the steroid receptor superfamily. Proc. Natl. Acad. Sci. USA 91:6040-6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins, L. L., Y. F. Lee, C. A. Heinlein, N. C. Liu, Y. T. Chen, C. R. Shyr, C. K. Meshul, H. Uno, K. A. Platt, and C. Chang. 2004. Growth retardation and abnormal maternal behavior in mice lacking testicular orphan nuclear receptor 4. Proc. Natl. Acad. Sci. USA 101:15058-15063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeChiara, T. M., R. Vejsada, W. T. Poueymirou, A. Acheson, C. Suri, J. C. Conover, B. Friedman, J. McClain, L. Pan, and N. Stahl. 1995. Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell 83:313-322. [DOI] [PubMed] [Google Scholar]
- 8.Dussault, I., D. Fawcett, A. Matthyssen, J. A. Bader, and V. Giguere. 1998. Orphan nuclear receptor RORα-deficient mice display the cerebellar defects of staggerer. Mech. Dev. 70:147-153. [DOI] [PubMed] [Google Scholar]
- 9.Engelkamp, D., P. Rashbass, A. Seawright, and V. van Heyningen. 1999. Role of Pax6 in development of the cerebellar system. Development 126:3585-3596. [DOI] [PubMed] [Google Scholar]
- 10.Fryer, J. D., J. W. Taylor, R. B. DeMattos, K. R. Bales, S. M. Paul, M. Parsadanian, and D. M. Holtzman. 2003. Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J. Neurosci. 23:7889-7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldowitz, D., and K. Hamre. 1998. The cells and molecules that make a cerebellum. Trends Neurosci. 21:375-382. [DOI] [PubMed] [Google Scholar]
- 12.Hamilton, B. A., W. N. Frankel, A. W. Kerrebrock, T. L. Hawkins, W. FitzHugh, K. Kusumi, L. B. Russell, K. L. Mueller, V. van Berkel, B. W. Birren, L. Kruglyak, and E. S. Lander. 1996. Disruption of the nuclear hormone receptor RORα in staggerer mice. Nature 379:736-739. [DOI] [PubMed] [Google Scholar]
- 13.Harding, H. P., G. B. Atkins, A. B. Jaffe, W. J. Seo, and M. A. Lazar. 1997. Transcriptional activation and repression by RORα, an orphan nuclear receptor required for cerebellar development. Mol. Endocrinol. 11:1737-1746. [DOI] [PubMed] [Google Scholar]
- 14.Herrup, K. 1983. Role of staggerer gene in determining cell number in cerebellar cortex. I. Granule cell death is an indirect consequence of staggerer gene action. Brain Res. 313:267-274. [DOI] [PubMed] [Google Scholar]
- 15.Herrup, K., and R. J. Mullen. 1979. Staggerer chimeras: intrinsic nature of Purkinje cell defects and implications for normal cerebellar development. Brain Res. 178:443-457. [DOI] [PubMed] [Google Scholar]
- 16.Herrup, K., and R. J. Mullen. 2002. Regional variation and absence of large neurons in the cerebellum of the staggerer mouse. Annu. Rev. Biochem. 71:405-434. [DOI] [PubMed] [Google Scholar]
- 17.Hirasawa, M., T. Ohshima, S. Takahashi, G. Longenecker, Y. Honjo, Veeranna, H. C. Pant, K. Mikoshiba, R. O. Brady, and A. B. Kulkarni. 2004. Perinatal abrogation of Cdk5 expression in brain results in neuronal migration defects. Proc. Natl. Acad. Sci. USA 101:6249-6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horie, M., K. Sango, K. Takeuchi, S. Honma, N. Osumi, K. Kawamura, and H. Kawano. 2003. Subpial neuronal migration in the medulla oblongata of Pax-6-deficient rats. Eur. J. Neurosci. 17:49-57. [DOI] [PubMed] [Google Scholar]
- 19.Hwang, S. B., J. P. Burbach, and C. Chang. 1998. TR4 orphan receptor crosstalks to chicken ovalbumin upstream protein-transcription factor and thyroid hormone receptor to induce the transcriptional activity of the human immunodeficiency virus type 1 long-terminal repeat. Endocrine 8:169-175. [DOI] [PubMed] [Google Scholar]
- 20.Inouye, M., and U. Murakami. 1980. Temporal and spatial patterns of Purkinje cell formation in the mouse cerebellum. J. Comp. Neurol. 194:499-503. [DOI] [PubMed] [Google Scholar]
- 21.Inui, S., Y. F. Lee, A. R. Haake, L. A. Goldsmith, and C. Chang. 1999. Induction of TR4 orphan receptor by retinoic acid in human HaCaT keratinocytes. J. Investig. Dermatol. 112:426-431. [DOI] [PubMed] [Google Scholar]
- 22.Kim, E., S. Xie, S. D. Yeh, Y. F. Lee, L. L. Collins, Y. C. Hu, C. R. Shyr, X. M. Mu, N. C. Liu, Y. T. Chen, P. H. Wang, and C. Chang. 2003. Disruption of TR4 orphan nuclear receptor reduces the expression of liver apolipoprotein E/C-I/C-II gene cluster. J. Biol. Chem. 278:46919-46926. [DOI] [PubMed] [Google Scholar]
- 23.Lee, Y. F., W. J. Young, J. P. Burbach, and C. Chang. 1998. Negative feedback control of the retinoid-retinoic acid/retinoid X receptor pathway by the human TR4 orphan receptor, a member of the steroid receptor superfamily. J. Biol. Chem. 273:13437-13443. [DOI] [PubMed] [Google Scholar]
-
24.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the
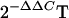 method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar] - 25.Magdaleno, S., L. Keshvara, and T. Curran. 2002. Rescue of ataxia and preplate splitting by ectopic expression of Reelin in reeler mice. Neuron 33:573-586. [DOI] [PubMed] [Google Scholar]
- 26.Magdaleno, S. M., and T. Curran. 2001. Brain development: integrins and the Reelin pathway. Curr. Biol. 11:R1032-R1035. [DOI] [PubMed] [Google Scholar]
- 27.Mikoshiba, K., S. Terada, K. Takamatsu, K. Shimai, and Y. Tsukada. 1983. Histochemical and immunohistochemical studies of the cerebellum from the reeler mutant mouse. Dev. Neurosci. 6:101-110. [DOI] [PubMed] [Google Scholar]
- 28.Mu, X., Y. F. Lee, N. C. Liu, Y. T. Chen, E. Kim, C. R. Shyr, and C. Chang. 2004. Targeted inactivation of testicular nuclear orphan receptor 4 delays and disrupts late meiotic prophase and subsequent meiotic divisions of spermatogenesis. Mol. Cell. Biol. 24:5887-5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nuovo, G. J. 1996. The foundation of successful RT in situ PCR. Front. Biosci. 1:c4-c15. [DOI] [PubMed] [Google Scholar]
- 30.Ohshima, T., E. C. Gilmore, G. Longenecker, D. M. Jacobowitz, R. O. Brady, K. Herrup, and A. B. Kulkarni. 1999. Migration defects of cdk5−/− neurons in the developing cerebellum is cell autonomous. J. Neurosci. 19:6017-6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oliver, T. G., L. L. Grasfeder, A. L. Carroll, C. Kaiser, C. L. Gillingham, S. M. Lin, R. Wickramasinghe, M. P. Scott, and R. J. Wechsler-Reya. 2003. Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proc. Natl. Acad. Sci. USA 100:7331-7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaefer, M. L., S. T. Wong, D. F. Wozniak, L. M. Muglia, J. A. Liauw, M. Zhuo, A. Nardi, R. E. Hartman, S. K. Vogt, C. E. Luedke, D. R. Storm, and L. J. Muglia. 2000. Altered stress-induced anxiety in adenylyl cyclase type VIII-deficient mice. J. Neurosci. 20:4809-4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmahl, W., M. Knoedlseder, J. Favor, and D. Davidson. 1993. Defects of neuronal migration and the pathogenesis of cortical malformations are associated with Small eye (Sey) in the mouse, a point mutation at the Pax-6-locus. Acta Neuropathol. (Berlin) 86:126-135. [DOI] [PubMed] [Google Scholar]
- 34.Sharpe, C., and K. Goldstone. 2000. The control of Xenopus embryonic primary neurogenesis is mediated by retinoid signalling in the neurectoderm. Mech. Dev. 91:69-80. [DOI] [PubMed] [Google Scholar]
- 35.Takeda, Y., K. Akasaka, S. Lee, S. Kobayashi, H. Kawano, S. Murayama, N. Takahashi, K. Hashimoto, M. Kano, M. Asano, K. Sudo, Y. Iwakura, and K. Watanabe. 2003. Impaired motor coordination in mice lacking neural recognition molecule NB-3 of the contactin/F3 subgroup. J. Neurobiol. 56:252-265. [DOI] [PubMed] [Google Scholar]
- 36.Uzman, L. L. 1960. The histogenesis of the mouse cerebellum as studied by its tritiated thymidine uptake. J. Comp. Neurol. 114:137-159. [DOI] [PubMed] [Google Scholar]
- 37.van Schaick, H. S., J. G. Rosmalen, D. S. Lopes, C. Chang, and J. P. Burbach. 2000. Expression of the orphan receptor TR4 during brain development of the rat. Brain Res. Mol. Brain Res. 77:104-110. [DOI] [PubMed] [Google Scholar]
- 38.Wallace, V. A. 1999. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr. Biol. 9:445-448. [DOI] [PubMed] [Google Scholar]
- 39.Wang, V. Y., and H. Y. Zoghbi. 2001. Genetic regulation of cerebellar development. Nat. Rev. Neurosci. 2:484-491. [DOI] [PubMed] [Google Scholar]
- 40.Wechsler-Reya, R. J., and M. P. Scott. 1999. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22:103-114. [DOI] [PubMed] [Google Scholar]
- 41.Young, W. J., S. M. Smith, and C. Chang. 1997. Induction of the intronic enhancer of the human ciliary neurotrophic factor receptor (CNTFRα) gene by the TR4 orphan receptor. A member of steroid receptor superfamily. J. Biol. Chem. 272:3109-3116. [DOI] [PubMed] [Google Scholar]
- 42.Zhou, C., Y. Qiu, F. A. Pereira, M. C. Crair, S. Y. Tsai, and M. J. Tsai. 1999. The nuclear orphan receptor COUP-TFI is required for differentiation of subplate neurons and guidance of thalamocortical axons. Neuron 24:847-859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.