

Abstract
The c-Jun NH2-terminal kinase (JNK)-interacting protein (JIP) group of scaffold proteins (JIP1, JIP2, and JIP3) can interact with components of the JNK signaling pathway and potently activate JNK. Here we describe the identification of a fourth member of the JIP family. The primary sequence of JIP4 is most closely related to that of JIP3. Like other members of the JIP family of scaffold proteins, JIP4 binds JNK and also the light chain of the microtubule motor protein kinesin-1. However, the function of JIP4 appears to be markedly different from other JIP proteins. Specifically, JIP4 does not activate JNK signaling. In contrast, JIP4 serves as an activator of the p38 mitogen-activated protein (MAP) kinase pathway by a mechanism that requires the MAP kinase kinases MKK3 and MKK6. The JIP4 scaffold protein therefore appears to be a new component of the p38 MAP kinase signaling pathway.
Seminal studies of the yeast Saccharomyces cerevisiae have established the concept that mitogen-activated protein (MAP) kinase (MAPK) signaling pathways can be assembled into functional signaling modules by protein-protein interactions (5). Examples include studies on the role of the Ste5p scaffold protein in the mating MAP kinase signaling pathway (5) and the role of Pbs2p in the osmosensing MAP kinase signaling pathway (22). Furthermore, it has been demonstrated that an artificial scaffold in yeast can direct both activation and signaling specificity within a MAP kinase signaling module (8, 21).
Recent studies of mammalian cells have demonstrated that scaffolding interactions can participate in the regulation of MAP kinase signaling modules (19). Thus, the KSR scaffold protein (20) and the MP1 scaffold protein, together with the accessory proteins Morg1 and p14 (14, 23, 28, 32, 35), can organize signaling by the ERK group of MAP kinases. Similarly, the OSM scaffold protein can coordinate signaling by the p38 MAP kinase signaling pathway (30). Furthermore, a number of potential scaffold proteins have been implicated in the regulation of JNK signaling modules (19), including members of the JIP protein family (37).
JIP1 is the founding member of the c-Jun NH2-terminal kinase (JNK)-interacting protein (JIP) group of scaffold proteins and is structurally related to JIP2 (4, 33, 37). Gene disruption studies with mice (9, 29, 34) have demonstrated that JIP1 contributes to JNK activation in neurons following exposure to an anoxic insult (9, 34) and is also required in adipose tissue for JNK activation in models of type II diabetes (11). Together, these data provide strong genetic evidence that the JIP proteins can function to regulate JNK signaling. However, and very importantly, these data also establish that JIP proteins are required for the ability of selective stimuli to activate JNK and are not core components of the JNK signaling pathway that are essential for multiple stimuli to activate JNK (19).
More recently, a new member of the JIP protein kinase group, JIP3, was identified (10, 13). JIP3 is structurally distinct from JIP1 and JIP2, but it shares many of the biochemical properties of JIP1 and JIP2. Thus, all three JIP proteins bind JNK, the MAP kinase kinase (MAP2K) MKK7, and members of the mixed-lineage protein kinase group of MAP kinase kinase kinases (MAP3Ks), and all three JIP proteins can strongly activate JNK signaling (10, 13). In addition, all three JIP proteins interact with the tetratricopeptide repeat (TPR) domain of the light chain of the microtubule motor protein kinesin-1 and can be transported as cargo molecules along microtubule networks within cells (1, 31, 34). Targeted disruption of the Jip3 gene in mice causes major defects in the development of the brain, including defects in the telencephalic commissure, and death immediately following birth because of a failure to breathe (12).
Sequences of cDNA related to JIP3, including JIP3β, JIP3γ, Syd-1, SPAG9, and JLP (1, 13, 15, 26), have been identified by searching libraries and databases. Here we describe a new JIP3-related protein (JIP4) that shares many of the properties of JIP3, such as its ability to bind to JNK and kinesin light chain. However, the function of JIP4 appears to differ from those of the other members of the JIP protein family because it does not activate JNK. In contrast, we found that JIP4 can activate p38 MAP kinase.
MATERIALS AND METHODS
Molecular cloning of JIP4.
The JIP4 cDNA was isolated from a mouse testis λZAPII library (Stratagene) by plaque hybridization using a 32P-labeled JIP3 cDNA probe. The largest clone obtained (3,711 bp) included the complete open reading frame of JIP4 (1,142 amino acids) with in-frame termination codons in the 5′ and 3′ noncoding regions. Sequence analysis was performed using an Applied Biosystems machine.
Plasmids.
The expression vectors for MAPK, MAP2K, MAP3K, JIP1, JIP2, JIP3, and KLC were described previously (13, 34). Expression vectors for JIP4 were constructed by subcloning PCR fragments in the plasmids pCDNA3 (Invitrogen Inc.) and pGEX-5X1 (Amersham Pharmacia Biotech Inc.).
Antibodies.
The antibodies to the Flag epitope tag (M2; Sigma Chemical Co.), the hemagglutinin (HA) epitope tag (12CA5; Roche), the T7 epitope tag (Novagen Inc.), the V5 epitope tag (Invitrogen Inc.), glutathione S-transferase (GST; Pharmacia-LKB Biotech Inc.), α-tubulin (Sigma Chemical Co.), and protein disulfide isomerase (Stressgen) were purchased from indicated suppliers. Antibodies to Grasp 65, STT3, and KLC were provided by C. Suetterlin, R. Gilmore, and L. S. Goldstein, respectively. Antibodies to p38α MAP kinase (Pharmingen), JNK1 (Santa Cruz), and ERK2 (Santa Cruz) were used for immune complex kinase assays. Antibodies to MKK3 (Santa Cruz) and MKK6 (Cell Signaling) were used for immunoblot analysis.
Cell culture.
Murine embryo fibroblasts (MEF) were prepared from wild-type, Mkk3−/− (16, 36), Mkk6−/− (27), and Mkk3−/− Mkk6−/− (2) embryos. These MEF and COS7 cells (obtained from the American Type Culture Collection) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum in an incubator with humidified air (5% CO2) at 37°C. Transfection with plasmid expression vectors was performed using the Lipofectamine reagent (Invitrogen). PC12 cells (obtained from the American Type Culture Collection) were cultured at 37°C on collagen-coated plastic dishes in DMEM supplemented with 0.5 mM glutamine, 10% horse serum, and 5% fetal bovine serum (Invitrogen) in a humidified incubator (10% CO2). The cells were differentiated by incubation in DMEM supplemented with 1% horse serum, 0.5 mM glutamine, and 50 ng of nerve growth factor per ml.
Biochemical assays.
Cultured cells were extracted in Triton lysis buffer (20 mM Tris-Cl [pH 7.4], 137 mM sodium chloride, 2 mM EDTA, 25 mM β-glycerophosphate, 2 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 μg of leupeptin/ml, 5 μg of aprotinin/ml, 10% glycerol, 1% Triton X-100). Proteins were immunoprecipitated by incubation with antibodies immobilized on protein G-Sepharose beads (Amersham Pharmacia Biotech Inc.) for 3 h at 4°C. Immunoblot analysis was carried out by enhanced chemical luminescence detection (Kirkegaard & Perry Inc.). GST fusion proteins were purified by incubation with glutathione-agarose beads (Amersham Pharmacia Biotech Inc.). Methods for binding assays using in vitro-translated proteins and immune complex kinase assays were described earlier (13, 33, 37). The effect of scaffold proteins on JNK activity was examined in COS7 cell transfection assays using the in-gel method with 250 μg of GST-c-Jun per ml polymerized in the gel (13).
Subcellular fractionation.
Cultured cells were fractionated into membrane and soluble fractions by suspension in hypotonic lysis buffer (5 mM Tris-Cl [pH 7.5], 1 mM MgCl2, 0.1 mM EDTA, 1 mM EGTA, 5 μg of aprotinin/ml, 10 μg of leupeptin/ml) by incubation on ice and passage through a 21-gauge needle. The extracts were clarified by centrifugation at a low speed (1,000 × g) and loaded onto a 20% (wt/vol) sucrose cushion (in lysis buffer) and centrifuged at a relative centrifugal force of 350,000 in an airfuge (Beckman Instruments). The membrane fraction was dissolved in 1% sodium dodecyl sulfate (SDS). The proteins (75 μg) were examined by immunoblot analysis.
Immunofluorescence studies.
Cells were grown on glass coverslips, fixed by incubation at −20°C in cold methanol (10 min), and permeabilized with 0.2% Triton X-100 in phosphate-buffered saline (PBS). After blocking by incubation in 3% (wt/vol) bovine serum albumin (BSA) in PBS (1 h), the coverslips were incubated at 4°C (16 h) in primary antibodies diluted in 3% BSA plus 2 U of RNase One (Promega) per ml. The coverslips were washed in 0.5% Tween 20 in PBS and incubated in tetramethyl rhodamine isocyanate-conjugated anti-rabbit immunoglobulin and fluorescein-conjugated anti-mouse immunoglobulin secondary antibodies (Jackson ImmunoResearch Inc.) and nucleic acid marker TOTO3 (Molecular Probes) in 3% BSA-PBS (1 h). The coverslips were mounted in Vectashield (Vector Laboratories Inc.). Fluorescence microscopy was performed with a Leica confocal microscope.
Nucleotide sequence accession number.
The sequence of the full-length cDNA encoding JIP4 was deposited in GenBank with accession number AY823270.
RESULTS
Molecular cloning of JIP4.
We have previously reported the molecular cloning of the scaffold protein JIP3 (13). Database analysis indicated the presence of partial cDNA fragments corresponding to a JIP3-related gene (13). We therefore screened a cDNA library to obtain a full-length clone. The largest clone obtained (3,711 bp) included the complete open reading frame for a large protein (1,142 amino acids) that is related to JIP3, with in-frame termination codons in the 5′ and 3′ untranslated regions. We have named this protein JIP4. The sequence alignment of JIP3 and JIP4 indicated similarities that extended from the NH2 terminus to the COOH terminus (Fig. 1A; see Fig. S1 in the supplemental material). The major difference between JIP3 and JIP4 was that JIP3 contained an NH2-terminal extension (170 amino acids) that was not present in JIP4. A computer-assisted sequence analysis of JIP3 and JIP4 revealed the presence of an extended coiled-coil domain and a leucine zipper in the NH2-terminal region together with a JNK binding domain, a JNK phosphorylation domain, and a putative transmembrane domain that are conserved in JIP3 and JIP4 (Fig. 1A; see Fig. S1 in the supplemental material).
FIG. 1.

Primary structures and expression levels of JIP3 and JIP4. (A) Schematic representation of the domain structures of JIP3 and JIP4. We have previously reported the primary sequence of JIP3 (GenBank accession numbers AF178636 and AF178637) (13). We now report the primary sequence of JIP4 (GenBank accession number AY823270). TM, transmembrane. (B) Northern blot analysis of 2 μg of poly(A)+ RNA isolated from several murine tissues. The blot was probed with double-stranded probes derived from JIP3, JIP4, and β-actin. The migration of RNA standards is indicated on the left.
A comparison of the deduced sequence of the JIP4 mRNA and the mouse genomic sequence (GenBank) revealed that Jip4 is a large gene which is located on chromosome 11 and that this gene consists of 31 exons (Fig. 2). The Jip4 gene encodes at least three proteins (JIP4, JLP, and SPAG9) that differ in utilization of 5′ exons but share common 3′ exons. The 5′ untranslated region of the JIP4 mRNA includes sequences derived from exons 2 through 5 (with an in-frame termination codon in exon 3 and an initiation codon in exon 5) that are fused to sequences derived from exons 7 through 31. In contrast, the 5′ region of the JLP mRNA (15) is encoded by exon 1 (with no in-frame termination codon but with an initiation codon in exon 1) that is fused to sequences derived from exons 2, 4, 5, and 7 through 31. Finally, the sperm-associated antigen 9 protein (SPAG9) mRNA (26) is encoded by exon 6 (with no in-frame termination codon but with an initiation codon in exon 6) that is fused to sequences derived from exons 7 to 31. These data indicate that the JIP4 protein is a novel product of the Jip4 gene and shares some similarities with both JLP and SPAG9.
FIG. 2.

Several proteins are encoded by different transcripts derived from the Jip4 gene by alternative splicing. The gene that encodes JIP4 is located on mouse chromosome 11 and consists of a total of 31 exons. The structure of the gene is illustrated schematically. The gene encodes three proteins (JIP4, JLP, and SPAG9) that are encoded by the selective inclusion of different 5′ exons of the Jip4 gene. The JIP4 mRNA is encoded by exons 2, 3, 4, 5, and 7 through 31 and includes an in-frame termination codon in the 5′ untranslated region. The JLP mRNA is encoded by exons 1, 2, 4, 5, and 7 through 31. The SPAG9 mRNA is encoded by exons 6 and 7 through 31. In-frame termination codons in the 5′ untranslated region of JLP and SPAG9 are not present. The GenBank entries for the genes that encode JIP1, JIP2, and JIP3 are designated Mapk8ip1, Mapk8ip2, and Mapk8ip3. This nomenclature suggests that the GenBank entry for the gene that encodes JIP4 should be designated Mapk8ip4.
Expression of JIP4 in murine tissues.
The expression of JIP4 mRNA in different murine tissues was examined by Northern blot analysis (Fig. 1B). Two major transcripts, of 4.8 and 8.9 kb, were detected. The 4.8-kb RNA is most abundant in the testis, while the larger transcript of 8.9 kb is more abundant in the brain, kidney, liver, heart, and other tissues. This broad tissue distribution of JIP4 differs from the selective expression of JIP3 in the brain and at low levels in the heart and testis.
JIP3 and JIP4 are soluble cytoplasmic proteins.
We have previously identified JIP3 as a soluble cytoplasmic protein (13). However, this conclusion has been questioned in a recent study that reported the identification of JIP3 as a transmembrane protein (1). Furthermore, the JIP4-related protein SPAG9 has been reported to be a cell surface membrane protein (26). Indeed, hydropathy plots indicate that JIP4 contains a putative transmembrane domain (residues 820 through 836) (Fig. 1A; see Fig. S1 in the supplemental material). The sequence of the corresponding region of JIP3 (residues 985 through 1001) is similar but is significantly less hydrophobic because one of the residues in this region of JIP4 (Ile) is replaced with a charged residue in JIP3 (Arg) (Fig. 1A; see Fig. S1 in the supplemental material). The primary sequence of JIP4 and possibly that of JIP3 therefore suggest that these proteins could span a membrane.
We examined whether JIP3 and JIP4 are integral membrane proteins and whether these proteins are soluble in the cytoplasm. Membrane and soluble fractions of cultured cells were prepared. Immunoblot analysis indicated that JIP3 and JIP4 were present in the soluble fractions (Fig. 3A and B). Similar results were obtained when the endogenous JIP3 and JIP4 proteins were examined (Fig. 3B) and when ectopically expressed JIP3 and JIP4 were examined (Fig. 3A). Control studies confirmed that the microsomal membrane protein STT3 was detected in the membrane fraction (Fig. 3B). This analysis indicated that while JIP4 has the potential to be a membrane protein, both JIP3 and JIP4 were detected in cultured cells as soluble proteins.
FIG. 3.

The cytoplasmic proteins JIP3 and JIP4 interact with kinesin light chain. (A) JIP3 and JIP4 are cytoplasmic proteins. Flag-tagged JIP proteins were expressed in COS cells. Cytosolic (C) and membrane (M) fractions of the cells were prepared and examined by an immunoblot analysis using antibodies to the Flag-tagged JIP3 and JIP4. (B) Nerve growth factor-differentiated PC12 cells were separated into cytosolic (C) and membrane (M) fractions. Endogenously expressed JIP3 and JIP4 were examined by immunoblot analysis using antibodies to JIP3 and JIP4. An immunoblot analysis using an antibody to the microsomal membrane protein STT3 was used as a control for cell fractionation. (C) Flag-tagged JIP3 and JIP4, together with V5 epitope-tagged kinesin light chain 1 (KLC), were expressed in COS cells. The cell lysates were examined by immunoprecipitation (IP) and immunoblot (IB) analyses using the indicated epitope tag antibodies. (D) COS cells expressing epitope-tagged JIP1, JIP2, JIP3, or JIP4 were incubated in serum or were serum starved (24 h). Cell lysates were prepared and incubated with the bacterially expressed TPR domain of KLC fused to GST. Protein complexes were collected on glutathione-agarose, and bound Flag-tagged JIP proteins were detected by immunoblot analysis. (E and F) The leucine zipper regions of JIP3 and JIP4 interact with the TPR domain of KLC. Bacterially expressed GST and GST-TPR domain of KLC (GST-KLC) were immobilized on glutathione-agarose and incubated with equal amounts of in vitro-translated and [35S]methionine-labeled JIP3 or JIP4. Control experiments were performed using [35S]methionine-labeled MLK3 and luciferase. The bound proteins were detected by SDS-PAGE and autoradiography.
To confirm that JIP3 and JIP4 are soluble cytoplasmic proteins, we examined the subcellular distributions of JIP3 and JIP4 by immunofluorescence analysis. The JIP4 antibody we prepared was useful for an immunoblot analysis of the endogenous expression of JIP4 (Fig. 3B), but it was not useful for an immunocytochemical analysis because of nonspecific background staining (data not shown). We therefore performed an immunofluorescence analysis using transfected cultured cells that express recombinant JIP3 and JIP4. Immunofluorescence microscopy demonstrated that JIP3 and JIP4 were distributed throughout the cytoplasm (Fig. 4). Disruption of microtubules with nocodazole caused a more punctate staining pattern for both JIP3 and JIP4, but the staining remained restricted to the cytoplasm (Fig. 4A). To test whether JIP3 or JIP4 might colocalize with an organelle, we compared the distributions of JIP3 and JIP4 with markers for mitochondria (mitotracker dye), Golgi (Grasp 65), and the endoplasmic reticulum (protein disulfide isomerase). This analysis did not demonstrate an association of JIP3 and JIP4 with these organelles (Fig. 4B). Together, these data indicate that JIP3 and JIP4 are broadly distributed cytoplasmic proteins.
FIG. 4.

Subcellular localization of JIP3 and JIP4. (A) COS cells transfected with Flag-tagged JIP3 or JIP4 were fixed and processed for confocal microscopy. Control or nocodazole-treated cells were stained (green) with antibodies to Flag-tagged JIP3, Flag-tagged JIP4, or α-tubulin. The nucleus was identified by staining DNA with 4,6-diamidino-2-phenylindole (blue). (B) The possible colocalization of JIP3 or JIP4 (green) with organelles was examined by staining (red) with mitotracker red (mitochondria) or antibodies to protein disulfide isomerase (endoplasmic reticulum) or Grasp 65 (Golgi).
JIP4 interacts with kinesin light chain.
JIP3 is known to interact with kinesin light chain (KLC1), a component of the microtubule motor protein kinesin-1 (1). To test whether JIP4 also interacts with kinesin-1, we performed a coimmunoprecipitation analysis. These assays demonstrated that KLC1 coimmunoprecipitated with both JIP3 and JIP4 (Fig. 3C). Interestingly, the interaction of KLC1 with JIP4 was greater than that that detected for KLC1 with JIP3.
The TPR domain of KLC1 has been suggested to be responsible for cargo binding, and this domain of KLC1 does interact with JIP1, JIP2, and JIP3 (1, 31, 34). To test whether JIP4 also binds to the TPR domain of KLC1, we examined the interactions of JIP3 and JIP4 with KLC1 in vitro. These data demonstrated that the TPR domain of KLC1 binds to both JIP3 and JIP4 (Fig. 3E). Binding of JIP3 and JIP4 to other domains of KLC1 was not observed (data not shown). To identify the region of JIP3 and JIP4 that interacts with the TPR domain of KLC1, we examined the effect of deletion mutations in JIP3 and JIP4 on their binding to KLC1 (Fig. 3F and data not shown). This analysis demonstrated that the leucine zipper domain of JIP3 and JIP4 was required for their binding to KLC1 (Fig. 3F). Together, these data demonstrate that binding to the TPR domain of KLC1 is mediated by the leucine zipper domain of JIP3 and JIP4. This interaction contrasts with those of JIP1 and JIP2, which bind to the TPR domain of KLC1 by an interaction that is mediated by the COOH terminus of JIP1 and the COOH terminus of JIP2 (31, 34). Thus, although the TPR domain of KLC1 binds to all four JIP proteins, the modes of binding by the TPR domain to JIP1 and JIP2 and to JIP3 and JIP4 are distinct.
To examine whether the interaction of KLC1 with JIP proteins might be regulated by extracellular stimuli, we examined the effect of serum starvation on the coimmunoprecipitation of JIP proteins with KLC1 (Fig. 3D). Serum starvation (24 h) was found to cause decreased interactions of JIP3 and JIP4 with KLC1. Similarly, a small decrease in KLC1 binding to JIP1 was detected, but no change in the interaction of JIP2 with KLC1 was observed. These observations confirm that the regulation of the interaction of JIP4 and KLC1 is similar to the regulation of the interaction of KLC1 and other JIP proteins, particularly JIP3.
JIP4 binds JNK but does not interact with MKK7 or MLK3.
The sequence similarity between JIP3 and JIP4 indicates that these proteins may have similar functions. It is established that the JNK binding domain of JIP3 interacts with JNK (13). The JNK binding domain of JIP3 is conserved in JIP4 (Fig. 1A) and, as expected, similar amounts of JNK were found to coimmunoprecipitate with JIP3 and JIP4 (Fig. 5A). Like JIP3 (13), JIP4 was not found to coimmunoprecipitate with other members of the MAP kinase family, including ERK2 and p38 MAP kinase (Fig. 5A). These observations confirm that JIP3 and JIP4 may have similar functions.
FIG. 5.

Comparison of protein interactions of JIP3 and JIP4. T7-tagged JIP3 or JIP4 was expressed in COS cells together with HA-tagged JNK1, p38α MAP kinase, and ERK2 (A), Flag-tagged MKK7 (B), HA-tagged MLK3 (C), or HA-tagged ASK1 (D). The JIP proteins were isolated from the cells by immunoprecipitation (IP) with the T7 antibody. JNK, MLK3, and MKK7 binding were examined by immunoblot (IB) analysis.
JIP3 also interacts with the MAP2K isoform MKK7 and the MAP3K isoform MLK3 (13). Comparative studies confirmed the interaction of JIP3 with MKK7 and MLK3, but no interaction of JIP4 with these MAP2K and MAP3K isoforms was detected (Fig. 5B and C).
Coimmunoprecipitation assays demonstrated that JIP4 did interact with the MAP3K ASK1 (Fig. 5D). We have previously reported that JIP3 does not bind ASK1 in a GST pull-down assay (13). To confirm this conclusion, we compared the interaction of ASK1 with JIP3 to the interaction of ASK1 with JIP4 by coimmunoprecipitation analysis (Fig. 5D). These assays demonstrated that ASK1 binds JIP3 more strongly than JIP4. It is most likely that our previous failure to detect an interaction between ASK1 and JIP3 was caused by the use of a GST-fusion protein strategy in our earlier experiments. Our coimmunoprecipitation analysis demonstrates that ASK1 can bind both JIP3 and JIP4 (Fig. 5D).
JIP4 is not a scaffold for the JNK-MKK7-MLK3 signaling module.
The absence of binding of JIP4 to MKK7 and MLK3 suggested that JIP4 may not function to assemble a functional JNK signaling module. To test this hypothesis, we compared the abilities of JIP3 and JIP4 to potentiate the activation of JNK by MLK3. This analysis demonstrated that, as expected, JIP3 strongly increased MLK3-stimulated JNK activation (Fig. 6). However, JIP4 was ineffective as an activator of JNK in this assay (Fig. 6).
FIG. 6.
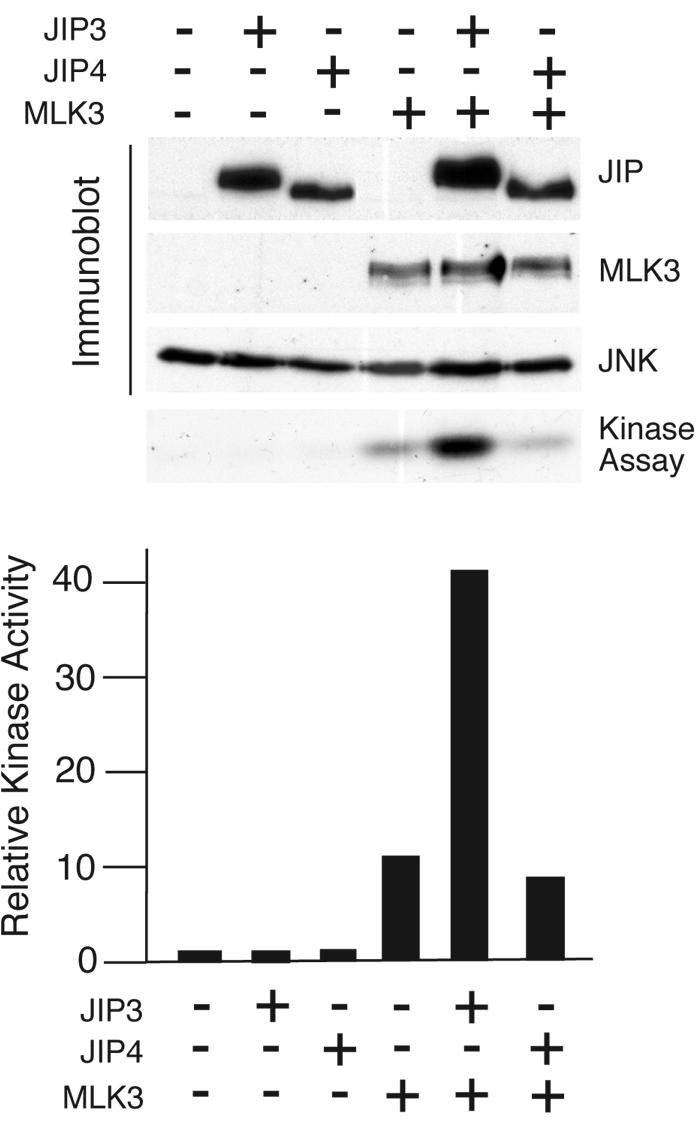
JIP4 is not a scaffold for the MLK3-MKK7-JNK module. The effect of JIP3 and JIP4 on MLK3-stimulated JNK1 activity was examined in a cotransfection assay using COS cells. The levels of expression of JIP3, JIP4, MLK3, and JNK were examined by immunoblot analysis (upper panel). JNK1 activity was measured by using an immune complex kinase assay with [γ-32P]ATP and c-Jun as the substrates. The phosphorylated c-Jun was detected following SDS-PAGE by autoradiography (middle panel) and quantitated by PhosphorImager (Molecular Dynamics) analysis (lower panel). The data presented were derived from one experiment. Similar data were obtained in three independent experiments.
We have previously reported that JIP proteins function as oligomers (most likely dimers) (13, 37). If JIP4 is a nonfunctional JNK scaffold protein, it is possible that JIP4 might act as a natural antagonist of the JNK scaffold activity of other JIP proteins. JIP1 and JIP2 form homo- and hetero-oligomers (37). JIP3 can form homo-oligomers and can also interact with JIP2 (13). Coimmunoprecipitation assays demonstrated that JIP4 can also form homo-oligomers, but no interaction of JIP4 with JIP1, JIP2, or JIP3 was detected (Fig. 7). The lack of an interaction between JIP4 and other JIP proteins suggests that JIP4 most likely does not act as an antagonist but is probably inactive as a JNK scaffold. Indeed, direct measurements of JIP-mediated potentiation of MLK3-stimulated JNK signaling demonstrated that the coexpression with JIP4 did not alter JNK activation (Fig. 6).
FIG. 7.
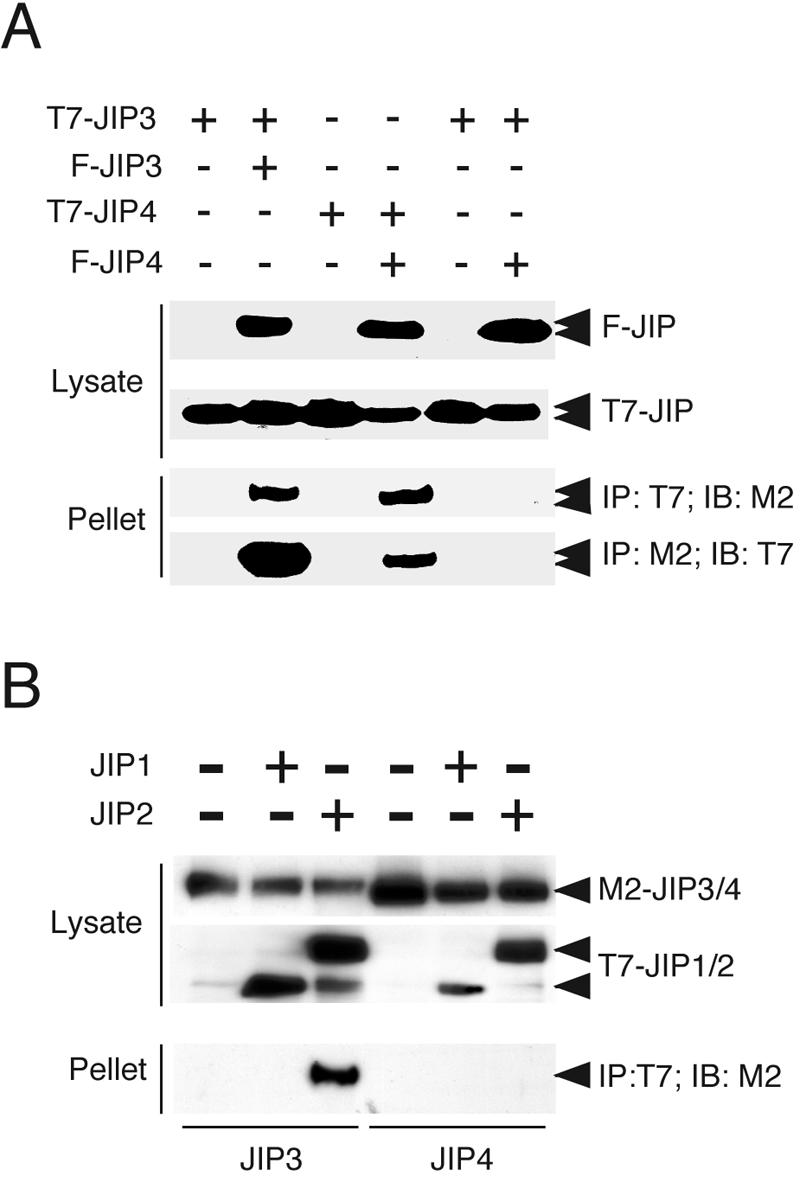
JIP4 forms homo-oligomers but does not interact with other JIP proteins. The interactions between JIP proteins were examined. Flag-tagged JIP3 and JIP4 proteins were coexpressed with T7-tagged JIP3 or JIP4 (A) and JIP1 or JIP2 (B). The JIP3 and JIP4 proteins were immunoprecipitated (IP) with the Flag (M2) or T7 antibody. The coimmunoprecipitation of other JIP proteins was examined by immunoblot (IB) analysis. JIP3/4, JIP3 and JIP4; JIP1/2, JIP1 and JIP2.
JIP4 is phosphorylated by JNK and p38 MAP kinase.
It is established that JNK phosphorylates JIP3 on three sites and that JIP3 is not a substrate for ERK2 or p38α MAP kinase (13). Two of these three phosphorylation sites on JIP3 (Thr-267 and Thr-288) are conserved in JIP4 (Thr-97 and Thr-113), and JIP4 also has two additional potential phosphorylation sites (Thr-52 and Thr-61) (Fig. 1A; see Fig. S1 in the supplemental material). The presence of these possible phosphorylation sites suggests that the functional significance of the interaction of JIP4 with JNK (Fig. 5A) is that JIP4 is a JNK substrate. To test this hypothesis, we examined JIP4 phosphorylation in an in vitro kinase assay. This analysis demonstrated that JIP4 is a substrate for JNK (Fig. 8A). JIP4 is also phosphorylated by p38α MAP kinase but not by ERK2 (Fig. 8A). A mutational analysis demonstrated that point mutations in any one of the four potential phosphorylation sites did not prevent JIP4 phosphorylation by JNK or p38α MAP kinase (data not shown). In contrast, the simultaneous replacement of all four phosphorylation sites with Ala residues prevented the phosphorylation of JIP4 by JNK and p38α MAP kinase (Fig. 8B). Together, these data indicate that the sites of JNK phosphorylation are conserved in JIP3 and JIP4. Furthermore, JIP4, but not JIP3, is also phosphorylated on these sites by p38α MAP kinase.
FIG. 8.
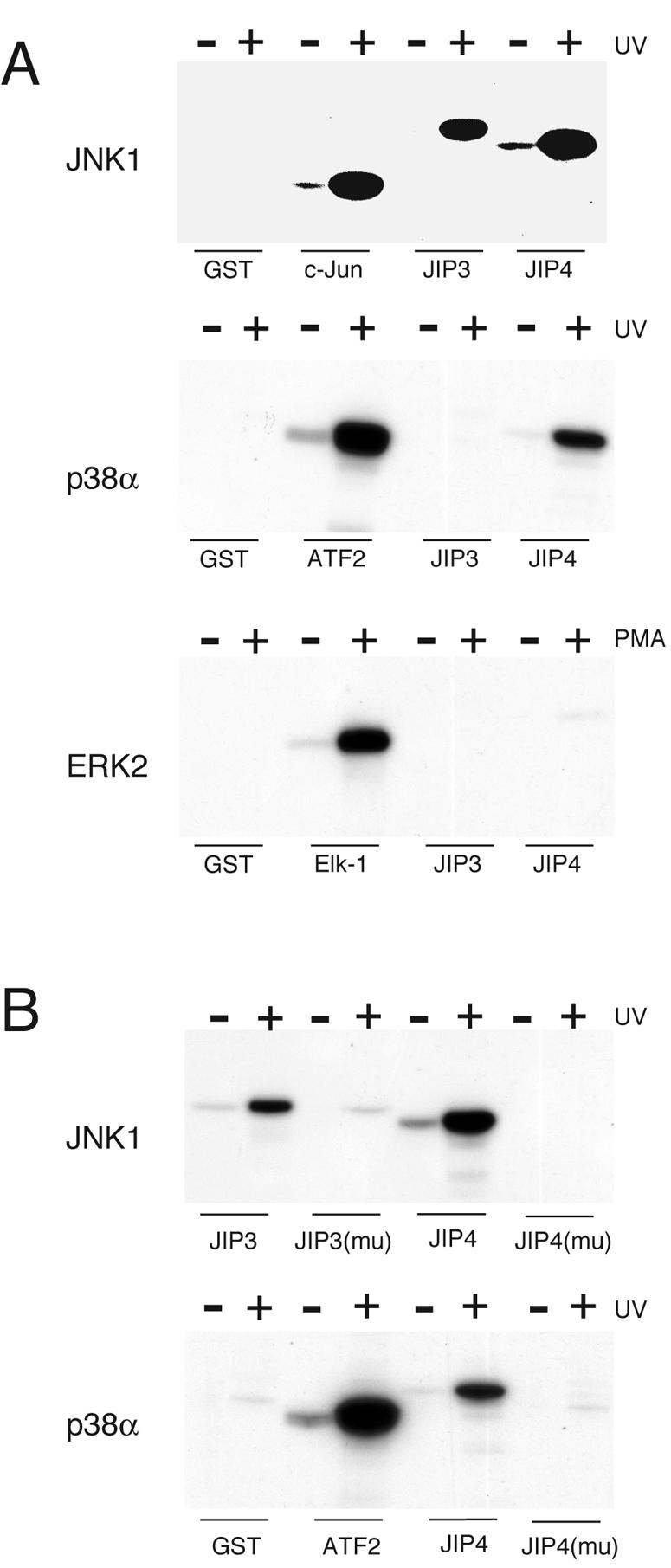
JIP4 is a substrate for JNK and p38 MAP kinase but not ERK2. (A) JIP4 is a substrate for JNK and p38α MAPK but not ERK2. UV-stimulated JNK1 (upper panel) and p38α MAPK (middle panel), together with phorbol myristate acetate (PMA)-stimulated ERK2 (lower panel), were employed in immune complex kinase assays using [γ-32P]ATP and GST-JIP3 or GST-JIP4 as the substrates. Control experiments were performed using known substrates for JNK (c-Jun), p38α MAPK (ATF2), and ERK2 (Elk-1). The phosphorylation of the substrates was examined by SDS-PAGE and autoradiography. (B) Immune complex kinase assays were performed using wild-type JIP3 and a mutated JIP3 [JIP3(mu)] protein in which the three sites of phosphorylation by JNK were replaced with Ala (Thr-266, Thr-276, and Thr-287). These phosphorylation sites are conserved in JIP4 (Thr-52, Thr-61, Thr-96, and Thr-112). These Thr phosphorylation sites were replaced with Ala residues. Wild-type JIP4 and the mutant JIP4 protein [JIP4(mu)] were examined as substrates for JNK and p38α MAP kinase. Control experiments were performed using wild-type and mutant JIP3 proteins for JNK assays (upper panel) and ATF2 for p38α MAP kinase assays (lower panel).
JIP4 can potentiate the activation of p38 MAP kinase.
Although coimmunoprecipitation analysis indicates that complexes of JIP4 with p38α MAP kinase were not detected (Fig. 5A), the finding that JIP4 is a p38α MAP kinase substrate demonstrates that JIP4 does interact with p38 MAP kinase (Fig. 8A). It is most likely that the failure to detect this interaction by coimmunoprecipitation analysis is a consequence of low affinity. To test this hypothesis, we examined the interactions of JNK and p38 MAP kinase with JIP3 and JIP4 by using [35S]methionine-labeled in vitro-translated protein kinases and GST-JIP fusion proteins in a more sensitive pull-down assay. This analysis demonstrated the binding of JNK1, JNK2, and JNK3 to JIP3 and JIP4 (Fig. 9A). Interestingly, the binding of JNK to JIP4 was greater than that of JNK to JIP3. No interaction between JIP3 and any of the four p38 MAP kinase isoforms was detected. However, low levels of JIP4 binding to p38α and p38β MAP kinases were observed (Fig. 9A). No specific binding of p38γ or p38δ MAP kinase to JIP4 was detected, although some nonspecific binding of p38δ MAP kinase to GST was observed. Together, these data demonstrate that JIP4, unlike JIP3, can interact with some p38 MAP kinase isoforms.
FIG. 9.

JIP4 can potentiate p38α MAP kinase activation. (A) JIP4 can interact with p38 MAP kinase. JNK and p38α MAP kinases were in vitro translated in the presence of [35S]methionine and quantitated. Equal amounts of these proteins were incubated with bacterially expressed GST, GST-JIP3 (residues 190 through 380), and GST-JIP4 (residues 1 through 150) immobilized on glutathione-agarose beads. The beads were washed, and bound proteins were detected by SDS-PAGE and autoradiography. (B) The effect of JIP3 and JIP4 on ASK1-stimulated p38 activity was examined in cotransfection assays in COS7 cells by using HA epitope-tagged p38α MAPK. The effect of the expression of ASK1, JIP3, or JIP4 was examined. The levels of expression of ASK1, JIP3, JIP4, and p38α MAP kinase were examined by immunoblot analysis. The p38α MAP kinase was immunoprecipitated, and its activity was measured in an immune complex kinase assay with [γ-32P]ATP and ATF2 as substrates. The phosphorylated ATF2 was detected by SDS-PAGE and autoradiography and was quantitated by a PhosphorImager analysis. (C) The effects of JIP3 and JIP4 on ASK1-stimulated JNK activity were examined in a similar assay as shown in panel B, using HA-tagged JNK1 and c-Jun as the substrates.
The interaction of JIP4 with p38α MAP kinase, along with the apparent lack of a role for JIP4 as a scaffold for the JNK signaling pathway, suggests that JIP4 might function to potentiate p38α MAP kinase activation. To test this hypothesis, we compared the effects of JIP3 and JIP4 on JNK and p38α MAP kinase activation in cotransfection assays with the MAP3K ASK1. We found that ASK1-stimulated JNK activity was not potentiated by JIP3 or JIP4 (Fig. 9C). This observation is consistent with the finding that JIP3 does not form a functional JNK signaling complex with ASK1 (13) and the conclusion that JIP4 is not a JNK scaffold (this study). In contrast, JIP4 (but not JIP3) did potentiate ASK1-stimulated p38α MAP kinase activation (Fig. 9B). These data suggest that JIP4 may function as a scaffold for the p38α MAP kinase signaling pathway.
MKK3 and MKK6 are required for p38 MAP kinase activation by JIP4.
Coimmunoprecipitation assays and pull-down assays using in vitro-translated protein kinases failed to demonstrate an interaction of JIP4 with MAP2Ks that activate JNK or p38 MAP kinase (MKK3, MKK4, MKK6, and MKK7) (Fig. 5B and data not shown). These data suggest that JIP4 does not form stable complexes of p38 MAP kinase together with a MAP2K. Previous studies indicate that p38 MAP kinase can be activated by a MAP2K-independent mechanism (6, 7). We therefore considered the possibility that JIP4 may activate p38 MAP kinase by a MAP2K-independent mechanism. To test this hypothesis, we compared the ability of JIP4 to regulate p38 MAP kinase activity in fibroblasts prepared from wild-type mouse embryos to that ability in fibroblasts prepared from MAP2K-deficient mouse embryos (Fig. 10A). We found that JIP4, but not JIP3, potentiated ASK1-stimulated p38α MAP kinase activation in wild-type cells (Fig. 10B). Similarly, JIP4 was found to potentiate p38 MAP kinase activation in both Mkk3−/− fibroblasts and Mkk6−/− fibroblasts (data not shown). However, JIP4 failed to potentiate p38α MAP kinase activation in Mkk3−/− Mkk6−/− compound mutant fibroblasts (Fig. 10C). Together, these data indicate that while the MAP2Ks MKK3 and MKK6 do not form stable complexes with JIP4, these MAP2Ks are required for the ability of JIP4 to regulate p38α MAP kinase activation.
FIG. 10.

JIP4 requires MKK3 and MKK6 for the potentiation of p38 MAP kinase activation. (A) Total protein extracts were prepared from wild-type MEF, Mkk3−/− MEF, Mkk6−/− MEF, and Mkk3−/− Mkk6−/− compound mutant MEF. The expression levels of MKK3, MKK6, and tubulin were examined by immunoblot analysis. (B) The effects of JIP3 and JIP4 on ASK1-stimulated p38 MAPK activity in wild-type MEF were examined by cotransfection assays. The activity of HA-tagged p38α MAP kinase was examined in an immune complex kinase assay using the substrate ATF2. (C) The effects of JIP3 and JIP4 on ASK1-stimulated p38α MAPK activity in an Mkk3−/− Mkk6−/− compound mutant MEF were examined by cotransfection assays. The activity of HA-tagged p38α MAP kinase was examined in an immune complex kinase assay using the substrate ATF2.
DISCUSSION
We report the identification of JIP4, a new member of the JIP group of scaffold proteins. The structural organization of JIP4 is most similar to that of JIP3. Like other members of the JIP protein group, JIP4 binds to JNK and to the TPR domain of kinesin light chain. However, unlike other JIP proteins, JIP4 does not activate JNK. In contrast, JIP4 causes MKK3- and MKK6-dependent activation of p38 MAP kinase.
The murine gene that encodes JIP4 encodes at least three different proteins: SPAG9, JLP, and JIP4 (Fig. 2). SPAG9, the sperm-associated antigen 9 protein, was reported to be a transmembrane protein with a large extracellular domain and a short cytoplasmic tail (26). SPAG9 is present on the surface of sperm and is selectively expressed by spermatids (26). The function of SPAG9 is unknown. The JLP protein was identified in a yeast two-hybrid screen using Max, the partner of the c-Myc transcription factor, as the bait (15). The binding of JLP to the leucine zipper protein Max is mediated by an NH2-terminal leucine zipper present in JLP but absent in JIP4 and SPAG9. JLP binds to Max monomers and can therefore cause the dissociation of Max homodimers, but JLP is unable to interact with c-Myc-Max heterodimers and cannot cause the dissociation of Max from complexes with Myc because the affinity of the leucine zipper interaction between c-Myc and Max is greater than the affinity of the interaction between JLP and Max (15). JLP binds multiple protein kinases (JNK, p38 MAP kinase, MKK4, and MEKK3) and has been reported to act as a scaffold that activates JNK and also recruits potential transcription factor substrates (15). Although Max is not an established MAPK substrate, it is possible that other leucine zipper domain transcription factors that are MAPK substrates might be recruited by JLP. Interestingly, JIP4 has properties that are distinct from both JLP and SPAG9. Thus, SPAG9 is a cell surface transmembrane protein (26) and JLP can interact with nuclear leucine zipper transcription factors via a unique NH2-terminal domain (15). In contrast, JIP4 is a cytoplasmic protein that is largely excluded from the nucleus (Fig. 3 and 4).
The coiled-coil domains of JIP4 can mediate oligomerization. Interestingly, although JIP3 and JIP4 can both form homo-oligomers (most likely dimers), these JIP proteins are unable to form hetero-oligomers (Fig. 7). However, it is possible that JIP4 may function as a hetero-oligomer with either SPAG9 or JLP in vivo. The interaction of JIP4 and either SPAG9 or JLP may be functionally significant.
Studies of JIP4 indicate that it does not appear to function as a scaffold for the JNK signaling pathway; for example, JIP4 does not increase JNK activation in transfection assays (Fig. 6 and 9). However, JIP4 was found to potentiate the activation of p38 MAP kinase (Fig. 9). This analysis suggests that JIP4 may function as a scaffold for the p38 MAP kinase pathway. This is plausible because JIP4 can bind both p38 MAP kinase and the MAP3K ASK1. However, an interaction between JIP4 and a MAP2K that can activate p38 was not detected. Gene disruption studies demonstrated that the MAP2Ks MKK3 and MKK6 were required for the ability of JIP4 to activate p38 MAP kinase (Fig. 10). In future studies, it will be important to distinguish between the possibilities that (i) MAP2K binding to the JIP4 complex is not required for p38 activation, (ii) the affinity of MAP2K for JIP4 is low but sufficient to form functional complexes with JIP4 in vivo, and (iii) the interaction with MAP2K is indirect. The last possibility is consistent with previous studies of the β-arrestin-2 scaffold protein that interacts with ASK1 and JNK3 directly and with MKK4 indirectly by interactions mediated by both ASK1 and JNK3 (18).
JIP4 may act as a scaffold for the p38 MAP kinase pathway in vivo. However, the role of the strong interaction of JIP4 with JNK is unclear. We can speculate on three possibilities. First, the interaction of JIP4 with JNK may have no physiological significance. Second, JNK may compete with p38 MAP kinase for binding to JIP4. Since JNK activation increases binding to JIP4, it is possible that JNK activation could act to inhibit JIP4-stimulated p38 MAP kinase activation. Third, JNK binding to JIP4 is associated with increased phosphorylation of JIP4 on several sites. It is possible that the phosphorylation of JIP4 by JNK occurs on sites that regulate JIP4 function. Further studies will be required to distinguish between these possibilities.
A critical goal for future studies will be to determine the physiological significance of JIP4 by using a loss-of-function analysis. A knock-down of JIP4 expression using RNA interference-based methods represents one approach. However, we have not found this procedure to be effective for JIP4, perhaps because of the long half-life of the JIP4 protein (unpublished observations). We have therefore initiated the construction of Jip4−/− mice. We have previously reported the phenotype of Jip3−/− mice (12), and given the similarities between JIP3 and JIP4, the phenotype of Jip3−/− Jip4−/− compound mutant animals may be informative. For example, recent studies have suggested a role for the JIP3 scaffold protein in JNK activation caused by lipopolysaccharide and mediated by Toll-like receptor 4 (17). Indeed, JIP3 has been reported to bind the cytoplasmic domain of Toll-like receptor 4 (17). It is possible that JIP4 may have a related function in the activation of the p38 MAP kinase signaling pathway.
In conclusion, we report the identification of JIP4, a novel putative scaffold protein for the p38 MAP kinase pathway. We propose that JIP4 may regulate p38 MAP kinase activation in response to specific stimuli in vivo. A similar role for the scaffold proteins OSM (30) and IB2/JIP2 (3, 24, 25) in the activation of the p38 MAP kinase pathway has also been proposed. It will be interesting to discover how these scaffold proteins contribute to the activation of the p38 MAP kinase pathway by specific stimuli in vivo.
Supplementary Material
Acknowledgments
We thank C. Suetterlin, R. Gilmore, and L. S. Goldstein for providing antibodies and Kathy Gemme for providing expert administrative assistance. R.J.D. is an investigator of the Howard Hughes Medical Institute.
This study was supported, in part, by a grant from the National Cancer Institute.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Bowman, A. B., A. Kamal, B. W. Ritchings, A. V. Philp, M. McGrail, J. G. Gindhart, and L. S. Goldstein. 2000. Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell 103:583-594. [DOI] [PubMed] [Google Scholar]
- 2.Brancho, D., N. Tanaka, A. Jaeschke, J. J. Ventura, N. Kelkar, Y. Tanaka, M. Kyuuma, T. Takeshita, R. A. Flavell, and R. J. Davis. 2003. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 17:1969-1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buchsbaum, R. J., B. A. Connolly, and L. A. Feig. 2002. Interaction of Rac exchange factors Tiam1 and Ras-GRF1 with a scaffold for the p38 mitogen-activated protein kinase cascade. Mol. Cell. Biol. 22:4073-4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dickens, M., J. S. Rogers, J. Cavanagh, A. Raitano, Z. Xia, J. R. Halpern, M. E. Greenberg, C. L. Sawyers, and R. J. Davis. 1997. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science 277:693-696. [DOI] [PubMed] [Google Scholar]
- 5.Elion, E. A. 2001. The Ste5p scaffold. J. Cell Sci. 114:3967-3978. [DOI] [PubMed] [Google Scholar]
- 6.Ge, B., H. Gram, F. Di Padova, B. Huang, L. New, R. J. Ulevitch, Y. Luo, and J. Han. 2002. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science 295:1291-1294. [DOI] [PubMed] [Google Scholar]
- 7.Ge, B., X. Xiong, Q. Jing, J. L. Mosley, A. Filose, D. Bian, S. Huang, and J. Han. 2003. TAB1beta (transforming growth factor-beta-activated protein kinase 1-binding protein 1beta), a novel splicing variant of TAB1 that interacts with p38alpha but not TAK1. J. Biol. Chem. 278:2286-2293. [DOI] [PubMed] [Google Scholar]
- 8.Harris, K., R. E. Lamson, B. Nelson, T. R. Hughes, M. J. Marton, C. J. Roberts, C. Boone, and P. M. Pryciak. 2001. Role of scaffolds in MAP kinase pathway specificity revealed by custom design of pathway-dedicated signaling proteins. Curr. Biol. 11:1815-1824. [PubMed] [Google Scholar]
- 9.Im, J. Y., K. W. Lee, M. H. Kim, S. H. Lee, H. Y. Ha, I. H. Cho, D. Kim, M. S. Yu, J. B. Kim, J. K. Lee, Y. J. Kim, B. W. Youn, S. D. Yang, H. S. Shin, and P. L. Han. 2003. Repression of phospho-JNK and infarct volume in ischemic brain of JIP1-deficient mice. J. Neurosci. Res. 74:326-332. [DOI] [PubMed] [Google Scholar]
- 10.Ito, M., K. Yoshioka, M. Akechi, S. Yamashita, N. Takamatsu, K. Sugiyama, M. Hibi, Y. Nakabeppu, T. Shiba, and K. I. Yamamoto. 1999. JSAP1, a novel Jun N-terminal protein kinase (JNK)-binding protein that functions as a scaffold factor in the JNK signaling pathway. Mol. Cell. Biol. 19:7539-7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaeschke, A., M. P. Czech, and R. J. Davis. 2004. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes Dev. 18:1976-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelkar, N., M. H. Delmotte, C. R. Weston, T. Barrett, B. J. Sheppard, R. A. Flavell, and R. J. Davis. 2003. Morphogenesis of the telencephalic commissure requires scaffold protein JNK-interacting protein 3 (JIP3). Proc. Natl. Acad. Sci. USA 100:9843-9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelkar, N., S. Gupta, M. Dickens, and R. J. Davis. 2000. Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol. Cell. Biol. 20:1030-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurzbauer, R., D. Teis, M. E. de Araujo, S. Maurer-Stroh, F. Eisenhaber, G. P. Bourenkov, H. D. Bartunik, M. Hekman, U. R. Rapp, L. A. Huber, and T. Clausen. 2004. Crystal structure of the p14/MP1 scaffolding complex: how a twin couple attaches mitogen-activated protein kinase signaling to late endosomes. Proc. Natl. Acad. Sci. USA 101:10984-10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, C. M., D. Onesime, C. D. Reddy, N. Dhanasekaran, and E. P. Reddy. 2002. JLP: A scaffolding protein that tethers JNK/p38MAPK signaling modules and transcription factors. Proc. Natl. Acad. Sci. USA 99:14189-14194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu, H. T., D. D. Yang, M. Wysk, E. Gatti, I. Mellman, R. J. Davis, and R. A. Flavell. 1999. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J. 18:1845-1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuguchi, T., A. Masuda, K. Sugimoto, Y. Nagai, and Y. Yoshikai. 2003. JNK-interacting protein 3 associates with Toll-like receptor 4 and is involved in LPS-mediated JNK activation. EMBO J. 22:4455-4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDonald, P. H., C. W. Chow, W. E. Miller, S. A. Laporte, M. E. Field, F. T. Lin, R. J. Davis, and R. J. Lefkowitz. 2000. Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290:1574-1577. [DOI] [PubMed] [Google Scholar]
- 19.Morrison, D. K., and R. J. Davis. 2003. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 19:91-118. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen, A., W. R. Burack, J. L. Stock, R. Kortum, O. V. Chaika, M. Afkarian, W. J. Muller, K. M. Murphy, D. K. Morrison, R. E. Lewis, J. McNeish, and A. S. Shaw. 2002. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol. Cell. Biol. 22:3035-3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park, S. H., A. Zarrinpar, and W. A. Lim. 2003. Rewiring MAP kinase pathways using alternative scaffold assembly mechanisms. Science 299:1061-1064. [DOI] [PubMed] [Google Scholar]
- 22.Posas, F., and H. Saito. 1997. Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science 276:1702-1705. [DOI] [PubMed] [Google Scholar]
- 23.Schaeffer, H. J., A. D. Catling, S. T. Eblen, L. S. Collier, A. Krauss, and M. J. Weber. 1998. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 281:1668-1671. [DOI] [PubMed] [Google Scholar]
- 24.Schoorlemmer, J., and M. Goldfarb. 2002. Fibroblast growth factor homologous factors and the islet brain-2 scaffold protein regulate activation of a stress-activated protein kinase. J. Biol. Chem. 277:49111-49119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schoorlemmer, J., and M. Goldfarb. 2001. Fibroblast growth factor homologous factors are intracellular signaling proteins. Curr. Biol. 11:793-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shankar, S., B. Mohapatra, and A. Suri. 1998. Cloning of a novel human testis mRNA specifically expressed in testicular haploid germ cells, having unique palindromic sequences and encoding a leucine zipper dimerization motif. Biochem. Biophys. Res. Commun. 243:561-565. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka, N., M. Kamanaka, H. Enslen, C. Dong, M. Wysk, R. J. Davis, and R. A. Flavell. 2002. Differential involvement of p38 mitogen-activated protein kinase kinases MKK3 and MKK6 in T-cell apoptosis. EMBO Rep. 3:785-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teis, D., W. Wunderlich, and L. A. Huber. 2002. Localization of the MP1-MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev. Cell 3:803-814. [DOI] [PubMed] [Google Scholar]
- 29.Thompson, N. A., J. A. Haefliger, A. Senn, T. Tawadros, F. Magara, B. Ledermann, P. Nicod, and G. Waeber. 2001. Islet-brain1/JNK-interacting protein-1 is required for early embryogenesis in mice. J. Biol. Chem. 276:27745-27748. [DOI] [PubMed] [Google Scholar]
- 30.Uhlik, M. T., A. N. Abell, N. L. Johnson, W. Sun, B. D. Cuevas, K. E. Lobel-Rice, E. A. Horne, M. L. Dell'Acqua, and G. L. Johnson. 2003. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat. Cell Biol. 5:1104-1110. [DOI] [PubMed] [Google Scholar]
- 31.Verhey, K. J., D. Meyer, R. Deehan, J. Blenis, B. J. Schnapp, T. A. Rapoport, and B. Margolis. 2001. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 152:959-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vomastek, T., H. J. Schaeffer, A. Tarcsafalvi, M. E. Smolkin, E. A. Bissonette, and M. J. Weber. 2004. Modular construction of a signaling scaffold: MORG1 interacts with components of the ERK cascade and links ERK signaling to specific agonists. Proc. Natl. Acad. Sci. USA 101:6981-6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitmarsh, A. J., J. Cavanagh, C. Tournier, J. Yasuda, and R. J. Davis. 1998. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science 281:1671-1674. [DOI] [PubMed] [Google Scholar]
- 34.Whitmarsh, A. J., C. Y. Kuan, N. J. Kennedy, N. Kelkar, T. F. Haydar, J. P. Mordes, M. Appel, A. A. Rossini, S. N. Jones, R. A. Flavell, P. Rakic, and R. J. Davis. 2001. Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev. 15:2421-2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wunderlich, W., I. Fialka, D. Teis, A. Alpi, A. Pfeifer, R. G. Parton, F. Lottspeich, and L. A. Huber. 2001. A novel 14-kilodalton protein interacts with the mitogen-activated protein kinase scaffold mp1 on a late endosomal/lysosomal compartment. J. Cell Biol. 152:765-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wysk, M., D. D. Yang, H. T. Lu, R. A. Flavell, and R. J. Davis. 1999. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for tumor necrosis factor-induced cytokine expression. Proc. Natl. Acad. Sci. USA 96:3763-3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yasuda, J., A. J. Whitmarsh, J. Cavanagh, M. Sharma, and R. J. Davis. 1999. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol. 19:7245-7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.