

Abstract
The mammalian circadian regulatory proteins PER1 and PER2 undergo a daily cycle of accumulation followed by phosphorylation and degradation. Although phosphorylation-regulated proteolysis of these inhibitors is postulated to be essential for the function of the clock, inhibition of this process has not yet been shown to alter mammalian circadian rhythm. We have developed a cell-based model of PER2 degradation. Murine PER2 (mPER2) hyperphosphorylation induced by the cell-permeable protein phosphatase inhibitor calyculin A is rapidly followed by ubiquitination and degradation by the 26S proteasome. Proteasome-mediated degradation is critically important in the circadian clock, as proteasome inhibitors cause a significant lengthening of the circadian period in Rat-1 cells. CKIɛ (casein kinase Iɛ) has been postulated to prime PER2 for degradation. Supporting this idea, CKIɛ inhibition also causes a significant lengthening of circadian period in synchronized Rat-1 cells. CKIɛ inhibition also slows the degradation of PER2 in cells. CKIɛ-mediated phosphorylation of PER2 recruits the ubiquitin ligase adapter protein β-TrCP to a specific site, and dominant negative β-TrCP blocks phosphorylation-dependent degradation of mPER2. These results provide a biochemical mechanism and functional relevance for the observed phosphorylation-degradation cycle of mammalian PER2. Cell culture-based biochemical assays combined with measurement of cell-based rhythm complement genetic studies to elucidate basic mechanisms controlling the mammalian clock.
Diverse organisms from prokaryotes to mammals coordinate behavioral and physiological rhythms with the daily dark-light cycle by means of a circadian clock. In mammals, the master circadian clock is located in the suprachiasmatic nucleus of the brain, and it entrains peripheral cell-autonomous clocks throughout the body. In mice, a positively acting heterodimeric transcription factor composed of the PAS-bHLH proteins CLOCK (CLK) and BMAL1 drives transcription of tissue-specific circadian output genes, as well as its own negative regulators, the Period (denoted mPer1, mPer2, and mPer3), and Cryptochrome (mCry1 and mCry2) genes. The mammalian PER and CRY proteins form multimeric complexes that enter the nucleus and repress the transcriptional activity of CLK/BMAL1, modulating circadian output (reviewed in references 29 and 41). Additional stabilizing feedback loops, including inhibition of Bmal1 transcription by REV-ERBα (37), further contribute to the timing and robustness of the cycle. The daily rhythmic degradation of PERIOD proteins leading to derepression of CLK/BMAL1 is postulated to be critical to the proper functioning of the clock. Therefore, the mechanism and control of this process are of great interest.
Genetic studies have identified CKIɛ (casein kinase Iɛ) as a key regulator of metazoan circadian rhythm and both genetic and biochemical studies suggest that the PER proteins are important substrates (reviewed in reference 10). CKIɛ was first implicated as a circadian regulator in Drosophila, when several alleles of the CKIɛ homolog double-time (dbt) were uncovered in flies with either long- or short-period phenotypes (21, 39). Hamsters with a semidominant mutation in the substrate recognition domain of CKIɛ exhibit shortened behavioral rhythms in vivo and decreased kinase activity in vitro (28). In humans, a point mutation in a putative CKIɛ phosphorylation site within the kinase binding domain of human PER2 is sufficient to substantially shorten the free-running period (44). While these genetic observations of mammals suggest that decreased CKIɛ activity shortens circadian period length, there are several alleles of Drosophila dbt that decrease kinase activity yet lengthen the period (38). CKIɛ is also a regulator of circadian rhythm in mammals. CKIɛ and CKIδ bind to and phosphorylate mPER proteins on multiple sites (7, 19, 47). Inhibitors of CKI delay phosphorylation of endogenous human PER1 (hPER1) after serum shock (33). A number of other protein kinases, including casein kinase II (3, 27), mitogen-activated protein (MAP) kinase (1, 6, 35) and glycogen synthase kinase 3 (31) have also been implicated as important regulators of circadian rhythm. The mechanism by which these kinases control the clock is not known. PER is a direct target of all of these kinases, with the exception of MAP kinase. Phosphorylation of PER2 by multiple kinases might therefore play multiple regulatory roles.
In invertebrates and fungi, genetic and biochemical data suggest that circadian rhythm is controlled by phosphorylation-regulated ubiquitination and proteasome-mediated proteolysis. A null mutation in dbt results in the accumulation of Drosophila PER (dPER) protein to high levels (39). Phosphorylation is likely to facilitate recognition by a ubiquitin ligase and subsequent proteasome-mediated degradation. dPER physically interacts with Slimb, the fly homolog of the ubiquitin ligase adapter protein, β-TrCP, and this interaction is enhanced by its phosphorylation by the DBT kinase (22). Flies null for Slimb have increased abundance of phosphorylated dPER and arrhythmic behavior rhythms. Similarly, in Neurospora, FRQ protein phosphorylation (52, 53) leads to the recruitment of FWD-1 (15), an F-box protein homologous to the human SCF adapter protein β-TrCP. Involvement of the proteasome is suggested by the fact that dPER expressed in insect cells is stabilized by the presence of proteasome inhibitors (22).
There are several biochemical observations that suggest that phosphorylation also regulates PER stability in mammals. In cultured cells, proteasome inhibition increases the abundance of endogenous hPER1 and mPER2 (33, 50). Consistent with this finding, the overexpression of CKIɛ or CKIδ promotes mPER ubiquitination (2). Thus, CKIɛ and CKIδ activity play a role in the phosphorylation and regulation of mPER stability, although the specific mechanism has not yet been identified. These results suggest that phosphorylation-dependent protein degradation is important for circadian rhythm. However, the mechanistic links among kinase activity, proteasome function, and proper clock timing in mammalian cells have not been firmly established.
In this report, the signaling events that target mPER2 for degradation are investigated. The data indicate that mPER2 stability is regulated by a dynamic cycle in which phosphorylation precedes its degradation. Pharmacological inhibition of endogenous serine/threonine phosphatases leads to hyperphosphorylation, polyubiquitination, and proteasome-mediated degradation of mPER2. Phosphorylated mPER2 requires binding of a ubiquitin ligase for degradation, since overexpression of a dominant negative β-TrCP blocks both mPER2 ubiquitination and degradation. CKIɛ phosphorylation of mPER2 also regulates its half-life, as mutation of putative CKIɛ binding sites stabilizes mPER2 and blocks both its phosphorylation and its interaction with β-TrCP. Consistent with these findings, CKIɛ phosphorylation of mPER2 is required for its interaction with β-TrCP in vitro. Similarly, mutation of a conserved β-TrCP binding site in mPER2 blocks the interaction. The data suggest a model in which phosphorylation of mPER2 by CKIɛ leads to its recognition by β-TrCP, followed by polyubiquitination and subsequent proteasome-mediated degradation. These findings are relevant to the function of the clock in intact cells, since pharmacological inhibition of either CKIɛ or the 26S proteasome in a Rat-1 fibroblast reporter cell line led to a significant lengthening of the circadian period, consistent with the cooperative role of CKIɛ and the 26S proteasome in regulating mPER2 degradation.
MATERIALS AND METHODS
Plasmids and generation of mutant cDNAs.
For expression in mammalian tissue culture cells, mPER2 and its smaller fragments were subcloned into pCS2+MT, which adds five Myc epitopes to the amino terminus of mPER2. The PCR was used to engineer NcoI and SalI sites at the amino and carboxyl termini, respectively, of mPer2. The PCR product was digested with NcoI and SalI, and CS2+MT was digested with NcoI and XhoI, resulting in the loss of one of its six Myc epitopes. The two products were then ligated. To generate Myc epitope-tagged mPer2, amino acid positions 1 to 904 [mPer2(1-904)], the above cDNA encoding full-length mPer2 was used as a template, and a stop codon was introduced that results in a truncation of mPER2 at position 904 by using a QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's directions. FLAG epitope-tagged mPER2 was generated by PCR cloning that engineered a BglII site at its amino terminus and a SalI site at its carboxyl terminus. pFLAG-CMV-2 (Sigma) and the PCR product were digested with BglII and SalI and were ligated. β-TrCP(ΔF-box) was also cloned into pCS2+MT by a PCR-based strategy that resulted in an NcoI site at its amino terminus and XhoI at its carboxyl terminus. Both the PCR product and pCS2+MT were subsequently cut with NcoI and XhoI and ligated together. mPER2 point mutations and internal deletions were generated by using a QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's directions.
Cell culture maintenance and transfection.
Human embryonic kidney 293 (HEK293) cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum and maintained in a humidified incubator at 37°C and 5% CO2. For transient transfections, cells were seeded in individual wells of a six-well dish treated with poly-l-lysine (Sigma). When the cells were 70% confluent, they were transiently transfected by using PLUS reagent and Lipofectamine (Invitrogen) according to the manufacturer's directions. For inhibitor and immunoprecipitation experiments, 30 and 300 ng, respectively, of mPer2 expression plasmid were used. When the β-TrCP expression construct was used, 2 μg was transfected. For analysis of mPER2 ubiquitination, 300 ng of a construct expressing eight copies of hemagglutinin (HA) epitope-tagged ubiquitin was used (45).
Analysis of mPER2 degradation.
About 20 h after transfection, cells were treated with inhibitors as indicated in each figure. In brief, each inhibitor was applied as follows. For stability assays, cycloheximide (25 μg/ml; Biomol) was diluted in DMEM and added 25 min prior to the addition of phosphatase or kinase inhibitors. To induce mPER2 degradation, calyculin A (80 nM; Calbiochem) was diluted in DMEM and added to the cells. The kinase inhibitors IC261 (a gift from ICOS Corp.) and U0126 (Calbiochem) were diluted in DMEM and applied at a concentration of 50 and 30 μM, respectively. After treatment with inhibitors, cell extracts were prepared as described below at the times indicated in each figure.
To confirm that mPER2 gel mobility shifts were due to phosphorylation, cell extracts from cells incubated with calyculin A were treated with either calf intestinal alkaline phosphatase (CIP; see Fig. 1 and 4B) or lambda phosphatase (see Fig. 8C). For CIP, 50 μg of protein in each sample was diluted in 100 mM NaCl, 70 mM Tris (pH 7.5), and 10 mM MgCl2. Forty units of CIP (New England BioLabs) was added, and the samples were incubated at 37°C for 1 h. The reactions were stopped by the addition of Laemmli sample buffer. For lambda phosphatase (New England BioLabs) treatment, 50 μg of protein was added to the 1× phosphatase buffer supplied by the manufacturer supplemented with 2 mM MnCl2 and incubated with 10 U of lambda phosphatase at 30°C for 30 min. The reactions were stopped by the addition of Laemmli sample buffer.
FIG. 1.

mPER2 stability is regulated by protein phosphorylation. (A) Phosphatase inhibition leads to rapid loss of full-length mPER2 protein. HEK293 cells transiently expressing Myc epitope-tagged full-length mPER2 protein were treated with cycloheximide (25 μg/ml) and either vehicle (lanes 1 to 3) or calyculin A (80 nM, lanes 4 to 6) for the length of time indicated. The abundance of mPER2 was assessed by Western blotting of lysates with 9E10 anti-Myc monoclonal antibodies. As a loading control, the blot was stripped and reprobed with anti-actin polyclonal antibodies. (B) Definition of a minimal domain required for phosphatase-induced destabilization of mPER2. The indicated fragments of mPER2 were expressed, treated, and analyzed as for panel A. Cal A, calyculin A.
FIG. 4.

Dominant negative β-TrCP inhibits mPER2 degradation. (A) Cells coexpressing Myc epitope-tagged mPER2(450-763) and β-TrCP(ΔF-box) (lanes 4 to 9) were treated with vehicle (lanes 4 to 6) or calyculin A (80 nM) (lanes 7 to 9) and harvested at the indicated times. mPER2(450-763) abundance was analyzed as in Fig. 1, with anti-Myc antibodies. (B) DN-β-TrCP protects a subset of phosphorylation sites. Lysates from cells coexpressing mPER2(450-763) and β-TrCP(ΔF-box) and treated without or with calyculin A for 40 min (from panel A, lanes 6 and 9) were further analyzed by incubation with CIP (lanes 2 and 4). Alterations in mPER2(450-763) electrophoretic mobility were assessed by Western blotting as described above. (C) DN-β-TrCP blocks the phosphorylation-stimulated polyubiquitination (Ubn) of mPER2. Myc-tagged mPER2(450-763) was coexpressed with HA epitope-tagged ubiquitin (HA-Ub; lanes 3 to 6) and β-TrCP(ΔF-box) (lanes 5 and 6) and treated with calyculin A and MG132 as in Fig. 2A. mPER2(450-763) was immunoprecipitated, and the degree of mPER2(450-763) ubiquitination was analyzed by Western blotting with 12CA5 anti-HA monoclonal antibodies (upper panel). To confirm the presence of mPER2(450-763) in the immunoprecipitation pellets, the blot was stripped and reprobed with 9E10 anti-Myc monoclonal antibodies (lower panel).
FIG. 8.

CKIɛ regulates β-TrCP binding. (A) CKIɛ promotes β-TrCP binding to mPER2. Myc epitope-tagged mPER2 and β-TrCP(ΔF-box) were translated in vitro. To aid in its detection, β-TrCP(ΔF-box) was synthesized in the presence of [35S]methionine. The two synthesis reactions were added together without (lanes 1 and 3) or with (lanes 2 and 4) 300 ng of recombinant CKIɛ(Δ319). mPER2 was immunoprecipitated with 9E10 anti-Myc antibodies, and the presence of β-TrCP(ΔF-box) in the immunoprecipitation pellets was visualized by autoradiography. The presence of mPER2 in the immunoprecipitation pellets was confirmed by Western blotting with anti-Myc antibodies (lower panel). (B) Identification of the β-TrCP binding motif in mPER2. β-TrCP and the indicated mutants of mPER2(450-763) were incubated with recombinant CKIɛ(Δ319), and their interaction was analyzed exactly as for panel A. (C) β-TrCP binding mutations inhibit phosphorylation-dependent degradation of mPER2. mPER2(450-763) and mutants mPER2(Δ582-606) and mPER2(S477A/G479A) were expressed in tissue culture cells. mPER2 stability after the addition of cycloheximide and calyculin A to the culture medium was analyzed by Western blotting with anti-Myc antibodies. wt, wild type; Cal A, calyculin A.
Immunoprecipitation and Western blotting.
Cell-free lysates were prepared by standard methods that have been described previously (9). Myc epitope-tagged proteins were immunoprecipitated from cell-free lysates that contain 300 μg of protein with 2 μg of anti-Myc antibodies (9E10). Antibody-antigen complexes were allowed to form for 1 h on ice. The immune complexes were collected with 20 μl of protein A agarose beads (Invitrogen) for 1 h at 4°C and gentle agitation. The beads were subsequently washed three times with lysis buffer. For sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the beads were resuspended in lysis buffer and Laemmli sample preparation buffer and boiled for 5 min. The proteins were resolved on either a 10 or 12% acrylamide gel and transferred to a nitrocellulose membrane (Amersham). Afterwards, the membrane was soaked in blotting buffer (3% nonfat dry milk and Tris-buffered saline supplemented with 0.25% Tween 20). To detect the presence of proteins by Western blotting, antibodies were diluted in blotting buffer and incubated with the membranes for 1 h at room temperature. After being washed with wash buffer (Tris-buffered saline with 0.25% Tween 20) three times, the appropriate secondary antibodies were diluted 1:7,500 in blotting buffer and incubated with the membrane as described above. After being washed three times with wash buffer, the membranes were washed once with Tris-buffered saline, and the presence of proteins was detected with an ECL chemiluminescence detection kit (Amersham) according to the manufacturer's directions.
For the analysis of mPER2 ubiquitination in tissue culture cells, Z-Leu-Leu-Leu-CHO (MG132; Biomol) or N-acetyl-Leu-Leu-methional (ALLM; Peptides International) was applied to the cells for 5 h at a final concentration of 20 or 50 μM, respectively. As indicated in the figures, calyculin A (80 nM) was then applied and allowed to incubate with the cells for 60 min prior to lysis and immunoprecipitation as described above.
In vitro immunoprecipitation.
β-TrCP(ΔF-box) or CKIɛ and Myc epitope-tagged mPER2 were synthesized in vitro by using a TNT Coupled Reticulocyte Lysate system (Promega) according to the manufacturer's directions. To aid in their detection, β-TrCP(ΔF-box) or CKIɛ was synthesized in the presence of [35S]methionine. The binding assay was performed essentially as described previously (47). Briefly, 40 μl of each protein synthesis reaction was combined as indicated in Fig. 8. For β-TrCP/mPER2 interaction, the binding reactions were performed either without or with the addition of 300 ng of recombinant CKIɛ(Δ319). Prior studies have established that reticulocyte lysates have minimal CKI activity (11). The binding reactions were incubated at 30°C for 30 min. The binding reactions were diluted in immunoprecipitation buffer (100 mM KCl, 25 mM HEPES [pH 7.5], 12.5 mM MgCl2, 100 μM EDTA, 20% glycerol, 0.1% NP-40, 1 mM dithiothreitol, and 1× complete protease inhibitor cocktail [Roche]). mPER2 was immunoprecipitated with 1 μg of 9E10, and the beads were washed three times with immunoprecipitation buffer. After SDS-PAGE, the gel was cut horizontally. To confirm the presence of mPER2 in the immunoprecipitation pellets, proteins in the top half of the gel were transferred to a nitrocellulose membrane and mPER2 was detected by Western blotting with anti-Myc antibodies (9E10) as described above. The degree of β-TrCP(ΔF-box) or CKIɛ binding to mPER2 was assessed by fixing and drying the lower half of the gel. [35S]methionine-labeled proteins were visualized by autoradiography with a Storm 860 gel imaging system (Molecular Dynamics).
In vitro kinase assay.
mPER2(450-763) protein and the indicated mutations were synthesized by a TNT quick system (Promega) as described above. Five microliters of each synthesis reaction was added to kinase buffer (25 mM Tris-HCl [pH 7.5], 15% glycerol, 20 mM NaF, 10 mM β-glycerolphosphate, 1 mM dithiothreitol, and 150 μM ATP) without or with 50 ng of recombinant CKIɛ(Δ319). The kinase reactions were incubated at 30°C for 1 h and stopped by the addition of Laemmli sample buffer. The proteins were resolved on a 5 to 15% acrylamide gradient gel. After the gel was fixed and dried, mPER2(450-763) was visualized by autoradiography with a Storm 860 gel imaging system (Molecular Dynamics).
Circadian rhythm measurements in Rat-1 fibroblasts.
A Rat-1 cell line with stable integration of the 6.7-kb mPer1 promoter driving expression of firefly luciferase was used. For circadian rhythm measurements, the Rat-1 (mPer1::luc) fibroblasts were plated onto a Visiplate 24-well tissue culture plate (1450-603; Wallac) and maintained in DMEM supplemented with 10% fetal bovine serum, penicillin-streptomycin, and Zeocin (100 μg/ml) until cultures reached 100% confluence (about 24 h). Cultures were synchronized for 2 h with DMEM supplemented with forskolin (10 μM) (5, 17). The synchronization medium was replaced with 1 ml of DMEM without phenol red (Invitrogen) supplemented with 10% fetal bovine serum, luciferin (32 μg/ml; Promega), and pyruvate (110 mg/liter). Inhibitors (lactacystin, MG132, and ALLM [Biomol]) or IC261 (ICOS) were added at the same time, as indicated in Fig. 3 and 5, respectively, and were not replenished. The tissue culture plates were immediately sealed with SealPlate adhesive film (Rainin) and transferred to a FLUOstar Optima luminometer (BMG Labtech) with a heated chamber (37°C) modified to maximize light detection from the top of a 24-well plate. Light output from each well was measured every 12 min for 72 to 120 h. The data was baseline corrected by subtraction of the running 24-h average light output from the data at each time point. Each experiment was performed in quadruplicate on four or more separate occasions with similar results.
FIG. 3.

Proteasome inhibition lengthens the circadian period in Rat-1 fibroblasts. (A to D) Rat-1 (mPer1::luc) fibroblasts were synchronized with 10 μM forskolin for 2 h. The synchronization medium was removed and replaced with medium containing the indicated protease inhibitors. (A) Synchronous luciferase oscillations between individual wells. Four individual wells used as the DMSO vehicle control were monitored for luciferase expression over time, and baseline-corrected data from each well was plotted. (B to D) Proteasome inhibition lengthens the circadian period. The indicated protease inhibitors were analyzed for their effect on period length. The average luciferase activity of cosynchronized DMSO controls is plotted in black, and the inhibitor is plotted in grey. Three to four wells were tested per compound and compared with the DMSO control experiment performed in the same 24-well plate. The experiments were repeated on at least four separate occasions with similar results. Each line is the baseline-corrected average for multiple wells from a single 24-well plate. Statistical significance was calculated by using the unpaired t test, and * and ** denote P values of <0.0025 and >0.5, respectively. Lact, lactacystin.
FIG. 5.

CKIɛ regulates period length and mPER2 protein stability. (A) CKIɛ inhibition lengthens the circadian period in cultured cells. Rat-1 (mPer1::luc) cells were synchronized as in Fig. 3, and the CKIδ/ɛ-selective inhibitor IC261 (5 μM) was added to the growth medium. Luciferase expression was measured and analyzed as in Fig. 3. *, P < 0.035. (B) CKI inhibition delays the degradation of mPER2. Myc epitope-tagged mPER2(450-763) was transiently expressed in HEK293 cells. After pretreatment with either IC261 (50 μM) (lanes 7 to 9) or U0126 (30 μM) (lanes 10 to 12), DMSO (lanes 1 to 3) or calyculin A (Cal A; 80 nM) (lanes 4 to 12) was added. The cells were harvested, and the amount of mPER2(450-763) was determined as in Fig. 1A, with anti-Myc antibodies.
Determination of cycling period.
The cycling period was determined by searching for a period that maximizes the power spectrum density function of the data. The power spectrum density function is the complex modulus of the discrete Fourier transform of the data and is widely used to identify dominant, periodic signals in time series data. The power spectrum density function of a time series data set can be written as
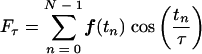 |
Here, Fτ is the power spectrum density at a period τ, and f(tn) is the smoothed activity measure at time point tn. Periods that maximized the power spectrum density were found by using a gradient optimization algorithm with an array of starting positions. To identify significant changes in period between two conditions, we calculated P values by using a one-tailed t test. Conditions with P values of ≤0.05 were considered significant.
RESULTS
Phosphorylation-dependent mPER2 degradation.
Several lines of evidence suggest that reversible phosphorylation regulates PER protein stability in both insects and mammals. Mammalian genetic models of circadian rhythm do not lend themselves to facile mechanistic studies, but the discovery that virtually all mammalian tissues have circadian rhythms provides a rational biological basis for biochemical studies of circadian regulators in cultured cells. To induce mPER2 hyperphosphorylation in tissue culture cells, endogenous phosphatases were inhibited with calyculin A, a cell-permeable inhibitor of type 1 and type 2A serine/threonine phosphatases (16). Cells were transiently transfected with plasmids expressing epitope-tagged full-length mPER2. Twenty hours posttransfection, the cells were treated with cycloheximide (to inhibit de novo protein synthesis) and calyculin A. At various times after the addition of calyculin A to the growth medium, the amount of mPER2 present in the cells was analyzed by immunoblotting. In the absence of phosphatase inhibition, mPER2 was stable for the duration of the assay (Fig. 1A, lanes 1 to 3), consistent with previous studies showing that it has a half-life of 6 to 12 h in transfected cells (7). However, after only 30 min of calyculin A treatment, mPER2 was almost completely degraded (Fig. 1A, lanes 4 to 6), suggesting that hyperphosphorylation of mPER2 targeted it for degradation. Multiple attempts were made to visualize the effects of phosphatase inhibitors and other interventions on endogenous PER2 in these cells. While PER2 mRNA was seen in multiple cell lines, we were unable to identify endogenous PER2 by immunoblotting with available antibodies.
To identify the minimal mPER2 sequence element sufficient for its calyculin A-regulated degradation, various fragments of mPER2 were analyzed for their sensitivity to phosphatase inhibition (Fig. 1B and data not shown). Fragments of mPER2 that include amino acids 450 through 763 were sufficient for calyculin A-induced protein degradation (Fig. 1B). This region has been previously shown to bind CKIɛ (44), consistent with a role for CKIɛ in the phosphorylation-regulated degradation of mPER2.
Phosphorylated mPER2 is degraded by the 26S proteasome.
Degradation of clock regulators by the 26S proteasome has been reported in Drosophila (13, 22, 34), Neurospora (15), and mammalian cells. Inhibitors of the 26S proteasome block degradation of endogenous mPER1 in tissue culture cells (2, 33) and increase the steady-state abundance of endogenous mPER2 protein in mouse embryonic fibroblasts (50). To determine if phosphorylation-regulated degradation of mPER2 was also proteasome dependent, transfected cells were pretreated with MG132, a specific inhibitor of the 26S proteasome, prior to the addition of calyculin A. After a 90-min exposure to calyculin A, mPER2(450-763) was almost completely degraded (Fig. 2A, lane 4). However, following pretreatment with MG132, a substantial amount of the mPER2 fragment remained after calyculin A treatment, despite its apparent hyperphosphorylation (Fig. 2A, lane 5). ALLM, a calpain II inhibitor, had no detectable effect on mPER2 stability in this assay (Fig. 2A, lane 6). Hence, under these conditions, proteasome function is required for mPER2 degradation following phosphorylation.
FIG. 2.

Phosphorylated mPER2 is degraded by the 26S proteasome. (A) Proteasome inhibition delays mPER2 degradation. Myc epitope-tagged mPER2(450-763) was transiently expressed in HEK293 cells. Five hours after the addition of the proteasome inhibitor MG132 (20 μM) (lane 5) or the calpain inhibitor ALLM (50 μM) (lane 6) to the growth medium, cells were treated with vehicle (lanes 1 and 2) or with calyculin A (Cal A; 80 nM) (lanes 3 to 6) and harvested at the indicated times, as in Fig. 1. As a loading control, the blot was stripped and reprobed with polyclonal antibodies that recognize mSin3A (lower panel). (B) Phosphorylation stimulates the polyubiquitination (Ubn) of mPER2. Myc epitope-tagged mPER2(450-764) was coexpressed with HA epitope-tagged ubiquitin and treated with combinations of calyculin A (80 nM) and MG132 (20 μM) as indicated in the figure. mPER2(450-764) was immunoprecipitated with 9E10 anti-Myc monoclonal antibodies, and the degree of ubiquitination was assessed by Western blotting with anti-HA monoclonal antibodies.
Phosphorylation-regulated proteasome-mediated protein degradation is usually preceded by polyubiquitination. To test if mPER2 was in fact conjugated to ubiquitin in a phosphorylation-dependent manner, mPER2(450-763) was coexpressed with HA epitope-tagged ubiquitin. mPER2 ubiquitination was assessed by immunoprecipitation, followed by immunoblotting for bound HA-ubiquitin. In the absence of calyculin A and MG132, minimal amounts of ubiquitin were coimmunoprecipitated with mPER2(450-763) (Fig. 2B, lane 1). Treatment of cells with calyculin A 60 min prior to lysis caused a substantial increase in mPER2 ubiquitination, which was more readily detectable after concomitant proteasome inhibition (Fig. 2B, lanes 2 and 3). Taken together, these results suggest that calyculin A promotes hyperphosphorylation of multiple sites in mPER2. A subset of these phosphorylation sites is likely to be required for subsequent polyubiquitination and degradation by the 26S proteasome.
The effect of mutations and drugs on circadian rhythm has traditionally been analyzed in whole animals. However, Schibler and coworkers demonstrated that cultured cell lines, including Rat-1 fibroblasts, have an intrinsic circadian rhythm of gene expression that can be analyzed when cells are synchronized in vitro (4, 5). Others have shown that circadian promoters, especially mPer1, can drive circadian expression of reporter genes, such as green fluorescent protein and luciferase in organ explants, zebra fish, and fibroblasts (17, 23, 46, 51). To directly assess the importance of proteasome-mediated protein degradation in regulating circadian rhythm, a Rat-1 (mPer1::luc) fibroblast cell line was synchronized with forskolin following published methods (4, 5, 17) and the effect of proteasome inhibition on the circadian expression of luciferase was assessed. As previously reported, 2 h of forskolin treatment lead to rhythmic luciferase expression, with a circadian period of approximately 24.38 h and high reproducibility between wells in any individual experiment (Fig. 3A). To test if the proteolytic activity of the 26S proteasome is in fact required for clock function, proteasome catalytic activity was inhibited by a single application of the test compound after synchronization of cells with forskolin. Inhibition of the 26S proteasome with either MG132 or lactacystin caused a measurable period lengthening of about 2 h (Fig. 3B and C) whereas ALLM, an inhibitor of calpain II, had no significant effect on the circadian period (Fig. 3D). We have consistently observed that the effect of proteasome inhibitors on the circadian period tends to diminish with time. This is presumably due to their chemical instability once they are placed in the culture medium. No consistent effect of the inhibitors on the average luciferase activity was seen.
Identification of an mPER2 E3 ubiquitin ligase.
In order for a protein destined for degradation by the 26S proteasome to be polyubiquitinated, it must first be recognized by an E3 ubiquitin ligase (18). One class of ubiquitin ligases, the SCF (Skp-Cullin-F box), is a multimeric complex. Substrates are recognized by an adapter subunit that contains the conserved F-box motif. The F-box protein recognizes and binds a specific sequence within its cognate protein substrate (36). In flies, Slimb binds phosphorylated dPER and targets it for ubiquitin-mediated degradation (13, 22). Since β-TrCP substrates require phosphorylation prior to recognition by the F-box adapter, we tested if β-TrCP had any effect on phosphorylation-induced mPER2 degradation.
For these studies, β-TrCP lacking the F-box motif [β-TrCP(ΔF-box)] was utilized (14). This protein can still bind phosphorylated substrates (e.g., β-catenin) but since it cannot interact with the other components of the ubiquitination pathway, it acts as a dominant negative that can protect the phosphorylation site and prevent protein degradation. When β-TrCP(ΔF-box) was coexpressed with mPER2(450-763), mPER2 protein remained stable even after 40 min of calyculin A treatment (Fig. 4A, lanes 7 to 9). Notably, the presence of β-TrCP(ΔF-box) led to the appearance of two distinct phosphorylated mPER2 species. First, in the absence of calyculin A, a fraction of the mPER2 fragment shifted upwards (Fig. 4A, lanes 4 to 6). Second, in the presence of calyculin A, nearly all of the mPER2 fragment was shifted to phosphorylated forms (Fig. 4A, lanes 7 to 9). To confirm that these mobility shifts were due to phosphorylation, extracts were treated with CIP (Fig. 4B). In all cases, incubation with CIP caused mPER2 electrophoretic mobility to return to baseline (Fig. 4B, lanes 2 and 4). The accumulation of hyperphosphorylated mPER2 in the presence of β-TrCP (Fig. 4A, compare lane 1 to lane 4) may be due to the basal turnover of specific phosphoryl groups on mPER2. As specific residues on mPER2 become phosphorylated, they may be recognized and bound by the dominant negative β-TrCP, thereby stabilizing the phosphorylated species. In the presence of calyculin A, the stabilized mPER2 is further phosphorylated on additional sites that may not be involved in regulating its stability (Fig. 4A, compare lanes 4 to 6 to lanes 7 to 9).
Since β-TrCP lacking the F-box motif stabilized phosphorylated mPER2, we predicted that it would also inhibit the ubiquitination of mPER2. To test this hypothesis, mPER2 was coexpressed with HA-ubiquitin and β-TrCP(ΔF-box) (Fig. 4C). To inhibit the degradation of ubiquitinated mPER2, the cells were also treated with MG132 (Fig. 4C, lanes 2, 4, and 6). mPER2 was then immunoprecipitated from cell extracts, and the degree of mPER2 ubiquitination was assessed by immunoblotting for bound ubiquitin. As expected, the inhibition of the 26S proteasome promoted an accumulation of ubiquitinated mPER2 (Fig. 4C, compare lane 3 to lane 4). However, when the dominant negative β-TrCP was coexpressed with mPER2, detectable polyubiquitination was almost completely blocked (Fig. 4C, compare lanes 3 and 4 to lanes 5 and 6). These results suggest that overexpressed β-TrCP(ΔF-box) binds phosphorylated mPER2, thereby excluding endogenous β-TrCP and inhibiting mPER2 ubiquitination.
CKIɛ regulates circadian rhythm and mPER2 stability.
These and other observations are consistent with the idea that phosphorylation of mPER2 at specific sites initiates its proteasome-mediated degradation. To test the effect of CKIɛ inhibition on the circadian period, the CKIδ/ɛ-selective inhibitor IC261 (32) was added to synchronized Rat-1 (mPer1::luc) cells. Notably, inhibiting CKIɛ kinase activity led to about a 1.2-h increase in period length compared to the dimethyl sulfoxide (DMSO) vehicle control (Fig. 5A). It is notable that both IC261 and proteasome inhibitors lengthen the period. The effect decreases with time, presumably due to breakdown of the inhibitors during the course of the experiment. Given the instability of the inhibitors, a phase shift and a short-term change in period length may appear similar. The lengthening of the period by IC261 is in contrast to the short period seen with the tau hamster with mutant CKIɛ, although similar in direction to several period-lengthening alleles of double-time in Drosophila (see Discussion).
Although numerous studies have indicated that CKIɛ regulates circadian rhythm (8, 9, 28, 40, 44), the specific mechanism has remained elusive. One emerging possibility is that CKIɛ destabilizes PER proteins (2, 7, 19, 22, 33, 39). To assess what effect CKIɛ inhibition has on mPER2 protein stability, tissue culture cells overexpressing mPER2(450-763) were treated with IC261, followed by calyculin A. The presence of IC261 markedly delayed the phosphorylation-induced degradation of mPER2(450-763) (Fig. 5B, lanes 7 to 9), whereas treatment with U0126, an inhibitor of the p42/p44 MAP kinases, had no discernible effect on mPER2 stability (Fig. 5B, lanes 10 to 12). Although CKIɛ inhibition slowed mPER2(450-763) degradation, it still existed as a hyperphosphorylated species. This finding is most likely due to either incomplete inhibition of CKIɛ and/or phosphorylation of mPER2 by unrelated kinases on additional sites that are not involved in regulating mPER2 stability.
Identification of the mPER2 stability regulatory domain.
To further define the specific sequence within mPER2 responsible for its phosphorylation-dependent degradation, we engineered a series of internal deletions that systematically removed 25 amino acids at a time throughout the region between residues 450 and 763 (Fig. 6A and B). Their sensitivity to calyculin A was tested in the degradation assay described above (Fig. 1 and 3B). Of eight deletion mutants tested, two (deletions of amino acids 582 to 606 and 731 to 756) were not degraded after calyculin A treatment (Fig. 6A and B and data not shown). In addition, the deletion of a larger section between amino acids 450 to 549 significantly decreased the rate of mPER2 degradation (data not shown). The stabilization of mPER2 by these deletions was not due to misfolding of the protein, because the presence of either deletion in the context of full-length mPER2 had no effect on mCRY1 binding (data not shown) and did not block calyculin-induced hyperphosphorylation. Notably, the stabilized mPER2 still underwent a phosphorylation-dependent mobility shift (Fig. 6, lanes 4 and 12). As detailed below (Fig. 7), the data suggest that the stabilized mPER2 is no longer phosphorylated by CKIɛ and hence not degraded. We suspect that the stabilized mPER2 is additionally phosphorylated by other cellular kinases, and that this is only apparent when the CKI-dependent degradation is eliminated.
FIG. 6.

Identification of mPER2 sequences necessary for phosphorylation-dependent degradation. (A) Two specific internal deletions in mPER2 block the calyculin A-induced degradation. The indicated internal deletions were engineered in the mPER2(450-763) background and transiently expressed in HEK293 cells. Cells were treated with vehicle (−) or calyculin A (+; 80 nM) for 60 min, and the amount of mPER2 was assessed as in Fig. 1A, with anti-Myc antibodies. (B) Summary of the constructs used and the results obtained in panel A. wt, wild type; Cal. A, calyculin A.
FIG. 7.

Stabilizing deletions in mPER2 block its interaction with β-TrCP(ΔF-box) and CKIɛ. (A) Deletions that block CKIɛ binding also block β-TrCP binding. Myc epitope-tagged mPER2(450-763) with or without the indicated internal deletions were coexpressed with FLAG epitope-tagged β-TrCP(ΔF-box) in HEK 293 cells. mPER2 was immunoprecipitated (IP) with 9E10 anti-Myc monoclonal antibodies, and the presence of β-TrCP(ΔF-box) or endogenous CKIɛ in the immunoprecipitation pellet was determined by Western blotting (WB) with anti-CKIɛ polyclonal antibodies (upper right panel) or anti-FLAG monoclonal antibodies (middle right panel). To confirm the presence of mPER2 in the immunoprecipitation pellet, the blot was stripped and reprobed with anti-Myc antibodies (lower right panel). The asterisk indicates mPER2 mobility shift due to binding of β-TrCP(ΔF-box), as described in the legend to Fig. 4A and B. (B) Deletion of the CKIɛ binding site prevents mPER2 phosphorylation. In vitro synthesized and [35S]methionine-labeled mPER2 or mPER2 with internal deletions was incubated with 50 ng of recombinant CKIɛ(Δ319) (+). Mobility shifts were analyzed after SDS-PAGE and PhosphorImager analysis. (C) Loss of CKIɛ binding sites prevents mPER2 polyubiquitination. Myc epitope-tagged mPER2(450-763) (lanes 1 and 2) or mPER2(450-764) (Δ582-606) (lanes 3 and 4) was coexpressed with HA epitope-tagged ubiquitin (HA-Ub). mPER2 was immunoprecipitated with 9E10 anti-Myc antibodies, and ubiquitination was determined by Western blotting with anti-HA antibodies.
Stabilizing deletions in mPER2 may block interaction of CKIɛ and β-TrCP with mPER2. To test this hypothesis, the binding of mutant mPER2(450-763) to β-TrCP(ΔF-box) and endogenous CKIɛ was assessed (Fig. 7A). The deletion of amino acids 582 to 606 or 731 to 756 eliminated mPER2 binding to both CKIɛ and β-TrCP (Fig. 7A, upper right panel, lanes 3′ and 4′). Deletion of amino acids 450 to 549 blocked binding of β-TrCP to mPER2 without affecting its interaction with endogenous CKIɛ (Fig. 7A, lanes 1′ and 2′). These results indicate that CKIɛ and β-TrCP bind distinct regions of mPER2 and are consistent with a requirement for ordered binding of CKIɛ, first followed by β-TrCP.
Since the stabilizing deletions eliminate the binding of CKIɛ, they should also abolish mPER2 phosphorylation by CKIɛ. mPER2 synthesized in reticulocyte lysates (that normally do not phosphorylate the protein) was incubated with recombinant CKIɛ in an in vitro phosphorylation assay. The addition of CKIɛ to the reticulocyte lysates caused a distinct gel shift in mPER2, indicating its phosphorylation (Fig. 7B, lanes 1 and 1′). When a stabilizing deletion was present [mPER2(Δ582-606)], there was no detectable mPER2 phosphorylation (Fig. 7B, lanes 2 and 2′), whereas mPER2 with a deletion that had no effect on stability (Δ607-631) was phosphorylated by CKIɛ nearly as well as the wild-type protein (Fig. 7B, lanes 3 and 3′).
If CKIɛ binding and phosphorylation precedes β-TrCP binding and ubiquitination, then loss of a CKIɛ binding site should block mPER2 ubiquitination. To test this hypothesis, mPER2(450-763) with amino acids 582 to 606 deleted was coexpressed with HA-ubiquitin in the absence or presence of MG132. The deletion of the CKIɛ binding site abolished the polyubiquitination of mPER2 (Fig. 7C, lanes 3 and 4). These results indicate that CKIɛ binding to mPER2 is a prerequisite for its interaction with β-TrCP and subsequent degradation by the 26S proteasome.
To directly test if CKIɛ promotes binding of β-TrCP to mPER2, full-length mPER2 and β-TrCP(ΔF-box) were synthesized separately in vitro in reticulocyte lysates and then incubated together either without or with added recombinant CKIɛ (Fig. 8A). mPER2 was then immunoprecipitated, and the binding of β-TrCP(ΔF-box) was assessed. In the absence of added CKIɛ, only a small amount of β-TrCP(ΔF-box) coprecipitated with mPER2 (Fig. 8A, upper panel, lane 3). However, when CKIɛ was included in the binding reaction, the amount of β-TrCP interacting with mPER2 was greatly increased (Fig. 8A, upper panel, lane 4). mPER2 also exhibits slower mobility in the presence of added CKIɛ (Fig. 8A, lower panel, compare lane 3 to lane 4), consistent with its phosphorylation by the added kinase.
The presence of distinct domains in mPER2 mediating interaction with β-TrCP and CKIɛ is consistent with a model in which CKIɛ first binds and phosphorylates mPER2, creating a β-TrCP recognition site. To further define the sequence within mPER2 that directly interacts with β-TrCP, a series of 25 amino acid internal deletions were created between residues 450 and 550. Their effect on CKIɛ-dependent binding was then assessed as in Fig. 8A. Initial experiments indicated that deletion of amino acids 475 to 499 abolished the binding of β-TrCP to mPER2 (Fig. 8B, lane 3 and data not shown). Many β-TrCP substrates contain the consensus recognition motif DpSGφXpS, where pS is phosphoserine, φ is a hydrophobic residue, and X is any amino acid (49). Both the aspartic acid and glycine are thought to be invariant residues (49). Further inspection of the sequence between residues 475 and 499 revealed the presence of a group of amino acids with the sequence 477SSGYGS482, which is similar to the consensus β-TrCP recognition motif. To determine if this sequence represents a β-TrCP recognition motif, an internal deletion that removed amino acids 477 through 482 was engineered [mPER2(Δ477-482)], and its CKIɛ-dependent β-TrCP binding was tested as described above (Fig. 8A). mPER2(Δ477-482) failed to interact with β-TrCP despite the presence of CKIɛ, indicating that the amino acids between 477 and 482 are required for β-TrCP binding. Since these residues are conserved in other PER2 proteins and are in the characteristic positions as dictated by the consensus sequence, their relevance for β-TrCP binding to mPER2 was tested. Mutation of serine 477 and glycine 479 to alanine [mPER2(S477A/G479A)] abolished almost all detectable CKIɛ-dependent β-TrCP binding to mPER2 (Fig. 8B, lane 5), indicating that these residues function as a novel β-TrCP binding motif.
Next, we asked if the absence of β-TrCP binding inhibits phosphorylation-dependent mPER2 degradation. mPER2(450-763) or mPER2(450-763) with CKI and β-TrCP binding mutations [mPER2(Δ582-606) and mPER2(S477A/G479A)] were expressed in cultured cells. Since mPER2 becomes hyperphosphorylated after phosphatase inhibition with calyculin A (Fig. 4B), cell-free lysates were treated with lambda phosphatase prior to SDS-PAGE to better visualize total protein. Both mPER2(Δ582-606) and mPER2(S477A/G479A) were degraded slower than wild-type protein after calyculin A was added to the growth medium (Fig. 8C). Although mutation of S477 and G479 slows mPER2 degradation, the effect on stability is less than that observed when amino acids 582 to 606 are deleted. This result may be due to the presence of other β-TrCP binding sites in mPER2 or residual β-TrCP binding sufficient to cause low-level turnover of mPER2. Taken together, these results suggest that CKIɛ regulates mPER2 stability by first binding and phosphorylating mPER2, thereby creating a β-TrCP binding site(s). β-TrCP is then recruited to mPER2, which leads to its polyubiquitination and destruction by the 26S proteasome.
DISCUSSION
Proper timing of circadian rhythm requires stable oscillations in regulatory protein abundance and activity within a transcription-translation negative feedback loop that drives expression of circadian regulators and output genes. While their molecular functions are not fully understood, mPER1 and mPER2 have the highest amplitude oscillations of all the known core clock proteins, with almost complete degradation near the end of the subjective night in the suprachiasmatic nucleus (25). mPER2 also undergoes temporal changes in phosphorylation that reaches a zenith just prior to its destruction (25).
To study phosphorylation-mediated mPER2 degradation, a combination of cell-based assays were used. Calyculin A treatment of cultured cells resulted in the rapid phosphorylation and degradation of mPER2, suggesting that both a kinase and phosphatase regulate mPER2 net phosphorylation and stability. The present study further defines the role of CKIɛ in regulating mPER2 protein stability in mammals. Inhibition of endogenous CKIɛ activity with IC261 decreased the rate of calyculin A-induced mPER2 degradation. Most notably, internal deletions in mPER2 that affected CKIɛ binding significantly decreased mPER2 degradation and blocked calyculin A-induced ubiquitination. The location of these deletions at distinct sites in mPER2 suggest either that these domains are near each other in the folded protein or that different regions of CKIɛ contact distinct domains of mPER2. Neither of the internal deletions appear to substantially alter the folding of the protein, since they did not block the binding of mCRY1 (data not shown) nor did they inhibit the ability of other kinases to phosphorylate mPER2.
In Drosophila, targeted proteolysis of dPER is mediated by the phosphorylation-dependent binding of Slimb, the fly homolog of the human F-box protein β-TrCP (13, 22). Likewise, in Neurospora, the F-box protein FWD-1 regulates the stability of the circadian regulator FRQ (15). In the present study, we have shown that β-TrCP is a potential regulator of mammalian circadian rhythm. β-TrCP bound a region just amino-terminal to the CKIɛ binding domain, and CKIɛ regulated the binding of β-TrCP to mPER2. A dominant negative form of β-TrCP inhibited calyculin A-induced degradation of mPER2. The data are consistent with a phosphorylation-regulated β-TrCP binding site between amino acids 450 to 550 of mPER2. β-TrCP is known to interact with the motif DpSGφXpS following serine phosphorylation in human immunodeficiency virus Vpu (30), IκBα (54), and β-catenin (14). Within amino acids 450 and 550, mPER2 contains a related sequence, 477SSGYGS482. Mutation of serine 477 and glycine 479 prevented CKIɛ-dependent β-TrCP binding and slowed the degradation of phosphorylated mPER2. We hypothesize that phosphoserine 477 can substitute for the aspartic acid by mimicking its charged character. Recent observations suggest that the β-TrCP consensus sequence may be somewhat degenerate. A variant of the consensus sequence in which the spacing of the serines is increase by one position, DSGxxxS, has been identified in the transcription factor ATF4 (24). Other novel variations in the sequence recognized by β-TrCP have been recently identified. For example, the phosphorylation-dependent degradation of the mitotic regulatory kinase Wee1 is mediated by β-TrCP despite the lack of a recognizable β-TrCP recognition motif (48). It was postulated that a glutamic acid may mimic the conserved aspartic acid normally found in the β-TrCP consensus sequence. Interestingly, Drosophila PER has a similar SSGYX4S motif in its DBT binding domain, and its stability is also regulated by a CKI/β-TrCP-dependent mechanism (22).
The ability of IC261 and calyculin A to markedly alter the rate of phosphorylation and degradation of mPER2 suggests that intracellular mPER2 phosphorylation is a dynamic process. Furthermore, CKIɛ and the calyculin A-sensitive phosphatase are likely to be targeting the same residues since both IC261 and deletions that block CKIɛ binding inhibited the calyculin A-dependent degradation of mPER2. The net phosphorylation state and, hence, the stability of mPER2 may therefore be determined by alterations in either CKI or a protein phosphatase activity.
Using a cell-based assay system, we found that pharmacological inhibition of either CKI or the proteasome resulted in a significant change in the circadian period. Additionally, overexpression of CKIɛ(K38R) in synchronized fibroblasts lengthens the circadian period (E. L. Vielhaber and D. M. Virshup, unpublished data). This finding is in contrast to the tau hamster bearing a mutant CKIɛ with decreased kinase activity and a shortened circadian period. These opposing changes in period are consistent with data from Drosophila, where different double-time alleles, all of which decrease kinase activity (38), alternatively lengthen and shorten the period (39). We have shown previously that CKIɛ is also capable of phosphorylating Cryptochrome proteins and BMAL1 and have speculated that subtle changes in phosphorylation of different regulators may cause various effects on rhythm (9). The finding that proteasome inhibition and kinase inhibition both prolong the circadian period suggests while CKI may have diverse targets, its rate-limiting function, at least in Rat-1 fibroblasts, is to promote the degradation of mPER2 (or other labile circadian regulators). Prolonging the survival of mPER2 may prolong its repression of CLK/BMAL1, thereby delaying the next cycle of transcription.
Calyculin A inhibits the catalytic subunit of several phosphatases, including PP1, PP2A, and PP5 (16). In Drosophila, two forms of PP2A may regulate dPER protein abundance. mRNA transcripts for the PP2A regulatory subunits twins (tws) and widerborst (wdb) exhibit daily changes in abundance. The overexpression of either TWS or WDB blunts the amplitude of dPER protein expression and lengthens the period (42). In addition, a PP2A-related catalytic subunit has been implicated in the timing of flowering in Arabidopsis (20). Although these reports indicate that PP2A can be a regulator of circadian rhythm in plants and invertebrates, further study is required to identify the specific phosphatase acting on mPER2 in the mammalian clock and to determine if phosphatase activity may be regulated in a circadian manner.
Notably, CKIɛ activity itself is also regulated by phosphorylation (12, 43). CKIɛ is a positive regulator of Wnt signaling. When Wnt ligand was expressed in tissue culture cells, CKIɛ activity increased and inhibitory autophosphorylation decreased (43). CKIɛ can be similarly activated in brain slices and in cultured neurons by calcineurin, a cellular phosphatase insensitive to calyculin A (26). Hence, CKIɛ and perhaps the mPER2 phosphatase themselves may be regulated to ultimately control mPER2 phosphorylation and stability at different times in the circadian period.
Acknowledgments
We thank Dirk Bohmann and Richard Benarous for ubiquitin and β-TrCP expression constructs and Mariko Izumo and Carl Johnson for help with cell-based circadian rhythm assays. We also acknowledge Don Ayer for anti-mSin3A antibodies.
These studies were funded by NIH grant R01 GM60387 and the Huntsman Cancer Foundation. Oligonucleotide synthesis and DNA sequencing were supported in part by grant P04CA42104.
REFERENCES
- 1.Akashi, M., and E. Nishida. 2000. Involvement of the MAP kinase cascade in resetting of the mammalian circadian clock. Genes Dev. 14:645-649. [PMC free article] [PubMed] [Google Scholar]
- 2.Akashi, M., Y. Tsuchiya, T. Yoshino, and E. Nishida. 2002. Control of intracellular dynamics of mammalian period proteins by casein kinase I ɛ (CKIɛ) and CKIδ in cultured cells. Mol. Cell. Biol. 22:1693-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akten, B., E. Jauch, G. K. Genova, E. Y. Kim, I. Edery, T. Raabe, and F. R. Jackson. 2003. A role for CK2 in the Drosophila circadian oscillator. Nat. Neurosci. 6:251-257. [DOI] [PubMed] [Google Scholar]
- 4.Balsalobre, A., F. Damiola, and U. Schibler. 1998. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 93:929-937. [DOI] [PubMed] [Google Scholar]
- 5.Balsalobre, A., L. Marcacci, and U. Schibler. 2000. Multiple signaling pathways elicit circadian gene expression in cultured Rat-1 fibroblasts Curr. Biol. 10:1291-1294. [DOI] [PubMed] [Google Scholar]
- 6.Butcher, G. Q., H. Dziema, M. Collamore, P. W. Burgoon, and K. Obrietan. 2002. The p42/44 mitogen-activated protein kinase pathway couples photic input to circadian clock entrainment. J. Biol. Chem. 277:29519-29525. [DOI] [PubMed] [Google Scholar]
- 7.Camacho, F., M. Cilio, Y. Guo, D. Virshup, K. Patel, O. Khorkova, S. Styren, B. Morse, Z. Yao, and G. A. Keesler. 2001. Human casein kinase Iδ phosphorylation of human circadian clock proteins period 1 and 2. FEBS Lett. 489:159-165. [DOI] [PubMed] [Google Scholar]
- 8.Doi, M., T. Okano, I. Yujnovsky, P. Sassone-Corsi, and Y. Fukada. 2004. Negative control of circadian clock regulator E4BP4 by casein kinase Iɛ-mediated phosphorylation. Curr. Biol. 14:975-980. [DOI] [PubMed] [Google Scholar]
- 9.Eide, E. J., E. L. Vielhaber, W. A. Hinz, and D. M. Virshup. 2002. The circadian regulatory proteins BMAL1 and cryptochromes are substrates of casein kinase Iɛ. J. Biol. Chem. 277:17248-17254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eide, E. J., and D. M. Virshup. 2001. Casein kinase I: another cog in the circadian clockworks. Chronobiol. Int. 18:389-398. [DOI] [PubMed] [Google Scholar]
- 11.Gao, Z. H., J. Metherall, and D. M. Virshup. 2000. Identification of casein kinase I substrates by in vitro expression cloning screening. Biochem. Biophys. Res. Commun. 268:562-566. [DOI] [PubMed] [Google Scholar]
- 12.Gietzen, K. F., and D. M. Virshup. 1999. Identification of inhibitory autophosphorylation sites in casein kinase I ɛ. J. Biol. Chem. 274:32063-32070. [DOI] [PubMed] [Google Scholar]
- 13.Grima, B., A. Lamouroux, E. Chélot, C. Papin, B. Limbourg-Bouchon, and F. Rouyer. 2002. The F-box protein Slimb controls the levels of clock proteins Period and Timeless. Nature 420:178-182. [DOI] [PubMed] [Google Scholar]
- 14.Hart, M., J. P. Concordet, I. Lassot, I. Albert, R. del los Santos, H. Durand, C. Perret, B. Rubinfeld, F. Margottin, R. Benarous, and P. Polakis. 1999. The F-box protein β-TrCP associates with phosphorylated β-catenin and regulates its activity in the cell. Curr. Biol. 9:207-210. [DOI] [PubMed] [Google Scholar]
- 15.He, Q., P. Cheng, Y. Yang, H. Yu, and Y. Liu. 2003. FWD1-mediated degradation of FREQUENCY in Neurospora establishes a conserved mechanism for circadian clock regulation. EMBO J. 22:4421-4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honkanen, R. E., and T. Golden. 2002. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr. Med. Chem. 9:2055-2075. [DOI] [PubMed] [Google Scholar]
- 17.Izumo, M., C. H. Johnson, and S. Yamazaki. 2003. Circadian gene expression in mammalian fibroblasts revealed by real-time luminescence reporting: temperature compensation and damping. Proc. Natl. Acad. Sci. USA 100:16089-16094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson, P. K., A. G. Eldridge, E. Freed, L. Furstenthal, J. Y. Hsu, B. K. Kaiser, and J. D. Reimann. 2000. The lore of the RINGs: substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol. 10:429-439. [DOI] [PubMed] [Google Scholar]
- 19.Keesler, G. A., F. Camacho, Y. Guo, D. Virshup, C. Mondadori, and Z. Yao. 2000. Phosphorylation and destabilization of human period I clock protein by human casein kinase I ɛ. Neuroreport 11:951-955. [DOI] [PubMed] [Google Scholar]
- 20.Kim, D. H., J. G. Kang, S. S. Yang, K. S. Chung, P. S. Song, and C. M. Park. 2002. A phytochrome-associated protein phosphatase 2A modulates light signals in flowering time control in Arabidopsis. Plant Cell 14:3043-3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kloss, B., J. L. Price, L. Saez, J. Blau, A. Rothenfluh, C. S. Wesley, and M. W. Young. 1998. The Drosophila clock gene double-time encodes a protein closely related to human casein kinase Iɛ. Cell 94:97-107. [DOI] [PubMed] [Google Scholar]
- 22.Ko, H. W., J. Jiang, and I. Edery. 2002. Role for Slimb in the degradation of Drosophila Period protein phosphorylated by Doubletime. Nature 420:673-678. [DOI] [PubMed] [Google Scholar]
- 23.Kuhlman, S. J., J. E. Quintero, and D. G. McMahon. 2000. GFP fluorescence reports Period 1 circadian gene regulation in the mammalian biological clock. Neuroreport 11:1479-1482. [PubMed] [Google Scholar]
- 24.Lassot, I., E. Ségéral, C. Berlioz-Torrent, H. Durand, L. Groussin, T. Hai, R. Benarous, and F. Margottin-Goguet. 2001. ATF4 degradation relies on a phosphorylation-dependent interaction with the SCFβTrCP ubiquitin ligase. Mol. Cell. Biol. 21:2192-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee, C., J. P. Etchegaray, F. R. Cagampang, A. S. Loudon, and S. M. Reppert. 2001. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 107:855-867. [DOI] [PubMed] [Google Scholar]
- 26.Liu, F., D. M. Virshup, A. C. Nairn, and P. Greengard. 2002. Mechanism of regulation of casein kinase I activity by group I metabotropic glutamate receptors. J. Biol. Chem. 277:45393-45399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu, Y., J. Loros, and J. C. Dunlap. 2000. Phosphorylation of the Neurospora clock protein FREQUENCY determines its degradation rate and strongly influences the period length of the circadian clock. Proc. Natl. Acad. Sci. USA 97:234-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowrey, P. L., K. Shimomura, M. P. Antoch, S. Yamazaki, P. D. Zemenides, M. R. Ralph, M. Menaker, and J. S. Takahashi. 2000. Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science 288:483-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowrey, P. L., and J. S. Takahashi. 2000. Genetics of the mammalian circadian system: photic entrainment, circadian pacemaker mechanisms, and posttranslational regulation. Annu. Rev. Genet. 34:533-562. [DOI] [PubMed] [Google Scholar]
- 30.Margottin, F., S. P. Bour, H. Durand, L. Selig, S. Benichou, V. Richard, D. Thomas, K. Strebel, and R. Benarous. 1998. A novel human WD protein, h-βTrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1:565-574. [DOI] [PubMed] [Google Scholar]
- 31.Martinek, S., S. Inonog, A. S. Manoukian, and M. W. Young. 2001. A role for the segment polarity gene shaggy/GSK-3 in the Drosophila circadian clock. Cell 105:769-779. [DOI] [PubMed] [Google Scholar]
- 32.Mashhoon, N., A. J. DeMaggio, V. Tereshko, S. C. Bergmeier, M. Egli, M. F. Hoekstra, and J. Kuret. 2000. Crystal structure of a conformation-selective casein kinase-1 inhibitor. J. Biol. Chem. 275:20052-20060. [DOI] [PubMed] [Google Scholar]
- 33.Miyazaki, K., T. Nagase, M. Mesaki, J. Narukawa, O. Ohara, and N. Ishida. 2004. Phosphorylation of clock protein PER1 regulates its circadian degradation in normal human fibroblasts. Biochem. J. 380:95-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naidoo, N., W. Song, M. Hunter-Ensor, and A. Sehgal. 1999. A role for the proteasome in the light response of the timeless clock protein. Science 285:1737-1741. [DOI] [PubMed] [Google Scholar]
- 35.Obrietan, K., S. Impey, and D. R. Storm. 1998. Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nat. Neurosci. 1:693-700. [DOI] [PubMed] [Google Scholar]
- 36.Pickart, C. M. 2001. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70:503-533. [DOI] [PubMed] [Google Scholar]
- 37.Preitner, N., F. Damiola, L. Lopez-Molina, J. Zakany, D. Duboule, U. Albrecht, and U. Schibler. 2002. The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110:251-260. [DOI] [PubMed] [Google Scholar]
- 38.Preuss, F., J.-Y. Fan, M. Kalive, S. Bao, E. Schuenemann, E. S. Bjes, and J. L. Price. 2004. Drosophila doubletime mutations which either shorten or lengthen the period of circadian rhythms decrease the protein kinase activity of casein kinase I. Mol. Cell. Biol. 24:886-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Price, J. L., J. Blau, A. Rothenfluh, M. Abodeely, B. Kloss, and M. W. Young. 1998. double-time is a novel Drosophila clock gene that regulates PERIOD protein accumulation. Cell 94:83-95. [DOI] [PubMed] [Google Scholar]
- 40.Price, J. L., M. E. Dembinska, M. W. Young, and M. Rosbash. 1995. Suppression of PERIOD protein abundance and circadian cycling by the Drosophila clock mutation timeless. EMBO J. 14:4044-4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reppert, S. M., and D. R. Weaver. 2002. Coordination of circadian timing in mammals. Nature 418:935-941. [DOI] [PubMed] [Google Scholar]
- 42.Sathyanarayanan, S., X. Zheng, R. Xiao, and A. Sehgal. 2004. Posttranslational regulation of Drosophila PERIOD protein by protein phosphatase 2A. Cell 116:603-615. [DOI] [PubMed] [Google Scholar]
- 43.Swiatek, W., I.-C. Tsai, L. Klimowski, A. Pepler, J. Barnette, H. J. Yost, and D. M. Virshup. 2004. Regulation of casein kinase Iɛ activity by Wnt signaling. J. Biol. Chem. 279:13011-13017. [Online.] [DOI] [PubMed] [Google Scholar]
- 44.Toh, K. L., C. R. Jones, Y. He, E. J. Eide, W. A. Hinz, D. M. Virshup, L. J. Ptacek, and Y.-H. Fu. 2001. An hPER2 phosphorylation site mutation in familial advanced sleep-phase syndrome. Science 291:1040-1043. [DOI] [PubMed] [Google Scholar]
- 45.Treier, M., L. M. Staszewski, and D. Bohmann. 1994. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 78:787-798. [DOI] [PubMed] [Google Scholar]
- 46.Vallone, D., S. B. Gondi, D. Whitmore, and N. S. Foulkes. 2004. E-box function in a period gene repressed by light. Proc. Natl. Acad. Sci. USA 101:4106-4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vielhaber, E., E. Eide, A. Rivers, Z.-H. Gao, and D. M. Virshup. 2000. Nuclear entry of the circadian regulator mPER1 is controlled by mammalian casein kinase I ɛ. Mol. Cell. Biol. 20:4888-4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watanabe, N., H. Arai, Y. Nishihara, M. Taniguchi, T. Hunter, and H. Osada. 2004. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFβ-TrCP. Proc. Natl. Acad. Sci. USA 101:4419-4424. [Online.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu, G., G. Xu, B. A. Schulman, P. D. Jeffrey, J. W. Harper, and N. P. Pavletich. 2003. Structure of a β-TrCP1-Skp1-β-catenin complex: destruction motif binding and lysine specificity of the SCFβ-TrCP1 ubiquitin ligase. Mol. Cell 11:1445-1456. [DOI] [PubMed] [Google Scholar]
- 50.Yagita, K., F. Tamanini, M. Yasuda, J. H. Hoeijmakers, G. T. van der Horst, and H. Okamura. 2002. Nucleocytoplasmic shuttling and mCRY-dependent inhibition of ubiquitylation of the mPER2 clock protein. EMBO J. 21:1301-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamazaki, S., R. Numano, M. Abe, A. Hida, R. Takahashi, M. Ueda, G. D. Block, Y. Sakaki, M. Menaker, and H. Tei. 2000. Resetting central and peripheral circadian oscillators in transgenic rats. Science 288:682-685. [DOI] [PubMed] [Google Scholar]
- 52.Yang, Y., P. Cheng, Q. He, L. Wang, and Y. Liu. 2003. Phosphorylation of FREQUENCY protein by casein kinase II is necessary for the function of the Neurospora circadian clock. Mol. Cell. Biol. 23:6221-6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang, Y., P. Cheng, and Y. Liu. 2002. Regulation of the Neurospora circadian clock by casein kinase II. Genes Dev. 16:994-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yaron, A., A. Hatzubai, M. Davis, I. Lavon, S. Amit, A. M. Manning, J. S. Andersen, M. Mann, F. Mercurio, and Y. Ben-Neriah. 1998. Identification of the receptor component of the IκBα-ubiquitin ligase. Nature 396:590-594. [DOI] [PubMed] [Google Scholar]