

Abstract
Adenosine monophosphate–activated protein kinase (AMPK) activity is stimulated to promote metabolic adaptation upon energy stress. However, sustained metabolic stress may cause cell death. The mechanisms by which AMPK dictates cell death are not fully understood. We report that metabolic stress promoted receptor-interacting protein kinase 1 (RIPK1) activation mediated by TRAIL receptors, whereas AMPK inhibited RIPK1 by phosphorylation at Ser415 to suppress energy stress–induced cell death. Inhibiting pS415-RIPK1 by Ampk deficiency or RIPK1 S415A mutation promoted RIPK1 activation. Furthermore, genetic inactivation of RIPK1 protected against ischemic injury in myeloid Ampkα1-deficient mice. Our studies reveal that AMPK phosphorylation of RIPK1 represents a crucial metabolic checkpoint, which dictates cell fate response to metabolic stress, and highlight a previously unappreciated role for the AMPK-RIPK1 axis in integrating metabolism, cell death, and inflammation.
Editor’s summary
A critical phosphorylation event links the metabolic state of a cell with control of cell death and inflammation. Adenosine monophosphate–dependent protein kinase (AMPK) is a sensor of the nutrient status and energy state of the cell. Zhang et al. found that activated AMPK phosphorylates and inhibits receptor-interacting protein kinase 1 (RIPK1) in nutrient-deprived cells, thus inhibiting cell death and inflammation (see the Perspective by Hardie). Under more prolonged nutrient stress, such inhibition was lost, allowing cell death to ensue. These results also confirm RIPK1’s role in cell death caused by ischemia, thus implicating the AMPK-RIPK1 interaction as a potential therapeutic target. —L. Bryan Ray
Adenosine monophosphate (AMP)–activated protein kinase (AMPK) is an evolutionary conserved sensor of cellular nutrient status and regulator of energy homeostasis in eukaryotes (1). In response to increases in intracellular AMP that always accompany decreases in adenosine triphosphate (ATP), AMPK is activated and serves as a metabolic checkpoint (1–4). However, when extensive metabolic stress overrides AMPK-mediated adaptation, it activates cell death (5, 6). The mechanisms by which AMPK modulates cell survival under metabolic stress are not fully understood. Receptor-interacting protein kinase 1 (RIPK1) is a key mediator of cell death and inflammation (7, 8). Activated RIPK1 may mediate receptor-interacting protein kinase 3 (RIPK3) and mixed lineage kinase domain-like (MLKL)–dependent necroptosis or caspase-8–dependent apoptosis upon stimulation of tumor necrosis factor receptor 1 (TNFR1) by tumor necrosis factor–α (TNFα) (9–12). RIPK1 is also activated downstream of the death receptors of Fas (also called CD95), as well as TRAIL receptor 1 (TRAIL-R1, also called DR4) and TRAIL-R2 (also called DR5) in addition to TNFR (13–18). Notably, inhibition of RIPK1 activation is highly effective in protecting against ischemic damage (19, 20). However, it remains unknown whether and how RIPK1 may be activated under ischemic conditions.
Metabolic stress promotes the activation of RIPK1
To this end, we incubated cells with glucose-free medium for various lengths of time. Wild-type (WT) mouse embryonic fibroblasts (MEFs) died after 48 hours of glucose deprivation, whereas most MEFs expressing catalytically inactive RIPK1 (Ripk1D138N/D138N) survived (Fig. 1A and fig. S1A). Moreover, glucose deprivation increased phosphorylation of RIPK1 (S166), a biomarker of RIPK1 activation (21,22), as well as that of RIPK3 (T231/S232) and MLKL (S345), hallmarks of necroptosis (11), which were all blocked in Ripk1D138N/D138N MEFs (Fig. 1, B and C, and fig. S1, B and C). Glucose deprivation activates RIPK1 in multiple cell lines, including colon cancer HT-29 cells, more microglia BV2 cells, human T lymphoyam Jurkat cells, and human U2OS osteosarcoma cells (fig. S1, D to I). Increased insolubility is another hallmark of activated RIPK1, RIPK3, and MLKL (22). In glucose-starved MEFs, the amounts of RIPK1, RIPK3, and MLKL were decreased in a mild-detergent (NP-40)–soluble fraction but increased in an NP-40–insoluble and 6M urea–soluble fraction, which was largely blocked by the Ripk1D138N/D138N mutation (Fig. 1C and fig. S1, C, F, and G). We also observed interaction of RIPK1 and RIPK3 in MEFs after glucose starvation (fig. S1J). Glucose deprivation induced apoptosis, as marked by the cleavage of RIPK1 and caspase-3 (CC3) (Fig. 1B and fig. S1K). We exposed cells to various concentrations of glucose and found that only severe energy stress induced necroptosis and apoptosis (Fig. 1D). Prolonged treatment of cells with the glucose analog 2-deoxyglucose (2DG), which blocks cellular glucose utilization by indirectly inhibiting hexokinase, also activated RIPK1 to promote necroptosis (fig. S1L).
Fig. 1. Metabolic stress promotes cell death and inflammation in a RIPK1-dependent manner.
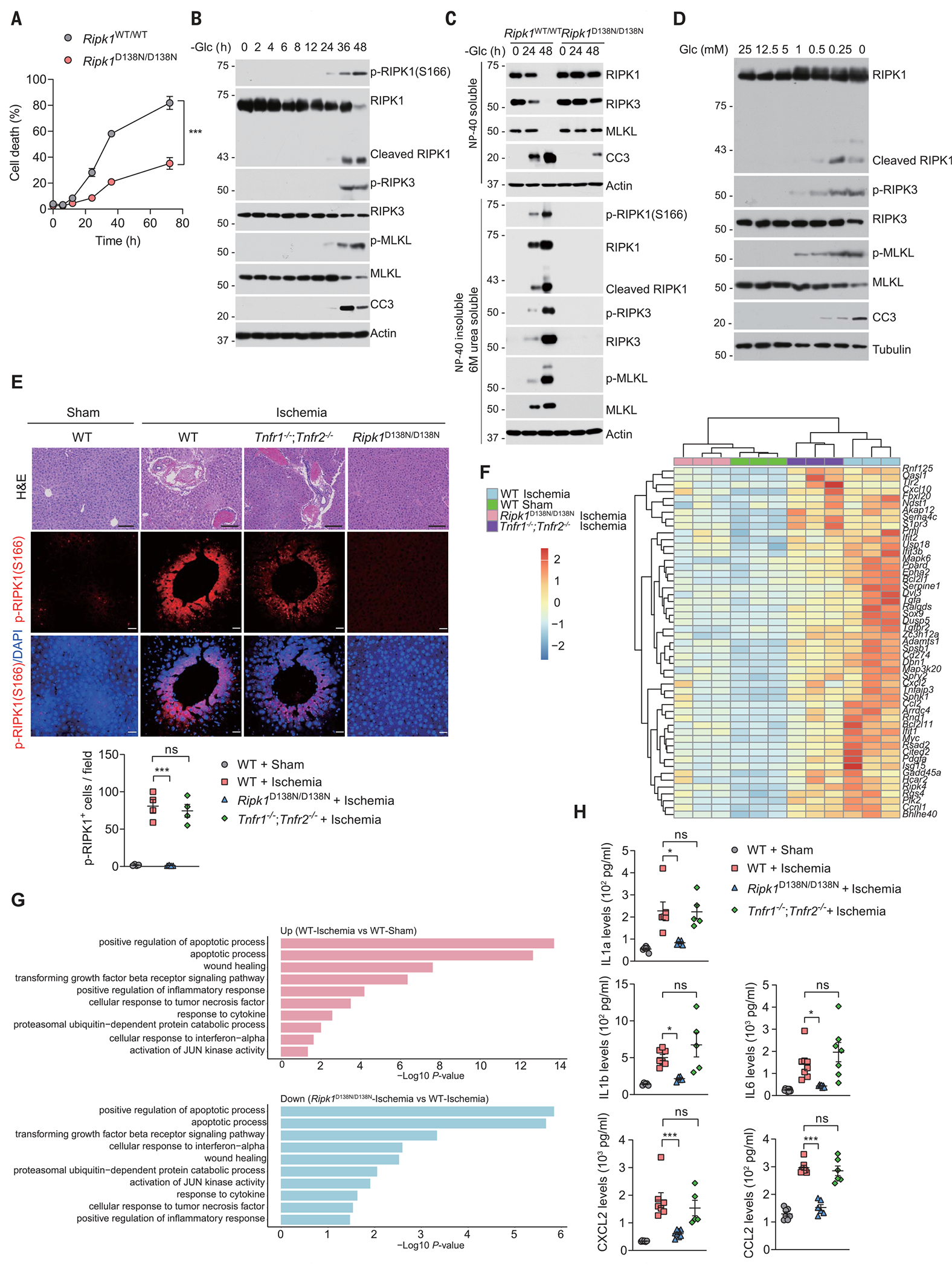
(A) WT or Ripk1D138N/D138N MEFs were cultured in glucose-free medium for the indicated times (hours), followed by cell viability analyses using propidium iodide (PI) uptake assay. Data are mean ± SD of n = 3 biological independent samples. Two-way analysis of variance (ANOVA); ***P < 0.001. (B) MEFs were cultured in glucose-free medium for the indicated times. The levels of proteins were determined by immunoblotting. n = 3 independent biological repeats. (C) WT or Ripk1D138N/D138N MEFs were cultured in glucose-free medium for the indicated times. The levels of proteins in mild-detergent (NP-40)–soluble fraction and 6 M urea–soluble fraction were determined by immunoblotting. n = 3 independent biological repeats. (D) MEFs were cultured in the medium with different glucose concentrations for 36 hours. The levels of proteins were determined by immunoblotting. n = 3 independent biological repeats. (E) WT, Tnfr1/2 DKO, and Rpik1D138N/D138N mice were subjected to a protocol of liver ischemia for 18 hours. Histological analysis and immunostaining for p-RIPK1(S166) were performed on liver sections (n = 4 mice in each group). DAPI (4’,6-diamidino-2-phenylindole) for nuclei. Representative images are shown. Quantification of p-RIPK1(S166)–positive cells is shown at the bottom. Data are mean ± SEM, one-way ANOVA, post hoc Dunnett’s test; ***P < 0.001; n.s., not significant. Scale bars, 100 μm. (F) Heatmap of genes differentially expressed in the whole livers derived of WT, Tnfr1/2 DKO, and Rpik1D138N/D138N mice subjected to liver ischemia for 18 hours. (G) Gene Ontology analysis of genes that are up-regulated in the livers of mice subjected to liver ischemia for 18 hours in a RIPK1-dependent manner. (H) ELISA analyses of the levels of indicated cytokines and chemokines in serum from WT, Tnfr1/2 DKO, and Rpik1D138N/D138N mice subjected to liver ischemia for 18 hours. Data are presented as mean ± SEM, and each dot represents one mouse. One-way ANOVA, post hoc Dunnett’s test; *P < 0.05, ***P < 0.001.
RIPK1 promotes cell death through its kinase activity, whereas it inhibits cell death through its scaffold functions (21, 23–25). Unlike Ripk1D138N/D138N MEFs, which showed resistance to glucose deprivation–induced cell death, Ripk1 knockout (KO) MEFs were more sensitive to cell death induced by glucose starvation than were WT MEFs (fig. S2A). However, in Jurkat cells, RIPK1 deficiency decreased cell death and the activation of RIPK3 and MLKL induced by glucose starvation (fig. S2, B and C), indicating that the effects of RIPK1 deficiency on cell death in response to glucose starvation depend on the cellular context. To investigate the role of necroptosis in glucose deprivation–induced cell death, we deprived WT, Ripk3, and Mlkl KO cells of glucose for various lengths of time. Deletion of either Ripk3 or Mlkl partially prevented cell death induced by glucose deprivation (fig. S2, D to F). Notably, the loss in viability and the degree of RIPK1 activation were comparable between WT and Tnfr1/2 double knockout (DKO) cells (fig. S2, G and H), which indicates that glucose deprivation–induced activation of RIPK1 and cell death is independent of the TNFα signaling pathway.
Next we subjected WT mice, Ripk1D138N/D138N mice, and Tnfr1/2 DKO mice to hepatic ischemia, a condition that causes mouse livers to become pale, enlarged, and damaged (fig. S3, A and B). Histological analysis showed severe inflammatory infiltration and cell death in ischemic WT and Tnfr1/2 DKO mice but not in Ripk1D138N/D138N mice (Fig. 1E). We detected activation of RIPK1 as determined by p-RIPK1 (S166) immunostaining and cell death as determined by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay in the livers of WT and Tnfr1/2 DKO mice but not in those of Ripk1D138N/D138N mice (Fig. 1E and fig. S3C). We analyzed the gene expression profiles of whole livers by RNA sequencing (RNA-seq). Compared to WT sham control mice, WT and Tnfr1/2 DKO mice showed increased expression of genes associated with an inflammatory response, including interferon alpha (Ifn-α), transforming growth factor beta (TGF-β), C-X-C motif chemokine ligand 10 (Cxcl10), C-X-C motif chemokine ligand 2 (Cxcl2), and C-C motif chemokine ligand 2 (Ccl2) (Fig. 1, F and G, and fig. S3, D and E); and down-regulation of genes involved in oxidation-reduction processes and the transport signaling pathway (fig. S3, F and G). These gene expression abnormalities were largely rescued by genetic inhibition of RIPK1 in Ripk1D138N/D138N mice (Fig. 1, F and G, and fig. S3, F and G). We confirmed the increased production of Ccl2, Cxcl2, interleukin 1 alpha (Il1a), interleukin 1 beta (Il1b), and interleukin 6 (Il6) by enzyme-linked immunosorbent assay (ELISA), all of which were reduced in the blood of Ripk1D138N mice but not in that of Tnfr1/2 DKO mice (Fig. 1H). Thus, these results indicate that ischemia promotes RIPK1-dependent cell death and inflammation in a TNFα-independent manner.
We next explored the possible roles for other death receptors including Fas, DR4, and DR5 in the regulation of RIPK1 activity upon glucose starvation. Deletion of DR4 or DR5 but not FAS reduced RIPK1 activation and cell death (fig. S4, A to F). Glucose deprivation activates TRAIL receptors through an activating transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP)–dependent mechanism, which promotes cell death in a ligand-independent manner (26). Consistently, we observed increased up-regulation of TRAIL receptors after glucose starvation (fig. S4, D and F). We used the liver ischemia injury mouse model to examine the role of Dr5, which is the single TRAIL receptor expressed in mice (27). Dr5 deficiency suppressed RIPK1 activation, cell death, and inflammation (fig. S4, G and H). Thus, our data demonstrate that TRAIL receptors function as upstream drivers for RIPK1 activation and cell death under glucose deprivation conditions.
AMPK phosphorylates RIPK1 in response to metabolic stress
AMPK is a highly conserved sensor of cellular energy status, which is activated under conditions of low cellular energy such as glucose deprivation (28). RIPK1 derived from cells exposed to glucose deprivation showed a profound RIPK1 mobility shift on standard SDS–polyacrylamide gel electrophoresis (SDS-PAGE), which was diminished by the treatment of the protein with lambda-phosphatase. Thus, glucose deprivation appears to induce RIPK1 phosphorylation (Fig. 2, A and B, and fig. S5, A and B). Moreover, this glucose deprivation–induced RIPK1 mobility shift was not reversed by RIPK1-specific inhibitor necrostatin-1s (Nec-1s) (19) (fig. S5, C to E). These data indicate that glucose deprivation–induced RIPK1 phosphorylation is not entirely dependent on RIPK1 autophosphorylation. In keeping with this notion, cells expressing RIPK1 catalytically inactive mutant (K45M) (29) showed a strong upshifted band, which was diminished in AMPK DKO cells, indicating that AMPK might target RIPK1 for phosphorylation (fig. S5F). We conducted mass spectrometry analysis of RIPK1 phosphorylation under glucose starvation conditions and found that Ser416 of RIPK1 is likely a specific phosphorylation site in response to glucose deprivation (fig. S6, A and B). Further sequence analysis showed that the RIPK1 Ser416 residue conforms to the AMPK substrate motif validated in some previously identified AMPK substrates (30) (fig. S6C) and represents an evolutionarily conserved residue in human, rat, and mouse (fig. S6C).
Fig. 2. AMPK phosphorylates RIPK1 at S416 in response to metabolic stress.
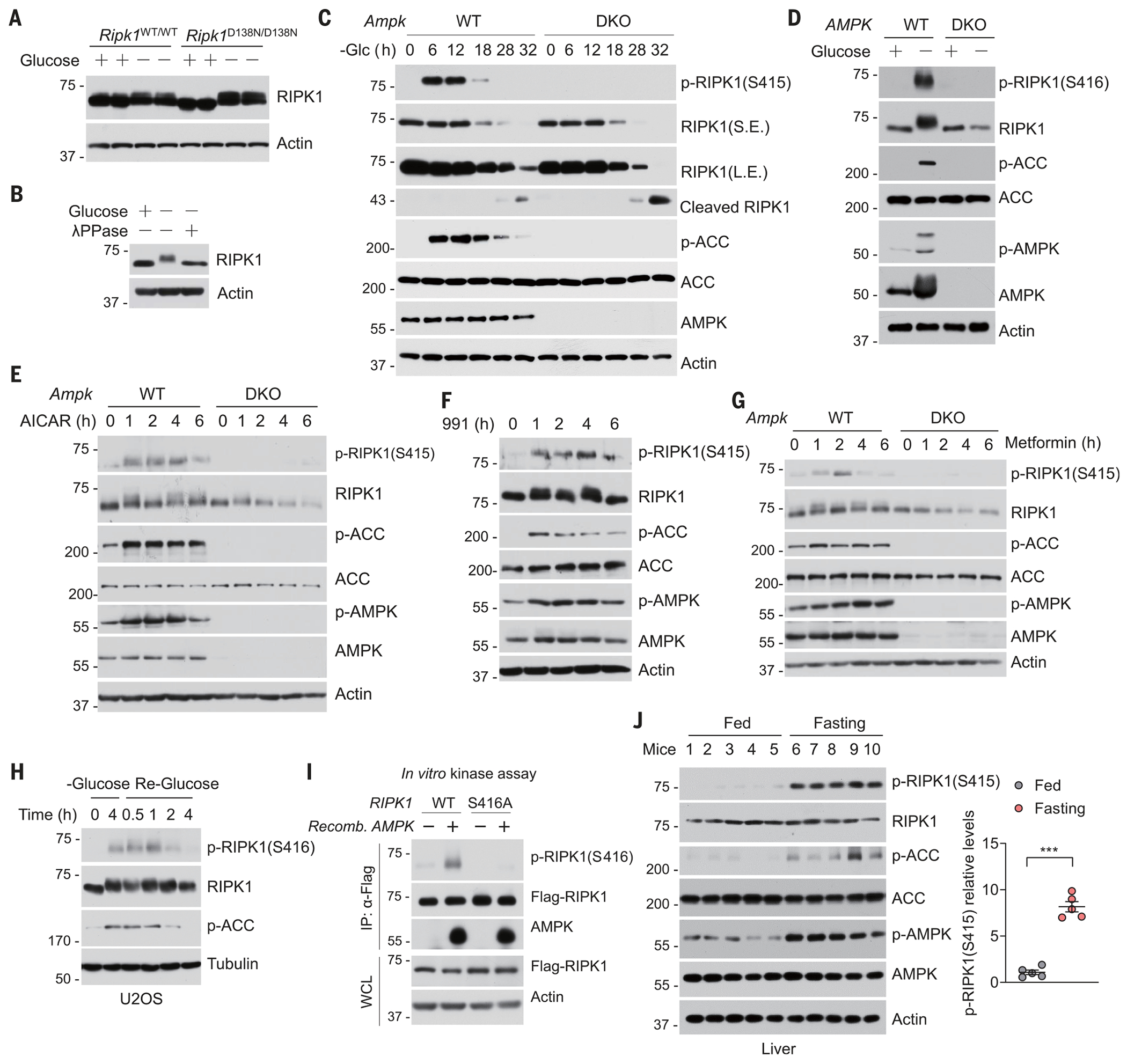
(A) WT or Ripk1D138N/D138N MEFs were cultured in glucose-free medium for 12 hours. Cell lysates were then subjected to immunoblotting using anti-RIPK1 antibody. n = 3 independent biological repeats. (B) U2OS were cultured in glucose-free medium for 4 hours. Cell lysates were treated with lambda-phosphatase (λPPase), as indicated, for 30 min and then subjected to immunoblotting using anti-RIPK1 antibody. (C) WT and Ampk DKO MEFs were cultured in glucose-free medium for the indicated times. The levels of proteins were determined by immunoblotting. n = 3 independent biological repeats. (D) WT and AMPK DKO HT-29 cells were cultured in glucose-free medium for 12 hours. The levels of proteins were determined by immunoblotting. n = 3 independent biological repeats. (E) WT and Ampk DKO MEFs were treated with 2 mM AICAR in the presence of glucose for the indicated times. The levels of proteins were determined by immunoblotting. n = 3 independent biological repeats. (F) MEFs were treated with 50 μM 991 in the presence of glucose for the indicated times. The levels of proteins were determined by immunoblotting. n = 3 independent biological repeats. (G) WT and Ampk DKO MEFs were treated with 2 mM metformin in the presence of glucose for various times. The levels of proteins were determined by immunoblotting. n = 3 independent biological repeats. (H) U2OS were starved of glucose (–Glucose) for 4 hours, and then the culture was switched to glucose-containing (25 mM) medium for indicated times (Re-Glucose), and samples were harvested. n = 3 independent biological repeats. (I) Flag-RIPK1 purified from human embryonic kidney 293T cells expressing Flag-tagged WT or S416A mutant of RIPK1 for 24 hours was combined with recombinant (Recomb.) active AMPK as indicated in an in vitro kinase reaction. The amounts of p-RIPK1 (S416) were determined by immunoblotting. n = 3 independent biological repeats. (J) Western blot analysis of p-RIPK1 (S415) in livers from fed and fasted (16 hours) animals (n = 5). Quantification of p-RIPK1(S415) is shown on the right. Data are presented as mean ± SEM, and each dot represents one mouse. Unpaired two-tailed t test; ***P < 0.001.
To examine endogenous RIPK1 phosphorylation, we developed a phosphospecific antibody against p-S416 of human RIPK1, which corresponds to p-S415 of mouse RIPK1 (fig. S6, D and E). p-S416 of RIPK1 was increased in response to a constitutively active AMPKα1 allele (31) (fig. S7A). We observed increased endogenous p-S415 of murine RIPK1 or p-S416 of human RIPK1 in WT but not in Ampk DKO MEFs and HT29 cells, respectively, after glucose starvation (Fig. 2, C and D). We tested whether direct pharmacological activation of AMPK was sufficient to induce phosphorylation of RIPK1. Phosphorylation of RIPK1 at Ser415 was induced by the treatment with AMP-mimetic aminoimidazole carboxamide riboside (AICAR) in an AMPK-dependent manner (Fig. 2E and fig. S7B). Similarly, treatment with compound 991, a small molecule that directly binds to and activates AMPK (32), was sufficient to induce RIPK1 phosphorylation at Ser415 (Fig. 2F). Consistently, phosphorylation of RIPK1 was also increased in response to treatment with metformin, a widely prescribed drug for type 2 diabetes that is known to activate AMPK both in vitro and in vivo (33) (Fig. 2G and fig. S7, C and D).
Liver kinase B1 (LKB1) is the major kinase that activates AMPK under conditions of energy stress (34). Treatment with metformin or glucose starvation resulted in the phosphorylation of RIPK1 at Ser415, which was abolished in Lkb1 KO MEFs (fig. S7, E and F). The phosphorylation of RIPK1S415 in these cells paralleled the phosphorylation of a well-established AMPK substrate, acetyl–coenzyme A carboxylase (ACC) (Fig. 2, C to G, and fig. S7, C and D) (35). Phosphorylation quickly returned to basal levels when cells were re-stimualted with 25 mM glucose after glucose deprivation for 4 hours (Fig. 2H and fig. S7G). We therefore tested whether AMPK might directly phosphorylate RIPK1. We detected a physical interaction between overexpressed Flag-tagged RIPK1 and hemagglutinin (HA)–tagged AMPK (fig. S7H). Recombinant AMPK induced phosphorylation of Flag-tagged RIPK1 at Ser416 or a myelin basic protein fusion with a RIPK1 fragment (390–436) in an in vitro kinase assay (Fig. 2I and fig. S7I). Furthermore, we used 32P-labeled ATP in an in vitro kinase assay (36). The inactive Flag-tagged RIPK1 K45M mutant or glutathione S-transferase (GST) fusion of RIPK1 fragment (390–436) was phosphorylated by AMPK (fig. S7, J and K). The stoichiometry of the phosphorylation of RIPK1 at Ser416 by AMPK was estimated to be 0.51 mol phosphate per mol of RIPK1 fragment (390–436). AMPK-mediated incorporation of 32P into RIPK1 was not observed for the RIPK1 fragment containing the S416A mutation (fig. S7K), indicating that AMPK predominantly phosphorylates RIPK1 at Ser416. Furthermore, we detected RIPK1 p-S415 in mouse tissues including lung and brain (fig. S7L). Moreover, in mice subjected to fasting, which causes low glucose in vivo (fig. S7M) (37), RIPK1 p-S415 was increased in liver and pancreas, two organs that are sensitive to fasting, a condition in which AMPK is activated (Fig. 2J and fig. S7N).
Ampk deficiency promotes RIPK1-driven cell death and inflammation
We tested whether phosphorylation of RIPK1 by AMPK represents a survival mechanism in response to glucose deprivation. Ampk deficiency sensitized cells to glucose deprivation–induced cell death (Fig. 3A and fig. S8A). Moreover, genetic deletion of Ampk in MEFs promoted activation of RIPK1 (Fig. 3B). Accordingly, glucose deprivation induced greater association of RIPK3 with immunoprecipitated RIPK1 in Ampk DKO MEFs than in WT MEFs, indicating that Ampk deficiency promotes RIPK1-driven necroptosis (Fig. 3C). Similar results were obtained in AMPK DKO HT-29 cells (fig. S8, B to E). Because the activation of RIPK1 by glucose deprivation induces both necroptosis and apoptosis, and RIPK1-induced apoptosis is caspase-8 dependent (38), we explored the contribution of necroptosis and apoptosis to the glucose deprivation-induced cell death in Ampk DKO MEFs. Cell death induced by glucose deprivation in Ampk DKO MEFs was reduced by 10 μM pan-caspase inhibitor quinoline-Val-Asp-difluorophenoxymethylketone (QVD-oph) or 3 μM RIPK3 inhibitor GSK’872 (fig. S8, F and G).
Fig. 3. AMPK deficiency promotes RIPK1-driven cell death and inflammation in vitro and in vivo.
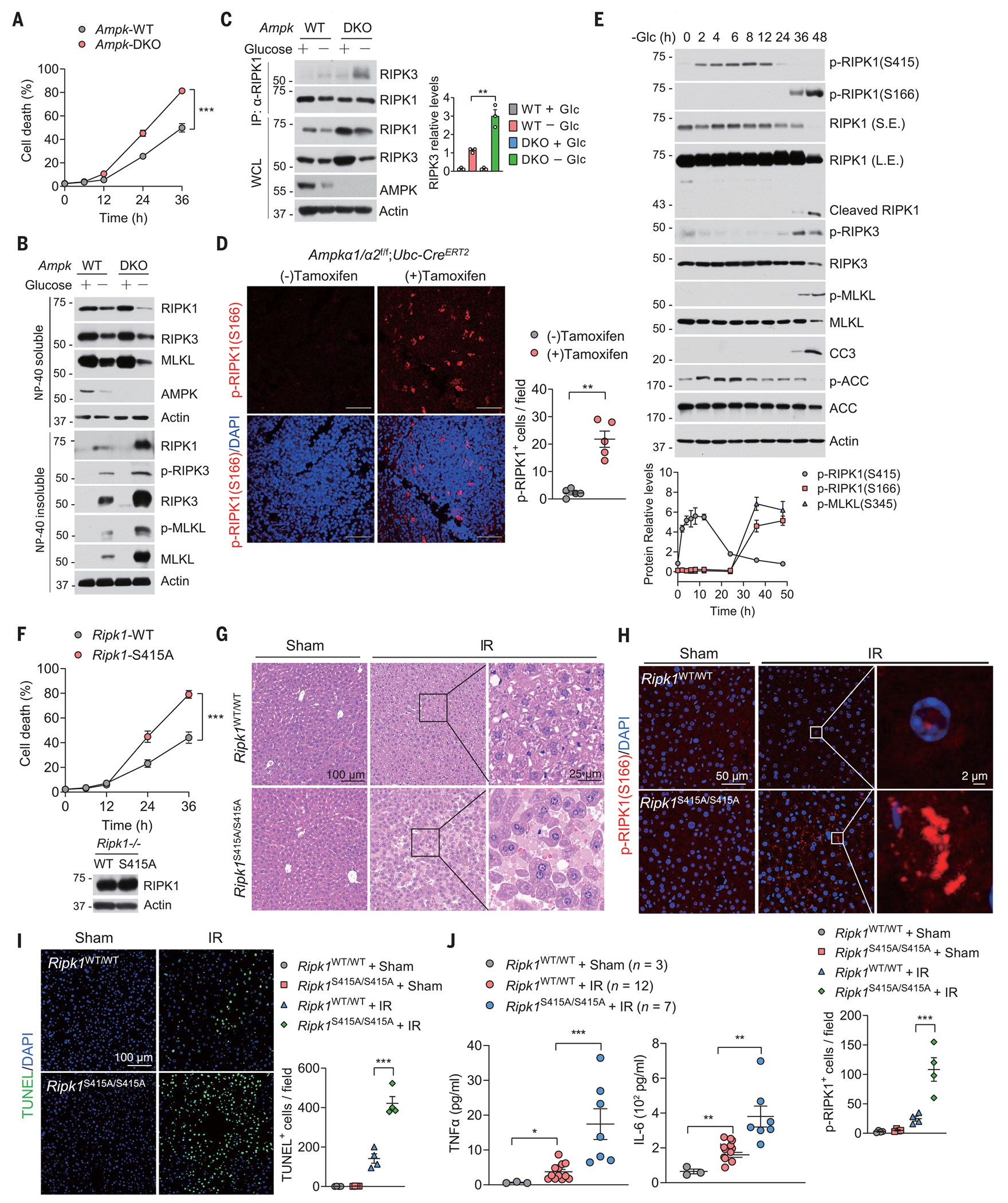
(A) WT or Ampk DKO MEFs were cultured in glucose-free medium for the indicated times followed by cell viability analyses using PI uptake assay. Data are mean ± SD of n = 3 biologically independent samples. Two-way ANOVA; ***P < 0.001. (B) WT or Ampk DKO MEFs were cultured in glucose-free medium for 30 hours. The levels of proteins in mild-detergent (NP-40)–soluble fraction and 6 M urea–soluble fraction were determined by immunoblotting. n = 3 independent biological repeats. (C) WT or Ampk DKO MEFs were cultured in glucose-free medium for 24 hours. Afterward, cell lysates were immunoprecipitated with anti-RIPK1 antibody, and the immunocomplexes were analyzed by immunoblotting using anti-RIPK3 antibody. n = 3 independent biological repeats. Quantified values for Western blot images are shown on the right. **P < 0.01. (D) Ampka1/a2f/f;Ubc-CreER mice were treated with or without tamoxifen (4 mg per day, for 5 days) to induce Ampk deletion. Spleens were harvested 8 weeks after tamoxifen treatment, and then immunostaining for p-RIPK1(S166) was performed on spleen sections (n = 5 mice in each group). DAPI for nuclei. Representative images are shown. Data are presented as mean ± SEM, and each dot represents one mouse. Unpaired two-tailed t test; **P < 0.01. Scale bars, 100 μm. (E) MEFs were cultured in glucose-free medium for the indicated times. The levels of proteins were determined by immunoblotting. n = 3 independent biological repeats. Quantified values for Western blot images are shown at the bottom. (F) Ripk1−/− MEFs reconstituted with WT or S415A mutant of RIPK1 were cultured in glucose-free medium for the indicated times followed by cell viability analyses using PI uptake assay. Data are mean ± SD of n = 3 biological independent samples. Two-way ANOVA; ***P < 0.001. The expression of RIPK1 was determined by immunoblotting and is shown at the bottom. (G) Ripk1S415A/S415A and control WT littermate mice were subjected to liver IR injury. Histological analysis on liver sections was performed (n = 4 mice in each group). (H) Ripk1S415A/S415A and control WT littermate mice were subjected to liver IR injury. Immunostaining for p-RIPK1(S166) on liver sections was performed (n = 4 mice in each group). DAPI for nuclei. Representative images are shown. Microscopic quantification of p-RIPK1(S166)–positive cells is shown at the bottom. Data are presented as mean ± SEM, and each dot represents one mouse. One-way ANOVA, post hoc Dunnett’s test; ***P < 0.001. (I) TUNEL assays were performed on liver sections from Ripk1S415A/S415A and control WT littermate mice subjected to liver IR injury (n = 4 mice in each group). DAPI for nuclei. Representative images are shown. Microscopic quantification of TUNEL-positive cells was shown on the right. Data are presented as mean ± SEM, and each dot represents one mouse. One-way ANOVA, post hoc Dunnett’s test; ***P < 0.001. (J) ELISA analyses of the concentrations of indicated cytokines and chemokines in serum from Ripk1S415A/S415A and control WT littermate mice subjected to liver IR injury (n = 4 mice each group). Data are presented as mean ± SEM, and each dot represents one mouse. One-way ANOVA, post hoc Dunnett’s test; **P < 0.01, ***P < 0.001.
To determine the role of AMPK in the regulation of RIPK1 activity in vivo, we measured RIPK1 activation by p-S166 RIPK1 immunostaining in a diverse set of tissues from Ampkα1α2f/f;Ubc-CreERT2 mice that were treated with tamoxifen to induce the deletion of both Ampkα1 and Ampkα2. Loss of Ampkα1 and Ampkα2 induced RIPK1 activation and increased cell death in multiple tissues, including spleen, kidney, and intestine (Fig. 3D and fig. S9, A to G). We did not observe increased RIPK1 activity in the livers of Ampkα1 and Ampkα2–deficient mice (fig. S9, H and I). Thus, there may be tissue-specific activation of RIPK1 in mice lacking Ampkα1 and Ampkα2. Notably, prolonged glucose deprivation or AICAR activation of AMPK overcame the adaptive response and triggered RIPK1 activation and cell death (Fig. 3E and fig. S10A). We also treated MEFs with 2-DG for various lengths of time. Short-term treatment of cells with 2-DG activated AMPK (39) and increased phosphorylation of RIPK1 at Ser415 (fig. S10B). However, phosphorylation of RIPK1 at Ser415 decreased after prolonged treatment of cells with 2-DG, which caused RIPK1 activation, as determined by p-RIPK1 (S166) (fig. S10B). Thus, modified AMPK-mediated phosphorylation of RIPK1 may contribute to the switch from adaptive homeostasis to cellular demise in cells exposed to prolonged metabolic stress.
Consistent with the notion that AMPK-mediated phosphorylation of RIPK1 directly inhibits its activity, overexpression of RIPK1 S416A mutant led to higher amounts of p-S166 RIPK1 (fig. S11A). We reintroduced WT and S415A mutant of RIPK1 into Ripk1 KO MEFs. Ripk1 KO MEFs reconstituted with the RIPK1 S415A mutant were more sensitive to cell death induced by glucose deprivation than were the cells reconstituted with WT RIPK1 (Fig. 3F and fig. S11B). Treatment of cells with either caspase inhibitor QVD-oph or RIPK3 inhibitor GSK’872 reduced cell death (fig. S11, C and D). To further investigate the physiological importance of AMPK-mediated phosphorylation of RIPK1 in vivo, we generated Ripk1S415A/S415A knock-in mice by the CRISPR-Cas9 technology (fig. S11, E and F). Ripk1S415A/S415A mutant mice were born in normal Mendelian ratios, and their growth appeared normal (fig. S11, G and H). However, the RIPK1 S415A mutation increased cell death and inflammation (Fig. 3, G to J, and fig. S11I). These results indicate that RIPK1 S415 phosphorylation inhibits RIPK1 activity and blocks cell death in response to energy stress.
We also investigated whether AMPK has a role in restraining RIPK1 activity in the context of TNFα-induced cell death. Ampk deficiency had no effect on RIPK1 activation and inflammation induced by TNFα alone or TNFα-SM-164 (TS) or TNFα-SM-164-zVAD.fmk (TSZ) (fig. S12). Moreover, the RIPK1 S415A mutation had no effect on RIPK1 activation, cell death, or inflammation induced by TNFα in combination with SM-164 or zVAD.fmk (fig. S13). Thus AMPK-mediated RIPK1 phosphorylation appears not to affect TNFα-induced cell death and inflammation.
Inhibition of RIPK1 protects against liver ischemia-reperfusion injury in myeloid Ampk KO mice
AMPK exerts anti-inflammatory effects, especially in macrophages, as metabolic activity and inflammatory status are directly linked in these cells (40). We conditionally deleted Ampkα1, which is the only catalytic subunit isoform expressed in macrophages (41), in myeloid cells using Ampkα1fl/fl mice expressing LysM-Cre and then crossed these mice with Ripk1D138N/D138N mice to generate Ampkα1f/f;LysM-Cre;Ripk1D138N/D138N mice. Ampkα1 expression was blocked in bone marrow–derived macrophages (BMDMs) isolated from Ampkα1fl/fl;LysM-Cre and Ampkα1f/f;LysM-Cre;Ripk1D138N/D138N mice (fig. S14A). Ampkα1fl/fl;LysM-Cre mice showed normal liver and body weight, normal amounts of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST), and normal liver morphology (fig. S14, B to E). Similar to MEFs, Ampk-deficient BMDMs showed increased RIPK1 activation when deprived of glucose (fig. S14, F to H).
Next we subjected these mice to liver ischemia-reperfusion (IR) injury (Fig. 4A). Ampkα1fl/fl;LysM-Cre mice showed increased abnormal ALT and AST in serum compared with WT mice (Fig. 4B). Moreover, Ampkα1fl/fl;LysM-Cre mice showed increased infiltration of inflammatory cells, which was reduced in Ampkα1fl/fl;LysM-Cre;Ripk1D138N/D138N mice (Fig. 4C and fig. S15, A and B). We also detected increased numbers of p-S166 RIPK1+ and TUNEL+ cells as well as increased expression of proinflammatory cytokines in the livers of Ampkα1fl/fl;LysM-Cre mice compared with those of WT mice (Fig. 4, D to H). We immunostained liver tissues after IR injury with a phosphospecific RIPK3 antibody validated in previous studies (42). Although we detected a substantial amount of TUNEL+ cells in livers of Ampkα1fl/fl;LysM-Cre mice after hepatic IR injury, none of them showed phosphorylation of T231/S232 of RIPK3, indicating the absence of necroptosis in IR-injured livers (fig. S15C). These data indicate that RIPK1-dependent apoptosis, more so than necroptosis, mediated cell death and inflammation in response to ischemic stress (fig. S15).
Fig. 4. Inhibition of RIPK1 activity protects against liver IR injury in myeloid Ampk KO mice.
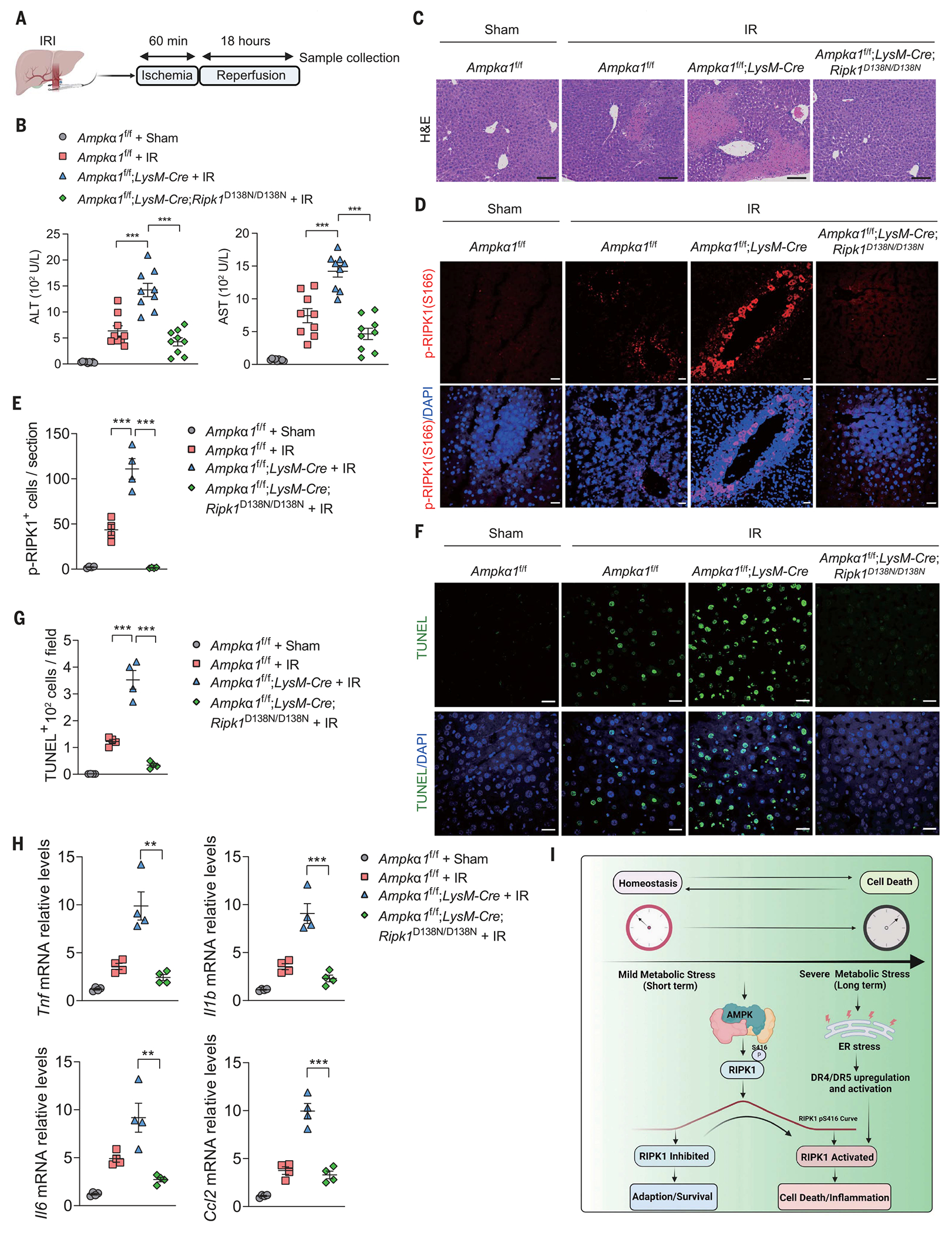
(A) Schematic of liver IR injury involving 60 min of global ischemia followed by an 18-hour reperfusion period. (B) Serum ALT and AST levels were measured from control (n = 9), Ampkα1f/f;LysM Cre (n = 9), and Ampkα1f/f;LysMCre;Ripk1D138N/D138N (n = 9) mice subjected to liver IR injury. Data are presented as mean ± SEM, and each dot represents one mouse. One-way ANOVA, post hoc Dunnett’s test; ***P < 0.001. (C) Histological analyses were performed on liver sections of Ampkα1f/f;LysM Cre, Ampkα1f/f;LysM Cre;Rpk1D138N/D138N, and control littermate mice subjected to liver IR injury (n = 4). Representative images are shown. Scale bars, 100 μm. (D) Immunostaining for p-RIPK1(S166) was performed on liver sections of Ampkα1f/f;LysM Cre, Ampkα1f/f;LysM Cre;Ripk1D138N/D138N, and control littermate mice subjected to liver IR injury (n = 4). DAPI for nuclei. Representative images are shown. Scale bars, 100 μm. (E) Quantification of p-RIPK1(S166)–positive cells on liver sections from (D). Data are presented as mean ± SEM, and each dot represents one mouse. One-way ANOVA, post hoc Dunnett’s test; ***P < 0.001. (F) TUNEL assays were performed on liver sections of Ampkα1f/f;LysM Cre, Ampkα1f/f;LysM Cre;Ripk1D138N/D138N, and control littermate mice subjected to liver IR injury (n = 4). DAPI for nuclei. Representative images were shown. Scale bars, 100 μm. (G) Quantification of TUNEL-positive cells on liver sections from (F). Data are presented as mean ± SEM, and each dot represents one mouse. One-way ANOVA, post hoc Dunnett’s test; ***P < 0.001. (H) Quantitative reverse transcription polymerase chain reaction analysis of the mRNA expression of cytokines and chemokines in livers from control (n = 4), Ampkα1f/f;LysM Cre (n = 4), and Ampkα1f/f;LysM Cre;Ripk1D138N/D138N (n = 4) mice subjected to liver IR injury. Data are presented as mean ± SEM, and each dot represents one mouse. One-way ANOVA, post hoc Dunnett’s test; **P < 0.01, ***P < 0.001. (I) A schematic model to illustrate a delicate and temporal cellular response of RIPK1 to metabolic stress: Cells activate AMPK to suppress RIPK1 activation, allowing survival under energy stress in the short term, whereas over longer time periods, as the AMPK phosphorylation of RIPK1 is lost, the inhibition is relieved, promoting a switch to activated RIPK1-mediated cell death and inflammation. Moreover, long-term glucose starvation causes the ATF4/CHOP-dependent up-regulation and activation of TRAIL receptors DR4 and DR5, which promotes RIPK1 activation, cell death, and inflammation.
Next we transplanted bone marrow from mice in which Ampk was deleted in myeloid cells with or without Ripk1D138N/D138N into WT, Ripk1D138N/D138N, and Ripk1S415A/S415A mice (fig. S16A) (43). Lethally irradiated WT recipient mice reconstituted with Ampkα1f/f;LysM-Cre bone marrow cells showed increased concentrations of ALT and AST in serum compared with WT recipient mice reconstituted with Ampkα1f/f bone marrow cells in response to liver IR injury (fig. S16B). Thus, Ampkα1 deficiency in myeloid cells appears to promote liver damage after hepatic IR injury in a non–cell-autonomous manner. Consistently, Ripk1D138N/D138N recipient mice reconstituted with Ampkα1fl/fl;LysM-Cre bone marrow cells showed decreased concentrations of ALT and AST in serum compared with WT recipient mice reconstituted with Ampkα1fl/fl;LysM-Cre bone marrow cells (fig. S16B). Thus, Ampkα1 deficiency in myeloid cells facilitates RIPK1 activation by IR injury, which promotes liver proinflammatory immune activation and contributes to the development of hepatocellular damage.
To further link the effects of AMPK on cell death and inflammation to RIPK1 phosphorylation at Ser415, we crossed the Ampkα1fl/fl;LysM-Cre mice into the Ripk1S415A/S415A background. Ampkα1fl/fl;LysM-Cre;Ripk1S415A/S415A mice did not show additional increase in IR injury–induced RIPK1 activation, cell death, or inflammation as compared with that of Ripk1S415A/S415A mice (fig. S16, C to E). Thus, the effects of AMPK on cell death and inflammation appear to be predominantly through AMPK-mediated RIPK1 Ser415 phosphorylation in the liver IR injury model we used.
Discussion
Our results indicate that phosphorylation-regulated control of RIPK1 activity enables cellular metabolic control of cell death and inflammation in response to metabolic stress (Fig. 4I). In parallel with other substrates of AMPK, AMPK directly inhibits RIPK1 by phosphorylation, which in turn suppresses energy stress–induced cell death and inflammation (fig. S16F). We revealed a delicate and temporal cellular response of RIPK1 to metabolic stress: Cells activate AMPK to suppress RIPK1 activation, allowing survival in the short term under energy stress, whereas over the longer term, as AMPK phosphorylation of RIPK1 is lost, the inhibition is relieved, promoting a switch to activated RIPK1–mediated cell death and inflammation. The work presented here expands our understanding of the interplay between metabolism and cell death regulation, which may help inform the development of therapeutic drugs aimed at preventing ischemia-induced cell death and tissue damage.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Kelliher (University of Massachusetts) and M. Pasparakis (University of Cologne, Germany) for providing Ripk1D138N/D138N mice and V. Dixit (Genentech) for RIPK3 KO mice and the antibodies recognizing phosphorylated RIPK3 Thr231/Ser232 (GEN135-35-9).
Funding:
This work was supported in part by NIH grants [CA177910 and R35CA253027 (W.W.); R00 CA194314 (C.C.D.); RO1 AA017729m and R01 AA011576 (G.Sz.); CA236226, CA239660, CA202634, P01 CA250959, and P01 CA120964 (P.S.); and RO1DK124328 and RO1DK133331 (J.M.)], the National Key R&D Program of China (2022YFC2303102 to L.P.; 2022ZD0213200 to D.X.), and the National Natural Science Foundation of China (92253301 to L.P.; 32070737 to D.X.). J.Y. was supported in part by the China National Natural Science Foundation (82188101, 21837004, 91849204, and 92049303). D.X. and J.Y. were supported in part by the Shanghai Municipal Science and Technology Major Project (grant no. 2019SHZDZX02), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB39030200 to J.Y.; XDB39030600 to D.X.). Y.-R.L. was supported in part by a Career Development Award, Academia Sinica Taiwan (AS-CDA-110-L07), and the Ministry of Science and Technology Taiwan (110-2320-B-001-029-MY2).
Footnotes
Competing interests: W.W. is a cofounder and consultant for ReKindle Therapeutics. P.S. has been a consultant at Novartis, Genovis, Guidepoint, The Planning Shop, ORIC Pharmaceuticals, Cedilla Therapeutics, Syros Pharmaceuticals, Exo Therapeutics, Curie Bio Operations, Exscientia, Ligature Therapeutics, and Redesign Science; and his laboratory receives research funding from Novartis. G.Sz. is a paid consultant for Cyta Therapeutics, Durect Co, Evive, Merck, Pfizer, Surrozen, Terra Firma, Pandion Therapeutics, LabCorp, Glympse Bio, Satellite Bio, and Zomagen. G.Sz. has additional financial interests in Glympse Bio, Satellite Bio, and Zomagen. The other authors declare that they have no competing financial interests.
Data and materials availability:
RNA-seq data used to support the present study have been deposited in the Gene Expression Omnibus with an access number of GSE173938. The biological material is available upon request to W.W. All data are available in the main text or the supplementary materials.
REFERENCES AND NOTES
- 1.Hardie DG, Nat. Rev. Mol. Cell Biol 8, 774–785 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Hardie DG, Ross FA, Hawley SA, Nat. Rev. Mol. Cell Biol 13, 251–262 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.González A, Hall MN, Lin SC, Hardie DG, Cell Metab. 31, 472–492 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Lin SC, Hardie DG, Cell Metab. 27, 299–313 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Green DR, Galluzzi L, Kroemer G, Science 345, 1250256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw RJ et al. Cancer Cell 6, 91–99 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Tummers B. et al. Immunity 52, 994–1006.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan J, Amin P, Ofengeim D, Nat. Rev. Neurosci 20, 19–33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu D. et al. Cell 174, 1477–1491.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun L. et al. Cell 148, 213–227 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Wang H. et al. Mol. Cell 54, 133–146 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Green DR, Cell 177, 1094–1107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan G. et al. Science 276, 111–113 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Pan G. et al. Science 277, 815–818 (1997). [DOI] [PubMed] [Google Scholar]
- 15.Kavuri SM et al. J. Biol. Chem 286, 16631–16646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cullen SP et al. Mol. Cell 49, 1034–1048 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Hartwig T. et al. Mol. Cell 65, 730–742.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henry CM, Martin SJ, Mol. Cell 65, 715–729.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Degterev A. et al. Nat. Chem. Biol 1, 112–119 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Linkermann A. et al. Proc. Natl. Acad. Sci. U.S.A 110, 12024–12029 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Degterev A. et al. Nat. Chem. Biol 4, 313–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ofengeim D. et al. Cell Rep. 10, 1836–1849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dillon CP et al. Cell 157, 1189–1202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green DR, Oberst A, Dillon CP, Weinlich R, Salvesen GS, Mol. Cell 44, 9–16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinlich R, Green DR, Mol. Cell 56, 469–480 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iurlaro R. et al. Mol. Cell. Biol 37, e00479–16 (2017).28242652 [Google Scholar]
- 27.Chaudhary PM et al. Immunity 7, 821–830 (1997). [DOI] [PubMed] [Google Scholar]
- 28.Kim J, Kundu M, Viollet B, Guan K-L, Nat. Cell Biol 13, 132–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng H. et al. Proc. Natl. Acad. Sci. U.S.A 115, E2001–E2009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardie DG, Schaffer BE, Brunet A, Trends Cell Biol. 26, 190–201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crute BE, Seefeld K, Gamble J, Kemp BE, Witters LA, J. Biol. Chem 273, 35347–35354 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Stein BD et al. Cell Rep. 29, 3331–3348.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howell JJ et al. Cell Metab. 25, 463–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaw RJ et al. Proc. Natl. Acad. Sci. U.S.A 101, 3329–3335 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gowans GJ, Hawley SA, Ross FA, Hardie DG, Cell Metab. 18, 556–566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hawley SA et al. Biochem. J 459, 275–287 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang CS et al. Nature 548, 112–116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newton K. et al. Nature 574, 428–431 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Hawley SA et al. Cell Metab. 11, 554–565 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly B, O’Neill LA, Cell Res. 25, 771–784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mounier R. et al. Cell Metab. 18, 251–264 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Heger K. et al. Nature 559, 120–124 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Dey A. et al. Science 337, 1541–1546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data used to support the present study have been deposited in the Gene Expression Omnibus with an access number of GSE173938. The biological material is available upon request to W.W. All data are available in the main text or the supplementary materials.