

Abstract
The filamentous fungus Cochliobolus carbonum produces endo-α1,4-polygalacturonase (endoPG), exo-α1,4-polygalacturonase (exoPG), and pectin methylesterase when grown in culture on pectin. Residual activity in a pgn1 mutant (lacking endoPG) was due to exoPG activity, and the responsible protein has now been purified. After chemical deglycosylation, the molecular mass of the purified protein decreased from greater than 60 to 45 kDa. The gene that encodes exoPG, PGX1, was isolated with PCR primers based on peptide sequences from the protein. The product of PGX1, Pgx1p, has a predicted molecular mass of 48 kDa, 12 potential N-glycosylation sites, and 61% amino acid identity to an exoPG from the saprophytic fungus Aspergillus tubingensis. Strains of C. carbonum mutated in PGX1 were constructed by targeted gene disruption and by gene replacement. Growth of pgx1 mutant strains on pectin was reduced by ca. 20%, and they were still pathogenic on maize. A double pgn1/pgx1 mutant strain was constructed by crossing. The double mutant grew as well as the pgx1 single mutant on pectin and was still pathogenic despite having less than 1% of total wild-type PG activity. Double mutants retained a small amount of PG activity with the same cation-exchange retention time as Pgn1p and also pectin methylesterase and a PG activity associated with the mycelium. Continued growth of the pgn1/pgx1 mutant on pectin could be due to one or more of these residual activities.
Many bacteria and fungi synthesize extracellular enzymes that can degrade pectin or its demethylesterified form, polygalacturonic acid, and a number of such enzymes and their encoding genes have been isolated from plant pathogenic microorganisms and studied for their role in the disease process. Extracellular pectin-degrading enzymes clearly contribute to symptom development in soft rot diseases and can induce defense responses in a number of plants (32), but their exact roles in the process of pathogenesis, especially in diseases not characterized by soft rotting, are largely unknown. A major barrier to a more precise definition of the role of pectin degradation in the disease process is the presence in most microorganisms of multiple pectin-degrading enzymes and -encoding genes (14, 26, 32).
Cochliobolus carbonum, the causal agent of Northern leaf spot of corn, attacks mainly foliar tissue but also the stalks and ears of susceptible maize. This filamentous fungus produces a number of extracellular cell wall-degrading enzymes including xylanases, cellulases, proteases, mixed-linked glucanases, exo-β1,3-glucanases, α-arabinosidase, and β-xylosidase (32). It produces at least three pectin-degrading enzymes, i.e., endo-α1,4-polygalacturonase (endoPG), exo-α1,4-polygalacturonase (exoPG), and pectin methylesterase (PME) (25, 26, 33). Strains of C. carbonum mutated in the gene encoding endoPG, PGN1, grow normally on pectin and are still pathogenic. Total PG activity is reduced by ca. 60%, and residual activity is due to one or more exo-acting activities that appear as a set of peaks of variable sizes when fractionated by cation-exchange chromatography (26). Although these earlier results demonstrated that PGN1 by itself is not required for pathogenicity, they leave unanswered the more general question of the requirement for any pectin-degrading ability in pathogenesis by C. carbonum. In order to address this, it is necessary to create a strain of the fungus that completely lacks all pectin-degrading activity. As a step in this direction, we report here the purification of the exoPG activity of C. carbonum and the cloning of its gene, PGX1. exoPGs have previously been identified and purified from plant pathogenic fungi (8, 22), and exoPG-encoding genes have been isolated from the bacterial pathogens Erwinia chrysanthemi and Ralstonia (Pseudomonas) solanacearum (9, 11). exoPG contributes to growth of E. chrysanthemi on polygalacturonic acid but not to tissue maceration (9), whereas the exoPG of R. solanacearum is a virulence factor (11). To the best of our knowledge, no exoPG-encoding gene has previously been isolated from a plant pathogenic fungus.
MATERIALS AND METHODS
Fungal cultures and enzyme purification.
C. carbonum was grown and maintained as previously described (34). PG activity was determining the production of reducing sugars from sodium polygalacturonic acid (Sigma) at 30°C. Reducing sugars were detected with p-hydroxybenzoic acid hydrazide (18, 33). endoPG activity was distinguished from exoPG activity by comparing activity as measured by viscometry and activity as measured by the release of reducing sugars (26, 33).
exoPG activity was purified from culture filtrates of strain PG1, in which the major endoPG gene, PGN1, is disrupted (26). The fungus was grown in still culture at 21°C on modified Fries’ medium (31) with 125 ml per 1-liter flask, in which 1% pectin (Sigma P-9135) was substituted for sucrose. After 14 days of growth, the medium (typically 500 ml) was collected by filtration through four layers of cheesecloth and Whatman no. 1 filter paper. Residual undegraded pectin was precipitated by the addition of 50 mM calcium chloride followed by incubation on ice for 1 h. The precipitated material was removed by centrifugation, and the filtrate was concentrated by rotary evaporation to 1/10 of its original volume. After dialysis, the filtrate was applied to a column of DEAE-cellulose (20), and material not binding to the DEAE-cellulose column was pooled and concentrated by ultrafiltration through an Amicon YM30 membrane. The DEAE-cellulose column was on several occasions washed with 25 mM acetate (pH 5) plus 0.8 M NaCl, but no pectin- or polygalacturonic acid-degrading activity was ever observed in the eluted material (25). Samples were fractionated by cation-exchange high-performance liquid chromatography (HPLC) on a sulfoethylaspartamide cation-exchange HPLC column (The Nest Group, Southboro, Mass.) with a linear gradient of 25 mM sodium acetate (pH 5) to 25 mM sodium acetate (pH 5) plus 0.4 M KCl over 30 min. The flow rate was 1 ml/min, and 1-ml fractions were collected. Hydrophobic interaction chromatography was done on a TSK phenyl column (BioRad, Richmond, Calif.) with a linear gradient from 0.1 M KH2PO4 plus 1.7 M ammonium sulfate (pH 7) to distilled water in 30 min. One-milliliter fractions were collected (20, 26).
Washed pectin was prepared by soaking citrus pectin in 70% (vol/vol) ethanol plus 0.1 N HCl for 30 min at 23°C, followed by filtration through a Whatman no. 1 filter and washing with additional 70% ethanol until all traces of Cl− were gone. The pectin was finally washed with 95% (vol/vol) ethanol and dried at 70°C.
Glycoproteins were detected by periodic acid-Schiff staining (25). Deglycosylation was done with trifluoromethanesulfonic acid with a GlycoFree Deglycosylation kit (Oxford GlycoSystems, Rosedale, N.Y.). Deglycosylated protein samples were digested with trypsin or proteinase Asp-N (Boehringer-Mannheim), and the resulting peptides were separated and sequenced by automated Edman degradation at the Michigan State University (MSU) Macromolecular Facility.
Nucleic acid manipulations.
Genomic DNA and RNA isolation, DNA and RNA analysis, and library screening were performed as described elsewhere (24, 26). PCR was carried out in a Perkin-Elmer thermocycler under the following conditions: 3 min of denaturation at 94°C; 35 cycles of 1 min of denaturation at 94°C, 2 min of annealing at 54°C, and 3 min of primer extension at 72°C; and 10 min of primer extension at 72°C. The PCR primers used were CARTAYCCNGGNGARGT and CCNGGCCANACYTTHAT, corresponding to the exoPG amino acid sequences QYPGEV and IKVWPG, respectively (R represents A or G, Y represents C or T, H represents A, T, or C, and N represents any base). A third oligonucleotide, TTYTCNACHATRTC, corresponding to the sequence DIVEN, was end labelled with polynucleotide kinase and used as a probe to select the correct PCR product. A 99-bp DNA fragment was identified, cloned, sequenced, and used to screen genomic and cDNA libraries as described elsewhere (17, 23). cDNA and genomic clones were sequenced by using nested deletions (26). Sequencing was done by automated fluorescent sequencing at the MSU-Department of Energy Plant Research Laboratory Plant Biochemistry Facility. PGN1 and PGX1 were chromosome mapped by DNA hybridization to a DNA blot of C. carbonum chromosomes separated by pulsed-field gel electrophoresis (1).
For in planta expression and virulence studies, 2-week-old plants were inoculated until runoff by using an atomizer with a suspension of 105 conidia in 0.1% Tween 20 and were maintained in a greenhouse. Under these conditions, lesions become visible within 48 h and were approximately 5 mm in diameter after 5 days, and the plants were killed and mostly dessicated after 10 days. Poly(A)+ RNA was extracted from infected plants and analyzed as described elsewhere (4). Fifteen micrograms of RNA per lane was loaded. The same blot was probed sequentially with PGN1, PGX1, and GPD1 (encoding glyceraldehyde-3-phosphate dehydrogenase). The blot was stripped between hybridizations.
Transformation-mediated gene disruptions and replacements.
Gene disruption mutants were obtained with vectors containing internal fragments from the coding region of each gene. For PGN1, an internal 655-bp BamHI fragment and a 4-kb KpnI/SalI fragment containing the amdS gene for acetamide utilization from Aspergillus nidulans (12) were cloned into vector pSP72 (Promega) to make plasmid pPGNK1. For PGX1, an internal 1.5-kb HindIII/BamHI fragment was cloned into the corresponding sites in pHyg1, which contains the hph1 gene for resistance to hygromycin driven by the P1 promoter of C. heterostrophus (28) to make plasmid pPGXK1. Plasmids were linearized by cutting at unique restriction sites in the cloned segments of C. carbonum DNA prior to transformation (26). Transformants were purified by seven rounds of single-spore isolation.
Gene replacement mutants of both PGN1 and PGX1 were obtained with vectors utilizing the Escherichia coli hygromycin resistance gene (hph-1) driven by the P1 promoter of C. heterostrophus from plasmid pHyg1 (28). The coding region of each gene was replaced by the selectable marker such that 0.5 to 1.5 kb of colinear DNA remained on each side. The vectors for replacement of PGN1 and PGX1 were called pPGNGR1 and pPGXGR1, respectively. The fragments were released from vector sequences by using two restriction endonucleases with differing recognition sites to prevent recircularization (35). Transformants were purified by four rounds of single-spore isolation. A double pgn1/pgx1 gene replacement mutant was obtained by crossing the PGN1 and PGX1 mutants (36).
Nucleotide sequence accession number.
The sequence of PGX1 was deposited in GenBank in November 1995 under accession number L48982.
RESULTS
Isolation of exoPG.
pgn1 mutants of C. carbonum have residual PG activity due to one or more exo-acting enzymes (26). Similarly to endoPG activity, production of exoPG activity is stimulated by the presence of pectin and is suppressed by 2% sucrose (33). In contrast to endoPG activity, which reaches a maximum after 7 days of growth on pectin and then declines to almost undetectable levels by 14 days (33), exoPG activity continues to increase until at least day 14 (25). After concentration and passage through a column of DEAE-cellulose to remove pigments and anionic proteins, culture filtrates of the C. carbonum pgn1 mutant strain grown for 14 days on pectin were analyzed by cation-exchange HPLC. exoPG activity appears as multiple peaks of activity of variable sizes and numbers, with a major broad peak of activity eluting just before Pgn1p, the product of PGN1 (26). These multiple exoPG activity peaks could be due to multiple exoPG-encoding genes and/or to variable posttranslational modifications of a single gene product.
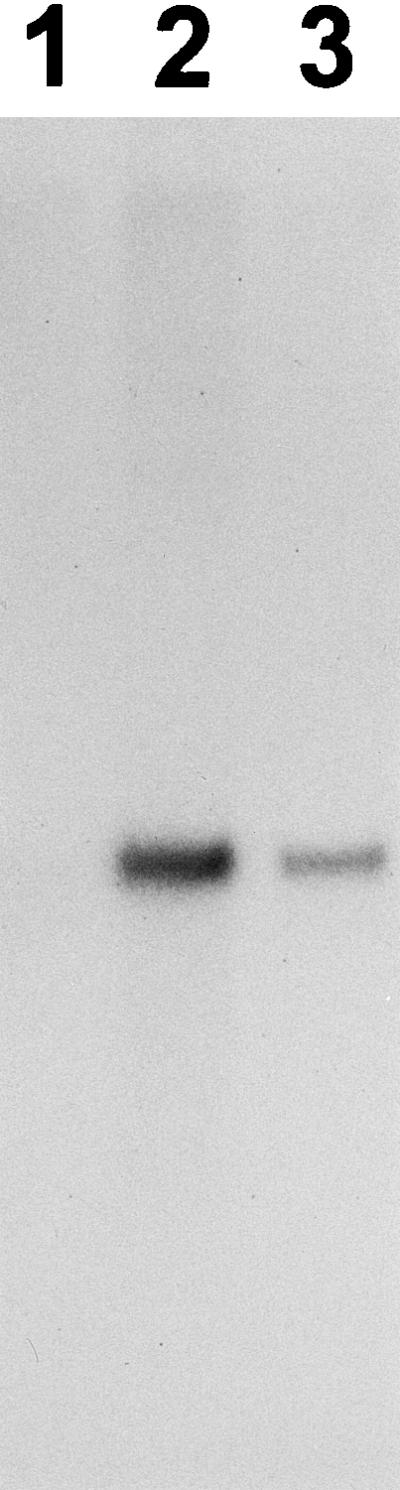
The major peak of activity (eluting at 20 to 25 min from a cation-exchange HPLC column) was further purified by hydrophobic-interaction HPLC, and the peak fraction containing exoPG activity was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). This fraction contained a single protein band which ran as a smear with a molecular mass of 60 to 95 kDa (Fig. 1). Staining with periodic acid indicated that this protein was glycosylated (29). Efforts to obtain N-terminal or internal amino acid sequences from this preparation after digestion with various proteases were unsuccessful. Following chemical deglycosylation, the protein smear was reduced to a single compact band of 45 kDa (Fig. 1), indicating that the smear was due to a single heavily glycosylated protein. The exoPGs of Fusarium oxysporum f. sp. lycopersici, Aspergillus tubingensis, and Alternaria mali are also glycosylated (8, 15, 22). Several peptide sequences were purified from the deglycosylated enzyme by reverse-phase microbore HPLC fractionation following proteolytic digestion. The following internal peptides were obtained: no. 1, DGARIKVWPGAS; no. 2, GQYPGEVDIVEN; no. 3, DYAI(E/I)(L/I)(T/L); and no. 4, DITIKNFKGTT (amino acids in parentheses indicate uncertainty).
FIG. 1.

SDS-PAGE of purified exoPG before (lane 2) and after (lane 3) chemical deglycosylation. Molecular mass markers (lane 1) are in kilodaltons. Lane 2 was loaded with 2 μg of protein, and lane 3 was loaded with what was left of 2 μg of protein after it had been deglycosylated.
Cloning of PGX1.
Oligonucleotides based on portions of peptide sequences 1 (IKVWPG) and 2 (QYPGEV) were synthesized, and a 99-bp fragment of PGX1 was amplified by PCR. This fragment hybridized with an oligonucleotide based on DIVEN in peptide sequence 2. The PCR product was used as a probe to isolate cDNA and genomic copies of the encoding gene, PGX1, and both were sequenced on both strands. The DNA sequence of PGX1 contains an open reading frame of 1,341 bases that is interrupted by three introns of 51, 63, and 70 bp (Fig. 2). The predicted protein product of PGX1 contains 446 amino acids with a molecular mass of 47.8 kDa, which is in good agreement with the molecular mass (45 kDa) of the deglycosylated gene product determined by SDS-PAGE (Fig. 1). The product of PGX1 (called Pgx1p) has 12 potential N-glycosylation sites (Fig. 2). The N terminus of the mature protein could not be experimentally determined, but the SignalP program predicted a signal peptide cleavage site between amino acids 19 and 20 (21). The dipeptide KR at amino acid 33 could be a second processing site for a Kex-2-like protease (6). Pgn1p is also likely to undergo a second proteolytic processing event based on comparison of its predicted signal peptide cleavage site and the known N terminus of the mature protein (SwissProt P26215) (26).
FIG. 2.

DNA and deduced amino acid sequences of PGX1. Amino acids are shown below the corresponding codons. The 12 potential N-glycosylation sites (NXS/T) are underlined. The two peptides used for the design of PCR primers are doubly underlined. The three introns are indicated by lowercase letters. ∗, stop codon; +, polyadenylation site.
TBLASTN and BLASTP analysis (2) indicated that the primary amino acid sequence of Pgx1p is highly similar (61% identity) to that of an exoPG from the saprophytic filamentous fungus A. tubingensis (15). This exoPG also has 12 predicted N-glycosylation sites. Pgx1p is also related, but much less so, to other bacterial, fungal, and plant endoPGs and exoPGs. The amino acid similarity of Pgx1p to other PGs, including Pgn1p of C. carbonum, is strong only in the region surrounding the His residue at the active site (amino acid 273 in Fig. 2) (7). The sequence surrounding this site in Pgx1p is in agreement with the PG signature motif defined in PROSITE (5), except for the substitution of Ser for Gly adjacent to the His residue. The only other known PG in which Ser replaces Gly at this position is the closely related exoPG of A. tubingensis (15).
The genomic locations of PGN1 and PGX1 were mapped by hybridization to chromosomes separated by pulsed-field gel electrophoresis. In strain SB111, PGN1 is on a chromosome of 1.9 MB and PGX1 is on a chromosome of 3.2 MB. CEL1 (28) and XYL1 (3) also hybridize to a chromosome of 3.2 MB, but it is not known if these three genes are on the same chromosome or on different chromosomes of the same size.
Expression of PGX1 in culture and in planta.
RNA was extracted from mycelial mats grown with sucrose, pectin, or purified maize cell walls as the carbon source. PGX1 mRNA was abundant in 7-day-old fungal mycelium grown on pectin, was present in mycelium grown on maize cell walls, and was absent in mycelium grown on sucrose (Fig. 3). During the course of infection of maize leaves, PGX1 mRNA could be detected starting 3 days after inoculation (Fig. 4). At this point, infection was well developed, with large, expanding lesions. PGN1 was also expressed during infection and could be detected before either PGX1 or GPD1 (Fig. 4).
FIG. 3.
RNA blot of total RNA (10 μg) extracted from wild-type strain 367-2 and probed with the PGX1 cDNA. The fungus was grown in still culture for 7 days on modified Fries’ medium salts with 2% sucrose (lane 1), 1% pectin (lane 2), or 1% maize cell walls (lane 3). Equal loading of each lane was confirmed by staining of the blot with methylene blue (10). The size of the PGX1 mRNA is 1.4 kb.
FIG. 4.

RNA blot of expression of PGN1, PGX1, and GPD1 during infection of maize plants. Insofar as the expression of GPD1 is constitutive, it serves as an indicator of fungal mass. Numbers indicate days after inoculation (day 0 is immediately after inoculation). Fifteen micrograms of poly(A)+ RNA per lane was loaded. The blot was first probed with PGN1, stripped, probed with PGX1, stripped, and probed with GPD1. Approximately equal loading of mRNA in each lane, including days 0 and 1, was confirmed by staining of the gel with ethidium bromide.
Gene disruptions and replacements of pgx1 and pgn1.
Initially, pgx1 and pgn1 were mutated by single-crossover gene disruption (26). In the pgx1 mutant, the major peak as well as the minor peaks of exoPG activity eluting from the cation-exchange HPLC column disappeared (Fig. 5). One explanation for this result is that the multiple peaks of exoPG activity are all encoded by PGX1 and represent glycosylation isoforms of Pgx1p. An alternative explanation is that the other peaks are the products of other genes whose expression is dependent on expression of PGX1; for example, the action of Pgx1p on pectin may release essential inducers of these genes.
FIG. 5.

Cation-exchange HPLC analysis of PG activity of wild-type, pgn1, and pgx1 single mutants and a pgn1/pgx double mutant made by gene disruption. Culture filtrates were harvested after 7 days of growth on 1% pectin as the carbon source. Approximately equal amounts of protein were loaded onto the HPLC column in each case. One-milliliter fractions were collected. Of each fraction, 25 μl was assayed in a total volume of 300 μl, and, at the end of a 30-min incubation at 30°C, 25 μl was sampled for total reducing sugars. Enzyme activity is expressed as change in the optical density at 410 nm (OD410); an OD410 of 1.0 represents 7.5 μg of galacturonic acid.
As previously reported, disruption of PGN1 resulted in the almost complete disappearance of the single peak of endoPG activity eluting at 32 min (Fig. 5). In the double pgn1/pgx1 mutant, both exoPG and endoPG activities were greatly reduced (Fig. 5).
pgn1 mutants grew on pectin to the same extent as the wild-type strain (26). pgx1 mutants still grew on pectin as the carbon source, although pgx1 and pgn1/pgx1 strains consistently grew approximately 20% less well on pectin than the wild-type strain (see below). The most striking phenotype of pgn1 and pgn1/pgx1 strains grown in vitro was that their concentrated culture filtrates were very viscous, and the addition of 50 mM calcium chloride caused formation of a large precipitate. Because pectin was the only polymer added to the culture and because polygalacturonic acid can be precipitated by calcium ions, this residual, viscous material was most likely deesterified pectin (polygalacturonic acid) that remained polymerized due to the near total absence of endoPG in strains lacking a wild-type PGN1 allele.
pgx1 mutants were still pathogenic, as judged by the numbers, sizes, and appearances of primary lesions on young maize plants over a 2-week period, at which point the plants were completely killed.
Despite the fact that endoPG activity was greatly reduced in pgn1 strains, pgn1 strains consistently showed a small peak of PG activity that had an elution time of 31 min, the same as that of Pgn1p. Due to its low level, it could be reliably detected only in the double-knockout strain (Fig. 5).
There are several possible explanations for the peak of residual PG activity eluting in the same position as Pgn1p. One is that the integrating disruption plasmid was excised in a subset of the nuclei in the pgn1 mutant strain by recombination of the flanking duplicated DNA. Although we have never observed this phenomenon in any of our other gene disruption mutants, to test this possibility, we made new pgn1 and pgx1 mutants by the technique of gene replacement, which, because it results in the irreversible loss of a major portion of the mutated gene, precludes the possibility of reversion.
In transformation vectors for gene replacements, the central portions of both the PGX1 and the PGN1 genes were removed and replaced by the hph1 gene driven by the P1 promoter from Cochliobolus heterostrophus (Fig. 6). A double pgn1/pgx1 mutant was constructed by crossing the two single mutants and screening the random ascospore progenies first for hygromycin resistance and then by Southern blotting (Fig. 7). DNA from the wild-type strain 367-2 hybridized to both PGN1 and PGX1 but not to hph1. DNAs from the 12 progenies of a cross between 639-2 (pgn1) and 640-11 (pgx1) all hybridized to hph1 and also to either PGX1 (lanes 4, 8, and 10 to 13), PGN1 (lanes 2, 3, 5, 7, and 9), or to neither (lane 6). One progeny was a double pgn1/pgx1 mutant (lane 6).
FIG. 6.

Genomic restriction maps and strategies for gene replacement mutation of PGN1 and PGX1. Dashed lines, DNA that was deleted and the DNA that was inserted. (Upper diagram) PGN1 replacement vector pPGNGR1; (lower diagram) PGX1 replacement vector pPGXGR1.
FIG. 7.

DNA blot analysis of progenies from a cross between PGN1 and PGX1 replacement mutants made as diagrammed in Fig. 6. DNA was cut with HindIII in all lanes. (Top panel) DNA blot probed with hph1 encoding hygromycin phosphotransferase; (bottom panel) the same blot after stripping and reprobing with PGN1 and PGX1 simultaneously (the faint bands are residual hph1 hybridization). In each lane of the bottom panel, the upper band, when present, is PGN1, and the bottom band, when present, is PGX1. Lanes: 1, DNA from 367-2 (wild type); 2 through 13: strains 655-1 through 655-12, respectively, which are progenies from a cross between 639-2 (pgn1) and 640-11 (pgx1).
The growth, PG activities, and pathogenic phenotypes of the single and double mutants constructed by gene replacement were indistinguishable from the phenotypes of the mutants constructed by gene disruption. Typical data for the growth of the wild type and of single- and double-gene replacement mutants on pectin and sucrose are shown in Table 1. The small peak of PG activity eluting at the same position as Pgn1p was still present in the gene replacement mutants. The reality of this small peak was clear when the assay sensitivity was increased by 200-fold (Fig. 8).
TABLE 1.
Growth of pgn1 and pgx1 mutants of C. carbonum on pectin or sucrosea
| Strain | Genotype | Growth (g [dry wt]/mat ± 1 SD)b
|
Precipitatec | |
|---|---|---|---|---|
| Pectin (% of wild type) | Sucrose (% of wild type) | |||
| SB111 | Wild type | 0.56 ± 0.03 (100) | 0.76 ± 0.04 (100) | − |
| 367-2 | Wild type | 0.54 ± 0.01 (100) | 0.81 ± 0.04 (100) | − |
| 639-3 | pgn1 | 0.60 ± 0.02 (109) | 0.75 ± 0.05 (96) | + |
| 640-11 | pgx1 | 0.45 ± 0.01 (82) | 0.82 ± 0.01 (104) | − |
| 655-5 | pgn1/pgx1 | 0.43 ± 0.04 (78) | 0.71 ± 0.08 (90) | + |
Cultures were harvested after 13 days.
n = 4 (pectin) or n = 2 (sucrose).
Presence (+) or absence (−) of large precipitate after addition of 50 mM calcium chloride to the culture filtrate.
FIG. 8.

Cation-exchange chromatography analysis of wild type (SB111) and the double mutant pgn1/pgx1 (655-5) made by gene replacement. Cultures were harvested after 7 days of growth on 1% pectin. One-milliliter fraction were collected. (Top panel) Short-term assay (25 μl of each fraction assayed in a total volume of 300 μl for 30 min at 30°C, followed by 25 μl of each reaction removed for measurement of released reducing sugars); (bottom panel) extended duration assay (50 μl of each fraction assayed in a total volume of 300 μl for 25 h at 30°C, followed by 50 μl of each reaction removed for measurement of released reducing sugars). Enzyme activity is expressed as change in the OD410; an OD410 of 1.0 represents 8 μg of galacturonic acid.
DISCUSSION
A strain of C. carbonum mutated both in PGN1, which encodes endoPG, and in PGX1, which encodes exoPG, had less than 0.8% of wild-type levels of total PG activity but was still pathogenic and grew almost as well as the wild-type strain in culture on pectin.
The simplest explanation for the unaltered pathogenicity of the double mutant is that Pgx1 and Pgn1 have no (or only a minor) role in pathogenesis. However, the importance of pectin degradation in pathogenesis is still an open question, because in vitro studies indicate that the double mutant is still able to utilize pectin for growth.
There are several possible explanations for why C. carbonum without its two major extracellular PGs can still grow on pectin. Residual heterokaryosis in the transformants was excluded by multiple rounds of single-spore isolation and by putting the mutant strains through a cross (17). Heterokaryosis due to reversion of a small percentage of the mutant genes in strains created by gene disruption was excluded by the construction of gene replacements in which reversion cannot occur.
Utilization of low-molecular-weight contaminants in the commercial pectin source is unlikely, because the fungus grows just as well on pectin that has been extensively washed with acidified ethanol. Utilization of nonpectinaceous high-molecular-weight contaminants also seems unlikely, because the same pectin was used for growth and for enzyme assays, and, therefore, any additional pectin-degrading enzymes contributing to growth would have been detected in the enzyme assays.
The double pgn1/pgx1 mutant probably grows on pectin by degrading pectin itself, and, if so, C. carbonum must have additional pectin-degrading enzymes. On the one hand, these enzymes could be ones that have avoided identification because they are not detectable by any of the assays that we have used (including different pHs and addition of divalent cations), are expressed more transiently than Pgn1p and Pgx1p, or are not stable during culture and purification. On the other hand, C. carbonum has three pectin-degrading activities that, alone or together, could account for the growth of the double mutant on pectin. The first of these is pectin methylesterase, which in addition to its role in converting pectin to polygalacturonic acid, the preferred substrate for Pgn1p and Pgx1p, releases methanol. C. carbonum can grow to a slight extent on methanol as the sole carbon source if it is added to cultures periodically in small amounts (25). However, even if the pectin used in these experiments were 100% methoxylated, and even if 100% of the released methanol were incorporated into fungal dry weight, this could account for less than half of the growth of the double mutant on pectin (Table 1). The role of pectin methylesterase in supporting growth of C. carbonum on pectin is currently being investigated by isolation and disruption of its encoding gene (25).
The second pectin-degrading activity in the double pgn1/pgx1 mutant is the small residual peak that is coeluted from a cation-exchange column with Pgn1p (Fig. 5). Based on its continued presence in gene replacement mutants that have been genetically purified by multiple rounds of single-spore isolation and crossing, it might be the product of a new PG-encoding gene. If so, this gene is not closely related to PGN1 or PGX1, because low-stringency hybridization to DNA blots of total genomic DNA with these genes as probes does not detect any related genes (25). We are purifying this residual peak of PG activity in order to characterize it further and to obtain amino acid sequence data for comparison with Pgn1p.
A third pectin-degrading activity that might support growth of the double mutant is an enzyme(s) that remains bound to the mycelium of C. carbonum. Wall-bound xylanase, cellulase, and pectinase have previously been reported from prokaryotes (19, 27). The major PG of the filamentous fungus Venturia inaequalis, an apple pathogen, is present in the mycelium, and, based on its extractability with high salt, is probably present in fungal cell walls (30). Mycelial extracts of the double mutant of C. carbonum do, in fact, contain pectin-degrading activity that is distinct from that of either Pgn1 or Pgx1 because it is optimal at pH 6, prefers methoxylated pectin over polygalacturonic acid, is stimulated by calcium ions, and is anionic in character (25). At this point, the presence of the mycelial pectinase seems the most likely of the three possible explanations for the continued ability of the double mutant to grow on pectin.
Although the two residual activities in the double mutant are weak compared with those due to Pgn1p and Pgx1p, they might be sufficient to support near-normal growth. If so, then C. carbonum in culture must normally produce approximately 100-fold more PG activity than is necessary. Strains of Saccharomyces cerevisiae with as little as 0.6% of their wild-type levels of invertase can still grow on sucrose (13, 16).
ACKNOWLEDGMENTS
The financial support of the U.S. Department of Energy Division of Energy Biosciences and the U.S. Department of Agriculture NRICGP (to J.D.W.) and of the NATO Collaborative Research Program (to J.D.W. and F.C.) is gratefully acknowledged.
We thank Carol Weiss and Fabienne Hamburger for technical assistance and Joe Leykam, MSU Macromolecular Facility, for peptide sequencing and oligonucleotide synthesis.
Footnotes
Present address: Monsanto Corporation, St. Louis, MO 63167.
REFERENCES
- 1.Ahn J-H, Walton J D. Chromosomal organization of TOX2, a complex locus controlling host-selective toxin biosynthesis in Cochliobolus carbonum. Plant Cell. 1996;8:887–897. doi: 10.1105/tpc.8.5.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 3.Apel P, Panaccione D G, Holden F R, Walton J D. Cloning and targeted gene disruption of XYL1, a β1,4-xylanase gene from the maize pathogen Cochliobolus carbonum. Mol Plant-Microbe Interact. 1993;6:467–473. doi: 10.1094/mpmi-6-467. [DOI] [PubMed] [Google Scholar]
- 4.Apel-Birkhold P, Walton J D. Cloning, disruption, and expression of two endo-β1,4-xylanase genes, XYL2 and XYL3, from Cochliobolus carbonum. Appl Env Microbiol. 1996;62:4129–4135. doi: 10.1128/aem.62.11.4129-4135.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bairoch A, Bucher P, Hofmann K. The PROSITE datatase, its status in 1995. Nucleic Acids Res. 1995;24:189–196. doi: 10.1093/nar/24.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brenner C. Biochemical and genetic methods for analyzing specificity and activity of a precursor-processing enzyme: yeast Kex2 protease, kexin. Methods Enzymol. 1994;244:152–167. doi: 10.1016/0076-6879(94)44013-1. [DOI] [PubMed] [Google Scholar]
- 7.Caprari C, Mattei B, Basile M L, Salvi G, Crescenzi V, De Lorenzo G, Cervone F. Mutagenesis of endopolygalacturonase from Fusarium moniliforme: histidine residue 234 is critical for enzymatic and macerating activities and not for binding to polygalacturonase-inhibiting protein (PGIP) Mol Plant-Microbe Interact. 1996;9:617–624. doi: 10.1094/mpmi-9-0617. [DOI] [PubMed] [Google Scholar]
- 8.Di Pietro A, Roncero M I G. Purification and characterization of an exopolygalacturonase from the tomato vascular wilt pathogen Fusarium oxysporum f.sp. lycopersici. FEMS Microbiol Lett. 1996;145:295–299. doi: 10.1111/j.1574-6968.1996.tb08592.x. [DOI] [PubMed] [Google Scholar]
- 9.He S Y, Collmer A. Molecular cloning, nucleotide sequence, and marker exchange mutagenesis of the exo-poly-α-d-galacturonosidase-encoding pehX gene of Erwinia chrysanthemi EC16. J Bacteriol. 1990;172:4988–4995. doi: 10.1128/jb.172.9.4988-4995.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herrin D L, Schmidt G W. Rapid, reversible staining of Northern blots prior to hybridization. BioTechniques. 1988;6:196–200. [PubMed] [Google Scholar]
- 11.Huang Q, Allen C. An exo-poly-α-d-galacturonosidase, PehB, is required for wild-type virulence in Ralstonia solanacearum. J Bacteriol. 1997;179:7369–7378. doi: 10.1128/jb.179.23.7369-7378.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hynes M J, Corrick C M, King J A. Isolation of genomic clones containing the amdS gene of Aspergillus nidulans and their use in the analysis of structural and regulatory mutations. Mol Cell Biol. 1983;3:1430–1439. doi: 10.1128/mcb.3.8.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaiser C A, Preuss D, Grisafi P, Botstein D. Many random sequences functionally replace the secretion signal sequence of yeast invertase. Science. 1987;235:312–317. doi: 10.1126/science.3541205. [DOI] [PubMed] [Google Scholar]
- 14.Kelemu S, Collmer A. Erwinia chrysanthemi EC16 produces a second set of plant-inducible pectate lyase isozymes. Appl Environ Microbiol. 1993;59:1756–1761. doi: 10.1128/aem.59.6.1756-1761.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kester H C M, Kusters-Van Someren M A, Müller Y, Visser J. Primary structure and characterization of an exopolygalacturonase from Aspergillus tubingensis. Eur J Biochem. 1996;240:738–746. doi: 10.1111/j.1432-1033.1996.0738h.x. [DOI] [PubMed] [Google Scholar]
- 16.Klein R D, Gu Q, Goddard A, Rosenthal A. Selection for genes encoding secreted proteins and receptors. Proc Natl Acad Sci USA. 1996;93:7108–7113. doi: 10.1073/pnas.93.14.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leach J, Yoder O C. Heterokaryosis in Cochliobolus heterostrophus. Exp Mycol. 1982;6:364–374. [Google Scholar]
- 18.Lever M. A new reaction for colorimetric determination of carbohydrates. Anal Biochem. 1972;47:273–279. doi: 10.1016/0003-2697(72)90301-6. [DOI] [PubMed] [Google Scholar]
- 19.Mateos P F, Jimenez-Zurdo J I, Chen J, Squartini A S, Haack S K, Martinez-Molina E, Hubbell D H, Dazzo F B. Cell-associated pectinolytic and cellulolytic enzymes in Rhizobium leguminosarum biovar trifolii. Appl Environ Microbiol. 1992;58:1816–1822. doi: 10.1128/aem.58.6.1816-1822.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy J M, Walton J D. Three extracellular proteases from Cochliobolus carbonum: cloning and targeted disruption of ALP1. Mol Plant-Microbe Interact. 1996;9:290–297. doi: 10.1094/mpmi-9-0290. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Prot Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 22.Nozaki K, Miyairi K, Hozumi S, Fukui Y, Okuno T. Novel exopolygalacturonases produced by Alternaria mali. Biosci Biotechnol Biochem. 1997;61:75–80. [Google Scholar]
- 23.Panaccione D G, Scott-Craig J S, Pocard J-A, Walton J D. A cyclic peptide synthetase gene required for pathogenicity of the fungus Cochliobolus carbonum on maize. Proc Natl Acad Sci USA. 1992;89:6590–6594. doi: 10.1073/pnas.89.14.6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pitkin J W, Panaccione D G, Walton J D. A putative cyclic peptide efflux pump encoded by the TOXA gene of the plant pathogenic fungus Cochliobolus carbonum. Microbiology. 1996;42:1557–1565. doi: 10.1099/13500872-142-6-1557. [DOI] [PubMed] [Google Scholar]
- 25.Scott-Craig, J. S., and J. D. Walton. Unpublished results.
- 26.Scott-Craig J S, Panaccione D G, Cervone F, Walton J D. Endopolygalacturonase is not required for pathogenicity of Cochliobolus carbonum on maize. Plant Cell. 1990;2:1191–1200. doi: 10.1105/tpc.2.12.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shao W, DeBlois S, Wiegel J. A high-molecular-weight, cell-associated xylanase isolated from exponentially growing Thermoanaerobacterium sp. strain JW/SL-YS485. Appl Environ Microbiol. 1995;61:937–940. doi: 10.1128/aem.61.3.937-940.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sposato P, Ahn J-H, Walton J D. Characterization and disruption of a gene in the maize pathogen Cochliobolus carbonum encoding a cellulase lacking a cellulose binding domain and hinge region. Mol Plant-Microbe Interact. 1995;8:602–609. doi: 10.1094/mpmi-8-0602. [DOI] [PubMed] [Google Scholar]
- 29.Strömqvist M, Gruffman H. Periodic acid/Schiff staining of glycoprotein immobilized on a blotting matrix. BioTechniques. 1992;13:744–746. [PubMed] [Google Scholar]
- 30.Valsangiacomo C, Gessler C. Purification and characterization of an exopolygalacturonase produced by Venturia inaequalis, the causal agent of apple scab. Physiol Mol Plant Pathol. 1992;40:63–77. [Google Scholar]
- 31.Van Hoof A, Leykam J, Schaeffer H J, Walton J D. A single β1,3-glucanase secreted by the maize pathogen Cochliobolus carbonum acts by an exolytic mechanism. Physiol Mol Plant Pathol. 1991;39:259–267. [Google Scholar]
- 32.Walton J D. Deconstructing the cell wall. Plant Physiol. 1994;104:1113–1118. doi: 10.1104/pp.104.4.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walton J D, Cervone F. Endopolygalacturonase from the maize pathogen Cochliobolus carbonum. Physiol Mol Plant Pathol. 1990;36:351–359. [Google Scholar]
- 34.Walton J D, Holden F R. Properties of two enzymes involved in the biosynthesis of the fungal pathogenicity factor HC-toxin. Mol Plant-Microbe Interact. 1988;1:128–134. [Google Scholar]
- 35.Wirsel S, Turgeon B G, Yoder O C. Deletion of the Cochliobolus heterostrophus mating-type (MAT) locus promotes the function of MAT transgenes. Curr Genet. 1996;29:241–249. [PubMed] [Google Scholar]
- 36.Yoder O C. Cochliobolus heterostrophus, cause of Southern corn leaf blight. Adv Plant Pathol. 1988;6:95–112. [Google Scholar]