

Summary
Activation of the MUC1-C protein promotes lineage plasticity, epigenetic reprogramming, and the cancer stem cell (CSC) state. The present studies performed on enriched populations of triple-negative breast cancer (TNBC) CSCs demonstrate that MUC1-C is essential for integrating activation of glycolytic pathway genes with self-renewal and tumorigenicity. MUC1-C further integrates the glycolytic pathway with suppression of mitochondrial DNA (mtDNA) genes encoding components of mitochondrial Complexes I–V. The repression of mtDNA genes is explained by MUC1-C-mediated (i) downregulation of the mitochondrial transcription factor A (TFAM) required for mtDNA transcription and (ii) induction of the mitochondrial transcription termination factor 3 (mTERF3). In support of pathogenesis that suppresses mitochondrial ROS production, targeting MUC1-C increases (i) mtDNA gene transcription, (ii) superoxide levels, and (iii) loss of self-renewal capacity. These findings and scRNA-seq analysis of CSC subpopulations indicate that MUC1-C regulates self-renewal and redox balance by integrating activation of glycolysis with suppression of oxidative phosphorylation.
Subject areas: Molecular biology, Cell biology, Omics, Transcriptomics
Graphical abstract
Highlights
-
•
Enriched CSCs are dependent on MUC1-C for self-renewal capacity and tumorigenicity
-
•
MUC1-C upregulates genes encoding GLUT1, HK2, PGAM1, and other glycolytic enzymes
-
•
MUC1-C represses mitochondrial DNA genes encoding Complexes I–V of the ETC
-
•
CSCs maintain redox balance by MUC1-C-driven regulation of aerobic glycolysis and OXPHOS
Molecular biology; Cell biology; Omics; Transcriptomics
Introduction
The MUC1 gene evolved in mammals to protect barrier tissues from the external environment.1,2 MUC1 encodes two subunits that form a heterodimer at the apical cell membrane.1,2 The MUC1 N-terminal (MUC1-N) subunit extends beyond the glycocalyx into a mucous gel that functions as a physical barrier.1,2 The transmembrane MUC1 C-terminal (MUC1-C) subunit is activated by loss of homeostasis and induces inflammatory, remodeling, and repair responses associated with wound healing.1,2 As a result, MUC1-C is imported from the cell membrane to the nucleus, where it interacts with proinflammatory transcription factors (TFs), such as NF-κB and STAT3, and contributes to the regulation of their target genes.3,4 MUC1-C thereby induces the epithelial-mesenchymal transition (EMT) by driving expression of the ZEB1, TWIST1, and SNAIL EMT TFs.5 Nuclear MUC1-C also interacts with NF-κB, MYC, and E2F in driving PRC1/2 complexes and epigenetic reprogramming.6 In addition, nuclear MUC1-C interacts with E2F1 in activating the SWI/SNF BAF and PBAF chromatin remodeling complexes7,8 and global changes in chromatin architecture.9,10 In principle, these changes are reversible with reestablishment of homeostasis; however, prolonged activation of MUC1-C in settings of chronic inflammation promotes pan-cancer progression.1,2,10 Along these lines, in TNBC and other types of recalcitrant carcinomas, MUC1-C induces lineage plasticity, self-renewal capacity, and tumorigenicity in concert with driving the cancer stem cell (CSC) state.5,11,12,13,14,15,16 Consistent with these findings, MUC1 expression associates poor clinical outcomes across pan-cancers.17
TNBCs are enriched in CSCs that contribute to DNA damage resistance, immune evasion, and poor clinical outcomes.16,18,19 TNBC progression is driven at least in part by metabolic reprogramming20,21; however, little is known in regard to the regulation of TNBC CSC metabolism. Certain TNBC tumors exhibit elevated glucose uptake and a glycolytic gene signature.22 In addition, TNBC cell lines grown as monolayers in two-dimensional (2D) culture are dependent on high levels of glycolysis.23 As substantiated by this metabolic profile, TNBC tumors and cell lines exhibit upregulation of the GLUT1 glucose transporter, whereas expression of other effectors in the glycolytic pathway is largely heterogeneous.24 Downstream to GLUT1, hexokinase 2 (HK2) functions as the rate-limiting enzyme that catalyzes the first step of glycolysis.25 Less is known about HK2 expression in TNBC cells, although HK2 is often upregulated in other human cancers and drives their reprogramming to aerobic glycolysis.25 Cancer cells are not exclusively dependent on aerobic glycolysis in that mitochondria also play important roles in growth and survival.26,27 Indeed, TNBC cells are at least in part dependent on mitochondrial function.20,28 Oxidative phosphorylation (OXPHOS) is upregulated in TNBC cells with RB deficiency or induction of E2F1,29 indicating that oncogenic signaling pathways may dictate reliance on glycolytic vs. OXPHOS activities.
MUC1-C is a pan-cancer oncogenic protein1,2,10 that has been linked to certain pathways of metabolic regulation.30,31,32,33 As one example, MUC1-C activates TP53-inducible regulator of glycolysis and apoptosis (TIGAR) and the pentose phosphate pathway (PPP).34 These studies were performed on TNBC and other types of cancer cells grown in 2D culture and thus may not reflect involvement associated with the CSC state. In this way, enriched TNBC CSCs selected by serial passage of mammospheres represent a potential model for identifying MUC1-C dependencies that integrate metabolic reprogramming with self-renewal capacity. The present studies demonstrate that MUC1-C is necessary for activation of the aerobic glycolytic pathway and suppression of OXPHOS in driving the TNBC CSC state.
Results
Serially passaged TNBC CSCs are dependent on MUC1-C for self-renewal
Studies of TNBC CSCs have been hampered by challenges using cell surface markers in purifying this cell population.35,36 Nonetheless, isolation of CSCs as defined functionally has been advanced by growth in serum-free conditions to maintain the undifferentiated CSC state.37,38 Accordingly, we enriched TNBC CSC populations based on the functional capacity to form mammospheres. Using this approach, BT-549 TNBC cells growing in 2D culture were established as serial passage 1 (S1) mammospheres (Figure 1A). These mammosphere cells (>100 μm) were then isolated and serially passaged for up to S10 mammospheres (Figure 1A). With each passage, we observed progressive increases in sphere size (Figure 1A), sphere forming efficiency (SFE) as the percentage of functional CSCs (Figure 1B), and proliferation rate (Figure S1A). As a second model, similar results were obtained with MDA-MB-436 TNBC cells; that is, increases in mammosphere size (Figure 1C), SFE (Figure 1D), and proliferation rate (Figure S1B). BT-549 cells are recognized for lacking the capacity to form tumors in immunocompromised mice. The basis for this deficiency has remained unclear. Nonetheless, implanting BT-549 S8 sphere cells in 3 of 3 NSG mice resulted in the formation of tumor xenografts (Figure 1E), indicating that enrichment of BT-549 cells for anchorage-independent growth increases their propensity for forming tumors, albeit not as efficiently as in other cancer models. In addition, serial passage of fragments from these founding xenografts readily formed tumors for further study (Figure S1C). Unlike BT-549 cells, MDA-MB-436 cells grown as 2D monolayers form tumors in nude mice. We therefore examined the effects of serial dilutions on the capacity of MDA-MB-436 2D and S8 cells to establish tumors. The results showed that MDA-MB-436 S8 sphere cells are more efficient in forming tumors (Figure S1D), further supporting the selection of TNBC CSCs as defined functionally in serially passaged mammospheres. MUC1-C promotes lineage plasticity and self-renewal of TNBC cells grown in 2D culture5; whereas, it is not known if serially passaged TNBC CSCs have similar dependencies. We found that silencing MUC1-C in BT-549 and MDA-MB-436 cells suppresses self-renewal capacity as evidenced by decreases in mammosphere formation (Figures 1F, S1E, and S1F). In confirming that MUC1-C is necessary for CSC self-renewal, we used a second MUC1shRNA#2 which similarly decreased mammosphere formation (Figures S1G and S1H). In addition, rescuing MUC1-C expression with a tet-MUC1-C cytoplasmic domain (MUC1-C/CD) expressing vector (Figure S1I) reestablished the capacity for forming mammospheres (Figure S1J). MUC1-C/CD includes a CQC motif that is required for MUC1-C dimerization, nuclear localization and oncogenic function.1 Treatment of BT-549 and MDA-MB-436 cells with the GO-203 inhibitor, which blocks the CQC motif, markedly suppressed mammosphere formation (Figures 1G and S1K), further indicating that MUC1-C is necessary for CSC self-renewal. Along these lines, MUC1-C expression was increased in lysates from BT-549 3D (S1) vs. 2D cells (Figure 1H). With serial passage (S3), MUC1-C levels declined to those observed in 2D cells (Figure 1H) coincident with increases in the chromatin fraction. Subsequent studies are therefore underway to assess the functional significance of MUC1-C localization to chromatin in association with progression of the CSC state. In regard to CSC markers, CD44high/CD24low cell surface expression has been used to isolate BC CSCs39; however, more recent work has indicated this population is a heterogeneous spectrum of EMT phenotypes.36 Compared to 2D cells, serially passaged 3D CSCs exhibited upregulation of the (i) ZEB1 and TWIST1 EMT-TFs that contribute to the CSC state,36 (ii) BMI1/PRC1 stemness marker,40 and (iii) SOX2 and MYC, which have also been linked to driving TNBC CSCs41,42(Figure 1H). Significantly, expression of these effectors in CSCs was suppressed by silencing MUC1-C (Figure 1I, left) and by targeting MUC1-C with GO-203, which blocks MUC1-C function in the absence of having an effect on MUC1-C levels (Figure 1I, right). Moreover, GO-203 treatment of mice bearing passaged BT-549 CSC xenografts inhibited tumor growth (Figures 1J and S1L), confirming that MUC1-C is necessary for self-renewal and tumorigenicity.
Figure 1.

Serial passage of TNBC mammospheres selects for MUC1-C-dependent CSC populations
(A and B) BT-549 cells growing as monolayers were seeded in mammosphere culture medium. After 10 days, the sphere 1 (S1) cells were isolated and reseeded for selection of S2 cells. Photomicrographs are shown for the serially passaged mammospheres up to S10. Scale bar: 100 μm. (A). The sphere forming efficiency (SFE) was determined by the percentage of cells that formed mammospheres as a function of the number of seeded cells. SFE is expressed as the % (mean ± SD of three determinations) (B).
(C and D) MDA-MB-436 cells serially passaged as mammospheres are shown in the indicated S1-S10 photomicrographs. Scale bar: 100 μm. (C). SFE is expressed as the % (mean ± SD of three determinations) (D).
(E) BT-549 2D and S8 mammosphere cells (10 × 106) were implanted into left and right flanks, respectively, of NSG mice. Shown are tumors at 5 months after implantation.
(F) BT-549/tet-MUC1shRNA 3D cells treated with vehicle or DOX for 7 days were analyzed for sphere formation. Shown are photomicrographs of representative mammospheres. Scale bar: 100 μm. (left). The relative SFE is expressed as the mean ± SD of three determinations as compared to that obtained for vehicle-treated cells (assigned a value of 1) (right). Asterisks represent ∗∗∗p ≤ 0.001.
(G) BT-549 3D cells were treated with vehicle or 2.5 μM GO-203 for 3 days. Shown are photomicrographs of representative mammospheres. Scale bar: 100 μm (left). The relative SFE is expressed as the mean ± SD of three determinations as compared to that obtained for vehicle-treated cells (assigned a value of 1) (right). Asterisks represent ∗∗∗p ≤ 0.001.
(H) Lysates from BT-549 cells grown in 2D culture and as serially passage mammospheres (S1-S3) were immunoblotted with antibodies against the indicated proteins.
(I) Lysates from BT-549/tet-MUC1shRNA 3D cells treated with vehicle or DOX for 10 days were immunoblotted with antibodies against the indicated proteins (left). Lysates from BT-549 3D cells were treated with vehicle or 2.5 μM GO-203 for 3 days were immunoblotted with antibodies against the indicated proteins (right).
(J) NSG mice with established BT-549 3D cell tumors were treated intraperitoneally with PBS or GO-203 (12 μg/gm body weight) each day for 70 days. Tumor volumes are expressed as the mean ± SEM for 6 tumors.
MUC1-C regulates glycolytic genes in TNBC CSCs
Having established MUC1-C dependency of TNBC CSCs, we then investigated how MUC1-C promotes TNBC cell stemness by performing RNA-seq on BT-549 S6 3D cells. Comparison of global transcriptional profiles in 3D vs. 2D cells demonstrated broad changes in gene expression (3070 DEGs; false discovery rate (FDR) < 0.10, fold change (FC) > 1.5), with 1,701 downregulated (DN) and 1,369 upregulated (UP) genes (Figure 2A). Furthermore, silencing MUC1-C in 3D cells demonstrated 1,678 DEGs with 604 downregulated and 1,074 upregulated genes (Figure 2A). Among 1,369 upregulated genes in 3D and 604 downregulated genes in MUC1-C silenced 3D cells, we identified 186 shared genes (Figure 2B, left). Within the 186 inversely regulated and overlapping genes, there was notable representation of genes related to the glycolysis pathway (Figure 2B, right). Accordingly, we assessed the top affected pathways (i) enriched in 3D cells and (ii) depleted in both MUC1-C-silenced or GO-203-treated 3D cells by gene set enrichment analysis (GSEA) and gene set variation analysis (GSVA). We found that the gene sets related to glycolysis, as well as the MYC signaling pathway, are significantly enriched in 3D and conversely repressed with MUC1-C targeting under 3D conditions (Figures 2C, S2A, and S2B). We initially focused on glycolysis and further identified by GSEA of the BT-549 3D cell datasets that multiple glycolysis-related signatures are suppressed by targeting MUC1-C genetically and pharmacologically (Figure 2D). TNBC cells have been associated with upregulation of SLC2A1; whereas expression of HK2 and other genes encoding effectors in the glycolytic pathway has been largely heterogeneous.25 Here, we identified multiple glycolytic genes upregulated in 3D vs. 2D cells and downregulated by targeting MUC1-C, which included (i) SLC2A1 encoding the GLUT1 glucose transporter, (ii) PFKM/L/P that encode isoforms of 6-phosphofructokinase necessary for generating fructose 1,6-bisphosphate, (iii) ALDOA/C, and (iv) ENO1/2 encoding enzymes that catalyze the penultimate step in glycolysis43 (Figures 2E and S2C). In addition, we found that phosphoglycerate mutase 1 (PGAM1), an important effector of glycolysis, the oxidative PPP, and the serine biosynthesis pathway,44 is upregulated in 3D cells by a MUC1-C-mediated mechanism (Figures 2E and S2C). Similar results were obtained for LDHA and LDHB that are also deregulated in TNBC, but not for LDHC, which is expressed at low levels in BT-549 cells45(Figure S2C). MetaPhOR analysis of metabolic networks to further identify transcriptional dysregulation confirmed that the glycolysis pathway is upregulated in 3D (Figure 2F) and downregulated in MUC1-C silenced 3D (Figure 2G) cells.
Figure 2.

MUC1-C is necessary for activation of glycolysis gene signatures in CSCs
(A) Volcano plots showing downregulated (left) and upregulated (right) genes in BT-549 cells grown in S6 3D vs. 2D culture (left; 3070 DEGs; <0.1 padj (Benjamini-Hochberg adjusted p value), log2fold change (FC) > 1.5), with 1,369 upregulated and 1,701 downregulated genes). Volcano plots of downregulated (left) and upregulated (right) genes in BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle for 10 days (right; 1678 DEGs; <0.1 padj (Benjamini-Hochberg adjusted p value), log2fold change ((FC) > 1.5), with 1,074 upregulated and 604 downregulated genes).
(B) Overlap of DEGs upregulated in 3D BT-549 cells and downregulated in 3D BT-549/tet-MUC1 shRNA cells treated with DOX (left). The differential expression of these shared genes (red) is shown for the two conditions (right). Glycolytic genes are labeled within the shared set.
(C) GSEA of genes in BT-549 cells grown in 3D vs. 2D culture (left) and BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle control (right) using the HALLMARK_GLYCOLYSIS gene signature. NES: Normalized Enrichment Score.
(D) GSEA lollipop plots of multiple glycolysis-related gene sets for (i) BT-549 cells grown in 3D vs. 2D culture and (ii) BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle control.
(E) Heatmaps of glycolytic genes in (i) BT-549 cells grown in 3D vs. 2D culture, (ii) BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle, and (iii) BT-549 3D cells treated with GO-203 vs. vehicle.
(F and G) MetaPhOR analysis identifying transcriptional dysregulation of metabolic pathways in 3D vs. 2D BT-549 cells (F) and 3D BT-549/tet-MUC1shRNA cells treated with DOX vs. vehicle (G).
MUC1-C drives expression of SLC2A1/GLUT1 and HK2 in TNBC CSCs
Based on the importance of GLUT1 and HK2 in driving glycolysis and tumorigenicity,25,46 we confirmed their (i) upregulation in 3D vs. 2D cells (Figure 3A) and (ii) suppression by targeting MUC1-C with silencing and GO-203 treatment (Figures 3B and S3A). GLUT1 and HK2 have been recognized as MYC target genes.47 Along these lines, we found that targeting MUC1-C in 3D cells results in the downregulation of MYC expression (Figure 3B). Analysis of the HALLMARK_MYC_TARGETS_V1 signature further revealed significant enrichment in 3D vs. 2D cells (Figure S2A) that was abrogated by MUC1-C targeting (Figure 3C). RNA-seq data from MYC silenced 3D cells also showed significant loss of enrichment in the REACTOME_GLYCOLYSIS gene set (Figure S3B). Moreover, Landscape In Silico deletion Analysis (LISA) demonstrated enrichment of multiple MYC cistromes for many of the upregulated genes in 3D vs. 2D cells (Figure S3C). These findings supported the potential involvement of MUC1-C→MYC signaling in activating GLUT1 and HK2 in CSCs. In accordance with this notion and, like MUC1, silencing MYC in 3D cells suppressed expression of GLUT1 and HK2 (Figures 3D, S3D, and S3E). MUC1-C associates with MYC in total lysates obtained from cells grown in 2D culture.48 Here, we found that the interaction between MUC1-C and MYC in the nucleus is increased in 3D vs. 2D cells (Figure 3E, left), consistent with the observation that MUC1-C localizes to the chromatin fraction of 3D cells. The MUC1-C CQC motif binds directly to the MYC HLH-LZ domain.48 In support of this interaction, treatment of 3D cells with GO-203, which blocks the CQC motif, disrupted the formation of nuclear MUC1-C/MYC complexes (Figure 3E, right). The GLUT1 promoter region includes E-boxes for potential MYC binding in a (i) promoter-like signature (PLS), (ii) proximal enhancer-like signature (pELS), and (iii) distal enhancer-like signature (dELS) (Figure 3F). ChIP studies performed on the GLUT1 PLS, pELS, and dELS regions demonstrated increased MYC occupancy in 3D vs. 2D cells (Figure 3F). Analysis of the HK2 promoter further identified PLS and dELS regions with E-boxes (Figure 3G). MYC occupancy was detectable on the PLS, but not the dELS, in 3D vs. 2D cells (Figure 3G). MUC1-C promotes MYC occupancy on MYC target genes in 2D cells.48 Here, we found that silencing MUC1-C in 3D cells decreases MYC occupancy of the (i) GLUT1 PLS and pELS regions (Figure 3H) and (ii) HK2 PLS (Figure 3I), in support of MUC1-C dependency in the activation of GLUT1 and HK2 expression. Notably, in contrast to SLC2A1 and HK2, other MUC1-C-dependent glycolysis genes, such as PFKL, ALDOA, and ENO2, were upregulated by silencing MYC (Figures S3E and S3F), indicating that MUC1-C also induces genes in the glycolytic pathway by MYC-independent mechanisms. As substantiated by the involvement of MUC1-C in inducing GLUT1 and HK2, silencing MUC1-C decreased glucose uptake in CSCs (Figure 3J). Moreover, treatment with the 2-deoxy-D-glucose (2DG) HK2 inhibitor49 markedly decreased mammosphere formation in concordance with dependence of CSCs on MUC1-C-induced activation of the glycolytic pathway (Figure 3K).
Figure 3.

MUC1-C is necessary for induction of GLUT1 and HK2 in TNBC CSCs
(A) Lysates from 2D and S1-S3 cells were immunoblotted with antibodies against the indicated proteins.
(B) Lysates from BT-549/tet-MUC1shRNA S6 3D cells treated with vehicle or DOX for 10 days (left) and BT-549 cells treated with vehicle or 2.5 μM GO-203 for 2 days (right) were immunoblotted with antibodies against the indicated proteins.
(C) GSEA of genes in (i) BT-549/tet-MUC1shRNA S6 cells treated with DOX vs. vehicle and (ii) BT-549 cells treated with GO-203 vs. vehicle using the HALLMARK_MYC_TARGETS_V1 gene signature.
(D) Lysates from BT-549/tet-MYCshRNA S6 3D cells treated with vehicle or DOX for 10 days were immunoblotted with antibodies against the indicated proteins.
(E) Nuclear lysates from BT-549 2D and 3D cells were immunoprecipitated with anti-MUC1-C or a control IgG (left). Nuclear lysates from BT-549 3D cells treated with vehicle or 2.5 μM GO-203 for 3 days were immunoprecipitated with anti-MUC1-C or a control IgG (right). The precipitates were immunoblotted with antibodies against the indicated proteins.
(F) Schema of GLUT1 with positioning of the PLS, pELS, and dELS regions. Soluble chromatin from BT-549 2D and 3D cells was precipitated with a control IgG or anti-MYC antibody. The DNA samples were amplified by qPCR with primers for the GLUT1 PLS, pELS, and dELS regions (Table S2). The results (mean ± SD of 3 determinations) are expressed as fold enrichment relative to that obtained with the IgG control (assigned a value of 1). Asterisks represent ∗p ≤ 0.05.
(G) Schema of HK2 with positioning of the PLS, dELS1, and dELS2 regions. Soluble chromatin from BT-549 2D and 3D cells was precipitated with a control IgG or anti-MYC antibody. The DNA samples were amplified by qPCR with primers for the HK2 PLS, dELS1, and dELS2 regions (Table S2). The results (mean ± SD of 3 determinations) are expressed as fold enrichment relative to that obtained with the IgG control (assigned a value of 1). Asterisks represent ∗∗p ≤ 0.01.
(H and I) Soluble chromatin from BT-549/tet-MUC1shRNA 3D cells treated with vehicle or DOX was precipitated with a control IgG or anti-MYC antibody. The DNA samples were amplified by qPCR with primers for the GLUT1 PLS and pELS regions (H), and HK2 PLS region (I). The results (mean ± SD of 3 determinations) are expressed as fold enrichment relative to that obtained with the IgG control (assigned a value of 1). Asterisks represent ∗p ≤ 0.05, ∗∗p ≤ 0.01.
(J) Glucose uptake was measured in BT-549 3D cells treated with vehicle or 2.5 μM GO-203 for 3 days. The results (mean ± SD of 3 determinations) are expressed as luminescence (relative light unit, RLU). Asterisks represent ∗∗∗p ≤ 0.001.
(K) BT-549 3D cells were treated with vehicle or 5 μM 2DG for 7 days. The relative SFE is expressed as the mean ± SD of three determinations as compared to that obtained for vehicle-treated cells (assigned a value of 1). Asterisks represent ∗p ≤ 0.05.
MUC1-C differentially regulates expression of nuclear genes encoding mitochondrial Complex I–V proteins
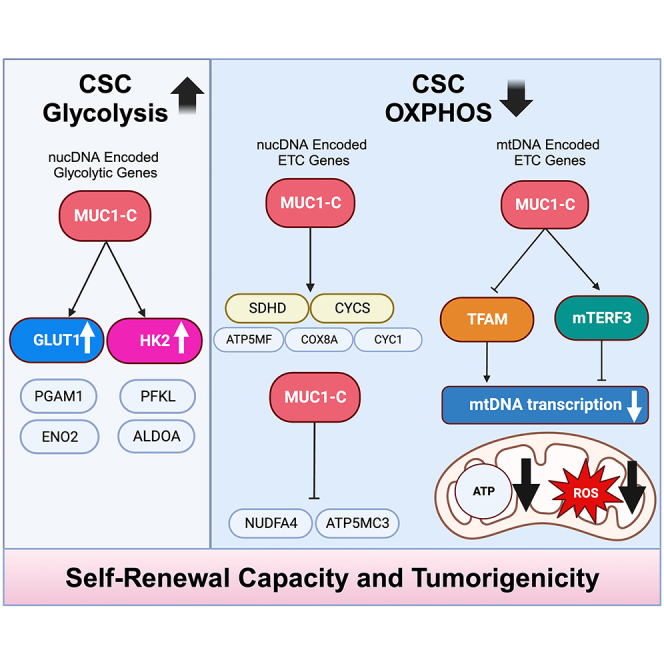
In extending the aforementioned observation that MUC1-C plays a role in regulating glycolysis in TNBC CSCs, GSEA using the WP_ETC_OXPHOS_MITOCHONDRIA signature demonstrated enrichment in BT-549 3D vs. 2D cells (Figure 4A). We also found that silencing MUC1-C in 3D CSCs both represses and activates OXPHOS genes (Figure 4A). Furthermore, heatmaps of genes activated in BT-549 3D vs. 2D cells and regulated in (i) BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle and (ii) BT-549 3D cells treated with GO-203 vs. vehicle identified specific nuclear genes encoding components of the electron transfer chain (ETC) (Figure 4B). Among these, we confirmed that silencing MUC1-C suppresses expression of (i) the succinate dehydrogenase A (SDHA) catalytic subunit of Complex II, (ii) ubiquinol-cytochrome c reductase complex cytochrome c1 (CYC1) component of Complex III, (iii) COX8A, an essential component of Complex IV cytochrome c oxidase and (iv) the ATP5MF component of Complex V ATP synthase50,51,52 (Figure 4C). In contrast, silencing MUC1-C increased expression of (i) the NDUFA4 component of Complex I that also associates with Complex IV53 and (ii) ATP5MC3, a subunit of Complex V ATP synthase54 (Figure 4C), indicating that MUC1-C differentially regulates nuclear genes encoding the ETC. MYC activates nuclear genes that play roles in mitochondrial replication and biogenesis; however, less is known about MYC function in regulating nuclear genes that encode ETC components in CSCs.55 We found that silencing MYC in CSCs suppresses the WP_ETC_OXPHOS_MITOCHONDRIA signature (Figure S4A). Heatmaps of BT-549/tet-MYCshRNA 3D cells treated with DOX vs. vehicle further identified nuclear genes encoding ETC components that are upregulated in 3D vs. 2D cells and suppressed by MYC silencing (Figures 4B and S4B). Among selected genes, we found that silencing MUC1-C and MYC downregulates SDHD (Figure 4D). A similar dependency on MUC1-C and MYC was identified for the CYCS gene that encodes cytochrome c, an essential carrier of electrons from Complex III to Complex IV (Figure 4E). By extension, MUC1-C and MYC were necessary for expression of the SDHD and cytochrome c proteins (Figure 4F). The SDHD promoter region includes an E-box for potential MYC binding in a pELS (Figure 4G). ChIP studies performed on the SDHD pELS region demonstrated increases in MYC occupancy in 3D vs. 2D cells (Figure 4G). Analysis of the CYCS promoter also identified a pELS with an E-box that was occupied by MYC at increased levels in 3D vs. 2D cells (Figure 4H). In addition, we found that silencing MUC1-C in 3D cells decreases MYC occupancy of the SDHD pELS (Figure 4I) and CYCS pELS (Figure 4J) regions. These results indicated that MUC1-C regulates nuclear genes encoding proteins necessary for function of mitochondrial Complexes I–V and that these MUC1-C-driven genes are regulated by MYC-independent and -dependent pathways.
Figure 4.

MUC1-C is necessary for expression of nuclear genes encoding components of the mitochondrial ETC
(A) GSEA of genes in BT-549 cells grown in 3D vs. 2D culture (left) and BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle control (right) using the WP_ETC_OXPHOS_MITOCHONDRIA gene signature.
(B) WP_ETC_OXPHOS_SYSTEM IN MITOCHONDRIA signature heatmaps of ETC encoding genes in (i) BT-549 cells grown in 3D vs. 2D culture, (ii) BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle, (iii) BT-549 3D cells treated with GO-203 vs. vehicle, and (iv) BT-549/tet-MYCshRNA 3D cells treated with DOX vs. vehicle. The row indicator shows gene origins, nuclear DNA (black) and mtDNA (yellow).
(C) Expression of the indicated nuclear genes in BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle for 7 days was determined by qRT-PCR. The results (mean ± SD of 3 determinations) are expressed as relative mRNA levels compared to that obtained for vehicle-treated cells (assigned a value of 1). Asterisks represent ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001.
(D–F) BT-549 3D vs. 2D cells and BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle for 7 days were analyzed for SDHD (D) and CYCS (E) expression. The qRT-PCR results (mean ± SD of 3 determinations) are expressed as relative mRNA levels compared to that obtained for 2D cells or vehicle-treated cells (assigned a value of 1). Lysates were immunoblotted with antibodies against the indicated proteins (F). Asterisks represent ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001.
(G) Schema of SDHD with positioning of an E-box in the pELS region. Soluble chromatin from BT-549 2D and 3D cells was precipitated with a control IgG or anti-MYC antibody. The DNA samples were amplified by qPCR with primers for the SDHD pELS region (Table S2). The results (mean ± SD of 3 determinations) are expressed as fold enrichment relative to that obtained with the IgG control (assigned a value of 1). Asterisks represent ∗p ≤ 0.05.
(H) Schema of CYCS with positioning of an E-box in the pELS region. Soluble chromatin from BT-549 2D and 3D cells was precipitated with a control IgG or anti-MYC antibody. The DNA samples were amplified by qPCR with primers for the CYCS PLS and pELS regions (Table S2). The results (mean ± SD of 3 determinations) are expressed as fold enrichment relative to that obtained with the IgG control (assigned a value of 1). Asterisks represent ∗p ≤ 0.05.
(I and J) Soluble chromatin from BT-549/tet-MUC1shRNA 3D cells treated with vehicle or DOX for 5 days was precipitated with a control IgG or anti-MYC antibody. The DNA samples were amplified by qPCR with primers for the SDHD pELS (I) and HK2 pELS regions (J). The results (mean ± SD of 3 determinations) are expressed as fold enrichment relative to that obtained with the IgG control (assigned a value of 1). Asterisks represent ∗p ≤ 0.05, ∗∗∗p ≤ 0.001.
MUC1-C suppresses mtDNA gene expression
Complex I interacts with Complexes III and IV to form the “respirasome”.51 Complex I consists of 45 subunits, which include the essential ND1-6 components of the ETC that are encoded by mtDNA genes.50,56 mtDNA includes (i) the light strand promoter (LSP), which generates transcripts encoding ND6 and 8 tRNAs, (ii) the heavy strand promoter 1 (HSP1) that controls transcription of 2 tRNAs and 2 ribosomal RNAs, and (iii) HSP2, which regulates expression of the additional 12 protein-encoding transcripts (ND1-ND5, COX1/2/3, ATP6/8, and CYB) and 12 tRNAs.57 Analysis of the WP_ETC_OXPHOS_MITOCHONDRIA gene signature, which includes nuclear- and mtDNA-encoding genes, indicated that expression of ND1-6, ND4L, COX1/2/3, ATP6/8, and CYB is downregulated in 3D vs. 2D cells (Figure 4B). By extension, we confirmed uniform suppression of these 13 mtDNA genes in 3D vs. 2D cells by qRT-PCR (Figure 5A), which supported repression of the mtDNA heavy and light strands. These observed effects in 3D vs. 2D cells were unrelated to decreases in mtDNA copy number, as evidenced by the mtDNA/B2M gDNA ratio (Figure S5A). MitoTracker Green staining of 3D vs. 2D cells further demonstrated similar levels of mitochondria (Figure S5B), in accordance with other mechanisms responsible for the suppression of mtDNA gene transcription. The WP_ETC_OXPHOS_SYSTEM IN MITOCHONDRIA gene heatmap also revealed that MUC1-C is involved in mtDNA gene regulation (Figure 4B). Accordingly, we confirmed that targeting MUC1-C with silencing (Figure 5B) and GO-203 treatment (Figure 5C) induces ND1-6, ND4L, COX1/2/3, ATP6/8, and CYB expression. Targeting MUC1-C also increased (i) expression of the ND1 and COX2 proteins (Figure 5D) and (ii) ATP levels (Figure 5E), indicating that the MUC1-C-induced suppression of mtDNA gene expression is coupled with downregulation of ETC function. Mitochondrial transcription factor A (TFAM) is a nuclear gene-encoded protein that binds mtDNA and is essential for mtDNA packaging and transcription.57 TFAM regulates mtDNA transcription in complexes with the TF2BM transcription factor.56 In association with the induction of mtDNA gene transcription, we found that targeting MUC1-C induces TFAM, but not TFB2M, expression (Figures 5F, 5G, and S5C), indicating that TFAM is downregulated by a MUC1-C-mediated mechanism. Silencing MYC in 3D cells was also associated with induction of mtDNA genes (Figures S5D and S5E) and increases in ATP production (Figure S5F); however, in contrast to MUC1-C, silencing MYC clearly decreased TFAM transcripts and protein (Figures 5H and S5G). In addition, silencing MYC decreased TFB2M transcripts and had variable effects on expression of the TFB2M protein (Figures 5H and S5G). Surprisingly, we also found that silencing MUC1-C, as well as MYC, downregulates the mitochondrial transcription termination factor 3 (mTERF3) (Figures 5I and 5J), which represses the mtDNA light and heavy strands.58 These results supported a model in which mtDNA gene transcription is systematically repressed by (i) MUC1-C-mediated suppression of TFAM and (ii) MUC1-C/MYC-induced mTERF3 expression.
Figure 5.

MUC1-C represses mtDNA gene expression
(A) Expression of the indicated mtDNA genes in 3D vs. 2D BT-549 cells was determined by qRT-PCR. The results (mean ± SD of 3 determinations) are expressed as relative mRNA levels compared to that obtained for 2D cells (assigned a value of 1). Asterisks represent ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001.
(B and C) Expression of the indicated mtDNA genes in (B) BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle for 10 days, and (C) BT-549 3D cells treated with 5 μM GO-203 vs. vehicle for 36 h was determined by qRT-PCR. The results (mean ± SD of 3 determinations) are expressed as relative mRNA levels compared to that obtained for vehicle-treated cells (assigned a value of 1). Asterisks represent ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001.
(D) Lysates from BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle for 7 days (left) and BT-549 3D cells treated with 2.5 μM GO-203 vs. vehicle for 2 days (right) were immunoblotted with antibodies against the indicated proteins.
(E) BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle for 4 days (left) and BT-549 3D cells treated with 5 μM GO-203 vs. vehicle for 2 days (right) were analyzed for ATP levels. The results (mean ± SD of 3 determinations) are expressed as relative ATP levels compared to that obtained for vehicle-treated cells (assigned a value of 1). Asterisks represent ∗∗p ≤ 0.01, ∗∗∗∗p ≤ 0.0001.
(F) BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle for 5 days were analyzed for TFAM expression by qRT-PCR. The results (mean ± SD of 3 determinations) are expressed as relative mRNA levels compared to that obtained for vehicle-treated cells (assigned a value of 1). Asterisks represent ∗∗∗∗p ≤ 0.0001.
(G) Lysates from BT-549/tet-MUC1shRNA 3D cells treated with DOX vs. vehicle for 7 days (left) and BT-549 3D cells treated with 2.5 μM GO-203 vs. vehicle for 2 days (right) were immunoblotted with antibodies against the indicated proteins.
(H) Lysates from BT-549/tet-MYCshRNA 3D cells treated with vehicle or DOX were immunoblotted with antibodies against the indicated proteins.
(I) BT-549/tet-MUC1shRNA (left) and BT-549/tet-MYCshRNA (right) 3D cells treated with vehicle or DOX were analyzed for mTERF3 expression by qRT-PCR. The results (mean ± SD of 3 determinations) are expressed as relative mRNA levels compared to that obtained for vehicle-treated cells (assigned a value of 1). Asterisks represent ∗∗p ≤ 0.01, ∗∗∗∗p ≤ 0.0001.
(J) Lysates from BT-549/tet-MUC1shRNA and BT-549/tet-MYCshRNA 3D cells treated with vehicle or DOX for 7 days were immunoblotted with antibodies against the indicated proteins.
MUC1-C suppresses mitochondrial ROS production in CSCs
Superoxides are largely produced by the ETC in association with ATP production.59 The demonstration that MUC1-C suppresses mtDNA gene expression and ATP production indicated that MUC1-C plays a role in regulating CSC mitochondrial redox balance. In support of such involvement, mitochondrial superoxide levels as assessed by MitoSOX staining were markedly increased by silencing MUC1-C in BT-549 mammosphere cells (Figure 6A). Flow cytometry of individual BT-549 3D cells confirmed significant increases in superoxide production and loss of viability (Figure 6B). Silencing MUC1-C in MDA-MB-436 mammosphere cells was also associated with induction of superoxide levels and cell death (Figure 6C). MUC1-C-driven decreases in ROS could potentially be associated with increases in expression of the mitochondrial superoxide dismutase 2 (SOD2) and peroxiredoxin 3 (PRDX3) ROS scavengers. However, silencing MUC1-C had little if any effect on expression of these anti-oxidant enzymes (Figure S6). Targeting MUC1-C with GO-203 treatment of BT-549 mammospheres further demonstrated increases in superoxides by MitoSOX staining (Figure 6D) that were confirmed in individual CSCs by flow cytometry (Figure 6E). Similar results were obtained from MDA-MB-436 mammospheres treated with GO-203 and stained with MitoSOX (Figure 6F). Excessive ROS production is associated with disruption of the mitochondrial membrane potential (MMP) and cell death.60 Consistent with that association, staining with the membrane-permeant JC-1 dye demonstrated that targeting MUC1-C with GO-203 in BT-549 (Figure 6G) and MDA-MB-436 (Figure 6H) 3D cells decreases the red/green fluorescence intensity ratio as evidence of mitochondrial depolarization. Moreover, GO-203-induced loss of the MMP was associated with CSC death (Figures 6I and 6J), in support of CSC dependence on MUC1-C to maintain mitochondrial function and thereby self-renewal capacity. These findings indicate that TNBC CSCs are largely dependent on MUC1-C for suppression of mtDNA gene expression and superoxide production to maintain redox balance and survival; however, we do not exclude the possibility that they could also be attributed to dysregulation of uncoupling protein expression in mitochondria.
Figure 6.

MUC1-C suppresses superoxide production in CSCs
(A) BT-549/tet-MUC1shRNA mammospheres treated with vehicle or DOX for 10 days were stained with MitoSOX Red. Shown are representative fluorescence microscopy images.
(B and C) BT-549/tet-MUC1shRNA (B) and MDA-MB-436/tet-MUC1shRNA (C) mammospheres were treated with vehicle or DOX for 10 days. MitoSOX Red flow cytometry data (mean ± SD of 3 determinations) are expressed as relative geometric mean fluorescence intensity (gMFI) compared to that obtained for vehicle-treated cells (assigned a value of 1)(left). Trypan blue staining results (mean ± SD of 3 determinations are expressed as the % cell death (right). (C) MitoSOX Red flow cytometry data in MDA-MB-436/tet-MUC1shRNA mammospheres treated with vehicle or DOX for 10 days. The results (mean ± SD of 3 determinations) are expressed as fold-change of gMFI for the mammosphere cells (left). MDA-MB-436/tet-MUC1shRNA mammospheres treated with vehicle or DOX for 10 days were monitored for cell death by trypan blue staining. The results are expressed as the % cell death (mean ± SD of 3 determinations)(right). Asterisks represent ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001.
(D) BT-549 mammospheres treated with vehicle or 5 μM GO-203 for 3 days were stained with MitoSOX Red. Shown are representative fluorescence microscopy images.
(E and F) BT-549 (E) and MDA-MB-436 (F) mammospheres were treated with vehicle or 5 μM GO-203 for 3 days. MitoSOX Red flow cytometry data (mean ± SD of 3 determinations) are expressed as relative geometric mean fluorescence intensity (gMFI) compared to that obtained for vehicle-treated cells (assigned a value of 1). The results (mean ± SD of 3 determinations) are expressed as fold-change of gMFI for the mammophere cells. Asterisks represent ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001.
(G) BT-549 mammospheres treated with vehicle or the indicated GO-203 concentrations for 3 days were stained with JC-1. Fluorescence images are shown for the control and GO-203-treated cells (upper panels). The JC-1 stained mammosphere cells were analyzed by flow cytometry for assessment of JC-1 red vs. green emission as a measure of the MMP (lower panels). The results (mean ± SD of 3 determinations) are expressed as the % depolarized mitochondria (JC-1 monomers). Asterisks represent ∗∗p ≤ 0.01.
(H) MDA-MB-436 mammospheres treated with vehicle or the indicated GO-203 concentrations for 3 days were stained with JC-1. The JC-1 stained mammosphere cells were analyzed by flow cytometry for assessment of JC-1 red vs. green emission. The results (mean ± SD of 3 determinations) are expressed as the % depolarized mitochondria (JC-1 monomers). Asterisks represent ∗∗∗∗p ≤ 0.0001.
(I and J) BT-549 (I) and MDA-MB-436 (J) mammospheres treated with vehicle or 5 μM GO-203 for 3 days were monitored for cell death by trypan blue staining. The results (mean ± SD of 3 determinations) are expressed as the % cell death. Asterisks represent ∗∗∗∗p ≤ 0.0001.
MUC1-C regulates distinct subpopulations of glycolytic- and OXPHOS-enriched CSCs
To determine if MUC1-C dictates glycolytic and OXPHOS pathways within the same or different CSCs, we performed scRNA-seq on isolated BT-549 mammospheres. The scRNA-seq data were filtered to remove dying cells and predicted cell doublets (Figure S7A). Consistent with bulk RNA-seq findings, knockdown of MUC1-C in BT-549 mammosphere cells drives substantial transcriptomic remodeling as indicated by nearly discrete clustering of MUC1-C silenced versus control single cells (Figure S7B). Graph-based nearest neighbor clustering identified 20 unique clusters with distinct transcriptomic signatures across both samples (Figures 7A and S7C). Furthermore, differential gene expression comparing MUC1-C silenced and control conditions displayed significant concordance between bulk- and scRNA-seq analyses (Figure S7D). Expression of MUC1-C was heterogeneous across control cells compared with consistent loss in MUC1-C knockdown cells (Figure 7B). As observed in the bulk RNA-seq studies, silencing MUC1-C downregulated key glycolysis genes (HK2, SLC2A1/GLUT1, and PFKL) (Figures 7B and 7C). Downregulation of MUC1-C also resulted in notable, uniform suppression of the integral glycolysis PGAM1 enzyme (Figures 7B and 7C). Moreover, silencing MUC1-C increased TFAM and decreased mTERF3 expression (Figures 7B and 7C). Notably, in MUC1-C-low cells, the combination of TFAM-high/mTERF3-low expression coincided with upregulation of mtDNA encoded genes, including ND1 and COX2 (Figure 7C). Normalized expression confirmed that these effects of silencing MUC1-C on critical components of glycolysis and the ETC are highly significant and associated with decreases in expression of the CD44 and ALDH2 stemness markers (Figure 7C). The differential single-cell gene expression profiles provided sufficient granularity to identify distinct, and mutually exclusive, functional clusters by GSEA (Figures 7A and S7E). Clusters 5 and 6 were specifically enriched for HALLMARK glycolysis, mTORC1 signaling, and hypoxia signatures; whereas, clusters 1, 3, 7, 12, and 19 represent cells with high OXPHOS and MYC regulated HALLMARK signatures (Figures 7D and S7E). Although cluster 11 is highly enriched for a glycolytic signature, further analysis suggested that these cells may be dying as evidenced by high expression of heat shock protein, HSPA5, and long non-coding RNAs, NEAT1, and MALAT1 (Figure S7C). Consistent with the bulk RNA-seq data, loss of MUC1-C expression was associated with significant downregulation of the HALLMARK_GLYCOLYSIS signature (Figure S7F). Moreover, glycolysis and OXPHOS signatures were inversely correlated and this tendency was conserved with alterations of MUC1 expression (Figures 7D and 7E). Along these lines, further analysis revealed a significant positive association between MUC1 and HK2 expression that drives enrichment of the HALLMARK_GLYCOLYSIS signature in MUC1-high and HK2-high cells (Figure 7F). We also found that the signature of mtDNA encoding ETC genes is significantly elevated upon MUC1 knockdown in association with upregulation of TFAM and downregulation of mTERF3 expression (Figures 7G and 7H). These findings indicate that CSCs are metabolically heterogeneous and MUC1-C is a key regulator of metabolic reprogramming between the glycolysis and OXPHOS phenotypes.
Figure 7.

Single-cell RNA-seq identifies metabolic heterogeneity in mammosphere CSCs that associates with MUC1 expression
(A) UMAP visualization of cells and clusters identified.
(B) Expression feature plots of select genes associated with glycolysis (HK2, SLC2A1, and PGAM1) and transcriptional regulators of mitochondrial DNA (TFAM and MTERF3) visualized by UMAP representation reveal metabolic heterogeneity within passaged sphere cells.
(C) Expression of glycolytic genes (HK2, SLC2A1, PFKL, and PGAM1), mitochondrial transcriptional regulators (TFAM and MTERF3), mitochondrial encoded components of ETC (MT-ND1 and MT-CO2), nuclear encoded components of ETC (SDHD and CYCS), and stemness genes (CD44 and ALDH2) associate with MUC1 status in passaged sphere cells.
(D) Metabolic signature scores and MUC1 expression levels across clusters.
(E) Comparison of single-cell HALLMARK_GLYCOLYSIS and HALLMARK_OXIDATIVE_PHOSPHORYLATION signature scores within individual cells. Color represents MUC1 expression within individual cells.
(F) Correlation of HK2 and MUC1 expression across CTL or MUC1 knockdown passaged sphere cells with color intensity representing HALLMARK_GLYCOLYSIS signature scores.
(G) Comparison of TFAM and MUC1 expression in CTL or MUC1 knockdown passaged sphere cells. Color represents overall signature of ETC genes encoded by the mitochondrial genome.
(H) Correlation of MTERF3 expression and MUC1 expression with color intensity indicating HALLMARK_GLYCOLYSIS enrichment score.
Discussion
Aerobic glycolysis and mitochondrial metabolism have been extensively studied in cancer cells growing as monolayers in 2D culture and as tumors27,61,62; whereas, less is known about the intersection of glycolytic and mitochondrial pathways in 3D cells. In the present work, serially passaged TNBC mammospheres were isolated as an approach for performing such studies on enriched CSCs. In this way, we found that TNBC CSC 3D vs. 2D cells are dependent on MUC1-C for (i) induction of genes encoding EMT-TFs, BMI1, SOX2, and MYC, (ii) self-renewal capacity, and (iii) tumorigenicity. MUC1-C is activated in barrier tissues by loss of homeostasis.2 As a result, MUC1-C induces inflammatory, remodeling, and repair responses associated with wound healing that, if prolonged as in settings of chronic inflammation, are established in promoting carcinogenesis.2 The wound healing response involves activation of glycolysis to maintain redox balance.63 We found that MUC1-C activates the glycolytic pathway in CSCs, which may relate to irreversible establishment of this pathway in the progression of chronic inflammation to cancer.2 Specifically, targeting MUC1-C genetically and pharmacologically in CSCs downregulated expression of SLC2A1/GLUT1, HK2, and other glycolytic genes. MUC1-C drives the CSC state in part by interacting with the MYC HLH-LZ domain that regulates MYC-mediated gene transcription.48 As a consequence, MUC1-C promotes binding of MYC to target genes involved in epigenetic reprogramming and chromatin remodeling.2,10,48,64 The present studies demonstrate that MUC1-C is necessary for MYC occupancy on the SLC2A1/GLUT1 and HK2 promoter regions and thereby induction of GLUT1 and HK2 expression. Direct binding of MUC1-C to MYC is dependent on the redox-regulated CQC motif in the MUC1-C cytoplasmic domain,2,48 indicating that MUC1-C and MYC could function in concert in the response of CSCs to changes in redox balance. Notably, however, MUC1-C-dependent, MYC-independent signaling was identified for induction of other genes, such as PFKL, ALDOA, and ENO2, that are essential for driving glycolysis and are upregulated in cancer.43 Additionally, scRNA-seq revealed that loss of MUC1-C results in marked suppression of PGAM1, which is essential for glycolytic flux and anabolic pathways of nucleotide and amino acid synthesis necessary for cancer cell biogenesis.65
Reports of CSC metabolic properties have been conflicting.66 CSCs have been associated with dependence on glycolysis and decreases in ROS levels.67,68,69 By contrast, CSCs have also been shown to display higher rates of oxygen consumption and ROS production compared to non-CSCs.70,71 Increases in mtROS, largely in the form of superoxides, result from leakage of electrons at Complexes I, III, and IV.72 The present results demonstrate that, analogous to the glycolytic pathway, MUC1-C regulates nuclear genes encoding effectors of Complexes I–V that are essential for OXPHOS.50,51,52 These findings supported a role for MUC1-C in controlling electron transfer and thereby potentially increasing mtROS levels. Intriguingly, however, we found that MUC1-C suppresses the expression of mtDNA genes encoding components of Complexes I–V. This imbalanced MUC1-C function in regulating nuclear genes encoding ETC components and repressing mtDNA genes could lead to complex misassembly, which has been linked to longevity and cancer progression.72 MUC1-C-dependent repression of mtDNA genes involved the light and heavy strands, consistent with a mechanism involving the mtDNA promoter region. Indeed, we found that MUC1-C suppresses expression of TFAM, a member of the HMGB family of DNA binding factors that controls mtDNA transcription initiation.57 Adding to the potential complexity in the regulation of effectors that control mtDNA transcription, our results further demonstrate that MUC1-C is necessary for expression of the mTERF3 terminating factor, which could cooperate with the downregulation of TFAM in repressing mtDNA gene transcription. Subsequent studies will now be needed to determine if these findings in TNBC CSCs extend to other types of cancers.
Activation of the glycolytic pathway in cancer cells has been associated with the capacity to attenuate the accumulation of ROS and thereby alleviate oxidative and replicative stress that confer resistance to treatment.73 In other studies of cancer cells, abrogation of the glycolytic pathway has indicated that OXPHOS is sufficient for sustaining redox and energetic requirements.27,74,75 Despite these apparent discrepancies, one notion is that aerobic glycolysis and OXPHOS are both of importance for cancer progression.26,51,76 Along this line of thinking, we found that the glycolytic and OXPHOS pathways are differentially regulated in 3D vs. 2D cells, which could hold important implications for interpreting studies of metabolic reprogramming in cancer cell models. In addition, the present findings provide support for a model in which MUC1-C regulates glycolysis and OXPHOS in distinct clusters of CSCs. In this regard, we found that CSCs are dependent on MUC1-C-induced glycolysis as evidenced by the demonstration that 2DG treatment inhibits self-renewal. We also found that CSCs are dependent on MUC1-C-driven repression of the ETC, as targeting MUC1-C with silencing and GO-203 treatment suppresses self-renewal by inducing superoxide production, loss of the MMP, and death. In summary, our findings in TNBC cells indicate that MUC1-C drives CSC self-renewal by integrating the activation of glycolysis in certain subpopulations with suppression of OXPHOS in others. Expression of MUC1 in TNBCs associates with depletion of immune effectors in the tumor microenvironment (TME).77,78 Aerobic glycolysis has been linked to dysregulation of the TME.79 Therefore, additional studies will be needed to determine if the effects of MUC1-C on glycolysis and OXPHOS contribute to immunosuppression or other alterations in the TME. Additional studies will also be needed to further assess the role of MUC1-C in coupling the CSC state with metabolic reprogramming, which may also contribute to understanding involvement of the Warburg effect in cancer progression.61,80
Limitations of the study
These studies were performed on enriched populations of TNBC CSCs as defined by serial passage of mammospheres with increasing capacity for self-renewal and tumorigenicity. One limitation of this work is that the enriched CSCs may not be representative of purified CSC populations. To address this limitation in part, we performed scRNA-seq on serially passaged mammosphere cells and confirmed our results from bulk RNA-seq studies of enriched mammospheres. Another limitation is that MUC1-C-dependent upregulation of aerobic glycolysis and suppression of OXPHOS in these enriched TNBC CSC populations may not extend to CSCs selected by different methods and from other tumor types. MUC1-C has been shown to drive the CSC state across pan-cancers. Subsequent studies are needed to determine if this dependency on MUC1-C in other settings is associated with integration of aerobic glycolysis and OXPHOS to maintain CSC redox balance and thereby self-renewal capacity.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-MUC1-C | Thermo Fisher Scientific | Cat # MA5-11202; RRID:AB_11000874 |
| Anti-ZEB1 | Cell Signaling Technology | Cat #3396; RRID:AB_1904164 |
| Anti-TWIST1 | Abcam | Cat #ab50887; RRID:AB_883294 |
| Anti-BMI1 | Cell Signaling Technology | Cat #6964; RRID:AB_10828713 |
| Anti-MYC | Abcam | Cat #ab32072; RRID:AB_731658 |
| Anti-SOX2 | Cell Signaling Technology | Cat #3579; RRID:AB_2195767 |
| Anti-β-actin | Sigma-Aldrich | Cat #A5441; RRID:AB_476744 |
| Anti-GLUT1 | Abcam | Cat #115730; RRID:AB_10903230 |
| anti-HK2 | Cell Signaling Technology | Cat #2867; RRID:AB_2232946 |
| anti-SDHD | NOVUS Biologicals | Cat #NBP2-83506 |
| anti-cytochrome c | Proteintech | Cat #10993-1-AP; RRID:AB_2090467 |
| anti-ND1 | Proteintech | Cat #19703-1-AP; RRID:AB_10637853 |
| anti-COX2 | Proteintech | Cat #55070-1-AP; RRID: AB_10859832 |
| anti-TFAM | Cell Signaling Technology | Cat #8076S; RRID:AB_1094911 |
| anti-TFB2M | Proteintech | Cat #24411-1-AP; RRID:AB_2879530 |
| anti-mTERF3 | Abcam | Cat #230232 |
| anti-SOD2 | Cell Signaling Technology | Cat #13141; RRID: AB_2636921 |
| anti-PRDX3 | Proteintech | Cat #10664-1-AP; RRID: AB_2284207 |
| Bacterial and virus strains | ||
| Lentiviral particles for MUC1 shRNA | This paper | N/A |
| Lentiviral particles for MYC shRNA | This paper | N/A |
| Lentiviral particles for MUC1 shRNA#2 | This paper | N/A |
| Lentiviral particles for control shRNA | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine 3000 Reagent | Invitrogen | Cat #L3000008 |
| Trizol Reagent | Invitrogen | Cat #15596018 |
| Puromycin | InvivoGen | Cat #ant-pr-1 |
| Geneticin | Invitrogen | Cat #10131035 |
| GO-203 | This paper | N/A |
| 2-DG | Selleck | Cat #S4701 |
| Doxycycline | Millipore Sigma | Cat #24390-14-5 |
| TrypLE Express | Thermo Scientific | Cat #12604013 |
| TRIzol | Invitrogen | Cat #15596-018 |
| Halt™ Protease Inhibitor Cocktail (100X) | Thermo Fisher Scientific | Cat #78430 |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat #10003D |
| Heparin Solution | Stemcell Technologies | Cat #ST-07980 |
| Critical commercial assays | ||
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat #4367659 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat #4368814 |
| DNeasy Blood & Tissue Kit | QIAGEN | Cat #69504 |
| MammoCult Human Medium Kit | Stemcell Technologies | Cat #ST-05620 |
| AlamarBlue Cell Viability Reagent | Thermo Scientific | Cat #DAL1100 |
| PureLink HiPure Plasmid DNA Purification Kit | Invitrogen | Cat #K210007 |
| TruSeq Stranded mRNA Kit | Illumina | Cat #20020594 |
| Glucose Uptake-Glo Assay | Promega | Cat #J1341 |
| Luminescent ATP Detection Assay Kit | Abcam | Cat #ab113849 |
| MitoTracker Green | Cell Signaling Technology | Cat #9074 |
| MitoSOX Red | Thermo Fisher Scientific | Cat #M36008 |
| MitoProbe JC-1 Assay Kit | Thermo Fisher Scientific | Cat #M34152 |
| Zombie Aqua™ | BioLegend | Cat #423101 |
| MycoAlert Mycoplasma Detection Kit | Lonza | Cat #LT07-118 |
| Deposited data | ||
| RNA-seq | NCBI GEO | GSE222377 |
| scRNA-seq | NCBI GEO | GSE230308 |
| Experimental models: Cell lines | ||
| BT-549 | ATCC | CVCL_1092 |
| MDA-MB-436 | ATCC | CVCL_0623 |
| HEK293T | ATCC | CVCL_0063 |
| BT-549/tet-CshRNA | This paper | N/A |
| BT-549/tet-MUC1shRNA | This paper | N/A |
| BT-549/CshRNA | This paper | N/A |
| BT-549/MUC1shRNA | This paper | N/A |
| MDA-MB-436/tet-CshRNA | This paper | N/A |
| MDA-MB-436/tet-MUC1shRNA | This paper | N/A |
| BT-549/tet-MYCshRNA | This paper | N/A |
| Experimental models: Organisms/strains | ||
| NSG mice | Jackson Laboratory | N/A |
| Nude mice | Jackson Laboratory | N/A |
| Oligonucleotides | ||
| MUC1shRNA | SIGMA | TRCN0000122938 |
| MYCshRNA | SIGMA | TRCN0000039642 |
| MUC1shRNA#2 | SIGMA | TRCN0000430218 |
| CshRNA | Millipore Sigma | N/A |
| Primers for real-time PCR, see Table S1 | This paper | N/A |
| Primers for ChIP-PCR, see Table S2 | This paper | N/A |
| Software and algorithms | ||
| FlowJo v10.6.2 | BD Biosciences | N/A |
| GraphPad Prism9 | Dotmatics | N/A |
| R package | CRAN | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Prof. Donald Kufe, Dana-Farber Cancer Institute, 450 Brookline Ave., Boston, MA 02215, USA. E-mail: donald_kufe@dfci.harvard.edu.
Materials availability
The study did not generate new unique reagents and there are no restrictions to availability.
Experimental model and study participant details
Cell and culture conditions
Human BT-549 BRCA1 wild-type TNBC (CVCL_1092, ATCC) cells were cultured in RPMI1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS; GEMINI BioProducts, West Sacramento, CA, USA), 100 μg/ml streptomycin, 100 U/ml penicillin and 10 μg/ml insulin. MDA-MB-436 BRCA1 mutant TNBC (CVCL_0623, ATCC) cells were cultured in RPMI1640 medium containing 10% FBS, 100 μg/ml streptomycin and 100 U/ml penicillin. Cells were treated with the MUC1-C inhibitor GO-203.16,19 Cell authentication was performed using short tandem repeat analysis every 3–4 months. The cells were monitored for mycoplasma contamination every 3–4 months using the MycoAlert Mycoplasma Detection Kit (Lonza, Rockland, MA, USA). Cells were maintained in culture for 3–4 months for performing experiments.
Method details
Mammosphere formation assay
Cells growing in 2D culture were seeded at 2.5–5×103/well in 6-well ultra-low attachment culture plates (Corning, Glendale, AZ, USA) using the MammoCult Human Medium Kit (Stemcell Technologies, Cambridge, MA, USA).19 Mammospheres with diameters >100 μm were counted in triplicate under an inverted microscope. Mammospheres with diameters >100 μm were isolated and disrupted in the presence of TrypLE Express (Thermo Scientific, Rockford, IL, USA) into single-cell suspensions, which were counted and then reseeded into mammocult medium for serial passage. Sphere forming efficiency (SFE) was determined as the percentage of seeded cells that formed mammospheres. Cell viability was assessed by AlamarBlue (Thermo Scientific) staining and fluorescence intensity (560 nm excitation/590 nm emission).
Gene silencing and rescue
MUC1shRNA (MISSION shRNA TRCN0000122938), MYCshRNA (MISSION shRNA TRCN0000039642) and a control scrambled shRNA (CshRNA)(Millipore Sigma) were inserted into pLKO-tet-puro (Plasmid #21915; Addgene, Cambridge, MA, USA; RRID:Addgene_32017).19 CshRNA, MUC1shRNA, MYCshRNA, MUC1shRNA#2 (MISSION shRNA TRCN0000430218) were produced in HEK293T cells (RRID:CVCL_0063).19 Flag-tagged MUC1-CD was inserted into pInducer20 (plasmid #44012, Addgene).19 Vector-transduced cells were selected in the presence of 1–2 μg/ml puromycin. Cells were treated with 0.1% DMSO as the vehicle control or 500 ng/ml doxycycline (DOX; Millipore Sigma) for inducible gene silencing.
Real-time quantitative reverse-transcription PCR (qRT-PCR)
Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA, USA). cDNAs were synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Grand Island, NY, USA). Samples were ampli- fied using the Power SYBR Green PCR Master Mix (Applied Biosystems) and the CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). Primers used for qRT-PCR and gPCR are listed in Table S1.
Immunoblot analysis
Whole cell lysates were prepared in RIPA buffer containing protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, USA).19 Immunoblotting was performed with anti-MUC1-C (#MA5-11202, 1:100; Thermo Fisher Scientific), anti-ZEB1 (#3396, 1:1000 dilution; Cell Signaling Technology (CST), Danvers, MA, USA), anti-TWIST1 (ab50887, 1:1000 dilution; Abcam, Waltham, MA, USA), anti-BMI1 (#6964, 1:1000 dilution; CST), anti-MYC (#ab32072, 1:1000 dilution; Abcam), anti-SOX2 (#3579, 1:1000 dilution; CST), anti-GLUT1 (#ab115730, 1:100000 dilution; Abcam), anti-HK2 (#2867, 1:1000 dilution; CST), anti-SDHD (#NBP2-83505, 1:1000, NOVUS Biologicals, Centennial, CO, USA), anti-cytochrome c (#10993, 1:4000 dilution; Proteintech; Rosemont, IL, USA), anti-ND1 (#19703, 1:1000 dilution; Proteintech), anti-COX2 (#55070, 1:5000 dilution; Proteintech), anti-TFAM (#8076, 1:1000 dilution; CST), anti-TFB2M (#24411, 1:500 dilution; Proteintech), anti-mTERF3 (#ab230232, 1:1000 dilution; Abcam), anti-SOD2 (#13141, 1:1000 dilution; CST), anti-PRDX3 (#10664, 1:2000 dilution; Proteintech), and anti-β-actin (A5441; 1:50000 dilution; Sigma, St. Louis, MO, USA).
Mouse tumor model studies
Six-week-old female NSG mice (Jackson Laboratory, Bar Harbor, ME, USA) were injected subcutaneously into the flank with the indicated numbers of BT-549 2D cultured and passaged mammosphere cells in 100 μl of a 1:1 solution of medium and Matrigel (BD Biosciences). The mice were monitored for the appearance of palpable tumors and sacrificed when tumor volumes reached 1000 mm3. Alternatively, when the mean BT-549 passaged mammosphere-derived tumor volume reached ∼250 mm3, the mice were pair-matched into groups and treated intraperitoneally with PBS or GO-203 (12 μg/gm body weight) daily for 70 days. Tumor measurements and body weights were recorded twice per week. For serial dilution tumor initiation assays, the indicated numbers of MDA-MB-436 2D and passaged mammosphere cells were implanted into the left and right flanks, respectively, of six-week-old female nude mice (Jackson Laboratory). These studies were conducted in accordance with the ethical regulations required for approval by the Dana-Farber Cancer Institute Animal Care and Use Committee (IACUC) under protocol 03–029.
RNA-seq analysis
Total RNA from cells cultured in triplicate was used to generate RNA-seq datasets.19 Briefly, TruSeq stranded paired-end sequencing was perfomed on an Illumina NovaSeq 6000. Reads were aligned to the human reference genome (GRCh38) using the STAR aligner (version 2.7.9a)81 and transcripts were quantified by featureCounts from the Subread package (version 2.0.1).82 Normalization and differential expression analysis were completed with DESeq2 in the R computing environment.83 Differential expression rank order for Gene Set Enrichment Analysis (GSEA) was performed using the fgsea package in R.84 Gene set variation analysis (GSVA) was performed using the GSVA package.19 Hallmark and Wikipedia (WP) Gene Signatures were obtained from the Molecular Signatures Database (MSigDB). Transcriptional regulator and motif enrichment analyses were performed using epigenetic Landscape in Silico deletion Analysis (LISA).85
MetaPhOR analysis
Transcriptional dysregulation of metabolic pathways was assessed with the R package, MetaPhOR, using bulk RNA-seq DEG lists as input.86 Briefly, MetaPhOR scores the direction and magnitude of dysregulation for key metabolic pathways from Kyoto Encyclopedia of Genes and Genomes (KEGG) and Pathway Studio pathways using expression data. A bootstrapping method with random selection of 100,000 comparator scores was employed to assess statistical significance of dysregulated pathways.
Co-immunoprecipitation of nuclear proteins
Cells were washed with PBS and incubated in cell lysis buffer (10 mmol/L HEPES, pH 8.0, 1.5 mmol/L MgCl2, 0.5% NP40, and 10 mmol/L KCl) for 10 minutes at 4°C. The total cell lysates were centrifuged at 4,000 rpm for 5 minutes at 4°C and the pellets were incubated in nuclear lysis buffer (10 mmol/L HEPES pH 8.0, 1.6 mmol/L MgCl2, 0.5% NP40, 420 mmol/L NaCl, 0.2 mmol/L EDTA, and 25% glycerol) for 20 minutes at 4°C, and then sheared by passage through 20 to 26 gauge needles. After centrifugation at 13,000 rpm for 10 minutes, the supernatants were collected as nuclear lysates.19 Nuclear proteins were incubated with anti-MUC1-C (#MA5-11202; Thermo Fisher Scientific) at 4°C overnight and then precipitated with Dynabeads Protein G (10003D; ThermoFisher Scientific) for 2 hours at 4°C. Beads were washed twice with washing buffer (20 mmol/L Tris-HCl, pH 8.0, 0.2 mmol/L EDTA, 1.5 mmol/L MgCl2, 0.5% NP40, and 150 mmol/L NaCl) and once with 10% TE buffer (BM-304A; Boston BioProducts), and then resuspended in sample loading buffer.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed on cells crosslinked with 1% formaldehyde for 5 min at 37°C, quenched with 2 M glycine, washed with PBS, and sonicated in a Covaris E220 sonicator to generate 300–600 bp DNA fragments.19 Immunoprecipitation was performed using a control IgG (#3900; CST), and anti-MYC (#ab32072; Abcam). Precipitated DNAs were detected by PCR using primers listed in Table S2. The immunoprecipitated DNA was quantified using SYBR-green and the CFX96 Touch Real-Time PCR Detection System (Bio-Rad).19 Data are reported as fold-enrichment relative to IgG levels.
Glucose uptake assays
Analysis of glucose uptake in CSCs was performed using the Glucose Uptake-Glo Assay according to the manufacturer’s instructions (Promega, Madison, WI, USA).
ATP assays
Measurement of intracellular ATP levels was performed using the Luminescent ATP Detection Assay Kit (#ab113849; Abcam) according to the manufacturer’s instructions.
Mitochondrial DNA quantification
Total DNA was extracted using a DNeasy Blood & Tissue Kit according to the manufacturer’s protocol (QIAGEN, Hilden, Germany). Relative mtDNA:nDNA ratio was calculated using the ΔΔCt method for the mitochondrial-encoded human tRNA-Leu(UUR) (Fwd: CACCCAAGAACAGGGTTTGT;_Rev: TGGCCATGGGTATGTTGTTA) and nuclear-encoded human B2M (Fwd: TGCTGTCTCCATGTTTGATGTATCT; Rev: TCTCTGCTCCCCACCTCTAAGT) genes.
Analysis of mitochondrial mass
MitoTracker Green stock solutions were diluted into prewarmed (37°C) PBS to a working concentration of 100 nM. Dead cells were stained with Zombie Aqua™ (BioLegend). Cells were analyzed by MACSQuant Analyzer 10 Flow Cytometer (Miltenyi Biotec, Waltham, MA). A total of 20,000 events were acquired for each sample. Data were analyzed with FlowJo v10.6.2 (BD Biosciences) software. Fluorescence was measured using the standard emission filters for green (FL-1 channel) fluorescence photomultipliers.
Analysis of mitochondrial superoxide levels
Cells were incubated with pre-warmed MitoSOX Red staining solution diluted in HBSS to a final concentration of 5 μM for 15 min at 37°C. MitoSOX Red stained mammospheres were mixed with Matrigel, placed on 35 mm glass-bottom dishes and analyzed using a Leica THUNDER Imager 3D Cell Culture microscope. For flow cytometry analysis, MitoSOX Red-stained mammosphere cells were washed and labeled with Zombie Aqua™ (BioLegend). Cells were analyzed by MACSQuant Analyzer 10 Flow Cytometer (Miltenyi Biotec). A total of 20,000 events were acquired for each sample. Data were analyzed with FlowJo v10.6.2 (BD Biosciences) software. Fluorescence was measured using the standard emission filters for red (FL-2 channel) fluorescence photomultipliers.
Analysis of mitochondrial membrane potential
Mammospheres were stained with 2 μM JC-1 (Thermo Fisher Scientific) for 30 min at 37°C. JC-1 stained mammospheres were mixed with Matrigel, placed on 35 mm glass-bottom dishes and analyzed using a Leica THUNDER Imager 3D Cell Culture microscope. For flow cytometry analysis, JC-1-stained mammosphere cells were washed and labeled with Zombie Aqua™ (BioLegend). Emission spectral overlap was corrected by compensation using carbonylcyanide m-chlorophenylhydrazone (CCCP). Cells were analyzed by MACSQuant Analyzer 10 Flow Cytometer (Miltenyi Biotec). A total of 20,000 events were acquired for each sample. Data were analyzed with FlowJo v10.6.2 (BD Biosciences) software. Fluorescence was measured using the standard emission filters for green (FL-1 channel) red (FL-2 channel) fluorescence photomultipliers.
Analysis of mammosphere CSC populations by scRNA-seq
Pelleted single-cell suspensions were washed in 1,000 ul of PBS plus 0.4% BSA, transferred to low-retention microcentrifuge tubes (Fisher Scientific, Hampton, NH, USA), and then centrifuged for 10 minutes at 300 g at 4°C. Pellets were resuspended in 800 ul of PBS plus 0.4% BSA, and cells were counted by eye using INCYTO C-Chip Neubauer Improved Disposable Hemacytometers (VWR International Ltd., Radnor, PA, USA). A total of > 10,000 cells per sample were loaded per channel of the Chromium Next GEM Chip K for processing on the 10x Chromium Controller (10x Genomics, Pleasanton, CA, USA) followed by cDNA generation and library construction, as per manufacturer’s instructions (Chromium Next GEM Single Cell 5ʹ Reagent Kits v2 User Guide, Rev E). Libraries were normalized and pooled for sequencing on an Illumina NextSeq 500 system using a 150 cycle Mid-Output flow cell (Illumina, Inc., San Diego, CA, USA) with run parameters 26, 10, 10, 90. Sequenced libraries were demultiplexed using cellranger mkfastq (Cell Ranger v6.1.1). As part of the cellranger count pipeline, demultiplexed libraries were aligned to the human transcriptome (excluding introns) based on the 10X Genomics pre-computed human reference, GRCh38-2020-A. Both raw and cell-associated gene expression feature matrices (cell barcodes x gene counts) were output for use in downstream analyses.
scRNA-seq raw data processing, quality control, and subsequent analyses
Raw sequence data demultiplexing, barcode processing, alignment (GRCh38) and filtering for true cells were performed using the Cell Ranger Single-Cell Software Suite (v6.0.0), yielding 23,535 cells (shCTL: 8,561 cells, shMUC1: 14,974 cells) with a mean of 22,239 reads/cell (90.15% mapping rate), median of 1,175 genes/cell, 19,831 total unique detectable genes, and 2,697 median UMI counts/cell. Subsequent filtering for high quality cells, and downstream analyses were performed using Seurat (v4)87 (Figure S7A). Genes expressed in less than 3 cells and cells that express less than 300 genes were excluded from further analyses. Additional filtering of cells was determined based on the overall distributions of total RNA counts (<80,000) and the proportion of mitochondrial genes (<10%) detected to eliminate potential doublets and dying cells, respectively. Additional detection of doublets was performed using Scrublet.88 Thresholding for doublet detection was set based on total distribution of doublet scores (doublet threshold = 0.25). Quantification of mitochondrial and ribosomal gene expression was calculated using the PercentageFeatureSet function, using gene sets compiled from the HUGO Gene Nomenclature Committee database. Ultimately, 6,806 cells were removed (28.92% of total cells) after quality control assessment, and 16,729 high quality cells (shCTL: 5,837 cells, shMUC1: 10,892 cells) were included in downstream analyses. Normalization and variance stabilization were conducted using regularized negative binomial regression (sctransform) implemented with Seurat. Principle component analysis (PCA) was performed on normalized data and optimal dimensionality of the dataset was decided by examination of the Elbow plot, as the total number of PCs where gain in cumulative variation explained was greater than 0.1% (PCs = 50). The FindNeighbors function was utilized that implements a graph based nearest neighbor clustering approach, and the FindClusters function was used to identify final cell clusters (n = 20) using a resolution of 0.08. UMAP was applied for non-linear dimensional reduction to obtain a low dimensional representation of cellular states. Differential expression between clusters or samples was determined using the MAST method89 via the FindMarkers function, using a minimum expression proportion of 25% and a minimum log fold change of 0.25 after correction for mitochondrial and ribosomal gene proportions. Cluster specific gene set enrichment was determined using enrichR, using the collection of HALLMARK pathway signatures.90 Single-cell gene set enrichment was performed by UCell,91 using gene sets reflecting glycolytic (HALLMARK_GLYCOLYSIS) and oxidative phosphorylation (WP_ETC_OXPHOS_MITOCHONDRIA) activity. Gene expression imputation was performed using MAGIC,92 with parameters knn = 15, t = 3.
Quantification and statistical analysis
Statistical analysis
Each experiment was performed at least three times. Unpaired two-tailed Student’s t-tests were used to assess differences between the mean ± SD of two groups. p-values were considered significant at p < 0.05. Adjusted p values derived from the Benjamini-Hochberg method identified differentially expressed genes at a false discovery rate (FDR) < 0.1 for bulk RNA sequencing data. GraphPad Prism9 was used for all statistical analyses. Asterisks represent ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 with CI = 95%.
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under grant numbers CA97098, CA1293701, and CA233084 awarded to DK.
Author contributions
Conceptualization: N.Y., H.W., M.D.L., and D.K.; Methodology: N.Y., H.W., Y.M., A.B., A.T., E.I., S.R., and M.D.L.; Investigation: N.Y., H.W., Y.M., A.B., N.H., T.D., A.F., A.N., E.I., S.R., and M.D.L; Writing – original draft: N.Y., H.W., M.D.L., and D.K.; Writing – review & editing: N.Y., H.W., A.T., M.D.L., and D.K.; Supervision: N.Y., H.W., M.D.L., and D.K.; Funding acquisition: M.D.L. and D.K.
Declaration of interests
DK has equity interests in Genus Oncology and Hillstream Biopharma and is a paid consultant to Reata and CanBas.
Published: October 11, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108168.
Contributor Information
Mark D. Long, Email: mark.long@roswellpark.org.
Donald Kufe, Email: donald_kufe@dfci.harvard.edu.
Supplemental information
Data and code availability
-
•
Total RNA-seq and Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
Total RNA-seq and scRNA-seq data reported here are available from the NCBI Gene Expression Omnibus (GEO) under accessions GSE222377 and GSE230308, respectively.
References
- 1.Kufe D.W. MUC1-C in chronic inflammation and carcinogenesis; emergence as a target for cancer treatment. Carcinogenesis. 2020;41:1173–1183. doi: 10.1093/carcin/bgaa082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kufe D.W. Emergence of MUC1 in mammals for adaptation of barrier epithelia Cancers. Basel) 2022;14:4805. doi: 10.3390/cancers14194805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmad R., Raina D., Joshi M.D., Kawano T., Ren J., Kharbanda S., Kufe D. MUC1-C oncoprotein functions as a direct activator of the NF-kappaB p65 transcription factor. Cancer Res. 2009;69:7013–7021. doi: 10.1182/blood-2011-07-369686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmad R., Rajabi H., Kosugi M., Joshi M.D., Alam M., Vasir B., Kawano T., Kharbanda S., Kufe D. MUC1-C oncoprotein promotes STAT3 activation in an auto-inductive regulatory loop. Sci. Signal. 2011;4:ra9. doi: 10.1126/scisignal.2001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hata T., Rajabi H., Yamamoto M., Jin C., Ahmad R., Zhang Y., Kui L., Li W., Yasumizu Y., Hong D., et al. Targeting MUC1-C inhibits TWIST1 signaling in triple-negative breast cancer. Mol. Cancer Ther. 2019;18:1744–1754. doi: 10.1158/1535-7163.MCT-19-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajabi H., Hiraki M., Kufe D. MUC1-C activates polycomb repressive complexes and downregulates tumor suppressor genes in human cancer cells. Oncogene. 2018;37:2079–2088. doi: 10.1038/s41388-017-0096-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagiwara M., Yasumizu Y., Yamashita N., Rajabi H., Fushimi A., Long M.D., Li W., Bhattacharya A., Ahmad R., Oya M., et al. MUC1-C activates the BAF (mSWI/SNF) complex in prostate cancer stem cells. Cancer Res. 2021;81:1111–1122. doi: 10.1158/0008-5472.CAN-20-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagiwara M., Fushimi A., Yamashita N., Bhattacharya A., Rajabi H., Long M.D., Yasumizu Y., Oya M., Liu S., Kufe D. MUC1-C activates the PBAF chromatin remodeling complex in integrating redox balance with progression of human prostate cancer stem cells. Oncogene. 2021;40:4930–4940. doi: 10.1038/s41388-021-01899-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhattacharya A., Fushimi A., Yamashita N., Hagiwara M., Morimoto Y., Rajabi H., Long M.D., Abdulla M., Ahmad R., Street K., et al. MUC1-C dictates JUN and BAF-mediated chromatin remodeling at enhancer signatures in cancer stem cells. Mol. Cancer Res. 2022;20:556–567. doi: 10.1158/1541-7786.MCR-21-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kufe D.W. Chronic activation of MUC1-C in wound repair promotes progression to cancer stem cells. J. Cancer Metastasis Treat. 2022;8 doi: 10.20517/2394-4722.2022.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yasumizu Y., Rajabi H., Jin C., Hata T., Pitroda S., Long M.D., Hagiwara M., Li W., Hu Q., Liu S., et al. MUC1-C regulates lineage plasticity driving progression to neuroendocrine prostate cancer. Nat. Commun. 2020;11:338. doi: 10.1038/s41467-019-14219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W., Zhang N., Jin C., Long M.D., Rajabi H., Yasumizu Y., Fushimi A., Yamashita N., Hagiwara M., Zheng R., et al. MUC1-C drives stemness in progression of colitis to colorectal cancer. JCI Insight. 2020;5 doi: 10.1172/jci.insight.137112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luan Z., Morimoto Y., Fushimi A., Yamashita N., Suo W., Bhattacharya A., Hagiwara M., Jin C., Kufe D. MUC1-C dictates neuroendocrine lineage specification in pancreatic ductal adenocarcinomas. Carcinogenesis. 2022;43:67–76. doi: 10.1093/carcin/bgab097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fushimi A., Morimoto Y., Ishikawa S., Yamashita N., Bhattacharya A., Daimon T., Rajabi H., Jin C., Hagiwara M., Yasumizu Y., et al. Dependence on the MUC1-C oncoprotein in classic, variant and non-neuroendocrine small cell lung cancer. Mol. Cancer Res. 2022;20:1379–1390. doi: 10.1158/1541-7786.MCR-22-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morimoto Y., Fushimi A., Yamashita N., Hagiwara M., Bhattacharya A., Cheng J., Frost T.C., Ahmad R., Daimon T., Huang L., et al. Addiction of Merkel cell carcinoma to MUC1-C identifies a potential new target for treatment. Oncogene. 2022;41:3511–3523. doi: 10.1038/s41388-022-02361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamashita N., Kufe D. Addiction of cancer stem cells to MUC1-C in triple-negative breast cancer progression. Int. J. Mol. Sci. 2022;23:8219. doi: 10.3390/ijms23158219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu F., Liu F., Zhao H., An G., Feng G. Prognostic significance of mucin antigen MUC1 in various human epithelial cancers: a meta-analysis. Medicine (Baltim.) 2015;94 doi: 10.1097/MD.0000000000002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bianchini G., De Angelis C., Licata L., Gianni L. Treatment landscape of triple-negative breast cancer - expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2022;19:91–113. doi: 10.1038/s41571-021-00565-2. [DOI] [PubMed] [Google Scholar]
- 19.Yamashita N., Morimoto Y., Fushimi A., Ahmad R., Bhattacharya A., Daimon T., Haratake N., Inoue Y., Ishikawa S., Yamamoto M., et al. MUC1-C dictates PBRM1-mediated chronic induction of interferon signaling, DNA damage resistance and immunosuppression in triple-negative breast cancer. Mol Canc Res. 2023;21:274–289. doi: 10.1158/1541-7786.MCR-22-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun X., Wang M., Wang M., Yu X., Guo J., Sun T., Li X., Yao L., Dong H., Xu Y. Metabolic reprogramming in triple-negative breast cancer. Front. Oncol. 2020;10:428. doi: 10.3389/fonc.2020.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z., Jiang Q., Dong C. Metabolic reprogramming in triple-negative breast cancer. Cancer Biol. Med. 2020;17:44–59. doi: 10.20892/j.issn.2095-3941.2019.0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palaskas N., Larson S.M., Schultz N., Komisopoulou E., Wong J., Rohle D., Campos C., Yannuzzi N., Osborne J.R., Linkov I., et al. 18F-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer Res. 2011;71:5164–5174. doi: 10.1158/0008-5472.CAN-10-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lanning N.J., Castle J.P., Singh S.J., Leon A.N., Tovar E.A., Sanghera A., MacKeigan J.P., Filipp F.V., Graveel C.R. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017;5:6. doi: 10.1186/s40170-017-0168-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oh S., Kim H., Nam K., Shin I. Glut1 promotes cell proliferation, migration and invasion by regulating epidermal growth factor receptor and integrin signaling in triple-negative breast cancer cells. BMB Rep. 2017;50:132–137. doi: 10.5483/bmbrep.2017.50.3.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciscato F., Ferrone L., Masgras I., Laquatra C., Rasola A. Hexokinase 2 in cancer: a prima donna playing multiple characters. Int. J. Mol. Sci. 2021;22:4716. doi: 10.3390/ijms22094716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vyas S., Zaganjor E., Haigis M.C. Mitochondria and cancer. Cell. 2016;166:555–566. doi: 10.1016/j.cell.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cassim S., Vučetić M., Ždralević M., Pouyssegur J. Warburg and beyond: the power of mitochondrial metabolism to collaborate or replace fermentative glycolysis in cancer. Cancers. 2020;12:1119. doi: 10.3390/cancers12051119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guha M., Srinivasan S., Raman P., Jiang Y., Kaufman B.A., Taylor D., Dong D., Chakrabarti R., Picard M., Carstens R.P., et al. Aggressive triple negative breast cancers have unique molecular signature on the basis of mitochondrial genetic and functional defects. Biochim. Biophys. Acta, Mol. Basis Dis. 2018;1864:1060–1071. doi: 10.1016/j.bbadis.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zacksenhaus E., Shrestha M., Liu J.C., Vorobieva I., Chung P.E.D., Ju Y., Nir U., Jiang Z. Mitochondrial OXPHOS induced by RB1 deficiency in breast cancer: implications for anabolic metabolism, stemness, and metastasis. Trends Cancer. 2017;3:768–779. doi: 10.1016/j.trecan.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Kosugi M., Ahmad R., Alam M., Uchida Y., Kufe D. MUC1-C oncoprotein regulates glycolysis and pyruvate kinase M2 activity in cancer cells. PLoS One. 2011;6 doi: 10.1371/journal.pone.0028234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hasegawa M., Takahashi H., Rajabi H., Alam M., Suzuki Y., Yin L., Tagde A., Maeda T., Hiraki M., Sukhatme V.P., Kufe D. Functional interactions of the cystine/glutamate antiporter, CD44v and MUC1-C oncoprotein in triple-negative breast cancer cells. Oncotarget. 2016;7:11756–11769. doi: 10.18632/oncotarget.7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shukla S.K., Purohit V., Mehla K., Gunda V., Chaika N.V., Vernucci E., King R.J., Abrego J., Goode G.D., Dasgupta A., et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32:71–87.e7. doi: 10.1016/j.ccell.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goode G., Gunda V., Chaika N.V., Purohit V., Yu F., Singh P.K. MUC1 facilitates metabolomic reprogramming in triple-negative breast cancer. PLoS One. 2017;12 doi: 10.1371/journal.pone.0176820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmad R., Alam M., Hasegawa M., Uchida Y., Al-Obaid O., Kharbanda S., Kufe D. Targeting MUC1-C inhibits the AKT-S6K1-elF4A pathway regulating TIGAR translation in colorectal cancer. Mol. Cancer. 2017;16:33. doi: 10.1186/s12943-017-0608-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thiagarajan P.S., Hitomi M., Hale J.S., Alvarado A.G., Otvos B., Sinyuk M., Stoltz K., Wiechert A., Mulkearns-Hubert E., Jarrar A., et al. Development of a fluorescent reporter system to delineate cancer stem cells in triple-negative breast cancer. Stem Cell. 2015;33:2114–2125. doi: 10.1002/stem.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lambert A.W., Fiore C., Chutake Y., Verhaar E.R., Strasser P.C., Chen M.W., Farouq D., Das S., Li X., Eaton E.N., et al. DeltaNp63/p73 drive metastatic colonization by controlling a regenerative epithelial stem cell program in quasi-mesenchymal cancer stem cells. Dev. Cell. 2022;57:2714–2730.e8. doi: 10.1016/j.devcel.2022.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nandi A., Chakrabarti R. Assessment of breast cancer stem cell activity using a spheroid formation assay. Methods Mol. Biol. 2022;2429:485–500. doi: 10.1007/978-1-0716-1979-7_33. [DOI] [PubMed] [Google Scholar]
- 38.Deshmukh A.P., den Hollander P., Kuburich N.A., Vasaikar S., Joseph R., Mani S.A. Enrichment of cancer stem cells in a tumorsphere assay. Methods Mol. Biol. 2022;2429:501–507. doi: 10.1007/978-1-0716-1979-7_34. [DOI] [PubMed] [Google Scholar]
- 39.Mani S.A., Guo W., Liao M.J., Eaton E.N., Ayyanan A., Zhou A.Y., Brooks M., Reinhard F., Zhang C.C., Shipitsin M., et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. S0092-8674(08)00444-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hiraki M., Maeda T., Bouillez A., Alam M., Tagde A., Hinohara K., Suzuki Y., Markert T., Miyo M., Komura K., et al. MUC1-C activates BMI1 in human cancer cells. Oncogene. 2017;36:2791–2801. doi: 10.1038/onc.2016.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukherjee P., Gupta A., Chattopadhyay D., Chatterji U. Modulation of SOX2 expression delineates an end-point for paclitaxel-effectiveness in breast cancer stem cells. Sci. Rep. 2017;7:9170. doi: 10.1038/s41598-017-08971-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng C.C., Shi L.H., Wang X.J., Wang S.X., Wan X.Q., Liu S.R., Wang Y.F., Lu Z., Wang L.H., Ding Y. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int. J. Oncol. 2018;53:339–348. doi: 10.3892/ijo.2018.4399. [DOI] [PubMed] [Google Scholar]
- 43.Fukushi A., Kim H.D., Chang Y.C., Kim C.H. Revisited metabolic control and reprogramming cancers by means of the Warburg effect in tumor cells. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms231710037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hitosugi T., Zhou L., Elf S., Fan J., Kang H.B., Seo J.H., Shan C., Dai Q., Zhang L., Xie J., et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. 2012;22:585–600. doi: 10.1016/j.ccr.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arundhathi J.R.D., Mathur S.R., Gogia A., Deo S.V.S., Mohapatra P., Prasad C.P. Metabolic changes in triple negative breast cancer-focus on aerobic glycolysis. Mol. Biol. Rep. 2021;48:4733–4745. doi: 10.1007/s11033-021-06414-w. [DOI] [PubMed] [Google Scholar]
- 46.Patra K.C., Wang Q., Bhaskar P.T., Miller L., Wang Z., Wheaton W., Chandel N., Laakso M., Muller W.J., Allen E.L., et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24:213–228. doi: 10.1016/j.ccr.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolpaw A.J., Dang C.V. MYC-induced metabolic stress and tumorigenesis. Biochim. Biophys. Acta. Rev. Cancer. 2018;1870:43–50. doi: 10.1016/j.bbcan.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 48.Hata T., Rajabi H., Takahashi H., Yasumizu Y., Li W., Jin C., Long M.D., Hu Q., Liu S., Fushimi A., et al. MUC1-C activates the NuRD complex to drive dedifferentiation of triple-negative breast cancer cells. Cancer Res. 2019;79:5711–5722. doi: 10.1158/0008-5472.CAN-19-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pajak B., Siwiak E., Sołtyka M., Priebe A., Zieliński R., Fokt I., Ziemniak M., Jaśkiewicz A., Borowski R., Domoradzki T., Priebe W. 2-Deoxy-d-Glucose and its analogs: from diagnostic to therapeutic agents. Int. J. Mol. Sci. 2019;21:234. doi: 10.3390/ijms21010234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Signes A., Fernandez-Vizarra E. Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem. 2018;62:255–270. doi: 10.1042/EBC20170098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grasso D., Zampieri L.X., Capelôa T., Van de Velde J.A., Sonveaux P. Mitochondria in cancer. Cell Stress. 2020;4:114–146. doi: 10.15698/cst2020.06.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Geldon S., Fernández-Vizarra E., Tokatlidis K. Redox-mediated regulation of mitochondrial biogenesis, dynamics, and respiratory chain assembly in yeast and human cells. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.720656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kadenbach B. Complex IV - the regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion. 2021;58:296–302. doi: 10.1016/j.mito.2020.10.004. [DOI] [PubMed] [Google Scholar]
- 54.Yan W.L., Lerner T.J., Haines J.L., Gusella J.F. Sequence analysis and mapping of a novel human mitochondrial ATP synthase subunit 9 cDNA (ATP5G3) Genomics. 1994;24:375–377. doi: 10.1006/geno.1994.1631. [DOI] [PubMed] [Google Scholar]
- 55.Morrish F., Hockenbery D. MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 2014;4:a014225. doi: 10.1101/cshperspect.a014225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barshad G., Marom S., Cohen T., Mishmar D. Mitochondrial DNA transcription and its regulation: an evolutionary perspective. Trends Genet. 2018;34:682–692. doi: 10.1016/j.tig.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 57.Campbell C.T., Kolesar J.E., Kaufman B.A. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta. 2012;1819:921–929. doi: 10.1016/j.bbagrm.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 58.Park C.B., Asin-Cayuela J., Cámara Y., Shi Y., Pellegrini M., Gaspari M., Wibom R., Hultenby K., Erdjument-Bromage H., Tempst P., et al. MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell. 2007;130:273–285. doi: 10.1016/j.cell.2007.05.046. [DOI] [PubMed] [Google Scholar]
- 59.Sena L.A., Chandel N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suski J., Lebiedzinska M., Bonora M., Pinton P., Duszynski J., Wieckowski M.R. Relation between mitochondrial membrane potential and ROS formation. Methods Mol. Biol. 2018;1782:357–381. doi: 10.1007/978-1-4939-7831-1_22. [DOI] [PubMed] [Google Scholar]
- 61.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 62.Jacquet P., Stéphanou A. Searching for the metabolic signature of cancer: A review from Warburg's time to now. Biomolecules. 2022;12:1412. doi: 10.3390/biom12101412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vinaik R., Barayan D., Auger C., Abdullahi A., Jeschke M.G. Regulation of glycolysis and the Warburg effect in wound healing. JCI Insight. 2020;5 doi: 10.1172/jci.insight.138949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rajabi H., Kufe D. MUC1-C oncoprotein integrates a program of EMT, epigenetic reprogramming and immune evasion in human carcinomas. BBA Rev. Cancer. 2017;1868:117–122. doi: 10.1016/j.bbcan.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang X., Sun Q., Li H., Li K., Ren X. The role of phosphoglycerate mutase 1 in tumor aerobic glycolysis and its potential therapeutic implications. Int. J. Cancer. 2014;135:1991–1996. doi: 10.1002/ijc.28637. [DOI] [PubMed] [Google Scholar]
- 66.Sebastian C., Ferrer C., Serra M., Choi J.E., Ducano N., Mira A., Shah M.S., Stopka S.A., Perciaccante A.J., Isella C., et al. A non-dividing cell population with high pyruvate dehydrogenase kinase activity regulates metabolic heterogeneity and tumorigenesis in the intestine. Nat. Commun. 2022;13:1503. doi: 10.1038/s41467-022-29085-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Skoda J., Borankova K., Jansson P.J., Huang M.L.H., Veselska R., Richardson D.R. Pharmacological targeting of mitochondria in cancer stem cells: an ancient organelle at the crossroad of novel anti-cancer therapies. Pharmacol. Res. 2019;139:298–313. doi: 10.1016/j.phrs.2018.11.020. [DOI] [PubMed] [Google Scholar]
- 68.Payandeh Z., Pirpour Tazehkand A., Barati G., Pouremamali F., Kahroba H., Baradaran B., Samadi N. Role of Nrf2 and mitochondria in cancer stem cells; in carcinogenesis, tumor progression, and chemoresistance. Biochimie. 2020;179:32–45. doi: 10.1016/j.biochi.2020.09.014. [DOI] [PubMed] [Google Scholar]
- 69.García-Heredia J.M., Carnero A. Role of mitochondria in cancer stem cell resistance. Cells. 2020;9:1693. doi: 10.3390/cells9071693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ding S., Li C., Cheng N., Cui X., Xu X., Zhou G. Redox regulation in cancer stem cells. Oxid. Med. Cell. Longev. 2015;2015 doi: 10.1155/2015/750798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Snyder V., Reed-Newman T.C., Arnold L., Thomas S.M., Anant S. Cancer stem cell metabolism and potential therapeutic targets. Front. Oncol. 2018;8:203. doi: 10.3389/fonc.2018.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raimondi V., Ciccarese F., Ciminale V. Oncogenic pathways and the electron transport chain: a dangeROS liaison. Br. J. Cancer. 2020;122:168–181. doi: 10.1038/s41416-019-0651-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nathansen J., Meyer F., Müller L., Schmitz M., Borgmann K., Dubrovska A. Beyond the double-strand breaks: the role of DNA repair proteins in cancer stem-cell regulation. Cancers. 2021;13:4818. doi: 10.3390/cancers13194818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ždralević M., Brand A., Di Ianni L., Dettmer K., Reinders J., Singer K., Peter K., Schnell A., Bruss C., Decking S.M., et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the "Warburg effect" restricting tumor growth to oxidative metabolism. J. Biol. Chem. 2018;293:15947–15961. doi: 10.1074/jbc.RA118.004180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Olszewski K., Barsotti A., Feng X.J., Momcilovic M., Liu K.G., Kim J.I., Morris K., Lamarque C., Gaffney J., Yu X., et al. Inhibition of glucose transport synergizes with chemical or genetic disruption of mitochondrial metabolism and suppresses TCA cycle-deficient tumors. Cell Chem. Biol. 2022;29:423–435.e10. doi: 10.1016/j.chembiol.2021.10.007. [DOI] [PubMed] [Google Scholar]
- 76.Wallace D.C. Mitochondria and cancer. Nat. Rev. Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamashita N., Long M., Fushimi A., Yamamoto M., Hata T., Hagiwara M., Bhattacharya A., Hu Q., Wong K.K., Liu S., Kufe D. MUC1-C integrates activation of the IFN-gamma pathway with suppression of the tumor immune microenvironment in triple-negative breast cancer. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2020-002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamashita N., Fushimi A., Morimoto Y., Bhattacharya A., Hagiwara M., Yamamoto M., Hata T., Shapiro G.I., Long M.D., Liu S., Kufe D. Targeting MUC1-C suppresses chronic activation of cytosolic nucleotide receptors and STING in triple-negative breast cancer. Cancers. 2022;14:2580. doi: 10.3390/cancers14112580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou D., Duan Z., Li Z., Ge F., Wei R., Kong L. The significance of glycolysis in tumor progression and its relationship with the tumor microenvironment. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.1091779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Faubert B., Solmonson A., DeBerardinis R.J. Metabolic reprogramming and cancer progression. Science. 2020;368 doi: 10.1126/science.aaw5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 83.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast Gene Set Enrichment Analysis. bioRxiv. 2021 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 85.Qin Q., Fan J., Zheng R., Wan C., Mei S., Wu Q., Sun H., Brown M., Zhang J., Meyer C.A., Liu X.S. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol. 2020;21:32. doi: 10.1186/s13059-020-1934-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rosario S.R., Long M.D., Affronti H.C., Rowsam A.M., Eng K.H., Smiraglia D.J. Pan-cancer analysis of transcriptional metabolic dysregulation using The Cancer Genome Atlas. Nat. Commun. 2018;9:5330. doi: 10.1038/s41467-018-07232-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wolock S.L., Lopez R., Klein A.M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M., et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Andreatta M., Carmona S.J. UCell: robust and scalable single-cell gene signature scoring. Comput. Struct. Biotechnol. J. 2021;19:3796–3798. doi: 10.1016/j.csbj.2021.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van Dijk D., Sharma R., Nainys J., Yim K., Kathail P., Carr A.J., Burdziak C., Moon K.R., Chaffer C.L., Pattabiraman D., et al. Recovering gene interactions from single-cell data using data diffusion. Cell. 2018;174:716–729.e27. doi: 10.1016/j.cell.2018.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Total RNA-seq and Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
Total RNA-seq and scRNA-seq data reported here are available from the NCBI Gene Expression Omnibus (GEO) under accessions GSE222377 and GSE230308, respectively.