

Abstract
Heterogeneity in sepsis and acute respiratory distress syndrome (ARDS) is increasingly being recognised as one of the principal barriers to finding targeted therapies that are efficacious. The advent of multiple high-throughput biological data (‘omics’) coupled with the widespread access to increased computational power has led to the emergence of phenotyping in critical care. Phenotyping aims to use a multitude of data to identify homogeneous subgroups within an otherwise heterogeneous population. Increasingly, phenotyping schema are being applied to sepsis and ARDS to help better understand these clinical conditions and identify potential therapies. Here we present a selective review of the biological phenotyping schema applied to sepsis and ARDS. Further, we outline some of the challenges that face the field when translating these conceptual findings to bedside clinical decision-making tools.
Keywords: Phenotypes, Subphenotypes, Endotypes, Sepsis, ARDS, Precision Medicine
Introduction
Modern critical care medicine has a heterogeneity problem. Over the last three decades, critical care has relied on broad and overly-sensitive diagnostic criteria for two of its mostly frequently encountered clinical syndromes, sepsis and acute respiratory distress syndrome (ARDS). These rigid clinical diagnostic criteria were introduced (sepsis-1 and AECC) because it was widely perceived at the time that the lack of a unifying definitions for these syndromes had led to significant heterogeneity.(1; 2) Paradoxically, when developing these criteria, the emphasis on sensitivity over specificity has resulted in capturing myriad aetiologies and pathophysiologies and has instead served to compound heterogeneity. Consequently, not only have randomised controlled trials (RCTs) using these definitions overwhelmingly failed to find effective therapies,(3–6) frequently the intervention’s observed effect on outcomes was opposite to those anticipated.(7) Although the reasons for the divergence between the predicted and observed outcomes are likely multifactorial, a highly plausible principal hypothesis is that the heterogeneity of RCT populations in which the interventions are tested far exceed those where the putative benefits of the intervention were discovered. For example, most experimental models of sepsis and ARDS focus on phenotypically and genotypically uniform organisms using uniform injury models. In contrast, in sepsis RCTs, it is entirely plausible an octogenarian with bowel perforation and a teenager with meningitis are recruited to the same trial, illustrating a disconnect between knowledge gathering and its subsequent implementation.
The recent explosion in computational power and its democratisation, combined with a plethora of high throughput biological data, broadly referred to as “omics”, has seen the emergence of a new science called Phenotyping.(8; 9) Phenotyping seeks to untangle heterogeneity in clinical conditions through its use of clinical, physiologic, and/or molecular data in computational algorithms leading to identify more homogeneous subgroups (Figure 1). At its loftiest ambitions, the field proposes to match the right therapies to the right patients at the right time. In other medical fields, such as asthma and oncology, great strides have been made towards translating biologically-derived phenotypes to inform therapeutic decision-making.(10; 11) In critical care, phenotyping offers an intuitive approach to deconvolute the heterogeneity subsumed within our non-specific syndromes. In this review, we present the latest advances in biological phenotyping in sepsis and ARDS, highlighting the advances and specific challenges facing the field.
Figure 1.
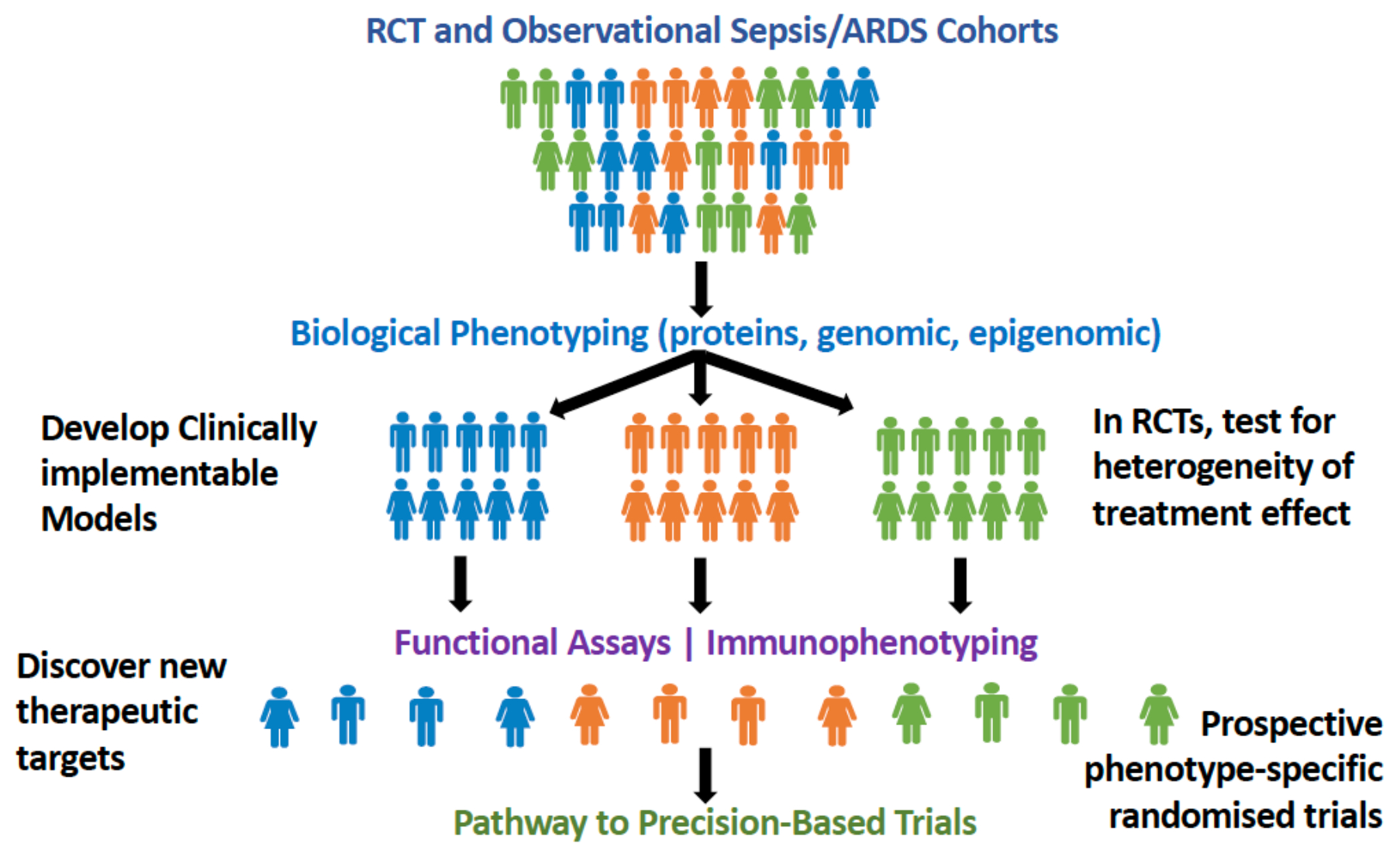
Schematic of approach to phenotyping where secondary analyses of biological data collected in randomised controlled trials (RCTs) and observational cohorts can be used to identify more homogeneous subgroups. Studying immune response in these phenotypes may allow more specific therapies to be discovered-leading the way to precision-based trials.
Endotypes, Phenotypes, Subphenotypes-what’s in a name?
As the field of phenotyping matures, a settled pattern of nomenclature is emerging. At its simplest, phenotyping could represent subgroups of a clinical syndrome or disease derived using a single variable to stratify. For example, ARDS has been categorised by severity of PaO2/FiO2 and has been shown to be effective in stratifying patients when enriching RCTs.(12; 13) For the purposes of this article, however, we mostly limited our review to phenotyping schema that have applied unbiased approaches for phenotype discovery using either multivariate and/or high throughput biological data in human sepsis and ARDS (Figure 2). The added value of biological data for discovering phenotype is a largely unanswered question. A recent study highlighted the contribution of plasma biomarkers to phenotype prediction, demonstrating a diminished ability to detect heterogeneous treatment effect in ARDS RCTs when phenotyping relied on clinical data alone.(14) Nevertheless, numerous excellent studies have leveraged multivariate physiological and clinical data, including the electronic health record (EHR), to phenotype patients in critical care. Such approaches, though important, are beyond the scope of our review and have been covered elsewhere.(15–17)
Figure 2.

A summary of the high-throughput biological data that have been used in sepsis and acute respiratory distress syndrome (ARDS) and the names of the key phenotypes that have been identified using each data type. Note only those studies that have been validated in a second dataset have been included in the figures. Ref = reference. *Represent phenotypes were differential treatment responses to interventions have been observed.
If a phenotype is defined by a set of features shared by a group of patients, e.g. sepsis and ARDS, then a subphenotype describes a set of features that distinguishes subgroups within that phenotype. Endotype describes subgroups that are defined by common intrinsic biology within groups of patients. In other words, “endotyping” refers to a mechanistically-informed subgrouping schema, and although this represents the ultimate goal for phenotyping, so far it proved elusive in critical care.
Biological Phenotypes in Sepsis
Transcriptomics
In their seminal work, Wong and colleagues evaluated the blood transcriptional profile of 98 paediatric patients admitted to the ICU with septic shock.(18) They identified three subphenotypes based exclusively on gene expression pattern. Compared to Subclasses B and C, Subclass A had the highest severity of illness and mortality and was associated with relative downregulation of genes corresponding to the adaptive immune system, glucocorticoid receptor signalling and zinc homeostasis/metabolism. To enable translation of these subclasses clinically, these investigators derived and validated a classifier model based on gene-expression intensity imaging 100 differentially expressed genes.(19; 20) Using their model to classify, based on quantification from a rapid high throughput multiplex messenger RNA (mRNA) platform, they observed that Subclass A was again associated with worse outcomes in the validation set. Further validation of the 100-gene visual classifier was performed in two paediatric septic shock cohorts (n = 168 and 132). In these cohorts, only Subclasses A and B were identifiable using this model and no subjects were adjudicated as subclass C.(21) Notably, Subclass A was associated with increased adjusted mortality with corticosteroid therapy, whereas no treatment effect was observed in Subclass B. Although this was the first study to suggest sepsis heterogeneous treatment effect based on gene expression profile, corticosteroid therapy in these cohorts were neither randomised nor protocolised, which constitutes an important limitation.
Several investigators have sought phenotypes in adult patients with sepsis using bulk transcriptomic data either from whole blood or leucocytes. Davenport and colleagues, using unsupervised clustering algorithms, described two sepsis response transcriptomic signatures (SRS1 and SRS2) in 265 adult septic patients admitted to the ICU with community-acquired pneumonia; they then validated this approach in 106 patients.(22) SRS1 was enriched with transcripts of cell death, apoptosis, necrosis, and cytotoxicity pathways. Leveraging work from other groups, they cross referenced gene expression in their study with prior datasets and found transcriptional patterns for endotoxin tolerance and immune exhaustion were also comparatively overexpressed in SRS1. Unsurprisingly, SRS1 was associated with worse clinical outcomes than SRS2. The authors developed a 7-gene parsimonious classifier that showed good performance metrics in a validation set, mapping a potential route to prospective identification of the SRS groups. The same investigators, using the same protocol, studied transcriptomics signatures in adult faecal peritonitis (N=117).(23) They again identified two SRS signatures in the faecal peritonitis patients and using the above-mentioned 7-gene classifier established high overlap with their previously described community-acquired pneumonia phenotypes, including clinical outcomes. An additional insight from the Davenport group was to combine DNA genotyping with whole blood mRNA expression to identify genetic variants associated with altered expression of specific transcripts, which are termed expression quantitative trait loci (eQTL).(22) Approximately a third of eQTL were only seen in the sepsis population but not in a large control datasets of ambulatory subjects, arguing for context-specific genetic regulation of gene expression. Sepsis eQTL were enriched for endotoxin tolerant genes and differentially enriched between SRS groups, providing more molecular insight to why individuals may respond differently to the same pathogen.
Using the 7-gene classifier, Antcliffe and colleagues asked retrospectively whether SRS interacted with randomized treatment effects in a small subset of the VANISH trial who had mRNA available (n=176 from the trial population of 409 subjects).(24) The authors detected significant interaction between randomized assignment to hydrocortisone (HC) and SRS, but surprisingly, they observed dramatically higher mortality in the SRS2 subjects randomized to HC (42% compared to 8%) without changing mortality for the SRS1 group (33% vs 37%). Although the results are provocative, the small sample size of SRS2 (n=55) and the unusually high mortality for this population may indicate an unstable effect estimate associated with small samples.
One challenge of transcriptional phenotyping is that different clusters of genes may be detected using different methods and different populations. Scicluna and colleagues used unsupervised clustering of whole blood bulk mRNA microarray data to identify four phenotypes (Mars1-4) in septic patients admitted to the ICU.(25) Their discovery cohort comprised 306 patients and two validation cohorts of 216 and 265 patients, respectively. Mars1 was associated with lower expression of genes relating to adaptive and innate immune response such as Toll-like receptor and T-cell receptor signalling and was consistently associated with higher mortality. MARS2, 3, and 4 were consistently identified in all datasets with distinctive patterns of gene expression; however, their relationship with outcomes was variable. Taking a slightly different approach, Sweeney and colleagues used unsupervised clustering in combined datasets of multiple publicly-available transcriptomics studies of patients with sepsis to identify three phenotypes of sepsis (derivation N=900; validation N=600) that have been validated using 33 transcript classifier.(26) By using a method designed for pooling studies, these investigators may have overcome limitations that were population-specific. Based on gene ontology and pathway analyses, they named their clusters “Inflammopathic”, “Adaptive”, and “Coagulopathic.” They observed divergent clinical characteristics in the phenotypes, including clinical outcomes. The Adaptive cluster had the lowest mortality, compared to the other two clusters whose mortality was similar to each other). Sweeney and colleagues also published their algorithm to classify subjects, a true model of data sharing that has advanced the field.(26) Their classifier identified the same 3 clusters in a population of hospitalized subjects with coronavirus disease 2019 (COVID-19), again highlighting the favourable prognosis for subjects with Adaptive expression pattern and high mortality for those with either Inflammopathic or Coagulopathic.(27) Yao and colleagues applied the Sweeney classifier to first classify subjects from 13 sepsis datasets into the 3 groups and then re-derived parsimonious model using fewer transcripts to identify each cluster, renaming the gene expression patterns Innate, Adaptive, Coagulant. Hypothesizing that corticosteroids have no reported effect on the coagulation cascade,(28) they confined their reanalysis of publicly available hydrocortisone trials to classify for Innate and Adaptive only. In a reanalysis of the corticosteroid effect of the VANISH trial based on 117 subjects with gene expression, they confirmed a higher mortality in subjects with high Adaptive scores that were treated with corticosteroids.(28) The reason for the difference in sample size (117 vs 176) compared to the Antcliffe analysis is unclear(24). With similar caveats as described above for the Antcliffe reanalysis regarding small sample size, these analyses suggest that there may be treatment implications for these transcriptomics-derived phenotypes.
Taking a more targeted approach, Cohen and colleagues assessed the relationship between leukocyte expression of adrenocortical candidate genes and corticosteroid response during sepsis using whole blood RNA-sequencing in a sub study of the ADRENAL (N=697) trial that tested the efficacy of corticosteroid therapy versus placebo in septic shock.(29) They found no association with the candidate gene expression levels and corticosteroid treatment for the outcomes of mortality or shock reversal. However, in a logistic regression model they observed a significant treatment interaction between expression of two genes from their proposed adrenocortical gene set (GLCCI1 and BHSD1) and corticosteroid treatment with shock reversal as the outcome. Although potentially powerful, given that the observed effect was an interaction, the dimensionality of therapy effect modification according to gene expression is daunting, and potentially difficult to scale up.
RNA-sequencing from single-cells (scRNA-seq), specifically, leucocytes, is a promising line of investigation for description of the landscape of immune cell responses, in comparison to bulk RNA-sequencing studies which report the overall transcript counts from many contributing cells. Reyes and colleagues performed scRNA-seq of peripheral blood mononuclear cells (PBMCs) from 29 sepsis patients and described four monocyte states (MS1-4).(30) MS1 were CD14+ cells characterised by high expression of RETN, ALOX5AP, and IL1R2. MS2 cells were characterized by high expression of class II MHC, MS3 corresponded to non-classical CD16-high monocytes, and MS4 were remaining CD14+ cells with low class II MHC and cytokine expression.(30) Sepsis was associated with expanded MS1 compared to healthy controls. Notably, expression of gene sets for mitochondrial respiration in MS1 state was inversely correlated with SOFA score, suggesting energy failure as pathophysiological feature leading to immune paralysis in sepsis. MS1 cells were also less responsive to ex vivo lipopolysaccharide (LPS) stimulation than peripheral monocytes from healthy volunteers, adding face validity to their hypothesis. Currently, the additional processing time and costs has limited the widespread use of scRNA-seq.
Immunophenotyping
While scRNA-seq clearly offers several advantages, cheaper and faster methods of studying immune cells, such as flow cytometry, have been leveraged to better understand subgroups in sepsis. Deficits in mHLA-DR recovery had previously been associated with sepsis mortality (31) and with ex vivo endotoxin tolerance.(32) Bodinier and colleagues studied the evolution of monocyte (m)HLA-DR surface protein expression in two adult sepsis cohorts (discovery: n=276 and verification: n=102) using daily measurements in the first week of ICU admission.(33) Using unsupervised clustering on trajectories data, they described four mHLA-DR phenotypes: Non-improvers, Decliners, Improvers, and High Expressors; of note, they determined the number of phenotypes a priori. The non-improvers and decliners had worse clinical outcomes compared to Improvers and High Expressors. Similarly, Leijte and colleagues identified three distinct trajectories of mHLA-DR expression in 241 patients with septic shock: ‘early improvers’, ‘delayed or non-improvers’ and ‘decliners,’ with worse clinical outcomes for the latter two phenotypes.(34)
Several attempts have been made to combine immune phenotyping across multiple cell populations into recognizable patterns and thus infer drivers of host response. The COVID-19 pandemic has accelerated this work. Mathew and colleagues profiled PBMCs collected early in hospitalization from subjects with COVID-19 and used high dimensional flow cytometry to identify roughly 200 cellular populations in the circulating blood.(35) Three patterns of immune expression, which the authors termed ‘immunotypes,’ were identified. Immunotype 1 was characterized by highly activated and proliferating CD8+ and CD4+ T cells, high expression of plasmablasts, and altered T follicular helper cells. Higher immunotype 1 score associated with higher severity of illness score and mortality. Immunotype 2 revealed more evidence of proliferating memory B cells and T-bet transcription factor-expressing memory B cells and T-bet+ effector T cells, whereas immunotype 3 demonstrated no discernible B or T cell activation despite COVID-19 status. Liang and colleagues integrated cytometric, transcriptomic, and plasma cytokine analysis from subjects with COVID-19 to identify a signature associated with severe COVID-19 and poor prognosis.(36) Whereas some features seemed COVID-19-specific, others including IL-8 upregulation, CD5+ B cell depletion, plasmablast activation (predominantly in viruses), reduced mHLA-DR expression, and selective T cell subset cytopenia are features that have also been reported in prior sepsis reports.(31; 37)
Another exciting, yet underdeveloped, approach to identifying subgroups is through phenotyping of immune cells based on ex vivo immune cell stimulation in patients with sepsis. Mazer and colleagues used ELISpot to measure interferon (IFN)-γ and tumour necrosis factor (TNF)-α in PBMCs following ex-vivo stimulation with anti-CD3/anti-CD28 antibodies and LPS respectively.(38) They compared PBMC responses in 19 septic patients and 20 healthy volunteers. They noted that after CD3/CD28 stimulation, in septic patients the number of IFN-γ producing cells were similar to healthy volunteers, however, the number of IFN-γ responsive cells were markedly lower in septic non-survivors compared to survivors. These patterns were similar with TNF-α producing cells following LPS stimulation. Notably, cells treated with IL-7, a potent T-cell activator, had restored IFN-γ production in septic non-survivors, but the same effect was not observed for TNF-α production.(38) These findings suggest a potential for phenotype-specific therapies based on functional immune assays. Similarly, Wang and colleagues identified two phenotypes in PBMCs derived from severe septic patients following ex-vivo stimulation: a hypo- and a hyper-responsive phenotype.(39) Notably, other markers of inflammation (C-reactive protein), infection (white cell count and procalcitonin), and disease severity (SOFA and APACHE III scores) were similar between the groups and unable to identify these phenotypes, emphasising the added value of functional assays over static measures.
Phenotyping with protein biomarkers
Individual biomarkers are clearly important in understanding biology and have been shown to be useful in subgrouping patients with sepsis, including signals toward treatment benefit in secondary analyses of RCTs.(40–42) However, given the underlying biological complexity of critical illness, it seems unlikely that phenotypes derived using single biomarkers can truly capture its underlying heterogeneity. Multivariate phenotyping based on protein biomarkers, however has been relatively sparse in sepsis, especially given the recent advances made in ARDS (discussed later).
In a secondary analysis of three ICU sepsis observational cohorts, Kudo and colleagues used baseline platelet counts, PT-INR, fibrinogen, fibrinogen/fibrin degradation products (FDP), D-dimer, and antithrombin activity to derive and validate phenotypes using k-mean clustering.(43) The authors identified four clusters (dA-dD), of which dA was characterized by severe coagulopathy, organ dysfunction, and higher mortality. Cluster dB also had severe organ dysfunction with moderate coagulopathy with high prevalence of abdominal sepsis. Clusters dC and dD had mild to moderate organ failure with and without coagulopathy, respectively. Notably, recombinant thrombomoulin treatment was associated with decreased risk of mortality on in cluster dA, albeit with the caveats of non-protocolised and non-randomised use of this treatment.
Using latent class analysis (LCA) with clinical data and protein biomarkers as class-defining variables, Shankar-Hari and colleagues identified three subphenotypes in the LeoPARDS trial that tested the efficacy of levosimendan versus placebo in adult patients with sepsis.(44) Subphenotype 3 was associated with the highest values for proinflammatory cytokines, organ dysfunction, and had the highest mortality. The authors observed no heterogeneity of treatment effect in the identified subphenotypes. Notably, their phenotyping schema did not replicate in the VANISH trial with the same approach, albeit with a sample size in the latter trial that was insufficiently powered for LCA. Wiersema and colleagues also performed an LCA in 301 patients with sepsis and acute kidney injury using clinical and protein biomarkers.(45) They identified two subphenotypes in their population, with subphenotype 2 associated with higher markers of endothelial dysfunction and inflammation and worse clinical outcomes. Madushani and colleagues similarly identified two clusters using hierarchical clustering using clinical and biological data in 157 adult surgical patients with sepsis.(46) Cluster I (n=18) was associated with higher levels of proinflammatory cytokines, proteins associated with endothelial dysfunction, organ dysfunction and worse outcomes. Both these studies lacked external validation of their phenotyping schema. The latter study’s limitations included a small overall sample size and unbalanced clusters.
Collectively, these data suggest that there may be prognostically informative subphenotypes in adult septic patients with potential therapeutic implications. However, none of these findings have been prospectively validated, and further investigations in studies with larger sample size are warranted.
Biological Phenotypes in ARDS
Transcriptomics
Unlike sepsis where transcriptomic data has led the way in identifying phenotypes, in ARDS, transcriptomic derived phenotyping lags behind. Yeyha and colleagues used K-means clustering of whole blood microarray data to identify three subphenotypes (CATS 1-3) in 96 paediatric patients with ARDS.(47) CATS 1 was enriched for adaptive immune and T-cell pathways, CATS 2 for complement pathways, and CATS 3 was upregulated for G-protein receptor signalling. CATS 1 had the worst probability of survival and CAT 3 the best. Although the study lacked external validation, it represents the first attempt at discovering de novo transcriptional phenotypes in ARDS.
In adult ARDS, to the best of our knowledge, no substantial phenotyping schema have been described using mRNA data. However, transcriptomic studies have provided insights into the heterogeneity of ARDS host response. Kangelaris and colleagues studied differences in mRNA expression in whole blood between critically-ill septic patients with (N=29) and without ARDS (N=28).(48) Neutrophil-related genes (Olfactomedin 4 (OLFM4), CD24, Lipocalin 2, and Matrix metallopeptidase 8) were consistently overexpressed in septic patients with ARDS compared to non-ARDS septic patients. Several of these candidate genes are also overexpressed in paediatric sepsis non-survivors, and the transcript OLFM4 may serve as a marker for an activated, highly granular neutrophil subset that associates with sepsis mortality.(18; 49) Neutrophils from subjects with ARDS are not only transcriptionally distinct from healthy patients’, but also are functionally distinct, with a primed, activated phenotype with delayed apoptosis.(50) Furthermore, the neutrophils collected from the circulation in ARDS subjects are distinct from those collected from bronchoalveolar lavage (BAL). Morell and colleagues extended this lung versus blood investigation and compared transcriptional profiles of alveolar macrophages (AMs) isolated from BAL to transcriptional profiles of PBMC in patients with ARDS.(51) They observed differential gene expression profiles between the two cell populations and noted that inflammatory gene sets were associated with better outcomes if upregulated in AMs but worse outcomes if upregulated in PBMCs. The same investigators in a follow up study noted that upregulation of inflammatory gene sets in AMs from day 1 was associated with better outcomes; however, the same gene sets were associated with worse outcomes at Days 4 and 8.(52)
Phenotyping with protein biomarkers
Plasma protein biomarkers have been studied in ARDS for over 3 decades, going back to early studies of von Willebrand Factor antigen as a predictor of ARDS in non-pulmonary sepsis (53); thus, it is perhaps unsurprising that they have been an appealing avenue for subphenotyping approaches in this field. LCA has been applied to clinical data and plasma protein biomarker data in 5 RCTs and 2 observational cohorts, and in all 7 of these datasets has identified a two class, or two phenotype, model as a better fit for the data than a one class/phenotype model.(54–58) The variables that characterize these two phenotypes have been quite consistent over these 7 datasets – namely, one phenotype that comprises approximately 30% of the patients is characterized by high plasma levels of inflammatory biomarkers (e.g. IL-6, IL-8, TNFr-1), lower plasma Protein C levels, a higher prevalence of shock, and lower serum bicarbonate.(54–58) Consequently, this phenotype has been labelled “hyper-inflammatory” and the other phenotype, comprising approximately 70% of patients, “hypo-inflammatory”, though in truth it is unclear if inflammation is the dominant biological difference between these two groups. The hyper-inflammatory phenotype has consistently worse clinical outcomes, with 60-day mortality typically at least 2-fold higher than the hypo-inflammatory phenotype. Perhaps of most interest, in secondary analysis of completed RCTs, the two LCA-defined phenotypes appear to respond differently to randomly assigned positive end-expiratory pressure (PEEP),(58) fluid management,(56) and simvastatin.(57) These findings suggest that they may represent treatment-responsive subgroups, though this hypothesis has not been prospectively evaluated. These phenotypes do appear to have prognostic value in COVID-19 ARDS and respond differently to corticosteroids in one analysis,(59) though importantly steroid treatment was not randomized in this dataset, in contrast to the analyses of PEEP, fluids, and simvastatin. These phenotypes have been since identified in paediatric ARDS,(60) and classification to the hyper-inflammatory phenotype was associated with worse survival in the LUNG-SAFE dataset of nearly 3000 patients with ARDS,(61) in other smaller ARDS cohorts,(62; 63) and even in acute hypoxemic respiratory failure,(64) suggesting they may have at least prognostic value beyond ARDS.
Another approach to utilizing plasma protein biomarkers to identify ARDS phenotypes has focused on application of unbiased clustering methods to plasma biomarker data alone, independent of clinical data. Bos and colleagues measured 20 plasma biomarkers of inflammation, coagulation and endothelial activation in a cohort of 454 patients with ARDS and identified 2 clusters, which they termed “uninflamed” and “reactive”.(65) The reactive cluster, characterized by higher levels of IL-6, IFN-gamma, Ang2/Ang1, and PAI-1, had significantly worse clinical outcomes in both training and test cohorts. Notably, as with the LCA-defined phenotypes, severity of hypoxemia as measured by P/F ratio was not associated with phenotype membership, indicating that the phenotypes provide additive information beyond ARDS severity. This group has since reported that the reactive and uninflamed groups are characterized by differential expression of 29% of transcripts in whole blood; notably, the reactive group was characterized by increased expression of genes related to neutrophil function, oxidative phosphorylation, and mitochondrial dysfunction, relative to the uninflamed group.(64) Classification into the reactive group has been associated with poor clinical outcomes in patients with acute hypoxemic respiratory failure, indicating potential prognostic value in other forms of critical illness. The precise correspondence between the LCA-defined phenotypes and the reactive vs uninflamed phenotypes remains unknown.
Next steps for biological phenotypes (Figure 3)
Figure 3.

Overview of the challenges facing the bedside implementation of biological phenotypes identified in sepsis and acute respiratory distress syndrome. ICU = Intensive Care Unit.
Clinical Implementation of phenotypes
While biological phenotyping is rapidly expanding, it must be emphasised that no biological phenotyping schema in critical care has been translated to clinical decision-making. To achieve this milestone, phenotype-specific treatment efficacy needs prospective evaluation in clinical trials. Before that, however, bedside identification of biological phenotypes is required, which is one of the biggest challenges in the field. The attempts to identify the above-described inflammatory phenotypes in the clinical setting, illustrates potential approaches to overcoming these challenges. The ARDS LCA models are comprised of 30-40 variables standardised to the population mean and therefore impractical for prospective use. We developed 3-or 4-variable parsimonious models comprising two protein biomarkers and clinical variables, all in their original scale, that accurately classify the phenotypes and substantially reduced model complexity. In a recent, proof-of-concept study, we showed feasibility of prospective bedside classification of the phenotypes in a small cohort of patients with COVID-19-ARDS (N=39).(62) The ongoing PHIND study (NCT04009330) will expand the findings of our preliminary study to a larger cohort.
As an alternative strategy to rapid protein biomarker measurement, we developed and validated machine-learning models that use only readily-available clinical variables to identify the ARDS phenotypes in secondary analysis of RCTs.(66) Further, we reported that these models retained their high performance metrics even when EHR-derived clinical variables were used to classify in an observational cohort of ARDS.(61) Importantly, the phenotypes identified using these clinical classifier models captured the biological and clinical characteristics of the LCA-derived phenotypes, including the differential treatment responses described above.
Temporal Kinetics
Most phenotyping schemas described in this review lean on data from cross-sectional time points. To implement phenotypes clinically, we must understand their temporal kinetics and the implications of changing phenotypes over time. Using inflammatory phenotypes as an example, the natural course of their disease states is unknown, as is the impact therapies have on changing phenotypes. The best data we have would suggest that at Day 3 two phenotypes are also identifiable and 94% of the patients transitioned to their corresponding class at the later time point.(67) However, whether the inflammatory burden and disease implications of the phenotypes identified at baseline and day 3 are the same is unknown. These knowledge gaps are critical because it may be that for phenotype-specific interventions, resolution of a dysfunctional phenotype may be a more valid outcome to study than mortality, which may have several unrelated mediators.(68) However, without knowledge of the “normal” disease course and anticipated timeframes in which interventions can alter phenotype classification, interpreting phenotype temporal kinetics in the context on an intervention’s optimal timing and response would be challenging. Moreover, temporally dynamic assessment of phenotypes may identify patients that switch to a phenotype where an efficacious therapy is known.
Another important time-related factor to consider is the inherent stability of the biological measurements that we use for phenotyping. Critical care is uniquely challenging in that patient biology can change rapidly. Burnham et al, found that SRS phenotypes were highly unstable, with 46% of the subjects switching SRS phenotypes between days 1, 3 and 7.(23) These findings are in keeping with other studies that have shown temporally unstable gene expression patterns in sepsis that can change over periods as short as 4 hours.(69; 70) Together these studies undergird the rationale for longitudinal studies of biological phenotypes.
Understanding Mechanism
A fundamental limitation in translating phenotypic observations to precision therapy has been our limited understanding of their biological significance. For example, DNA variants implicated in disease states do not necessarily manifest as differentially expressed at an mRNA or protein level. In turn, the downstream functional protein production in relation to mRNA expression is also largely unstudied in critical care. The evoked regulation of proteins, metabolites, transcripts, or cell populations is far from understood, and will require patient-oriented and model system approaches to better elucidate drivers of pathology and therapeutic response.
Even with protein biomarker-derived phenotypes, the mechanistic role of said biomarkers to the identified disease states is largely unknown. In all likelihood, our understanding of current phenotypes is too rudimentary and proposed interventions too simplistic for the complexity observed in critical illness.(41) An important consideration for future studies is how best to further subdivide phenotypes and how small a phenotype can realistically be tolerated in the clinical setting. The answer would likely be determined by the specificity of a phenotype balanced against the need to test interventions in realistic sample sizes sufficiently powered to detect a benefit. In order to optimise this balance, biomarkers that are specific to disease states, yet orthogonal to each other, are likely to be the highest yield for new phenotype discovery.
Finally, to truly understand the mechanistic principles underlying a phenotype, developing experimental models of the phenotypes will be crucial. Such models will also be imperative to test plausibility of novel therapeutic interventions that are phenotype-specific. Although several phenotype-specific models have been described in the literature, they remain under-studied and should evolve as human phenotypes are validated.
Conclusions
Biological phenotypes in sepsis and ARDS offer a new and intuitive way to spilt heterogeneous patient populations that maximise our understanding from human studies, are prognostically informative, and may have treatment implications. Future studies need to gather knowledge that deepen our understanding of existing phenotypes while broadening to discover new phenotypes. Most importantly, the clinical utility of biological phenotypes urgently needs demonstrating in prospective clinical trials.
Summary statements.
Critical care syndromes such as sepsis and acute respiratory distress syndrome (ARDS) are highly heterogeneous, which has led to numerous failed trials.
Phenotyping involves using multivariate data to try and identify more uniform subgroups within heterogeneous populations.
Phenotyping using biological high-throughput data are increasingly being used to identify biologically uniform subphenotypes in sepsis and ARDS.
In critically-ill sepsis patients, subphenotypes derived using transcriptional data are leading the way in identifying homogeneous phenotypes that are prognostically and biologically informative.
In ARDS, a composite of protein biomarkers and clinical data have used to consistently identify the Hypo- and Hyperinflammatory phenotypes with divergent outcomes and treatment responses.
While promising, most phenotype schemas lack prospective validation and identifying them at the bedside has proved challenging.
To realise their potential for delivering precision medicine, prospective phenotype-specific trials are needed.
References:
- 1.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, et al. 1994. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 149:818–24 [DOI] [PubMed] [Google Scholar]
- 2.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, et al. 1992. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 101:1644–55 [DOI] [PubMed] [Google Scholar]
- 3.Marshall JC. 2014. Why have clinical trials in sepsis failed? Trends Mol Med 20:195–203 [DOI] [PubMed] [Google Scholar]
- 4.Bone RC. 1996. Why sepsis trials fail. JAMA 276:565–6 [PubMed] [Google Scholar]
- 5.Matthay MA, McAuley DF, Ware LB. 2017. Clinical trials in acute respiratory distress syndrome: challenges and opportunities. Lancet Respir Med 5:524–34 [DOI] [PubMed] [Google Scholar]
- 6.Laffey JG, Kavanagh BP. 2018. Negative trials in critical care: why most research is probably wrong. Lancet Respir Med 6:659–60 [DOI] [PubMed] [Google Scholar]
- 7.Harhay MO, Wagner J, Ratcliffe SJ, Bronheim RS, Gopal A, et al. 2014. Outcomes and statistical power in adult critical care randomized trials. Am J Respir Crit Care Med 189:1469–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drawnel FM, Zhang JD, Kung E, Aoyama N, Benmansour F, et al. 2017. Molecular Phenotyping Combines Molecular Information, Biological Relevance, and Patient Data to Improve Productivity of Early Drug Discovery. Cell Chem Biol 24:624–34 e3 [DOI] [PubMed] [Google Scholar]
- 9.Shivade C, Raghavan P, Fosler-Lussier E, Embi PJ, Elhadad N, et al. 2014. A review of approaches to identifying patient phenotype cohorts using electronic health records. J Am Med Inform Assoc 21:221–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, et al. 2011. Lebrikizumab treatment in adults with asthma. N Engl J Med 365:1088–98 [DOI] [PubMed] [Google Scholar]
- 11.Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr., et al. 2005. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 353:1673–84 [DOI] [PubMed] [Google Scholar]
- 12.Guerin C, Reignier J, Richard JC, Beuret P, Gacouin A, et al. 2013. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med 368:2159–68 [DOI] [PubMed] [Google Scholar]
- 13.Papazian L, Forel JM, Gacouin A, Penot-Ragon C, Perrin G, et al. 2010. Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med 363:1107–16 [DOI] [PubMed] [Google Scholar]
- 14.Sinha P, Spicer A, Delucchi KL, McAuley DF, Calfee CS, Churpek MM. 2021. Comparison of machine learning clustering algorithms for detecting heterogeneity of treatment effect in acute respiratory distress syndrome: A secondary analysis of three randomised controlled trials. EBioMedicine 74:103697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinha P, Calfee CS. 2019. Phenotypes in acute respiratory distress syndrome: moving towards precision medicine. Curr Opin Crit Care 25:12–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reddy K, Sinha P, O’Kane CM, Gordon AC, Calfee CS, McAuley DF. 2020. Subphenotypes in critical care: translation into clinical practice. Lancet Respir Med 8:631–43 [DOI] [PubMed] [Google Scholar]
- 17.Sinha P, Bos LD. 2021. Pathophysiology of the Acute Respiratory Distress Syndrome: Insights from Clinical Studies. Crit Care Clin 37:795–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong HR, Cvijanovich N, Lin R, Allen GL, Thomas NJ, et al. 2009. Identification of pediatric septic shock subclasses based on genome-wide expression profiling. BMC Med 7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong HR, Cvijanovich NZ, Allen GL, Thomas NJ, Freishtat RJ, et al. 2011. Validation of a gene expression-based subclassification strategy for pediatric septic shock. Crit Care Med 39:2511–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong HR, Wheeler DS, Tegtmeyer K, Poynter SE, Kaplan JM, et al. 2010. Toward a clinically feasible gene expression-based subclassification strategy for septic shock: proof of concept. Crit Care Med 38:1955–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong HR, Cvijanovich NZ, Anas N, Allen GL, Thomas NJ, et al. 2015. Developing a clinically feasible personalized medicine approach to pediatric septic shock. Am J Respir Crit Care Med 191:309–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, et al. 2016. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med 4:259–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burnham KL, Davenport EE, Radhakrishnan J, Humburg P, Gordon AC, et al. 2017. Shared and Distinct Aspects of the Sepsis Transcriptomic Response to Fecal Peritonitis and Pneumonia. Am J Respir Crit Care Med 196:328–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antcliffe DB, Burnham KL, Al-Beidh F, Santhakumaran S, Brett SJ, et al. 2019. Transcriptomic Signatures in Sepsis and a Differential Response to Steroids. From the VANISH Randomized Trial. Am J Respir Crit Care Med 199:980–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scicluna BP, van Vught LA, Zwinderman AH, Wiewel MA, Davenport EE, et al. 2017. Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. Lancet Respir Med 5:816–26 [DOI] [PubMed] [Google Scholar]
- 26.Sweeney TE, Azad TD, Donato M, Haynes WA, Perumal TM, et al. 2018. Unsupervised Analysis of Transcriptomics in Bacterial Sepsis Across Multiple Datasets Reveals Three Robust Clusters. Crit Care Med 46:915–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sweeney TE, Liesenfeld O, Wacker J, He YD, Rawling D, et al. 2021. Validation of Inflammopathic, Adaptive, and Coagulopathic Sepsis Endotypes in Coronavirus Disease 2019. Crit Care Med 49:e170–e8 [DOI] [PubMed] [Google Scholar]
- 28.Yao L, Rey DA, Bulgarelli L, Kast R, Osborn J, et al. 2022. Gene Expression Scoring of Immune Activity Levels for Precision Use of Hydrocortisone in Vasodilatory Shock. Shock 57:384–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen J, Blumenthal A, Cuellar-Partida G, Evans DM, Finfer S, et al. 2021. The relationship between adrenocortical candidate gene expression and clinical response to hydrocortisone in patients with septic shock. Intensive Care Med 47:974–83 [DOI] [PubMed] [Google Scholar]
- 30.Reyes M, Filbin MR, Bhattacharyya RP, Billman K, Eisenhaure T, et al. 2020. An immune-cell signature of bacterial sepsis. Nat Med 26:333–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monneret G, Lepape A, Voirin N, Bohe J, Venet F, et al. 2006. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med 32:1175–83 [DOI] [PubMed] [Google Scholar]
- 32.Heftrig D, Sturm R, Oppermann E, Kontradowitz K, Jurida K, et al. 2017. Impaired Surface Expression of HLA-DR, TLR2, TLR4, and TLR9 in Ex Vivo-In Vitro Stimulated Monocytes from Severely Injured Trauma Patients. Mediators Inflamm 2017:2608349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bodinier M, Peronnet E, Brengel-Pesce K, Conti F, Rimmele T, et al. 2021. Monocyte Trajectories Endotypes Are Associated With Worsening in Septic Patients. Front Immunol 12:795052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leijte GP, Rimmele T, Kox M, Bruse N, Monard C, et al. 2020. Monocytic HLA-DR expression kinetics in septic shock patients with different pathogens, sites of infection and adverse outcomes. Crit Care 24:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, et al. 2020. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laing AG, Lorenc A, Del Molino Del Barrio I, Das A, Fish M, et al. 2020. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat Med 26:1623–35 [DOI] [PubMed] [Google Scholar]
- 37.Dunning J, Blankley S, Hoang LT, Cox M, Graham CM, et al. 2018. Progression of whole-blood transcriptional signatures from interferon-induced to neutrophil-associated patterns in severe influenza. Nat Immunol 19:625–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mazer MB, CC C, Hanson J, Mannion D, Turnbull IR, et al. 2021. A Whole Blood Enzyme-Linked Immunospot Assay for Functional Immune Endotyping of Septic Patients. J Immunol 206:23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Gloss B, Tang B, Dervish S, Santner-Nanan B, et al. 2021. Immunophenotyping of Peripheral Blood Mononuclear Cells in Septic Shock Patients With High-Dimensional Flow Cytometry Analysis Reveals Two Subgroups With Differential Responses to Immunostimulant Drugs. Front Immunol 12:634127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panacek EA, Marshall JC, Albertson TE, Johnson DH, Johnson S, et al. 2004. Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody F(ab’)2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels. Crit Care Med 32:2173–82 [DOI] [PubMed] [Google Scholar]
- 41.Meyer NJ, Reilly JP, Anderson BJ, Palakshappa JA, Jones TK, et al. 2018. Mortality Benefit of Recombinant Human Interleukin-1 Receptor Antagonist for Sepsis Varies by Initial Interleukin-1 Receptor Antagonist Plasma Concentration. Crit Care Med 46:21–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janz DR, Bastarache JA, Rice TW, Bernard GR, Warren MA, et al. 2015. Randomized, placebo-controlled trial of acetaminophen for the reduction of oxidative injury in severe sepsis: the Acetaminophen for the Reduction of Oxidative Injury in Severe Sepsis trial. Crit Care Med 43:534–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kudo D, Goto T, Uchimido R, Hayakawa M, Yamakawa K, et al. 2021. Coagulation phenotypes in sepsis and effects of recombinant human thrombomodulin: an analysis of three multicentre observational studies. Crit Care 25:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shankar-Hari M, Santhakumaran S, Prevost AT, Ward JK, Marshall T, et al. 2021. In Defining phenotypes and treatment effect heterogeneity to inform acute respiratory distress syndrome and sepsis trials: secondary analyses of three RCTs. Southampton (UK). Number of. [PubMed] [Google Scholar]
- 45.Wiersema R, Jukarainen S, Vaara ST, Poukkanen M, Lakkisto P, et al. 2020. Two subphenotypes of septic acute kidney injury are associated with different 90-day mortality and renal recovery. Crit Care 24:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madushani R, Patel V, Loftus T, Ren Y, Li HJ, et al. 2022. Early Biomarker Signatures in Surgical Sepsis. J Surg Res 277:372–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yehya N, Varisco BM, Thomas NJ, Wong HR, Christie JD, Feng R. 2020. Peripheral blood transcriptomic sub-phenotypes of pediatric acute respiratory distress syndrome. Crit Care 24:681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kangelaris KN, Prakash A, Liu KD, Aouizerat B, Woodruff PG, et al. 2015. Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am J Physiol Lung Cell Mol Physiol 308:L1102–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kangelaris KN, Clemens R, Fang X, Jauregui A, Liu T, et al. 2021. A neutrophil subset defined by intracellular olfactomedin 4 is associated with mortality in sepsis. Am J Physiol Lung Cell Mol Physiol 320:L892–L902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Juss JK, House D, Amour A, Begg M, Herre J, et al. 2016. Acute Respiratory Distress Syndrome Neutrophils Have a Distinct Phenotype and Are Resistant to Phosphoinositide 3-Kinase Inhibition. Am J Respir Crit Care Med 194:961–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morrell ED, Radella F 2nd, Manicone AM, Mikacenic C, Stapleton RD, et al. 2018. Peripheral and Alveolar Cell Transcriptional Programs Are Distinct in Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 197:528–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morrell ED, Bhatraju PK, Mikacenic CR, Radella F 2nd, Manicone AM, et al. 2019. Alveolar Macrophage Transcriptional Programs Are Associated with Outcomes in Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 200:732–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubin DB, Wiener-Kronish JP, Murray JF, Green DR, Turner J, et al. 1990. Elevated von Willebrand factor antigen is an early plasma predictor of acute lung injury in nonpulmonary sepsis syndrome. J Clin Invest 86:474–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinha P, Delucchi KL, Chen Y, Zhuo H, Abbott J, et al. 2022. Latent class analysis-derived subphenotypes are generalisable to observational cohorts of acute respiratory distress syndrome: a prospective study. Thorax 77:13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sinha P, Delucchi KL, Thompson BT, McAuley DF, Matthay MA, et al. 2018. Latent class analysis of ARDS subphenotypes: a secondary analysis of the statins for acutely injured lungs from sepsis (SAILS) study. Intensive Care Med 44:1859–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, et al. 2017. Acute Respiratory Distress Syndrome Subphenotypes Respond Differently to Randomized Fluid Management Strategy. Am J Respir Crit Care Med 195:331–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, et al. 2018. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med 6:691–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, et al. 2014. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med 2:611–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sinha P, Furfaro D, Cummings MJ, Abrams D, Delucchi K, et al. 2021. Latent Class Analysis Reveals COVID-19-related Acute Respiratory Distress Syndrome Subgroups with Differential Responses to Corticosteroids. Am J Respir Crit Care Med 204:1274–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dahmer MK, Yang G, Zhang M, Quasney MW, Sapru A, et al. 2022. Identification of phenotypes in paediatric patients with acute respiratory distress syndrome: a latent class analysis. Lancet Respir Med 10:289–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maddali MV, Churpek M, Pham T, Rezoagli E, Zhuo H, et al. 2022. Validation and utility of ARDS subphenotypes identified by machine-learning models using clinical data: an observational, multicohort, retrospective analysis. Lancet Respir Med 10:367–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sinha P, Calfee CS, Cherian S, Brealey D, Cutler S, et al. 2020. Prevalence of phenotypes of acute respiratory distress syndrome in critically ill patients with COVID-19: a prospective observational study. Lancet Respir Med 8:1209–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sinha P, Delucchi KL, McAuley DF, O’Kane CM, Matthay MA, Calfee CS. 2020. Development and validation of parsimonious algorithms to classify acute respiratory distress syndrome phenotypes: a secondary analysis of randomised controlled trials. Lancet Respir Med 8:247–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heijnen NFL, Hagens LA, Smit MR, Cremer OL, Ong DSY, et al. 2021. Biological Subphenotypes of Acute Respiratory Distress Syndrome Show Prognostic Enrichment in Mechanically Ventilated Patients without Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 203:1503–11 [DOI] [PubMed] [Google Scholar]
- 65.Bos LD, Schouten LR, van Vught LA, Wiewel MA, Ong DSY, et al. 2017. Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax 72:876–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sinha P, Churpek MM, Calfee CS. 2020. Machine Learning Classifier Models Can Identify Acute Respiratory Distress Syndrome Phenotypes Using Readily Available Clinical Data. Am J Respir Crit Care Med 202:996–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delucchi K, Famous KR, Ware LB, Parsons PE, Thompson BT, et al. 2018. Stability of ARDS subphenotypes over time in two randomised controlled trials. Thorax 73:439–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mayr VD, Dunser MW, Greil V, Jochberger S, Luckner G, et al. 2006. Causes of death and determinants of outcome in critically ill patients. Crit Care 10:R154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Talwar S, Munson PJ, Barb J, Fiuza C, Cintron AP, et al. 2006. Gene expression profiles of peripheral blood leukocytes after endotoxin challenge in humans. Physiol Genomics 25:203–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cazalis MA, Lepape A, Venet F, Frager F, Mougin B, et al. 2014. Early and dynamic changes in gene expression in septic shock patients: a genome-wide approach. Intensive Care Med Exp 2:20. [DOI] [PMC free article] [PubMed] [Google Scholar]