

ABSTRACT
Compelling epidemiological and animal experimental data demonstrate that cardiometabolic and neuropsychiatric diseases originate in a suboptimal intrauterine environment. Here, we review evidence suggesting that altered placental function may, at least in part, mediate the link between the maternal environment and changes in fetal growth and development. Emerging evidence indicates that the placenta controls the development and function of several fetal tissues through nutrient sensing, modulation of trophoblast nutrient transporters and by altering the number and cargo of released extracellular vesicles. In this Review, we discuss the development and functions of the maternal-placental-fetal interface (in humans and mice) and how cross-talk between these compartments may be a mechanism for in utero programming, focusing on mechanistic target of rapamycin (mTOR), adiponectin and O-GlcNac transferase (OGT) signaling. We also discuss how maternal diet and stress influences fetal development and metabolism and how fetal growth restriction can result in susceptibility to developing chronic disease later in life. Finally, we speculate how interventions targeting placental function may offer unprecedented opportunities to prevent cardiometabolic disease in future generations.
Keywords: Maternal-fetal exchange, Fetal development, Programming, Prenatal, Extracellular vesicles, Epigenetics
Summary This Review summarizes emerging evidence that cardiometabolic diseases in offspring arise from changes in placental development and function and discusses mechanisms of cross-talk between the mother and fetus via the placenta.
Introduction
A person's susceptibility to developing diseases such as diabetes and hypertension was previously thought to be determined by the interaction between genes and environment in adult life. However, this model has since been revised because compelling epidemiological evidence and animal experimental data demonstrate a strong link between the intrauterine environment and the risk for developing diseases later in life, including cardiometabolic and neuropsychiatric disorders (Gluckman et al., 2008; Khambadkone et al., 2020; Fernandez-Twinn et al., 2019). For example, ∼50% of Type 2 diabetes in young individuals can be attributed to intrauterine exposure to gestational diabetes or maternal obesity (Dabelea et al., 2008). Development of such disorders is strongly linked with fetal growth restriction (FGR), often a result of placental insufficiency and impaired nutrient and oxygen delivery to the fetus, as well as fetal overgrowth, which is associated with increased nutrient delivery to the fetus (Gluckman et al., 2008; Khambadkone et al., 2020; Fernandez-Twinn et al., 2019). This concept is known as the developmental origins of health and disease (DOHaD) and has profound public health consequences. This concept of intrauterine exposure determining life-long health is likely to transform the way we practice medicine in the future. Moreover, this paradigm offers unprecedented opportunities to reduce the risk of major diseases across the life course by early preventive interventions.
The placenta constitutes the primary interface between mother and fetus, performing a multitude of functions essential for fetal growth and development. In humans, changes in placental structure have been associated with the development of chronic disease later in life (Burton et al., 2016), including an inverse correlation between placental surface area at delivery and hypertension and heart failure in adulthood (Barker et al., 2010a,b). Moreover, placental thickness has been reported to be inversely associated with sudden cardiac death later in life (Barker et al., 2012). It is widely believed that these alterations in structure reflect functional changes in the placenta, which mediate the link between perturbations in the maternal compartment and programming of adult disease (Burton et al., 2016; Jansson and Powell, 2013; Myatt, 2006; Jansson et al., 2009; Staud and Karahoda, 2018). In support of a link between placental function and cardiometabolic outcomes in children, we reported that the activity of placental AMP-activated protein kinase (AMPK), a cellular energy sensor, is negatively associated with adiposity in children and the activity of placental mechanistic target of rapamycin (mTOR) signaling, a cellular nutrient sensor, was positively correlated to systolic blood pressure at 4-6 years of age (Fig. 1) (Keleher et al., 2020). Moreover, placental insulin signaling is positively associated with triglyceride levels and adiposity at 4-6 years of age, and markers of placental inflammation predict adiposity in children (Keleher et al., 2021). These observations strongly suggest that placental signaling can provide crucial information on which infants have a high risk to develop cardiometabolic diseases later in life. It may also be possible in the future to develop treatments that improve placental function to prevent such disorders in the next generation.
Fig. 1.
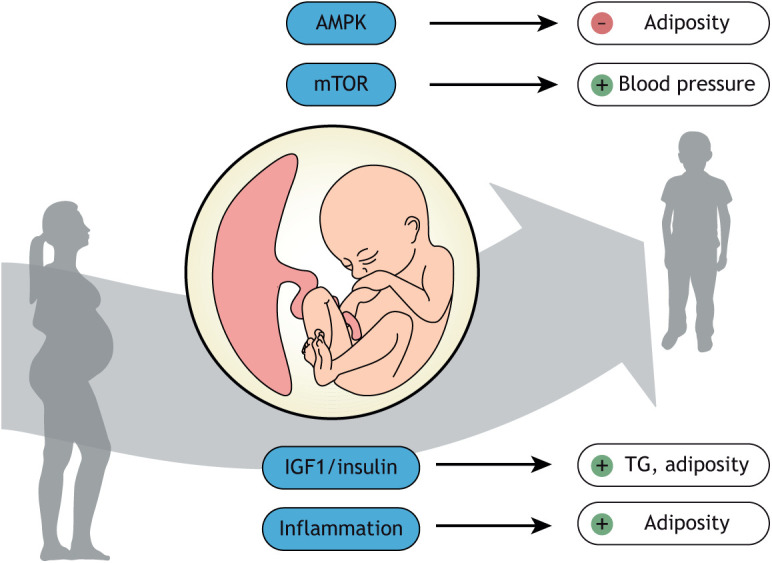
Key placental metabolic signaling pathways and their associations with cardiometabolic outcomes in children. Placental AMP-activated protein kinase (AMPK) signaling (a cellular energy sensor) is negatively associated with adiposity in children, whereas the activity of placental mechanistic target of rapamycin (mTOR) signaling (a cellular nutrient sensor) is positively correlated to systolic blood pressure at 4-6 years of age. Moreover, placental IGF1/insulin signaling is positively associated with triglyceride (TG) levels and adiposity at 4-6 years of age, and placental inflammation predicts adiposity in children.
In this Review, we focus on recent work that has not been comprehensively covered by previous excellent reviews (such as Burton et al., 2016), with a particular focus on data linking placental function to the intrauterine environment, long-term outcomes in children and emerging mechanistic evidence in animal models.
Placental development and primary functions
Placental development requires the coordinated interactions between trophoblast cell lineages of the placenta and maternal endometrium in order to carry out its primary function of supporting fetal development. The mouse is a widely accepted animal model for placental studies and, although there are developmental and structural differences between the human and mouse placenta (Fig. 2), mRNA and protein expression profiles are relatively conserved across the two species, making this a useful model to study the many aspects of human placental development and function (Hemberger et al., 2020; Cox et al., 2009). In mice, the placenta has three layers of trophoblast cells separating the maternal sinusoids from the fetal capillary (haemotrichorial), including a mononuclear layer of trophoblast giant cells and a bilayer of syncytiotrophoblast cells. Meanwhile, humans have a monolayer of syncytiotrophoblast cells separating maternal and fetal blood (haemomonochorial) (Hemberger et al., 2020; Georgiades et al., 2002) (Fig. 2). The placenta is primarily composed of fetal-derived trophoblast cells, which are the first cells to undergo differentiation to form the trophectoderm at the blastocyst stage. The trophectoderm is the outer-most layer of the blastocyst and ultimately forms all placental layers, except the maternal decidua (Rossant and Cross, 2001). During early pregnancy, extravillous trophoblast cells invade the decidua and the spiral arteries to initiate vascular remodeling, which is believed to be a prerequisite for establishment of uteroplacental blood flow at the end of the first trimester in humans and in mid-gestation in mice, allowing blood flow to progressively increase across gestation (Adamson et al., 2002). In humans, placental villi are in direct contact with maternal blood in the intervillous space and are made up of a syncytiotrophoblast layer and villous cytotrophoblast cells, which are anchored by the extravillous cytotrophoblast to the maternal decidua (Carter, 2007). In the mouse, there are two major zones of the placenta, the labyrinth and junctional zones. The labyrinth contains cytotrophoblast cells surrounded by a multinucleated syncytiotrophoblast layer that is responsible for compartmentalizing maternal and fetal blood and transporting oxygen and nutrients from the maternal to the fetal circulation. The junctional zone, an endocrine region, separates the labyrinth from the maternal decidua along a line of trophoblast giant cells and spongiotrophoblast cells (Hemberger et al., 2020) (Fig. 2).
Fig. 2.

Comparison of mouse and human placental structures. (A,B) Both mouse and human placentas are hemochorial (trophoblast bathed in maternal blood) and discoid in shape. (A) The murine placental structure includes the labyrinth and junctional zones. The box depicts the maternal-fetal interface of the labyrinth, demonstrating the haemotrichorial or three trophoblast layers (a mononuclear trophoblast cell layer and syncytiotrophoblast layers I and II). (B) The human placenta has analogous layers: a decidual layer with spiral arteries and the placental villi. The box details a section of a chorionic villus illustrating the haemomonochorial or monolayer of syncytiotrophoblast cells separating maternal and fetal blood. The apical brush border of the human syncytiotrophoblast in contact with maternal blood is referred to as microvillous membrane (MVM) and the opposing plasma membrane juxtaposed to the fetal capillary is referred to as basal membrane (BM). The corresponding apical membrane in the mouse placenta is referred to as the trophoblast plasma membrane (TPM) and is localized to syncytial layer II.
Transfer of nutrients, respiratory gases and waste products across the placental barrier is achieved by passive and facilitated diffusion, active transport and endo- and exocytosis. Macromolecules are transported via specialized transporting proteins, channels and exchangers in the syncytiotrophoblast plasma membranes (Lager and Powell, 2012). Transport of nutrients by the placenta is modulated by maternal nutrient availability, as well as in response to fetal demands (Watson and Cross, 2005). Therefore, the placenta adapts its capacity for delivering nutrients in response to both fetal and maternal signals, ultimately impacting fetal growth trajectory. One signal is fetal sex; there is growing evidence demonstrating that the placenta is mediated by sex-specific fetal adaptations in response to an adverse in utero environment. As such, female fetuses adapt more readily by altering placental growth and reducing fetal growth, whereas male fetuses undergo fewer placental changes and maintain normal fetal growth (Clifton, 2010; Brown et al., 1987; Larson et al., 2001; Cvitic et al., 2013). The placenta is not only a conduit for nutrient and gas exchange but also actively secretes a variety of important factors, such as hormones, proteins, lipids and extracellular vesicles (EVs) (Jin and Menon, 2018). EVs and, in particular, small EVs (sEVs, or exosomes) are now gaining traction as a key communication mechanism to facilitate fetal-maternal cross-talk. Release of these placental signals are crucial for the adaptation of both the maternal physiology to pregnancy and fetal development and growth (Fig. 3).
Fig. 3.

Human placental morphology and transport across the maternal-fetal interface. Transport across the syncytiotrophoblast occurs via passive and facilitated diffusion, active transport and endo- and exocytosis. Macromolecules and waste products are transported across the syncytiotrophoblast via specialized transporting proteins, channels and exchangers located in the plasma membranes. Extracellular vesicles (EVs) and hormones released by the syncytiotrophoblast cells are released to the maternal and fetal circulations, thereby participating in maternal-fetal cross-talk. BM, basal plasma membrane; E, estrogen; P4, progesterone; hCG, human chorionic gonadotropin; MVM, microvillous membrane; plGF, placental growth factor; sFLT1, soluble fms-like tyrosine kinase.
Placental function and fetal growth
Intrauterine growth restriction (IUGR) occurs when fetuses are unable to reach their genetically determined growth potential due to limitations in the mother and the placenta to deliver nutrients and oxygen to support normal growth. As a result, these infants are born with a low birth weight for gestational age (typically defined as below the 10th or 5th percentile), often with additional signs of compromise such as abnormal uteroplacental and umbilical blood flows and oligohydramnios (low amniotic fluid volume) (Gordijn et al., 2016). On the opposite end of the birth weight spectrum is fetal overgrowth, resulting in the delivery of a large-for-gestational age (LGA) baby with birth weight above the 90th percentile for gestational age or macrosomia, defined as a birth weight greater than 4 kg. Abnormal fetal growth patterns are associated with increased perinatal morbidity and mortality along with increased risk of developing obesity, diabetes, cardiovascular and neuropsychiatric disorders in childhood and adult life (Gluckman et al., 2008; Khambadkone et al., 2020). Changes in placental function contribute to both reduced and accelerated fetal growth, as well as changes in body composition at birth. Therefore, the placenta plays a crucial role in the developmental programming of risk for disease in later life (Jansson and Powell, 2007; Burton et al., 2016).
Placental blood flow
Placental gas exchange (transfer of oxygen to the fetus and removal of carbon dioxide from the fetal compartment) is vital for optimal fetal growth and is dependent on adequate blood flow on both sides of the placental barrier. Maternal blood flow to the placenta increases when uterine immune cells and extravillous trophoblast cells transform the spiral arteries in the decidua and radial arteries in the myometrium, converting tightly coiled spiral arteries to large unresponsive conduits that open into the placental intervillous space (Fig. 2) (Ives et al., 2020; Chau et al., 2017). Failure to remodel maternal spiral arteries results in ischemia, oxidative stress, low nitric oxide production and release of anti-angiogenic soluble factors, such as soluble fms-like tyrosine kinase 1 (sFLT1) and endoglin (ENG) (Levine et al., 2006). Pro-angiogenic factors like placental growth factor (plGF; also known as PGF) and vascular endothelial growth factor A (VEGFA) are antagonized by these factors (Shibata et al., 2005), leading to endothelial dysfunction, inhibition of angiogenesis and increased vasoconstriction.
The increase in uteroplacental blood flow across gestation is paralleled by increased feto-placental (umbilical) blood flow, regulated by growth factors, oxygen, vascular tone and angiogenesis (Burton et al., 2009). IUGR pregnancies are often associated with signs of poor placental angiogenesis; however, the mechanisms regulating this process are complex and not well understood. Impaired fetal vascular development has been linked to reduced prostaglandin E2 reactivity (Luria et al., 2012) and higher endothelin 1 levels (Li et al., 2018); the net result of these alterations is an imbalance in branching and non-branching angiogenesis and longer unbranched capillary loops in the villous vascular tree (reviewed by Li et al., 2018), promoting vasoconstriction and reducing fetal blood flow.
Placental nutrient transfer and fetal growth
Glucose, the primary substrate for fetal energy metabolism, is transferred across the placental barrier by facilitated diffusion, and as a result glucose concentrations in the umbilical vein are lower than maternal blood glucose and are subject to fluctuations according to maternal glycemic status (Sibiak et al., 2022). An array of facilitative glucose transporters [GLUTs, or solute carrier 2 (SLC2) family] are expressed in the human placenta, including isoforms that are insensitive to insulin (GLUT1, 3 and 9) and those that are regulated by insulin (GLUT4 and 12) (Stanirowski et al., 2018). The apical brush border or microvillous plasma membrane (MVM) of the human syncytiotrophoblast has a very high abundance of GLUT1 transporters, allowing for high-capacity uptake of glucose from maternal blood (Fig. 3) (Jansson et al., 1993; Sibiak et al., 2022). GLUTs in the fetal facing basal plasma membrane (BM) allow glucose to diffuse down its concentration gradient to the fetal capillary. The BM is believed to be the rate-limiting step for glucose transfer due to the lower expression of GLUT proteins and smaller surface area compared with the opposing MVM (Jansson et al., 2002b). Human IUGR does not appear to be associated with significant changes in MVM or BM GLUT1 expression or activity (Jansson et al., 2002b; Johansson et al., 2002; Chassen and Jansson, 2020). However, pregnancies complicated with fetal overgrowth, e.g. due to diabetes and/or maternal obesity, show increased expression and activity of BM GLUT1 (Acosta et al., 2015). Moreover, the expression of GLUT isoforms 1, 4 and 9 have been positively correlated with fetal growth in pregnancies complicated by gestational diabetes mellitus (GDM) (Stanirowski et al., 2019). Glucose entering the fetal circulation stimulates the fetal pancreas to release insulin, which promotes fetal growth (Boehmer et al., 2017). In pregnancies complicated by GDM and/or maternal hyperglycemia, increased placental flux of glucose to the fetus likely contributes to accelerated fetal growth (Catalano et al., 2012; Stanirowski et al., 2022).
Amino acids are taken up by the syncytiotrophoblast through secondary active transport, which is dependent on the sodium gradient generated by Na+K+ATPase or through exchange transporters (Pizzagalli et al., 2021). This results in amino acid concentrations that are higher in the fetal circulation compared with the mother. System A (SLC38 family) is an amino acid transporter in the MVM responsible for uptake of non-essential amino acids against their concentration gradient, energized by the inwardly directed Na+ gradient (Mahendran et al., 1993). Meanwhile, system L (SLC7 family) exchanges the high levels of non-essential amino acids in the syncytiotrophoblast for essential amino acids, such as leucine, in the maternal circulation (Gaccioli et al., 2015). Therefore, high concentrations of essential amino acids are generated in the syncytiotrophoblast, allowing them to diffuse passively across the BM to enter the fetal blood, mediated by efflux transporters and exchangers (Cleal et al., 2011). IUGR in human pregnancy has consistently been associated with reduced placental amino acid transport activity and protein expression (reviewed by Chassen and Jansson, 2020), whereas increased placental amino acid transporter activity and/or protein expression has been reported in pregnancies with LGA babies in most (Jansson et al., 2002a, 2013; Shang and Wen, 2018), but not all (Kuruvilla et al., 1994) studies.
Normal fetal development is also dependent on the transfer of lipids across the placental barrier (Lauritzen et al., 2016). Microvillous lipoprotein receptors allow complex maternal lipids to bind to the syncytiotrophoblast, where MVM-localized lipases then release non-esterified fatty acids that can be taken up into the placenta by fatty acid transporters (Duttaroy and Basak, 2021). Lipase activity has been demonstrated to be lower in pregnancies complicated by IUGR and higher in diabetic pregnancies delivering LGA babies (Magnusson et al., 2004). Fatty acid transporting proteins (FATPs, or SLC27) and CD36 fatty acid translocase are localized to both plasma membranes of the syncytiotrophoblast (Lager et al., 2016). MVM protein expression of several FATPs was reported to be increased in IUGR pregnancies (Chassen et al., 2018), suggesting a compensatory mechanism for delivering lipids to the IUGR fetus. Deficiencies of omega 3 long chain polyunsaturated fatty acids, such as docosahexaenoic acid (22:6, n-3), during development are associated with learning, memory, motor and visual impairments, and their transport from the maternal circulation to the fetus is essential, as the fetus and placenta have a limited ability to synthesize these lipids (Lauritzen et al., 2016). However, although studies of the uptake of fatty acids from the maternal circulation have been reported, the specific mechanisms for release of crucial fatty acids from the placenta to support fetal growth and development remain understudied.
Mechanistic studies in animal models
Associative studies in humans suggest that placental nutrient transport capacity and blood flow are related to fetal growth trajectories, but causality is difficult to establish in human pregnancy (Huang et al., 2018; Dumolt et al., 2021; Garcia-Santillan et al., 2022). Using rodent animal models of pregnancy, mechanistic studies have provided insights into how the placenta may mediate changes in fetal growth. Early studies demonstrated that reduction in blood flow to the uterus in rats resulted in FGR (Wigglesworth, 1964). Maternal undernutrition, such as calorie or protein restriction, also resulted in FGR (Young and Widdowson, 1975). Conversely, exposing pregnant animals to high fat (40%) and high sugar diets results in fetal overgrowth (Aye et al., 2015), although extreme high fat diet (60%) or excessive weight gain in rodents has also been reported to cause FGR (Taylor et al., 2003). In line with human IUGR, utero-placental blood flow and placental nutrient transfer capacity are decreased in many animal models of restricted fetal growth (Gaccioli et al., 2013). By employing emerging technologies for placental-specific gene manipulation in mice, it was recently reported that reductions in the expression of the system A amino acid transporter isoform Snat2 (SLC38A2) caused IUGR (Vaughan et al., 2021) and overexpression of the system L amino acid transporter isoform Lat1 (SLC7A5) resulted in fetal overgrowth (F. Rosario, K. Barentsen, T.L.P., J. Urschitz and T.J., unpublished). Although these experimental and animal studies indicate that alterations in blood flow and placental nutrient transfer are sufficient to cause changes in fetal growth, they do not provide adequate information on the regulatory events that lead to these phenotypes. A number of global regulatory systems have been identified in the human placenta to date and many of these have been confirmed in animal models and are discussed below.
Placental mTOR signaling and fetal growth
mTOR is a serine/threonine kinase regulating cellular metabolism, growth and proliferation in response to a range of cues, including nutrients and growth factor signaling. Placental mTOR is inhibited in IUGR in both humans (Yung et al., 2008; Chen et al., 2015b; Dimasuay et al., 2017) and animal models (Rosario et al., 2011; Kavitha et al., 2014). Conversely, placental mTOR signaling activity is increased in pregnancies complicated by fetal overgrowth (Jansson et al., 2013; Aye et al., 2015). Mechanistically, trophoblast mTOR is activated by insulin/IGF1, glucose and amino acids (Roos et al., 2009), fatty acids (Lager et al., 2013) and folate (Rosario et al., 2017). Meanwhile, it is inhibited by cortisol (Vaughan et al., 2015), adiponectin (Rosario et al., 2012; Aye et al., 2015), infection (Dimasuay et al., 2017) and reduced uteroplacental blood flow (Chen et al., 2015b). mTOR signaling is a positive regulator of trophoblast amino acid (Rosario et al., 2013, 2016a) and folate (Rosario et al., 2016b) transport, protein synthesis and mitochondrial respiration (Rosario et al., 2019), while inhibiting autophagy (Hung et al., 2017).
This body of literature suggests that trophoblast mTOR signaling functions as an important hub linking maternal nutrition and metabolism and utero-placental blood flow to placental function, fetal growth and developmental programming (Fig. 4A). This model is supported by recent mechanistic studies in mice demonstrating that placental-specific knockdown of mTOR causes intrauterine growth restriction and programs the offspring for obesity and diabetes (Akhaphong et al., 2021). Emerging evidence also suggests that mTOR signaling in primary human trophoblast cells regulates the synthesis and/or release of secreted factors that have profound effects on the phosphorylation of insulin-like growth factor binding protein 1 (IGFBP1) in fetal liver cells (Rosario et al., 2023). Because phosphorylation of IGFBP1 regulates its binding affinity for IGF1, the primary fetal growth factor, these data are consistent with the possibility that placental mTOR indirectly regulates fetal growth by also modulating the bioactivity of fetal IGF1.
Fig. 4.

Roles of placental mTOR and OGT in maternal and fetal health. (A) Mechanistic target of rapamycin (mTOR) signaling is one of several key placental sensors integrating maternal signals and conveying information on the ability of the maternal supply line to deliver nutrients and oxygen to the placenta. mTOR is a master regulator of placental function including mitochondrial respiration, nutrient transport, protein synthesis and hormone secretion, thereby regulating fetal growth and development and impacting the long-term health of the offspring. Importantly, these regulatory loops can function in response to both maternal overnutrition and undernutrition to regulate fetal growth according to the available resources. (B) Placental OGT serves as a key cellular mechanism that senses available energy levels and dynamically alters placental function via multiple mechanisms to broadly regulate maternal homeostasis and impact transplacental signals important for fetal development. Importantly, OGT controls local trophoblast responses to a changing maternal environment where maternal stress hormones activate the glucocorticoid receptor (GR), reducing OGT levels. OGT is a key regulator of transcriptomic pathways via stabilization of the H3K27 methyl transferase EZH2. Reduced OGT results in decreases in EZH2 and the transcriptional repressive histone mark, H3K27me3. As OGT is X chromosome-linked, this transcriptomic regulation is much tighter in female XX trophoblast cells than male XY cells, resulting in dynamic placental responses and transplacental signals to the male fetus. A separate cellular signaling pathway links OGT to activation of annexin A1, an essential component in extracellular vesicle (EV) loading and secretion. EVs secreted by the placenta into maternal circulation contribute to homeostatic regulation, including maternal glucose levels in pregnancy. OGT, O-linked N-acetylglucosamine (O-GlcNAc) transferase; EZH2, enhancer of zeste homolog 2.
From the perspective of fetal homeostatic regulation, the above model of placental mTOR signaling and its link to fetal growth and programming may appear to be counterintuitive. For example, in response to decreased uteroplacental blood flow, a common cause of FGR, the model predicts that placental mTOR signaling activity is decreased, inhibiting an array of important placental functions, such as amino acid transport. Indeed, placental mTOR is inhibited (Chen et al., 2015a) and amino acid transport activity is decreased (Mahendran et al., 1993; Paolini et al., 2001) in human pregnancies complicated by IUGR due to placental insufficiency. In contrast, homeostatic regulation predicts that signals from a fetus deprived of nutrients would tend to upregulate nutrient transport in the placenta, largely a fetal organ. Thus, our model (Fig. 1) based on published data (for example Dabelea et al., 2008; Fisk and Atun, 2008; Gaccioli et al., 2013; Hemberger et al., 2020; Jansson et al., 2008; Aye et al., 2014; James-Allan et al., 2020) suggests that, with respect to regulation of placental function, maternal supply signals dominate over fetal demand signals. We speculate that this regulatory system involving placental mTOR signaling has developed in response to the evolutionary pressure from maternal undernutrition or starvation. Matching fetal growth to maternal resources in situations of significant maternal undernutrition will produce an offspring that is smaller in size but, in most instances, can survive and reproduce. Thus, rather than extracting additional nutrients from the already deprived mother, which will jeopardize the survival of both the mother and her fetus, restricted fetal growth might be the preferred strategy. Importantly, these regulatory loops may also function in the opposite direction in response to maternal overnutrition, which may explain the observed activation of placental mTOR signaling and amino acid transport in studies of fetal overgrowth (Jansson et al., 2008, 2013; James-Allan et al., 2020; Dumolt et al., 2022; Johansson et al., 2002; Pedrioli et al., 2021).
Adiponectin as an endocrine link between maternal nutrition and fetal growth
Adiponectin is an adipokine secreted by adipose tissue that acts on peripheral tissues, such as the liver and skeletal muscle, and has insulin-sensitizing effects (Yamauchi et al., 2003). Adiponectin binds to the adiponectin receptor (AdipoR) 1 and AdipoR2, and exerts its effects through activation of AMPK, p38 mitogen activated protein kinase (MAPK) and/or peroxisome proliferator-activated receptor alpha (PPARα) (Yoon et al., 2006). In contrast to its insulin-sensitizing effects in the liver and muscle, adiponectin inhibits insulin signaling in primary human trophoblast cells, through PPARα-mediated synthesis of ceramides (Aye et al., 2014). Ceramides phosphorylate insulin receptor substrate 1 (IRS1) at an inhibitory site, resulting in decreased insulin signaling (Fig. 5) (Aye et al., 2014). These findings were confirmed in vivo in normal pregnant mice, in which chronic infusion of adiponectin by osmotic pump during the last 4 days of pregnancy inhibited placental insulin signaling and caused IUGR (Rosario et al., 2012). In agreement with the model that maternal adiponectin limits fetal growth, knockout of the adiponectin gene in pregnant mice is associated with fetal overgrowth (Qiao et al., 2016). Furthermore, maternal pre-pregnancy body mass index (BMI) is positively correlated with birthweight (Hull et al., 2008). Similar to non-pregnant individuals, circulating concentrations of adiponectin are inversely correlated with BMI in pregnant mothers. In human IUGR, maternal adiponectin levels are elevated and when maternal adiponectin is low, as in obese mothers, there is an increased risk of fetal overgrowth. In support of this concept, negative correlations between maternal serum levels of adiponectin and birth weight have been reported (Mohamad et al., 2018; Jansson et al., 2008) (Fig. 5).
Fig. 5.

Maternal adiponectin informs the placenta about maternal nutrition status. High maternal adiponectin in low nutrient status mothers inhibits insulin signaling in the placenta and reduces nutrient transport capacity. The combination of high adiponectin and low insulin limits placental uptake and protects the mother from additional depletion, resulting in a smaller but potentially viable offspring. In the obese mother with low adiponectin and high insulin there is no inhibition of transport, leading to larger placentas, greater transport capacity and the excess supply of nutrients to the fetus, enhancing fetal growth and resulting in large-for-gestational age babies. These studies demonstrate that the placenta is responding to maternal nutrient status and altering delivery of nutrients to the fetus to match maternal stores. Correcting adiponectin in obese mice to normal levels prevents these changes in placental function, fetal overgrowth and developmental programming of cardiometabolic disease, suggesting that targeting the placenta is a therapeutic alternative that should be further investigated.
In an effort to further understand these effects of adiponectin, we developed a model of maternal obesity in C57Bl/6 mice by feeding a high fat (40%) diet supplemented with a 20% sucrose solution. This model of obesity shows extensive similarities to the human condition, including low levels of maternal adiponectin, glucose intolerance, activation of placental insulin and mTOR signaling, increased placental nutrient transport and fetal overgrowth (Rosario et al., 2015; Aye et al., 2015). Using this model, we recently reported that normalization of maternal circulating adiponectin in obese dams during the last 4 days of pregnancy prevented the activation of placental mTOR signaling and nutrient transport, fetal overgrowth and programming of metabolic and cardiovascular disease in the offspring (Aye et al., 2015; Paulsen et al., 2019; Vaughan et al., 2020). Collectively, these data suggest that maternal adiponectin limits fetal growth by inhibition of placental insulin, mTOR signaling and decreased placental nutrient transport. In the context of maternal obesity, low maternal adiponectin promotes placental function and fetal growth.
By regulating placental function and fetal growth, maternal adiponectin plays an important role in fetal programming of cardiac dysfunction, obesity and insulin resistance (Aye et al., 2015; Paulsen et al., 2019; Vaughan et al., 2020). The exact mechanisms occurring in the fetal compartment that are responsible for the programming of future disease remain unclear. Changes in the availability of nutrients to fetal tissues, both reduced and enhanced placental nutrient delivery, may result in epigenetic modifications leading to metabolic memory that impacts life-long health (Chen and Natarajan, 2022). Direct signaling from the placenta to the fetal organs that may contribute to programming of the fetal organs has been suggested and one signaling possibility is the release of placental sEVs that communicate with fetal organs. How specific maternal nutrient and hormonal states communicate across the placental barrier is an essential area of further research.
The placenta links maternal stress and fetal development
Perturbations in pregnancy such as maternal stress or dietary challenges are key risk factors for neurodevelopmental disorders (Bronson and Bale, 2016; Nugent and Bale, 2015; Sullivan et al., 2010). In human pregnancy, recent studies identified potential causal genes that linked pregnancy perturbations with fetal neurodevelopmental disease risk (Ursini et al., 2018, 2023). Homeostatic signals related to stress and nutrient availability are of particular importance for epigenetic programming in the placenta. The enzyme O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT), which catalyzes the addition of O-GlcNAc to serine and threonine residues on thousands of intracellular proteins, is at the crossroads of nutritional signals and chromatin regulation (Butkinaree et al., 2010). Identified almost 40 years ago in lymphocytes (Torres and Hart, 1984), the pervasiveness of this post-translational modification has been widely appreciated for its important role in regulating the activity of hundreds of nuclear, cytoplasmic and plasma membrane proteins. In fact, nuclear pore proteins, governing trafficking into and out of the nucleus, are the largest group of O-GlcNAcylated cellular proteins (Hart et al., 1989). This places OGT in a key position to control transcription (Kreppel et al., 1997). In addition, the monosaccharide O-GlcNAc competes with phosphorylation at serine/threonine residues, affording OGT enormous potential to dynamically alter cell signaling.
OGT is located on the X chromosome (Shafi et al., 2000) and escapes X-inactivation in placental tissue in both rodents and humans, resulting in higher OGT expression and O-GlcNAclyation in female (XX) tissues, adding an additional level of importance to its role in sex-specific signaling (Howerton et al., 2013; Howerton and Bale, 2014).
Similar to that discussed above for mTOR, placental OGT regulates transplacental signals known to influence fetal growth and development, including for the brain (Howerton and Bale, 2014). Therefore, significant changes in placental OGT levels and function resulting from maternal exposures (e.g. to stress, immune activation or obesity) during pregnancy can have profound effects on the fetus (Howerton et al., 2013; Howerton and Bale, 2014; Nugent and Bale, 2015). For example, dynamic placental transcriptional changes are tightly controlled by OGT via its epigenetic stabilization of the histone methyltransferase EZH2 and trimethylation of histone H3 at lysine 27 (H3K27me3) (Fig. 4B) (Nugent et al., 2018). In fact, higher levels of OGT are associated with significantly increased H3K27me3 in female (XX) human and mouse placentas (Nugent et al., 2018). As a ubiquitous transcriptional repressor, H3K27me3 ensures that XX trophoblast cells are less dynamically responsive to changes in metabolic hormones arising from maternal stress, obesity, diabetes and infection than XY cells. This likely alters the type and amount of transplacental signals being relayed to the fetal compartment and contributes to sex differences in developmental programming of disease, especially related to neurodevelopmental disorders (Nugent and Bale, 2015; Bale, 2016; Ursini et al., 2018, 2023; Bronson and Bale, 2016). OGT also coordinates with other key transcription factors, including the glucocorticoid receptor, and cellular regulators, such as mTOR (as discussed above), to tightly control cellular processes related to changes in maternal nutrient status (Kelly et al., 2020; Briffa et al., 2017; Pantaleon et al., 2017). In response to maternal stress, glucocorticoids can have significant and lasting effects on OGT and its downstream functions, including placental transcriptional regulation and nutrient transport (Howerton et al., 2013; Howerton and Bale, 2014; Nugent and Bale, 2015; Briffa et al., 2017; Pantaleon et al., 2017). In mice, chronic stress experienced early in pregnancy (days 1-7) significantly reduced OGT levels in both male and female placentas throughout the remainder of pregnancy (Howerton et al., 2013). However, although this effect of stress occurs in both sexes, the outcome is more profound in male (XY) placentas, as the starting level of OGT is so much lower to begin with in males. Therefore, males may be more vulnerable in utero to perturbations of maternal stress as their placental OGT ‘levels of protection’ are so greatly diminished, falling below a threshold of vulnerability for the male fetus. Interestingly, in human placental tissue, similar fetal sex differences in OGT expression have been confirmed (Howerton et al., 2013; Nugent et al., 2018), where higher perceived maternal stress and maternal immune status were associated with altered placental OGT levels at delivery (Cowell et al., 2020; Shallie et al., 2023).
Maternal-placental-fetal crosstalk
It is well established that the placenta secretes hormones, such as human chorionic gonadotropin hormone, estrogens and progesterone, into the maternal circulation to mediate the maternal cardiovascular (Sanghavi and Rutherford, 2014) and metabolic (Zeng et al., 2017) adaptation to pregnancy. Emerging evidence suggests that, beyond these classical placental hormones, the placenta secretes a myriad of factors into the maternal and fetal circulations, which appear to be part of an active maternal-placental-fetal crosstalk. However, for most of these factors, their physiological function remains to be determined; examples of factors with known functions include plGF and sFLT1 (Degnes et al., 2022). Both of these proteins regulate angiogenesis: plGF acts as a pro-angiogenic factor by enhancing the binding of VEGF to the VEGF receptor-2 and sFLT1 is anti-angiogenic by binding to free VEGF and plGF in the maternal circulation, thus reducing their bioavailability for their membrane receptor.
Both term (Michelsen et al., 2019) and preterm (Schreiner et al., 2022) human placentas secrete an array of proteins of unknown function into the fetal circulation. Some of these proteins are predicted to be involved in the development of the brain, heart and lung and we speculate that, when born prematurely, infants are deprived of these important developmental regulating placental proteins, which may contribute to their poor outcomes (Schreiner et al., 2022).
Recent studies suggest that EVs are a primary mechanism by which the placenta secretes such factors. EVs are secreted from many cell types and contain bioactive molecules including proteins, messenger RNAs (mRNAs) and microRNAs (miRNAs). sEVs range from ∼50-150 nm in size, are produced from the late endosomal pathway and released into the extracellular compartment upon fusion of multivesicular bodies (MVBs) with the plasma membrane (Jeppesen et al., 2023). sEVs are believed to play a central role in cell-to-cell communication in normal physiology, including in pregnancy. The human placenta secretes sEVs into the maternal (Mincheva-Nilsson and Baranov, 2010) and fetal (Miranda et al., 2018; Chang et al., 2017) circulations. Although the physiological role of placental sEVs on fetal growth and development remains to be established, numerous physiological roles have been proposed for placental sEVs released into the maternal circulation, mostly based on in vitro studies. Currently, the strongest evidence suggests a role of placental sEVs regulating maternal glucose homeostasis in pregnancies with and without GDM. For example, GDM is associated with changes in the concentration and bioactivity of placenta-derived sEVs in maternal circulation across gestation (Salomon et al., 2016) and human placental sEVs in GDM carry a specific set of miRNAs associated with skeletal muscle insulin sensitivity (Nair et al., 2018). Moreover, healthy mice infused for 4 days with sEVs isolated from plasma of non-diabetic pregnant women became hyperinsulinemic owing to moderate peripheral insulin resistance and increased islet glucose-stimulated insulin secretion (James-Allan et al., 2020). In contrast, mice infused with sEVs isolated from plasma of women with GDM developed glucose intolerance due to marked peripheral insulin resistance and an inability to increase insulin secretion to maintain glucose tolerance (James-Allan et al., 2020). Thus, sEVs, including those from the placenta, may represent a mechanism regulating maternal glucose homeostasis in pregnancy and our data support the hypothesis that altered placental sEV content contributes to the development of GDM.
Recent work has identified placental OGT as an important regulator of EV secretion. Interestingly, OGT modifies annexin A1, a key protein involved in EV packaging and secretion. Consistent with the decreased levels of placental OGT in response to maternal stress, levels of O-GlycNAcylated annexin A1 were reduced by maternal stress in mice (Howerton et al., 2013). As a sensor for maternal glucose levels, placental OGT is positioned to dynamically alter the composition and the total number of EVs secreted into the maternal circulation (Zierden et al., 2023; Cui et al., 2023; Moore et al., 2021). For example, placental OGT promotes an increase in the number of EVs secreted into the maternal circulation in mice, and experimentally elevating circulating EV concentration via tail vein injection reduces the maximal rise in blood glucose in response to a glucose tolerance test (Zierden et al., 2023). Another model to study the influence of placental EVs uses a transgenic mouse model in which membrane-targeted, red fluorescent protein tdTomato and enhanced green fluorescent protein cyclic recombinase-reporter constructs are expressed only in fetal tissues (Sheller-Miller et al., 2019). This model allows monitoring of sEV trafficking across the placenta and the authors reported that sEVs can be trafficked from the mother to the fetus and from the fetus to the mother (Sheller-Miller et al., 2019). These findings were in line with a previous study using a transgenic mouse line with placental expression of the human C19 miRNA cluster (Chang et al., 2017). Because this miRNA cluster is expressed in the primate placenta only, this approach allowed the investigators to use C19 miRNA as a unique marker of sEVs secreted from the placenta and they demonstrated trafficking of C19 miRNA-carrying sEVs between maternal, placental and fetal compartments. The mechanisms by which sEVs are transported across the placental barrier and the physiological role of the sEV-mediated crosstalk between the maternal and fetal compartments remain largely unknown. Moreover, whether sEVs are trafficked between the maternal and fetal compartments across the placental barrier in humans has not yet been fully established.
Targeting the placenta to alleviate abnormal fetal development and programming
Impaired placental development often results in poor remodeling of uterine spiral arteries and inadequate increase in uteroplacental blood flow across gestation, which leads to placental insufficiency, characterized by complex, coordinated and highly regulated changes in placental signaling and nutrient transport capacity (Chassen and Jansson, 2020). Placental insufficiency is believed to result in or contribute to the development of a number of important pregnancy complications, such as IUGR, preeclampsia, stillbirth, placental abruption and premature birth (Pepe and Albrecht, 2021).
Because of potential risks to the mother and the developing fetus, only a handful of drugs are currently approved for use during pregnancy (Fisk and Atun, 2008). In rodent and sheep models, systemic adenoviral gene or protein therapy has been shown to improve utero-placental blood flow; however, the impact on human pregnancy is unknown (Li et al., 2007; Carr et al., 2016). These data, along with the current lack of drugs that can be used in pregnant people, have shifted research toward the development of delivery systems to specifically target the placenta using non-viral and viral vectors, nanoparticles, microbubbles and EVs. Viral vectors have been used in animal models for placental gene delivery. For example, in animal models of IUGR due to placental insufficiency, delivery of an IGF1 overexpression construct to the placenta using viral vectors has been shown to prevent the decreased fetal body, liver and musculoskeletal weight associated with IUGR (Jones et al., 2013, 2014). Similarly, another study examined the impact of gene delivery of VEGF using adenovirus vectors to the uterine artery in sheep pregnancies complicated by IUGR (Carr et al., 2014). VEGF gene therapy increased ultrasonographic fetal growth velocity at 3-4 weeks post-injection and decreased the number of severely growth restricted lambs close to term (Carr et al., 2014). Together, these studies demonstrate that viral vectors are potentially applicable in humans due to the targeted placental effects observed without adverse side effects (Pepe and Albrecht, 2021). Another technique developed involves affinity-based peptide targeting of nanoparticles. Pregnant mice injected with liposomes affixed with specific peptide sequences were found to be bound only to placental-derived cells and were absent from maternal and other fetal tissues (King et al., 2016). This method was used to deliver IGF2 to the placenta, which resulted in increased placental weights in wild-type animals and restored normal fetal weights in an IUGR model (King et al., 2016). More recently, nanoparticles have been engineered to directly target trophoblast cells; engineered nanoparticles are being used to investigate the effects of different molecules, such as methotrexate (a chemotherapy and immunosuppressive drug) and IGF1, and have demonstrated specificity for placental cells without affecting maternal or other fetal tissues (Pepe and Albrecht, 2021).
In addition, the use of microbubbles and ultrasound is being developed to allow for local release of drugs and may assist in intracellular drug delivery. This approach involves adding plasmid vectors to the cationic shells of lipid microbubble ultrasound contrast agents, 3-6 μm in size. These microbubbles are then administered intravenously. Delivery of the plasmids from the microbubbles in circulation is mediated through the application of low energy ultrasound, leading to microbubble cavitation and a transient membrane pore formation in the cells exposed to the microbubbles (Lentacker et al., 2014). These pores range in size from nanometers to several micrometers, allowing for the uptake of gene delivery vectors in highly targeted regions. Microbubbles are currently being used for visualization and identification of anatomical or blood flow related placental abnormalities (Roberts and Frias, 2020). However, other fields are using microbubbles to deliver their contents locally, as well as increase drug uptake at the ultrasound treated site (reviewed by Chowdhury et al., 2020; Jangjou et al., 2021). This drug delivery system has the potential to reduce the required dose of the administered drug and provide localized effects (Lentacker et al., 2014). These are just a few examples of how therapeutic cargo delivery techniques are currently being employed and research is ongoing to improve specific delivery of gene constructs and pharmacological agents to the placenta to alleviate abnormal placental and fetal development and the long-term consequences for the child.
Conclusions and future perspectives
What we have learned
Although the specific mechanisms involved remain to be fully established, major pregnancy complications such as preeclampsia, IUGR and stillbirth are believed to be caused by abnormal placental development and changes in placental function. Herein, with the support of compelling association studies in humans and emerging cause-and-effect studies in mice, we highlight the novel aspect that changes in placental function may also be mechanistically linked to the development of childhood and adult disease. Placental mTOR signaling responds to an array of maternal nutritional and metabolic signals to regulate placental function, programming offspring susceptibility for obesity and diabetes. Maternal adiponectin acts directly on the placenta to regulate insulin signaling and nutrient transport and serves as an endocrine link between maternal nutrition, fetal growth and offspring health. Inhibition of placental OGT constitutes a link between maternal stress, fetal hypothalamic gene expression and offspring neurocognitive development. Emerging evidence suggest that an array of factors, including proteins and sEVs containing miRNA and other signaling molecules, participate in an intense maternal-placental-fetal crosstalk that is not only essential for the physiological changes in maternal cardiometabolic adaptation to pregnancy, but may also be crucial for the normal development of specific fetal tissues. Thus, disruptions or modulations in the maternal-placental-fetal signaling may program the infant for future disease. We speculate that readouts of placental function may provide crucial information on which infants are at highest risk to develop adult disease and that interventions targeting placental function may offer opportunities to prevent cardiometabolic and neuropsychiatric disease in future generations.
What we need to understand better
One of the most urgent gaps of knowledge is the mechanisms underpinning the placental origins of disease, a major road-block to developing specific intervention strategies. This is an important area for future research requiring collaborative science involving a number of disciplines including developmental biologists, physiologists, molecular biologists, bioengineers and clinical scientists who care for pregnant people and their infants. Rigorous studies determining associations between changes in placental function and long-term outcomes in children, in combination with mechanistic studies in animals using approaches for placenta-specific gene targeting, will move the field forward. We speculate that there are multiple mechanisms mediating the link between changes in placental function and fetal programming of adult disease, including proteins and sEVs released into the fetal circulation that modulate the development and function of specific fetal tissues. It is also imperative that we further develop minimally invasive techniques and approaches that effectively deliver gene constructs, miRNA, proteins and pharmacological agents specifically to the placenta without negative effects on the mother or direct effects on the fetus. These challenges need to be addressed to develop treatments for major pregnancy complications and to alleviate the growing epidemic of cardiometabolic and neuropsychiatric disorders likely resulting from in utero developmental programming.
Acknowledgements
We thank Kimen Design4Research for developing the figures for this paper.
Footnotes
Funding
Supported by grants from the National Institutes of Health [HD065007, HD068370, HD105701, HD104644 and HD097093]. Deposited in PMC for release after 12 months.
References
- Acosta, O., Ramirez, V. I., Lager, S., Gaccioli, F., Dudley, D. J., Powell, T. L. and Jansson, T. (2015). Increased glucose and placental GLUT-1 in large infants of obese nondiabetic mothers. Am. J. Obstet. Gynecol. 212, e1-e7. 10.1016/j.ajog.2014.08.009 [DOI] [PubMed] [Google Scholar]
- Adamson, S. L., Lu, Y., Whiteley, K. J., Holmyard, D., Hemberger, M., Pfarrer, C. and Cross, J. C. (2002). Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev. Biol. 250, 358-373. 10.1006/dbio.2002.0773 [DOI] [PubMed] [Google Scholar]
- Akhaphong, B., Baumann, D. C., Beetch, M., Lockridge, A. D., Jo, S., Wong, A., Zemanovic, T., Mohan, R., Fondevilla, D. L., Sia, M.et al. (2021). Placental mTOR complex 1 regulates fetal programming of obesity and insulin resistance in mice. JCI Insight 6, e149271. 10.1172/jci.insight.149271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye, I. L., Gao, X., Weintraub, S. T., Jansson, T. and Powell, T. L. (2014). Adiponectin inhibits insulin function in primary trophoblasts by PPARα-mediated ceramide synthesis. Mol. Endocrinol. 28, 512-524. 10.1210/me.2013-1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye, I. L., Rosario, F. J., Powell, T. L. and Jansson, T. (2015). Adiponectin supplementation in pregnant mice prevents the adverse effects of maternal obesity on placental function and fetal growth. Proc. Natl. Acad. Sci. USA 112, 12858-12863. 10.1073/pnas.1515484112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale, T. L. (2016). The placenta and neurodevelopment: sex differences in prenatal vulnerability. Dialogues Clin. Neurosci. 18, 459-464. 10.31887/DCNS.2016.18.4/tbale [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker, D. J. P., Gelow, J., Thornburg, K., Osmond, C., Kajantie, E. and Eriksson, J. G. (2010a). The early origins of chronic heart failure: impaired placental growth and initiation of insulin resistance in childhood. Eur. J. Heart Fail 12, 819-825. 10.1093/eurjhf/hfq069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker, D. J. P., Thornburg, K. L., Osmond, C., Kajantie, E. and Eriksson, J. G. (2010b). The surface area of the placenta and hypertension in the offspring in later life. Int. J. Dev. Biol. 54, 525-530. 10.1387/ijdb.082760db [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker, D. J., Larsen, G., Osmond, C., Thornburg, K. L., Kajantie, E. and Eriksson, J. G. (2012). The placental origins of sudden cardiac death. Int. J. Epidemiol. 41, 1394-1399. 10.1093/ije/dys116 [DOI] [PubMed] [Google Scholar]
- Boehmer, B. H., Limesand, S. W. and Rozance, P. J. (2017). The impact of IUGR on pancreatic islet development and beta-cell function. J. Endocrinol. 235, R63-R76. 10.1530/JOE-17-0076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briffa, J. F., Hosseini, S. S., Tran, M., Moritz, K. M., Cuffe, J. S. M. and Wlodek, M. E. (2017). Maternal growth restriction and stress exposure in rats differentially alters expression of components of the placental glucocorticoid barrier and nutrient transporters. Placenta 59, 30-38. 10.1016/j.placenta.2017.09.006 [DOI] [PubMed] [Google Scholar]
- Bronson, S. L. and Bale, T. L. (2016). The placenta as a mediator of stress effects on neurodevelopmental reprogramming. Neuropsychopharmacology 41, 207-218. 10.1038/npp.2015.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M. J., Cook, C. L., Henry, J. L. and Schultz, G. S. (1987). Levels of epidermal growth factor binding in third-trimester and term human placentas: elevated binding in term placentas of male fetuses. Am. J. Obstet. Gynecol. 156, 716-720. 10.1016/0002-9378(87)90085-8 [DOI] [PubMed] [Google Scholar]
- Burton, G. J., Charnock-Jones, D. S. and Jauniaux, E. (2009). Regulation of vascular growth and function in the human placenta. Reproduction 138, 895-902. 10.1530/REP-09-0092 [DOI] [PubMed] [Google Scholar]
- Burton, G. J., Fowden, A. L. and Thornburg, K. L. (2016). Placental origins of chronic disease. Physiol. Rev. 96, 1509-1565. 10.1152/physrev.00029.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butkinaree, C., Park, K. and Hart, G. W. (2010). O-linked beta-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim. Biophys. Acta 1800, 96-106. 10.1016/j.bbagen.2009.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr, D. J., Wallace, J. M., Aitken, R. P., Milne, J. S., Mehta, V., Martin, J. F., Zachary, I. C., Peebles, D. M. and David, A. L. (2014). Uteroplacental adenovirus vascular endothelial growth factor gene therapy increases fetal growth velocity in growth-restricted sheep pregnancies. Hum. Gene. Ther. 25, 375-384. 10.1089/hum.2013.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr, D. J., Wallace, J. M., Aitken, R. P., Milne, J. S., Martin, J. F., Zachary, I. C., Peebles, D. M. and David, A. L. (2016). Peri- and postnatal effects of prenatal adenoviral VEGF gene therapy in growth-restricted sheep. Biol. Reprod. 94, 142. 10.1095/biolreprod.115.133744 [DOI] [PubMed] [Google Scholar]
- Carter, A. M. (2007). Animal models of human placentation--a review. Placenta 28 Suppl. A, S41-S47. 10.1016/j.placenta.2006.11.002 [DOI] [PubMed] [Google Scholar]
- Catalano, P. M., Mcintyre, H. D., Cruickshank, J. K., Mccance, D. R., Dyer, A. R., Metzger, B. E., Lowe, L. P., Trimble, E. R., Coustan, D. R., Hadden, D. R.et al. (2012). The hyperglycemia and adverse pregnancy outcome study: associations of GDM and obesity with pregnancy outcomes. Diabetes Care 35, 780-786. 10.2337/dc11-1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, G., Mouillet, J. F., Mishima, T., Chu, T., Sadovsky, E., Coyne, C. B., Parks, W. T., Surti, U. and Sadovsky, Y. (2017). Expression and trafficking of placental microRNAs at the feto-maternal interface. FASEB J. 31, 2760-2770. 10.1096/fj.201601146R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassen, S. and Jansson, T. (2020). Complex, coordinated and highly regulated changes in placental signaling and nutrient transport capacity in IUGR. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165373. 10.1016/j.bbadis.2018.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassen, S. S., Ferchaud-Roucher, V., Gupta, M. B., Jansson, T. and Powell, T. L. (2018). Alterations in placental long chain polyunsaturated fatty acid metabolism in human intrauterine growth restriction. Clin. Sci. (Lond.) 132, 595-607. 10.1042/CS20171340 [DOI] [PubMed] [Google Scholar]
- Chau, K., Hennessy, A. and Makris, A. (2017). Placental growth factor and pre-eclampsia. J. Hum. Hypertens. 31, 782-786. 10.1038/jhh.2017.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. Q., Cheung, L. S., Feng, L., Tanner, W. and Frommer, W. B. (2015a). Transport of sugars. Annu. Rev. Biochem. 84, 865-894. 10.1146/annurev-biochem-060614-033904 [DOI] [PubMed] [Google Scholar]
- Chen, Y. Y., Rosario, F. J., Shehab, M. A., Powell, T. L., Gupta, M. B. and Jansson, T. (2015b). Increased ubiquitination and reduced plasma membrane trafficking of placental amino acid transporter SNAT-2 in human IUGR. Clin. Sci. (Lond.) 129, 1131-1141. 10.1042/CS20150511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. and Natarajan, R. (2022). Epigenetic modifications in metabolic memory: What are the memories. and can we erase them? Am. J. Physiol. Cell Physiol. 323, C570-c582. 10.1152/ajpcell.00201.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury, S. M., Abou-Elkacem, L., Lee, T., Dahl, J. and Lutz, A. M. (2020). Ultrasound and microbubble mediated therapeutic delivery: Underlying mechanisms and future outlook. J. Control. Release 326, 75-90. 10.1016/j.jconrel.2020.06.008 [DOI] [PubMed] [Google Scholar]
- Cleal, J. K., Glazier, J. D., Ntani, G., Crozier, S. R., Day, P. E., Harvey, N. C., Robinson, S. M., Cooper, C., Godfrey, K. M., Hanson, M. A.et al. (2011). Facilitated transporters mediate net efflux of amino acids to the fetus across the basal membrane of the placental syncytiotrophoblast. J. Physiol. 589, 987-997. 10.1113/jphysiol.2010.198549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton, V. L. (2010). Review: Sex and the human placenta: mediating differential strategies of fetal growth and survival. Placenta 31 Suppl, S33-S39. 10.1016/j.placenta.2009.11.010 [DOI] [PubMed] [Google Scholar]
- Cowell, W., Deyssenroth, M., Chen, J. and Wright, R. J. (2020). Maternal stress in relation to sex-specific expression of placental genes involved in nutrient transport, oxygen tension, immune response, and the glucocorticoid barrier. Placenta 96, 19-26. 10.1016/j.placenta.2020.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, B., Kotlyar, M., Evangelou, A. I., Ignatchenko, V., Ignatchenko, A., Whiteley, K., Jurisica, I., Adamson, S. L., Rossant, J. and Kislinger, T. (2009). Comparative systems biology of human and mouse as a tool to guide the modeling of human placental pathology. Mol. Syst. Biol. 5, 279. 10.1038/msb.2009.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, Y., Cruz, M., Palatnik, A. and Olivier-Van, S. (2023). O-GlcNAc transferase contributes to sex-specific placental deregulation in gestational diabetes. Placenta 131, 1-12. 10.1016/j.placenta.2022.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvitic, S., Longtine, M. S., Hackl, H., Wagner, K., Nelson, M. D., Desoye, G. and Hiden, U. (2013). The human placental sexome differs between trophoblast epithelium and villous vessel endothelium. PLoS One 8, e79233. 10.1371/journal.pone.0079233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabelea, D., Mayer-Davis, E. J., Lamichhane, A. P., D'agostino, R. B., Jr, Liese, A. D., Vehik, K. S., Narayan, K. M., Zeitler, P. and Hamman, R. F. (2008). Association of intrauterine exposure to maternal diabetes and obesity with type 2 diabetes in youth: the SEARCH Case-Control Study. Diabetes Care 31, 1422-1426. 10.2337/dc07-2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnes, M. L., Westerberg, A. C., Zucknick, M., Powell, T. L., Jansson, T., Henriksen, T., Roland, M. C. P. and Michelsen, T. M. (2022). Placenta-derived proteins across gestation in healthy pregnancies-a novel approach to assess placental function? BMC Med. 20, 227. 10.1186/s12916-022-02415-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimasuay, K. G., Aitken, E. H., Rosario, F., Njie, M., Glazier, J., Rogerson, S. J., Fowkes, F. J., Beeson, J. G., Powell, T., Jansson, T.et al. (2017). Inhibition of placental mTOR signaling provides a link between placental malaria and reduced birthweight. BMC Med. 15, 1. 10.1186/s12916-016-0759-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumolt, J. H., Powell, T. L. and Jansson, T. (2021). Placental function and the development of fetal overgrowth and fetal growth restriction. Obstet. Gynecol. Clin. North Am. 48, 247-266. 10.1016/j.ogc.2021.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumolt, J., Powell, T. L., Jansson, T. and Rosario, F. J. (2022). Normalization of maternal adiponectin in obese pregnant mice prevents programming of impaired glucose metabolism in adult offspring. FASEB J. 36, e22383. 10.1096/fj.202200326R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttaroy, A. K. and Basak, S. (2021). Maternal fatty acid metabolism in pregnancy and its consequences in the feto-placental development. Front. Physiol. 12, 787848. 10.3389/fphys.2021.787848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Twinn, D. S., Hjort, L., Novakovic, B., Ozanne, S. E. and Saffery, R. (2019). Intrauterine programming of obesity and type 2 diabetes. Diabetologia 62, 1789-1801. 10.1007/s00125-019-4951-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk, N. M. and Atun, R. (2008). Market failure and the poverty of new drugs in maternal health. PLoS Med. 5, e22. 10.1371/journal.pmed.0050022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaccioli, F., Lager, S., Powell, T. L. and Jansson, T. (2013). Placental transport in response to altered maternal nutrition. J. Dev. Orig. Health Dis. 4, 101-115. 10.1017/S2040174412000529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaccioli, F., Aye, I. L., Roos, S., Lager, S., Ramirez, V. I., Kanai, Y., Powell, T. L. and Jansson, T. (2015). Expression and functional characterisation of System L amino acid transporters in the human term placenta. Reprod. Biol. Endocrinol. 13, 57. 10.1186/s12958-015-0054-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Santillan, J. A., Lazo-De-La-Vega-Monroy, M. L., Rodriguez-Saldaña, G. C., Solis-Barbosa, M. A., Corona-Figueroa, M. A., Gonzalez-Dominguez, M. I., Gomez-Zapata, H. M., Malacara, J. M. and Barbosa-Sabanero, G. (2022). Placental nutrient transporters and maternal fatty acids in SGA, AGA, and LGA newborns from mothers with and without obesity. Front. Cell Dev. Biol. 10, 822527. 10.3389/fcell.2022.822527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades, P., Ferguson-Smith, A. C. and Burton, G. J. (2002). Comparative developmental anatomy of the murine and human definitive placentae. Placenta 23, 3-19. 10.1053/plac.2001.0738 [DOI] [PubMed] [Google Scholar]
- Gluckman, P. D., Hanson, M. A., Cooper, C. and Thornburg, K. L. (2008). Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 359, 61-73. 10.1056/NEJMra0708473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordijn, S. J., Beune, I. M., Thilaganathan, B., Papageorghiou, A., Baschat, A. A., Baker, P. N., Silver, R. M., Wynia, K. and Ganzevoort, W. (2016). Consensus definition of fetal growth restriction: a Delphi procedure. Ultrasound Obstet. Gynecol. 48, 333-339. 10.1002/uog.15884 [DOI] [PubMed] [Google Scholar]
- Hart, G. W., Haltiwanger, R. S., Holt, G. D. and Kelly, W. G. (1989). Glycosylation in the nucleus and cytoplasm. Annu. Rev. Biochem. 58, 841-874. 10.1146/annurev.bi.58.070189.004205 [DOI] [PubMed] [Google Scholar]
- Hemberger, M., Hanna, C. W. and Dean, W. (2020). Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 21, 27-43. 10.1038/s41576-019-0169-4 [DOI] [PubMed] [Google Scholar]
- Howerton, C. L. and Bale, T. L. (2014). Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 111, 9639-9644. 10.1073/pnas.1401203111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howerton, C. L., Morgan, C. P., Fischer, D. B. and Bale, T. L. (2013). O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc. Natl. Acad. Sci. USA 110, 5169-5174. 10.1073/pnas.1300065110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X., Anderle, P., Hostettler, L., Baumann, M. U., Surbek, D. V., Ontsouka, E. C. and Albrecht, C. (2018). Identification of placental nutrient transporters associated with intrauterine growth restriction and pre-eclampsia. BMC Genomics 19, 173. 10.1186/s12864-018-4518-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull, H. R., Dinger, M. K., Knehans, A. W., Thompson, D. M. and Fields, D. A. (2008). Impact of maternal body mass index on neonate birthweight and body composition. Am. J. Obstet. Gynecol. 198, 416.e1-6. 10.1016/j.ajog.2007.10.79 [DOI] [PubMed] [Google Scholar]
- Hung, T. H., Hsieh, T. T., Wu, C. P., Li, M. J., Yeh, Y. L. and Chen, S. F. (2017). Mammalian target of rapamycin signaling is a mechanistic link between increased endoplasmic reticulum stress and autophagy in the placentas of pregnancies complicated by growth restriction. Placenta 60, 9-20. 10.1016/j.placenta.2017.10.001 [DOI] [PubMed] [Google Scholar]
- Ives, C. W., Sinkey, R., Rajapreyar, I., Tita, A. T. N. and Oparil, S. (2020). Preeclampsia-pathophysiology and clinical presentations: JACC state-of-the-art review. J. Am. Coll. Cardiol. 76, 1690-1702. 10.1016/j.jacc.2020.08.014 [DOI] [PubMed] [Google Scholar]
- James-Allan, L. B., Rosario, F. J., Barner, K., Lai, A., Guanzon, D., Mcintyre, H. D., Lappas, M., Powell, T. L., Salomon, C. and Jansson, T. (2020). Regulation of glucose homeostasis by small extracellular vesicles in normal pregnancy and in gestational diabetes. FASEB J. 34, 5724-5739. 10.1096/fj.201902522RR [DOI] [PubMed] [Google Scholar]
- Jangjou, A., Meisami, A. H., Jamali, K., Niakan, M. H., Abbasi, M., Shafiee, M., Salehi, M., Hosseinzadeh, A., Amani, A. M. and Vaez, A. (2021). The promising shadow of microbubble over medical sciences: from fighting wide scope of prevalence disease to cancer eradication. J. Biomed. Sci. 28, 49. 10.1186/s12929-021-00744-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson, T. and Powell, T. L. (2007). Role of the placenta in fetal programming: underlying mechanisms and potential interventional appraoches. Clin. Sci. 113, 1-13. 10.1042/CS20060339 [DOI] [PubMed] [Google Scholar]
- Jansson, T. and Powell, T. L. (2013). Role of placental nutrient sensing in developmental programming. Clin. Obstet. Gynecol. 56, 591-601. 10.1097/GRF.0b013e3182993a2e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson, T., Wennergren, M. and Illsley, N. P. (1993). Glucose transporter protein expression in human placenta throughout gestation and in intrauterine growth retardation. J. Clin. Endocrinol. Metab. 77, 1554-1562. 10.1210/jcem.77.6.8263141 [DOI] [PubMed] [Google Scholar]
- Jansson, T., Ekstrand, Y., Björn, C., Wennergren, M. and Powell, T. L. (2002a). Alterations in the activity of placental amino acid transporters in pregnancies complicated by diabetes. Diabetes 51, 2214-2219. 10.2337/diabetes.51.7.2214 [DOI] [PubMed] [Google Scholar]
- Jansson, T., Ylvén, K., Wennergren, M. and Powell, T. L. (2002b). Glucose transport and system A activity in syncytiotrophoblast microvillous and basal membranes in intrauterine growth restriction. Placenta 23, 392-399. 10.1053/plac.2002.0826 [DOI] [PubMed] [Google Scholar]
- Jansson, N., Nilsfelt, A., Gellerstedt, M., Wennergren, M., Rossander-Hulthén, L., Powell, T. L. and Jansson, T. (2008). Maternal hormones linking maternal body mass index and dietary intake to birth weight. Am. J. Clin. Nutr. 87, 1743-1749. 10.1093/ajcn/87.6.1743 [DOI] [PubMed] [Google Scholar]
- Jansson, T., Myatt, L. and Powell, T. L. (2009). The role of trophoblast nutrient and ion transporters in the development of pregnancy complications and adult disease. Curr. Vasc. Pharmacol. 7, 521-533. 10.2174/157016109789043982 [DOI] [PubMed] [Google Scholar]
- Jansson, N., Rosario, F. J., Gaccioli, F., Lager, S., Jones, H. N., Roos, S., Jansson, T. and Powell, T. L. (2013). Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J. Clin. Endocrinol. Metab. 98, 105-113. 10.1210/jc.2012-2667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen, D. K., Zhang, Q., Franklin, J. L. and Coffey, R. J. (2023). Extracellular vesicles and nanoparticles: emerging complexities. Trends Cell Biol. 33, 667-681. 10.1016/j.tcb.2023.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, J. and Menon, R. (2018). Placental exosomes: a proxy to understand pregnancy complications. Am. J. Reprod. Immunol. 79, e12788. 10.1111/aji.12788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson, M., Glazier, J. D., Sibley, C. P., Jansson, T. and Powell, T. L. (2002). Activity and protein expression of the Na+/H+ exchanger is reduced in syncytiotrophoblast microvillous plasma membranes isolated from preterm intrauterine growth restriction pregnancies. J. Clin. Endocrinol. Metab. 87, 5686-5694. 10.1210/jc.2002-020214 [DOI] [PubMed] [Google Scholar]
- Jones, H. N., Crombleholme, T. and Habli, M. (2013). Adenoviral-mediated placental gene transfer of IGF-1 corrects placental insufficiency via enhanced placental glucose transport mechanisms. PLoS One 8, e74632. 10.1371/journal.pone.0074632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, H. N., Crombleholme, T. and Habli, M. (2014). Regulation of amino acid transporters by adenoviral-mediated human insulin-like growth factor-1 in a mouse model of placental insufficiency in vivo and the human trophoblast line BeWo in vitro. Placenta 35, 132-138. 10.1016/j.placenta.2013.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavitha, J. V., Rosario, F. J., Nijland, M. J., Mcdonald, T. J., Wu, G., Kanai, Y., Powell, T. L., Nathanielsz, P. W. and Jansson, T. (2014). Down-regulation of placental mTOR, insulin/IGF-I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. FASEB J. 28, 1294-1305. 10.1096/fj.13-242271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keleher, M. R., Erickson, K., Kechris, K., Yang, I. V., Dabelea, D., Friedman, J. E., Boyle, K. E. and Jansson, T. (2020). Associations between the activity of placental nutrient-sensing pathways and neonatal and postnatal metabolic health: the ECHO Healthy Start cohort. Int. J. Obes. (Lond 44, 2203-2212. 10.1038/s41366-020-0574-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keleher, M. R., Erickson, K., Smith, H. A., Kechris, K. J., Yang, I. V., Dabelea, D., Friedman, J. E., Boyle, K. E. and Jansson, T. (2021). Placental insulin/IGF-1 signaling, PGC-1alpha, and inflammatory pathways are associated with metabolic outcomes at 4–6 years of age: the ECHO healthy start cohort. Diabetes 70, 745-751. 10.2337/db20-0902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, A. C., Powell, T. L. and Jansson, T. (2020). Placental function in maternal obesity. Clin. Sci. (Lond.) 134, 961-984. 10.1042/CS20190266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khambadkone, S. G., Cordner, Z. A. and Tamashiro, K. L. K. (2020). Maternal stressors and the developmental origins of neuropsychiatric risk. Front. Neuroendocrinol. 57, 100834. 10.1016/j.yfrne.2020.100834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, A., Ndifon, C., Lui, S., Widdows, K., Kotamraju, V. R., Agemy, L., Teesalu, T., Glazier, J. D., Cellesi, F., Tirelli, N.et al. (2016). Tumor-homing peptides as tools for targeted delivery of payloads to the placenta. Sci. Adv. 2, e1600349. 10.1126/sciadv.1600349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreppel, L. K., Blomberg, M. A. and Hart, G. W. (1997). Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J. Biol. Chem. 272, 9308-9315. 10.1074/jbc.272.14.9308 [DOI] [PubMed] [Google Scholar]
- Kuruvilla, A. G., D'souza, S. W., Glazier, J. D., Mahendran, D., Maresh, M. J. and Sibley, C. P. (1994). Altered activity of the system A amino acid transporter in microvillous membrane vesicles from placentas of macrosomic babies born to diabetic women. J. Clin. Invest 94, 689-695. 10.1172/JCI117386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lager, S., Gaccioli, F., Ramirez, V. I., Jones, H. N., Jansson, T. and Powell, T. L. (2013). Oleic acid stimulates system A amino acid transport in primary human trophoblast cells mediated by toll-like receptor 4. J. Lipid Res. 54, 725-733. 10.1194/jlr.M033050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lager, S. and Powell, T. L. (2012). Regulation of nutrient transport across the placenta. J. Pregnancy 2012, 179827. 10.1155/2012/179827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lager, S., Ramirez, V. I., Gaccioli, F., Jang, B., Jansson, T. and Powell, T. L. (2016). Protein expression of fatty acid transporter 2 is polarized to the trophoblast basal plasma membrane and increased in placentas from overweight/obese women. Placenta 40, 60-66. 10.1016/j.placenta.2016.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, M. A., Kimura, K., Kubisch, H. M. and Roberts, R. M. (2001). Sexual dimorphism among bovine embryos in their ability to make the transition to expanded blastocyst and in the expression of the signaling molecule IFN-tau. Proc. Natl. Acad. Sci. USA 98, 9677-9682. 10.1073/pnas.171305398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen, L., Brambilla, P., Mazzocchi, A., Harsløf, L. B., Ciappolino, V. and Agostoni, C. (2016). DHA effects in brain development and function. Nutrients 8, 6. 10.3390/nu8010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentacker, I., De Cock, I., Deckers, R., De Smedt, S. C. and Moonen, C. T. (2014). Understanding ultrasound induced sonoporation: definitions and underlying mechanisms. Adv. Drug Deliv. Rev. 72, 49-64. 10.1016/j.addr.2013.11.008 [DOI] [PubMed] [Google Scholar]
- Levine, R. J., Lam, C., Qian, C., Yu, K. F., Maynard, S. E., Sachs, B. P., Sibai, B. M., Epstein, F. H., Romero, R., Thadhani, R.et al. (2006). Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N. Engl. J. Med. 355, 992-1005. 10.1056/NEJMoa055352 [DOI] [PubMed] [Google Scholar]
- Li, Z., Zhang, Y., Ying Ma, J., Kapoun, A. M., Shao, Q., Kerr, I., Lam, A., O'young, G., Sannajust, F., Stathis, P.et al. (2007). Recombinant vascular endothelial growth factor 121 attenuates hypertension and improves kidney damage in a rat model of preeclampsia. Hypertension 50, 686-692. 10.1161/HYPERTENSIONAHA.107.092098 [DOI] [PubMed] [Google Scholar]
- Li, Y., Lorca, R. A. and Su, E. J. (2018). Molecular and cellular underpinnings of normal and abnormal human placental blood flows. J. Mol. Endocrinol. 60, R9-r22. 10.1530/JME-17-0139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria, O., Bar, J., Barnea, O., Golan, A. and Kovo, M. (2012). Reactivity of blood vessels in response to prostaglandin E2 in placentas from pregnancies complicated by fetal growth restriction. Prenat. Diagn. 32, 417-422. 10.1002/pd.3827 [DOI] [PubMed] [Google Scholar]
- Magnusson, A. L., Waterman, I. J., Wennergren, M., Jansson, T. and Powell, T. L. (2004). Triglyceride hydrolase activities and expression of fatty acid binding proteins in human placenta in pregnancies complicated by IUGR and diabetes. J. Clin. Endocrinol. Metab. 89, 4607-4614. 10.1210/jc.2003-032234 [DOI] [PubMed] [Google Scholar]
- Mahendran, D., Donnai, P., Glazier, J. D., D'souza, S. W., Boyd, R. D. and Sibley, C. P. (1993). Amino acid (system A) transporter activity in microvillous membrane vesicles from the placentas of appropriate and small for gestational age babies. Pediatr. Res. 34, 661-665. 10.1203/00006450-199311000-00019 [DOI] [PubMed] [Google Scholar]
- Michelsen, T. M., Henriksen, T., Reinhold, D., Powell, T. L. and Jansson, T. (2019). The human placental proteome secreted into the maternal and fetal circulations in normal pregnancy based on 4-vessel sampling. FASEB J. 33, 2944-2956. 10.1096/fj.201801193R [DOI] [PubMed] [Google Scholar]
- Mincheva-Nilsson, L. and Baranov, V. (2010). The role of placental exosomes in reproduction. Am. J. Reprod. Immunol. 63, 520-533. 10.1111/j.1600-0897.2010.00822.x [DOI] [PubMed] [Google Scholar]
- Miranda, J., Paules, C., Nair, S., Lai, A., Palma, C., Scholz-Romero, K., Rice, G. E., Gratacos, E., Crispi, F. and Salomon, C. (2018). Placental exosomes profile in maternal and fetal circulation in intrauterine growth restriction - Liquid biopsies to monitoring fetal growth. Placenta 64, 34-43. 10.1016/j.placenta.2018.02.006 [DOI] [PubMed] [Google Scholar]
- Mohamad, M., Loy, S. L., Lim, P. Y., Wang, Y., Soo, K. L. and Mohamed, H. J. J. (2018). Maternal serum and breast milk adiponectin: the association with infant adiposity development. Int. J. Environ. Res. Public Health 15, 1250. 10.3390/ijerph15061250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, M., Avula, N., Jo, S., Beetch, M. and Alejandro, E. U. (2021). Disruption of O-Linked N-acetylglucosamine signaling in placenta induces insulin sensitivity in female offspring. Int. J. Mol. Sci. 22, 6918. 10.3390/ijms22136918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myatt, L. (2006). Placental adaptive responses and fetal programming. J. Physiol. 572, 25-30. 10.1113/jphysiol.2006.104968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair, S., Jayabalan, N., Guanzon, D., Palma, C., Scholz-Romero, K., Elfeky, O., Zuñiga, F., Ormazabal, V., Diaz, E., Rice, G. E.et al. (2018). Human placental exosomes in gestational diabetes mellitus carry a specific set of miRNAs associated with skeletal muscle insulin sensitivity. Clin. Sci. (Lond.) 132, 2451-2467. 10.1042/CS20180487 [DOI] [PubMed] [Google Scholar]
- Nugent, B. M. and Bale, T. L. (2015). The omniscient placenta: metabolic and epigenetic regulation of fetal programming. Front. Neuroendocrinol. 39, 28-37. 10.1016/j.yfrne.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent, B. M., O'donnell, C. M., Epperson, C. N. and Bale, T. L. (2018). Placental H3K27me3 establishes female resilience to prenatal insults. Nat. Commun. 9, 2555. 10.1038/s41467-018-04992-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleon, M., Steane, S. E., Mcmahon, K., Cuffe, J. S. M. and Moritz, K. M. (2017). Placental O-GlcNAc-transferase expression and interactions with the glucocorticoid receptor are sex specific and regulated by maternal corticosterone exposure in mice. Sci. Rep. 7, 2017. 10.1038/s41598-017-01666-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolini, C. L., Meschia, G., Fennessey, P. V., Pike, A. W., Teng, C., Battaglia, F. C. and Wilkening, R. B. (2001). An in vivo study of ovine placental transport of essential amino acids. Am. J. Physiol. Endocrinol. Metab. 280, E31-E39. 10.1152/ajpendo.2001.280.1.E31 [DOI] [PubMed] [Google Scholar]
- Paulsen, M. E., Rosario, F. J., Wesolowski, S. R., Powell, T. L. and Jansson, T. (2019). Normalizing adiponectin levels in obese pregnant mice prevents adverse metabolic outcomes in offspring. FASEB J. 33, 2899-2909. 10.1096/fj.201801015R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrioli, G., Piovesana, E., Vacchi, E. and Balbi, C. (2021). Extracellular vesicles as promising carriers in drug delivery: considerations from a cell biologist's perspective. Biology (Basel) 10, 376. 10.3390/biology10050376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepe, G. J. and Albrecht, E. D. (2021). Novel technologies for target delivery of therapeutics to the placenta during pregnancy: a review. Genes (Basel) 12, 280-304. 10.3390/genes12020280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzagalli, M. D., Bensimon, A. and Superti-Furga, G. (2021). A guide to plasma membrane solute carrier proteins. FEBS J. 288, 2784-2835. 10.1111/febs.15531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, L., Wattez, J. S., Lee, S., Guo, Z., Schaack, J., Hay, W. W., Jr, Zita, M. M., Parast, M. and Shao, J. (2016). Knockout maternal adiponectin increases fetal growth in mice: potential role for trophoblast IGFBP-1. Diabetologia 59, 2417-2425. 10.1007/s00125-016-4061-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, V. H. and Frias, A. E. (2020). Contrast-enhanced ultrasound for the assessment of placental development and function. BioTechniques 69, 392-399. 10.2144/btn-2020-0069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos, S., Lagerlöf, O., Wennergren, M., Powell, T. L. and Jansson, T. (2009). Regulation of amino acid transporters by glucose and growth factors in cultured primary human trophoblast cells is mediated by mTOR signaling. Am. J. Physiol. Cell Physiol. 297, C723-C731. 10.1152/ajpcell.00191.2009 [DOI] [PubMed] [Google Scholar]
- Rosario, F. J., Chopra, A., Biggar, K., Powell, T. L., Gupta, M. B. and Jansson, T. (2023). Placental remote control of fetal metabolism: trophoblast mTOR signaling regulates liver IGFBP-1 phosphorylation and IGF-1 bioavailability. Int. J. Mol. Sci. 24, 7273. 10.3390/ijms24087273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario, F. J., Jansson, N., Kanai, Y., Prasad, P. D., Powell, T. L. and Jansson, T. (2011). Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down-regulates placental amino acid transporters. Endocrinology 152, 1119-1129. 10.1210/en.2010-1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario, F. J., Schumacher, M. A., Jiang, J., Kanai, Y., Powell, T. L. and Jansson, T. (2012). Chronic maternal infusion of full-length adiponectin in pregnant mice down-regulates placental amino acid transporter activity and expression and decreases fetal growth. J. Physiol. 590, 1495-1509. 10.1113/jphysiol.2011.226399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario, F. J., Kanai, Y., Powell, T. L. and Jansson, T. (2013). Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J. Physiol. 591, 609-625. 10.1113/jphysiol.2012.238014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario, F. J., Kanai, Y., Powell, T. L. and Jansson, T. (2015). Increased placental nutrient transport in a novel mouse model of maternal obesity with fetal overgrowth. Obesity (Silver Spring) 23, 1663-1670. 10.1002/oby.21165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario, F. J., Dimasuay, K. G., Kanai, Y., Powell, T. L. and Jansson, T. (2016a). Regulation of amino acid transporter trafficking by mTORC1 in primary human trophoblast cells is mediated by the ubiquitin ligase Nedd4-2. Clin. Sci. (Lond.) 130, 499-512. 10.1042/CS20150554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario, F. J., Powell, T. L. and Jansson, T. (2016b). Mechanistic target of rapamycin (mTOR) regulates trophoblast folate uptake by modulating the cell surface expression of FR-α and the RFC. Sci. Rep. 6, 31705. 10.1038/srep31705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario, F. J., Powell, T. L. and Jansson, T. (2017). mTOR folate sensing links folate availability to trophoblast cell function. J. Physiol. 595, 4189-4206. 10.1113/JP272424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario, F. J., Gupta, M. B., Myatt, L., Powell, T. L., Glenn, J. P., Cox, L. and Jansson, T. (2019). Mechanistic target of rapamycin complex 1 promotes the expression of genes encoding electron transport chain proteins and stimulates oxidative phosphorylation in primary human trophoblast cells by regulating mitochondrial biogenesis. Sci. Rep. 9, 246. 10.1038/s41598-018-36265-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant, J. and Cross, J. C. (2001). Placental development: lessons from mouse mutants. Nat. Rev. Genet. 2, 538-548. 10.1038/35080570 [DOI] [PubMed] [Google Scholar]
- Salomon, C., Scholz-Romero, K., Sarker, S., Sweeney, E., Kobayashi, M., Correa, P., Longo, S., Duncombe, G., Mitchell, M. D., Rice, G. E.et al. (2016). Gestational diabetes mellitus is associated with changes in the concentration and bioactivity of placenta-derived exosomes in maternal circulation across gestation. Diabetes 65, 598-609. 10.2337/db15-0966 [DOI] [PubMed] [Google Scholar]
- Sanghavi, M. and Rutherford, J. D. (2014). Cardiovascular physiology of pregnancy. Circulation 130, 1003-1008. 10.1161/CIRCULATIONAHA.114.009029 [DOI] [PubMed] [Google Scholar]
- Schreiner, C., Powell, T. L., Palmer, C. and Jansson, T. (2022). Placental proteins with predicted roles in fetal development decrease in premature infants. Pediatr. Res. 92, 1316-1324. 10.1038/s41390-022-01942-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafi, R., Iyer, S. P., Ellies, L. G., O'donnell, N., Marek, K. W., Chui, D., Hart, G. W. and Marth, J. D. (2000). The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl. Acad. Sci. USA 97, 5735-5739. 10.1073/pnas.100471497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shallie, P. D., Naicker, T. and Nayak, N. R. (2023). Stress-sensitive regulators of fetal neurodevelopment in HIV and preeclampsia: an immunocytochemical appraisal of placental OGT and T4 Levels. Arch. Immunol. Ther. Exp. (Warsz) 71, 3. 10.1007/s00005-023-00668-x [DOI] [PubMed] [Google Scholar]
- Shang, M. and Wen, Z. (2018). Increased placental IGF-1/mTOR activity in macrosomia born to women with gestational diabetes. Diabetes Res. Clin. Pract. 146, 211-219. 10.1016/j.diabres.2018.10.017 [DOI] [PubMed] [Google Scholar]
- Sheller-Miller, S., Choi, K., Choi, C. and Menon, R. (2019). Cyclic-recombinase-reporter mouse model to determine exosome communication and function during pregnancy. Am. J. Obstet. Gynecol. 221, 502.e1-502.e12. 10.1016/j.ajog.2019.06.010 [DOI] [PubMed] [Google Scholar]
- Shibata, E., Rajakumar, A., Powers, R. W., Larkin, R. W., Gilmour, C., Bodnar, L. M., Crombleholme, W. R., Ness, R. B., Roberts, J. M. and Hubel, C. A. (2005). Soluble fms-like tyrosine kinase 1 is increased in preeclampsia but not in normotensive pregnancies with small-for-gestational-age neonates: relationship to circulating placental growth factor. J. Clin. Endocrinol. Metab. 90, 4895-4903. 10.1210/jc.2004-1955 [DOI] [PubMed] [Google Scholar]
- Sibiak, R., Ozegowska, K., Wender-Ozegowska, E., Gutaj, P., Mozdziak, P. and Kempisty, B. (2022). Fetomaternal expression of glucose transporters (GLUTs)-biochemical, cellular and clinical aspects. Nutrients 14, 2025. 10.3390/nu14102025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanirowski, P. J., Szukiewicz, D., Pazura-Turowska, M., Sawicki, W. and Cendrowski, K. (2018). Placental expression of glucose transporter proteins in pregnancies complicated by gestational and pregestational diabetes mellitus. Can. J. Diabetes 42, 209-217. 10.1016/j.jcjd.2017.04.008 [DOI] [PubMed] [Google Scholar]
- Stanirowski, P. J., Szukiewicz, D., Pyzlak, M., Abdalla, N., Sawicki, W. and Cendrowski, K. (2019). Analysis of correlations between the placental expression of glucose transporters GLUT-1, GLUT-4 and GLUT-9 and selected maternal and fetal parameters in pregnancies complicated by diabetes mellitus. J. Matern. Fetal. Neonatal. Med. 32, 650-659. 10.1080/14767058.2017.1387897 [DOI] [PubMed] [Google Scholar]
- Stanirowski, P. J., Szukiewicz, D., Majewska, A., Wątroba, M., Pyzlak, M., Bomba-Opoń, D. and Wielgoś, M. (2022). Placental expression of glucose transporters GLUT-1, GLUT-3, GLUT-8 and GLUT-12 in pregnancies complicated by gestational and type 1 diabetes mellitus. J. Diabetes Investig. 13, 560-570. 10.1111/jdi.13680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staud, F. and Karahoda, R. (2018). Trophoblast: The central unit of fetal growth, protection and programming. Int. J. Biochem. Cell Biol. 105, 35-40. 10.1016/j.biocel.2018.09.016 [DOI] [PubMed] [Google Scholar]
- Sullivan, E. L., Grayson, B., Takahashi, D., Robertson, N., Maier, A., Bethea, C. L., Smith, M. S., Coleman, K. and Grove, K. L. (2010). Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J. Neurosci. 30, 3826-3830. 10.1523/JNEUROSCI.5560-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, P. D., Khan, I. Y., Lakasing, L., Dekou, V., O'brien-Coker, I., Mallet, A. I., Hanson, M. A. and Poston, L. (2003). Uterine artery function in pregnant rats fed a diet supplemented with animal lard. Exp. Physiol. 88, 389-398. 10.1113/eph8802495 [DOI] [PubMed] [Google Scholar]
- Torres, C. R. and Hart, G. W. (1984). Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 259, 3308-3317. 10.1016/S0021-9258(17)43295-9 [DOI] [PubMed] [Google Scholar]
- Ursini, G., Punzi, G., Chen, Q., Marenco, S., Robinson, J. F., Porcelli, A., Hamilton, E. G., Mitjans, M., Maddalena, G., Begemann, M.et al. (2018). Convergence of placenta biology and genetic risk for schizophrenia. Nat. Med. 24, 792-801. 10.1038/s41591-018-0021-y [DOI] [PubMed] [Google Scholar]
- Ursini, G., Di Carlo, P., Mukherjee, S., Chen, Q., Han, S., Kim, J., Deyssenroth, M., Marsit, C. J., Chen, J., Hao, K.et al. (2023). Prioritization of potential causative genes for schizophrenia in placenta. Nat. Commun. 14, 2613. 10.1038/s41467-023-38140-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan, O. R., Fisher, H. M., Dionelis, K. N., Jefferies, E. C., Higgins, J. S., Musial, B., Sferruzzi-Perri, A. N. and Fowden, A. L. (2015). Corticosterone alters materno-fetal glucose partitioning and insulin signalling in pregnant mice. J. Physiol. 593, 1307-1321. 10.1113/jphysiol.2014.287177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan, O. R., Rosario, F. J., Powell, T. L. and Jansson, T. (2020). Normalisation of circulating adiponectin levels in obese pregnant mice prevents cardiac dysfunction in adult offspring. Int. J. Obes. (Lond) 44, 488-499. 10.1038/s41366-019-0374-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan, O. R., Maksym, K., Silva, E., Barentsen, K., Anthony, R. V., Brown, T. L., Hillman, S. L., Spencer, R., David, A. L., Rosario, F. J.et al. (2021). Placenta-specific Slc38a2/SNAT2 knockdown causes fetal growth restriction in mice. Clin. Sci. (Lond.) 135, 2049-2066. 10.1042/CS20210575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, E. D. and Cross, J. C. (2005). Development of structures and transport functions in the mouse placenta. Physiology (Bethesda). 20, 180-193. 10.1152/physiol.00001.2005 [DOI] [PubMed] [Google Scholar]
- Wigglesworth, J. S. (1964). Experimental growth retardation in the foetal rat. J. Pathol. Bacteriol. 88, 1-13. 10.1002/path.1700880102 [DOI] [PubMed] [Google Scholar]
- Yamauchi, T., Kamon, J., Ito, Y., Tsuchida, A., Yokomizo, T., Kita, S., Sugiyama, T., Miyagishi, M., Hara, K., Tsunoda, M.et al. (2003). Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423, 762-769. 10.1038/nature01705 [DOI] [PubMed] [Google Scholar]
- Yoon, M. J., Lee, G. Y., Chung, J. J., Ahn, Y. H., Hong, S. H. and Kim, J. B. (2006). Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes 55, 2562-2570. 10.2337/db05-1322 [DOI] [PubMed] [Google Scholar]
- Young, M. and Widdowson, E. M. (1975). The influence of diets deficient in energy, or in protein, on conceptus weight, and the placental transfer of a non-metabolisable amino acid in the guinea pig. Biol. Neonate 27, 184-191. 10.1159/000240775 [DOI] [PubMed] [Google Scholar]
- Yung, H. W., Calabrese, S., Hynx, D., Hemmings, B. A., Cetin, I., Charnock-Jones, S. and Burton, G. J. (2008). Evidence of translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am. J. Pathol. 173, 311-314. 10.2353/ajpath.2008.080412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z., Liu, F. and Li, S. (2017). Metabolic adaptations in pregnancy: a review. Ann. Nutr. Metab. 70, 59-65. 10.1159/000459633 [DOI] [PubMed] [Google Scholar]
- Zierden, H. C., Marx-Rattner, R., Rock, K. D., Montgomery, K. R., Anastasiadis, P., Folts, L. and Bale, T. L. (2023). Extracellular vesicles are dynamic regulators of maternal glucose homeostasis during pregnancy. Sci. Rep. 13, 4568. 10.1038/s41598-023-31425-x [DOI] [PMC free article] [PubMed] [Google Scholar]