

SUMMARY
Alzheimer’s disease (AD) is the most prevalent cause of dementia; microglia have been implicated in AD pathogenesis, but their role is still matter of debate. Our study showed that single systemic wild-type (WT) hematopoietic stem and progenitor cell (HSPC) transplantation rescued the AD phenotype in 5xFAD mice and that transplantation may prevent microglia activation. Indeed, complete prevention of memory loss and neurocognitive impairment and decrease of β-amyloid plaques in the hippocampus and cortex were observed in the WT HSPC-transplanted 5xFAD mice compared with untreated 5xFAD mice and with mice transplanted with 5xFAD HSPCs. Neuroinflammation was also significantly reduced. Transcriptomic analysis revealed a significant decrease in gene expression related to “disease-associated microglia” in the cortex and “neurodegeneration-associated endothelial cells” in the hippocampus of the WT HSPC-transplanted 5xFAD mice compared with diseased controls. This work shows that HSPC transplant has the potential to prevent AD-associated complications and represents a promising therapeutic avenue for this disease.
Graphical Abstract
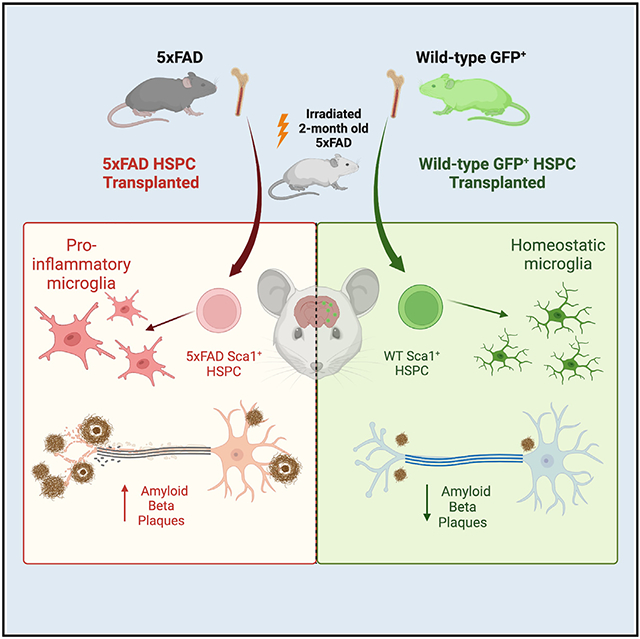
In brief
Mishra et al. demonstrate that single systemic wild-type hematopoietic stem and progenitor cell transplantation leads to complete preservation of the neurocognitive performance, partial preservation of blood-brain barrier integrity, and reduction of the Aβ plaque, microgliosis, and neuroinflammation in 5xFAD mice. In contrast, 5xFAD HSPC transplantation has no to limited impact.
INTRODUCTION
Alzheimer’s disease (AD) is the most prevalent cause of dementia and the most common age-related neurodegenerative disorder, projected to affect over 13 million people in the US by 2050.1 With an increasingly aging population, 3%–5% of individuals 65 years or older may be suffering from AD, adding 5–7 million new cases annually.2 As a result of AD, dementia develops into severe disability throughout the disease progression, resulting in death generally within 5–12 years after onset.3 Significant neuronal degradation, inflammation, progressive memory, and behavioral decline result from an accumulation of extracellular β-amyloid (Aβ) plaques and hyperphosphorylated tau in neurofibrillary tangles (NFTs) throughout the brain, predominantly in the hippocampus and the cortex. These major hallmarks of AD lead to increased oxidative stress, inflammation, synapse loss, and cholinergic dysfunction, and they elicit neuronal dystrophy.4 Current treatments approved by the FDA assist in the management of cognitive impairment, yet there are no effective disease-modifying treatments available.4,5 Given the complex pathobiology of AD, any potential treatment must target multiple pathological pathways to improve the disease state.
For a long time, reactive microglia have been considered a consequence of AD pathology; however, they are now regarded as potentially playing a role in disease progression and maybe initiation.6-9 Sustained microglia inflammation has been identified as a contributor to AD pathogenesis, as the release of inflammatory cytokines, chemokines, and complement proteins increases Aβ production.10,11 In addition, microglia have been shown to be involved in the clearance of Aβ plaque, which is impaired in AD due to mutations in microglia-related genes, including P2ry12, Apoe, and Trem2.12 Furthermore, with impaired microglia clearance, debris and other byproducts are diverted for clearance to other brain cells, such as endothelial cells, which do not proliferate efficiently and exhibit similar dysfunction, resulting in cell death and impaired blood flow.13 Thus, targeting microglia offers a potential therapeutic opportunity for AD.
We have previously demonstrated that a single systemic transplant of wild-type hematopoietic stem and progenitor cells (HSPCs) led to long-term rescue in both mouse models for cystinosis, a lysosomal storage disease, and Friedreich’s ataxia, a neurodegenerative disease.14-16 In Friedreich’s ataxia mouse model, transplanted HSPCs engrafted and differentiated into microglia in the brain and spinal cord, and into macrophages in the dorsal root ganglions (DRGs), resulting in the preservation of the neurons and locomotor function.15 Efficient replacement of microglia in the central nervous system (CNS) by bone marrow stem cell transplantation has previously been described.17 Therefore, because microglia may play an important role in AD, we hypothesized that wild-type (WT) HSPC transplantation could result in the generation of healthy microglia that may have a beneficial impact on AD.
To explore this hypothesis, we used the 5xFAD transgenic mouse model, which expresses mutant human Aβ (A4) precursor protein 695 (APP) with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) familial AD (FAD) mutations and human presenilin 1 (PSEN1) harboring two FAD mutations (M146L and L286V). Aβ plaque accumulation is observed in the hippocampus and the cortex of 5xFAD mice by 2–4 months of age.18,19 These mice exhibit significant neurocognitive impairment and altered anxiety behavior between 3 and 6 months of age.20 Moreover, 5xFAD mice demonstrate major features of amyloid plaque pathology of AD, including proliferation and activation of microglia.18,21
Here, we demonstrate that single systemic WT HSPC transplantation into adult 5xFAD mice led to the complete preservation of memory and neurocognitive performance. Both WT and 5xFAD HSPCs differentiated into microglia-like cells, but only WT HSPC transplantation led to the reduction of the Aβ plaques in the hippocampus and the cortex, as well as microgliosis and neuroinflammation, and to the preservation of blood-brain barrier (BBB) integrity. As therapeutic perspectives, this work supports that HSPC transplant could represent a promising avenue for the treatment of AD.
RESULTS
Transplantation of WT HSPCs results in normal neurocognitive performance in 5xFAD mice
To investigate the potential of WT HSPC transplantation for treating AD, we intravenously injected lethally irradiated 2-month-old 5xFAD mice with Sca1+ HSPCs isolated from WT green fluorescent protein (GFP) transgenic mice (5xFAD/WT HSPC; n = 19, 11 female [F] and 8 male [M]). Donor-derived HSPC engraftment, expressed as a percentage of GFP+ blood cells in peripheral blood, was measured by flow cytometry at 4 months post-transplant and ranged from 25% to 90% (Figure 1A; Table S1). As controls, we analyzed WT littermates (WT; n = 9, 5 F and 4 M), lethally irradiated WT littermates transplanted with Sca1+ WT GFP+ HSPCs (WT/WT HSPC; n = 9, 5 F and 4 M), non-treated 5xFAD littermates (5xFAD; n = 15, 8 F and 7 M), and lethally irradiated 5xFAD mice transplanted with Sca1+ 5xFAD HSPCs (5xFAD/5xFAD HSPC; n = 13, 7 F and 6 M). The difference between 5xFAD and WT HSPCs is the presence of the human mutated APP/PSEN1 transgenes (hAPP/PSEN1) exclusively in 5xFAD HPSCs. These transgenes are expressed under the control of elements of the mouse Thy-1 gene promoter.22,23 Thy1 is expressed in hematopoietic stem cells,24 and qPCR analysis confirmed expression of hAPP/PSEN1 in the murine Sca1+ HSPCs isolated from 5xFAD mice (Figure S1A). To determine the impact of WT HSPC transplantation on the neurocognitive function in 5xFAD mice, we performed a series of well-characterized behavioral testing in 6-month-old mice, i.e., 4 months post-transplantation. No difference between males and females was observed in any of the tests (Figures 1B-1D), as previously reported for this mouse model at 6 months of age.25 In addition, the myeloablative irradiation did not have any impact on activity in the WT and 5xFAD mice, as no difference was observed in any of the tests when compared with WT/WT HSPC and 5xFAD/5xFAD HSPC mice, respectively (Figures 1B-1D). Memory function of 5xFAD mice begins to deteriorate between 4 and 5 months of age.26,27 To determine whether WT HSPC transplantation prevents memory function loss, we used a hippocampus-dependent novel object recognition task (NORT).28,29 On the test day, the non-treated 5xFAD and 5xFAD/5xFAD HSPC mice showed no significant changes in the time exploring the novel or familiar objects (Figure 1B), indicating impaired memory function. In contrast, WT and WT/WT HSPC, but also 5xFAD/WT HSPC, mice spent significantly more time exploring the novel object, indicating that 5xFAD/WT HSPC-transplanted mice remembered the familiar object and had a normal ability to discriminate between the novel and familiar objects (Figure 1B). Altered anxiety is also a significant complication of AD and is correlated with cognitive impairment in patients.30,31 Alteration in anxiety-like behavior was also reported in 5xFAD mice in the elevated plus maze (EPM), with the mice spending more time in the open arms,26 whereas rodents usually prefer the closed arms, as they provide protection from potential predation.32 Both 5xFAD and 5xFAD/5xFAD HSPC mice spent more time within the open exposed arm (Figure 1C). In contrast, WT controls but also 5xFAD/WT HSPC mice spent more time in the closed arms at a similar level (Figure 1C), indicating rescue of the perception of risks in 5xFAD mice treated with WT HSPCs. Note that there was no significant difference in the distance between 5xFAD/WT HSPC mice and the two disease control groups (Figure S1B). Next, we tested the locomotor activity of the mice in an open field. General motor function as expressed as speed and distance was significantly elevated in 5xFAD and 5xFAD/5xFAD HSPC mice compared with WT, WT/WT HSPC, and 5xFAD/WT HSPC mice in the open field (Figures 1D and S1B). Altogether, these results demonstrate that transplantation of WT HSPCs in 5xFAD mice led to the preservation of their memory, perception of risks, normal anxiety level, and locomotor activity.
Figure 1. Transplantation of WT HSPCs prevents neurocognition impairments in 5xFAD mice.

(A) Schematic representation of the experimental design and timeline.
(B) Memory recognition test evaluated by discrimination index and preference index in mice at 6 months of age. A representative tracking plot of the 2 day test is shown for 5xFAD/5xFAD HSPC and 5xFAD/WT HSPC mice in the right panels.
(C) Elevated plus maze test in the different mouse groups with time expressed in seconds (s) spent in the closed arms and in the open arms and the ratio of time spent in the open arms versus the total time of the test. A representative tracking plot of the test is shown for 5xFAD/5xFAD HSPC and 5xFAD/WT HSPC mice in the right panels.
(D) Open field test with the total distance and periphery distance expressed in meters (m) traveled by the different mouse groups, as well as the average speed over a duration of 5 min. A representative tracking plot of the test is shown for 5xFAD/5xFAD HSPC and 5xFAD/WT HSPC mice in the right panels.
All data are indicated as mean ± SEM. **p < 0.005 and ***p < 0.0005 determined by one-way ANOVA followed by Tukey’s multiple comparisons, and each dot represent individual samples.
WT HSPC transplantation leads to the reduction of amyloid-β plaques in the brain of 5xFAD mice
Aβ plaques are most abundant in the hippocampus and cortex areas of the brain in patients with AD and 5xFAD mice.20,33 We quantified the Aβ plaques in the hippocampus and the cortex of our 5xFAD mice after immunohistochemical analysis using the 6E10 antibody, which is directed against the amino acids 1–16 of Aβ. The level of Aβ in 5xFAD mice transplanted with 5xFAD HSPCs was equivalent to the non-treated 5xFAD mice in both the hippocampus and the cortex (Figures 2A and 2B). In contrast, 5xFAD mice transplanted with WT HSPCs exhibited significant reduction in 6E10+ plaque number, size, and total area in both the cortex and the hippocampus compared with non-treated 5xFAD mice and in size and total area in the cortex and in number and size in the hippocampus compared with 5XFAD/5XFAD HSPC mice (Figures 2A and 2B). We also measured the levels of soluble (Triton fraction) and insoluble (guanidine fraction) Aβ protein fragment 1–42 (Aβ1–42), which is the most commonly found in AD.34 A significant reduction in the levels of soluble and insoluble Aβ1–42 was observed in 5xFAD mice transplanted with WT HSPC compared with both untreated and 5xFAD/5xFAD mice (Figure 2C). These results demonstrate that transplantation of WT HSPCs had a beneficial impact on reducing the Aβ plaque burden in the cortex and hippocampus areas in 5xFAD mice.
Figure 2. Transplantation of WT HSPCs led to the decrease of Aβ plaque deposition in the cortex and hippocampus in 5xFAD mice.

(A) Representative sagittal sections of the cortex and hippocampus stained against the Aβ plaque marker 6E10 of 5xFAD/5xFAD HSPC and 5xFAD/WT HSPC mice. Scale bars, 500 μm.
(B) Quantification of the plaque number, area occupied by the plaques, and percentage of total area occupied in the cortex and hippocampus in 5xFAD, 5xFAD/5xFAD HSPC, and 5xFAD/WT HSPC mice.
(C) Triton-X-soluble and guanidine insoluble of Aβ1–42 (ELISA) in the brain of WT, 5xFAD, 5xFAD/5xFAD HSPC, and 5xFAD/WT HSPC mice.
All data are indicated as mean ± SEM. **p < 0.005, ***p < 0.0005, and ****p < 0.0001 determined by one-way ANOVA followed by Bonferroni’s multiple comparisons, and each dot represent individual samples.
Engrafted WT and 5xFAD HSPCs differentiate into microglia-like cells, but only WT HSPCs correct microgliosis and neuroinflammation
We then characterized the engraftment and differentiation of WT GFP+ HSPCs in the brain of 5xFAD mice and found substantial engraftment of GFP+ HSPC-derived cells in the whole brain including the cortex and the hippocampus of all treated mice (Figure 3A). These cells were immunoreactive with Iba1 and CD11b, characterizing these cells as microglia-like cells (Figures 3B and 3C). In addition, the presence of 6E10+ aggregates near and within GFP+ microglia suggest active engulfment of the amyloid plaques by the WT HSPC-derived microglia (Figures 3C and S1C). Using flow cytometry analysis of whole brain, we determined the proportion of GFP+ microglia-like cells and found that an average of 28.19% ± 10.19% (n = 3) cells were derived from the transplanted GFP+ WT HSPCs (Figures 3D and 3E; Table 1). Note that one 5xFAD/WT HSPC mouse had a low blood cell engraftment (<20%) and exhibited a low percentage of GFP+ microglia (Table 1). The proportion of Iba1+ CD11b+ microglia derived from the GFP+ HSPCs was also determined by immunostaining in the whole brain, hippocampus, and cortex showing no significant difference in the quantity of HSPC-derived microglia-like cells in the different brain regions (Figure 3F). As control, to prove that 5xFAD HSPCs could also differentiate into microglia-like cells, we ex vivo transduced 5xFAD HSPCs with a lentiviral vector containing the GFP reporter gene to follow the HSPCs after transplant. We confirmed the differentiation of the GFP+ 5xFAD HSPCs into microglia-like cells by immunofluorescence and flow cytometry (Figures S2A-S2C).
Figure 3. Infiltration of transplanted GFP+ WT HSPCs into the brain of 5xFAD mice and differentiation into microglia-like cells.

(A) Representative immunohistochemistry image of a sagittal section of the brain from 5xFAD/WT HSPC mice at 4 months post-transplantation showing GFP+ cells; insets show GFP+ cells in hippocampus and cortex. Scale bars, 500 μm (whole brain) and 100 μm (insets).
(B) Representative immunofluorescent image of the brain showing engrafted GFP+ HSPC-derived cells (green) and anti-Iba1 (red) and anti-Cd11b (magenta) antibodies. Scale bars, 100 μm. Inset showing HSCP-derived microglia-like cells. Scale bar, 20 μm (inset).
(C) Representative 3D reconstitution of immunofluorescence image of brain sections from 5xFAD/5xFAD HSPC (top panel) and 5xFAD/WT HSPC mice (bottom panel) stained with anti-GFP (green), anti-Iba1 (red), and anti-6E10 (magenta) antibodies. Scale bars, 10 μm. The top inset shows inflamed active Iba1+ in close proximity to 6E10+ plaques of 5xFAD/5xFAD HSPC mice. The bottom inset shows ramified GFP+ Iba1+ microglia around 6E10+ plaques of 5xFAD/WT HSPC mice. Scale bar, 2 μm.
(D) Flow cytometry plots of live and GFP+ cells in the whole brain of WT, 5xFAD, 5xFAD/5xFAD HSPC, and 5xFAD/WT HSPC mice. Brain cells were labeled with Zombie Aqua for live-/dead-cell populations.
(E) Flow cytometry of microglia (CD45+ CD11bhigh) and of GFP+ microglia in 5xFAD/WT HSPC mice.
(F) Percentage of Iba1+ Cd11b+ microglia derived from the GFP+ HSPCs quantified in the whole brain, hippocampus, and cortex. Each dot represents individual samples.
Table 1.
Characterization of GFP+ HSPC engraftment and microglia population
| Mice | Genotype | Blood engraftment (%) | GFP+ cells in the brain (% of live cells) |
Microglia (CD45lowCD11bhigh) (% of live cells) |
GFP+ microglia (% of microglia) |
|---|---|---|---|---|---|
| 1 | WT | – | – | 28.7 | – |
| 2 | WT | – | – | 24.3 | – |
| 3 | WT/WT HSPC | 43.4 | 7.23 | 12.8 | 28.8 |
| Mean ± SEM | 21.93 ± 4.74 | ||||
| 1 | 5xFAD | – | – | 33.7 | – |
| 2 | 5xFAD | – | – | 49.4 | – |
| 3 | 5xFAD | – | – | 43.5 | – |
| Mean ± SEM | 42.2 ± 4.58a | ||||
| 1 | 5xFAD/5xFAD HSPC | – | – | 15.9 | – |
| 2 | 5xFAD/5xFAD HSPC | – | – | 30.5 | – |
| 3 | 5xFAD/5xFAD HSPC | – | – | 32.3 | – |
| Mean ± SEM | 26.23 ± 5.19 | ||||
| 1 | 5xFAD/WT HSPC | 35.8 | 26.4 | 26.4 | 35.4 |
| 2 | 5xFAD/WT HSPC | 78.5 | 25.6 | 26.5 | 41.1 |
| 3 | 5xFAD/WT HSPC | 14.9 | 4.28 | 11.7 | 8.07 |
| Mean ± SEM | 21.57 ± 2.17b | 28.19 ± 10.19 |
p < 0.05 compared with WT mice.
p <0.05 compared with 5xFAD mice.
Phenotypic difference of microglia-like cells was confirmed by immunochemistry. Both 5xFAD and 5xFAD/5xFAD HSPC mice displayed a significantly higher density and area covered by Iba1+ microglia in the hippocampus and cortex compared with WT mice but also compared with 5xFAD/WT HSPC mice (Figures 4A, 4B, S1D, and S1E). In addition, microglia were mostly ameboid and activated, suggesting ongoing inflammation in the hippocampus and the cortex of 5xFAD and 5xFAD/5xFAD mice (Figures 4A and S1D) as previously described for AD.9,35 In contrast, Iba1+ microglia in 5xFAD/WT HSPC mice displayed a more ramified “resting” phenotype (Figures 4A and S1D). Similarly, dual chromogenic staining of Iba1/6E10 showed that plaque-associated Iba1+ cells were active and inflamed in 5xFAD mice transplanted with 5xFAD HSPCs, whereas they had a ramified and multipolar morphology in the brain of WT HSPC-transplanted 5xFAD mice (Figure 4C). To ascertain these observations, we evaluated microglia morphology via Sholl analysis, a well-established technique to quantitatively analyze microglia processes and branching complexity.36 A Sholl analysis plot clearly illustrates that the branching profile of microglia in the hippocampus and the cortex of 5xFAD and 5xFAD/5xFAD HSPC mice is shifted to the left, an indication of decreased branching and ramification as compared with WT and 5xFAD/WT HSPCs (Figure 4D). Together, these data suggest decreased microgliosis in the brain of the WT HSPC-treated mice.
Figure 4. Transplantation of WT HSPCs led to microglia activation and neuroinflammation reduction.

(A) Representative images of hippocampus sections immunostained for the microglial marker Iba1; inset shows dentate gyrus region of the hippocampus. Scale bars, 100μm.
(B) Quantification of the area occupied by Iba1+ cells, as well as Iba1+ cell density, in the hippocampus.
(C) Representative chromogenic image showing Iba1+ microglia (green) and 6E10+ plaques (purple). Scale bars, 50 μm.
(D) Representative images of Imaris rendering of Iba1+ microglia. Sholl analysis quantification of number of Sholl intersections in the cortex and hippocampus brain sections in the different groups of mice.
(E) Quantitative PCR quantification of murine cytokine (Ifnγ, Tnfα, and Il6) mRNA expression in whole brains from WT, 5xFAD, 5xFAD/5xFAD HSPC, and 5xFAD/WT HSPC mice. Data are represented as fold change relative to WT and were normalized with GAPDH.
(F) Level of IFN-γ, TNF-α, and IL-6 proteins measured by ELISA.
Data are means ± SEM. **p < 0.005, ***p < 0.0005, and ****p < 0.0001 determined by one-way ANOVA, and each dot represent individual samples.
To confirm the reduction of neuroinflammation in 5xFAD/WT HSPC mice, we measured the expression of inflammatory cytokines released from microglia that are known to be elevated in AD brains.37 By qPCR, we found interferon γ (IFN-γ), tumor necrosis factor α (TNF-α), and interleukin-6 (IL-6) to be elevated in the non-treated and 5xFAD/5xFAD HSPC mice compared with WT mice and to be significantly decreased in 5xFAD/WT HSPC mice (Figure 4E). These results were confirmed by ELISA (Figure 4F). IFN-γ was also found to be decreased in 5xFAD/WT HSPC mice compared with controls by RNA sequencing (RNA-seq) analysis (data not shown). Altogether, these data demonstrate that WT HSPC transplantation decreased neuroinflammation in 5xFAD mice.
RNA-seq analysis reveals major differences in the transcriptome profile of microglia in cortex and of endothelial cells in hippocampus after WT HSPC transplantation in 5xFAD mice
To decipher the relative contributions of WT HSPC transplantation to AD disease mitigation, we examined the transcriptome profile by RNA-seq of the cortex and the hippocampus isolated from WT (n = 2), 5xFAD (n = 3), 5xFAD/5xFAD HSPC (n = 3), and 5xFAD/WT HSPC (n = 3) mice. Recent genome-wide association studies (GWASs) in patients with AD have identified genes that are differentially expressed in microglia related to inflammatory state and endo-lysosomal function.8,12,38,39
Since we observed that transplanted WT HSPCs differentiated into microglia in the brain, leading to decreased microgliosis in the cortex and the hippocampus in 5xFAD mice, we analyzed the transcriptome profile by composite gene score analysis on a previously published “disease-associated microglia” (DAM) gene set associated with AD.8 Two distinct stages of DAM have been described: stage 1 represents the first stage of DAM activation and corresponds to downregulation in the expression of microglia homeostatic genes and the upregulation of phagocytic pathway-related genes, including multiple known AD risk genes (Apoe, Cstb), and stage 2, corresponding to fully activated DAM, is Trem2 dependent and includes induction of lipid metabolism and phagocytic pathway-related genes (Figure S3A). In addition, we also assess previously published universal macrophage makers for human and mice tissues (Tyorbp, Fcer1g), which were also found to be differentially expressed in AD.40 Composite gene analysis revealed that stage 1 and stage 2 DAM and universal macrophage markers have higher expression levels in the cortex of 5xFAD and 5xFAD/5xFAD HSPC mice compared with those of 5xFAD/WT HSPC and WT mice; because of an outlier in the 5xFAD/5xFAD HSPC group, the difference was significant only with 5xFAD mice (Figure S3A). These data confirmed that microglia inflammatory activation associated with AD in 5xFAD mice is rescued by WT HSPC transplantation. This was confirmed by differentially expressed gene (DEG) analysis, which revealed that out of 61 genes differentially expressed (adjusted p value < 0.1 and log2 fold change > 1) in the cortex between 5xFAD and 5xFAD/WT HSPC mice, 49 were upregulated in 5xFAD compared with in 5xFAD/WT HSPC mice, and 24 of them were related to microglia (Figure S3B). Some DEGs were validated by qPCR (Figure S4D). Additionally, ApoE, which is implicated in the fibrilization of Aβ,41 was found to be increased in 5xAFD and 5xFAD/5xFAD HSPC mice by RNA-seq and qPCR compared with in WT and 5xFAD/WT HSPC mice (Figures S3 and S4D). Gene Ontology (GO) Biological Process pathway analysis as well as the protein-protein interaction (PPI) network were used to identify biological pathways that were significantly altered between 5xFAD and 5xFAD/WT HSPC mice, revealing innate immunity changes in the cortex such as leukocyte activation, cytokine stimulus, and antigen pathways that were significantly increased in the 5xFAD cortex compared with the 5xFAD/WT HSPC cortex (Figures S4A-S4C).
Interestingly, the microglia profile was not different between the different groups in the hippocampus. Further gene score analysis revealed a significant increase in expression of neurodegeneration-associated endothelial cell genes in the hippocampus of 5xFAD mice and 5xFAD/5xFAD HSPC mice compared with 5xFAD/WT HSPC and WT mice (Figure S3A). DEG analyses also revealed that the top 7 DEGs in the hippocampus of 5xFAD/WT HSPC mice compared with 5xFAD controls are associated with endothelial cells (Figure S3B); some of the genes were validated by qPCR (Figure S4E). This phenomenon is specific to the hippocampus, as the endothelial cell gene profile was not different in the cortex between the different groups. These data suggest an endothelial cell-specific impact of the WT HSPC transplant in the hippocampus of 5xFAD mice. GO and PPI analyses revealed that, in addition to innate immunity changes in hippocampus, pathways involving cell adhesion and extracellular matrix organization were also significantly increased in the 5xFAD hippocampus compared with the 5xFAD/WT HSPC hippocampus (Figures S4A-S4C).
Based on our RNA-seq findings, we investigated potential structural changes in endothelial cell organization and BBB integrity in the hippocampus of the different 5xFAD groups. We first assessed expression of VE-cadherin (Cdh5), an endothelial-specific transmembrane protein essential for BBB integrity,42 which was differentially expressed in the diseased groups compared with WT and 5xFAD/WT HSPC mice. Quantitative image analysis revealed a significant decrease of Cdh5+ vessels and vessel diameter in the hippocampus of 5xFAD and 5xFAD/5xFAD HSPC mice compared with WT and 5xFAD/WT HSPC mice (Figures 5A and 5B). In addition, we examined the co-localization of the platelet endothelial cell adhesion molecule (PECAM1) involved in endothelial cell junction and BBB integrity,43 relative to VE-cadherin+ vessels. Reduction in co-staining was observed in 5xFAD and 5xFAD/5xFAD mice (42.40% and 43.6%, respectively) compared with WT and 5xFAD/WT HSPC mice (59.67% and 55%, respectively; Figure 5B). GFP+ WT HSPC-derived microglia-like cells could be observed in close proximity to cerebral microvessels (Figure 5C).
Figure 5. Transplantation of WT HSPCs led to preservation of neurovascular integrity in the hippocampus of 5xFAD mice.

(A) Representative immunofluorescence image of brain sections from WT, 5xFAD, 5XFAD/5XFAD HSPC, and 5XFAD/WT HSPC mice stained with anti-Cdh5 (red) antibodies and anti-PECAM1 (magenta). Scale bars, 50 μm.
(B) Quantification of total number of Cdh5+ vessels, vessel density, area, and vessel diameter in the hippocampus in the different groups of mice. Quantification of vascular PECAM1 coverage (percentage of total PECAM1 staining area co-localizing with Cdh5). Data are means ± SEM. *p < 0.05 and **p < 0.005 determined by one-way ANOVA followed by Tukey’s multiple comparisons.
(C) Representative immunofluorescence image of an 5xFAD/WT HSPC hippocampus showing an HSPC-derived microglia-like cell (arrow) in close proximity to a cerebral vessel from mice. Anti-GFP is shown in green, anti-PECAM1 in red, and anti-Cdh5 in magenta. Scale bars, 20 μm. Each dot represent individual samples.
DISCUSSION
With an increasingly aging population, AD is projected to affect 131.5 million people globally by 2050 and to have a devastating impact. The costs of AD are accelerating from 1 trillion to a projected 2 trillion in 2030, which results in a heavy burden on patients, families, and the public health system.44,45 Significant efforts have been made over the last several decades to create disease-ameliorating medicines that stop, prevent, or delay AD. Unfortunately, while hundreds of therapies from a variety of pharmacological classes have shown preclinical efficacy in animal models for alleviating cognitive impairment and disease load, none have yet proven effective in human clinical trials.45 Therefore, there is a pressing unmet medical need for the treatment of AD.
Our study represents direct evidence that microglia play a key role in disease progression and that preventing microgliosis via single systemic WT HPSC transplantation in 5xFAD mice led to complete rescue of the neurocognitive impairment in the mice. We showed that WT HSPCs differentiated into microglia-like cells by immunofluorescence and flow cytometry analysis at a proportion that could reach >40% of the microglia. Microglia-like cells originate from the infiltration of myeloid cell populations and are reported to have a long lifespan.46-49 These cells form a network with embryonically derived microglia and play an important role in maintaining homeostasis and controlling neuroinflammation.46 In addition, it is important to note that differentiation of HSPCs into microglia-like cells is not only a consequence of irradiation, as chemotherapy agents such as busulfan have a more positive effect on promoting the repopulation of bone marrow-derived microglia in the CNS.50 A very recent article showed that chemotherapy leads to the senescence of resident microglia and their replacement by HSPC-derived microglia-like cells.51 This critical feature has been used to rescue neurological diseases using allogenic HSPC transplantation as well as in the gene therapy field using autologous gene-modified hematopoietic stem cells in both preclinical and clinical settings. The first successful clinical outcome of autologous genetically modified HSPC was reported in X-linked adrenoleukodystrophy in 2009, and a long-term beneficial effect was later confirmed in phase 2–3 clinical trials and is believed to be mediated by the HSPC-derived replacement of the microglia.52,53 In metachromatic leukodystrophy (MLD), differentiation of gene-modified HSPCs into functionally corrected microglia-like cells was shown following transplantation of gene-modified HSPCs, leading to cross-correction of the neighboring cells in murine models.54,55 They showed that myeloablative conditioning by both irradiation and busulfan, which have an ablative effect on CNS-resident myeloid precursors, was essential for repopulation of the recipient myeloid compartment, including microglia by the transplanted HSPCs.56 These results have been confirmed in clinical trials for MLD in which patients are going through myeloablation using busulfan and autologous transplantation of ex vivo gene-modified HSPCs, resulting in prevention of neurodegeneration and neurocognitive preservation in patients when treated early enough.57,58 Furthermore, as control, we used 5xFAD mice receiving 5xFAD HPSCs to prove that irradiation was not responsible for this beneficial impact. Some reduction in the plaque deposits was observed, which could be explained by the replacement of some of the exhausted resident microglia with newly formed HSPC-derived microglia-like cells. Indeed, it was shown that a high Aβ burden led to the exhaustion of microglia for Aβ clearance, whereas young 5xFAD microglia had normal clearance capacity in hippocampal slice cultures.59 However, the difference between untreated 5xFAD and 5xFAD/5xFAD HSPC mice was limited, showing that transplanted 5xFAD HSPCs do not have the capacity to prevent amyloid deposits, microgliosis, neuroinflammation, or the neurocognitive phenotype.
Proliferation and activation of microglia in the brain is an important characteristic in AD,59 and we confirmed these features in 5xFAD mice untreated or transplanted with 5xFAD HPSCs. Remarkably, there was a distinct reduction in the density and morphology of Iba1+ microglia in WT HSPC-trans-planted mice. This key feature plays a direct role in neurocognitive preservation in 5xFAD mice. Indeed, chronically activated microglia continuously release inflammatory mediators and amplify the chronic inflammatory environment in AD, contributing to synapse and neuronal loss by engulfment of synapses and secretion of inflammatory factors that injure neurons.60-62 We showed that transplanting WT HSPCs led to the reduction of microgliosis and inflammatory state in the brain. We also showed that preventing microgliosis was sufficient to prevent neurocognitive impairments in 5xFAD mice, strongly suggesting that microglia-associated neuroinflammation in AD is an important feature of neurodegeneration in AD, and modulating this phenotype represents an important therapeutic target.
In addition, we observed the reduction of amyloid deposits in 5xFAD mice transplanted with WT HSPCs. In line with previous reports correlating Aβ reduction with functional and cognitive improvement, the effect of WT HSPC transplantation on behavior was accompanied by a reduction in the level of 6E10+ plaques and soluble and insoluble Aβ1–42 in the hippocampus and cortex. Similarly, it was previously shown that GFP+ bone marrow cells transplanted in the APPSWE/PS1 mouse model could infiltrate the CNS and differentiate into microglia-like cells with the capacity to reduce Aβ plaque deposits.63 Previous studies displayed that in amyloidosis mouse models and in patients with AD, plaques form over time64,65 and are surrounded by microglial cells, which seem to be reactive and inefficient in phagocytosing and clearing Aβ.60,66-68 In contrast, microglia derived from the transplanted WT HSPCs displayed a more ramified “resting” phenotype, but co-localized with 6E10+ plaques that appear smaller, suggesting engulfment of the Aβ plaques by the WT microglia in 5xFAD/WT HSPC mice. However, it is also possible that the reduction in amyloid deposits could be attributed to a decrease in the formation of plaques. Indeed, it was shown that activated microglia may participate in the spreading of Aβ aggregates to form new plaques by carrying and releasing phagocytosed Aβ seeds within the brain.69 In addition, it was shown that persistent neuroinflammation exacerbates amyloid deposits by recruiting more microglia to plaques, resulting in a characteristic halo of activated microglia surrounding plaques.70,71 TNF-α and IL-6, which we found to be increased in the 5xFAD disease groups compared with 5xFAD/WT HSPC mice, were also shown to be directly associated with increased Aβ plaques.72,73 Therefore, decreased microgliosis and neuroinflammation after WT HSPC transplantation may lead to the reduction of Aβ aggregate formation and spreading.
To further evaluate the impact of WT HPSC transplantation on the gene expression profile levels in the two main areas of the brain impacted by the plaque Aβ accumulation, we analyzed the cortex and hippocampus of 5xFAD mice using RNA-seq analysis. It has been shown that microglia associated with plaques activate specific signaling pathways, and Keren-Shaul et al. identified DAMs, which increase with disease progression in 5xFAD mice8 but also in postmortem brains from patients with AD. Indeed, many of the DAM genes were found in human GWASs including TREM2, a receptor required for DAM activation but also for Aβ clearance.8,74,75 Similarly, it was shown that plaque-containing microglia show increased DAM and phagocytosing capability.76 In later stages of AD, DAMs are significantly overexpressed, resulting in increased phagocytic and inflammatory activities, which lead to more damage to the surrounding cell populations. We believe our analysis of 5xFAD mice occurs during this late stage of AD, when increased DAM is present in the untreated and 5xFAD HSPC-treated mice, along with enrichment for genes related to immune response and cytokine activity, as observed in the GO analysis of DEGs. Increased cytokines and chemokines in the brain of 5xFAD and 5xFAD/5xFAD HSPC mice were also shown by qPCR and ELISA. In contrast, 5xFAD mice transplanted with WT HSPCs exhibited a significant decrease of DAMs in the cortex as well as neuroinflammatory-related genes, reaching a similar level to that in WT mice. The universal macrophage markers that increase in neurological diseases, including AD,40 also decreased in 5xFAD/WT HSPC mice. Overall, these data proved that WT HSPC transplantation rescues disease microglia activation and dysfunction as well as neuroinflammation in the cortex of 5xFAD mice.
Surprisingly, the microglia transcriptomic profile in the hippocampus did not differ between the different groups. But interestingly, the neurodegeneration-associated endothelial cell gene expression profiling that has been reported to be increased in other neurodegenerative disorders77 has been also found to increase in this tissue compartment in 5xFAD mice compared with in WT mice, and this was corrected in 5xFAD/WT HSPC mice. In AD, age-dependent deterioration of the BBB occurs in the human hippocampus, and severe impairment of the BBB transport mechanism has been reported in an advanced phase of the disease, leading to hippocampus-dependent cognitive impairment.78-81 VE-cadherin (Cdh5) mRNA was found to be increased in the hippocampus of the disease 5xFAD groups compared with the WT and 5xFAD mice treated with WT HSPCs, which may compensate for the decrease of VE-cadherin+ vessels seen in the hippocampus of the disease 5xFAD and 5xFAD/5xFAD HSPC mice. Because VE-cadherin is an important component of the brain vascular endothelial cell adhesion system modulating BBB integrity and function,42 these data suggest BBB impairment in the hippocampus of the 5xFAD diseased groups. BBB disruption has been previously reported in 5xFAD mice as early as 4 months of age.82,83 Because PECAM1 and VE-cadherin are the anatomical basis of the BBB,84 we also investigated PECAM1 expression on VE-cadherin+ vessels and found that this cell adhesion protein decreased in the 5xFAD disease groups compared with in the WT and 5xFAD/WT HSPC mice. BBB disruption may be caused by neuroinflammation.85 Indeed, proinflammatory cytokines such as TNF-α, which we found to be increased in the disease 5xFAD groups, have been reported to destabilize VE-cadherin and reduce PECAM1 from the endothelial cell junctions.42,86 Thus, reduction of neuroinflammation by WT HSPC transplant may lead to the conservation of BBB integrity. In addition, some GFP+ WT HSPC-derived microglia-like cells were observed adjacent to brain vasculature in 5xFAD/WT HSPC mice, potentially contributing to the maintenance of BBB integrity. Indeed, in vivo studies previously demonstrated that perivascular microglia may directly supply critical proteins to the BBB endothelium.87
The exact mechanism behind the significant therapeutic impact of transplanting WT HPSCs into 5xFAD mice remains unclear. WT HSPC transplant fully prevents microgliosis, neuroinflammation, and BBB disruption, whereas partial microglia are replaced by HSPC-derived microglia-like cells, suggesting a systemic beneficial impact of the WT HSPCs. In contrast, HSPCs isolated from 5xFAD mice have limited to no impact. One reason could be that chronic inflammation in 5xFAD mice has a long-lasting effect on the HSPCs that makes them reactive to the inflammatory environment when re-transplanted into 5xFAD mice. Indeed, it was shown that chronic inflammation could cause a shift in hematopoietic stem cell fate, resulting, in particular, in the higher production of activated monocytes secreting cytokines such as IFN-γ, which further propagates inflammation.88,89 As such, we found IFN-γ to be increased by RNA-seq, qPCR, and ELISA in the brain of 5xFAD and 5xFAD/5xFAD HSPC brains as opposed to 5xFAD/WT HSPC brains. Similarly, bone marrow transplantation experiments in hypercholesterolemia and Duchenne muscular dystrophy mice models revealed that the diseased bone marrow microenvironment activates the HSPCs and skews their development toward myeloid lineage, which maintained heightened responsiveness to endogenous stimuli and exhibited a proinflammatory state.90 In contrast, WT HSPC transplant may stop the inflammatory cascade by generating non-reactive monocytes and microglia-like cells in 5xFAD mice. One mechanism of this is “trained immunity,” which involves epigenetic modifications that promote strong expression of immune genes in hematopoietic stem cells, which persists after transplant.91 Thus, genetic susceptibility may exist in 5xFAD HSPCs as opposed to WT HSPCs, and epigenomic and transcriptomic assessments have previously identified risk alleles that function as active enhancers of monocytes, macrophages, and microglia in AD.38 Alternatively, human mutated APP/PSEN1 expression in HSPCs may trigger a proinflammatory state of the immune progeny. Indeed, it was shown that APP could act as a proinflammatory receptor on immune cells and that hAPP knockout in APP/PSEN1 transgenic mice resulted in decreased microgliosis, despite abundant Aβ deposits.92 Although additional research will be necessary to fully understand the advantageous therapeutic impact of the WT HSPC transplant in 5xFAD mice, this study demonstrates that HSPC could be a potential target for treating AD.
In conclusion, our findings demonstrate that transplantation of WT HSPCs into the 5xFAD mouse model resulted in the engraftment and differentiation of these cells into microglia-like cells and in the rescue of AD. Specifically, WT HSPC transplant reduced Aβ accumulation and prevented microgliosis, neuroinflammation, and vasculature impairment, all of which play crucial roles in preventing neurocognitive decline in AD. Therefore, our study serves as a proof of concept for the use of HSPC transplantation as a therapeutic target for this untreatable disorder.
Limitations of the study
A limitation of this study is that the 5xFAD mouse model expresses both mutated human APP and PSEN1 transgenes. Consequently, if there is a direct impact of mutated APP and/or PSEN1 in the HSPCs that causes neuroinflammation, it is unclear whether it is caused by the mutated APP or PSEN1 or by both. A single-mutation mouse model will have to be utilized to resolve this question. In addition, we evaluated the impact of WT HSPC transplant in 5xFAD mice prior to the onset of neurocognitive phenotype and found that we could prevent these complications. It will be important to examine whether WT HSPC transplantation could halt or improve neurocognitive function after onset by transplanting older 5xFAD mice. Finally, comprehensive understanding of the mechanism underlying the behavioral rescue in AD by WT HSPCs remains to be discovered.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr Stephanie Cherqui (scherqui@ucsd.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The data used for generation of figures are available from the corresponding authors upon reasonable request.
RNA sequencing data that support the findings of this study have been deposited in NCBI GEO with the accession number GSE217885. The codes used to conduct RNA sequencing analysis are publicly available at the following links: https://github.com/sahoo00/BoNE; https://github.com/sahoo00/Hegemon.
All data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All the mice were on a C57BL6 background. We used the 5xFAD transgenic mouse model, (Tg6799, C57BL6-Tg (APPSwFlLon, PSEN1*M146L*L286V) 6799 Vas, Jackson Laboratory).18 Standard PCR reactions were carried out to identify the mice expressing all 5 mutations using the primers listed in Table S2. GFP transgenic mice, ubiquitously expressing enhanced GFP complementary DNA under the control of the chicken beta-actin promoter, were used as donors for the HSPC transplantation experiments (C57Bl/6-Tg (CAG-EGFP)1Osb/J; 003291, Jackson Laboratory). Mice were maintained in a temperature- and humidity-controlled animal facility, with a 12-h light/dark cycle and free access to water and food. Both male and female mice were used in all experiments. Each control and test group consisted of 9–19 mice and were analyzed at 6 months of age; the sex and number in each group are listed in the result section. All mice were bred at the University of California, San Diego (UCSD) vivarium, and all protocols were approved by the UCSD Institutional Animal Care and Use Committee.
METHOD DETAILS
HSPC isolation, transduction, transplantation, and engraftment
Bone marrow cells were flushed from the femurs of 6- to 8-week-old 5xFAD, GFP transgenic mice or 5xFAD mice. HSPCs were isolated by immunomagnetic separation using an anti-Sca1 antibody conjugated to magnetic beads (Miltenyi Biotec). Sca1+ HSPCs were directly transplanted by tail vein injection of 2 × 106 cells resuspended in 100 μL of phosphate-buffered saline (PBS) into lethally irradiated (7 Gy; X-Rad 320, PXi) 5xFAD mice. For mice receiving WT GFP+ HSPCs, bone marrow cell engraftment of the transplanted cells was measured in peripheral blood 2 months after transplantation; blood samples freshly harvested from the tails were treated with red blood cell lysis buffer (eBioscience) and subsequently analyzed by flow cytometry (BD Accuri C6, BD Biosciences) to determine the proportion of GFP+ cells. To follow 5xFAD HSPC differentiation potential within the brain, 5xFAD Sca1+ HSPC were transduced with the lentiviral vector containing eGFP as previously described.96 Cells were plated at a density of 2 × 106 cells in 0.5 mL of media per well in a 24-well plate pretreated with 200 μL of RetroNectin (Takara Bio USA, Madison, WI) and transduced using a multiplicity of infection of 10 and incubated for 16–18 h at 37°C, 5% CO2. Cells from each well were collected and washed twice in PBS and then resuspended in 100 μL of PBS. Tail vein injection of 100 μL containing ~2 × 106 cells was performed in each 5xFAD mice lethally irradiated (7Gy) on the previous day. Blood engraftment of eGFP+ cells was determined at 1-month post-transplant, mice were euthanized, and the brains were harvested for further analysis by flow cytometry and immunofluorescence staining.
Behavioral tests
Mice were tested at 6 months of age using three main behavioral tests to evaluate the neurocognitive and locomotor activity. Prior to behavioral tests, the mice were exposed to the testing apparatus and testing room in order to acclimate to the environment. We then performed and analyzed NORT as previously published.97,98 In brief, at the day of habituation, the mice were brought in the testing room and allowed to acclimate in the room for 30 min. Then the mice were placed in the arena to explore for 5 min as previously described.97,98 Next day, all mice were freely exposed to two replicates of the same object for 10 min during the familiarization phase before returning to their home cage. 24 h later, one object was kept as a familiar object, while another object was replaced with a novel object. The entire process was recorded via an overhead video camera, and object investigation time (discrimination index and preference index) was recorded manually by blinded researchers within a set area of exploration of 2.5 cm surrounding the object. Discrimination Index (DI), allows discrimination between the novel and familiar objects, i.e., it uses the difference in exploration time (T) for novel objective (N), and dividing this value by the total amount of exploration of the novel (N) and familiar objects (F) [DI = (TN − TF)/(TN + TF)]. This result can vary between +1 and −1, where a positive score indicates more time spent with the novel object, a negative score indicates more time spent with the familiar object, and a zero score indicates a null preference. The Preference Index is a ratio of the amount of time spent exploring the novel objective (N) in test phase over the total time spent exploring both objects, TN/(TN + TF) × 100 (%).99-101 The elevated plus maze test is one of the most widely used tests for measuring anxiety-like behavior in mice. We performed this EPM used the protocol as published before.102 In brief, the elevated plus maze has the shape of a “+” with two alternate open and two alternate closed arms extending from a central platform. Each mouse was placed onto the center field and was allowed to explore the maze for 5 min. Time spent in the open and closed arms was recorded. Each animal was placed in the center of an open field arena (50 cm × 50 cm) and with movement being recorded via overhead camera and recorded by an automated tracking system (ANY-Maze).
Immunohistochemistry analyses
Brains were collected from euthanized mice and fixed using 10% formalin, embedded in paraffin wax, and sectioned at 5 μm thickness by standard methods. Sections were deparaffinized and stained with the mouse anti-6E10 (1:30,000; 803001, Biolegend), rabbit anti-GFP (1:600; ab183734, Abcam) and rabbit anti-Iba-1 (1:3000; 019–19741, Wako) antibodies. Slides were stained on a Ventana Discovery Ultra (Ventana Medical Systems). Antigen retrieval was performed using CC1 (Tris-EDTA based; pH 8.6) for 40 min at 95°C. The primary antibodies were incubated for 32 min at 37°C. For 6E10, an additional rabbit-anti mouse IgG antibody (1:6000; ab133469, Abcam) was utilized to minimize mouse-on-mouse non-specific staining. All rabbit antibodies were detected using an HRP-coupled goat anti-rabbit (OmniMap system; Ventana, catalog #760–4315) incubated on the sections for 12 min at 37°C and visualized using diaminobenzidine as a chromogen followed by hematoxylin as a counterstain. Slides were rinsed, dehydrated through alcohol and xylene, and cover slipped. Images were acquired by Keyence BZ-X710 digital microscope system for high-resolution stitching images of tissue sections. ImagePro Premier software (Media Cybernetics) was used for the quantification of Aβ plaque and Iba1.
Immunofluorescence, image acquisition and analysis
Sections were deparaffinized and are transferred to pre-warmed antigen retrieval solution at 95°C for 30 min and cooled at room temperature for 20 min and incubated in blocking solution (0.25% Triton X-100 and 3% BSA in tris-buffered saline). Sections were then incubated overnight at 4°C with the following primary antibodies: chicken anti-GFP (1:200; ab290, Abcam), rabbit anti-Iba1 (1:200; 234003, Synaptic Systems), rat anti-CD11b (1:100; HS-384117, Synaptic Systems), rabbit anti-PECAM1 (1:100; NB100-2284, Novus Bio), mouse anti-VE-Cadherin/Cdh5 (1; 100; sc-9989, Santa Cruz), mouse anti-6E10 (1:250; 805701, BioLegend),. The appropriate Alexa Fluor-conjugated secondary antibodies (Invitrogen) were used for the visualization of antigens. Images were acquired using a Leica SP8 confocal microscope for high-resolution stitching images of tissue sections. Confocal image stacks and 3D view image were analyzed with the Imaris software (BitPlane, Oxford Instruments). Total number of blood vessels were determined from whole hippocampus stitching image acquired by a Keyence microscope using ImagePro software with thresholding to remove vessels <3um in diameter. Blood vessel density was calculated by dividing the total number of blood vessels by the entire area of hippocampus per mouse. Average vessel diameter was determined using ImagePro software to obtain whole vessel mean diameter value from 50 to 100 vessels/mouse. Proportion of colocalization (Mander’s overlap coefficients) for PECAM1 in z-stacks was determined using ImageJ (n = 5 animals per group, average value from 50 to 100 vessels/mouse, five images taken per section).
Sholl analysis
First, Imaris was used to reconstruct the microglial surface. Unspecific background signals and microglia with incomplete somata were manually removed and not included in further analysis. Multiple microglia, which were fused and incorrectly recognized as a single cell, were manually separated using the cut function or excluded if separation was not possible. Fifteen microglia were randomly selected and analyzed from both cortex and hippocampus for each mouse. Sholl analysis was performed using Imaris in the filament reconstruction mode, and individual datasets were exported into separate Excel files for further analysis.
Aβ1–42 ELISA
Brain tissue was homogenized in 0.15% Triton in PBS containing protease inhibitor cocktail with a homogenizer and centrifuged at 200,000 × g for 20 min at 4°C. The supernatant was removed and defined as the soluble fraction. Guanidine–HCl was added to give 0.5 m (final concentration) before application to enzyme-linked immunosorbent assay (ELISA). The pellet was solubilized by sonication in a 6m guanidine–HCl buffer containing protease inhibitor cocktail. The solubilized pellet was centrifuged at 200,000 × g for 20 min at 4°C and used as the insoluble fraction. The amounts of Aβ1-42 in each fraction were determined by a sandwich amyloid beta 42 ELISA kit (KHB3441, Invitrogen).
Cytokine and chemokine ELISA
Brain tissue was homogenized in radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitor cocktail with a homogenizer and centrifuged at 200,000 × g for 20 min at 4°C. ELISA for TNF-α (MTA00B, R & D system), IFN-γ (ab100690, abcam) and IL6 (ab100713, abcam) where performed according to manufactory instruction.
RNA extraction and real time quantitative PCR
Whole RNA extraction was performed using RNeasy Mini Kit as per manufacturer protocol (Qiagen, Hilden, Germany, cat. 74104). cDNA synthesis occurred by reverse transcription (RT) of 200 ng tissue and cell RNA using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). RT-qPCR reaction assembly consisted of 5 μL iTaq Universal SYBR Green, 2.5 μL of 1:10 diluted cDNA (5 ng/ul), 0.4 μL forward and reverse primer (10 μM), and 2.3 μL H20, and ran on a LightCycler 480 ii (Roche, Basel, Switzerland) under the following conditions: 95°C (30 s); 40 cycles of 95°C (5 s) and 60°C (30 s); then 65°C (5 s); and 95°C (5 s). Gene expression was measured using the delta/delta CT method relative to WT and normalized to glyceraldehyde 3-phosphate dehydrogenase (Gapdh). All primer sequences are shown in Table S2.
Flow cytometric analysis of adult brain microglia
Mice were deeply anesthetized and perfused with cold PBS. Brain was collected and tissue from adult mice is dissociated into single-cell suspensions according to manufacture instruction using the adult brain dissociation kit (130-107-677, Miltenyi Biotec, Bergisch-Gladbach, Germany). For flow cytometry analysis, single cell suspensions were adjusted to a density of 2 × 106 cells/200μL and nonspecific binding was blocked by incubation with purified anti-CD16/CD32 (70-0161-U500, Tonbo Biosciences) for 10 min at 4°C. Then, cells were incubated for 20 min at 4°C with the following antibodies: anti-CD45-APC (103112, Biolegend), anti-CD11b-PE (101207, Biolegend), and Zombie Aqua Fixable Viability Kit were used for live cells staining (423101, Biolegend). Data were acquired on an Aria II and Aria fusion (BD Bioscience) and FlowJo software (BD Bioscience). Gating strategy was made according to previously publish protocol.62
RNA-seq
Sample preparation
Hippocampus and cortex were dissected from brains using previously publish protocol,103 and frozen at −80°c in RNA later. Total RNA was isolated using the RNeasy Tissue kit (Qiagen) according to the manufacturer’s instructions. RNA was assessed for quality using an Agilent Tapestation 4200, and 50 ng of RNA with an RNA Integrity Number (RIN) greater than 8.0 were used to generate RNA sequencing libraries using the Illumina Stranded mRNA Prep (Illumina) following manufacturer’s instructions. The resulting libraries were multiplexed and sequenced with 100 base pairs (bp) Paired End reads (PE100) to a depth of approximately 25 million reads per sample on an Illumina NovaSeq 6000. Samples were demultiplexed using bcl2fastq Conversion Software (Illumina).
RNA-seq data analysis
RNA-Seq data were processed using Kallisto (version 0.45.0), Mus musculus genome GRCm38 Ensembl version 94 annotation (Mus_musculus GRCm38.94 chr_patch_hapl_scaff.gtf). Gene-level TPM values and gene annotations were computed using tximport and the biomaRt R package. A custom python script was used to organize the data and log reduced using log2(TPM+1). The raw data and processed data are deposited in Gene Expression Omnibus under accession no. GSE217885. A Composite Signature Analysis was performed and to compute the composite score of a set of genes, genes were first normalized and averaged. Gene expression values were normalized according to a modified Z score approach centered around StepMiner104 threshold (formula = (expr - SThr)/3*stddev). The samples were ordered based on the final combined score. We performed differential expression analysis using DESeq2.94 The false discovery rate was controlled by setting an adjusted p value threshold of 0.1.
Gene Ontology enrichment and protein-protein interaction analysis
Gene Ontology (GO) enrichment analysis on differentially expressed genes (DEGs) in both the cortex and hippocampus was performed using ShinyGO v0.76 (http://bioinformatics.sdstate.edu/go/). Significance was set to FDR ≤0.05 and limited to the top 25 most significant categories for all analyses. For the hierarchical clustering tree (Figure S3) summarizing the correlation among significant pathways, the pathways with many shared genes are clustered together. Bigger dots indicate more significant p values. The Retrieval of Interacting Genes web-tool (STRING 11.5; https://string-db.org/) was used to analyze and construct the Protein-Protein Interaction (PPI) network of the group of proteins that were over-represented in the cortex and hippocampus.
QUANTIFICATION AND STATISTICAL ANALYSIS
We did not exclude any animals from our experiments. Experimenters were blinded to the genotype of the specific sample to every extent possible. In all experiments, cohorts were age-matched and sex-balanced. Non-objective computational measurements such as immunostaining quantification by scanning devices did not require blinding. All data are presented as the mean ± SEM, and the statistics and graphs were performed using GraphPad software Prism v.9.0. No statistical methods were used to predetermine sample sizes, but our sample sizes were similar to those reported in previous publications with 5xFAD model.69 One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons was used to compare multiple groups. Statistical significance was set at p < 0.05. Unpaired t test was used for flow cytometry population comparison, and for APP and PSEN1 qPCR analysis, data was log transformed to confirm the normality. For RNA-seq data, gene signature was used to classify sample categories and the performance of the multi-class classification is measured by ROC-AUC (Receiver Operating Characteristics Area Under The Curve) values. A color-coded bar plot is combined with a violin plot to visualize the gene signature-based classification. All statistical tests were performed using R version 3.2.3 (2015-12-10). Standard t-tests were performed using python scipy.stats.ttest_ind package (version 0.19.0) with Welch’s Two Sample t test (unpaired, unequal variance (equal_var = False), and unequal sample size) parameters. Multiple hypothesis correction were performed by adjusting p values with statsmodels.stats.multitest.multipletests (fdr_bh: Benjamini/Hochberg principles). The results were independently validated with R statistical software (R version 3.6.1; 2019-07-05). Violin, Swarm and Bubble plots are created using python seaborn package version 0.10.1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-6E10 (Biolegend, 803001) | Biolegend | 803001; RRID:AB_2564653 |
| rabbit anti-GFP (Abcam, ab183734) | Abcam | Ab183734; RRID:AB_2732027 |
| rabbit anti-Iba-1 (Wako, 019–19741) | Wako | 019–19741; RRID:AB_839504 |
| rabbit-anti mouse IgG antibody (ab133469, Abcam) | Abcam | Ab133469: RRID:AB_2910607 |
| HRP-coupled goat anti-rabbit (OmniMap system; Ventana, catalog #760–4315) | OmniMap | #760-4315 |
| rabbit anti-GFP (ab290, Abcam) | Abcam | Ab290; RRID:AB_303395 |
| chicken anti-Iba1 (234006, Synaptic Systems) | Synaptic Systems | 234006; RRID:AB_2619949 |
| mouse anti-6E10 (805701, BioLegend), | Biolegend | 805701; RRID:AB_2564982 |
| rat anti-CD11b (1:100; HS-384117, Synaptic Systems | Synaptic Systems | HS-384117; RRID:AB_2891269 |
| rabbit anti-PECAM1 (1:100; NB100-2284, Novus Bio) | Novus Bio | NB100-2284; RRID:AB_10002513 |
| mouse anti-VE-Cadherin/Cdh5 | Santa Cruz | Sc-9989; RRID:AB_2077957 |
| anti-CD45-APC | Biolegend | 103112; RRID:AB_312977 |
| anti-CD11b-PE | Biolegend | 101207; RRID:AB_312790 |
| anti-CD16/CD32 | Tonbo Biosciences | 70-0161-U500; RRID:AB_2621487 |
| Zombie Aqua™ Fixable Viability Kit | Biolegend | 423101 |
| Anti-Sca1 Microbead Kit | Miltenyi Biotec | 130-123-124 |
| Miltenyi QuadroMACS Separator | Miltenyi Biotec | 130-090-976 |
| Miltenyi LS columns | Miltenyi Biotec | 130-042-401 |
| Miltenyi gentleMACS Octo Dissociator with Heaters | Miltenyi Biotec | 130-096-427 |
| Miltenyi gentleMACS C Tubes | Miltenyi Biotec | 130-093-237 |
| Biological samples | ||
| Murine Sca1+ HSPCs | GFP-transgenic mice or 5XFAD mice (This Paper) | N/A |
| Critical commercial assays | ||
| Adult Brain Dissociation kit | Miltyeni Biotec | 130-107-677 |
| Human Amyloid beta 42 ELISA kit | Invitrogen | KHB3441 |
| Mouse TNF-α ELISA kit | R&D Systems | MTA00B |
| Mouse IFN-γ ELISA kit | Abcam | ab100690 |
| Mouse IL6 ELISA kit | Abcam | Ab100713 |
| RNeasy Mini kit | Qiagen | 74104 |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708891 |
| Illumina Stranded mRNA Prep | Illumina | 20040534 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO:GSE217885 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL6-Tg(APPSwFlLon, PSEN1*M146L*L286V)6799Vas | The Jackson Laboratory | #034848-JAX RRID:MMRRC_034848-JAX |
| Mouse: C57BL/6-Tg(CAG-EGFP)1Osb/J | The Jackson Laboratory | #:003291 RRID:IMSR_JAX:003291 |
| Oligonucleotides | ||
| Primers for 5xFAD genotyping, see Table S2 | This Paper | N/A |
| Primers for qPCR, see Table S2 | This Paper | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| ImagePro Premier | Media Cybernetics | N/A |
| Imaris V10 | Bitplane | N/A |
| Graphpad Prism v.9.0 | Graphpad | N/A |
| FlowJo | BD Bioscience | N/A |
| Bcl2fastq Conversion Software | Illumina | N/A |
| Kallisto (version 0.45.0) | Bray et al., 201693 | https://pachterlab.github.io/kallisto/ |
| R (v3.2.3) | https://www.R-project.org/ | |
| DESeq2 | Love et al.94 | N/A |
| ShinyGO v0.76 | Ge et al., 202095 | http://bioinformatics.sdstate.edu/go/ |
Highlights.
WT HSPC transplantation rescues memory and neurocognitive decline
WT HSPC transplantation reduces Aβ plaque density and partially preserves blood-brain integrity
WT HSPC transplantation may prevent microgliosis and neuroinflammation
ACKNOWLEDGMENTS
We gratefully acknowledge Dr. Thomas Hnasko and his team for sharing the behavioral room and helping in setting up the behavioral testing. We acknowledge Allen Wang and Janet Chen for their help with genotyping and immunofluorescent imaging. We acknowledge Samantha Ivezich and Jessica Diaz-Vigil for their help with the immunochemistry analysis. These studies were carried out with support from the Epstein Family Research Collaboration Fund and the Alzheimer’s Disease Cooperative Study (ADCS) at UCSD, the National Institutes of Health (NIH) R01-NS108965, the California Institute of Regenerative Medicine (CIRM, CLIN2-11478), the Cystinosis Research Foundation, and the Friedreich’s Ataxia Research Alliance (FARA). The UCSD Neuroscience Microscopy Shared Facility was funded by grant P30-NS047101. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a NIH SIG grant (#S10 OD026929). The graphical abstract was created with BioRender.com. A.S. received a fellowship from CIRM (CIRM Bridges to Stem Cell Research Program, EDUC2-08388). This publication includes data from a FACSAria II Cell Sorter that was purchased with funding from an NIH SIG grant (S10 RR027366).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
S.C. is a co-founder, shareholder, and member of both the scientific board and board of directors of Papillon Therapeutics, Inc. S.C. serves as a consultant for AVROBIO and receives compensation for these services. S.C. also serves as a member of the scientific review board and the board of trustees of Cystinosis Research Foundation. This work is covered in the patent entitled “Methods for treating Alzheimer’s Disease” (#114198-8190). The terms of this arrangement have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112956.
REFERENCES
- 1.Jia J, Wei C, Chen S, Li F, Tang Y, Qin W, Zhao L, Jin H, Xu H, Wang F, et al. (2018). The cost of Alzheimer’s disease in China and reestimation of costs worldwide. Alzheimers Dement. 14, 483–491. 10.1016/j.jalz.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 2.Robinson M, Lee BY, and Hane FT (2017). Recent Progress in Alzheimer’s Disease Research, Part 2: Genetics and Epidemiology. J. Alzheimers Dis. 57, 317–330. 10.3233/JAD-161149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vermunt L, Sikkes SAM, van den Hout A, Handels R, Bos I, van der Flier WM, Kern S, Ousset PJ, Maruff P, Skoog I, et al. (2019). Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. 15, 888–898. 10.1016/j.jalz.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Long JM, and Holtzman DM (2019). Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 179, 312–339. 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaz M, and Silvestre S (2020). Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol 887, 173554. 10.1016/j.ej-phar.2020.173554. [DOI] [PubMed] [Google Scholar]
- 6.McGeer PL, and Rogers J (1992). Anti-inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology 42, 447–449. 10.1212/wnl.42.2.447. [DOI] [PubMed] [Google Scholar]
- 7.Andreone BJ, Przybyla L, Llapashtica C, Rana A, Davis SS, van Lengerich B, Lin K, Shi J, Mei Y, Astarita G, et al. (2020). Alzheimer’s-associated PLCgamma2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat. Neurosci 23, 927–938. 10.1038/s41593-020-0650-6. [DOI] [PubMed] [Google Scholar]
- 8.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 9.Daria A, Colombo A, Llovera G, Hampel H, Willem M, Liesz A, Haass C, and Tahirovic S (2017). Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 36, 583–603. 10.15252/embj.201694591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fakhoury M (2018). Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol 16, 508–518. 10.2174/1570159X15666170720095240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai Z, Hussain MD, and Yan LJ (2014). Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci 124, 307–321. 10.3109/00207454.2013.833510. [DOI] [PubMed] [Google Scholar]
- 12.Franco-Bocanegra DK, Gourari Y, McAuley C, Chatelet DS, Johnston DA, Nicoll JAR, and Boche D (2021). Microglial morphology in Alzheimer’s disease and after Abeta immunotherapy. Sci. Rep 11, 15955. 10.1038/s41598-021-95535-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, Losada PM, Chen MB, Schaum N, Khoury N, et al. (2022). A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603, 885–892. 10.1038/s41586-021-04369-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rocca CJ, Rainaldi JN, Sharma J, Shi Y, Haquang JH, Luebeck J, Mali P, and Cherqui S (2020). CRISPR-Cas9 Gene Editing of Hematopoietic Stem Cells from Patients with Friedreich’s Ataxia. Mol. Ther. Methods Clin. Dev 17, 1026–1036. 10.1016/j.omtm.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rocca CJ, Goodman SM, Dulin JN, Haquang JH, Gertsman I, Blondelle J, Smith JLM, Heyser CJ, and Cherqui S (2017). Transplantation of wild-type mouse hematopoietic stem and progenitor cells ameliorates deficits in a mouse model of Friedreich’s ataxia. Sci. Transl. Med 9, eaaj2347. 10.1126/scitranslmed.aaj2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Syres K, Harrison F, Tadlock M, Jester JV, Simpson J, Roy S, Salomon DR, and Cherqui S (2009). Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood 114, 2542–2552. 10.1182/blood-2009-03-213934. [DOI] [PubMed] [Google Scholar]
- 17.Xu Z, Rao Y, Huang Y, Zhou T, Feng R, Xiong S, Yuan TF, Qin S, Lu Y, Zhou X, et al. (2020). Efficient Strategies for Microglia Replacement in the Central Nervous System. Cell Rep. 32, 108041. 10.1016/j.celrep.2020.108041. [DOI] [PubMed] [Google Scholar]
- 18.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci 26, 10129–10140. 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richard BC, Kurdakova A, Baches S, Bayer TA, Weggen S, and Wirths O (2015). Gene Dosage Dependent Aggravation of the Neurological Phenotype in the 5XFAD Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis 45, 1223–1236. 10.3233/JAD-143120. [DOI] [PubMed] [Google Scholar]
- 20.Jawhar S, Trawicka A, Jenneckens C, Bayer TA, and Wirths O (2012). Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 33, 196.e29–196.e40. 10.1016/j.neurobiolaging.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 21.Wendt S, Maricos M, Vana N, Meyer N, Guneykaya D, Semtner M, and Kettenmann H (2017). Changes in phagocytosis and potassium channel activity in microglia of 5xFAD mice indicate alterations in purinergic signaling in a mouse model of Alzheimer’s disease. Neurobiol. Aging 58, 41–53. 10.1016/j.neurobiolaging.2017.05.027. [DOI] [PubMed] [Google Scholar]
- 22.Moechars D, Lorent K, De Strooper B, Dewachter I, and Van Leuven F (1996). Expression in brain of amyloid precursor protein mutated in the alpha-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 15, 1265–1274. [PMC free article] [PubMed] [Google Scholar]
- 23.Vidal M, Morris R, Grosveld F, and Spanopoulou E (1990). Tissue-specific control elements of the Thy-1 gene. EMBO J. 9, 833–840. 10.1002/j.1460-2075.1990.tb08180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradley JE, Ramirez G, and Hagood JS (2009). Roles and regulation of Thy-1, a context-dependent modulator of cell phenotype. Biofactors 35, 258–265. 10.1002/biof.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oblak AL, Lin PB, Kotredes KP, Pandey RS, Garceau D, Williams HM, Uyar A, O’Rourke R, O’Rourke S, Ingraham C, et al. (2021). Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: A MODEL-AD Study. Front. Aging Neurosci 13, 713726. 10.3389/fnagi.2021.713726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jawhar S, Trawicka A, Jenneckens C, Bayer TA, and Wirths O (2012). Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 33, 196.e29–196.e40. 10.1016/j.neurobiolaging.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 27.Cotel MC, Jawhar S, Christensen DZ, Bayer TA, and Wirths O (2012). Environmental enrichment fails to rescue working memory deficits, neuron loss, and neurogenesis in APP/PS1KI mice. Neurobiol. Aging 33, 96–107. 10.1016/j.neurobiolaging.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 28.Reed JM, and Squire LR (1997). Impaired recognition memory in patients with lesions limited to the hippocampal formation. Behav. Neurosci 111, 667–675. 10.1037//0735-7044.111.4.667. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Ferguson GD, Pineda VV, Cundiff PE, and Storm DR (2004). Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat. Neurosci 7, 635–642. 10.1038/nn1248. [DOI] [PubMed] [Google Scholar]
- 30.Burke AD, Goldfarb D, Bollam P, and Khokher S (2019). Diagnosing and Treating Depression in Patients with Alzheimer’s Disease. Neurol. Ther 8, 325–350. 10.1007/s40120-019-00148-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaiser NC, Liang LJ, Melrose RJ, Wilkins SS, Sultzer DL, and Mendez MF (2014). Differences in anxiety among patients with early-versus late-onset Alzheimer’s disease. J. Neuropsychiatry Clin. Neurosci 26, 73–80. 10.1176/appi.neuropsych.12100240. [DOI] [PubMed] [Google Scholar]
- 32.Dawson GR, and Tricklebank MD (1995). Use of the elevated plus maze in the search for novel anxiolytic agents. Trends Pharmacol. Sci 16, 33–36. 10.1016/s0165-6147(00)88973-7. [DOI] [PubMed] [Google Scholar]
- 33.DeTure MA, and Dickson DW (2019). The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener 14, 32. 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Youmans KL, Leung S, Zhang J, Maus E, Baysac K, Bu G, Vassar R, Yu C, and LaDu MJ (2011). Amyloid-beta42 alters apolipoprotein E solubility in brains of mice with five familial AD mutations. J. Neurosci. Methods 196, 51–59. 10.1016/j.jneumeth.2010.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biber K. (2017). Reestablishing microglia function: good news for Alzheimer’s therapy? EMBO J. 36, 565–567. 10.15252/embj.201796627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrison HW, and Filosa JA (2013). A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J. Neuroinflammation 10, 4. 10.1186/1742-2094-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang WY, Tan MS, Yu JT, and Tan L (2015). Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med 3, 136. 10.3978/j.issn.2305-5839.2015.03.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Novikova G, Kapoor M, Tcw J, Abud EM, Efthymiou AG, Chen SX, Cheng H, Fullard JF, Bendl J, Liu Y, et al. (2021). Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat. Commun 12, 1610. 10.1038/s41467-021-21823-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szabo MP, Mishra S, Knupp A, and Young JE (2022). The role of Alzheimer’s disease risk genes in endolysosomal pathways. Neurobiol. Dis 162, 105576. 10.1016/j.nbd.2021.105576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dang D, Taheri S, Das S, Ghosh P, Prince LS, and Sahoo D (2020). Computational Approach to Identifying Universal Macrophage Biomarkers. Front. Physiol 11, 275. 10.3389/fphys.2020.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hori Y, Hashimoto T, Nomoto H, Hyman BT, and Iwatsubo T (2015). Role of Apolipoprotein E in beta-Amyloidogenesis: ISOFORM-SPECIFIC EFFECTS ON PROTOFIBRIL TO FIBRIL CONVERSION OF Abeta IN VITRO AND BRAIN Abeta DEPOSITION IN VIVO. J. Biol. Chem 290, 15163–15174. 10.1074/jbc.M114.622209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li W, Chen Z, Chin I, Chen Z, and Dai H (2018). The Role of VE-cadherin in Blood-brain Barrier Integrity Under Central Nervous System Pathological Conditions. Curr. Neuropharmacol 16, 1375–1384. 10.2174/1570159X16666180222164809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wimmer I, Tietz S, Nishihara H, Deutsch U, Sallusto F, Gosselet F, Lyck R, Muller WA, Lassmann H, and Engelhardt B (2019). PECAM-1 Stabilizes Blood-Brain Barrier Integrity and Favors Paracellular T-Cell Diapedesis Across the Blood-Brain Barrier During Neuroinflammation. Front. Immunol 10, 711. 10.3389/fimmu.2019.00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castro DM, Dillon C, Machnicki G, and Allegri RF (2010). The economic cost of Alzheimer’s disease: Family or public health burden? Dement. Neuropsychol 4, 262–267. 10.1590/S1980-57642010DN40400003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cummings J, Lee G, Ritter A, Sabbagh M, and Zhong K (2020). Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement. 6, e12050. 10.1002/trc2.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Varvel NH, Grathwohl SA, Baumann F, Liebig C, Bosch A, Brawek B, Thal DR, Charo IF, Heppner FL, Aguzzi A, et al. (2012). Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc. Natl. Acad. Sci. USA 109, 18150–18155. 10.1073/pnas.1210150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Greter M, Lelios I, and Croxford AL (2015). Microglia Versus Myeloid Cell Nomenclature during Brain Inflammation. Front. Immunol 6, 249. 10.3389/fimmu.2015.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silvin A, Uderhardt S, Piot C, Da Mesquita S, Yang K, Geirsdottir L, Mulder K, Eyal D, Liu Z, Bridlance C, et al. (2022). Dual ontogeny of disease-associated microglia and disease inflammatory macrophages in aging and neurodegeneration. Immunity 55, 1448–1465.e6. 10.1016/j.immuni.2022.07.004. [DOI] [PubMed] [Google Scholar]
- 49.Thome AD, Faridar A, Beers DR, Thonhoff JR, Zhao W, Wen S, Pascual B, Masdeu JC, and Appel SH (2018). Functional alterations of myeloid cells during the course of Alzheimer’s disease. Mol. Neurodegener 13, 61. 10.1186/s13024-018-0293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilkinson FL, Sergijenko A, Langford-Smith KJ, Malinowska M, Wynn RF, and Bigger BW (2013). Busulfan conditioning enhances engraftment of hematopoietic donor-derived cells in the brain compared with irradiation. Mol. Ther 21, 868–876. 10.1038/mt.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sailor KA, Agoranos G, López-Manzaneda S, Tada S, Gillet-Legrand B, Guerinot C, Masson JB, Vestergaard CL, Bonner M, Gagnidze K, et al. (2022). Hematopoietic stem cell transplantation chemotherapy causes microglia senescence and peripheral macrophage engraftment in the brain. Nat. Med 28, 517–527. 10.1038/s41591-022-01691-9. [DOI] [PubMed] [Google Scholar]
- 52.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, et al. (2009). Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326, 818–823. 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 53.Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ, Armant M, Dansereau C, Lund TC, Miller WP, et al. (2017). Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med 377, 1630–1638. 10.1056/NEJMoa1700554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biffi A, Capotondo A, Fasano S, del Carro U, Marchesini S, Azuma H, Malaguti MC, Amadio S, Brambilla R, Grompe M, et al. (2006). Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J. Clin. Invest 116, 3070–3082. 10.1172/JCI28873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biffi A, De Palma M, Quattrini A, Del Carro U, Amadio S, Visigalli I, Sessa M, Fasano S, Brambilla R, Marchesini S, et al. (2004). Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Invest 113, 1118–1129. 10.1172/JCI19205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Capotondo A, Milazzo R, Politi LS, Quattrini A, Palini A, Plati T, Merella S, Nonis A, di Serio C, Montini E, et al. (2012). Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc. Natl. Acad. Sci. USA 109, 15018–15023. 10.1073/pnas.1205858109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S, et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341, 1233158. 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 58.Sessa M, Lorioli L, Fumagalli F, Acquati S, Redaelli D, Baldoli C, Canale S, Lopez ID, Morena F, Calabria A, et al. (2016). Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 388, 476–487. 10.1016/S0140-6736(16)30374-9. [DOI] [PubMed] [Google Scholar]
- 59.Hellwig S, Masuch A, Nestel S, Katzmarski N, Meyer-Luehmann M, and Biber K (2015). Forebrain microglia from wild-type but not adult 5xFAD mice prevent amyloid-beta plaque formation in organotypic hippocampal slice cultures. Sci. Rep 5, 14624. 10.1038/srep14624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K, and Hol EM (2014). Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 35, 2746–2760. 10.1016/j.neurobiolaging.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 61.Chung WS, Welsh CA, Barres BA, and Stevens B (2015). Do glia drive synaptic and cognitive impairment in disease? Nat. Neurosci 18, 1539–1545. 10.1038/nn.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hansen DV, Hanson JE, and Sheng M (2018). Microglia in Alzheimer’s disease. J. Cell Biol 217, 459–472. 10.1083/jcb.201709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simard AR, Soulet D, Gowing G, Julien JP, and Rivest S (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49, 489–502. 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 64.Hefendehl JK, Wegenast-Braun BM, Liebig C, Eicke D, Milford D, Calhoun ME, Kohsaka S, Eichner M, and Jucker M (2011). Longterm in vivo imaging of beta-amyloid plaque appearance and growth in a mouse model of cerebral beta-amyloidosis. J. Neurosci 31, 624–629. 10.1523/JNEUROSCI.5147-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bittner T, Burgold S, Dorostkar MM, Fuhrmann M, Wegenast-Braun BM, Schmidt B, Kretzschmar H, and Herms J (2012). Amyloid plaque formation precedes dendritic spine loss. Acta Neuropathol. 124, 797–807. 10.1007/s00401-012-1047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hickman SE, Allison EK, and El Khoury J (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci 28, 8354–8360. 10.1523/JNEUROSCI.0616-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, and Hyman BT (2008). Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724. 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, Miller KR, Prokop S, Kettenmann H, and Heppner FL (2013). Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One 8, e60921. 10.1371/journal.pone.0060921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.d’Errico P, Ziegler-Waldkirch S, Aires V, Hoffmann P, Mezö C, Erny D, Monasor LS, Liebscher S, Ravi VM, Joseph K, et al. (2022). Microglia contribute to the propagation of Abeta into unaffected brain tissue. Nat. Neurosci 25, 20–25. 10.1038/s41593-021-00951-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baik SH, Kang S, Son SM, and Mook-Jung I (2016). Microglia contributes to plaque growth by cell death due to uptake of amyloid beta in the brain of Alzheimer’s disease mouse model. Glia 64, 2274–2290. 10.1002/glia.23074. [DOI] [PubMed] [Google Scholar]
- 71.Wisniewski HM, Moretz RC, and Lossinsky AS (1981). Evidence for induction of localized amyloid deposits and neuritic plaques by an infectious agent. Ann. Neurol 10, 517–522. 10.1002/ana.410100605. [DOI] [PubMed] [Google Scholar]
- 72.He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, and Shen Y (2007). Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol 178, 829–841. 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ringheim GE, Szczepanik AM, Petko W, Burgher KL, Zhu SZ, and Chao CC (1998). Enhancement of beta-amyloid precursor protein transcription and expression by the soluble interleukin-6 receptor/interleukin-6 complex. Brain Res. Mol. Brain Res 55, 35–44. 10.1016/s0169-328x(97)00356-2. [DOI] [PubMed] [Google Scholar]
- 74.Yeh FL, Hansen DV, and Sheng M (2017). TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med 23, 512–533. 10.1016/j.molmed.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 75.Dvir-Szternfeld R, Castellani G, Arad M, Cahalon L, Colaiuta SP, Keren-Shaul H, Croese T, Burgaletto C, Baruch K, Ulland T, et al. (2022). Alzheimer’s disease modification mediated by bone marrow-derived macrophages via a TREM2-independent pathway in mouse model of amyloidosis. Nat. Aging 2, 60–73. 10.1038/s43587-021-00149-w. [DOI] [PubMed] [Google Scholar]
- 76.Grubman A, Choo XY, Chew G, Ouyang JF, Sun G, Croft NP, Rossello FJ, Simmons R, Buckberry S, Landin DV, et al. (2021). Transcriptional signature in microglia associated with Abeta plaque phagocytosis. Nat. Commun 12, 3015. 10.1038/s41467-021-23111-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee SH, Haddick PCG, Ngu H, et al. (2018). Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 22, 832–847. 10.1016/j.celrep.2017.12.066. [DOI] [PubMed] [Google Scholar]
- 78.Kalaria RN, and Hedera P (1995). Differential degeneration of the cerebral microvasculature in Alzheimer’s disease. Neuroreport 6, 477–480. 10.1097/00001756-199502000-00018. [DOI] [PubMed] [Google Scholar]
- 79.Zlokovic BV (2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 80.Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, et al. (2015). Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zenaro E, Piacentino G, and Constantin G (2017). The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis 107, 41–56. 10.1016/j.nbd.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu Y, Huber CC, and Wang H (2020). Disrupted blood-brain barrier in 5xFAD mouse model of Alzheimer’s disease can be mimicked and repaired in vitro with neural stem cell-derived exosomes. Biochem. Biophys. Res. Commun 525, 192–196. 10.1016/j.bbrc.2020.02.074. [DOI] [PubMed] [Google Scholar]
- 83.Park JC, Baik SH, Han SH, Cho HJ, Choi H, Kim HJ, Choi H, Lee W, Kim DK, and Mook-Jung I (2017). Annexin A1 restores Abeta(1-42) -induced blood-brain barrier disruption through the inhibition of RhoA-ROCK signaling pathway. Aging Cell 16, 149–161. 10.1111/acel.12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ransohoff RM, Kivisäkk P, and Kidd G (2003). Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol 3, 569–581. 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- 85.Ye L, Huang Y, Zhao L, Li Y, Sun L, Zhou Y, Qian G, and Zheng JC (2013). IL-1beta and TNF-alpha induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J. Neurochem 125, 897–908. 10.1111/jnc.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Woodfin A, Voisin MB, and Nourshargh S (2007). PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler. Thromb. Vasc. Biol 27, 2514–2523. 10.1161/ATVBAHA.107.151456. [DOI] [PubMed] [Google Scholar]
- 87.Haruwaka K, Ikegami A, Tachibana Y, Ohno N, Konishi H, Hashimoto A, Matsumoto M, Kato D, Ono R, Kiyama H, et al. (2019). Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun 10, 5816. 10.1038/s41467-019-13812-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boettcher S, and Manz MG (2017). Regulation of Inflammation- and Infection-Driven Hematopoiesis. Trends Immunol. 38, 345–357. 10.1016/j.it.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 89.Pasquevich KA, Bieber K, Günter M, Grauer M, Pötz O, Schleicher U, Biedermann T, Beer-Hammer S, Bühring HJ, Rammensee HG, et al. (2015). Innate immune system favors emergency monopoiesis at the expense of DC-differentiation to control systemic bacterial infection in mice. Eur. J. Immunol 45, 2821–2833. 10.1002/eji.201545530. [DOI] [PubMed] [Google Scholar]
- 90.Bhattarai S, Li Q, Ding J, Liang F, Gusev E, Lapohos O, Fonseca GJ, Kaufmann E, Divangahi M, and Petrof BJ (2022). TLR4 is a regulator of trained immunity in a murine model of Duchenne muscular dystrophy. Nat. Commun 13, 879. 10.1038/s41467-022-28531-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, et al. (2018). Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172, 147–161.e12. 10.1016/j.cell.2017.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manocha GD, Floden AM, Rausch K, Kulas JA, McGregor BA, Rojanathammanee L, Puig KR, Puig KL, Karki S, Nichols MR, et al. (2016). APP Regulates Microglial Phenotype in a Mouse Model of Alzheimer’s Disease. J. Neurosci 36, 8471–8486. 10.1523/JNEUROSCI.4654-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 94.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ge SX, Jung D, and Yao R (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Harrison F, Yeagy BA, Rocca CJ, Kohn DB, Salomon DR, and Cherqui S (2013). Hematopoietic stem cell gene therapy for the multi-systemic lysosomal storage disorder cystinosis. Mol. Ther 21, 433–444. 10.1038/mt.2012.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Denninger JK, Smith BM, and Kirby ED (2018). Novel Object Recognition and Object Location Behavioral Testing in Mice on a Budget. J. Vis. Exp 10.3791/58593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lueptow LM (2017). Novel Object Recognition Test for the Investigation of Learning and Memory in Mice. J. Vis. Exp 55718 10.3791/55718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Antunes M, and Biala G (2012). The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn. Process 13, 93–110. 10.1007/s10339-011-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aubele T, Kaufman R, Montalmant F, and Kritzer MF (2008). Effects of gonadectomy and hormone replacement on a spontaneous novel object recognition task in adult male rats. Horm. Behav 54, 244–252. 10.1016/j.yhbeh.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Burke SN, Wallace JL, Nematollahi S, Uprety AR, and Barnes CA (2010). Pattern separation deficits may contribute to age-associated recognition impairments. Behav. Neurosci 124, 559–573. 10.1037/a0020893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Komada M, Takao K, and Miyakawa T (2008). Elevated plus maze for mice. J. Vis. Exp 10.3791/1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spijker S. (2011). Dissection of Rodent Brain Regions. In Neuroproteomics, Li K, ed. (Humana Press; ), pp. 13–26. 10.1007/978-1-61779-111-6_2. [DOI] [Google Scholar]
- 104.Sahoo D, Dill DL, Tibshirani R, and Plevritis SK (2007). Extracting binary signals from microarray time-course data. Nucleic Acids Res. 35, 3705–3712. 10.1093/nar/gkm284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used for generation of figures are available from the corresponding authors upon reasonable request.
RNA sequencing data that support the findings of this study have been deposited in NCBI GEO with the accession number GSE217885. The codes used to conduct RNA sequencing analysis are publicly available at the following links: https://github.com/sahoo00/BoNE; https://github.com/sahoo00/Hegemon.
All data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.