

Summary
O-GlcNAc transferase (OGT) acts in the development of various cancers, but its role in clear cell renal cell carcinoma (ccRCC) remains unclear. In this study, we found that OGT was upregulated in ccRCC and this upregulation was associated with a worse survival. Moreover, OGT promoted the proliferation, clone formation, and invasion of VHL-mutated ccRCC cells. Mechanistically, OGT increased the protein level of hypoxia-inducible factor-2α (HIF-2α) (the main driver of the clear cell phenotype) by repressing ubiquitin‒proteasome system-mediated degradation. Interestingly, the OGT/HIF-2α axis conferred ccRCC a high sensitivity to ferroptosis. In conclusion, OGT promotes the progression of VHL-mutated ccRCC by inhibiting the degradation of HIF-2α, and agents that can modulate the OGT/HIF-2α axis may exert therapeutic effects on mutated VHL ccRCC.
Subject areas: Molecular biology experimental approach, Molecular network, In vitro toxicology, Functional aspects of cell biology, Cancer
Graphical abstract
Highlights
-
•
OGT plays oncogenic role mainly in VHL-mutated ccRCC
-
•
HIF-2a, but not HIF-1a, is the main target of OGT VHL-mutated ccRCC
-
•
OGT promotes protein stability of HIF-2a in O-GlcNac independent manner
-
•
OGT increases the sensitivity of VHL-mutated ccRCC to ferroptosis
Molecular biology experimental approach; Molecular network; In vitro toxicology; Functional aspects of cell biology; Cancer
Introduction
Renal cell carcinoma (RCC), the most lethal urological tumor originating from renal tubular epithelial cells, accounts for 2%–3% of all adult malignant tumors. Of patients with RCC, about 70%–80% are diagnosed with clear cell carcinoma (ccRCC).1,2
VHL, an E3 ubiquitin ligase, targets hydroxylated hypoxia-inducible factor (HIF) for ubiquitination and degradation. Appearing in up to 90% of ccRCC cases, VHL inactivation leads to the accumulation of HIF1/2α, which can activate the HIF signaling pathway and reprogram tumor cell metabolism to promote the development of ccRCC.3,4 As the main oncogene in ccRCC, HIF-2α drives cancer cell angiogenesis, proliferation, and invasion through a series of target genes, including vascular growth factor (VEGFA) and Cyclin D1 (CCND1).5,6 Interestingly, previous studies have confirmed that HIF-2α confers a high sensitivity to ferroptosis, a new iron-dependent programmed cell death. During ferroptosis, divalent iron or ester oxygenase catalyzes the oxidation of unsaturated fatty acids, which are highly expressed on the cell membrane, to induce lipid peroxidation and subsequent cell death. HIF-2α-induced sensitivity to ferroptosis underscores a prominent oncogene-induced vulnerability in ccRCC.7,8
O-linked-N-acetylglucosamine (O-GlcNAc) modification is dynamic, reversible, posttranslational, and ubiquitous on protein serine/threonine residues. This modification, often happening in the cytoplasm and nucleus, is involved in the regulation of multiple cellular pathways.9,10 Two enzymes serve as major regulators in O-GlcNAc modification: O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA).11 OGT catalyzes the addition of a single N-acetylglucosamine in the O-glycosidic linkage to serine or threonine residues, and this process is reversed by OGA.12 O-GlcNAc modification is disrupted to favor tumor metabolism, invasion and metastasis in tumor cells. OGT targets many tumor activators and suppressors, including p53, c-Myc, HIF-1α, and Snail.13,14,15 There activation, stability, translocation, and transcription are regulated by OGT-mediated O-GlcNAcylation.14 For instance, OGT downregulation enhances cisplatin-induced autophagy via SNAP-29, resulting in cisplatin-resistant ovarian cancer.16 In papillary thyroid cancer, OGT, as an upstream regulator of YAP, coordinates with classic Hippo pathway kinases, thus promoting malignant phenotypes.12 In colorectal cancer, O-GlcNAcylation of XIAP suppresses colon cancer cell growth and invasion by promoting the proteasomal degradation of OGT.17
Glycolysis is active in the hypoxia environment of ccRCC, which suggests O-GlcNAc might be correspondingly activated. However, few studies have investigated the role of OGT/O-GlcNAc in ccRCC, which is expected to be clarified in the present research.
Results
O-N-acetylglucosamine transferase is upregulated in clear cell renal cell carcinoma and associated with cancer progression
To determine the roles of OGT, OGA, and O-GlcNac modification in ccRCC, we investigated their expression in ccRCC and paired nontumor renal tissues by Western Blotting (n = 5). OGT and O-GlcNac levels were significantly upregulated in ccRCC, compared with those in nontumor renal tissues; however, OGA showed no consistent differences (Figure 1A). Consistent with protein expression, the mRNA expression of OGT, but not OGA, was significantly upregulated in ccRCC (Figure 1B). Subsequently, we further performed IHC analysis of 92 ccRCC tissue samples (Table 1). The expression of OGT, but neither OGA nor O-GlcNac, increased significantly in tissues with a higher grade or TNM stage (Figures 1B, 1C, and S1A–S1D). Similar results were also acquired in the external data derived from GSE53757 and GSE73731 in the GEO database (Figures S1E–S1G).18,19 Moreover, the overexpression of OGT, but neither OGA nor O-GlcNac, was also associated with a worse survival in our 92-case ccRCC trial (p = 0.0159) and 516 patients with ccRCC derived from the TCGA database (p = 0.026, Figures 1D–1G).
Figure 1.

OGT is upregulated in ccRCC and associated with cancer progression
(A and B) The protein (A) and mRNA (B) expression of OGT, OGA, and O-GlcNac in ccRCC and paired non-tumor renal tissues (n = 5).
(C) The expression of OGT in different grades of ccRCC (Grade 1: n = 36, Grade 2: n = 42, Grade 3: n = 10, Grade 4: n = 4) detected by IHC staining.
(D) The expression of OGT in different pathological stages of ccRCC (Stage Ⅰ: n = 72, Stage Ⅱ: n = 6, Stage Ⅲ: n = 14) detected by IHC staining.
(E) The association between OGT and overall survival of 512 cases patients with ccRCC in the TCGA database (Kaplan-Meier analysis).
(F–H) The association between OGT (F), OGA (G), O-GlcNac (H) and overall survival of our 92 cases of patients with ccRCC (Kaplan-Meier analysis). Scale bar, 400 μm. Data are represented as mean ± SD. Statistical analysis of the data from 2 groups was performed using Student’s t test. Comparisons among multiple groups were performed by one-way ANOVA followed by Fisher’s LSD test. (∗p < 0.05, ∗∗∗p < 0.001).
Table 1.
The association between OGT, OGA, O-GlcNac and clinical features in patients with ccRCC
| Characteristics | Number | OGT |
OGA |
O-GlcNac |
|||
|---|---|---|---|---|---|---|---|
| Mean ± SD | p value | Mean ± SD | p value | Mean ± SD | p value | ||
| Gender | |||||||
| Male | 60 | 37.53 ± 11.9 | 50.05 ± 12.8 | 66.28 ± 24.3 | |||
| Female | 32 | 37.36 ± 13.0 | 0.412 | 60.45 ± 12.7 | 0.621 | 66.48 ± 19.3 | 0.660 |
| Age | |||||||
| ≤60 | 57 | 38.23 ± 12.3 | 58.02 ± 13.4 | 66.38 ± 21.4 | |||
| >60 | 35 | 38.43 ± 12.5 | 0.939 | 62.02 ± 11.2 | 0.152 | 68.15 ± 24.8 | 0.718 |
| Grade | |||||||
| 1 | 36 | 32.18 ± 7.8 | 60.17 ± 13.5 | 60.51 ± 22.1 | |||
| 2 | 42 | 39.52 ± 13.0 | 57.41 ± 12.5 | 72.44 ± 21.3 | |||
| 3 | 10 | 48.69 ± 10.1 | 64.19 ± 10.3 | 67.01 ± 27.2 | |||
| 4 | 4 | 54.78 ± 8.3 | 0.000∗∗∗ | 65.96 ± 13.6 | 0.331 | 69.55 ± 21.7 | 0.142 |
| Stage | |||||||
| Ⅰ | 72 | 37.27 ± 12.0 | 59.53 ± 13.0 | 66.95 ± 21.1 | |||
| Ⅱ | 6 | 34.61 ± 9.9 | 50.97 ± 10.2 | 86.92 ± 23.6 | |||
| Ⅲ | 14 | 45.23 ± 13.3 | 0.134 | 63.70 ± 10.7 | 0.128 | 60.80 ± 20.95 | 0.063 |
| Lymphatic metastasis | |||||||
| N | 89 | 37.95 ± 12.3 | 59.44 ± 12.8 | 67.40 ± 22.4 | |||
| Y | 3 | 47.03 ± 5.3 | 0.125 | 64.54 ± 8.6 | 0.579 | 57.47 ± 32.3 | 0.427 |
One-way Anova test was applied. ∗∗∗p < 0.001.
O-N-acetylglucosamine transferase promotes the invasion and proliferation of Von Hippel-Lindau -mutated clear cell renal cell carcinoma cells
To investigate the role of OGT in cancer progression, we overexpressed and knocked out OGT in 786-O, 786-P (VHL-mutated cells) and Caki1 (VHL-wild-type cells) cells (Figures 2A, 2H, S2A, and S2E). We found that OGT overexpression significantly promoted the proliferation and clone formation of 786-O and 786-P cells (Figures 2A–2E). Meanwhile, OGT overexpression also promoted the invasion of 786-O and 786-P cells (Figures 2F and 2G). In contrast, OGT knockout significantly inhibited O-GlcNac modification, proliferation and clone formation in 786-O and 786-P cells (Figures 2H–2L). Moreover, OGT knockout also repressed the invasion in 786-O and 786-P cells (Figures 2M and 2N). Notably, both overexpression and knockout of OGT showed no effects on the proliferation, clone formation or invasion of Caki1 cells (Figures S2A–S2H).
Figure 2.

OGT promotes the invasion and proliferation of ccRCC cells
(A) Overexpression of OGT in 786-O and 769-P cells validated by Western Blotting analysis.
(B) The proliferative abilities of 786-NC (786-O negative control) and 786-OGT (OGT overexpressed 786-O) detected by CCK8 assay.
(C) The proliferative abilities of 769-NC (769-P negative control) and 769-OGT (OGT overexpressed 769-P) detected by CCK8 assay.
(D and E) The clone formation ability of ccRCC cells.
(F and G) The invasive ability of ccRCC cells detected by Transwell assay. Scale bar, 100 μm.
(H) Knockout of OGT in 786-O and 769-P cells validated by Western Blotting analysis.
(I and J) The proliferative abilities OGT knockout ccRCC cells.
(K and L) The clone formation ability of OGT knockout ccRCC cells.
(M and N) The invasive abilities of OGT knockout ccRCC cells detected by Transwell assay. Scale bar, 100 μm. Data are represented as mean ± SD. Statistical analysis of the data from 2 groups was performed using Student’s t test. Comparisons among multiple groups were performed by one-way ANOVA followed by Fisher’s LSD test. (∗∗∗p < 0.001).
O-N-acetylglucosamine transferase regulates the proteasomal degradation of hypoxia-inducible factor-2α through direct binding
Caki1 cells are wild-type VHL cells, while 786-O and 769-P cells have mutated VHL. VHL binds to hypoxia-inducible factor (HIF) and promotes its degradation by the ubiquitin‒proteasome system. Therefore, we further investigated the expression of HIF-1α and HIF-2α, finding that OGT overexpression significantly upregulated the expression of HIF-2α, but OGT knockout significantly inhibited its expression (Figures 3A and 3B). It should be noted that OGT knockout also inhibited the expression of HIF-1α in 769-P cells. However, HIF-1α was not expressed in 786-O wild-type cells, and remained so even under OGT overexpression (Figure S3A). Previous reports have confirmed that O-GlcNAcylation can regulate breast cancer cell metabolism by regulating HIF-1α and its downstream target GLUT1.15 Given that OGT also promoted ccRCC progression in HIF-1α-null 786-O cells, we further focused on the OGT/HIF-2α axis, finding that OGT did not affect the mRNA expression of HIF-2α, which suggests that OGT regulates HIF-2α at the protein level, but not at the transcriptional level (Figure S3B). Subsequently, we investigated the protein stability of HIF-2α by treating ccRCC cells with the protein synthesis inhibitor cycloheximide (CHX) for 0–2 h. We found that OGT overexpression enhanced the stability of HIF-2α, whereas OGT knockout decreased the stability of HIF-2α (Figures 3C, 3D, and S3C). Given the leading role of ubiquitin‒proteasome system (UPS) in the stability and degradation of HIF-2α, we further investigated the ubiquitination of HIF-2α in the presence or absence of the proteasome inhibitor MG132. As expected, OGT overexpression significantly inhibited the ubiquitination of HIF-2α in ccRCC in the presence or absence of MG132, whereas OGT knockout significantly boosted the ubiquitination of HIF-2α (Figures 3E and 3F). Thus, we confirmed that OGT raised the protein level of HIF-2α by reducing its UPS-mediated degradation. We further confirmed the colocalization of OGT and HIF-2α in the nuclei of 786-O cells (Figure 3G). Notably, we observed a strong interaction between OGT and HIF-2α, and overexpression (OE) of OGT or HIF-2α increased this interaction (Figures 3H and 3I). However, we did not detect the O-GlcNac modification of HIF-2α through Co-IP analysis in ccRCC cells (Figure 3J) or LC/MS in ccRCC specimens (Table S1). O-GlcNac modification was detected in Vimentin proteins, which was detected by LC/MS in ccRCC and set as a positive control here (Figure S3D).
Figure 3.

OGT regulates proteasomal degradation of HIF-2α by direct binding
(A) Overexpression of OGT increased the protein level of HIF-2α.
(B) Knockout of OGT repressed the protein level of HIF-2α.
(C) Overexpression of OGT increased the protein stability of HIF-2α. Protein synthesis inhibitor Cycloheximide (CHX, 20 μg/mL).
(D) Knockout of OGT decreased the protein stability of HIF-2α.
(E) Overexpression of OGT inhibited the ubiquitination of HIF-2α. Proteasome inhibitor MG132 (10 μM, 6h).
(F) Knockout of OGT enhanced the ubiquitination of HIF-2α in 786-O cells.
(G) Immunofluorescence staining of OGT and HIF-2α in 786-O cells. Scale bar, 100 μm.
(H–J) Interaction between OGT and HIF-2α validated by GST pull down (H) and Co-IP (I and J) assays. EV: empty vector; OE: Overexpression.
(K) Transcriptional files of OGT (786-OGT) and HIF-2α (786- HIF2α) overexpressed 786-O cells. DEGs: Differentially expressed genes. |Log2FC|>1.5, adjust p value<0.05. Data are represented as mean ± SD. Statistical analysis of the data from 2 groups was performed using Student’s t test. Comparisons among multiple groups were performed by one-way ANOVA followed by Fisher’s LSD test. (∗∗∗p < 0.001).
We next investigated the transcriptional files of OGT-overexpressing (786-OGT) and HIF-2α overexpressing (786-HIF2a) 786-O cells using RNA-seq. Interestingly, OGT and HIF-2α shared over 50% of differentially expressed genes (DEGs), compared to negative control (786-NC), which further confirmed the interaction between OGT and HIF-2α (Figure 3K). The target genes of HIF-2α are associated with cancer progression (VEGF and CCND1) and contribute to angiogenesis and cell cycle.20 The transcriptional levels of VEGF and CCND1 showed a significantly positive correlation with OGT in the TCGA database (R = 0.57 and 0.2; p value < 0.000, Figure S3E). Moreover, OGT overexpression elevated the expression of VEGF and CCND1 (Figure S3F). Additionally, we investigated the transcriptional activity of HIF-2α with a luciferase reporter assay. As expected, OGT overexpression promoted the transcriptional activity of HIF-2α (Figure S3G). Taken together, we suggested that OGT might upregulate the expression of HIF-2α by reducing UPS-mediated degradation through direct binding, thereafter promoting cancer progression through the transcriptional activation of a bunch of target genes (VEGF, CCND1, and so forth).
O-N-acetylglucosamine transferase/hypoxia-inducible factor-2α axis regulates the invasion and proliferation of Von Hippel-Lindau-mutated clear cell renal cell carcinoma cells
We performed rescue assays to confirm whether HIF-2α contributes to OGT-regulated ccRCC progression. As expected, in HIF-2α-knockout cells, OGT overexpression showed no effect on the proliferation (Figures 4A and 4B), invasion and clone formation of HIF-2α-knockout cells (Figures 4C and 4D). Moreover, HIF-2α overexpression re-enhanced the proliferation (Figures 4E and 4F), invasion and clone formation of OGT-knockout cells (Figures 4G and 4H).
Figure 4.

OGT/HIF-2α axis regulates the invasion and proliferation of ccRCC cells
(A) Knockout of HIF-2α validated by Western Blotting analysis.
(B–D) Overexpression of OGT showed no effect on the proliferation (B), invasion (C), and clone formation (D) of HIF-2α knockout 786-O cells. Scale bar, 100 μm.
(E) Overexpression of HIF-2α validated by Western Blotting analysis (EV: Empty vector; OE: Overexpression).
(F–H) Overexpression of HIF-2α rescued the repressed proliferation (F), invasion (G), and clone formation (H) of OGT knockout 786-O cells. Scale bar, 100 μm. Data are represented as mean ± SD. Statistical analysis of the data from 2 groups was performed using Student’s t test. Comparisons among multiple groups were performed by one-way ANOVA followed by Fisher’s LSD test. (∗p < 0.05, ∗∗∗p < 0.001, ns: no significance).
O-N-acetylglucosamine transferase/hypoxia-inducible factor-2α axis endows Von Hippel-Lindau-mutated clear cell renal cell carcinoma with a high sensitivity to ferroptosis
HIF-2α selectively enriches polyunsaturated lipids, a group of rate-limiting substrates for lipid peroxidation, thus conferring a sensitivity to ferroptosis.7 We investigated whether the OGT/HIF-2α axis strengthens the response to ferroptosis in ccRCC. By investigating transcriptional files of OGT-overexpressing (786-OGT) and control (786-NC) 786-O cells through ssGSEA analysis, we found ferroptosis score of 786-OGT was significantly higher than that of 786-NC (Figure S3H). OGT overexpression did not directly induce cell death, but sensitized cells to the lethal effect of Erastin, an inducer of ferroptosis. Moreover, in HIF-2α-deficient 786-O cells, this sensitizing effect was eliminated (Figure 5A). In contrast, OGT knockout increase resistance to Erastin in 786-O cells, while HIF-2α overexpression repressed this effect (Figures 5B and 5C). OGT overexpression significantly promoted lipid peroxidation induced by Erastin in 786-O cells, whereas in HIF-2α-deficient 786-O cells, this promotion was eliminated (Figures 5D and 5G). Conversely, OGT knockout inhibited lipid peroxidation induced by Erastin, while HIF-2α overexpression restored this effect in 786-O cells (Figures 5E and 5F). Taken together, these data confirmed that the OGT/HIF-2α axis confers a high sensitivity to ferroptosis on ccRCC cells.
Figure 5.

OGT/HIF-2α axis confers a high sensitivity to ferroptosis on ccRCC cells
(A) Overexpression of OGT strengthened the killing effect of ferroptosis inducer Erastin (24 h) on 786-O cells (sgNC: HIF-2α wild type cells), but not HIF-2α knockout 786-O cells. The response curve of ccRCC cells to Erastin was determined by CCK8 assay.
(B) Knockout of OGT enhanced resistance to Erastin, whereas overexpression of HIF-2α restored the sensitivity. The response curve of ccRCC cells to Erastin was determined by CCK8 assay.
(C) Representative images for Figure 5B. Erastin (5μM, 24h). Scale bar, 200 μm.
(D and G) Overexpression of OGT promoted Erastin-induced lipid peroxidation in 786-O (sgNC: HIF-2α wild type cells) cells, but not HIF-2α knockout 786-O cells.
(E and F) Knockout of OGT repressed Erastin-induced lipid peroxidation, whereas overexpression of HIF-2α restored it. Lipid peroxidation of ccRCC cells was detected by C11 BODIPY 581/591 probes. Data are represented as mean ± SD. Statistical analysis of the data from 2 groups was performed using Student’s t test. Comparisons among multiple groups were performed by one-way ANOVA followed by Fisher’s LSD test. (∗∗∗p < 0.001, ns: no significance).
Ferroptosis inducer sulfasalazine increases O-N-acetylglucosamine transferase-mediated sensitivity in Von Hippel-Lindau -mutated clear cell renal cell carcinoma
Finally, we examined the role of OGT and its therapeutic potential in vivo. As expected, OGT overexpression promoted the growth of 786-O xenograft tumors in mice. In mice treated with sulfasalazine (an in vivo ferroptosis inducer that blocks the activity of SLC7A11), the growth curve was completely reversed (Figures 6A–6C).21 Mice transplanted with OGT-overexpressing 786-O xenograft tumors benefited more from sulfasalazine (Figure 6D). As expected, OGT-overexpressing 786-O xenograft tumors exhibited a higher level of ferroptosis in tumors (increased staining of lipid peroxidation marker 4-hydroxy-2-noneal [4-HNE] and increased cell death areas shown by H&E staining) treated with sulfasalazine (Figure 6E).
Figure 6.

Ferroptosis inducer sulfasalazine increases OGT-mediated sensitivity in ccRCC
(A–D) Representative images (A), growth curve (B), tumor weight (C) of 786-O xenograft tumors with or without treating ferroptosis inducer sulfasalazine (200 mg/kg, per day, i.p.).
(D) Survival time of 786-O xenograft tumors treated with or without sulfasalazine. Survival time in another group of mice was different from that of mice included into A–C.
(E) IHC of 786-O xenograft tumors. Scale bar, 50 μm. 4-HNE: lipid peroxidation marker. Data are represented as mean ± SD. Statistical analysis of the data from 2 groups was performed using Student’s t test. Comparisons among multiple groups were performed by one-way ANOVA followed by Fisher’s LSD test. (∗∗p < 0.01, ∗∗∗p < 0.001).
Discussion
OGT is close implicated in the progression and metabolism of various cancers.22 However, few studies have determined the role of OGT in ccRCC. In this study, we found that OGT expression was significantly upregulated in ccRCC, compared with paired nontumor renal tissues. Moreover, this upregulation showed a close association with a worse survival, an advanced stage and a higher grade, suggesting that OGT contributes to the progression of VHL-mutated ccRCC.
We found that OGT promoted the proliferation, invasion, and clone formation of VHL-mutated ccRCC cells, suggesting that OGT is oncogenic for VHL-mutated ccRCC. As HIF-2α may be a driver of ccRCC, we investigated whether OGT regulates HIF-2α. As expected, OGT inhibited UPS-mediated degradation of HIF-2α to increase its expression and stability. The target genes for HIF-2α, including VEGF and CCND1, were also upregulated by OGT. VEGF-induced tumor angiogenesis promotes the proliferation and metastasis of ccRCC.5,23 CCND1 is a checkpoint in the G1/S phase of cell cycle, and leads to rampancy of ccRCC.24
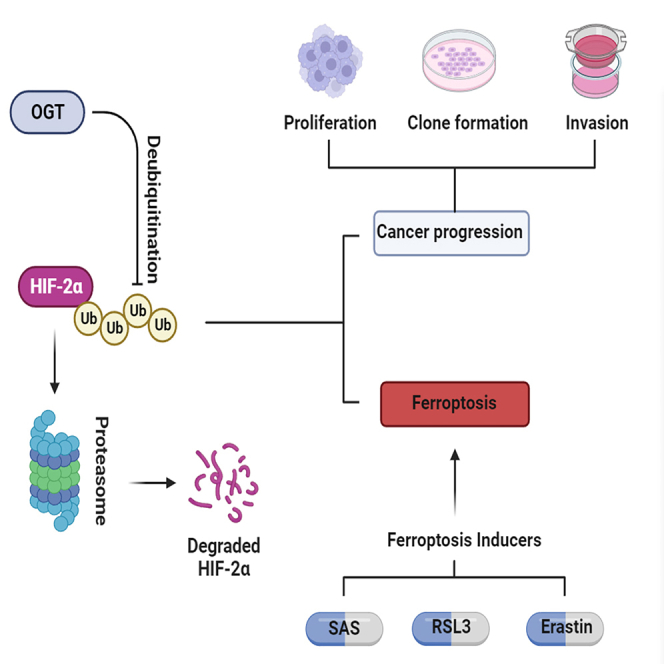
Recently, ferroptosis has garnered enormous interest in cancer research, partly because it is mechanistically and morphologically different from other forms of cell death and conceals great therapeutic potential for cancers.25,26 Previous research has confirmed that HIF-2α can confer sensitivity to ferroptosis by enriching polyunsaturated lipids in the cell membrane.7 We investigated whether the OGT/HIF-2α axis manipulates the response to ferroptosis in VHL-mutated ccRCC. Interestingly, OGT did not directly induce cell death, but sensitized cells to erastin-induced ferroptosis via HIF-2α. On the one hand, OGT increased the progression of ccRCC and rendered ccRCC uniquely vulnerable to ferroptosis. This interesting finding suggests that ferroptosis inducers could be promising therapeutic agents for OGT-overexpressing ccRCC. Finally, we further confirmed the vulnerability of OGT-overexpressing ccRCC to ferroptosis in vivo. Although OGT promoted the growth of ccRCC in vivo, and sulfasalazine, a ferroptosis inducer, successfully reversed this effect (Figure 7).
Figure 7.

Mechanism diagram for OGT-HIF-2α-ferroptosis axis in ccRCC
In conclusion, our research demonstrated that the overexpression of OGT in VHL-mutated ccRCC facilitated cancer progression. OGT promoted the proliferation, clone formation, and invasion of VHL-mutated ccRCC cells by inhibiting UPS-mediated HIF-2α degradation. The OGT/HIF-2α axis further conferred sensitivity to ferroptosis on ccRCC cells. Therefore, ferroptosis inducers may be a promising therapy for ccRCC.
Limitations of the study
In this study, we indicated that OGT increased the protein level of hypoxia-inducible factor-2α (HIF-2α) by repressing ubiquitin‒proteasome system-mediated degradation. Meanwhile, the OGT/HIF-2α axis conferred ccRCC sensitivity to ferroptosis. However, the role of O-GlcNac modification in ccRCC remains unclear. The association between O-GlcNac modification and ubiquitination was also poorly clarified.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| OGT Rabbit Polyclonal antibody | Cell Signaling Technology | Cat#24083 |
| OGA Rabbit Polyclonal antibody | Cell Signaling Technology | Cat#60406 |
| O-GlcNac Mouse Monoclonal antibody | Cell Signaling Technology | Cat#12938 |
| HIF-1α Rabbit Polyclonal antibody | Cell Signal Technology | Cat#36169 |
| HIF-2α Rabbit Polyclonal antibody | Cell Signal Technology | Cat#71565 |
| Actin Rabbit Polyclonal antibody | Cell Signal Technology | Cat#4970 |
| IgG Rabbit Polyclonal antibody | Proteintech | Cat#30000-0-AP |
| Ubiquitin mouse monoclonal antibody | Santa Cruz Biotechnology | Cat#sc-8017 |
| Chemicals, peptides, and recombinant proteins | ||
| DMSO | MedChemExpress | Cat#HY-100218A |
| Erastin | MedChemExpress | Cat#HY-15763 |
| sulfasalazine | MedChemExpress | Cat#HY-14655 |
| propidium iodide | Invitrogen | Cat#P3566 |
| C11 BODIPY 581/591 | Invitrogen | Cat#D3861 |
| TRIzol Reagent | Invitrogen | Cat#15596026 |
| Puromycin | Beyotime Biotechnology | Cat#ST551 |
| Protein A/G sepharose beads | Santa Cruz Biotechnology | Cat#sc-2003 |
| Critical commercial assays | ||
| CCK8 reagent | Dojindo Molecular Technologies | Cat#CK04 |
| PrimeScript™ RT Reagent Kit | Takara Bio | Cat#RR037A |
| SYBR Premix Ex Taq™ kit | Takara Bio | Cat#DRR081A |
| Dual-Glo Luciferase Assay system | Promega | Cat#E2920 |
| Experimental models: Cell lines | ||
| 786-O | Cell Technologies Resource Center in the University of Colorado Anschutz Medical Campus | RRID: SCR_021982 |
| 769-P | Cell Technologies Resource Center in the University of Colorado Anschutz Medical Campus | RRID: SCR_021982 |
| Caki-1 | Cell Technologies Resource Center in the University of Colorado Anschutz Medical Campus | RRID: SCR_021982 |
| Experimental models: Organisms/strains | ||
| BALB/c-nu mice | Beijing Vital River Laboratory Animal Technology | N/A |
| Oligonucleotides | ||
| sgNC: GCACTACCAGAGCTAACTCA | This paper | N/A |
| sgOGT#1: GCTTAAGTTTCAGAGCCGTG | This paper | N/A |
| sgOGT#2: TAGTTCGATAGCCCGCCTGT | This paper | N/A |
| sgHIF2a: AGATGGGAGCTCACACTGTG | This paper | N/A |
| Primer: HIF2a-F GAACGTCGAAAAGAAAAGTCTCG | This paper | N/A |
| Primer: HIF2a-R CCTTATCAAGATGCGAACTCACA | This paper | N/A |
| Primer: Actin-F GGGACCTGACTGACTACCTC | This paper | N/A |
| Primer: Actin-R TCATACTCCTGCTTGCTGAT | This paper | N/A |
| Recombinant DNA | ||
| CRISPR-V2 vector | Addgene Sanjana et al.27 |
Cat#52961 |
| pGL3-Basic vector | Addgene Chang et al.28 |
Cat#64784 |
| Software and algorithms | ||
| SPSS software v22.0 | IBM Corp | N/A |
| GraphPad Prism v8.0 | GraphPad Software | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Chao Qin (qinchao@njmu.edu.cn).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and study participant details
Patients and specimens
A total of 92 ccRCC and paired normal bowel tissues were collected from July 2008 to July 2010. Excluded were patients who had received adjuvant chemotherapy or radiotherapy prior to surgery or had additional cancers diagnoses. All patients were classified according to the 7th edition of the TNM staging system (23). Postoperative adjuvant therapies were performed, according to standard schedules and doses. All participants gave their written informed consent. This study was approved by the Ethics Committee of The Affiliated Hospital of Nanjing Medical University. The clinical data of all ccRCC patients are displayed in Table 1. No restrictions on sex were placed on the inclusion of patients. There were no restrictions on race or ethnicity, but all patients who were included were Chinese.
Animal studies
Six-week-old male Balb/c-nu mice were provided by Beijing Vital River Laboratory Animal Technology Co. Ltd. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University. Every 5×106 786-O cells suspended in 100 μl of PBS were injected subcutaneously through the axilla of each nude mice. After 7 d, 200 mg/kg sulfasalazine dissolved in 0.1M of NaOH was intraperitoneally injected. The long (L) and short (S) diameters of the tumor were measured every 3–4 days (tumor volume = L∗S2/2). In the growth curve analysis, all mice were euthanized with rapid cervical dislocation at five weeks after the injection of cells, then xenografts were removed and photographed. In the survival analysis, another group of mice was treated as described above. Once the diameter of any xenograft reached 20 mm, or the volume reached 2000 mm3, or the body weight lost by 20%, the mouse was considered dead and survival time was recorded.
Ethics approval and consent to participate
All procedures involving human participants were performed in accordance with the First Affiliated Hospital of Nanjing Medical University Ethics Committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. All patients provided written informed consent. The study protocol was approved by the Ethics Committee of The Affiliated Hospital of Nanjing Medical University.
Consent for publication
Written informed consent for publication was obtained from all the participants.
Method details
Immunohistochemical (IHC) staining
Paraffin-embedded sections were deparaffinized in xylene and hydrated with decreasing concentrations of ethanol (100, 90, 80, 75%) (3 min at each concentration) and microwave-heated in a sodium citrate buffer for antigen retrieval. Then, the sections were blocked in 5% BSA and incubated with anti-OGT, OGA rabbit polyclonal antibodies (1:200; Cell Signaling Technology, Inc., Danvers, MA, USA) and anti-O-GlcNac mouse monoclonal antibodies (1:100; Cell Signaling Technology, Inc., Danvers, MA, USA) at 4°C overnight, and then with horseradish peroxidase (HRP)-conjugated rabbit or mouse secondary antibodies (1:200; ProteinTech Group, Inc.) for 60 min at room temperature. Afterward, 3,3′-diaminobenzidine development (DAB Substrate Chromogen System; Dako, Denmark) and hematoxylin staining were performed. Finally, the sections were fixed, and images were caught with an inverted microscope (Olympus IX71, Japan).
An H-score was calculated using the following formula: H-score = ∑(PI×I)=(percentage of cells of weak intensity ×1) + (percentage of cells of moderate intensity ×2) + percentage of cells of strong intensity ×3).29 Here the H-score was recorded as a continuous variable.
Cell culture and reagents
Human ccRCC cell lines 786-O, 769-P and Caki1 were purchased from the University of Colorado Cancer Center Cell Bank, and cultured in RPMI-1640 medium supplemented with 10% FBS (Invitrogen, Carlsbad, CA, USA) at 37°C in a 5% CO2 atmosphere. Erastin (HY-15763) and Sulfasalazine (HY-14655) were purchased from MedChemExpress (Monmouth Junction, NJ, USA).
Measurement of cell viability, cell death and lipid peroxidation
Every 104 cells suspended in 100 μl of medium were seeded into 96-well plates overnight, and then treated with indicated concentrations of Erastin for 24 h. The cell viability was measured by injecting 10 μl of CCK8 reagent (Dojindo Molecular Technologies, Kumamoto, Japan) into each well. After incubating for two hours, the absorbance was measured at a wavelength of 450 nm (OD450).
For measurements of lipid peroxidation and cell death, every 106 cells suspended in 2 ml of medium were seeded into 6-well plates overnight, treated with indicated concentrations of Erastin for 24 h, and incubated with 2 μM C11 BODIPY 581/591 (Invitrogen, Carlsbad, CA, USA) for detection of lipid peroxidation, or 1 μg/mL propidium iodide (PI) (Invitrogen) in PBS for detection of cell death. Having been washed twice with PBS, the cells were collected and subjected to fluorescence activated cell sorting (FACS) analysis.
CRISPR/Cas9-mediated genome editing and Western Blotting analysis
For CRISPR/Cas9-mediated genome editing, single guide RNAs (sgOGT#1: GCTTAAGTTTCAGAGCCGTG; sgOGT#2: TAGTTCGATAGCCCGCCTGT; sgHIF2a: AGATGGGAGCTCACACTGTG) were designed using the http://crispr.mit.edu website, and cloned into the CRISPR-V2 vector (Addgene, #52961).30 Lentivirus package and infection were performed as described previously.23 Briefly, lipofectamine 3000 (Invitrogen, Inc.) was used to co-transfect the CRISPR-V2 vectors, psPAX2, PMG.2G into the HEK293T tool cells to obtain a lentivirus. Then, the lentivirus (multiplicity of infection, MOI = 10) was used to infect cells for 72 h. After screening of puromycin (2 μg/ml for 72h), a single cell was sorted into a well to acquire clone. Finally, the clones were validated by the Sanger sequence and Western Blotting analysis to acquire genome editing cells.
Western Blotting analysis was performed as described previously.23 Briefly, total cellular proteins from each group were extracted using RIPA lysis buffer (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS). Then, equal amounts (20 μg) of protein determined by BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA) were separated using 10% SDS-PAGE gels. The proteins were then transferred to PVDF membranes (0.45 mm, Millipore, Bedford, MA, USA). The membranes were blocked with 5% nonfat milk for 1 h at room temperature and then incubated with primary antibodies at 4°C for 12 h. The following antibodies were introduced: anti-OGT, anti-actin rabbit polyclonal antibodies, anti-O-GlcNac mouse monoclonal antibodies (1:1000, Cell Signaling Technology, Inc.) and anti-HIF-2α, HIF-1α rabbit polyclonal antibodies (1:1000, Cell Signaling Technology, Inc.). The secondary antibodies were anti-mouse or anti-rabbit antibody and conjugated to horseradish peroxidase (HRP) (1:4000, Proteintech Group. Inc). The secondary antibodies were used at a 1:4000 dilution and were incubated for approximately 1 h at room temperature. The bands were visualized with ECL reagents (Thermo Fisher Scientific) and developed by Omega Lum G (Aplegen, USA).
Co-immunoprecipitation (Co-IP) and GST pull down assays
For Co-IP assay, every 107 cells were harvested and lysed using the/a NP-40 buffer. Lysates were pre-cleared by 20 μL of Protein A/G sepharose beads (Santa Cruz Biotechnology, Dallas, TX, USA) and centrifuged for supernatant collection. The pre-cleared lysate was added with 2 μg of HIF-2α (Cell Signaling Technology, Inc.), OGT (Cell Signaling Technology, Inc.), or IgG rabbit polyclonal antibody (ProteinTech Group, Inc.), and incubated for 12 h at 4°C in rotating. Another 50 μl of Protein A/G sepharose beads was added into the lysate to capture the immunocomplex. After incubating for 4 h at 4°C, the beads were harvested by centrifugation at 3000g for 3 min, and washed for four times with NP-40 buffer. Protein was eluted by adding 2 x SDS loading buffer to the beads and boiling them for 5 min at 95°C. Subsequently, Western Blotting analysis was performed as described above using anti-ubiquitin mouse monoclonal antibodies (Santa Cruz Biotechnology).
For GST pull down assay, GST-OGT fusion protein was produced in BL21 cells and immobilized on glutathione sepharose beads. Then, 50 μg of purified GST or GST-OGT was added to 1 mL (1 mg protein) of total cell lysate of 786-O transfected with His-HIF-2α vector, and rotated for 1 h at 4°C. Subsequently, the beads were washed for five times with NP-40 buffer and eluted with 2 x SDS loading buffer. Western Blotting analysis was performed as described above.
Clone formation and cell proliferation assays
For clone formation assay, every 500 cells were seeded into 6-well plates and incubated at 37°C. Clone size was observed daily under a microscope, until the number of cells exceeded 50 in the majority of clones. Then, the medium was removed, and the cells were stained with 0.2% crystal violet for 30 min, washed thrice with PBS, and then photographed to count the clones.
For cell proliferation assay, every 3×103 cells suspended in 100 ul of RPMI-1640 medium were seeded into 96-well plates. The cell proliferation was assessed by the CCK8 (Dojindo Molecular Technologies, Japan). Then, 10 μl of CCK8 solution was injected into each well after different incubation periods: 0 h, 24 h, 48 h, and 72 h. Finally, we measured the absorbance at 450nm wavelength after two hours of incubation.
Cell invasion assay
Cell invasion was analyzed with Transwell plates (24-well insert, 8 μm pore size; BD Biosciences, Bedford, MA, USA). The filters (Corning Inc., NY, USA) were coated with 55 μL of Matrigel (1:8 dilution; BD Biosciences, Franklin Lakes, NJ, USA). Every 104 cells were suspended in 100 μl of RPMI-1640 medium without serum, and seeded into the upper chamber. Next, 600 μl of 90% RPMI-1640 supplement with 10% FBS was added into the bottom chamber. After incubation for 24 h, the chambers were fixed by 4% paraformaldehyde for 30 min, and then stained by 0.1% crystal violet for 30 min. Finally, a magnification microscope was used to count the cells having invaded into the bottom of the chamber.
Luciferase reporter assay
The hypoxia response elements (HREs) in the promoter of VEGF were constructed into luciferase reporter vector pGL3-Basic, followed by the Dual-Glo luciferase assay system (Promega Corp., Madison, WI, USA). After a 36-h transfection with reporter vector, the cells were lysed by passive lysis buffer. Firefly, Luciferase (F-luc) and Renilla Luciferase (R-luc) of lysis were detected, respectively. Transcription activity was represented as F-luc/R-luc, and translation efficiency was further normalized by the mRNA level of F-luc.
Detection of O-GlcNac modification in ccRCC
Three ccRCC specimens were lysed by RIPA lysis buffer. Then, the lysates were incubated overnight with O-GlcNAc [GlcNAc-S/T] Immunoaffinity Beads (Cell Signaling Technology, Inc. #95220) at 4°C. Immunoprecipitated complexes were eluted and dissolved in 1 mL of chilled methanol, acetonitrile and H2O mixture (2:2:1, v/v/v), and sonicated at a low temperature (30 min). The supernatant was dried in a vacuum centrifuge. For LC-MS analysis, the samples were re-dissolved in 100 μL of acetonitrile and water mixture (1:1, v/v), and subjectd to the Easy nLC 1200 system (Thermo Scientific) coupled to a timsTOF (Bruker) for DDA-PASEF analysis. Detection mode: positive ion; mother ion scanning range: 100-1700m/z; Intensity Threshold: 5000.00; Ion mobility: 0.6–1.6 Vs/cm2; Ramp Time: 100.0 ms; Accumulation Time: 2.0 ms. The dynamic elimination time for the mother ion to reach the target value of 20000 counts was 0.4 min. Quadrupole isolation width: 2 m/z for m/z < 700 to 3 m/z for m/z > 700. Each TIMS cycle time was 1.1 sec, including 1 Full MS and 10 PASEF MS/MS. The analysis of O-GlcNac-modified peptides in ccRCC is supplemented in Table S1.
RNA extraction, real-tie quantitative PCR (qPCR), and RNA-sequencing
Total RNA was extracted by Trizol Regent (Invitrogen) from CRC cells. cDNA was obtained from total RNA with PrimeScript™ RT reagent kit (Takara Bio, Inc., Otsu, Japan). The mRNA expression was assessed by qPCR carried out in triplicate by a SYBR Premix Ex Taq™ kit (Takara Bio) and ABI 7900HT Real-Time PCR system (Applied Biosystems Life Technologies, Foster City, CA, USA).
For RNA-seq, 1 μg of RNA per sample was used as input material for RNA preparation. Sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA), and index codes were added to attribute sequences to each sample. The index-coded samples were clustered on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia). After clustering, the library preparations were sequenced on an Illumina Novaseq6000 platform, and 150 bp paired-end reads were generated. Raw data (raw reads) of fastq format were first processed through in-house perl scripts. In this step, clean data (clean reads) were obtained by removing reads containing adapters, ploy-N or low-quality reads. STAR was used to align clean reads with reference genome. HTSeq v0.6.0 was used to count the reads mapped to each gene. Then, FPKM of each gene was calculated based on the length of gene and read count. DESeq2 algorithm was applied to filter the differentially expressed genes according to (i) |log2FC| >1.5 and (ii) P-value <0.05. RNA-seq profiles have been deposited in GEO database (GSE230386).
Analysis of data in the TCGA and GEO databases
TCGA-KIRC (https://cancergenome.nih.gov/) contained the basic information of 530 ccRCC cases, involving survival time, age, sex, race, history, type of diagnosis, tumor grade stage. The Kaplan-Meier analysis of ccRCC patients was performed using GEPIA website (http://gepia.cancer-pku.cn/). GSE53757 dataset provided transcriptional files of 72 cases of ccRCC samples and paired non-tumor samples. GSE73731 dataset presented transcriptional files of ccRCC samples from 265 cases. All transcriptional files accompanied by clinical data were downloaded by R-software for subsequent analysis. Then transcriptional files were normalized through R software “Limma” package.
Quantification and statistical analysis
All experiments were performed for at least three times. All the experimental data were processed by the SPSS software (version 19.0, IBM Corp., Armonk, NY, USA). GraphPad Prism (version 7, GraphPad Software, La Jolla, CA, USA) was used to determine the statistical results. All data were expressed as mean + standard deviation (mean + sd). Between-group comparison was performed using a t-test. The comparison among multiple groups was performed by one-way ANOVA and then an LSD-t test. p < 0.05 was considered statistically significant. ns: no significance, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Acknowledgments
Not applicable. Funding: This study was supported by the National Natural Science Foundation of China (Grant Numbers: 81972386, 81672531, 82071638 and 82002989).
Author contributions
ZY and XW contributed to the experiments. CJ and WS contributed to the statistical analysis of the data. RX was responsible for bioinformatics analysis. YW, Jingwan Zhou, Zheng Zhao, Pengcheng Zhou, and Kejie Zhao contributed to the specimen collection. CQ, NS and BY contributed to the design of the study. All authors read and approved the final version of the article.
Declaration of interests
The authors declare no competing interests.
Published: October 5, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108148.
Contributor Information
Bing Yao, Email: byao@njmu.edu.cn.
Ninghong Song, Email: songninghong@126.com.
Chao Qin, Email: qinchao@njmu.edu.cn.
Supplemental information
Data and code availability
-
•
This paper does not report any original code.
-
•
The data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
References
- 1.Hakimi A.A., Furberg H., Zabor E.C., Jacobsen A., Schultz N., Ciriello G., Mikklineni N., Fiegoli B., Kim P.H., Voss M.H., et al. An epidemiologic and genomic investigation into the obesity paradox in renal cell carcinoma. J. Natl. Cancer Inst. 2013;105:1862–1870. doi: 10.1093/jnci/djt310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capitanio U., Bensalah K., Bex A., Boorjian S.A., Bray F., Coleman J., Gore J.L., Sun M., Wood C., Russo P. Epidemiology of Renal Cell Carcinoma. Eur. Urol. 2019;75:74–84. doi: 10.1016/j.eururo.2018.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim H., Shim B.Y., Lee S.J., Lee J.Y., Lee H.J., Kim I.H. Loss of Von Hippel-Lindau (VHL) Tumor Suppressor Gene Function: VHL-HIF Pathway and Advances in Treatments for Metastatic Renal Cell Carcinoma (RCC) Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22189795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoefflin R., Harlander S., Schäfer S., Metzger P., Kuo F., Schönenberger D., Adlesic M., Peighambari A., Seidel P., Chen C.Y., et al. HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 2020;11:4111. doi: 10.1038/s41467-020-17873-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choueiri T.K., Kaelin W.G., Jr. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat. Med. 2020;26:1519–1530. doi: 10.1038/s41591-020-1093-z. [DOI] [PubMed] [Google Scholar]
- 6.Zhang C., Samanta D., Lu H., Bullen J.W., Zhang H., Chen I., He X., Semenza G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA. 2016;113:E2047–E2056. doi: 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou Y., Palte M.J., Deik A.A., Li H., Eaton J.K., Wang W., Tseng Y.Y., Deasy R., Kost-Alimova M., Dančík V., et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019;10:1617. doi: 10.1038/s41467-019-09277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singhal R., Mitta S.R., Das N.K., Kerk S.A., Sajjakulnukit P., Solanki S., Andren A., Kumar R., Olive K.P., Banerjee R., et al. HIF-2α activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J. Clin. Invest. 2021;131 doi: 10.1172/jci143691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slawson C., Hart G.W. O-GlcNAc signalling: implications for cancer cell biology. Nat. Rev. Cancer. 2011;11:678–684. doi: 10.1038/nrc3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatham J.C., Zhang J., Wende A.R. Role of O-Linked N-Acetylglucosamine Protein Modification in Cellular (Patho)Physiology. Physiol. Rev. 2021;101:427–493. doi: 10.1152/physrev.00043.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stephen H.M., Adams T.M., Wells L. Regulating the Regulators: Mechanisms of Substrate Selection of the O-GlcNAc Cycling Enzymes OGT and OGA. Glycobiology. 2021;31:724–733. doi: 10.1093/glycob/cwab005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X., Wu Z., He J., Jin Y., Chu C., Cao Y., Gu F., Wang H., Hou C., Liu X., Zou Q. OGT regulated O-GlcNAcylation promotes papillary thyroid cancer malignancy via activating YAP. Oncogene. 2021;40:4859–4871. doi: 10.1038/s41388-021-01901-7. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y.Y., Liu H.Y., Yu T.J., Lu Q., Zhang F.L., Liu G.Y., Shao Z.M., Li D.Q. O-GlcNAcylation of MORC2 at threonine 556 by OGT couples TGF-β signaling to breast cancer progression. Cell Death Differ. 2022;29:861–873. doi: 10.1038/s41418-021-00901-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sodi V.L., Khaku S., Krutilina R., Schwab L.P., Vocadlo D.J., Seagroves T.N., Reginato M.J. mTOR/MYC Axis Regulates O-GlcNAc Transferase Expression and O-GlcNAcylation in Breast Cancer. Mol. Cancer Res. 2015;13:923–933. doi: 10.1158/1541-7786.mcr-14-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrer C.M., Lynch T.P., Sodi V.L., Falcone J.N., Schwab L.P., Peacock D.L., Vocadlo D.J., Seagroves T.N., Reginato M.J. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol. Cell. 2014;54:820–831. doi: 10.1016/j.molcel.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou F., Yang X., Zhao H., Liu Y., Feng Y., An R., Lv X., Li J., Chen B. Down-regulation of OGT promotes cisplatin resistance by inducing autophagy in ovarian cancer. Theranostics. 2018;8:5200–5212. doi: 10.7150/thno.27806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seo H.G., Kim H.B., Yoon J.Y., Kweon T.H., Park Y.S., Kang J., Jung J., Son S., Yi E.C., Lee T.H., et al. Mutual regulation between OGT and XIAP to control colon cancer cell growth and invasion. Cell Death Dis. 2020;11:815. doi: 10.1038/s41419-020-02999-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei X., Choudhury Y., Lim W.K., Anema J., Kahnoski R.J., Lane B., Ludlow J., Takahashi M., Kanayama H.O., Belldegrun A., et al. Recognizing the Continuous Nature of Expression Heterogeneity and Clinical Outcomes in Clear Cell Renal Cell Carcinoma. Sci. Rep. 2017;7:7342. doi: 10.1038/s41598-017-07191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Roemeling C.A., Radisky D.C., Marlow L.A., Cooper S.J., Grebe S.K., Anastasiadis P.Z., Tun H.W., Copland J.A. Neuronal pentraxin 2 supports clear cell renal cell carcinoma by activating the AMPA-selective glutamate receptor-4. Cancer Res. 2014;74:4796–4810. doi: 10.1158/0008-5472.can-14-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palazon A., Tyrakis P.A., Macias D., Veliça P., Rundqvist H., Fitzpatrick S., Vojnovic N., Phan A.T., Loman N., Hedenfalk I., et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell. 2017;32:669–683.e5. doi: 10.1016/j.ccell.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao C., Liu X., Zhang Y., Lei G., Yan Y., Lee H., Koppula P., Wu S., Zhuang L., Fang B., et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–590. doi: 10.1038/s41586-021-03539-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan W., Cao M., Ruan X., Jiang L., Lee S., Lemanek A., Ghassemian M., Pizzo D.P., Wan Y., Qiao Y., et al. Cancer-cell-secreted miR-122 suppresses O-GlcNAcylation to promote skeletal muscle proteolysis. Nat. Cell Biol. 2022;24:793–804. doi: 10.1038/s41556-022-00893-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Z., Wang T., Wu D., Min Z., Tan J., Yu B. RNA N6-methyladenosine reader IGF2BP3 regulates cell cycle and angiogenesis in colon cancer. J. Exp. Clin. Cancer Res. 2020;39:203. doi: 10.1186/s13046-020-01714-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schödel J., Bardella C., Sciesielski L.K., Brown J.M., Pugh C.W., Buckle V., Tomlinson I.P., Ratcliffe P.J., Mole D.R. Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression. Nat. Genet. 2012;44:420–425. doi: 10.1038/ng.2204. s421-s422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lei G., Zhuang L., Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer. 2022;22:381–396. doi: 10.1038/s41568-022-00459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X., Comish P.B., Tang D., Kang R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.637162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang T.C., Wentzel E.A., Kent O.A., Ramachandran K., Mullendore M., Lee K.H., Feldmann G., Yamakuchi M., Ferlito M., Lowenstein C.J., et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azim H.A., Jr., Peccatori F.A., Brohée S., Branstetter D., Loi S., Viale G., Piccart M., Dougall W.C., Pruneri G., Sotiriou C. RANK-ligand (RANKL) expression in young breast cancer patients and during pregnancy. Breast Cancer Res. 2015;17:24. doi: 10.1186/s13058-015-0538-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper does not report any original code.
-
•
The data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.