

Abstract
The lack of direct targets for TATA-binding protein (TBP)-like factors (TLFs) confounds the understanding of their role in gene expression. Here we report that human TLF (also called TBP-related factor 2 [TRF2]) activates a number of different genes, including the neurofibromatosis type 1 (NF1) gene. The overexpression of TLF increases the amount of NF1 mRNA in cells. In vivo, TLF binds to and upregulates transcription from a fragment of the NF1 promoter. In vitro, purified TLF-TFIIA binds directly to the same NF1 promoter fragment that is required for TLF responsiveness in cells. Furthermore, targeted deletion of TLF in mice reduces NF1 levels. In contrast, TLF inhibits transcription driven by a fragment from the TATA-containing c-fos promoter by sequestering TFIIA. TBP affects the NF1 and c-fos promoters in a manner reciprocal to that of TLF, stimulating the c-fos promoter and inhibiting NF1 transcription. We conclude that TLF is a functional regulator of transcription with targets distinct from those of TBP.
TATA-binding protein (TBP) is a highly conserved and essential component in RNA polymerase I-, II-, and III-mediated transcription in eukaryotes (14, 35). TBP binding is the rate-limiting step in transcription from TATA-containing but not TATA-less, class II promoters (5, 7, 22). Although TBP was originally thought to be unique, homologs of TBP were recently identified. The first TBP-related factor (TRF1) is highly homologous to TBP and is found only in Drosophila (8). It activates transcription from some of the same RNA polymerase II promoters that are activated by TBP (13, 15) but is also involved in transcription mediated by RNA polymerase III in vitro (39). Various investigators identified a more distantly related member of the TBP family, TBP-like factor (TLF; also called TLP, TRF, TRF2, TRP, and STUD [GenBank accession number AF130312]) (2, 9, 18, 27, 29, 34, 40, 41). TLF exists in many species, including Drosophila, Caenorhabditis elegans, Xenopus, zebra fish, chick, mouse, and human (see above and reference 27). A third protein, TRF3/TBP2, which is highly related to TBP, was recently identified (1, 33).
The role of TLF in vertebrates is not well understood. Like TBP and dTRF1, TLF interacts with members of the basal transcription machinery (28, 34, 40), but disagreement persists about the function of TLF, particularly with respect to its promoter interactions. Initial reports proposed that TLF binds to the TATA element and substitutes for TBP in transcription from TATA-containing promoters (23). Subsequent findings suggested that TLF does not interact with the TATA box and in fact inhibits transcription from TATA-containing promoters in vitro and in vivo (26, 30, 34, 40). These observations motivated the suggestion that TLF acts as a “false face” (26, 34, 40). Recently, it was shown that TLF can stimulate transcription from transiently transfected reporter gene constructs containing a TATA-less promoter from either the mouse or the human terminal deoxynucleotidyl transferase genes (31).
The function of TLF during development is also incompletely understood. Experiments with C. elegans, zebra fish, and Xenopus indicated that proper embryonic development requires functional TBP and TLF, although the two proteins affect different subsets of genes (9, 18, 27, 41). Despite the high degree of amino acid conservation between Xenopus and mouse TLFs (>90% identity), TLF-deficient mice develop normally, exhibiting a deficit only in spermiogenesis, as far as has been determined (25, 43).
Here we show that human TLF and TBP can affect gene transcription in reciprocal and opposite fashions. We identify a number of human genes whose expression is upregulated in response to TLF overexpression in vivo, including the neurofibromatosis type 1 (NF1) gene. NF1 is a tumor suppressor, and many of the symptoms of neurofibromatosis, which include café-au-lait spots, benign peripheral neurofibromas, and malignant plexiform neurofibromas, may result from haploinsufficiency at the NF1 locus (6). We show that TLF binds to and increases transcription from a fragment of the NF1 promoter in vivo. In addition, we show that NF1 transcript levels are decreased in TLF knockout mice. Furthermore, affinity-purified TLF-TFIIA binds to an NF1 promoter fragment in vitro. The same sequence that is recognized by TLF-TFIIA in vitro is also sufficient for mediating TLF responsiveness of the NF1 promoter in vivo. In contrast, TBP does not bind to the NF1 promoter in cultured cells, and transcription from an NF1 promoter fragment is inhibited by TBP overexpression. Moreover, TBP stimulates transcription from the c-fos promoter, while TLF inhibits c-fos transcription by sequestering TFIIA. Thus, TBP and TLF regulate the NF1 and c-fos promoters in a reciprocal manner.
MATERIALS AND METHODS
Transfections.
Transfections of HEK-293 and COS-7 cells were performed by using either 100-mm dishes or six-well plates with matched controls. HEK-293 cells were transfected with Lipofectamine 2000 (Invitrogen) 24 h after being split 1:4. Cells were harvested 40 h after transfection. Nonfusion protein constructs of TLF and TBP were expressed in pTracerCMV-2 (Invitrogen). The TLF construct included the 5′ untranslated region (UTR) of the TLF gene to increase expression levels. Transfection efficiency was monitored on the basis of green fluorescent protein (GFP) production. 3T3 cells were transfected by using a Nucleofector device (Amaxa) in accordance with the manufacturer's protocol.
Subcellular localization.
Cells on glass coverslips were cotransfected with the entire coding sequence for human TLF and human TBP subcloned in frame into pEGFP-C2 (Clontech). pRFP-N1 was used for cotransfection (Clontech). Images were obtained with a Zeiss LSM 410 inverted confocal microscope. To verify that TLF was in the nucleus, a number of cells were stained with Hoescht 33342 (Molecular Probes).
Antibodies.
The polyclonal anti-TLF antibody was made against a peptide representing amino acids 178 to 186 (Zymed) and was used at a 1:200 dilution for Western blotting. The monoclonal anti-TBP antibody (Santa Cruz) was also diluted 1:200. The anti-TFIIB antibody (Promega) was used at a dilution of 1:500 for Western blotting. The anti-GFP polyclonal antibody (Molecular Probes) was diluted 1:1,000 for Western blotting. Four micrograms was used for each plasmid immunoprecipitation (PIP) sample. The polyclonal anti-TFIIA antibody (Santa Cruz) was used at a 1:200 dilution.
Western blotting.
Tissue from adult male mice was frozen on dry ice and lysed with 1% Triton X-100. The DNA was subsequently sheared by passage through a 27-gauge needle. Samples were normalized with a bicinchoninic acid (BCA) protein assay (Pierce) and subsequently electrophoresed on Nu-PAGE 4 to 12% polyacrylamide gels (Invitrogen).
Gene chip analysis.
Two 150-mm dishes of HEK-293 cells transfected with either pTracer-CMV2 or pTLF-Tracer-CMV2 were harvested with 1.5 ml of Tri-Reagent (Sigma). Isolated RNA was resuspended in nuclease-free water, quantified spectrophotometrically, and stored at −80°C. Approximately 24 μg of RNA was mixed with a d(T)24-T7 (GenSet) primer and reverse transcribed with SuperScript II reverse transcriptase (Invitrogen). Second-strand cDNA synthesis was performed, and the resulting double-stranded cDNA was subsequently purified. One-half of the purified, double-stranded cDNA served as the template for a MegaScript (Ambion) reaction which incorporated biotin-11-CTP and biotin-16-UTP into the cRNA (Sigma). The cRNA was purified by using an RNeasy kit (Qiagen). The purified cRNA was fragmented in accordance with the Affymetrix protocol, and Affymetrix Hu35K gene chips were screened.
PIP.
Cells were transfected for PIP with a plasmid containing a GFP, TBP-GFP fusion, or TLF-GFP fusion construct and pNF1-CAT. At 40 h after transfection, cells were harvested, cross-linked with 1% formaldehyde for 10 min at room temperature, rinsed with PBS, collected by centrifugation, and suspended in PBS containing 1% NP-40. Vigorous vortexing and sequential passages through 18- and 22-gauge needles disrupted the cells. Multiple passages through a 27-gauge needle sheared the DNA. Cells were diluted with 0.1% NP-40, and the debris was pelleted. Lysates were incubated with protein G-protein A-agarose beads that had been precoated with an anti-GFP polyclonal antibody for 2 h at room temperature. Beads were collected by centrifugation and washed with PBS containing 0.1% NP-40 and 0.05% sodium deoxycholate. The DNA was released by treatment with proteinase K followed by phenol-chloroform extraction and ethanol precipitation. The DNA then was used as a template for PCR with primers designed on the basis of the NF1 promoter (ATCGGAGGTCGTGTACCTTAT and GCGATCCTCCTGGAGGTGACGT). M13 forward and reverse primers were used to amplify sequences from pfosCAT. PCR products were resolved on agarose gels.
Primer extension assays.
The reverse primer (AAAAGCGATCCTCCTGGAG; positions 328 to 346 of the pNF1-CAT construct) was labeled with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP and used for primer extension analyses. A 100-μg sample of total RNA was used for the determination of endogenous start sites. When cells were transfected with the pNF1-CAT construct, 15 μg of total RNA was used. After the labeled primer was mixed with the RNA samples, the samples were heated to 58°C for 20 min and then slowly cooled to 42°C, and SuperScript II reverse transcriptase was added. Reaction mixtures were incubated for 50 min at 42°C, diluted with formamide loading buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol FF), and electrophoresed on 6% acrylamide-Tris-borate-EDTA (TBE)-urea sequencing gels. The start sites were verified by primer extension with two additional primers (CGGCCTCCGGGTTTGGATTGC and AGCATCCACTCCCATCCC).
Chloramphenicol acetyltransferase (CAT) assays.
HEK-293 cells were transfected with 6 μg of expression plasmid, 1 μg of reporter plasmid, and 1 μg of pAdvantage (Promega) in 100-mm plates. The NF1 reporter construct (pNF1-CAT) consisted of a 350-bp fragment of the published promoter sequence containing the putative start sites subcloned into pCAT3-Basic (see Fig. 6). Constructs used for deletion analysis were generated by PCR and were also subcloned into pCAT3-Basic. The c-fos reporter construct contained positions −75 to 109 of the 5′-flanking region (3). TLFN37Y-GFP was made by site-directed mutagenesis by using a QuikChange system (Stratagene) to change asparagine 37 of TLF-GFP to a tyrosine. The coding sequences for TFIIA α, β, and γ were obtained by PCR and subcloned into pBudCE4.1 (Invitrogen). For transfection with excess TFIIA, 1 μg of reporter, 1 μg of pAdvantage, 3 μg of TFIIA-Bud, and 3 μg of TLF-GFP or TBP-GFP were used.
FIG. 6.

Nucleotides 1 to 103 of pNF1-CAT respond to TLF binding. (A) Sequence of the NF1 promoter fragment from pNF1-CAT. This sequence differs slightly from the published sequence of the NF1 promoter (12) but is identical to genomic sequences in the public database that correspond to the NF1 locus. Base number 1 corresponds to the most 5′ base of the sequence. The responsive region is shaded in gray (nucleotides 1 to 103). Putative transcriptional start sites determined by primer extension analyses are underlined. Oligonucleotides 1 and 3, which contain potential TLF-binding sites, are indicated. (B) Effects of TLF on each reporter construct, quantified by fluorometry and compared to the results for pNF1-CAT transfected with eGFP. Error bars indicate the standard error of the mean (n = 6). (C) EMSAs showing that TLF binds directly to the 1-103 promoter fragment. The first lane shows the height of the radiolabeled probe in the absence of proteins. The second lane shows that the addition of approximately 10 ng of TLF-TFIIA caused retardation of some of the probe. This retardation was reduced by a 10-fold excess of unlabeled oligonucleotide 1 (third lane) and was eliminated by a 10-fold excess of unlabeled oligonucleotide 3 (fifth lane), suggesting that these oligonucleotides contained TLF-binding sites. Oligonucleotides 2 and 4 did not affect the shift (fourth and sixth lanes). (D) EMSAs showing that TLF binds directly to oligonucleotide 3. The first lane shows the radiolabeled probe in the absence of proteins. The addition of ∼10 ng of TLF-TFIIA retarded probe migration (second lane).
At 40 h after transfection, cells were rinsed with PBS, scraped into 500 μl of 50 mM Tris-HCl (pH 7.5), and lysed by three freeze-thaw cycles. Samples were heated to 65°C for 10 min to inactivate endogenous acetylases. Insoluble material was subsequently pelleted, and the protein concentration of the soluble portion was determined with a BCA protein assay. CAT activity was measured by using a FAST-CAT deoxy-green kit in accordance with the manufacturer's instructions (Molecular Probes). CAT activity was quantified by fluorometry with a Storm system (Molecular Dynamics).
RNase protection assays with transfected cells.
RNase protection assays were performed by using a HybSpeed RPA kit (Ambion) in accordance with the manufacturer's instructions. Briefly, nonradioactive probes were synthesized to correspond to a portion of the 3′ UTR of the NF1 message and control plasmid pTri-Actin (Ambion) by using T7 RNA polymerase in the presence of biotin-14-CTP and biotin-16-UTP. The purified probes were coprecipitated with 50 μg of RNA isolated by using Tri-Reagent from HEK-293 cells transiently transfected with either pTracer-CMV2 or TLF. Control tubes contained probes mixed with yeast RNA. The reaction mixtures were resuspended in 10 μl of HybSpeed buffer, incubated at 95°C with vigorous vortexing for 2 min, and incubated at 65°C for 10 min. After digestion with RNase A/T1, the reaction mixtures were precipitated and electrophoresed on 6% acrylamide-TBE-urea gels. After electrophoresis, the products were transferred to positively charged nylon membranes by electroblotting. Signals were detected by using a BrightStar Biodetect kit (Ambion). Images were captured on X-ray film.
RNase protection assays with TLF−/− mice.
RNase protection assays were performed by using an RPA III kit (Ambion) in accordance with the manufacturer's instructions. Briefly, radioactive probes were synthesized to correspond to a portion of the 3′ UTR of the NF1 message and control plasmid pTri-Actin by using T7 RNA polymerase in the presence of [α-33P]UTP. The purified probes were coprecipitated with 20 μg of total RNA from the brains of either TLF+/+ or TLF−/− juvenile male mice. TLF−/− mice were created in the strain SV129 background and backcrossed to the strain C57BL/6 background as previously described (43). Control tubes contained probes mixed with yeast RNA. The reaction mixtures were resuspended in 10 μl of hybridization buffer, incubated at 95°C for 4 min, and cooled to 42°C. Hybridization proceeded overnight. After digestion with RNase A or T1, the reaction mixtures were precipitated and electrophoresed on 6% acrylamide-TBE-urea gels. The gels were dried, and signals were detected by using a Storm system PhosphorImager (Molecular Dynamics).
Gel shift assays.
Digestion of a construct consisting of a 350-bp fragment of the published NF1 promoter (see Fig. 6) subcloned into pCRII-TOPO (Invitrogen) (pNF1-TOPO) with EcoRI yielded a 350-bp fragment that was purified from a 1% agarose gel. The 103 5′-most nucleotides of the NF1 promoter present in pNF1-TOPO (the promoter 1-103 promoter fragment) was amplified by PCR and subcloned into pCRII-TOPO. The 1-103 promoter fragment was excised by using EcoRI and isolated on a 1% agarose gel. To generate radiolabeled probes for electrophoretic mobility shift assays (EMSAs), we added [α-33P]dATP to the NF1 fragments in the presence of the Klenow fragment of Escherichia coli DNA polymerase I. Approximately 5 pmol of oligonucleotide 3 (TAAGCTGAGAGCACAGCCTCCCCA) was incubated with 20 U of T4 polynucleotide kinase in the presence of 50 pmol of [γ-33P]ATP. Following inactivation of the kinase, a complementary oligonucleotide was added to the reaction mixture; heating to 99°C and slowly cooling to room temperature annealed the two oligonucleotides. Ethanol precipitation removed unincorporated nucleotides. Binding reactions proceeded for 45 to 60 min at 30°C with approximately 10 ng of purified TLF and equimolar amounts of TFIIA in a buffer containing 20% glycerol, 60 mM KCl, 3 mM dithiothreitol, and 20 mM HEPES (pH 7.6). The addition of 20 μg of bovine serum albumin prevented the purified protein from adhering to the test tube. To block nonspecific protein-DNA interactions, 200 ng of poly(dG-dC) was included in the binding reaction mixtures. Reaction mixtures were resolved on 4% acrylamide gels (29:1) made with 0.5× TBE. Running the gels at 100 V and room temperature prevented excess heating of the samples. A 10-fold molar excess of unlabeled competitors was used in competition experiments. Approximately 200 ng of recombinant human TFIIA (Protein One) was used for control gel shift assays in the absence of TLF.
RESULTS
Tissue and subcellular distributions of TLF.
We identified human TLF by screening the human expressed sequence tag database with a sequence corresponding to the C-terminal half of TBP. As reported previously (29, 34, 40), Northern blot analysis revealed that TLF mRNA is expressed at moderately low levels ubiquitously and at high levels in the testes. The high levels of expression in the testes were reported to be from a testis-specific TLF transcript (38, 40). To determine the protein distribution, we made a polyclonal antipeptide antibody against the C-terminal region of TLF. This antibody recognizes both bacterially expressed TLF and epitope-tagged TLF in mammalian cells. In protein extracts from mouse tissues, the antibody recognized a 21-kDa band; this mass corresponds to the predicted molecular mass of TLF. The addition of excess peptide blocked this interaction (data not shown).
When multiple mouse and human tissues were probed on Western blots, a significant disparity between RNA and protein levels was apparent. TLF protein was expressed at the highest levels in the mouse liver and pancreas (Fig. 1A), and strong reactivity in the heart was seen at a slightly higher molecular mass. Longer exposures revealed a band in brain tissue corresponding to 21 kDa. This distribution differs from that reported by Perletti et al. (32), who observed a very high level of TLF protein in brain tissue. The differences in these results may be attributed to different solubilization conditions and normalization procedures.
FIG. 1.

TLF and TBP have different tissue and subnuclear localizations. (A) Western blot showing the presence of TLF in heart (He), kidneys (Ki), liver (Li), lungs (Lu), pancreas (Pa), and testes (Te) from an adult mouse. A longer exposure showed that TLF was also present in the brain (Br). (B) Western blots showing distinct amounts of TLF and TBP relative to TFIIB in various cell lines. Equal amounts of proteins from HEK-293, HeLa, and SK-N-SH cells were loaded. Parallel blots were stained for TFIIB, TLF, or TBP. HEK-293 cells have a lower TLF/TBP ratio than either SK-N-SH or HeLa cells. The amount of protein in each lane was normalized by a BCA protein assay and verified by Ponceau S staining. (C) eGFP does not localize to the nucleus. The upper cell, including the nucleus, is completely filled with eGFP (stained with Hoechst dye). The lower cell was untransfected, allowing visualization of the nucleus. (D) TLF-GFP localizes to the nucleus. HEK-293 cells were cotransfected with RFP and TLF-GFP. RFP fills the cell, while TLF-GFP is restricted to the nucleus. (E through H) TBP-GFP and TLF-GFP have different subnuclear localizations. Arrows mark the positions of the nucleoli. (E and F) Transmission differential interference contrast (DIC) and fluorescence images of COS-7 cells transfected with a TBP-GFP fusion construct. (G and H) Transmission DIC and fluorescence images of COS-7 cells transfected with a TLF-GFP fusion construct. (I and J) High-resolution images showing that TBP-GFP is excluded from, while TLF-GFP is concentrated in, the nucleoli. (Inset) Similar nucleolar exclusion in HEK-293 cells. Scale bars, 10 μm.
The distinctive TLF mRNA and protein distribution patterns indicate a high degree of posttranscriptional regulation. Posttranscriptional regulation, particularly obvious in the testes, is a feature of several basal transcription machinery proteins (10, 32, 36, 37). A comparison of our Western blot results with previously published data on the distribution of TBP indicates that the ratios of TBP to TLF vary from tissue to tissue. Western blot analysis of various human cell lines confirmed that the amounts of TLF and TBP varied with respect to one another, while the amounts of TFIIB were relatively constant (Fig. 1B).
Since our antibody does not recognize TLF in its native form, we used eGFP fusion constructs to determine the localizations of TLF and TBP in living, nonpermeabilized cells. TLF-GFP (Fig. 1D, G, H, and J), eGFP (Fig. 1C), GFP-TBP (Fig. 1E, F, and I), and red fluorescent protein (RFP) (Fig. 1D) were introduced into HEK-293 cells (Fig. 1C and D) and COS-7 cells (Fig. 1E to J). While GFP and RFP did not localize to any particular subcellular organelle (Fig. 1C and D), the TLF-GFP fusion localized to the nucleus in greater than 99% of the cells (Fig. 1D). Strikingly, TLF-GFP associated strongly with nucleoli (Fig. 1D, G, H, and J) in HEK-293 cells and even more noticeably in COS-7 cells. Similar results have been obtained with HeLa and COS-1 cells and an anti-TLF antibody (19). In contrast, 3T3 cells did not sequester TLF-GFP in nucleoli. In fact, diffuse TLF-GFP fluorescence could be seen throughout transfected cells (unpublished data [see http://clapham.tch.harvard.edu/publications.html]). As observed with FLAG-tagged TLF (19), TLF-GFP was expressed very poorly in 3T3 cells. These observations, together with the finding that native TLF is undetectable in 3T3 cells, suggest that TLF is subject to significant posttranscriptional regulation.
Like native TBP (17), TBP-GFP is extranucleolar in HEK-293, COS-7, and 3T3 cells. However, mild detergent treatment dramatically altered its localization. After 10 min of incubation with 0.2% Triton X-100 (reported to allow TBP visualization in the nucleolus) (17), TBP-GFP filled the entire nucleus, including the nucleolus (data not shown). This treatment did not change the distribution of TLF-GFP. TLF-GFP persistently localized to the nucleolus even during cytokinesis. Nucleoli of TLF-GFP-transfected cells rapidly recovered from photobleaching, indicating that TLF-GFP readily accesses this compartment (data not shown). Interestingly, recent reports indicated that the nucleolar localization of TLF is dependent on the presence of RNA and active polymerase I (19). The differential localizations of TLF and TBP in the nucleus indicate that they may fulfill different functions and highlight the possibility that TLF may regulate the transcription of rRNA. Since the subcellular localization of TLF depends on the cell type, TLF functions may differ between tissues.
Identification of TLF targets.
To determine whether TLF regulated a unique subset of genes within cells, we screened Affymetrix Hu35K gene chips representing approximately 35,000 different human expressed sequence tags. We probed the chips with biotinylated cRNA made from HEK-293 cells that transiently overexpressed both GFP and TLF or GFP alone. The data from the gene chip screening were normalized to overall intensity (unpublished data [http://clapham.tch.harvard.edu/publications.html]). Since TLF can indirectly inhibit transcription from viral TATA-containing promoters by sequestering essential factors (26, 40), we focused on genes that were upregulated. TLF overexpression increased the abundance of 125 (∼0.35%) of the transcripts represented on the chips by more than threefold. This value is likely to be a significant underestimate of TLF-regulated genes because the criteria used to determine upregulation were stringent. In addition, the concentration of TLF may not be the rate-limiting step in transcription from promoters that contain silencer- or repressor-binding sites.
We verified the upregulation of a number of targets, including NF1 (Fig. 2A), jumonji (data not shown), and ACF7 (data not shown), by RNase protection assays. We chose the NF1 promoter as a model to test the activity of TLF because it had been previously characterized (11, 12) and because its regulation has clinical significance (6).
FIG. 2.

TLF upregulates NF1 transcripts by using endogenous start sites. (A) RNase protection assay comparing levels of NF1 transcripts in HEK-293 cells expressing GFP and those expressing TLF. The probe generated against NF1 recognizes a 306-bp fragment of the 3′ UTR. A β-actin probe served as a control to ensure equivalent precipitation of the samples. (B) Primer extension analysis showing that TLF upregulates NF1 transcription from endogenous start sites. RNA from cells transfected with TLF-GFP or GFP alone was used for primer extension analysis with an antisense primer corresponding to bases 328 to 346 of the NF1 promoter. The start sites of the NF1 transcripts (arrows) were identical in samples transfected with TLF-GFP and those transfected with GFP, indicating that TLF regulates NF1 in a physiologically relevant manner. (C) Primer extension analysis showing that pNF1-CAT uses the same start sites as the endogenous NF1 transcripts. When cells were transfected with pNF1-CAT, 15 μg of RNA was used per reaction; 100 μg was used for cells transfected with GFP alone.
Primer extension assays showed that TLF stimulates transcription from the NF1 gene without altering its start sites (Fig. 2B). Unlike alternate TBPs in Drosophila that upregulate their targets from a subset of transcriptional start sites (13, 15), TLF displayed no obvious preference for start sites (its overexpression increased the expression of all transcripts to similar degrees). Comparison with a sequencing ladder indicated multiple transcriptional start sites (see Fig. 6A), a finding which is not uncommon for promoters that lack a TATA box (42). Interestingly, the start sites that we observed in HEK-293 cells differed from those reported for the brain (12), suggesting that tissue-specific factors lead to differential utilization of the NF1 promoter or promoters.
We subcloned a 350-bp fragment of the NF1 promoter into pCAT3-Basic and sought to determine whether the resulting reporter construct (termed pNF1-CAT) effectively mimicked the endogenous gene. Primer extension assays showed that this reporter gene construct utilized the same transcriptional start sites as the endogenous NF1 gene (Fig. 2C). In addition, TLF stimulated transcription from this promoter in a manner analogous to that of the endogenous gene, suggesting that pNF1-CAT is an appropriate model of promoter function.
We subsequently used PIP assays to test whether TLF upregulated NF1 transcription by binding to the NF1 basal promoter (Fig. 3A). This technique is a modified form of the chromatin immunoprecipitation technique that allows more specific determination of the TLF-NF1-binding site (21). HEK-293 cells were cotransfected with TLF-GFP, TBP-GFP, or parental enhanced GFP (eGFP) and pNF1-CAT. Treatment with 1% formaldehyde cross-linked protein to plasmid DNA; GFP-containing complexes were isolated by immunoprecipitation. From these immunoprecipitates, we were able to amplify the NF1 sequence when cells were transfected with TLF-GFP but not from samples containing either eGFP or TBP-GFP (Fig. 3C). We confirmed the expression of eGFP, TBP-GFP, and TLF-GFP by Western blotting with an anti-GFP antibody (Fig. 3B). pNF1-CAT was present in all inputs to the assay (Fig. 3C). We conclude that TLF-GFP but not TBP-GFP interacts with this fragment of the NF1 promoter. In contrast, the TATA-containing c-fos promoter coimmunoprecipitated with TBP-GFP but not TLF-GFP (Fig. 3D).
FIG. 3.

PIP showing that TLF binds directly to the pNF1-CAT reporter construct. (A) Diagrammatic representation of the PIP procedure. pNF1-CAT and GFP, TLF-GFP, or TBP-GFP were cotransfected into HEK-293 cells. After 40 h, treatment with 1% formaldehyde cross-linked the proteins to DNA. Subsequent immunoprecipitation with an anti-GFP antibody and proteinase K digestion allowed the identification of DNA regions that associate with fusion proteins. (B) The expression of GFP-containing TBP and TLF was verified by Western blot analysis with an anti-GFP antibody. (C) NF1 immunoprecipitates with TLF-GFP but not TBP-GFP or GFP. Cells transfected with the NF1 promoter plasmid and GFP, TLF-GFP, or TBP-GFP were subjected to PIP. A 350-bp band was detected by agarose gel electrophoresis (PIP products). The presence of the NF1 promoter plasmid in the cultures was verified by performing PCR with aliquots of the lysates removed just prior to mixing with agarose beads and purified by proteinase K digestion and phenol-chloroform extraction (PIP inputs). (D) The c-fos promoter associated with TBP-GFP but not TLF-GFP. Cells transfected with the c-fos promoter plasmid and TLF-GFP or TBP-GFP were subjected to PIP. A 2,100-bp band was detected by agarose gel electrophoresis (IP). The presence of the c-fos promoter plasmid in the cultures was verified by performing PCR with aliquots of the lysates (Input).
Reciprocal regulation of the NF1 and c-fos promoters by TBP and TLF.
Hypothesizing that TLF binding limits the rate of transcription from the NF1 promoter, we transfected pNF1-CAT into HEK-293 cells with GFP, TLF-GFP, or TBP-GFP. Compared to the results obtained with GFP alone, cotransfection with TLF-GFP increased the amount of CAT reporter activity from pNF1-CAT in a dose-dependent manner. At the highest concentration of DNA, TLF-GFP caused a fourfold increase in CAT activity compared to that obtained with GFP alone (n = 6) (Fig. 4A).
FIG. 4.
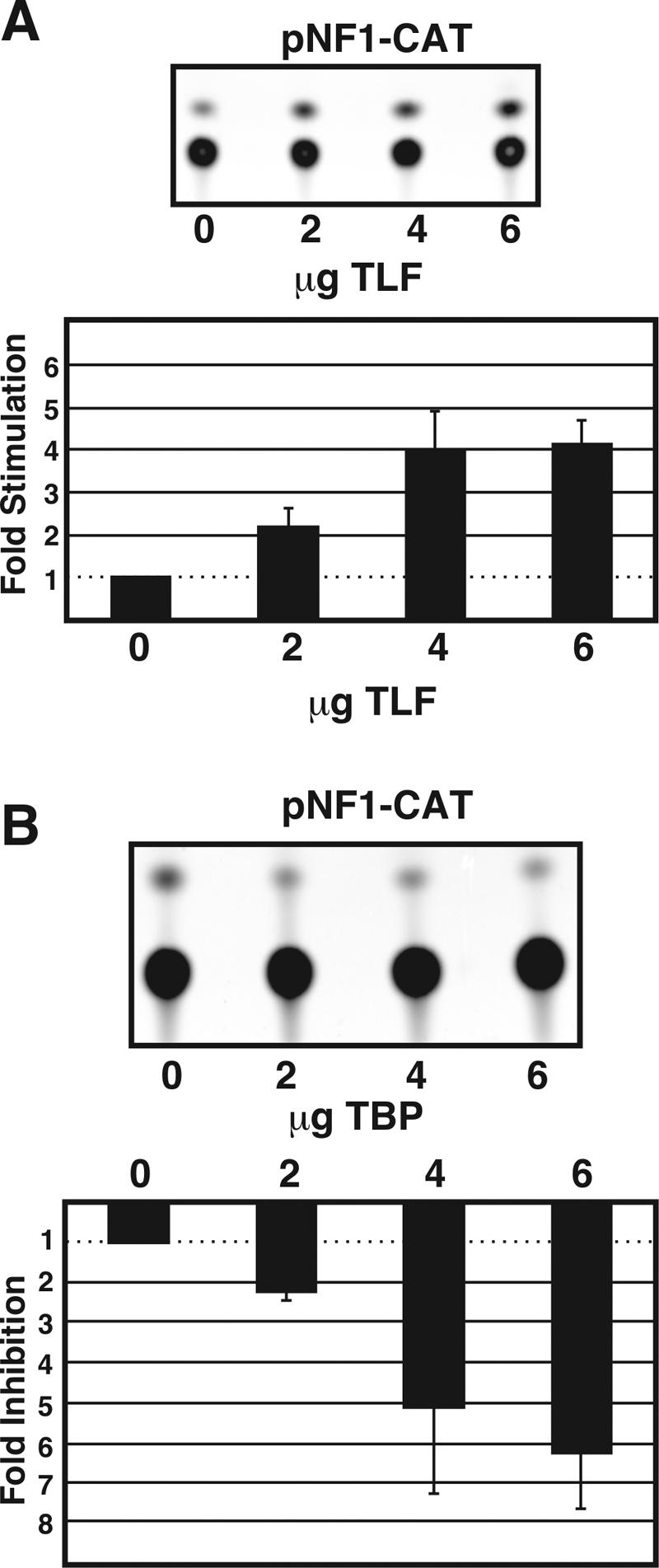
TLF but not TBP increased transcription from the NF1 promoter. (A) CAT assays showing that TLF stimulates transcription from an NF1 promoter fragment in a dose-dependent manner. (Top) Thin-layer chromatography (TLC) plate showing an increase in pNF1-CAT activity, measured by the acetylation of the FAST-CAT deoxy-green substrate, in response to TLF overexpression. (Bottom) Quantification of the effects of TLF on pNF1-CAT activity. The effects of TLF on pNF1-CAT were quantified by fluorometry and were compared to those for cells cotransfected with pNF1-CAT and eGFP. Error bars indicate the standard error of the mean (n = 6). Total DNA transfected in each condition was equalized to 6 μg by the addition of eGFP-C3. (B) TBP decreases pNF1-CAT activity in a dose-dependent manner. (Top) TLC plate showing a decrease in pNF1-CAT activity, measured by the acetylation of the FAST-CAT deoxy-green substrate, in response to TBP overexpression. (Bottom) Quantification of the effects of TBP on pNF1-CAT activity. The effects of TBP on pNF1-CAT were quantified by fluorometry and were compared to those for cells cotransfected with pNF1-CAT and eGFP. Error bars indicate the standard error of the mean (n = 6). Total DNA transfected in each condition was equalized to 6 μg by the addition of eGFP-C3.
To determine whether TLF-mediated stimulation of the NF1 promoter was specific, we introduced TBP-GFP into cells with the pNF1-CAT construct. Surprisingly, TBP-GFP overexpression decreased transcription from the NF1 promoter in a dose-dependent manner, with the highest plasmid concentration leading to more than fourfold repression (n = 6) (Fig. 4B). Since even low levels of TBP produced an inhibitory effect, it is unlikely that the observed inhibition was a result of squelching. Nonfusion protein versions of TLF and TBP yielded identical results. TBP-GFP-mediated stimulation of both the SV40 and the human c-fos promoters confirmed that this construct was functional (data not shown) (see Fig. 7). To assess the in vivo significance of TLF regulation of NF1 transcription, we compared the levels of NF1 transcripts in the brains of TLF−/− mice (43) to those in their wild-type littermates. RNase protection assays revealed that TLF−/− mice expressed 30% less NF1 transcripts than wild-type control mice (n = 3) (Fig. 5).
FIG. 7.
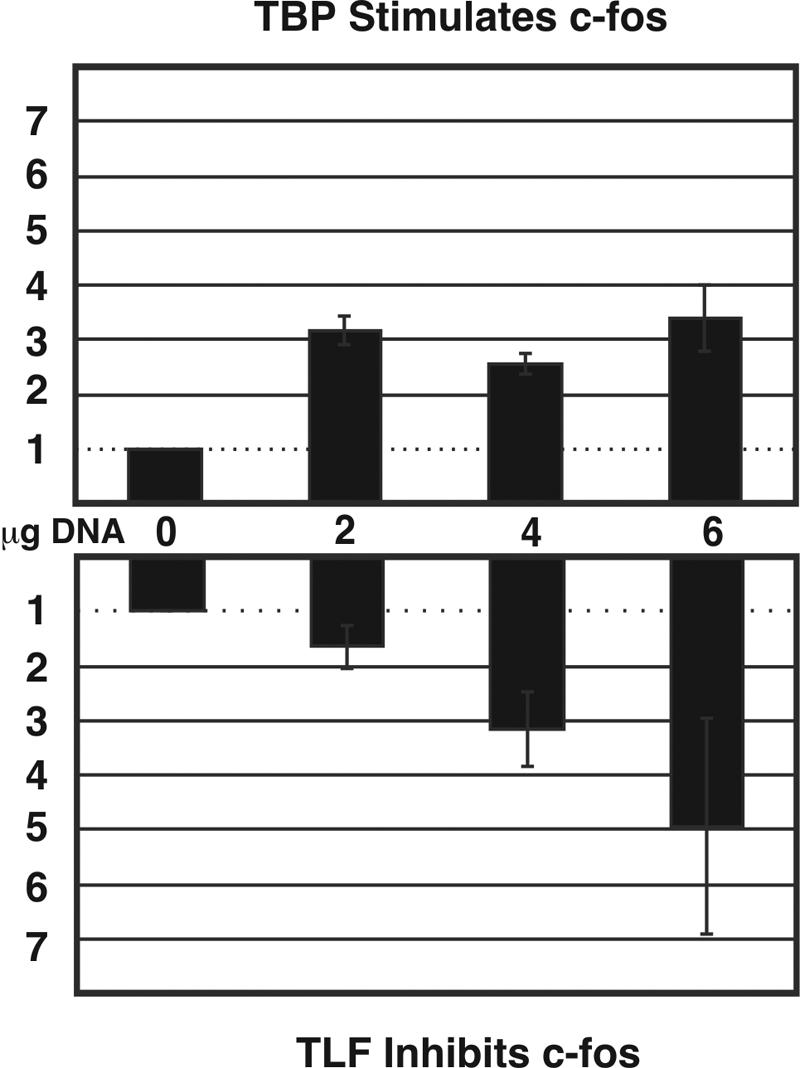
TLF and TBP have reciprocal effects on a fragment of the c-fos promoter. (Upper panel) Quantification of the effects of TBP on c-fos CAT activity. The effects of TBP on c-fos CAT activity were quantified by fluorometry and compared to those for cells cotransfected with the c-fos CAT construct and eGFP. Error bars indicate the standard error of the mean (n = 6). Total DNA transfected in each condition was equalized to 6 μg by the addition of eGFP-C3. (Lower panel) Quantification of the effects of TLF on c-fos CAT activity. The effects of TLF on c-fos CAT activity were quantified by fluorometry and compared to those for cells cotransfected with the c-fos CAT construct and eGFP. Error bars indicate the standard error of the mean (n = 6).
FIG. 5.
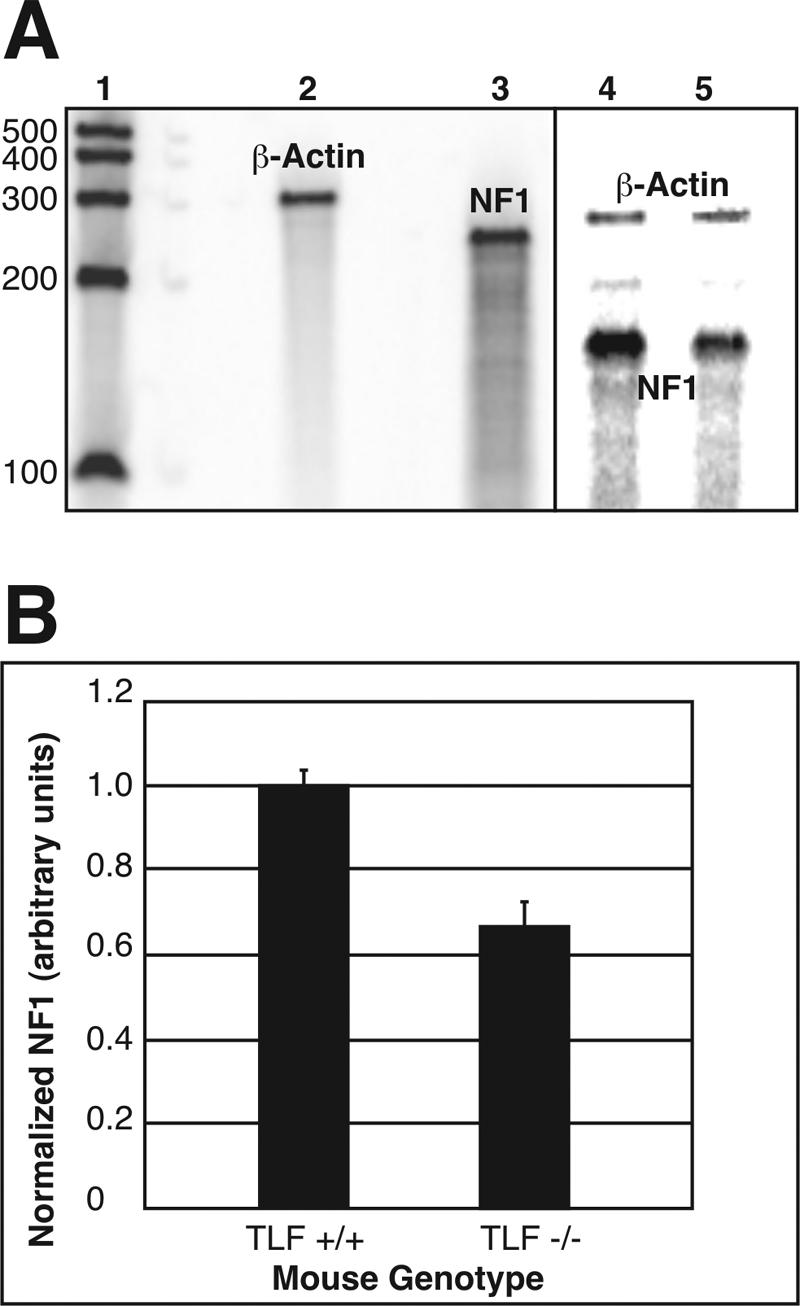
Endogenous NF1 transcripts were downregulated in the absence of TLF. (A) RNase protection assay. Lane 1 contains RNA Century markers (Ambion) labeled with [33P]UTP. Lanes 2 and 3 contain undigested probes for β-actin and NF1, respectively. β-Actin was chosen as an internal control for the amount of RNA loaded in each lane. Lanes 4 and 5 contain RNA from either a TLF+/+ mouse brain or a TLF−/− mouse brain hybridized with probes for both β-actin and NF1. (B) The amount of NF1 RNA in each lane was quantified by phosphorimaging and normalized to the amount of β-actin RNA in each lane. The amount of RNA in TLF+/+ mice was normalized to 1; error bars indicate the standard error of the mean (n = 3) (P < 0.05).
In order to better define the sites within the NF1 promoter that were required for the TLF-mediated upregulation of NF1 transcription, we performed NF1 promoter deletion analysis. A fragment containing bases 1 to 103 of pNF1-CAT was necessary and sufficient for TLF-mediated activation of NF1 transcription (Fig. 6B). This finding agrees well with our finding that TLF stimulates transcription from a group of start sites contained within this fragment (Fig. 6A).
To determine whether TLF also binds to the NF1 promoter in vitro, we performed EMSAs. For that purpose, we used a TLF protein preparation immunopurified from a HeLa cell line that stably expresses FLAG-tagged TLF (unpublished data [see http://clapham.tch.harvard.edu/publications.html]) (40). As previously reported (40), TLF copurifies with approximately stoichiometric quantities of the general transcription factor TFIIA. In the EMSAs, TLF specifically bound to a radiolabeled probe corresponding to the 350-bp promoter fragment from pNF1-CAT. Consistent with our deletion analysis, a 10-fold molar excess of unlabeled 1-103 promoter fragment abolished this interaction. In contrast, a 10-fold excess of the promoter fragment spanning nucleotides 103 to 350 did not affect retardation on the gel (data not shown).
To further delineate the TLF-binding site, we created four double-stranded oligonucleotides that covered positions 1 to 103 of the NF1 promoter. Incubation with an excess of oligonucleotide 3 but not oligonucleotide 2 or 4 abolished the TLF-TFIIA-mediated shift of the probe for positions 1 to 103 (Fig. 6C). Unlabeled oligonucleotide 1 also significantly reduced but did not eliminate this shift. The results of these binding experiments suggest the existence of multiple binding sites for TLF within the 1-103 promoter fragment. The presence of multiple binding sites for TLF is consistent with the large number of start sites that we observed. TLF-TFIIA also effectively retarded the migration of radiolabeled oligonucleotide 3 (Fig. 6D). Recombinant TFIIA in the absence of TLF had no effect on the mobility of this oligonucleotide. These findings suggest that the 24-bp sequence of oligonucleotide 3 is sufficient for TLF-TFIIA binding. In addition, TLF interacts with the same portions of the NF1 promoter in vivo and in vitro.
TLF expression and TBP expression affect the TATA-containing c-fos promoter in a reciprocal manner. As expected (5, 7, 26), the overexpression of TBP-GFP more than doubled CAT activity from the basal c-fos promoter. In contrast, transfection with TLF-GFP decreased the rate of transcription from the c-fos promoter in a dose-dependent fashion (Fig. 7), consistent with a repressive function for TLF in transcription from a TATA-containing promoter (26, 31, 40).
The reports cited above also showed that in vitro TLF-mediated inhibition could be overcome by the addition of excess TFIIA. To determine whether competition for TFIIA also inhibited TBP-regulated promoters in vivo, we mutated a single amino acid of the proposed TFIIA-binding site in TLF (TLFN37Y) (Fig. 8A). We chose to mutate this site based on its sequence similarity to the TFIIA-binding site in TBP (4). TLFN37Y-GFP and TLF-GFP had similar expression levels (Fig. 8C), and both localized to the nucleolus (Fig. 8B). To determine whether TLFN37Y was deficient in binding to TFIIA, we performed coimmunoprecipitation experiments with an anti-GFP antibody. As expected, TFIIA coimmunoprecipitated with TLF-GFP but not TLFN37Y-GFP. Interestingly, TLF-GFP stably associated with p55, the uncleaved precursor of TFIIA α and β, as well as the cleaved α subunit (Fig. 8D and E). TLFN37Ywas much less effective than TLF in inhibiting the c-fos promoter (Fig. 8F) and failed to stimulate the NF1 promoter (data not shown). Additionally, the overexpression of TFIIA alleviated the TLF-mediated inhibition of c-fos transcription (Fig. 8F). The simplest interpretation of these results is that TLF sequesters TFIIA to inhibit TBP-mediated transcription.
FIG. 8.

TLF inhibits c-fos reporter gene activity by sequestering TFIIA. (A) Asparagine 37 of TLF-GFP was replaced with tyrosine in the construct TLFN37Y-GFP to disrupt the TFIIA-binding site of TLF. (B) TLF-GFP and TLFN37Y-GFP localizations in COS-7 cells were similar. Scale bars, 10 μm. (C) Western blot analysis showing that TLF-GFP and TLFN37Y-GFP are expressed at similar levels. BCA protein assays were used to ensure equal amounts of total protein were loaded in the lanes. (D and E) Western blots of TLF-GFP and TLFN37Y-GFP immunoprecipitated with an anti-GFP antibody from transfected HEK-293 cells. Staining with the anti-GFP antibody indicates that TLF-GFP and TLFN37Y-GFP were immunoprecipitated with similar efficiencies (D). Analysis with an anti-TFIIA antibody shows that TFIIA did not coimmunoprecipitate withTLFN37Y-GFP, indicating that the mutation disrupted TFIIA-TLF interactions (E). Interestingly, the uncleaved p55 product (TFIIA α and β) was also efficiently immunoprecipitated with TLF. (F) Repression of the c-fos reporter by TLF-GFP, TLFN37Y-GFP, GFP plus TFIIA, and TLF-GFP plus TFIIA. The addition of excess TFIIA or mutation of the TFIIA-binding site of TLF alleviated the TLF-mediated inhibition of c-fos reporter gene activity. Error bars indicate the standard error of the mean (n = 6, 5, 3, and 5, respectively, from left to right). (G) Model for TLF function. TLF and TBP bind to the NF1 and c-fos promoters, respectively. In the model, TLF competes with TBP for TFIIA, thus inhibiting TBP-responsive promoters. TBP competes with TLF for an undetermined general transcription factor (GTF) to inhibit TLF-responsive promoters.
Mutating the TFIIA-binding site in TBP dramatically decreased transcription from the c-fos promoter and decreased cell viability. In contrast to TFIIA’s abolishment of TLF-mediated repression, overexpression of TFIIA did not relieve TBP-mediated repression of pNF1-CAT. This result may suggest that TBP competes with TLF for a factor other than TFIIA.
Our results suggest that TLF is not simply a false face but is a functional activator of transcription both in vitro and in vivo. However, TLF can inhibit transcription from promoters driven by TBP, and TBP can inhibit transcription from TLF-driven promoters.
DISCUSSION
In this study, we identified genes that are positively regulated by TLF/TRF2. We analyzed the regulation of the NF1 promoter by TLF and found that TLF and TBP play antagonistic roles in regulating transcription from this promoter. This finding shows that basal transcription factors such as TBP or TLF not only are involved in executing transcriptional programs determined by gene-specific activators but also are themselves gene-selective factors that are involved in the generation of transcriptional specificity.
Our results show that human TLF is a functional transcription factor that activates RNA polymerase II transcription from a subset of promoters. TLF increases transcription from the endogenous NF1 promoter from the same major start sites as those naturally used in vivo. TLF also binds to and increases the rate of transcription from an NF1 promoter-dependent reporter construct. Purified TLF-TFIIA binds to the NF1 promoter in vitro, and TLF-GFP associates with an NF1 promoter fragment in vivo. In addition, TLF−/− mice show significantly reduced levels of NF1 transcripts. The remaining NF1 transcripts may arise from initiator-dependent transcription or from an alternate promoter that does not require TLF. At present there is no evidence for an alternate promoter in the NF1 gene, but the gene is large (∼300 kb), and there are several known splice variants.
Despite reduced levels of NF1 mRNA, TLF−/− mice do not have a propensity for tumor formation, as do mice heterozygous for NF1 (16). The increased tumor formation rate in NF1 heterozygotes is a subtle phenotype (75% of heterozygotes and 15% of wild-type mice developed tumors over 27 months) (16). TLF−/− mice were generally sacrificed after 12 months, and it is possible that an increased tumor formation rate went unnoticed. Alternatively, this apparent discrepancy might have been the result of the complexities of tumorigenesis. Approximately half of the tumors isolated from animals heterozygous for NF1 deletion had also lost the wild-type NF1 allele (16), consistent with the two-hit model of inactivation of tumor suppressor genes (16, 20). Even if one copy of the gene had been inactivated, some NF1 still would have been produced in TLF−/− mice. It is possible that this low level of NF1 would have been sufficient to prevent tumor formation. Finally, it is possible that the decrease in NF1 transcript levels did not result in a decrease in neurofibromin protein levels in vivo.
In contrast to TLF, TBP does not bind to the NF1 promoter but does inhibit its transcription. For c-fos, TBP and TLF affect the TATA-containing promoter in a reciprocal manner: TBP increases c-fos promoter transcription, while TLF decreases transcription from this promoter. These findings are summarized in a model in which TLF and TBP compete for TFIIA and probably other general transcription factors (Fig. 8G). According to this model, TLF may bind to cognate promoter sequences and mediate the formation of a functional transcription initiation complex. We propose that TLF is a positively acting RNA polymerase II transcription factor. Also consistent with the model is a function for TLF in a promoter-bound form, or exerted independently of cognate promoter sequences, in which TLF competes with TBP for the association with other general transcription factors. The latter model, also referred to as the false-face hypothesis, may explain the ability of TLF to repress transcription from TBP-dependent promoters by competing for binding to TFIIA or other general transcription factors. Although it seems likely that TLF functions as a basal transcription factor given its similarity to TBP and its constitutive association with TFIIA, we cannot exclude the possibility that TLF acts as a specific activator of TATA-less promoters.
TBP and TLF protein levels are not correlated in many cell types (Fig. 1A and B) (32). In contrast to what was observed for TBP, we observed the highest levels of TLF protein in the liver and pancreas. Both TLF and TBP were expressed at low levels in the brain. The ratios of TLF to TBP differed in HeLa, HEK-293, and SK-N-SH human cell lines (Fig. 1B). These distinct expression patterns suggest that TLF and TBP may regulate distinct sets of genes.
In support of distinct roles for TLF and TBP in vivo, a large proportion of TLF-GFP strikingly localized to the nucleolus, while the majority of TBP-GFP was in the nucleus outside the nucleolus. The subcellular localization of TLF-GFP was cell type dependent, indicating that TLF likely serves different functions in different cells. The nucleolar localization of TLF in HEK-293 and COS-7 cells may indicate a function for TLF within the nucleolus such as RNA polymerase I transcription. Previous findings showed that TBP is associated with SL1 and rRNA transcription and that the majority of TBP is extranucleolar (reviewed in references 14 and 35). Further research will clarify whether TLF also participates in transcription by RNA polymerase I.
We have demonstrated that TLF-mediated repression of the c-fos promoter depends on TLF limitation of TFIIA availability. As suggested by the false-face hypothesis, TLF binds to TFIIA to decrease the amount of TFIIA available for TBP binding and thus indirectly regulates the c-fos promoter. Mutation of the TFIIA-binding site on TBP led to a significant decrease in the transcription of all reporter constructs tested, showing that the disruption of TBP-TFIIA interactions results in reduced transcription of a large majority of genes and in reduced cell viability. For the NF1 promoter, repression by TBP cannot be overcome by the addition of excess TFIIA. This finding indicates that TBP competes with TLF for the binding of another basal transcription factor, a transcriptional coactivator, or a transcriptional activator other than TFIIA.
Acknowledgments
We thank Jed Needle for technical assistance, Manlin Luo for gene chip screening, Di Zhang for the TLF−/− RNA, Anne West and Michael Greenberg for the c-fos CAT construct, Xunbin Wei for assistance with confocal microscopy, and Steve Buratowski for helpful advice. We also thank the entire Clapham Laboratory staff for helpful comments.
This work was supported by the Department of Cardiology at Children's Hospital, DOD neurofibromatosis research program NF01000149, and NIH grant CA42567.
REFERENCES
- 1.Bartfai, R., C. Balduf, T. Hilton, Y. Rathmann, Y. Hadzhiev, L. Tora, L. Orban, and F. Muller. 2004. TBP2, a vertebrate-specific member of the TBP family, is required in embryonic development of zebrafish. Curr. Biol. 14:593-598. [DOI] [PubMed] [Google Scholar]
- 2.Berk, A. J. 2000. TBP-like factors come into focus. Cell 103:5-8. [DOI] [PubMed] [Google Scholar]
- 3.Bonni, A., D. D. Ginty, H. Dudek, and M. E. Greenberg. 1995. Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol. Cell. Neurosci. 6:168-183. [DOI] [PubMed] [Google Scholar]
- 4.Bryant, G. O., L. S. Martel, S. K. Burley, and A. J. Berk. 1996. Radical mutations reveal TATA-box binding protein surfaces required for activated transcription in vivo. Genes Dev. 10:2491-2504. [DOI] [PubMed] [Google Scholar]
- 5.Chatterjee, S., and K. Struhl. 1995. Connecting a promoter-bound protein to TBP bypasses the need for a transcriptional activation domain. Nature 374:820-822. [DOI] [PubMed] [Google Scholar]
- 6.Cichowski, K., and T. Jacks. 2001. NF1 tumor suppressor gene function: narrowing the GAP. Cell 104:593-604. [DOI] [PubMed] [Google Scholar]
- 7.Colgan, J., and J. L. Manley. 1992. TFIID can be rate limiting in vivo for TATA-containing, but not TATA-lacking, RNA polymerase II promoters. Genes Dev. 6:304-315. [DOI] [PubMed] [Google Scholar]
- 8.Crowley, T. E., T. Hoey, J. K. Liu, Y. N. Jan, L. Y. Jan, and R. Tjian. 1993. A new factor related to TATA-binding protein has highly restricted expression patterns in Drosophila. Nature 361:557-561. [DOI] [PubMed] [Google Scholar]
- 9.Dantonel, J. C., S. Quintin, L. Lakatos, M. Labouesse, and L. Tora. 2000. TBP-like factor is required for embryonic RNA polymerase II transcription in C. elegans. Mol. Cell 6:715-722. [DOI] [PubMed] [Google Scholar]
- 10.Dikstein, R., S. Zhou, and R. Tjian. 1996. Human TAFII 105 is a cell type-specific TFIID subunit related to hTAFII130. Cell 87:137-146. [DOI] [PubMed] [Google Scholar]
- 11.Feigenbaum, L., K. Fujita, F. S. Collins, and G. Jay. 1996. Repression of the NF1 gene by Tax may expain the development of neurofibromas in human T-lymphotropic virus type 1 transgenic mice. J. Virol. 70:3280-3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hajra, A., A. Martin-Gallardo, S. A. Tarle, M. Freedman, S. Wilson-Gunn, A. Bernards, and F. S. Collins. 1994. DNA sequences in the promoter region of the NF1 gene are highly conserved between human and mouse. Genomics 21:649-652. [DOI] [PubMed] [Google Scholar]
- 13.Hansen, S. K., S. Takada, R. H. Jacobson, J. T. Lis, and R. Tjian. 1997. Transcription properties of a cell type-specific TATA-binding protein, TRF. Cell 91:71-83. [DOI] [PubMed] [Google Scholar]
- 14.Hernandez, N. 1993. TBP, a universal eukaryotic transcription factor? Genes Dev. 7:1291-1308. [DOI] [PubMed] [Google Scholar]
- 15.Holmes, M. C., and R. Tjian. 2000. Promoter-selective properties of the TBP-related factor TRF1. Science 288:867-870. [DOI] [PubMed] [Google Scholar]
- 16.Jacks, T., T. S. Shih, E. M. Schmitt, R. T. Bronson, A. Bernards, and R. A. Weinberg. 1994. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 7:353-361. [DOI] [PubMed] [Google Scholar]
- 17.Jordan, P., M. Mannervik, L. Tora, and M. Carmo-Fonseca. 1996. In vivo evidence that TATA-binding protein/SL1 colocalizes with UBF and RNA polymerase I when rRNA synthesis is either active or inactive. J. Cell Biol. 133:225-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaltenbach, L., M. A. Horner, J. H. Rothman, and S. E. Mango. 2000. The TBP-like factor CeTLF is required to activate RNA polymerase II transcription during C. elegans embryogenesis. Mol. Cell 6:705-713. [DOI] [PubMed] [Google Scholar]
- 19.Kieffer-Kwon, P., I. Martianov, and I. Davidson. 2004. Cell-specific nucleolar localization of TBP-related factor 2. Mol. Biol. Cell 15:4356-4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knudson, A. G., Jr. 1971. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 68:820-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuras, L., and K. Struhl. 1999. Binding of TBP to promoters in vivo is stimulated by activators and requires Pol II holoenzyme. Nature 399:609-613. [DOI] [PubMed] [Google Scholar]
- 22.Majello, B., G. Napolitano, P. De Luca, and L. Lania. 1998. Recruitment of human TBP selectively activates RNA polymerase II TATA-dependent promoters. J. Biol. Chem. 273:16509-16516. [DOI] [PubMed] [Google Scholar]
- 23.Maldonado, E. 1999. Transcriptional functions of a new mammalian TATA-binding protein-related factor. J. Biol. Chem. 274:12963-12966. [DOI] [PubMed] [Google Scholar]
- 24.Martianov, I., S. Brancorsini, A. Gansmuller, M. Parvinen, I. Davidson, and P. Sassone-Corsi. 2002. Distinct functions of TBP and TLF/TRF2 during spermatogenesis: requirement of TLF for heterochromatic chromocenter formation in haploid round spermatids. Development 129:945-955. [DOI] [PubMed] [Google Scholar]
- 25.Martianov, I., G. M. Fimia, A. Dierich, M. Parvinen, P. Sassone-Corsi, and I. Davidson. 2001. Late arrest of spermiogenesis and germ cell apoptosis in mice lacking the TBP-like TLF/TRF2 gene. Mol. Cell 7:509-515. [DOI] [PubMed] [Google Scholar]
- 26.Moore, P. A., J. Ozer, M. Salunek, G. Jan, D. Zerby, S. Campbell, and P. M. Lieberman. 1999. A human TATA binding protein-related protein with altered DNA binding specificity inhibits transcription from multiple promoters and activators. Mol. Cell. Biol. 19:7610-7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller, F., L. Lakatos, J. Dantonel, U. Strahle, and L. Tora. 2001. TBP is not universally required for zygotic RNA polymerase II transcription in zebrafish. Curr. Biol. 11:282-287. [DOI] [PubMed] [Google Scholar]
- 28.Nakadai, T., M. Shimada, D. Shima, H. Handa, and T. A. Tamura. 2004. Specific interaction with transcription factor IIA and localization of the mammalian TATA-binding protein-like protein (TLP/TRF2/TLF). J. Biol. Chem. 279:7447-7455. [DOI] [PubMed] [Google Scholar]
- 29.Ohbayashi, T., T. Kishimoto, Y. Makino, M. Shimada, T. Nakadai, T. Aoki, T. Kawata, S. Niwa, and T. Tamura. 1999. Isolation of cDNA, chromosome mapping, and expression of the human TBP-like protein. Biochem. Biophys. Res. Commun. 255:137-142. [DOI] [PubMed] [Google Scholar]
- 30.Ohbayashi, T., Y. Makino, and T. A. Tamura. 1999. Identification of a mouse TBP-like protein (TLP) distantly related to the drosophila TBP-related factor. Nucleic Acids Res. 27:750-755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohbayashi, T., M. Shimada, T. Nakadai, T. Wada, H. Handa, and T. Tamura. 2003. Vertebrate TBP-like protein (TLP/TRF2/TLF) stimulates TATA-less terminal deoxynucleotidyl transferase promoters in a transient reporter assay, and TFIIA-binding capacity of TLP is required for this function. Nucleic Acids Res. 31:2127-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perletti, L., J. C. Dantonel, and I. Davidson. 1999. The TATA-binding protein and its associated factors are differentially expressed in adult mouse tissues. J. Biol. Chem. 274:15301-15304. [DOI] [PubMed] [Google Scholar]
- 33.Persengiev, S. P., X. Zhu, B. L. Dixit, G. A. Maston, E. L. Kittler, and M. R. Green. 2003. TRF3, a TATA-box-binding protein-related factor, is vertebrate-specific and widely expressed. Proc. Natl. Acad. Sci. USA 100:14887-14891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rabenstein, M. D., S. Zhou, J. T. Lis, and R. Tjian. 1999. TATA box-binding protein (TBP)-related factor 2 (TRF2), a third member of the TBP family. Proc. Natl. Acad. Sci. USA 96:4791-4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roeder, R. G. 1996. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem. Sci. 21:327-335. [PubMed] [Google Scholar]
- 36.Schmidt, E. E., and U. Schibler. 1997. Developmental testis-specific regulation of mRNA levels and mRNA translational efficiencies for TATA-binding protein mRNA isoforms. Dev. Biol. 184:138-149. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt, E. E., and U. Schibler. 1995. High accumulation of components of the RNA polymerase II transcription machinery in rodent spermatids. Development 121:2373-2383. [DOI] [PubMed] [Google Scholar]
- 38.Sugiura, S., S. Kashiwabara, S. Iwase, and T. Baba. 2003. Expression of a testis-specific form of TBP-related factor 2 (TRF2) mRNA during mouse spermatogenesis. J. Reprod. Dev. 49:107-111. [DOI] [PubMed] [Google Scholar]
- 39.Takada, S., J. T. Lis, S. Zhou, and R. Tjian. 2000. A TRF1:BRF complex directs Drosophila RNA polymerase III transcription. Cell 101:459-469. [DOI] [PubMed] [Google Scholar]
- 40.Teichmann, M., Z. Wang, E. Martinez, A. Tjernberg, D. Zhang, F. Vollmer, B. T. Chait, and R. G. Roeder. 1999. Human TATA-binding protein-related factor-2 (hTRF2) stably associates with hTFIIA in HeLa cells. Proc. Natl. Acad. Sci. USA 96:13720-13725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veenstra, G. J., D. L. Weeks, and A. P. Wolffe. 2000. Distinct roles for TBP and TBP-like factor in early embryonic gene transcription in Xenopus. Science 290:2312-2315. [DOI] [PubMed] [Google Scholar]
- 42.Ye, K., C. A. Dinarello, and B. D. Clark. 1993. Identification of the promoter region of human interleukin 1 type I receptor gene: multiple initiation sites, high G+C content, and constitutive expression. Proc. Natl. Acad. Sci. USA 90:2295-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang, D., T. L. Penttila, P. L. Morris, M. Teichmann, and R. G. Roeder. 2001. Spermiogenesis deficiency in mice lacking the Trf2 gene. Science 292:1153-1155. [DOI] [PubMed] [Google Scholar]