

Abstract
Clinically, cardiac dysfunction is a key component of sepsis-induced multi-organ failure. Mitochondria are essential for cardiomyocyte homeostasis, as disruption of mitochondrial dynamics enhances mitophagy and apoptosis. However, therapies targeted to improve mitochondrial function in septic patients have not been explored. Transcriptomic data analysis revealed that the peroxisome proliferator-activated receptor (PPAR) signaling pathway in the heart was the most significantly decreased in the cecal ligation puncture-treated mouse heart model, and PPARα was the most notably decreased among the three PPAR family members. Male Pparafl/fl (wild-type), cardiomyocyte-specific Ppara-deficient (PparaΔCM), and myeloid-specific Ppara-deficient (PparaΔMac) mice were injected intraperitoneally with lipopolysaccharide (LPS) to induce endotoxic cardiac dysfunction. PPARα signaling was decreased in LPS-treated wild-type mouse hearts. To determine the cell type in which PPARα signaling was suppressed, the cell type-specific Ppara-null mice were examined. Cardiomyocyte- but not myeloid-specific Ppara deficiency resulted in exacerbated LPS-induced cardiac dysfunction. Ppara disruption in cardiomyocytes augmented mitochondrial dysfunction, as revealed by damaged mitochondria, lowered ATP contents, decreased mitochondrial complex activities, and increased DRP1/MFN1 protein levels. RNA sequencing results further showed that cardiomyocyte Ppara deficiency potentiated the impairment of fatty acid metabolism in LPS-treated heart tissue. Disruption of mitochondrial dynamics resulted in increased mitophagy and mitochondrial-dependent apoptosis in Ppara△CM mice. Moreover, mitochondrial dysfunction caused an increase of reactive oxygen species, leading to increased IL-6/STAT3/NF-κB signaling. 3-Methyladenine (3-MA, an autophagosome formation inhibitor) alleviated cardiomyocyte Ppara disruption-induced mitochondrial dysfunction and cardiomyopathy. Finally, pre-treatment with the PPARα agonist WY14643 lowered mitochondrial dysfunction-induced cardiomyopathy in hearts from LPS-treated mice. Thus, cardiomyocyte but not myeloid PPARα protects against septic cardiomyopathy by improving fatty acid metabolism and mitochondrial dysfunction, thus highlighting that cardiomyocyte PPARα may be a therapeutic target for the treatment of cardiac disease.
Keywords: sepsis, cardiac dysfunction, mitochondrial dysfunction, mitophagy, inflammation, peroxisome proliferator-activated receptor α
Introduction
Sepsis, defined as systemic inflammatory response syndrome (SIRS) in response to infection, is the most frequent cause of death in intensive care units. The increasing mortality rate of sepsis is mainly due to septic shock [1]. The cardiovascular system is constantly compromised by sepsis and cardiac dysfunction is a main cause of septic shock [2]. Therefore, novel mechanistic insights and effective treatments for sepsis-induced cardiac dysfunction are urgently needed.
Mitochondrial function and quality control are essential for cardiomyocyte homeostasis [3]. Previous studies have shown that mitochondria are the main targets of damage in the acute phase of sepsis [2]. Mitochondria form highly dynamic networks and stabilization of these intricate networks depends on the balance of mitochondrial dynamics due to mitochondrial fission and fusion [4]. However, therapies targeted to mitochondrial dysfunction in septic patients have not been explored.
Disturbed mitochondrial dynamics enhance mitophagy. Macroautophagy (hereafter referred to as autophagy) is a lysosome-dependent process that is characterized by the appearance of double-membrane vesicles, called autophagosomes, that contain cytosolic proteins and organelles that are degraded by lysosomal enzymes. When autophagy selectively degrades mitochondria, it is termed mitophagy [5]. Mitophagy is a protective process under normal physiological responses or mild stress, but excessive autophagic activity under severe or chronic stress leads to massive self-degradation or the accumulation of toxic products [6]. Moreover, enhanced mitochondrial fission results in cytochrome c (Cyto c) release, which further activates the mitochondrion-dependent apoptosis pathway (BCL-2/BAX-Caspase 9-Caspase 3 signaling pathway) [7]. Mitochondrial function, mitophagy and apoptosis are linked to mitochondrial reactive oxygen species (ROS) production. ROS signaling and oxidative stress have a pivotal role in sepsis [8]. Notably, high ROS results in increased oxidative stress and proinflammatory cytokines such as interleukin 6 (IL-6) and tumor necrosis factor (TNF) [9].
Peroxisome proliferator-activated receptor α (PPARα) is a nuclear receptor that regulates transcription of genes involved in energy metabolism following ligand activation [10]. PPARα also enhances anti-inflammatory activity and promotes antioxidant stress. PPARα is mainly found in tissues with high metabolic activity such as liver, heart, and skeletal muscle [10]. Previous studies revealed that PPARα is involved in the occurrence and development of cardiovascular disease [11]. Persistent PPARα activation induces insulin resistance, lipid accumulation and lipotoxicity, that mimics the hearts of diabetic patients [12]. By contrast, using an inducible PPARα transgenic mouse model and the PPARα agonist WY14643, revealed that short-term 2-week activation of PPARα maintained cardiac fatty acid oxidation (FAO), improved myocardial energetics, and partially prevented cardiac remodeling in pressure-overload heart failure [13]. During sepsis, the global whole body Ppara-null mouse had reduced cardiac performance and FAO [14]. A previous study revealed that cardiomyocyte-specific knockout of PPARα exacerbates transverse aortic constriction (TAC)-induced cardiac hypertrophy and fibrosis [15]. However, it remains elusive which cell type that is modulated by PPARα activation plays an important role in sepsis-induced cardiac dysfunction. In this study, cardiomyocyte-specific and myeloid cell-specific PPARα knockout mice were used to investigate which cell type plays a crucial role and the mechanism by which PPARα protects against sepsis-induced cardiac dysfunction.
Materials and methods
The manuscript does not contain clinical studies or patient data.
Animals and treatment
C57BL/6 J wild-type (WT) mice were purchased from Charlies River Company (Beijing, China). Pparafl/fl, cardiomyocyte-specific Ppara-deficient (PparaΔCM) [15], and myeloid-specific Ppara-deficient [16] (PparaΔMac) mice on a C57BL/6 J background were generated as described previously. All mice were kept on a standard 12 h light/dark cycle with free access to normal chow diet and water. All experimental procedures conformed to the US National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and under a project license (AEEI-2018-127) granted by the ethics board of Capital Medical University.
Eight- to twelve-week-old male mice were injected intraperitoneally with LPS (Sigma Aldrich, St. Louis, USA), (5 mg/kg, 10 mg/kg) dissolved in phosphate buffered saline (PBS) or PBS alone as their control mice as described previously [17]. Twelve hours later, the plasma and heart tissue were collected and stored at −80 °C for protein and RNA analysis. For WY14643 treatment, two days before LPS treatment, the WT mice were fed a diet containing 0.1% WY14643 (APExBIO Technology, Houston, USA).
Echocardiography
Left ventricular (LV) wall thickness, chamber dimensions, and cardiac function were analyzed by B-Mode and M-mode echocardiography. Scans were carried out by operators blinded to genotype and treatment using a 40 MHz linear transducer (Vevo 2100, VisaulSonics, Toronto, ON, Canada). Mice were placed on a heating pad under anesthesia with 0.5%-1.5% isoflurane inhalation. LV wall thickness was calculated as the mean of LV anterior wall thickness in diastole (LVAW;d) and LV posterior wall thickness in diastole (LVPW; d) acquired in the short axis view. Images were analyzed off-line by investigators blinded to mouse genotype and treatment using Vevo 2100 analysis software (VisualSonics).
Transmission electron microscopy (TEM)
Hearts were retrograde perfused with buffer containing 4% paraformaldehyde/1% glutaraldehyde/0.1 M Na cacodylate, pH 7.4. Small blocks of tissue from the midsection of the left ventricular wall were fixed (buffer: 2.5% glutaraldehyde/0.1 M Na cacodylate, pH 7.4). Sections (75–80 nm) were cut using a Leica ultramicrotome and examined under a JEM-1400plus electron microscope and a JEM-2100plus electron microscope (JEOL, Tokyo, Japan).
Histopathological analyses
Fresh heart tissues were fixed in 10% phosphate-buffered formalin, embedded in paraffin and then sectioned (4 μm). Hematoxylin and eosin (H&E) staining was performed on the sections using standard procedure as previously described [18]. TUNEL staining was performed by using In Situ Cell Death Detection Kit (Roche, Basel, Switzerland). The relative positive area was quantified with Image-J (NIH, Bethesda, MD, USA). For immunochemistry (IHC), sections were incubated in citrate buffer (pH 6.0) for 10 min at 120 °C, and the endogenous peroxidase was blocked by 0.3% H2O2 for 15 min. The slides were incubated with 5% bovine serum albumin (BSA) in PBS for 60 min to block the non-specific binding sites, followed by incubation with appropriate primary antibodies overnight at 4 °C, and then with horseradish peroxidase (HRP) anti-rabbit IgG antibodies for 1 h. Color was then developed by incubation with DAB Substrate kit (Gene technology, Shanghai, China). After washing in PBS, tissue sections were counterstained with hematoxylin. Major primary antibodies used in the study include CD45 (1:200, ab10558; Abcam, Cambridge, MA, USA), MPO (1:100, ab65871; Abcam, Cambridge, MA, USA), F4/80(1:50, 70076; CST, Boston, USA) and PPARα (1:200, PA1-822A; Invitrogen, Carlsbad, California, USA). Slide imaging was performed using a high-capacity digital slide scanner (Pannoramic SCAN, 3DHISTECH, Budapest, HUN) or viewed by a microscope (SZX7, Olympus, Japan).
Quantitative real-time PCR
Total RNA was extracted from heart tissues using the Trizol Reagent method (Invitrogen, New York, USA). Two µg of total RNA was reverse transcribed into cDNA using GoScriptTM reverse transcription system (Promega, Southampton, UK). Quantitative real-time PCR was performed using SYBR Green Master Mix (Takara, Tokyo, Japan) with CFX Connect Real-Time System (Bio-Rad, Hercules, CA). Amplification was performed at 95 °C for 3 min, 95 °C for 45 s, and 60 °C for 40 s for each step for 40 cycles. The expression of mRNA was normalized to the endogenous housekeeping gene Actb Level changes were calculated by the comparative cycle threshold (ΔΔCT) method. All primers are shown in the Table S1.
Western blot
Whole cell lysate was obtained from heart tissues by Tissue Protein Extraction Reagent (Thermo Scientific, Waltham, MA, USA) containing with protease inhibitors (Cocktail, Thermo Fisher Scientific, Waltham, MA, USA) and phosphatase inhibitors (Cocktail, Thermo Scientific, Waltham, MA, USA). Protein concentrations were measured using the PierceTM BCA Protein Assay Kit (Pierce, Thermo Scientific, Waltham, MA, USA). An equal amount of protein for all samples was separated by SDS-PAGE Gel and then transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA) using a Trans-Blot Turbo Transfer System (Bio-Rad, Hercules, CA). The membranes were precut and blocked with 5% (w/v) non-fat milk or BSA in TBS containing 0.1% (v/v) Tween-20 for 1 h and incubated at 4 °C overnight with primary antibodies against PPARα (1:1000, ab97609; Abcam, Cambridge, MA, USA), LC3I/II (1:1000, 4108 s; CST, Boston, USA), p-NF-κB (1:1000, 3033 s; CST, Boston, USA), T-NF-κB (1:1000, 8242 s; CST, Boston, USA), Parkin (1:1000, 2132 s; CST, Boston, USA), PINK1(1:1000, ab186303, Abcam, Cambridge, MA, USA), DRP 1 (1:1000, ab184247, Abcam, Cambridge, MA, USA), MFN1 (1:1000, ab221661; Abcam, Cambridge, MA, USA), IL-6 (1:1000, 12912 s; CST, Boston, USA), p-STAT3 (1:1000, 9131 s; CST, Boston, USA), STAT3 (1:1000, 8786 s; CST, Boston, USA), Caspase 3 (1:1000, 9662s; CST, Boston, USA), BAX (1:1000, ab32503; Abcam, Cambridge, MA, USA), BCL-2 (1:1000, ab182858; Abcam, Cambridge, MA, USA) followed by secondary antibody with anti-mouse or anti-rabbit HRP-conjugated (7076 s; Cell Signaling Technology, Danvers, MA) for 1 h at room temperature. Blots were developed using the FluorChem E imaging system (Proteinsimple, San Jose, CA, USA) and a chemiluminescence detection kit (Millipore, Billerica, MA, USA). The relative protein levels of other proteins were quantified with Image-J 6.0 and normalized to GAPDH (1:5000, 10494-1-AP, Proteintech, Chicago, USA) and ACTB (1:5000, 66009-1-Ig, Proteintech, Chicago, USA). The remaining Western blot images are shown in the supporting information.
Lactate dehydrogenase activity detection
Lactate dehydrogenase (LDH) activity was detected by a LDH activity detection kit (Solarbio, Beijing, China) according to manufacturer’s instructions. Plasma and reagents were added to 96-well plates in turn. The absorbance at 450 nm was determined. Each measuring tube should be provided with a control tube, ΔA = Ameasure- Acontrol. LDH activity was calculated according to the following formula: LDH(U/mg prot)= y × Vsample ÷ (Cpr × Vsample)÷T × 103 (Vsample: sample volume added to the reaction system; T: reaction time; Cpr: Protein concentration, mg/mL).
IL-1β detection
IL-1β levels in plasma were detected with a mouse IL-1β/IL-1F2 Quantikine ELISA Kit (MLB00C, R&D Systems, Minnesota, USA) according to manufacturer’s instructions. Plasma and reagents were added to 96-well plates in turn. The absorbance at 540 nm and 450 nm was determined. Each measuring sample has two absorbance values.ΔA=A540nm- A450nm.
Measurement of ATP levels
Heart tissue was harvested and lysed by ATP detection lysis solution (Beyotime, Shanghai, China) according to the manufacturer’s protocol. ATP detection working solution was added to the detection hole, then the sample or standard solution was also added to the detection hole and mixed quickly with a micropipette. After an interval of at least 2 s, a luminometer (Enspire, PerkinElmer, Waltham, MA, USA) was used to measure the RLU value.
Mitochondrial complex I detection
Heart tissue was harvested and lysed by mitochondrial complex I extraction buffer (Solarbio, Beijing, China) according to the manufacturer’s protocol. Then, ultrasonic crushing (20% power, ultrasonic 5 s, 10 s interval, repeat 15 times) was used for the determination of complex I enzyme activity and protein content detection. The samples and reagents were added in turn to the 96-well plate, quickly blown and mixed, and the absorbance value A1 was recorded at 10 s. Then the mixture was incubated at 37 °C for an accurate reaction of 2 min, the absorbance A2 was recorded at 2 min, and δ A = A1-A2 was calculated. Mitochondrial complex I activity was calculated according to the following formula: mitochondrial complex I activity(U/mg prot) = [ΔA × Vtotal ÷ (ε × d) × 109] ÷ (Vsample × Cpr) ÷ T (Vtotal: total volume of reaction system, 8.4 × 10−4L; ε: cytochrome c molar extinction coefficient, 1.91 × 104 L·mol-1·cm-1; D: 96 well plate, 0.658 cm; VSample: add sample volume, 0.04 ml; T: reaction time, 1 min; Cpr: sample protein concentration, mg/mL, should be self-determined; 109: unit conversion coefficient, 1 mol = 109 nmol).
Mitochondrial complex IV detection
Heart tissue was harvested and lysed by mitochondrial complex IV extraction buffer (Solarbio, Beijing, China) according to the manufacturer’s protocol. Then, ultrasonic crushing (20% power, ultrasonic 5 s, 10 s interval, repeat 15 times) was used for the determination of complex I enzyme activity and protein detection and quantification. The samples and reagents were added in turn to the 96-well plate, quickly blown and mixed. The initial absorbance value A1 at 550 nm and the absorbance value A2 after 1 min were recorded for the measured tube and blank tube respectively. The measured tube was denoted as A1 measured tube, A2 measured tube and blank tube as A1 blank tube and A2 blank tube. δ A1 = A1 test tube -A2 test tube, δ A2 = A1 blank tube -A2 blank tube, δ A= δ A1- δ A2. Mitochondrial complex IV activity was calculated according to the following formula: mitochondrial complex I activity(U/mg prot) = [ΔA × Vtotal ÷ (ε × d) × 109] ÷ (Vsample × Cpr) ÷ T = 1099 × ΔA ÷ Cpr (Vtotal: total volume of reaction system, 8.4 × 10−4 L; ε: cytochrome c molar extinction coefficient, 1.91 × 104 L·mol-1·cm-1; D: 96 well plate, 0.658 cm; V Sample: add sample volume, 0.04 ml; T: reaction time, 1 min; Cpr: sample protein concentration, mg/mL, should be self-determined; 109: unit conversion coefficient, 1 mol=109 nmol).
ROS detection
Heart tissue was harvested and lysed by homogenate buffer (biorab, Beijing, China) according to the manufacturer’s protocol. The homogenate was centrifuged at 1000 ×g for 10 min at 4 °C and thethe supernatant transferred to a new centrifuge tube. Homogenate supernatant and O13 probe were added into a 96-well plate and mixed. The mixed liquor was incubated for 30 min at 37 °C in the dark. The fluorescence intensity was detected after the excitation wavelength was 488–535 nm and the emission wavelength was 610 nm. The protein was quantified, and the tissue reactive oxygen species intensity was expressed by fluorescence intensity/mg protein.
Cytochrome c release detection
Heart tissue was harvested and processed by a cytochrome c release apoptosis detection kit (Genmed gene, Shanghai, China). Before the experiment, 10 μl GENMED active solution (Reagent F) and 1 ml GENMED purification solution (Reagent C) were added and mixed with 4 ml GENMED lysis buffer (Reagent B), and labeled as GENMED lysis working solution. Heart tissue (0.1 g) was added to 5 ml GENMED cleaning solution (Regent A) and put the minced tissue into a new 15 ml centrifuge tube. GENMED lysis working solution was added and vortexed for 5 s, and used for tissue homogenization. The tissue homogenate was transferred to a 15 ml centrifuge tube and centrifuged at 4 °C for 10 min at 1500 × g. The supernatant was transferred to a new centrifuge tube and centrifuged at 4 °C for 10 min at 10,000 × g. The supernatant was transferred to a new 15 ml centrifuge tube yieldingthe cytoplasm component while the pellet was mitochondria. GENMED solution (Regent G) 100 μL was added to the precipitated particles, followed by vortex oscillation for 15 s, transfer to an ice bath for 15 min, vortex oscillation for 60 s, centrifugation at 4 °C for 2 min at 16000 ×g, and the supernatant was transferred to a 1.5 ml centrifuge tube, which was designated as mitochondrial protein. Mitochondrial protein and cytoplasmic protein were quantitatively detected by Western blotting. Cytochrome c (1:1000, 11940s; CST, Boston, USA) protein levels in cytoplasm were normalized to GAPDH (1:5000, 10494-1-AP, proteintech, Chicago, USA) and in mitochondria was normalized to VDAC1 (1:2000, 4661s; CST, Boston, USA).
Transcriptome analysis
For RNA sequencing, total RNA was extracted from heart tissue of four biological replicates (n = 4 mice per group; i.e. Pparafl/fl/PBS, PparaΔCM/PBS, Pparafl/fl/LPS, PparaΔCM/LPS) using TRIzol reagent (Invitrogen, New York, USA). A total amount of 1 µg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s recommendations and index codes were added to attribute sequences to each sample. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5×). First strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H-). Second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3’ ends of DNA fragments, NEBNext Adaptor with hairpin loop structure was ligated to prepare for hybridization. In order to select cDNA fragments of preferentially 250 ~ 300 bp in length, the library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, USA). Then 3 µl USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min followed by 5 min at 95 °C before PCR. Then PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers and Index (X) Primer. At last, PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Novaseq platform and 150 bp paired-end reads were generated. Quality control Raw data (raw reads) of fastq format were firstly processed through in-house perl scripts. In this step, clean data (clean reads) were obtained by removing reads containing adapter, reads containing ploy-N and low quality reads from raw data. At the same time, Q20, Q30 and GC content the clean data were calculated. All the downstream analyses were based on the clean data with high quality. The raw data were uploaded to https://www.ncbi.nlm.nih.gov/geo/ (GSE229298). For anyone who would like to further analyze our data, please inform us.
Moreover, for analysis of the comprehensive insights into the septic cardiac dysfunction, published transcriptome data (SRA at the NCBI under the accession number SRP00123) were downloaded [19]. Hierarchical clustering g was performed based on differentially expressed mRNAs using R package heatmap (version:1.0.12). The Gene Ontology (GO) is a structured and controlled vocabulary of terms. The terms are subdivided in three non-overlapping ontologies, Molecular Function (MF), Biological Process (BP) and Cellular Component (CC). GO analysis was performed to analyze the primary function of the differential expression of mRNAs. Statistical analysis was performed using the two side Fisher’s exact test and Benjamini-Hochberg was used for multiple tests correction (FDR was used to adjust the P-values for multiple comparisons). The threshold set for significantly changed GOs was and P < 0.01 was considered as the significance threshold.
Kyoto Encyclopedia of Genes and Genomes (KEGG) is a knowledge base for systematic analysis of gene functions, linking genomic information with higher order functional information. Pathway analysis was used to find out the significant pathway of the differential genes according to the KEGG database. Statistical analysis was performed using the two side Fisher’s exact test and Benjamini-Hochberg was used for multiple tests correction (FDR was used to adjust the P-values for multiple comparisons). The threshold set for significantly changed pathways was and P < 0.05 was considered as the significance threshold.
Statistical analyses
Results are expressed as mean ± SD. All statistical analyses were performed using Graphpad Prism 8.0 (GraphPad Software, La Jolla, CA). Data were first subjected to the D’Agostino and Pearson omnibus normality test. If the normality test was passed, data were analyzed by Student’s t-test for experiments with two conditions and one variable, by one-way ANOVA followed by Bonferroni’s post-test for experiments with more than two conditions and one variable. Difference was considered significant at P < 0.05. The ‘n’ in figure legends indicates biological replicates.
Results
PPARα expression and signaling is significantly decreased in LPS-induced cardiac dysfunction
To obtain comprehensive insights into the septic cardiac dysfunction, published transcriptome data on CLP- and sham-mice (SRA number: SRP00123) were downloaded and analyzed [19]. A comparison of the transcription profiles revealed that sepsis resulted in an increase of 202 mRNAs and a decrease of 318 mRNAs in WT mice. Hierarchical clustering and principal component analysis (PCA) indicated a good separation between normalized read counts of all experimental groups (Fig. S1a, b). KEGG pathway analysis identified that PPAR signaling was significantly decreased in septic cardiac dysfunction. Heatmap analysis further revealed that Ppara mRNA is significantly decreased in sepsis-induced cardiac dysfunction which is companied by decreased PPARα target gene mRNA levels (Fig. 1a). To further explore the expression of PPARα in sepsis, an LPS (5 mg/kg or 10 mg/kg)-induced endotoxemia model was applied (Fig. S2a–c, Table S2). Ppara mRNA was the most significantly decreased among all three PPAR family members in LPS-treated hearts (Fig. 1b). PPARα protein was also significantly decreased in LPS-treated mouse hearts (Fig. 1c). Accordingly, the PPARα target gene Pgc1a, Cpt1b and Acox1 mRNAs were also decreased in LPS-treated hearts compared with control hearts (Fig. 1d). IHC staining showed that PPARα reduction mainly occurred in cardiomyocyte nuclei in LPS-treated hearts (Fig. 1e), indicating that cardiomyocyte PPARα might play an important role in LPS-induced cardiac dysfunction.
Fig. 1. The expression of PPARα and its target genes are decreased in LPS-treated heart tissue.

A published transcriptomics data of heart tissue from WT and CLP mice. a Ten KEGG pathway analysis of identified down- and up-regulated genes; Heatmap of differential gene in heart tissue from WT and CLP treatment mice. Wild-type (WT) mice were randomized to LPS (5 mg/kg, 10 mg/kg) or PBS treatment. b Expression of Ppara, Ppard, Pparg mRNA levels in heart tissue after PBS or LPS treatment (n = 2–4). c Expression of PPARα protein in heart tissue after PBS or LPS treatment, and quantitative analysis of WB (n = 3–4). d Expression of Cpt1b, Acox1, Cd36 and Pgc1a mRNAs in heart tissue after PBS or LPS treatment (n = 3–4). e Representative images of IHC staining of PPARα expression in heart tissue after PBS or LPS (5 mg/kg) treatment. Data are expressed as mean ± SD. *P < 0.05, ***P < 0.001.
Cardiomyocyte-specific Ppara deficiency but not myeloid cell-specific Ppara deficiency potentiates LPS-induced cardiac dysfunction
To explore which cell type that PPARα plays a predominant role in its protection against LPS-induced cardiomyopathy, Pparafl/fl mice and PparaΔMac mice were intraperitoneally injected with LPS to construct an endotoxemia model. Echocardiography showed that LPS induced a significant decrease of cardiac dysfunction, but there was no significant difference in Pparafl/fl mice and PparaΔMac mice after 10 h of LPS treatment (Fig. 2a, Table S3). Sepsis-induced mortality and cardiac dysfunction were previously ascribed to an increased inflammatory response [20]. Innate immune cells are the early responders to infection and tissue damage, especially neutrophils and macrophages [21]. There was no significant difference in the inflammatory response between Pparafl/fl mice and PparaΔMac mice as revealed by H&E or IHC staining and the mRNA levels of inflammatory or oxidative stress related genes (Fig. 2b, c, Fig. S3).
Fig. 2. Myeloid cell-specific Ppara deficiency has no significant effect in LPS-induced cardiac dysfunction.

Pparafl/fl and PparaΔMac mice were randomized to LPS (5 mg/kg) or PBS administration for 12 h. a Representative two-dimensional guided M-mode echocardiograms of the left ventricle in Pparafl/fl and PparaΔMac mice after PBS or LPS treatment and quantification of ejection fraction (EF%), fractional shortening (FS%) (n = 4–5). b Representative images of hematoxylin and eosin (H&E) and IHC (CD45+, MPO+, F4/80+) staining of heart sections. c Il1b, Il6, Tnf mRNA levels in Pparafl/fl and PparaΔMac mouse heart tissue after PBS or LPS treatment (n = 4–6). Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
To investigate the role of PPARα in cardiac parenchymal cells in septic cardiac dysfunction, the inducible cardiomyocyte-specific PPARα knockout (PparaΔCM) mice was constructed. PparaΔCM and Pparafl/fl mice were injected with LPS (5 mg/kg and 10 mg/kg) for 10 h. Echocardiography showed that LPS induced PparaΔCM and Pparafl/fl mouse cardiac dysfunction, EF% and FS%, that were further dose-dependently decreased in LPS-treated PparaΔCM mice compared with Pparafl/fl mice, but there was no difference between PparaΔCM and Pparafl/fl mice under basal conditions (Fig. 3a, Tables S4, S5). LDH activity and IL-1β levels in plasma of Pparafl/fl mice were elevated after LPS treatment and further increased in PparaΔCM mouse plasma (Fig. 3b, c). H&E and IHC staining showed that cardiac inflammatory cell infiltration was elevated in PparaΔCM mice compared with Pparafl/fl mice after LPS treatment, especially neutrophils, however, there was no significant difference between the macrophage in heart tissues from PparaΔCM mice and Pparafl/fl mice after LPS treatment (Fig. 3d, e). Moreover, quantification of Il1b, Il6, and Tnf mRNAs showed that the cardiac inflammatory response was elevated in PparaΔCM mice compared with Pparafl/fl mice after LPS treatment (Fig. 3f). These results show that PPARα deficiency in cardiomyocyte potentiated LPS induced-cardiac dysfunction in a dose-dependent manner, and thus low dose LPS was used in subsequent experiments. These data indicated that cardiomyocyte PPARα and not myeloid cell PPARα plays an important role in septic cardiac dysfunction.
Fig. 3. Cardiomyocyte-specific Ppara knockout potentiates LPS-induced cardiac dysfunction and cardiac inflammation.

Pparafl/fl and PparaΔCM mice were randomized to LPS (5 mg/kg, 10 mg/kg) or PBS administration for 12 h. a Representative two-dimensional guided M-mode echocardiograms of the left ventricle in Pparafl/fl and PparaΔCM mice after PBS or LPS treatment and quantification of ejection fraction (EF%), fractional shortening (FS%) (n = 5). b LDH activity detection in plasma from Pparafl/fl and PparaΔCM mice after PBS or LPS treatment (n = 5–10). c IL-1β protein in plasma from Pparafl/fl and PparaΔCM mice after PBS or LPS treatment (n = 4–6). d Representative images of hematoxylin and eosin (H&E) staining of heart sections. e Representative images of IHC (CD45+, MPO+, F4/80+) staining of heart sections in Pparafl/fl and PparaΔCM mice after PBS or LPS treatment (5 mg/kg), and quantitative analysis of IHC (n = 3). f Pro-inflammatory gene Il1b, Il6, and Tnf mRNA levels in Pparafl/fl and PparaΔCM mouse heart tissue after PBS or LPS treatment (n = 3–6). Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Cardiomyocyte-specific Ppara deficiency potentiates LPS-induced mitochondrial dysfunction and dynamic disorders
Next, the mechanism by which cardiomyocyte PPARα deficiency exacerbates septic cardiac dysfunction was explored. Mitochondria are essential for generating ATP and mitochondrial dysfunction is one of the important mechanisms leading to cardiac dysfunction in sepsis [22]. TEM imaging revealed that LPS treatment induced mitochondrial morphology variation and dysfunction, which is manifested by partial loss of mitochondrial cristae, vacuoles, and an irregular arrangement which was more pronounced in PparaΔCM mice as compared with Pparafl/fl mice. TEM also showed that mitochondrial fission and fusion were both markedly increased (Fig. 4a). Mitochondria are responsible for managing cell life and death by balanced homeostasis passing through a network of structures, regulated principally via fission and fusion [23]. The mitochondrial protein mitofusin 1 (MFN1) and dynamin-related protein (DRP1) levels were measured in heart tissue from Pparafl/fl and PparaΔCM mice treated with LPS or PBS, revealing that MFN1 and DRP1 were further elevated in PparaΔCM as compared with Pparafl/fl mice after LPS treatment (Fig. 4b). Interestingly, fission and fusion related-mRNAs were significantly decreased in PparaΔCM mice after LPS treatment, but Mfn2 mRNA level was not significantly different (Fig. S4), which is in contrast to the protein results. This result indicates that cardiomyocyte Ppara disruption may lead to reduced degradation of DRP1 and MFN1 proteins in LPS-treated heart tissue. The mRNAs results of mitochondrial proliferation-related genes indicated that mitochondrial proliferation in PparaΔCM mice was significantly inhibited after LPS treatment (Fig. 4c). For analysis of mitochondrial function, ATP production and mitochondrial complex activity results revealed that they were significantly decreased in PparaΔCM mice after LPS treatment as compared to Pparafl/fl mice (Fig. 4d, e). Inhibition of mitochondrial complex I, III, and IV of the electron transport chain is thought to promote ROS production [9]. Enhanced ROS levels are known to increase cardiac damage [24]. ROS production was elevated in LPS-treated hearts and further increased in PparaΔCM mice compared with Pparafl/fl mice (Fig. 4f). These results indicate that loss of cardiomyocyte PPARα exacerbates LPS-induced cardiac dysfunction by potentiating mitochondrial dysfunction and dynamic disorders.
Fig. 4. LPS-induced mitochondrial dysfunction is exacerbated in cardiac tissue of PparaΔCM mice.

Pparafl/fl and PparaΔCM mice were randomized to LPS (5 mg/kg) or PBS administration for 12 h. a Representative images of myocardial tissue ultra -structure and mitochondria from Pparafl/fl and PparaΔCM mouse heart tissue after PBS or LPS treatment, the area indicated by the arrow is the area positive for mitochondrial fusion and fission. b MFN1 and DRP1 protein levels in Pparafl/fl and PparaΔCM heart tissue after PBS or LPS treatment, and quantitative analysis of WB (n = 5–6). c The mitochondrial biosynthesis gene Pgc1a, Erra, and Nrf1 mRNA levels in heart tissue from Pparafl/fl and PparaΔCM mice after PBS or LPS treatment (n = 3–5). d ATP contents in heart tissue from Pparafl/fl and PparaΔCM heart tissue after PBS or LPS treatment (n = 5–6). e Mitochondria complex I/IV activity in heart tissue from Pparafl/fl and PparaΔCM heart tissue after PBS or LPS treatment (n = 4–8). f ROS production in heart tissue from Pparafl/fl and PparaΔCM heart tissue after PBS or LPS treatment (n = 3). Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Cardiomyocyte-specific Ppara deficiency potentiates LPS-induced fatty acid metabolism dysfunction
To elucidate the mechanism of how cardiomyocyte PPARα maintains mitochondrial homeostasis, next-generation sequencing of RNAs from heart tissue of PBS- or LPS-treated Pparafl/fl and PparaΔCM mice was performed. PCA showed an ideal separation of normalized read counts between normalized read counts of all experimental groups (Fig. 5a). Venn diagram revealed that LPS induced the downrelation of 1922 genes and upregulation of 1846 genes in Pparafl/fl mice. Cardiomyocyte PPARα knockout induced the downregulation of 232 genes and upregulation of 207 genes in LPS- treated PparaΔCMmice compared with LPS-treated Pparafl/fl mice. From the downregulated genes, only 16 genes overlapped and in the upregulated genes, only 14 genes overlapped (Fig. 5b, Table S6). Volcanoplot showed that cardiomyocyte PPARα deficiency significantly decreased fatty acid metabolism-related gene mRNAs such as Decr1, Acox1, Cpt2, Fabp3, Acsl1, Ucp3, Acot2, Cpt1b, and Acaa2, in heart tissue after LPS treatment (Fig. 5c). GO analysis revealed that fatty acid metabolic-related processes, including fatty acid metabolism, long-chain fatty acid metabolism, lipid metabolism, including fatty acid β-oxidation and fatty acid oxidation, were significantly decreased in LPS-treated PparaΔCM mice. However, genes involved in the negative regulation of protein kinase activity, negative regulation of JAK-STAT cascade, cytokine-mediated signaling, immune response and acute-phase response were increased in LPS treated PparaΔCM mice compared with Pparafl/fl mice (Fig. 5d). Hierarchical clustering further revealed decreased fatty acid-related genes in all PBS- and LPS-treated Pparafl/fl mice and PparaΔCM mice. The heatmap showed that cardiomyocytes PPARα knockout exacerbated LPS-induced fatty acid metabolism impairment (Fig. 5e, Table S7). Previous studies showed that fatty acid accumulation and metabolic disorders disrupt mitochondrial homeostasis [25]. Thus, these results suggest that cardiomyocyte PPARα maintains mitochondrial homeostasis through the regulation of fatty acids metabolism.
Fig. 5. Cardiomyocytes-specific Ppara disruption potentiates LPS-induced fatty acid metabolism dysfunction.

Pparafl/fl and PparaΔCM mice were administered PBS or LPS (5 mg/kg) for 12 h. a PCA performed using DESeq2 rlog-normalized RNA-seq data. Loadings for principal components (PC) 1, PC2 and PC3 are shown in graph on x-, y- and z-axis. b Venn diagrams showing the number of twofold upregulated (top panel) and two-fold downregulated (bottom panel) genes and their overlap in hearts of Pparafl/fl and PparaΔCM mice after PBS or LPS treatment. c Volcano Plot showing the upregulated and downregulated genes in heart tissue from PparaΔCM mice compared with Pparafl/fl mice after LPS treatment. d Selected enrichment of GO term analysis of upregulated or downregulated genes in hearts of LPS-treated PparaΔCM mice compared with Pparafl/fl mice. e Heatmap of normalized expression values of genes involved in fatty acid metabolism in hearts of Pparafl/fl and PparaΔCM mice after PBS or LPS treatment.
Cardiomyocyte-specific Ppara deficiency increases LPS-induced mitophagy
Mitophagy, a process of damaged mitochondria elimination, has been linked to recovery of cardiac function during sepsis in animal models, but excessive autophagic activity under severe or chronic stress leads to massive self-degradation or the accumulation of toxic products [7]. The expression of mitogen-related factors or degradation of mitofusion-related factors is necessary to induce mitophagy, and they regulate the process of mitophagy by interacting with LC3 adaptor proteins or LC3 receptors [26]. TEM imaging indicated that LPS induced autophagosomes were more prominent in PparaΔCM mice than in Pparafl/fl mice (Fig. 6a). Mitophagy is mainly a PINK1-Parkin-mediated autophagy process. When mitochondria are damaged, PINK1 accumulates in the outer membrane of mitochondria through the outer membrane translocation enzyme (TOM) to activate and recruit Parkin. Subsequently, VDAC1 and MFN1/2 proteins on the outer membrane of mitochondria, are ubiquitinated by Parkin to induce mitochondrial autophagy. After ubiquitination, receptor proteins including P62 accumulate in the outer membrane of mitochondria, resulting in the ubiquitin products being recruited into autophagosomes by binding to LC3. Mature autophagosomes fuse with lysosomes to form autophagolysosomes, and the captured mitochondria are subsequently degraded [27]. Parkin/PINK1 protein levels were elevated after LPS treatment, and further increased in heart tissue from PparaΔCM mice as compared to Pparafl/fl mice (Fig. 6b). Consistent with the above results, the protein levels of P62 and LC3II were elevated in both LPS-treated PparaΔCM and Pparafl/fl hearts, but more prominent in PparaΔCM mice (Fig. 6c). These results indicate that cardiomyocyte PPARα ameliorates LPS-induced mitochondrial dysfunction and dynamic disorders by inhibiting mitophagy.
Fig. 6. Cardiomyocytes-specific Ppara disruption potentiates LPS-induced mitophagy.

Pparafl/fl and PparaΔCM mice were randomized to LPS (5 mg/kg) or PBS administration for 12 h. a Representative images of mitophagy in heart tissue section from Pparafl/fl and PparaΔCM mice after PBS or LPS treatment, the area circled is the autophagosome. Magnification: top ×1500; bottom ×3000. b Parkin, PINK1 protein levels in heart tissues of Pparafl/fl and PparaΔCM mice after PBS or LPS treatment and quantitative analysis (n = 6–8). c P62 and LC3II/I protein levels in heart tissues from Pparafl/fl and PparaΔCM mice after PBS or LPS treatment and quantitative analysis (n = 5–8). Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
The autophagy inhibitor 3-MA improves LPS-induced cardiac and mitochondrial dysfunction in PparaΔCM mice
To further verify the role of mitophagy in exacerbation of cardiomyocyte-specific PPARα knockout mouse-induced septic cardiac dysfunction, 3-MA, a widely used inhibitor of autophagosome formation, was used. Echocardiography showed that 3-MA improved LPS-induced cardiac dysfunction in both Pparafl/fl and PparaΔCM mice; even EF% and FS% reached the same level as Pparafl/fl mice (Fig. S5a, 5b, Table S8). LDH activity was improved in the plasma of PparaΔCM mice after 3-MA treatment (Fig. S5c). H&E and IHC staining of CD45+ cells showed that 3-MA reduced inflammatory cell infiltration in Pparafl/fl and in PparaΔCM mouse heart tissues (Fig. S6a, b). Reduced p-NF-κB protein levels further confirmed that 3-MA alleviated LPS-induced augmentation of PparaΔCM mouse inflammation (Fig. S6c). Il1b, Il6 and Tnf mRNA levels were also decreased in 3-MA treated septic hearts, and were at similar levels in PparaΔCM and Pparafl/fl mice (Fig. S6d). Mitochondrial morphology, as detected by electron microscopy, revealed that 3-MA significantly improved LPS-induced mitochondrial derangement, cristae disappearance and mitochondria fusion in PparaΔCM mice which was similar to Pparafl/fl mice (Fig. S7a). 3-MA treatment improved cardiomyocyte-specific PPARα knockout-induced inhibition of mitochondria biogenesis (Fig. S7b). ATP contents were then measured further to confirm improvement of mitochondrial dysfunction (Fig. S7c). In summary, 3-MA improved LPS-induced cardiac and mitochondrial dysfunction in PparaΔCM mice and achieved the same level as Pparafl/fl mice. These results showed that inhibition of autophagosome formation improved cardiomyocyte PPARα knockout mouse-induced septic cardiac dysfunction and exacerbation of mitochondrial dysfunction.
Cardiomyocyte-specific Ppara knockout has increased mitochondrial dysfunction-induced inflammation and mitochondrial-dependent apoptosis
High production of ROS increases the level of proinflammatory cytokines such as IL-6 and TNF-α [9]. IL-6 protein was elevated after LPS treatment, and further increased in PparaΔCM mice (Fig. 7a). Consistent with this, p-STAT3 protein levels were increased. Enhanced IL-6 inflammatory cytokines activate NF-κB and the pro-inflammatory response [28, 29] (Fig. 7b). p-NF-κB protein levels were elevated after LPS treatment, and further increased in Ppara△CM mice (Fig. 7c). Mitochondria participate in apoptosis through releasing cytochrome c to promote caspase activation in cytoplasm, which is the result of the loss of the mitochondrial outer membrane caused by pro-apoptosis members of the Bcl-2 family. This process is always accompanied by mitochondrial fission [30]. After LPS treatment, cytochrome c was released from mitochondria to the cytoplasm in heart tissue. The cardiomyocyte-specific PPARα knockout exacerbated cytochrome c release (Fig. 8a). Following cytochrome c release, the expression of BAX was slightly increased, but BCL-2 was significantly decreased (Fig. 8b), accompanied by increased cleaved caspase 3 protein levels (Fig. 8c). TUNEL staining also verified these results (Fig. 8d). These results indicate that cardiomyocyte PPARα knockout increases LPS-induced mitochondrial-dependent apoptosis and ROS production-induced inflammation through the IL-6/STAT3/NF-κB signaling pathway.
Fig. 7. Cardiomyocyte-specific Ppara deletion increases mitochondrial dysfunction-induced inflammation.

Pparafl/fl and PparaΔCM mice were randomized to LPS (5 mg/kg) or PBS administration. a IL-6 protein level in heart tissue from Pparafl/fl and PparaΔCM mice after PBS or LPS treatment and quantitative analysis of Western blot (n = 6–8). b p-STAT3 and STAT3 protein levels and quantitative analysis of Western blot (n = 6–7). c p-NF-κB and T-NF-κB protein levels and quantitative analysis (n = 3–4). Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Fig. 8. Cardiomyocyte-specific Ppara deletion increased mitochondrial-dependent apoptosis.

Pparafl/fl and PparaΔCM mice were randomized to LPS (5 mg/kg) or PBS administration. a Cytochrome c protein levels in mitochondria and cytoplasm from Pparafl/fl and PparaΔCM mouse heart tissues after PBS or LPS treatment and quantitative analysis of Western blot (n = 3). b BCL-2 and BAX protein levels in heart tissue from Pparafl/fl and PparaΔCM mice after PBS or LPS treatment and BAX/BCL-2 quantitative analysis of Western blot (n = 3–4). c Cleaved caspase 3 and total caspase 3 protein levels and quantitative analysis of Western blots (n = 5–6). d Representative images of TUNEL staining of heart sections and quantitative analysis of positive area (n = 3-4). Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
PPARα activation alleviates LPS-induced cardiac dysfunction and mitochondrial dysfunction
To confirm the protective effect of PPARα activation in septic cardiac dysfunction, WT mice were administered WY14643 (0.1%) in the diet for two days before LPS treatment. Cpt1b mRNA levels were increased after WY14643 treatment thus verifying activation of PPARα signaling (Fig. S8a). Echocardiography results showed that EF% and FS% were improved in the WY14643 diet-fed group compared with the control diet-treated mice after LPS administration (Fig. 9a, Table S9). LDH activity was decreased in the WY14643 diet group compared with the control diet group after LPS treatment (Fig. 9b). ATP contents were elevated in WY14643 diet-fed mice compared with control diet-fed mice after LPS treatment (Fig. 9c). The mRNA levels of cardiac inflammatory factors were reduced in WY14643-treated mice compared with control diet-fed mice after LPS treatment (Fig. 9d). H&E staining and IHC staining of CD45+ cells proved that WY14643 treatment alleviated LPS induced- inflammatory cells infiltration (Fig. 9e). Moreover, p-NF-κB protein levels were also decreased after WY14643 treatment (Fig. 9f). To confirm that activation of PPARα protects the heart against septic cardiac dysfunction by alleviating mitochondrial dysfunction, levels of mRNAs encoding mitochondrial biogenesis and dynamics proteins were measured. WY14643 treatment improved LPS-induced mitochondrial biogenesis inhibition and dynamics disturbance (Fig. S8b, c). LC3II protein was elevated by LPS in control diet-treated mice, but this elevation was reduced by WY-14643 diet treatment (Fig. S8d). Thus, PPARα activation protects against septic cardiac dysfunction by alleviating mitochondrial dysfunction.
Fig. 9. PPARα activation improves LPS-induced cardiac dysfunction and mitochondrial dysfunction.

WT mice were given diet with or without WY14643 for two days before LPS (5 mg/kg) or PBS treatment for 12 h. a Representative two-dimensional guided M-mode echocardiograms of the left ventricule in WT mice fed with control or WY14643-supplemented diets and treated with LPS (5 mg/kg) or PBS and quantification of ejection fraction (EF%) and fractional shortening (FS%) in WT mice fed with control or WY14643-supplemented diets and treated with LPS (5 mg/kg) or PBS (n = 6-7). b LDH activity detection in plasma from WT mice fed with control or WY14643-supplemented diets and treated with LPS (5 mg/kg) or PBS (n = 6). c ATP contents in heart tissue from WT mice fed with control or WY14643-supplemented diets and treated with LPS (5 mg/kg) or PBS (n = 4). d Il1b, Il6, and Tnf mRNA levels in heart tissue from WT mice fed with control or WY14643-supplemented diets and treated with LPS (5 mg/kg) or PBS (n = 3–6). e Representative images of H&E staining and IHC staining for detecting CD45+ cells in heart sections. f p-NF-κB and T-NF-κB protein levels in heart tissue from WT mice fed with control or WY14643-supplemented diets and treated with LPS (5 mg/kg) or PBS and quantitative analysis (n = 4). Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
PPARα plays an important role in regulating energy metabolism in cardiomyocytes and has anti-inflammatory and anti-oxidative properties that improve pathologies associated with cardiovascular disease, including inflammation, metabolic disorders and cardiac dysfunction [31]. The current study demonstrated that PPARα expression was significantly inhibited in septic cardiac dysfunction as revealed by quantitative analysis of LPS-treated heart tissue and a transcriptome analysis of CLP-treated heart tissue. These results indicate that PPARα plays a major role in sepsis-induced cardiac dysfunction.
An earlier study revealed that absence of PPARα causes a reduction of cardiac performance and fatty acid oxidation in sepsis [14]. PPARα expression in nonhematopoietic tissues was found to have a critical role in determining the clinical outcome of experimental polymicrobial sepsis through a bone marrow transplant experiment [32]. However, which cell type has the primary role in the PPARα protection remained unclear. The current study proposed that cardiomyocyte PPARα plays a prominent role in septic cardiac dysfunction as cardiomyocyte-specific Ppara deficiency resulted in an exacerbation of LPS-induced cardiac dysfunction and inflammation. In addition, PPARα activation by WY14643 alleviated LPS-induced cardiac dysfunction and inflammation. In this study, myeloid-specific PPARα knockout did not significantly affect LPS-induced cardiac dysfunction, which is consistent with the previous study [32]. These results suggest that cardiomyocyte PPARα plays an important role in sepsis-induced cardiac dysfunction and thus could be a therapeutic target.
Mitochondrial dysfunction is a critical pathophysiological mechanism of septic cardiomyopathy. Cardiomyocytes have a high abundance of mitochondria that are essential for generating ATP [33]. Mitochondria form highly dynamic networks and the homeostasis of these intricate networks depends on the balance between the opposing processes of mitochondrial fission and fusion [34]. Regulators of mitochondrial dynamic are under the control of fusion and fission proteins. Mitochondrial fission and fusion work together to maintain the mitochondrial and cellular homeostasis [35]. In this study, the role of cardiomyocyte PPARα in LPS-induced mitochondrial dysfunction was investigated. Disruption of cardiomyocyte Ppara exacerbates LPS-induced mitochondrial damage associated with swelling, increased quantity, disappearance of palate and appearance of vacuoles as revealed by TEM. The majority of mitochondrial ATP production is derived from fatty acid oxidation [36]. The present study revealed that ATP contents and mitochondrial complexes I and IV were significantly reduced in PparaΔCM mice compared with Pparafl/fl mice after LPS treatment.
PPARα is well-known for controlling the expression of numerous genes involved in a plethora of lipid metabolic pathways, including mitochondrial fatty acid oxidation (FAO), synthesis and breakdown of triglycerides (TGs), formation of lipid droplets, lipoprotein metabolism, gluconeogenesis, and various other metabolic pathways [10]. Previous studies showed that fatty acid accumulation and metabolic disorders disrupt mitochondrial homeostasis [25]. To explore the mechanism of how cardiomyocyte PPARα regulates mitochondrial function, RNA sequencing was performed. RNA sequencing results showed that cardiomyocyte Ppara deletion resulted in a significant inhibition of fatty acid metabolism in hearts from LPS-treated mice. Furthermore, the interaction between PPARα and members of the PGC-1 family of regulated coactivators (PGC-1α, PGC-1β, and PRC) is crucial for controlling nuclei-mitochondria interactions. PPARα target genes are involved in the respiratory chain, notably mitochondrial transcription, translation and replication machinery, and protein import and assembly apparatus among others. These factors are in turn activated directly or indirectly by the PGC-1 family coactivators [37]. In the present study, cardiomyocytes-specific PPARα knockout mice had augmented LPS-induced inhibition of PGC-1α expression. Overall, PPARα maintains mitochondrial homeostasis through keeping the balance of fatty acid metabolism and transcription factor activity. Moreover, TEM, qPCR, and Western blot results showed that loss of PPARα in cardiomyocytes exacerbates mitochondrial fusion and fission, ultimately inducing loss of mitochondrial function. Furthermore, PPARα activation increased ATP levels that were markedly decreased upon LPS treatment, and increased the expression of genes involved in mitochondrial function and biogenesis. These findings highlight that cardiomyocyte PPARα could be a therapeutic target in mitochondrial dysfunction in the septic heart.
Mitophagy, a specialized form of autophagy that functions in regulating the turnover of dysfunctional mitochondria, has a pivotal role in maintaining mitochondrial dynamics and homeostasis [38]. Mitophagy is a selective autophagic process essential for cellular homeostasis. Numerous studies have shown that mitophagy is a “double-edged sword” and is protective under physiological responses or mild stress, but excessive autophagic activity under severe or chronic stress leads to mitochondria degradation or the accumulation of toxic products [39]. A previous study demonstrated that restoration of LPS-induced cardiac mitochondrial number treated with rosiglitazone was associated with mitophagy inhibition which was shown by decreased autophagy marker levels [39]. Moreover, in an ischemia-reperfusion-induced cardiac injury model, reducing excessive mitophagy could preserve mitochondrial integrity and protect against ischemia-reperfusion-induced cardiac injury [40]. Thus, excessive mitophagy causes mitochondrial damage and inhibition of mitophagy improves mitochondrial function. In general, mitophagy was divided into PRKN-independent and -dependent mitophagy pathways which are known as the best-studied pathway [27]. Parkin is an E3 ubiquitin ligase existing in cytoplasm. PINK1 is a serine/threonine kinase of mitochondria that contains a mitochondrial targeting sequence. When mitochondria are damaged and membrane potential depolarization occurs, PINK1 accumulates in the outer membrane of mitochondria under the effect of TOM and is not degraded. PINK1 is activated by autophosphorylation and phosphorylates the Ser65 of a nearby ubiquitin molecule. Phosphorylated ubiquitin molecules bind and recruit Parkin molecules, and PINK1 phosphorylates Parkin resulting in its activation. Activated Parkin can then be polyubiquitinated on a variety of mitochondrial protein substrates [41]. With the effect of LC3 connector protein which contains ubiquitin-binding protein P62 (Sequestosome-1), autophagosomes are targeted to mitochondria and induce autophagy [5]. In the present study, LPS induced mitophagy occurrence which was increased in cardiomyocyte-specific PPARα knockout mice. This was verified by increased autophagosomes which were observed by TEM and increased Parkin/PINK1/P62 signaling pathway and LC3II/I protein levels as shown by Western blotting. 3-MA is an autophagosome inhibitor via its effect on class III PI3K [42]. 3-MA treatment improved cardiac dysfunction, inflammation, and mitochondrial dysfunction in the septic hearts of PparaΔCM mice, thus establishing an important role for mitophagy in potentiating cardiac and mitochondrial dysfunction. In addition, the PPARα agonist WY14643 could reduce LC3II/I protein levels that were reduced in the septic heart. These results indicate that cardiomyocyte PPARα could inhibit mitophagy in septic hearts.
Enhanced mitochondrial fission results in the release of cytochrome c from mitochondria to cytosol, which further activates the mitochondrion-dependent apoptosis pathway [7]. Cardiomyocyte PPARα disruption increased mitochondrial-dependent apoptosis. Mitochondrial function, mitophagy and apoptosis are linked to ROS production. ROS signaling and oxidative stress have a pivotal role in septic conditions [8]. High production of ROS results in increased oxidative stress which leads to production of proinflammatory cytokines such as IL-6 and TNF-α [9]. Enhanced IL-6 is associated with activation of NF-κB and subsequently the pro-inflammatory response [28, 29]. IL-6 mRNA and protein levels were elevated after LPS treatment, and further increased in PparaΔCM mice. Consistent with this, p-STAT3 protein levels were increased. p-NF-κB protein levels were then elevated after LPS treatment and further increased in PparaΔCM mice. In addition, WY-14643 treatment reduced LPS-induced p-NF-κB protein levels and the inflammatory response. These results indicate that loss of cardiomyocyte PPARα signaling increases ROS production-induced inflammation through the activation of IL-6/STAT3/NF-κB signaling pathway.
In this study, PPARα agonist WY-14643 treatment improved LPS-induced mitochondrial dysfunction and cardiac dysfunction. However, an earlier study considered that administration of PPARα agonist could not prevent cardiac dysfunction that occurs in sepsis, which might be due to the profound reduction of PPARα levels and eventually to lower agonist activation of PPARα [43]. In the present study, PPARα was activated by pretreatment with WY-14643 for two days, followed by LPS treatment instead of LPS treatment first. Moreover, WY-14643 was added to the diet instead of administered by intraperitoneal injection with LPS. Thus, the present study indicates that PPARα activation can be used as a preventive strategy for septic cardiac dysfunction.
In conclusion, this study demonstrated that PPARα and the expression of its target genes is inhibited in the hearts of septic mice (Fig. 10). Cardiomyocyte PPARα, but not myeloid PPARα plays an important role in sepsis-induced cardiac dysfunction. Cardiomyocyte-specific Ppara deficiency results in an exacerbation of LPS-induced cardiac dysfunction. PPARα disruption in cardiomyocytes potentiates LPS-induced-mitochondrial damage, followed by increased mitophagy. Cardiomyocyte-specific PPARα deficiency results in an increasing mitochondrial-dependent apoptosis and ROS production-induced inflammation through activation of the IL-6/STAT3/NF-κB signaling pathway. PPARα activation improves septic cardiac dysfunction by ameliorating mitochondrial dysfunction and inflammation. Taken together, cardiomyocyte PPARα could be a preventive and therapeutic target in septic cardiac dysfunction.
Fig. 10. Cardiomyocyte PPARα protects against septic cardiac dysfunction by ameliorating mitochondrial dysfunction.
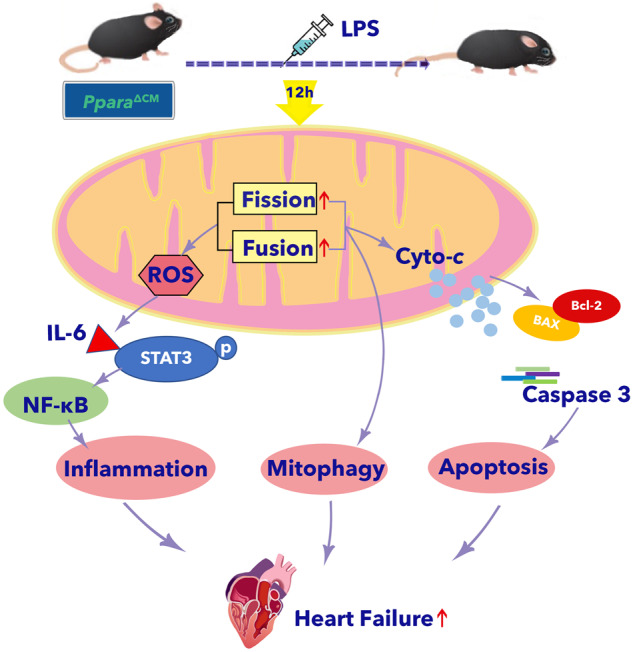
Cardiomyocyte PPARα but not myeloid PPARα plays an important role in sepsis-induced cardiac dysfunction. Cardiomyocyte-specific Ppara deficiency results in an exacerbation of LPS-induced cardiac dysfunction. PPARα disruption in cardiomyocytes potentiates LPS-induced-mitochondrial dynamic disturbance, followed by increased mitophagy. Cardiomyocyte-specific PPARα deficiency results in an increasing mitochondrial-dependent apoptosis and ROS production-induced inflammation through the activation of IL-6/STAT3/NF-κB signaling pathway.
Supplementary information
Acknowledgements
This study was supported by National Key R&D Program of China (2021YFA0805100), National Natural Science Foundation of China (82070474), Support Project of High-level Teachers in Beijing Municipal Universities in the Period of 13th Five-year Plan (CIT&TCD20190332), The Key Science and Technology Project of Beijing Municipal Institutions (KZ202010025032) to AJQ, National Natural Science Foundation of China (81800233) to XW. and the National Cancer Institute Intramural Research Program to FJG.
Author contributions
XXZ designed the study, performed the experiments, and wrote the manuscript. XW, SYJ, YL, LS, YEC, QZ, YTS, and MW performed the experiments and analyzed the data. QX participated in the echocardiography. JJW was in charge of mice. JD and BQY participated in the initial elaboration of the project. JF provided transcription data and involved in writing the manuscript. FJG designed the experimental plan, supervised the study and involved in writing the manuscript. AJQ conceived, supervised the study, and wrote the manuscript. All authors read and edited the manuscript.
Competing interests
The authors declare no competing interests.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-023-01107-5.
References
- 1.Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet. 2018;392:75–87. doi: 10.1016/S0140-6736(18)30696-2. [DOI] [PubMed] [Google Scholar]
- 2.Lelubre C, Vincent JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol. 2018;14:417–27. doi: 10.1038/s41581-018-0005-7. [DOI] [PubMed] [Google Scholar]
- 3.Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. 2019;51:162. doi: 10.1038/s12276-019-0355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Picca A, Mankowski RT, Burman JL, Donisi L, Kim JS, Marzetti E, et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol. 2018;15:543–54. doi: 10.1038/s41569-018-0059-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40:e104705. doi: 10.15252/embj.2020104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 7.Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85–100. doi: 10.1038/s41580-019-0173-8. [DOI] [PubMed] [Google Scholar]
- 8.Patoli D, Mignotte F, Deckert V, Dusuel A, Dumont A, Rieu A, et al. Inhibition of mitophagy drives macrophage activation and antibacterial defense during sepsis. J Clin Invest. 2020;130:5858–74. doi: 10.1172/JCI130996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hussain T, Tan B, Yin Y, Blachier F, Tossou MC, Rahu N. Oxidative stress and inflammation: What polyphenols can do for us? Oxid Med Cell Longev. 2016;2016:7432797. doi: 10.1155/2016/7432797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular Actions of PPARalpha in lipid metabolism and inflammation. Endocr Rev. 2018;39:760–802. doi: 10.1210/er.2018-00064. [DOI] [PubMed] [Google Scholar]
- 11.Barlaka E, Galatou E, Mellidis K, Ravingerova T, Lazou A. Role of pleiotropic properties of peroxisome proliferator-activated receptors in the heart: focus on the nonmetabolic effects in cardiac protection. Cardiovasc Ther. 2016;34:37–48. doi: 10.1111/1755-5922.12166. [DOI] [PubMed] [Google Scholar]
- 12.Young ME, Laws FA, Goodwin GW, Taegtmeyer H. Reactivation of peroxisome proliferator-activated receptor alpha is associated with contractile dysfunction in hypertrophied rat heart. J Biol Chem. 2001;276:44390–5. doi: 10.1074/jbc.M103826200. [DOI] [PubMed] [Google Scholar]
- 13.Kaimoto S, Hoshino A, Ariyoshi M, Okawa Y, Tateishi S, Ono K, et al. Activation of PPAR-alpha in the early stage of heart failure maintained myocardial function and energetics in pressure-overload heart failure. Am J Physiol Heart Circ Physiol. 2017;312:H305–H313. doi: 10.1152/ajpheart.00553.2016. [DOI] [PubMed] [Google Scholar]
- 14.Standage SW, Bennion BG, Knowles TO, Ledee DR, Portman MA, McGuire JK, et al. PPARalpha augments heart function and cardiac fatty acid oxidation in early experimental polymicrobial sepsis. Am J Physiol Heart Circ Physiol. 2017;312:H239–H249. doi: 10.1152/ajpheart.00457.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Zhu XX, Jiao SY, Qi D, Yu BQ, Xie GM, et al. Cardiomyocyte peroxisome proliferator-activated receptor alpha is essential for energy metabolism and extracellular matrix homeostasis during pressure overload-induced cardiac remodeling. Acta Pharmacol Sin. 2022;43:1231–42. doi: 10.1038/s41401-021-00743-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brocker CN, Yue J, Kim D, Qu A, Bonzo JA, Gonzalez FJ. Hepatocyte-specific PPARA expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am J Physiol Gastrointest Liver Physiol. 2017;312:G283–G299. doi: 10.1152/ajpgi.00205.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao H, Zhang M, Zhou F, Cao W, Bi L, Xie Y, et al. Cinnamaldehyde ameliorates LPS-induced cardiac dysfunction via TLR4-NOX4 pathway: The regulation of autophagy and ROS production. J Mol Cell Cardiol. 2016;101:11–24. doi: 10.1016/j.yjmcc.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 18.Qi D, Wei M, Jiao S, Song Y, Wang X, Xie G, et al. Hypoxia inducible factor 1alpha in vascular smooth muscle cells promotes angiotensin II-induced vascular remodeling via activation of CCL7-mediated macrophage recruitment. Cell Death Dis. 2019;10:544. doi: 10.1038/s41419-019-1757-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Busch K, Kny M, Huang N, Klassert TE, Stock M, Hahn A, et al. Inhibition of the NLRP3/IL-1beta axis protects against sepsis-induced cardiomyopathy. J Cachexia Sarcopenia Muscle. 2021;12:1653–68. doi: 10.1002/jcsm.12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W, et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019;24:101215. doi: 10.1016/j.redox.2019.101215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhagat A, Shrestha P, Kleinerman ES. The innate immune system in cardiovascular diseases and its role in doxorubicin-induced cardiotoxicity. Int J Mol Sci. 2022;23:14649. doi: 10.3390/ijms232314649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ouyang H, Li Q, Zhong J, Xia F, Zheng S, Lu J, et al. Combination of melatonin and irisin ameliorates lipopolysaccharide-induced cardiac dysfunction through suppressing the Mst1-JNK pathways. J Cell Physiol. 2020;235:6647–59. doi: 10.1002/jcp.29561. [DOI] [PubMed] [Google Scholar]
- 23.Abate M, Festa A, Falco M, Lombardi A, Luce A, Grimaldi A, et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin Cell Dev Biol. 2020;98:139–53. doi: 10.1016/j.semcdb.2019.05.022. [DOI] [PubMed] [Google Scholar]
- 24.Boengler K, Kosiol M, Mayr M, Schulz R, Rohrbach S. Mitochondria and ageing: role in heart, skeletal muscle and adipose tissue. J Cachexia Sarcopenia Muscle. 2017;8:349–69. doi: 10.1002/jcsm.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wajner M, Amaral AU. Mitochondrial dysfunction in fatty acid oxidation disorders: insights from human and animal studies. Biosci Rep. 2015;36:e00281. doi: 10.1042/BSR20150240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shirihai OS, Song M, Dorn GW., 2nd How mitochondrial dynamism orchestrates mitophagy. Circ Res. 2015;116:1835–49. doi: 10.1161/CIRCRESAHA.116.306374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun K, Jing X, Guo J, Yao X, Guo F. Mitophagy in degenerative joint diseases. Autophagy. 2021;17:2082–92. doi: 10.1080/15548627.2020.1822097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu L, Huang B, Bai S, Tan J, Liu Y, Chen H, et al. SO2 derivatives induce dysfunction in human trophoblasts via inhibiting ROS/IL-6/STAT3 pathway. Ecotoxicol Environ Saf. 2021;210:111872. doi: 10.1016/j.ecoenv.2020.111872. [DOI] [PubMed] [Google Scholar]
- 29.Zanders L, Kny M, Hahn A, Schmidt S, Wundersitz S, Todiras M, et al. Sepsis induces interleukin 6, gp130/JAK2/STAT3, and muscle wasting. J Cachexia Sarcopenia Muscle. 2022;13:713–27. doi: 10.1002/jcsm.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montaigne D, Butruille L, Staels B. PPAR control of metabolism and cardiovascular functions. Nat Rev Cardiol. 2021;18:809–23. doi: 10.1038/s41569-021-00569-6. [DOI] [PubMed] [Google Scholar]
- 32.Standage SW, Waworuntu RL, Delaney MA, Maskal SM, Bennion BG, Duffield JS, et al. Nonhematopoietic peroxisome proliferator-activated receptor-alpha protects against cardiac injury and enhances survival in experimental polymicrobial sepsis. Crit Care Med. 2016;44:e594–603. doi: 10.1097/CCM.0000000000001585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makrecka-Kuka M, Korzh S, Vilks K, Vilskersts R, Cirule H, Dambrova M, et al. Mitochondrial function in the kidney and heart, but not the brain, is mainly altered in an experimental model of endotoxaemia. Shock. 2019;52:e153–e162. doi: 10.1097/SHK.0000000000001315. [DOI] [PubMed] [Google Scholar]
- 34.Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. 2020;15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711. [DOI] [PubMed] [Google Scholar]
- 35.Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. 2020;21:204–24. doi: 10.1038/s41580-020-0210-7. [DOI] [PubMed] [Google Scholar]
- 36.Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res. 2021;128:1487–513. doi: 10.1161/CIRCRESAHA.121.318241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–38. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 38.Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12:689–702. doi: 10.1080/15548627.2016.1151580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doblado L, Lueck C, Rey C, Samhan-Arias AK, Prieto I, Stacchiotti A, et al. Mitophagy in human diseases. Int J Mol Sci. 2020;22:3903. doi: 10.3390/ijms22083903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng Y, Madungwe NB, da Cruz Junho CV, Bopassa JC. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br J Pharmacol. 2017;174:4329–44. doi: 10.1111/bph.14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 2016;26:733–44. doi: 10.1016/j.tcb.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 42.Miller S, Oleksy A, Perisic O, Williams RL. Finding a fitting shoe for Cinderella: Searching for an autophagy inhibitor. Autophagy. 2010;6:805–7. doi: 10.4161/auto.6.6.12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drosatos K, Khan RS, Trent CM, Jiang H, Son NH, Blaner WS, et al. Peroxisome proliferator-activated receptor-gamma activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ Heart Fail. 2013;6:550–62. doi: 10.1161/CIRCHEARTFAILURE.112.000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.