

Abstract
Kidney disease is highly prevalent and affects approximately 850 million people worldwide. It is also associated with high morbidity and mortality, and current therapies are incurable and often ineffective. Animal models are indispensable for understanding the pathophysiology of various kidney diseases and for preclinically testing novel remedies. In the last two decades, rodents continue to be the most used models for imitating human kidney diseases, largely because of the increasing availability of many unique genetically modified mice. Despite many limitations and pitfalls, animal models play an essential and irreplaceable role in gaining novel insights into the mechanisms, pathologies, and therapeutic targets of kidney disease. In this review, we highlight commonly used animal models of kidney diseases by focusing on experimental AKI, CKD, and diabetic kidney disease. We briefly summarize the pathological characteristics, advantages, and drawbacks of some widely used models. Emerging animal models such as mini pig, salamander, zebrafish, and drosophila, as well as human-derived kidney organoids and kidney-on-a-chip are also discussed. Undoubtedly, careful selection and utilization of appropriate animal models is of vital importance in deciphering the mechanisms underlying nephropathies and evaluating the efficacy of new treatment options. Such studies will provide a solid foundation for future diagnosis, prevention, and treatment of human kidney diseases.
Keywords: animal model, AKI, CKD, diabetic kidney disease, glomerular disease, kidney fibrosis, acute kidney failure, chronic nephropathy, chronic renal disease, cisplatin, diabetic nephropathy, fibrosis
Introduction
Kidney disease has become a public health problem on a global scale and is often associated with high morbidity and mortality. The prevalence of kidney diseases is continuing to increase, and currently there are approximately 850 million people having some kinds of kidney disorders worldwide.1 Based primarily on the duration of the disorders, kidney disease is generally classified into two distinct syndromes, AKI and CKD.2 Clinically, AKI and CKD are closely interconnected, and they share many common risk factors such as old age, diabetes mellitus, hypertension, and other preexisting medical conditions.2–4 Epidemiological studies have suggested that AKI and CKD are each a risk factor of the other.5,6 Although AKI is now recognized to pose an important risk leading to the development of CKD and even to ESKD, the extent to which AKI contributes to the incidence of CKD remains unclear. The pathologies of AKI-to-CKD transition or AKI-in-CKD coexistence, combining with many serious complications and comorbidities, pose a challenge in the modeling, diagnosis, and treatment of specific syndromes. Strategies to precisely target the underlying mechanisms and to halt or reverse the progression of CKD have been largely unsuccessful.
Animal models are indispensable in understanding the pathophysiology of kidney diseases and in providing reliable preclinical testing systems for new therapies. A variety of experimental models for AKI and CKD have been developed, and rodents, particularly mice, are the most used species. Despite the valuable insights into kidney disease gained from existing models, many results do not fruitfully translate into clinical treatments in humans, raising concerns about the applicability of these widely used models. In this context, it is prudent to determine which aspects of animal models recapitulate the relevant phenotypes of human kidney disease. In this review, we highlight commonly used animal models of kidney diseases by focusing on experimental AKI, CKD, and diabetic kidney disease (DKD). We briefly summarize the model types, pathological features, advantages, and drawbacks of some popular models. We also discuss current challenges for modeling human kidney diseases in animals and provide future perspectives on emerging experimental models.
AKI Models
As a form of acute and rapidly developing disorder, AKI is determined based on the sudden elevation of serum creatinine (SCr) and/or oliguria within a duration of 7 days.7 AKI affects one in five hospitalized patients worldwide and associates with high mortality.8,9 The sudden loss of kidney function generally results in the retention of metabolic wastes and imbalance of body fluid, with subsequent development of various complications and even endangering other organs.10 Although functional criteria present detailed definitions regarding urine and serum components,7 structural assessment is unavailable with existing techniques beyond biopsy. Of note, before the changes of urine and serum composition are detectable, the eGFR has greatly reduced, reflecting an abnormal kidney function. A series of AKI biomarkers for injury have been proposed and investigated in clinical trials over the past decade.11 However, these biomarkers have limitations with modest predictive performance and low sensitivity, which hinders their clinical usefulness.
AKI is encountered with a variety of pathologies and pathophysiological processes. The major causes of AKI in humans include renal hypoperfusion, direct nephrotoxicity, and sepsis.4,12 Thus, commonly used AKI models are induced by ischemia and reperfusion, drugs or nephrotoxins, and sepsis inducers in animals, which reflects the diverse etiologies of AKI in humans (Table 1).
Table 1.
The commonly used rodent models of AKI
| AKI Models | Induction Methods | Pathology | Pros and Cons | Refs. |
|---|---|---|---|---|
| Ischemic AKI | BIRI | • Mainly damage the S3 segments of proximal tubules. • Sublethal tubular injury and cell death. • Interstitial inflammation. |
• Kidney function can be monitored; species-independent and strain-independent; relevant to humans. • Severer tubular damage than that in human; significant animal loss; female mice are resistant. |
13,23,25,127,128 |
| UIRI | • Similar to BIRI. | • Enables longer observation time; no animal loss; develops more severe fibrosis. • Unable to monitor kidney function. |
||
| UIRI/UNX | • Similar to BIRI. | • SCr and BUN at 24 h after UNX can be monitored; enable AKI-to-CKD progression. • Additional surgical stress. |
||
| Nephrotoxic AKI | Cisplatin | • Mainly damage the proximal tubular cells. • Severe model with increased BUN and SCr, inflammation, fibrosis. |
• Simple and reproducible, many similarities to humans. • No standardized protocol. |
33,129 |
| Gentamicin | • Collecting duct epithelial cell death, necrosis. • Increased BUN and SCr, decreased GFR. • Inflammation, fibrosis. |
• Resemble aminoglycoside-induced AKI in humans. • Require high dose. |
36,37,130 | |
| Aristolochic acid | • Proximal tubular cell injury and death. • Increased BUN and SCr. • Progressive interstitial fibrosis. |
• Simulate clinical aristolochic acid nephropathy and AKI-to-CKD progression. • Require high-dose, systemic toxicity. |
38,39 | |
| Folic acid | • Disruption of tubular integrity. • Increased BUN and SCr. • Fibrotic lesions. |
• Reproducible and simple, simulate AKI-to-CKD progression. • High variable injury, less clinical relevance. |
40,131 | |
| Glycerol | • Tubular cell death. • Myoglobinuria. • Inflammation. |
• Simulate the rhabdomyolysis. • Moderate kidney injury and dysfunction. |
41 | |
| Septic AKI | LPS | • Tubular cell apoptosis. • Inflammatory cell infiltration. • Increased BUN and SCr. • Mitochondrial injury. |
• Simulate the hyperinflammatory state of sepsis. • Transient increases in inflammatory mediators, less clinical relevance. |
44,132 |
| CLP | • Much later and lower cytokine levels than LPS model. • Severely hypotensive. |
• Simulate human sepsis better than LPS model. • Difficult to control the severity of sepsis, differences in age and strain. |
47,133 |
BIRI, bilateral ischemia–reperfusion injury; UIRI, unilateral ischemia–reperfusion injury; UNX, uninephrectomy; SCr, serum creatinine; CLP, cecal ligation puncture.
Ischemic AKI Models
Ischemic AKI models in mice or rats mainly include bilateral ischemia–reperfusion injury (BIRI), unilateral ischemia–reperfusion injury (UIRI), and UIRI combined with uninephrectomy (UIRI/UNX).13 In the ischemic model, the proximal tubular cells in the S3 segments of the nephron are mostly affected, which is due to the blockage of oxygen and nutrient delivery and waste removal channels for the renal cells.14,15 Ischemia–reperfusion will trigger tubular cells to undergo sublethal injury or cell death by different modes such as apoptosis, necrosis, necroptosis, pyroptosis, and ferroptosis, which in turn leads to kidney dysfunction.16–18 The typical kidney tubular damage includes loss of brush border, contraction and flattening of the cells, cell death or detachment, and apparent debris in the lumen.15 In the same settings, the time points for assessment after reperfusion determine the injury phenotype and pathological findings because short time points within 3 days manifest tubular cell damage, various modes of cell death, and inflammation, whereas long time points within weeks present with fibrosis.17,19,20 Moreover, longer ischemia duration worsens kidney function and usually leads to progression to CKD.21,22 Among the three ischemia-reperfusion injury (IRI) models, BIRI has the most relevant renal hemodynamics to human pathophysiology, and the changes in kidney function could be readily monitored because of ischemia of both kidneys. UIRI does not allow for measuring kidney functional changes based on SCr and BUN because the contralateral uninjured kidney will compensate for the lost function of the injured one. Severe BIRI leads to the death of animals, whereas UIRI rarely cause animal loss and thus enables longer observation and develops more robust fibrosis than BIRI. These features of UIRI make it a suitable model for investigation of postischemic AKI-CKD transition.23,24 Compared with UIRI, UIRI/UNX allows one to assess kidney function by SCr and BUN for monitoring renal injury and repair.25
The pathological changes of ischemic AKI largely depend on the models used, as well as the animal species, strain, sex, and age.26 In general, female mice are more resistant to IRI than male counterparts. Therefore, longer duration of ischemia is often used in female animals to achieve a comparable degree of AKI. Age is another important factor in determining the severity after IRI. The aged mice typically exhibit an increased mortality and exacerbated kidney damage, compared with the young.27 Another variable is the body temperature during ischemia, and warm ischemia is used in most IRI models to increase the severity of renal injury, rather than cold one.23 Therefore, the body temperature of animals is usually kept at 37°C or above during ischemia.
It is worthwhile to point out that there are significant differences in histological changes after IRI between humans and animal models. Some investigators have questioned whether the murine models of ischemic AKI are truly relevant to humans, especially considering the more severe injury in animal models than that in patients.28 Furthermore, unlike animal models, the duration of ischemia in humans does not always correlate with the outcome.29 Nevertheless, substantial studies have demonstrated that murine IRI model exhibits significant similarity to human in short-term outcomes and characteristic features.21 In this context, one should keep in mind that translation of the experimental findings from murine models to humans has to be cautious. Attention should be paid to details including, but not limited to, duration of ischemia, body temperature during ischemia, and observation timing after reperfusion.
Nephrotoxic AKI Models
Nephrotoxins or medications with nephrotoxic effect are another common cause of AKI in hospitalized patients.12,30 The cellular mechanisms and signal pathways causing kidney dysfunction are different among various agents, mainly through direct tubular injury, renal inflammation, or intratubular obstruction leading to AKI.31 A variety of drugs and toxic agents, such as cisplatin, gentamicin, aristolochic acid, and folic acid, are used to induce nephrotoxic AKI models. Cisplatin-induced AKI is commonly used to model drug nephrotoxicity in humans because cisplatin, as an effective chemotherapeutic agent for a variety of cancers, incites adverse effects on renal proximal tubular cells and often causes AKI.32 Repeated-dosing protocol with cisplatin in mice has been demonstrated to recapitulate the nephropathy of those kidneys given cisplatin in humans.33,34 Gentamicin-induced AKI is considered as a promising model to study the clinical effects of aminoglycosides on kidney.35–37 Aristolochic acid and folic acid models are also widely used in AKI research field, which are particularly attractive for modeling the transition of AKI-to-CKD in mice because of their early fibrotic characteristics.38–40 In humans, rhabdomyolysis often causes AKI because of intrinsic muscle dysfunction. Accordingly, intramuscular injection of glycerol in rodents is the well-established model of rhabdomyolysis AKI.41,42
Septic AKI Models
Sepsis is the most common etiology of AKI in humans.12 Septic AKI is characterized by the excessive production of proinflammatory cytokines, which trigger kidney injury and dysfunction.43 LPS-induced animal model is the simplest one for septic AKI mimicking, which is frequently harnessed to study the pathophysiology, cellular mechanism, and therapeutic potentials of sepsis-associated AKI.44–46 Another commonly used model, the cecal ligation puncture model, is believed to resemble the progression and characteristics of human sepsis more accurately than the LPS model.47 However, cecal ligation puncture model is difficult to control the severity of sepsis and therefore results in poor reproducibility with significant animal loss.48,49
CKD Models
The prevalent and incurable CKD is posing a substantial burden and challenge to the world.3 CKD is characterized by progressive destruction of renal parenchyma and reduction of functional nephrons. Processes such as glomerular hypertension, renal inflammation, glomerulosclerosis, and tubulointerstitial fibrosis are associated with progression of CKD. Therapies for patients with CKD in clinic are often ineffective and cannot completely halt its progression to renal deficiency, although significant progresses have been made recently.50,51 Animal models are extremely valuable tools for investigating the pathological mechanisms of these diseases and for testing the therapeutic efficacy of potential remedies. In this section, we discuss some experimental CKD models that are widely used in the nephrology community (Table 2).
Table 2.
The commonly used rodent models of CKD
| CKD Models | Induction Methods | Models(Refs.) | Pathology | Pros and Cons |
|---|---|---|---|---|
| Interstitial fibrosis models | Surgical | UUO53,54 | • Tubular injury and atrophy. • Myofibroblast activation and interstitial fibrosis. • Infiltration of inflammatory cells. |
• Work in all species and strains. • Rapid and robust fibrosis. • No functional assessment. • Less clinically relevant. |
| Severe UIRI23 | • Tubular cell injury and atrophy. • Interstitial inflammation. • Long-term expression of tubular injury markers and cytokines. |
• Avoid animal loss. • Significant fibrosis. • No functional assessment. |
||
| Two-stage UIRI/UNX22 | • Increased BUN and SCr levels. • Myofibroblast activation, interstitial fibrosis. • Interstitial inflammation. |
• Avoid animal loss. • Significant fibrosis. • SCr and BUN assessing impaired compensation after UNX. • Additional surgery. |
||
| Drug-induced | High-dose folic acid134 | • Damage proximal tubular cells. • Interstitial inflammation. • Interstitial fibrosis, increased BUN and SCr. |
• Modeling intratubular obstruction. • Moderate fibrosis. |
|
| Repeated low-dose cisplatin34,58 | • Tubular cell injury and atrophy. • Interstitial fibrosis, increased BUN and SCr. • Long-term expression of cytokines. |
• Modeling clinical protocol. • Moderate fibrosis. |
||
| Remnant kidney model | Surgical | 5/6 subtotal nephrectomy60,61 | • Hypertension. • Proteinuria, glomerular sclerosis. • Tubulointerstitial inflammation and fibrosis. |
• Recapitulate most human CKD features. • Work well in rats. • Resistant in C57BL6 mice. |
| Hypertensive nephropathy model | Drug-induced | DOCA-salt63 | • Low-renin, neurogenic form of hypertension. • Renal sodium retention, aldosterone release, inflammation, and mild fibrosis. • Cardiovascular lesions. |
• Neurogenic model of hypertension. • Relevant to humans. |
| Ang II infusion62 | • Hypertension, lesions in kidney, and extrarenal organs. • Proteinuria and mild-to-moderate podocyte injury. • Renal inflammation and mild fibrosis. |
• Imitate systemic RAS activation. • Affects extrarenal organs such as heart. |
||
| Genetic | Spontaneously hypertensive rat135,136 | • Model of essential hypertension. • Mild proteinuria. • Renal inflammation and fibrosis. • Cardiovascular lesions. |
• Spontaneity. • Enigmatic origin. • Normotensive WKY rats as controls. |
|
| Glomerular disease models | Drug-induced | Adriamycin65,67 Puromycin66,68 |
• Podocytes injury, proteinuria. • Glomerulosclerosis. • Tubular injury and interstitial fibrosis. |
• Simple injection operation. • High reproducibility and relatively low mortality. • Resistant in C57BL6 mice. |
| Immunological | Lupus nephritis (NZB/W F1, MRL/lpr, or BXSB strains)73 | • Mesangial proliferation. • Proteinuria and glomerulonephritis. • Tubular atrophy and interstitial inflammation. |
• Rodent sex-related. • Variable in individual animals. |
|
| Thy-1 nephritis137 | • Mesangial cell proliferation. • Inflammatory cell infiltration. • Mesangial matrix expansion. • Proteinuria and hematuria. |
• Simple injection operation. | ||
| Anti-GBM nephritis138,139 | • Proteinuria and crescent formation. • Mesangial cell proliferation. • Inflammatory cell infiltration. • Interstitial fibrosis. |
• Simple injection operation. • Variable susceptibility among different strains. • Animal loss. |
||
| Polycystic kidney disease models | Hereditary model | Pcy mice75,76 Cy rats PCK rats |
• Renal cysts formation and grow. • Renal volume expansion. • Increased cell proliferation. |
• Relatively long lifespan. • Suitable for pharmacological experiments. • Variable severity of phenotypes. |
| Gene-modified model | Pkd1 knockout75,76 | • Renal cysts formation and grow. • Renal volume expansion. • Increased cell proliferation. • Inflammatory cell infiltration. • Renal fibrosis. |
• Relevant to humans. • Suitable for cystogenesis research. |
UUO, unilateral ureteral obstruction; UIRI, unilateral ischemia–reperfusion injury; UNX, uninephrectomy; SCr, serum creatinine; DOCA, deoxycorticosterone acetate and high-salt; GBM, glomerular basement membrane. PCK, polycystic kidney; RAT, renin-angiotensin system; WKY, Wistar Kyoto.
Interstitial Fibrosis Models
Kidney fibrosis is the common outcome of CKD and can serve as a strong predictor for CKD progression and prognosis.52 The unilateral ureteral obstruction (UUO) model has become increasingly popular and is a widely accepted in vivo model for studying kidney fibrosis because it recapitulates the fundamental pathological features of CKD, including tubular injury and atrophy, interstitial infiltration of inflammatory cells, myofibroblast activation, and deposition of the extracellular matrix leading to fibrotic lesions and microvascular rarefaction, in a relatively short time span.53 The UUO model is performed by ureteral ligation with a short duration ranging from 3 days to 2 weeks in mice and rats.54 The fibrotic lesions in this model are very robust and highly reproducible. Furthermore, this model shows no specific species, strain, and gender dependence, which are particularly suited for studies involved in genetically modified mice with C57BL/6J background. Despite these advantages, the limitations of this model should be carefully considered because it lacks a functional readout because of the compensation of the unobstructed contralateral kidney. Complete UUO is uncommon clinically, and therefore, its clinical relevance has been questioned. In this regard, partial UUO model has also been established using silastic tubing,55 but it is unpopular because of technical difficulty. Contrary to the irreversible model of UUO, reversible UUO model has been established to study the resolution of inflammation and fibrosis by relieving the ligation of the ureter.56
Epidemiological studies have shown that AKI is a significant risk factor leading to progressive CKD.57 Consistent with clinical studies, a variety of experimental models have demonstrated that AKI can result in chronic damage and lead to CKD. As such, those models that are initially used to incite AKI have been increasingly used for studying CKD progression in a long-term course. For example, CKDs are induced by severe UIRI,23 two-stage UIRI/UNX in which UIRI is followed by UNX at a later time point,22 and repeated low-dose cisplatin in a long-term protocol.24,58 Of note, renal function parameters in the two-stage UIRI/UNX model at the early time points such as 1 day after UNX may only reflect an impaired compensatory capacity of the injured kidney and do not truly indicate renal failure because SCr and BUN levels gradually return toward the baseline in several days after UNX in this model.59 Furthermore, owing to another surgery required to remove the uninjured kidney, this two-stage UIRI/UNX induces additional surgical stress to the animals. In recognition of the fact that the same etiologies such as IRI can induce AKI, CKD, and the AKI-CKD transition, careful attention should be paid on selection of the timing to administer therapeutic remedies. It should be noticed that a key limitation to current models of AKI-to-CKD progression is the lack of a sustained effect on renal function. Although fibrosis develops at later time points after AKI, SCr and BUN values typically return toward the baseline levels, which preclude the use of renal function as an end point in evaluating the therapeutic efficacy of potential remedies in these models.
Remnant Kidney and Other Hypertensive Models
The remnant kidney after 5/6 nephrectomy (5/6NX) in rodents is a classic and widely used model to mimic the CKD progression caused by nephron number loss, and it is also a model of hypertensive CKD. Ligation and ablation are the most common ways to produce the remnant kidney. Regarding phenotypic similarities, both methods produce hypertension, proteinuria, glomerular sclerosis, tubular atrophy, tubulointerstitial fibrosis, and possible ESKD.60 However, the ligation model develops more rapid-onset and more severe hypertension than the ablation model. The 5/6NX model develops hypertension and proteinuria, which could enable one to continuously monitor the disease progression. Although 5/6NX model in rats faithfully replicates the most pathological features of human CKD, one major drawback is that mouse genetic background exerts quite differential responses to renal mass reduction, and especially C57BL/6 strain seems highly resistant to the development of fibrosis and progressive CKD.61 Because most of the genetically modified (knockout or transgenic) mice are generated in C57BL/6 background, this drawback makes 5/6NX model increasingly less popular in mice.
There are several hypertension models, including the deoxycorticosterone acetate and high-salt diet rodent model, the spontaneously hypertensive rat model, and the chronic angiotensin II infusion model. These models recapitulate many aspects of human hypertensive nephropathy, including hypertension, proteinuria, and glomerulosclerotic and tubulointerstitial fibrotic lesions.62,63
Glomerular Disease Models
Glomerular disease is characterized by two major pathological features including podocyte injury leading to proteinuria and mesangial activation leading to glomerulosclerosis. FSGS is a common primary glomerular disorder, which can be induced by 5/6NX or numerous drugs such as Adriamycin and puromycin.64 As the commonly used antibiotics for the study of podocyte injury and glomerulosclerosis,65,66 these drugs exert direct toxic damage to podocytes, thereby impairing the glomerular filtration barrier leading to proteinuria and glomerulosclerosis. Pathology of kidney structure and function in these models largely resembles human FSGS. These models feature simple injection, high reproducibility, and relatively low mortality. However, the degree of renal injury in these models is heavily influenced by the strain, drug dosage and batches, and route of administration.67,68 In particular, BALB/c mice are very sensitive to Adriamycin, but C57BL/6 strain is resistant. Some investigators use a high dose of Adriamycin protocol in mice with C57BL/6 background and show only limited success.69,70 Other genetic models also have good performance for recapitulating the major features of human glomerular sclerotic disease, such as HIV-associated nephropathy, NPHS1−/− mice, NPHS2−/− mice, and Col4a3−/− mouse model of Alport syndrome.71 The albumin overload model,72 characterized by proteinuria and podocyte injury, also imitate some features of human glomerular diseases.
Another predominant cause of glomerulopathies leading to CKD and ESKD is glomerulonephritis. Lupus nephritis is a form of glomerulonephritis caused by systemic lupus erythematosus, and its spontaneous mouse models are available. There are three widely used genetically modified models including NZB/W F1 mice, MRL/lpr mice, and BXSB mice, exhibiting some similar features relevant to human lupus nephritis.73,74 Regarding sex, female NZB/W F1 and MRL/lpr mice are more severely affected; instead, BXSB mice are more severely affected in males.73 In addition to the lupus nephritis models, the nephrotoxic serum nephritis induced by antiglomerular basement membrane and anti–Thy-1 models are also common experimental models of glomerulonephritis. Unfortunately, there is no widely accepted animal model for human IgA nephropathy, despite this disease is highly prevalent.
Polycystic Kidney Disease Models
Polycystic kidney disease (PKD) is an inherited disorder and often leads to the development of CKD and ESKD. Two major types of animal model of PKD have been established: spontaneous hereditary models or gene-modified models.75 The spontaneous hereditary models include pcy mice, cy rats, and polycystic kidney rats, which are considered suitable for testing medicine efficacy because of their relatively long lifespan of more than half a year. The gene-modified models include human orthologous PKD1 transgenic mice and pkd1 gene knockout mice. Both types of animal models are used to study the mechanism of cystogenesis and the efficacy of drug therapy. However, disease progression is often variable in PKD animal models.76 It is necessary to apply a sufficient number of animals and multiple models to increase the credibility of the results.
DKD Models
DKD is a leading cause of CKD and kidney failure globally, particularly in the industrialized nations.77,78 In humans, DKD is characterized by glomerular hyperfiltration, progressive albuminuria, declining GFR, and ultimately ESKD.79 Currently, there is a variety of diabetic animal models that resemble pathophysiology of both type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM). However, these animal models could not recapitulate every aspect of DKD and often only represent the early stage of diabetic nephropathy and do not progress to glomerulosclerosis and renal failure. Therefore, it is necessary to assess the extent to which the pathophysiology in animal DKD resembles that in humans. In this section, we concisely summarize some commonly used rodent models of T1DM and T2DM and evaluate their utility in the study of DKD (Table 3).
Table 3.
The experimental models of diabetic kidney disease
| Disease | Induction Methods | Models(Refs.) | Pathology | Stage Simulating |
|---|---|---|---|---|
| T1DM | Drug-induced | STZ mice80 | • Mild-to-moderate albuminuria. • Mild glomerular and tubular damage. • No hypertension, glomerulosclerosis, or interstitial fibrosis. |
Early |
| STZ/eNOS−/− mice82 | • Albuminuria. • Glomerulosclerosis. • Interstitial fibrosis. |
Advanced | ||
| Spontaneous | Akita mice86 | • Hyperglycaemia. • Mild hypertension, modest albuminuria. • No glomerular or interstitial fibrosis. |
Early | |
| Ove26 mice87 | • Progressive albuminuria. • Glomerulosclerosis, interstitial fibrosis. • Poor viability. |
Advanced | ||
| T2DM | Spontaneous | Zucker diabetic fatty rat92 | • Hyperlipidemia. • Moderate hypertension and obesity. • Progressive renal injury. • High cost and slow progression to CKD. |
Advanced |
| db/db mice140 | • Glomerular and tubular hypertrophy. • Modest albuminuria. • Mesangial matrix expansion. • No glomerular and interstitial fibrosis. |
Early | ||
| db/db mice with UNX89,90 | • Proteinuria and tubular atrophy. • Interstitial fibrosis. • Inflammation. |
Advanced | ||
| db/db mice with eNOS−/−91 | • Albuminuria. • Arteriolar hyalinosis. • Glomerulosclerosis, interstitial fibrosis. |
Advanced | ||
| ob/ob mice141 | • Podocyte loss, GBM thickening. • Mesangial matrix expansion. • No mesangiolysis, glomerular sclerosis. |
Early to modest |
Early stage: mild-to-moderate albuminuria, mild mesangial expansion, reduced nephrin expression. Advanced stage: moderate to macroalbuminuria, severe mesangial expansion and thickened glomerular basement membrane, podocyte loss, glomerular sclerosis, and tubulointerstitial fibrosis. T1DM, type 1 diabetes mellitus; STZ, streptozotocin; eNOS, endothelial nitric oxide synthase; T2DM, type 2 diabetes mellitus; db/db, leptin receptor deficient; UNX, uninephrectomy; GBM, glomerular basement membrane; ob/ob, leptin deficient.
T1DM Models
T1DM is an autoimmune disease caused by the immune system destroying β cells in the pancreas. Streptozotocin (STZ), a compound that specifically causes β cell toxicity, is routinely used to induce T1DM in rodents. Pathological changes of STZ-induced diabetes include mild-to-moderate albuminuria, mild glomerular damage manifested by thickened glomerular basement membrane and mesangial expansion, and slight tubular changes, but do not develop hypertension, glomerulosclerosis, or tubulointerstitial fibrosis.80 Although C57BL/6 is the most commonly used strain in the studies involving genetically modified mice, CD-1, DBA/2J, and KK/HlJ mice are more susceptible to diabetic nephropathy.81 To accelerate the progression of DKD to an advanced stage, two approaches are often used. One is to inject STZ in the endothelial nitric oxide synthase (eNOS)–deficient mice because they sensitize to diabetic injury and produce a progressive DKD, characterized by advanced nephropathic changes with pronounced albuminuria, glomerulosclerosis, and interstitial fibrosis.82 Therefore, this model can be used to study changes in more advanced stages of DKD.83 Another approach is adding second or third injurious stimuli such as advanced oxidation protein products, high-fat diet, and/or UNX on STZ-treated mice, which produces a more advanced DKD.84,85 Spontaneous T1DM models due to genetic mutations, such as Akita and Ove26 mice, particularly combined with UNX, are also prevalent tools for DKD research.86,87
T2DM Models
T2DM is more commonly encountered in the clinic and represents a greater proportion of the worldwide burden.88 Models of T2DM typically use hereditary obesity which can be established by leptin-deficient (ob/ob) mice or inactivation of the leptin receptor (db/db) mice. Both models exhibit typical features resembling the early stage of human DKD including hyperinsulinemia, albuminuria, and hyperglycemia. However, they do not experience progressive kidney dysfunction and therefore fail to manifest advanced features of DKD. More advanced progressive models can be induced by manipulation with UNX or genetic modification. For example, early UNX can accelerate the development of advanced DKD in db/db mice, which display tubular atrophy, interstitial fibrosis, and inflammation.89 In addition, eNOS−/− db/db mice have also been created to accelerate DKD.90,91 It is worthwhile to mention that animal models of DKD are highly strain-specific. Besides the traditional rodent models, several studies have reported novel genetic models by crossbreeding. For example, the Zucker diabetic fatty rats, generated by crossbreeding between ZF rat and Wistar Kyoto rat, are widely used in the T2DM studies.92
Emerging Models
Rodent models remain to be the mainstay of experimental approaches in nephrology and have a profound effect on kidney disease modeling and drug discovery. However, the findings derived from traditional rodent models cannot always be extrapolated to humans. In addition, there are many limitations for using rodent models, such as the inability to perform high-throughput screening, the high cost, the time-consuming experimentation, and the difficulty for genetic manipulation. Therefore, in addition to common rodent models, other animal models as well as human-derived organoids and kidney-on-a-chip are emerging for the studies of kidney diseases (Figure 1).
Figure 1.
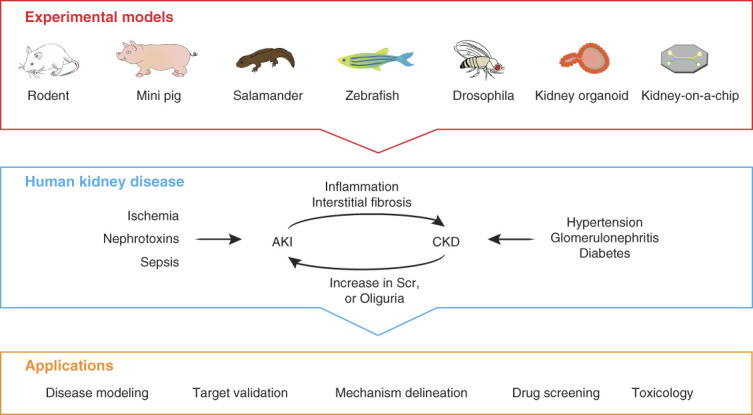
Schematic presentation of commonly used experimental models of kidney diseases and their applications. The experimental models used to study human kidney disease are diverse in species. Rodent models remain to be the mainstay of experimental approaches in nephrology and have a profound effect on kidney disease modeling, target validation, mechanism delineation, potential drug discovery, and toxicology evaluation. Although rodent models dominate kidney research, the use of large animals, such as mini pigs or simple and humanized models such as salamander, zebrafish, drosophila, human-derived kidney organoids, and kidney-on-a-chip, can also provide important insights into the pathogenesis of kidney disease and enable high-throughput screening for therapeutics to prevent and treat human kidney disorders. SCr, serum creatinine.
Large Animal Models
Several large laboratory animals, such as pigs, dogs, cats, sheep, and non-human primates, are also used for studying human kidney disease. The main advantage of these large animal models is that they are closer to humans in physical size, anatomic structure, and physiological features, compared with rodent models. The larger physical size enables the acquisition of sufficient amounts of blood and tissue material for experimental analysis. However, tools such as genetic modification, antibodies, and commercial test kits are not as advance in large animal models as they are in rodent models. Furthermore, large animals are costly to house and use and require specially trained surgeons.
Mini pigs, with kidney size resembling that of the human kidney, are the most commonly used large animal model in nephrology research. It has been reported that the AKI model induced by IRI in pigs exhibits significant impairment within 3 days after reperfusion and begins to recover gradually afterward.93,94 The nephrotoxic AKI induced by drugs, such as cisplatin and gentamicin, has also been developed in pigs.95 Compared with gentamicin, cisplatin-induced model is more reproducible. In addition to AKI models, researchers have successfully established porcine CKD models by performing nephrectomy,96 inducing bilateral renal artery stenosis,97 or by knocking out PKD1 gene.98 Because of the body size and anatomical resemblance to humans, surgery instruments such as laparoscope used in humans can be used in mini pigs. Like murine models, general attributes such as sex, strain, age, and body weight of pigs have an effect on some experimental results. Some important limitations of porcine models include the lack of elaborated data at the molecular level and the high cost of rearing.
Salamander Model
The salamander (axolotl) may be an underestimated tool for studying kidney injury and regeneration. A distinct advantage of the axolotl is its unique and powerful regenerative capability.99 The axolotl kidney shares remarkable structural, molecular, and functional similarities with the mammalian kidney.99,100 New tools for studying axolotl are improving with the development of their genome resources.101 Recent studies on single-cell RNA-seqencing profiles have provided many insights into understanding of the mechanism underlying axolotl organ regeneration.102 Using gentamicin-induced nephrotoxicity and doxorubicin-induced glomerular injury models, new studies have shown that axolotl nephrons including renal tubules and glomeruli can completely regenerate after severe renal injury.99 In this context, axolotl may be an invaluable model for understanding the mechanisms of kidney injury repair and regeneration. However, axolotl is inapplicable to the long-term CKD progression studies, owing to its ability to repair and regenerate the injured nephrons in several weeks.
Zebrafish Model
Zebrafish is an alternative animal model to study the pathophysiology and molecular mechanism of human kidney diseases because nephron structures of the fish recapitulate various aspects of mammalian kidney morphology and function.103 Zebrafish is often used for investigation of drug-induced AKI including cisplatin, gentamicin, and doxorubicin, which exhibit similar nephrotoxic response and injury biomarkers to those of mammals, suggesting applicability to pathological characterization.104 Moreover, the small size, low cost, and large quantity of zebrafish and the feasibility of live time-lapse imaging based on fluorescent tracers makes it possible for high-throughput drug screening and for disease progression monitoring. Genetic manipulations are also widely implemented in zebrafish to model glomerular disease and PKD.105 However, similar to axolotl, zebrafish is also inapplicable to CKD studies because of its regenerative ability. Moreover, although the kidneys of zebrafish and mammals have a close functional analogy, there are structural differences. Zebrafish kidney represents the pronephros rather than the metanephros in mammals. There is no bladder in zebrafish, making it difficult to detect changes in the urine for assessing renal function.
Drosophila Model
A wide range of models of kidney diseases have been developed in drosophila because many kidney-associated genes are shared between flies and mammals.106 Major advantages of the fly models include their low cost, short lifespan, simple renal system, and the availability of live imaging and easy genetic manipulation, as compared with mammalian system. Flies can be fed with chemical compounds before dissection for testing therapeutic efficacy. When combined with genetic manipulation, flies can be humanized by introducing human genes. Currently, drosophila systems have been taken in studying nephrolithiasis, glomerular disease, tubular disorders, diabetic nephropathy, and stone disease.107 Further applications of flies for large-scale screening may help to facilitate the discovery of therapeutic drugs. Nonetheless, some limitations need to be pointed out when using the drosophila model system. For example, the insect kidney is not vascularized and lacks a renin–angiotensin–aldosterone system, and dysregulation of which plays an important role in the pathogenesis of mammalian kidney diseases.
Human-Derived Kidney Organoids
The emergence of three-dimensional (3D) organoids originated from stem cells by means of self-organization has brought a tool innovation for human kidney development and disease interrogation.108 Organoids permit in vivo and in vitro investigation on human tissues and display near-physiologic cellular composition and behaviors. Therefore, organoids have been rapidly applied to human disease modeling and providing knowledge for translational medicine and personalized therapy.109 Human kidney organoids derived from human pluripotent stem cells are being harnessed to model kidney development and diseases. In comparison with other models, kidney organoids comprise several major advantages, such as human cell-derived, unlimited supply, high-throughput screening, and high recapitulation of injury response. To date, kidney organoids have enabled the pathophysiological validation of both AKI and CKD in a human context. Studies have demonstrated that exposing kidney organoids to cisplatin and gentamicin, two drugs that cause AKI in clinic, display substantial tubular injury and upregulation of AKI biomarkers.110,111 Extensive characterization of the kidney organoids undergoing drug-induced injury through single-cell sequencing has revealed detailed kidney-specific cell populations and underlying pathological pathways.112 In addition, knowledge gained from using kidney organoids has improved the understanding of disease progression from AKI to CKD.113 Leveraging the genetic manipulability of the organoid, the specific organoids with PKD mutation or podocalyxin-like deficiency have also been established and could serve as genetic kidney disease models.114 Although there are many merits of kidney organoid in deciphering kidney development and disease, limitations such as poor vascularization, absence of immune cells and lack of perfusion and glomerular filtration and tubular flow should be carefully considered.115
Kidney-on-a-Chip
With the advances in microelectromechanical systems and 3D printing techniques, kidney-on-a-chip technologies hold great potential to improve the in vitro kidney models that enable microfluidics and vasculature to cells.116 Kidney-on-a-chip devices can include single or multiple cell types inside a microchamber with continuous fluid flow recapitulating the tissue microenvironment. Various kidney-on-a-chip devices have been developed for kidney disease modeling and drug screening, such as glomerulus-on-a-chip117 and tubuloids-on-a-chip.118 The integration of sensors permits real-time monitoring of cell behavior and cell function.119 Still, most kidney-on-a-chip devices are composed of a single cell type or single tissue compartment. Further efforts need to focus on integrating multiple tissue compartments, such as the tubule, glomerulus, and interstitium, in the kidney-on-a-chip to better mimic overall kidney tissue function.
Challenges and Perspectives
Despite great efforts and advances in animal modeling of human kidney diseases, many results from animal studies have not successfully translated into clinical treatments in humans. For examples, while abundant studies have demonstrated the therapeutic efficiency of inhibition of TGF-β signaling in retarding the progression of CKD in animal models, such a therapy in the clinical trials for patients with CKD is disappointed.120,121 This disconnection underscores the vast difference between animal models and humans and raises concerns about the applicability of the findings from animal studies.
The reasons behind this discrepancy between animal models and humans are multifactorial and variable depending on the nature of the disease, genetic background, sex, age, and various comorbidities associated with patients. It should be stressed that there is no such a thing as a perfect animal model for human kidney disease, and every model has its own limitations and pitfalls. These limitations in each model make it a challenge to select appropriate model for a particular aspect of pathology and for the assessment of effective therapy.
Animal models are usually performed by using the inbred animals with the same or closely similar genetic background. However, it is well documented that the genetic makeup in different species and strains plays a determining role in the pathogenesis and trajectories of kidney disease. For example, remnant kidney model works well in rats, but the same procedure causes little nephropathy in C57BL/6 mice. Adriamycin causes severe podocyte injury and heavy proteinuria in BALB/c mice, whereas C57BL/6 mice are resistant to its damage. In future perspective, any therapy may need to be tested in multiple animal models with different strains to reflect the complex genetic backgrounds of human populations before proceeding to the clinical trials.
At present, the vast majority of animal models are performed in healthy young mice and rats, whereas kidney diseases in the clinical setting often occur in older patients with various comorbidities. In this context, the translational success of animal models requires consideration of comorbidities, such as old age, diabetes, hypertension, anemia, and preexisting CKD, to name a few, to establish models that could recapitulate the clinical situation more accurately. For example, comorbidity model of DKD has been developed to simulate complications such as hypertension or atherosclerosis.122 The comorbidity model of AKI with aged mice has been shown to better reflect the older patient population.27,123 AKI model with preexisting CKD confirms the clinical concept that loss of renal mass caused by prior disease adversely affects the outcome of superimposed AKI.124 In the future, developing animal models by multiple injurious stimuli to imitate the patients' conditions with various comorbidities should be carefully considered and increasingly used. Another potential issue is that all current animal studies are performed in the pathogen-free facility. Given the emerging role of an altered gut microbiome in the pathogenesis of kidney diseases,125 whether or to what extent the different housing environments between the experimental animals and humans contributes to the failure of clinical trials remains elusive.
Animal studies are an integral part of scientific inquiry in kidney disease and play a key role in preclinical development of therapeutic drugs. There are many issues needing to be considered, such as the selection of appropriate disease model and the choice of animal species and strains. Although mice are increasingly being used, thanks to the availability of genetically modified models, rats remain to be irreplaceable and extensively used in both AKI and CKD studies as well. Rats not only are an excellent model for studying CKD-associated comorbidities such as hypertension, vascular calcification, and anemia in remnant kidney after 5/6NX, but also have practical advantages because they enable multiple blood draws, metabolic cage use, and abundant tissue, blood, and urine for analysis. Another issue should be kept in mind that zebrafish and drosophila share only 70% and 60% identities of the same genes with human, respectively, suggesting that there is a significant evolutionary distance that may discourage the use of these models for drug development. Regardless of the animal type, studies with animal models should be adequately powered and reproducible within the same laboratory and other groups. Recently, the International Society of Nephrology has offered a set of consensus guidance for preclinical animal studies in translational nephrology.126 Hopefully, these recommendations will facilitate the optimal conduct of translational animal studies and help to accelerate the development of new drugs to treat kidney diseases.
Conclusion
Animal models are essential and irreplaceable for understanding the pathophysiology of kidney diseases and for evaluating the efficacy of new therapeutics in the preclinical setting. In the past several decades, rodents continue to be the most used species for modeling human kidney diseases. From countless animal studies, we have gained valuable knowledge and obtained novel insights into the mechanisms, pathologies, and therapeutic targets of kidney disease. At the same time, it should be stressed that there is no so-called perfect animal model for human disease. Furthermore, the different responses to the exact same injury in animals are relatively common and often influenced by the specific species, genetic background, age, and sex, making it extremely imperative to select the right animal model for the question asked. These shortcomings pose a great challenge in using animal models for imitating human kidney diseases and call for more cautious interpretation of the experimental data generated from animal studies.
Although mammalian models dominate kidney research, the use of simple or humanized models, such as salamander, zebrafish, and drosophila, as well as human-derived kidney organoids or kidney-on-a-chip, can also provide important insights and allow a high-throughput screening at affordable price. As for future perspective, multiple models with different genetic backgrounds and models of large animals, such as mini pigs, may be warranted before clinical trials in patients. Undoubtedly, careful selection and utilization of appropriate animal models will help to obtain accurate and reliable research results and provide a solid foundation for future diagnosis, prevention, and treatment of human kidney diseases.
Acknowledgments
We thank the anonymous reviewers for their insightful critiques and valuable suggestions on this manuscript.
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by National Natural Science Foundation of China from 81920108007 and 82230020 (Y. Liu), China Postdoctoral Science Foundation from 2022M721508 (J. Liang).
Author Contributions
Conceptualization: Youhua Liu.
Data curation: Jianqing Liang.
Funding acquisition: Jianqing Liang, Youhua Liu.
Investigation: Jianqing Liang.
Supervision: Youhua Liu.
Validation: Youhua Liu.
Writing – original draft: Jianqing Liang.
Writing – review & editing: Youhua Liu.
References
- 1.Foreman KJ, Marquez N, Dolgert A, Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet. 2018;392(10159):2052–2090. doi: 10.1016/S0140-6736(1831694-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014;371(1):58–66. doi: 10.1056/NEJMra1214243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalantar-Zadeh K, Jafar TH, Nitsch D, Neuen BL, Perkovic V. Chronic kidney disease. Lancet. 2021;398(10302):786–802. doi: 10.1016/S0140-6736(21)00519-5 [DOI] [PubMed] [Google Scholar]
- 4.Ronco C, Bellomo R, Kellum JA. Acute kidney injury. Lancet. 2019;394(10212):1949–1964. doi: 10.1016/S0140-6736(19)32563-2 [DOI] [PubMed] [Google Scholar]
- 5.See EJ, Jayasinghe K, Glassford N, Long-term risk of adverse outcomes after acute kidney injury: a systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019;95(1):160–172. doi: 10.1016/j.kint.2018.08.036 [DOI] [PubMed] [Google Scholar]
- 6.Pannu N. Bidirectional relationships between acute kidney injury and chronic kidney disease. Curr Opin Nephrol Hypertens. 2013;22(3):351–356. doi: 10.1097/MNH.0b013e32835fe5c5 [DOI] [PubMed] [Google Scholar]
- 7.Palevsky PM, Liu KD, Brophy PD, KDOQI US commentary on the 2012 KDIGO clinical practice guideline for acute kidney injury. Am J Kidney Dis. 2013;61(5):649–672. doi: 10.1053/j.ajkd.2013.02.349 [DOI] [PubMed] [Google Scholar]
- 8.Hoste EAJ, Kellum JA, Selby NM, Global epidemiology and outcomes of acute kidney injury. Nat Rev Nephrol. 2018;14(10):607–625. doi: 10.1038/s41581-018-0052-0 [DOI] [PubMed] [Google Scholar]
- 9.Susantitaphong P Cruz DN Cerda J, et al. . World incidence of AKI: a meta-analysis. Clin J Am Soc Nephrol. 2013;8(9):1482–1493. doi: 10.2215/CJN.00710113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kellum JA, Romagnani P, Ashuntantang G. Acute kidney injury. Nat Rev Dis Primers. 2021;7(1):52. doi: 10.1038/s41572-021-00284-z [DOI] [PubMed] [Google Scholar]
- 11.Ostermann M, Zarbock A, Goldstein S, Recommendations on acute kidney injury biomarkers from the acute disease quality initiative consensus conference: a consensus statement. JAMA Netw Open. 2020;3(10):e2019209. doi: 10.1001/jamanetworkopen.2020.19209 [DOI] [PubMed] [Google Scholar]
- 12.Hoste EA, Bagshaw SM, Bellomo R, Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. 2015;41(8):1411–1423. doi: 10.1007/s00134-015-3934-7 [DOI] [PubMed] [Google Scholar]
- 13.Shiva N, Sharma N, Kulkarni YA, Mulay SR, Gaikwad AB. Renal ischemia/reperfusion injury: an insight on in vitro and in vivo models. Life Sci. 2020;256:117860. doi: 10.1016/j.lfs.2020.117860 [DOI] [PubMed] [Google Scholar]
- 14.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7(4):189–200. doi: 10.1038/nrneph.2011.16 [DOI] [PubMed] [Google Scholar]
- 15.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121(11):4210–4221. doi: 10.1172/JCI45161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han SJ, Lee HT. Mechanisms and therapeutic targets of ischemic acute kidney injury. Kidney Res Clin Pract. 2019;38(4):427–440. doi: 10.23876/j.krcp.19.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang JR, Yao FH, Zhang JG, Ischemia-reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am J Physiol Renal Physiol. 2014;306(1):F75–F84. doi: 10.1152/ajprenal.00117.2013 [DOI] [PubMed] [Google Scholar]
- 18.Linkermann A, Skouta R, Himmerkus N, Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA. 2014;111(47):16836–16841. doi: 10.1073/pnas.1415518111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danelli L, Madjene LC, Madera-Salcedo I, Early phase mast cell activation determines the chronic outcome of renal ischemia-reperfusion injury. J Immunol. 2017;198(6):2374–2382. doi: 10.4049/jimmunol.1601282 [DOI] [PubMed] [Google Scholar]
- 20.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(5):535–543. doi: 10.1038/nm.2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soranno DE, Gil HW, Kirkbride-Romeo L, Matching human unilateral AKI, a reverse translational approach to investigate kidney recovery after ischemia. J Am Soc Nephrol. 2019;30(6):990–1005. doi: 10.1681/ASN.2018080808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao L, Zhou D, Tan RJ, Sustained activation of Wnt/β-catenin signaling drives AKI to CKD progression. J Am Soc Nephrol. 2016;27(6):1727–1740. doi: 10.1681/ASN.2015040449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Clef N, Verhulst A, D'Haese PC, Vervaet BA. Unilateral renal ischemia-reperfusion as a robust model for acute to chronic kidney injury in mice. PLoS One. 2016;11(3):e0152153. doi: 10.1371/journal.pone.0152153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu Y, Tang C, Cai J. Rodent models of AKI-CKD transition. Am J Physiol Renal Physiol. 2018;315(4):F1098–F1106. doi: 10.1152/ajprenal.00199.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan Q, Ren Q, Li L, A Klotho-derived peptide protects against kidney fibrosis by targeting TGF-beta signaling. Nat Commun. 2022;13(1):438. doi: 10.1038/s41467-022-28096-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bufi R, Korstanje R. The impact of genetic background on mouse models of kidney disease. Kidney Int. 2022;102(1):38–44. doi: 10.1016/j.kint.2022.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clements ME, Chaber CJ, Ledbetter SR, Zuk A. Increased cellular senescence and vascular rarefaction exacerbate the progression of kidney fibrosis in aged mice following transient ischemic injury. PLoS One. 2013;8(8):e70464. doi: 10.1371/journal.pone.0070464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parekh DJ, Weinberg JM, Ercole B, Tolerance of the human kidney to isolated controlled ischemia. J Am Soc Nephrol. 2013;24(3):506–517. doi: 10.1681/ASN.2012080786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kallingal GJ, Weinberg JM, Reis IM. Long-term response to renal ischaemia in the human kidney after partial nephrectomy: results from a prospective clinical trial. BJU Int. 2016;117(5):766–774. doi: 10.1111/bju.13192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu C Yan S Wang Y, et al. . Drug-induced hospital-acquired acute kidney injury in China: a multicenter cross-sectional survey. Kidney Dis. 2021;7(2):143–155. doi: 10.1159/000510455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perazella MA. Pharmacology behind common drug nephrotoxicities. Clin J Am Soc Nephrol. 2018;13(12):1897–1908. doi: 10.2215/CJN.00150118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perazella MA, Rosner MH. Drug-induced acute kidney injury. Clin J Am Soc Nephrol. 2022;17(8):1220–1233. doi: 10.2215/CJN.11290821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharp CN, Doll MA, Dupre TV, Repeated administration of low-dose cisplatin in mice induces fibrosis. Am J Physiol Renal Physiol. 2016;310(6):F560–F568. doi: 10.1152/ajprenal.00512.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu Y, Xiang Y, Wang Y, The STAT1/HMGB1/NF-κB pathway in chronic inflammation and kidney injury after cisplatin exposure. Theranostics. 2023;13(9):2757–2773. doi: 10.7150/thno.81406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki S, Takamura S, Yoshida J. Comparison of gentamicin nephrotoxicity between rats and mice. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol. 1995;112(1):15–28. doi: 10.1016/0742-8413(95)00075-5 [DOI] [PubMed] [Google Scholar]
- 36.Huang H, Jin WW, Huang M, Gentamicin-induced acute kidney injury in an animal model involves programmed necrosis of the collecting duct. J Am Soc Nephrol. 2020;31(9):2097–2115. doi: 10.1681/ASN.2019020204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balakumar P, Rohilla A, Thangathirupathi A. Gentamicin-induced nephrotoxicity: do we have a promising therapeutic approach to blunt it? Pharmacol Res. 2010;62(3):179–186. doi: 10.1016/j.phrs.2010.04.004 [DOI] [PubMed] [Google Scholar]
- 38.Wu JP. Aristolochic acid induces chronic kidney disease in ACE knockout mice. Int J Prev Med. 2021;12:151. PMID: 34912527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jadot I, Colombaro V, Martin B, Restored nitric oxide bioavailability reduces the severity of acute-to-chronic transition in a mouse model of aristolochic acid nephropathy. PLoS One. 2017;12(8):e0183604. doi: 10.1371/journal.pone.0183604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stallons LJ, Whitaker RM, Schnellmann RG. Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol Lett. 2014;224(3):326–332. doi: 10.1016/j.toxlet.2013.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Homsi E, Janino P, de Faria JB. Role of caspases on cell death, inflammation, and cell cycle in glycerol-induced acute renal failure. Kidney Int. 2006;69(8):1385–1392. doi: 10.1038/sj.ki.5000315 [DOI] [PubMed] [Google Scholar]
- 42.Geng Y, Zhang L, Fu B, Mesenchymal stem cells ameliorate rhabdomyolysis-induced acute kidney injury via the activation of M2 macrophages. Stem Cell Res Ther. 2014;5(3):80. doi: 10.1186/scrt469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest. 2009;119(10):2868–2878. doi: 10.1172/JCI39421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu L, Song Y, Zhao M, Yi Z, Zeng Q. Protective effects of edaravone, a free radical scavenger, on lipopolysaccharide-induced acute kidney injury in a rat model of sepsis. Int Urol Nephrol. 2015;47(10):1745–1752. doi: 10.1007/s11255-015-1070-5 [DOI] [PubMed] [Google Scholar]
- 45.Cunningham PN, Dyanov HM, Park P. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J Immunol. 2002;168(11):5817–5823. doi: 10.4049/jimmunol.168.11.5817 [DOI] [PubMed] [Google Scholar]
- 46.Cunningham PN, Holers VM, Alexander JJ. Complement is activated in kidney by endotoxin but does not cause the ensuing acute renal failure. Kidney Int. 2000;58(4):1580–1587. doi: 10.1046/j.1523-1755.2000.00319.x [DOI] [PubMed] [Google Scholar]
- 47.Toscano MG, Ganea D, Gamero AM. Cecal ligation puncture procedure. J Vis Exp. 2011;7(51):2860. doi: 10.3791/2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dejager L, Pinheiro I, Dejonckheere E, Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 2011;19(4):198–208. doi: 10.1016/j.tim.2011.01.001 [DOI] [PubMed] [Google Scholar]
- 49.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4(1):31–36. doi: 10.1038/nprot.2008.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR. Targeting the progression of chronic kidney disease. Nat Rev Nephrol. 2020;16(5):269–288. doi: 10.1038/s41581-019-0248-y [DOI] [PubMed] [Google Scholar]
- 51.Heerspink HJL, Stefansson BV, Correa-Rotter R, Dapagliflozin in patients with chronic kidney disease. N Engl J Med. 2020;383(15):1436–1446. doi: 10.1056/NEJMoa2024816 [DOI] [PubMed] [Google Scholar]
- 52.Li L, Fu H, Liu Y. The fibrogenic niche in kidney fibrosis: components and mechanisms. Nat Rev Nephrol. 2022;18(9):545–557. doi: 10.1038/s41581-022-00590-z [DOI] [PubMed] [Google Scholar]
- 53.Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009;75(11):1145–1152. doi: 10.1038/ki.2009.86 [DOI] [PubMed] [Google Scholar]
- 54.Song D, Shang J, Long Y, Insulin-like growth factor 2 mRNA-binding protein 3 promotes kidney injury by regulating beta-catenin signaling. JCI Insight. 2023;8(2):e162060. doi: 10.1172/jci.insight.162060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hesketh EE, Vernon MA, Ding P, A murine model of irreversible and reversible unilateral ureteric obstruction. J Vis Exp. 2014;94:52559. doi: 10.3791/52559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Narvaez Barros A, Guiteras R, Sola A. Reversal unilateral ureteral obstruction: a mice experimental model. Nephron. 2019;142(2):125–134. doi: 10.1159/000497119 [DOI] [PubMed] [Google Scholar]
- 57.Kurzhagen JT, Dellepiane S, Cantaluppi V, Rabb H. AKI: an increasingly recognized risk factor for CKD development and progression. J Nephrol. 2020;33(6):1171–1187. doi: 10.1007/s40620-020-00793-2 [DOI] [PubMed] [Google Scholar]
- 58.Fu Y, Cai J, Li F, Chronic effects of repeated low-dose cisplatin treatment in mouse kidneys and renal tubular cells. Am J Physiol Renal Physiol. 2019;317(6):F1582–F1592. doi: 10.1152/ajprenal.00385.2019 [DOI] [PubMed] [Google Scholar]
- 59.Skrypnyk NI, Harris RC, de Caestecker MP. Ischemia-reperfusion model of acute kidney injury and post injury fibrosis in mice. J Vis Exp. 2013;78:50495. doi: 10.3791/50495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adam RJ, Williams AC, Kriegel AJ. Comparison of the surgical resection and infarct 5/6 nephrectomy rat models of chronic kidney disease. Am J Physiol Renal Physiol. 2022;322(6):F639–F654. doi: 10.1152/ajprenal.00398.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leelahavanichkul A, Yan Q, Hu X, Angiotensin II overcomes strain-dependent resistance of rapid CKD progression in a new remnant kidney mouse model. Kidney Int. 2010;78(11):1136–1153. doi: 10.1038/ki.2010.287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao Y, Wang C, Wang C, An essential role for Wnt/β-catenin signaling in mediating hypertensive heart disease. Sci Rep. 2018;8(1):8996. doi: 10.1038/s41598-018-27064-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Basting T, Lazartigues E. DOCA-salt hypertension: an update. Curr Hypertens Rep. 2017;19(4):32. doi: 10.1007/s11906-017-0731-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fogo AB. Animal models of FSGS: lessons for pathogenesis and treatment. Semin Nephrol. 2003;23(2):161–171. doi: 10.1053/snep.2003.50015 [DOI] [PubMed] [Google Scholar]
- 65.Lee VW, Harris DC. Adriamycin nephropathy: a model of focal segmental glomerulosclerosis. Nephrology. 2011;16(1):30–38. doi: 10.1111/j.1440-1797.2010.01383.x [DOI] [PubMed] [Google Scholar]
- 66.Lowenborg EK, Jaremko G, Berg UB. Glomerular function and morphology in puromycin aminonucleoside nephropathy in rats. Nephrol Dial Transplant. 2000;15(10):1547–1555. doi: 10.1093/ndt/15.10.1547 [DOI] [PubMed] [Google Scholar]
- 67.Watanabe M, Hiura K, Sasaki H, Okamura T, Sasaki N. Genetic background strongly influences the transition to chronic kidney disease of adriamycin nephropathy in mice. Exp Anim. 2023;72(1):47–54. doi: 10.1538/expanim.22-0057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen GF, Moningka NC, Sasser JM, Arginine and asymmetric dimethylarginine in puromycin aminonucleoside-induced chronic kidney disease in the rat. Am J Nephrol. 2012;35(1):40–48. doi: 10.1159/000334740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mo H, Ren Q, Song D, CXCR4 induces podocyte injury and proteinuria by activating beta-catenin signaling. Theranostics. 2022;12(2):767–781. doi: 10.7150/thno.65948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dai C, Stolz DB, Kiss LP. Wnt/β-Catenin signaling promotes podocyte dysfunction and albuminuria. J Am Soc Nephrol. 2009;20(9):1997–2008. doi: 10.1681/ASN.2009010019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Katayama K, Nomura S, Tryggvason K, Ito M. Searching for a treatment for Alport syndrome using mouse models. World J Nephrol. 2014;3(4):230–236. doi: 10.5527/wjn.v3.i4.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tan RJ Zhou D Xiao L, et al. . Extracellular superoxide dismutase protects against proteinuric kidney disease. J Am Soc Nephrol. 2015;26(10):2447–2459. doi: 10.1681/ASN.2014060613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McGaha TL, Madaio MP. Lupus nephritis: animal modeling of a complex disease syndrome pathology. Drug Discov Today Dis Models. 2014;11:13–18. doi: 10.1016/j.ddmod.2014.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reddy PS, Legault HM, Sypek JP, Mapping similarities in mTOR pathway perturbations in mouse lupus nephritis models and human lupus nephritis. Arthritis Res Ther. 2008;10(6):R127. doi: 10.1186/ar2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagao S, Kugita M, Yoshihara D, Yamaguchi T. Animal models for human polycystic kidney disease. Exp Anim. 2012;61(5):477–488. doi: 10.1538/expanim.61.477 [DOI] [PubMed] [Google Scholar]
- 76.Nagao S, Yamaguchi T. Review of the use of animal models of human polycystic kidney disease for the evaluation of experimental therapeutic modalities. J Clin Med. 2023;12(2):668. doi: 10.3390/jcm12020668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2020;395(10225):709–733. doi: 10.1016/S0140-6736(20)30045-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bonner R, Albajrami O, Hudspeth J, Upadhyay A. Diabetic kidney disease. Prim Care. 2020;47(4):645–659. doi: 10.1016/j.pop.2020.08.004 [DOI] [PubMed] [Google Scholar]
- 79.Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol. 2017;12(12):2032–2045. doi: 10.2215/CJN.11491116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qi Z, Fujita H, Jin J, Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54(9):2628–2637. doi: 10.2337/diabetes.54.9.2628 [DOI] [PubMed] [Google Scholar]
- 81.Dai C, Yang J, Bastacky S. Intravenous administration of hepatocyte growth factor gene ameliorates diabetic nephropathy in mice. J Am Soc Nephrol. 2004;15(10):2637–2647. doi: 10.1097/01.ASN.0000139479.09658.ee [DOI] [PubMed] [Google Scholar]
- 82.Li F, Wang CH, Wang JG, Elevated tissue factor expression contributes to exacerbated diabetic nephropathy in mice lacking eNOS fed a high fat diet. J Thromb Haemost. 2010;8(10):2122–2132. doi: 10.1111/j.1538-7836.2010.03976.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oe Y, Miyazaki M, Takahashi N. Coagulation, protease-activated receptors, and diabetic kidney disease: lessons from eNOS-deficient mice. Tohoku J Exp Med. 2021;255(1):1–8. doi: 10.1620/tjem.255.1 [DOI] [PubMed] [Google Scholar]
- 84.Bai X, Li X, Tian J. A new model of diabetic nephropathy in C57BL/6 mice challenged with advanced oxidation protein products. Free Radic Biol Med. 2018;118:71–84. doi: 10.1016/j.freeradbiomed.2018.02.020 [DOI] [PubMed] [Google Scholar]
- 85.Chen X, Tan H, Xu J, Klotho-derived peptide 6 ameliorates diabetic kidney disease by targeting Wnt/β-catenin signaling. Kidney Int. 2022;102(3):506–520. doi: 10.1016/j.kint.2022.04.028 [DOI] [PubMed] [Google Scholar]
- 86.Gurley SB, Mach CL, Stegbauer J, Influence of genetic background on albuminuria and kidney injury in Ins2(+/C96Y) (Akita) mice. Am J Physiol Renal Physiol. 2010;298(3):F788–F795. doi: 10.1152/ajprenal.90515.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zheng S, Noonan WT, Metreveli NS, Development of late-stage diabetic nephropathy in OVE26 diabetic mice. Diabetes. 2004;53(12):3248–3257. doi: 10.2337/diabetes.53.12.3248 [DOI] [PubMed] [Google Scholar]
- 88.Kovesdy CP. Epidemiology of chronic kidney disease: an update 2022. Kidney Int Suppl (2011). 2022;12(1):7–11. doi: 10.1016/j.kisu.2021.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ninichuk V, Kulkarni O, Clauss S, Anders H. Tubular atrophy, interstitial fibrosis, and inflammation in type 2 diabetic db/db mice. An accelerated model of advanced diabetic nephropathy. Eur J Med Res. 2007;12(8):351–355. PMID: 17933712. [PubMed] [Google Scholar]
- 90.Zhao HJ, Wang S, Cheng H, Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17(10):2664–2669. doi: 10.1681/ASN.2006070798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mohan S Reddick RL Musi N, et al. . Diabetic eNOS knockout mice develop distinct macro- and microvascular complications. Lab Invest. 2008;88(5):515–528. doi: 10.1038/labinvest.2008.23 [DOI] [PubMed] [Google Scholar]
- 92.Hoshi S, Shu Y, Yoshida F, Podocyte injury promotes progressive nephropathy in Zucker diabetic fatty rats. Lab Invest. 2002;82(1):25–35. doi: 10.1038/labinvest.3780392 [DOI] [PubMed] [Google Scholar]
- 93.Favreau F, Rossard L, Zhang K, Expression and modulation of translocator protein and its partners by hypoxia reoxygenation or ischemia and reperfusion in porcine renal models. Am J Physiol Renal Physiol. 2009;297(1):F177–F190. doi: 10.1152/ajprenal.90422.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jayle C, Milinkevitch S, Favreau F, Protective role of selectin ligand inhibition in a large animal model of kidney ischemia-reperfusion injury. Kidney Int. 2006;69(10):1749–1755. doi: 10.1038/sj.ki.5000335 [DOI] [PubMed] [Google Scholar]
- 95.Wang SY, Zhang CY, Cai GY, Chen XM. Method used to establish a large animal model of drug-induced acute kidney injury. Exp Biol Med. 2021;246(8):986–995. doi: 10.1177/1535370220981756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu HF, Li H, Bai G, Establishment of renal failure models by laparoscopy in Bama pigs which underwent partial nephrectomy and radical contralateral nephrectomy. J Vet Res. 2019;63(3):447–455. doi: 10.2478/jvetres-2019-0052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chade AR, Williams ML, Engel J, Guise E, Harvey TW. A translational model of chronic kidney disease in swine. Am J Physiol Renal Physiol. 2018;315(2):F364–F373. doi: 10.1152/ajprenal.00063.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lian X, Zhao J, Wu X, The changes in glucose metabolism and cell proliferation in the kidneys of polycystic kidney disease mini-pig models. Biochem Biophys Res Commun. 2017;488(2):374–381. doi: 10.1016/j.bbrc.2017.05.060 [DOI] [PubMed] [Google Scholar]
- 99.Chen L, Li J, Ou Y, The axolotl kidney: a novel model to study kidney regeneration [published online ahead of print June 6, 2023]. Kidney Int. doi: 10.1016/j.kint.2023.05.020 [DOI] [PubMed] [Google Scholar]
- 100.Gross ML, Hanke W, Koch A. Intraperitoneal protein injection in the axolotl: the amphibian kidney as a novel model to study tubulointerstitial activation. Kidney Int. 2002;62(1):51–59. doi: 10.1046/j.1523-1755.2002.00402.x [DOI] [PubMed] [Google Scholar]
- 101.Nowoshilow S, Schloissnig S, Fei JF, The axolotl genome and the evolution of key tissue formation regulators. Nature. 2018;554(7690):50–55. doi: 10.1038/nature25458 [DOI] [PubMed] [Google Scholar]
- 102.Lust K, Maynard A, Gomes T, Single-cell analyses of axolotl telencephalon organization, neurogenesis, and regeneration. Science. 2022;377(6610):eabp9262. doi: 10.1126/science.abp9262 [DOI] [PubMed] [Google Scholar]
- 103.Fatma S, Nayak U, Swain RK. Methods to generate and evaluate zebrafish models of human kidney diseases. Int J Dev Biol. 2021;65(7-8-9):475–485. doi: 10.1387/ijdb.210041rs [DOI] [PubMed] [Google Scholar]
- 104.Kato Y, Tonomura Y, Hanafusa H. Adult zebrafish model for screening drug-induced kidney injury. Toxicol Sci. 2020;174(2):241–253. doi: 10.1093/toxsci/kfaa009 [DOI] [PubMed] [Google Scholar]
- 105.Outtandy P, Russell C, Kleta R, Bockenhauer D. Zebrafish as a model for kidney function and disease. Pediatr Nephrol. 2019;34(5):751–762. doi: 10.1007/s00467-018-3921-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dow JAT, Simons M, Romero MF. Drosophila melanogaster: a simple genetic model of kidney structure, function and disease. Nat Rev Nephrol. 2022;18(7):417–434. doi: 10.1038/s41581-022-00561-4 [DOI] [PubMed] [Google Scholar]
- 107.Gautam NK, Verma P, Tapadia MG. Drosophila malpighian tubules: a model for understanding kidney development, function, and disease. Results Probl Cell Differ. 2017;60:3–25. doi: 10.1007/978-3-319-51436-9_1 [DOI] [PubMed] [Google Scholar]
- 108.Clevers H. Modeling development and disease with organoids. Cell. 2016;165(7):1586–1597. doi: 10.1016/j.cell.2016.05.082 [DOI] [PubMed] [Google Scholar]
- 109.Li M, Izpisua Belmonte JC. Organoids—preclinical models of human disease. N Engl J Med. 2019;380(6):569–579. doi: 10.1056/NEJMra1806175 [DOI] [PubMed] [Google Scholar]
- 110.Morizane R, Lam AQ, Freedman BS. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol. 2015;33(11):1193–1200. doi: 10.1038/nbt.3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Digby JLM, Vanichapol T, Przepiorski A, Davidson AJ, Sander V. Evaluation of cisplatin-induced injury in human kidney organoids. Am J Physiol Renal Physiol. 2020;318(4):F971–F978. doi: 10.1152/ajprenal.00597.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gupta N, Matsumoto T, Hiratsuka K, Modeling injury and repair in kidney organoids reveals that homologous recombination governs tubular intrinsic repair. Sci Transl Med. 2022;14(634):eabj4772. doi: 10.1126/scitranslmed.abj4772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.De Chiara L, Xia Y. Modelling AKI in vitro: taking organoids to the next level. Kidney Int. 2022;102(3):465–468. doi: 10.1016/j.kint.2022.05.023 [DOI] [PubMed] [Google Scholar]
- 114.Safi W, Marco A, Moya D. Assessing kidney development and disease using kidney organoids and CRISPR engineering. Front Cell Dev Biol. 2022;10:948395. doi: 10.3389/fcell.2022.948395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu M, Cardilla A, Ngeow J, Gong X, Xia Y. Studying kidney diseases using organoid models. Front Cell Dev Biol. 2022;10:845401. doi: 10.3389/fcell.2022.845401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang D, Gust M, Ferrell N. Kidney-on-a-chip: mechanical stimulation and sensor integration. Sensors. 2022;22(18):6889. doi: 10.3390/s22186889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang L, Tao T, Su W. A disease model of diabetic nephropathy in a glomerulus-on-a-chip microdevice. Lab Chip. 2017;17(10):1749–1760. doi: 10.1039/c7lc00134g [DOI] [PubMed] [Google Scholar]
- 118.Schutgens F, Rookmaaker MB, Margaritis T, Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat Biotechnol. 2019;37(3):303–313. doi: 10.1038/s41587-019-0048-8 [DOI] [PubMed] [Google Scholar]
- 119.Cohen A, Ioannidis K, Ehrlich A, Mechanism and reversal of drug-induced nephrotoxicity on a chip. Sci Transl Med. 2021;13(582):eabd6299. doi: 10.1126/scitranslmed.abd6299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vincenti F, Fervenza FC, Campbell KN, A Phase 2, double-blind, placebo-controlled, randomized study of fresolimumab in patients with steroid-resistant primary focal segmental glomerulosclerosis. Kidney Int Rep. 2017;2(5):800–810. doi: 10.1016/j.ekir.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Trachtman H, Fervenza FC, Gipson DS, A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 2011;79(11):1236–1243. doi: 10.1038/ki.2011.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Luo W, Tang S, Xiao X, Translation animal models of diabetic kidney disease: biochemical and histological phenotypes, advantages and limitations. Diabetes Metab Syndr Obes. 2023;16:1297–1321. doi: 10.2147/DMSO.S408170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang Y, Li Q, Liu D, GDF11 improves tubular regeneration after acute kidney injury in elderly mice. Sci Rep. 2016;6(1):34624. doi: 10.1038/srep34624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Polichnowski AJ, Lan R, Geng H. Severe renal mass reduction impairs recovery and promotes fibrosis after AKI. J Am Soc Nephrol. 2014;25(7):1496–1507. doi: 10.1681/ASN.2013040359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhu H Cao C Wu Z, et al. . The probiotic L. casei Zhang slows the progression of acute and chronic kidney disease. Cell Metab. 2021;33(10):2091–2093. doi: 10.1016/j.cmet.2021.08.015 [DOI] [PubMed] [Google Scholar]
- 126.Nangaku M, Kitching AR, Boor P, International Society of Nephrology first consensus guidance for preclinical animal studies in translational nephrology. Kidney Int. 2023;104(1):36–45. doi: 10.1016/j.kint.2023.03.007 [DOI] [PubMed] [Google Scholar]
- 127.Wei Q, Dong Z. Mouse model of ischemic acute kidney injury: technical notes and tricks. Am J Physiol Renal Physiol. 2012;303(11):F1487–F1494. doi: 10.1152/ajprenal.00352.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zager RA, Johnson AC, Becker K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am J Physiol Renal Physiol. 2011;301(6):F1334–F1345. doi: 10.1152/ajprenal.00431.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Perse M, Veceric-Haler Z. Cisplatin-induced rodent model of kidney injury: characteristics and challenges. Biomed Res Int. 2018;2018:1–29. doi: 10.1155/2018/1462802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhan F, Wang X, Zhang J, Yi S, He P. Glutamine alleviates the renal dysfunction associated with gentamicin-induced acute kidney injury in Sprague-Dawley rats. Biotechnol Appl Biochem. 2022;69(1):323–329. doi: 10.1002/bab.2111 [DOI] [PubMed] [Google Scholar]
- 131.Perales-Quintana MM Saucedo AL Lucio-Gutierrez JR, et al. . Metabolomic and biochemical characterization of a new model of the transition of acute kidney injury to chronic kidney disease induced by folic acid. PeerJ. 2019;7:e7113. doi: 10.7717/peerj.7113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Legrand M, Bezemer R, Kandil A. The role of renal hypoperfusion in development of renal microcirculatory dysfunction in endotoxemic rats. Intensive Care Med. 2011;37(9):1534–1542. doi: 10.1007/s00134-011-2267-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu X, Wang N, Wei G, Consistency and pathophysiological characterization of a rat polymicrobial sepsis model via the improved cecal ligation and puncture surgery. Int Immunopharmacol. 2016;32:66–75. doi: 10.1016/j.intimp.2015.12.041 [DOI] [PubMed] [Google Scholar]
- 134.Yan LJ. Folic acid-induced animal model of kidney disease. Animal Model Exp Med. 2021;4(4):329–342. doi: 10.1002/ame2.12194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shinozaki Y, Fukui K, Maekawa M, Unilateral nephrectomized SHR/NDmcr-cp rat shows a progressive decline of glomerular filtration with tubular interstitial lesions. Physiol Res. 2023;72(2):209–220. doi: 10.33549/physiolres.934969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ansari A, Walton SL, Denton KM. Sex- and age-related differences in renal and cardiac injury and senescence in stroke-prone spontaneously hypertensive rats. Biol Sex Differ. 2023;14(1):33. doi: 10.1186/s13293-023-00519-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu N, Makino T, Nogaki F, Coagulation in the mesangial area promotes ECM accumulation through factor V expression in MsPGN in rats. Am J Physiol Renal Physiol. 2004;287(4):F612–F620. doi: 10.1152/ajprenal.00322.2003 [DOI] [PubMed] [Google Scholar]
- 138.Kim JH Ha IS Hwang CI, et al. . Gene expression profiling of anti-GBM glomerulonephritis model: the role of NF-κB in immune complex kidney disease. Kidney Int. 2004;66(5):1826–1837. doi: 10.1111/j.1523-1755.2004.00956.x [DOI] [PubMed] [Google Scholar]
- 139.Isome M, Fujinaka H, Adhikary LP, Important role for macrophages in induction of crescentic anti-GBM glomerulonephritis in WKY rats. Nephrol Dial Transplant. 2004;19(12):2997–3004. doi: 10.1093/ndt/gfh558 [DOI] [PubMed] [Google Scholar]
- 140.Sharma K, McCue P, Dunn SR. Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol. 2003;284(6):F1138–F1144. doi: 10.1152/ajprenal.00315.2002 [DOI] [PubMed] [Google Scholar]
- 141.Hudkins KL, Pichaiwong W, Wietecha T, BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J Am Soc Nephrol. 2010;21(9):1533–1542. doi: 10.1681/ASN.2009121290 [DOI] [PMC free article] [PubMed] [Google Scholar]