

Extracellular matrix stiffening drives bevacizumab resistance by stimulating hepatic stellate cells to provide fuel for mCRC cells in the liver, indicating a potential metabolism-based therapeutic strategy for overcoming resistance.
Abstract
Bevacizumab is an anti-VEGF monoclonal antibody that plays an important role in the combination treatment of advanced colorectal cancer. However, resistance remains a major hurdle limiting bevacizumab efficacy, highlighting the importance of identifying a mechanism of antiangiogenic therapy resistance. Here, we investigated biophysical properties of the extracellular matrix (ECM) related to metabolic processes and acquired resistance to bevacizumab. Evaluation of paired pre- and posttreatment samples of liver metastases from 20 colorectal cancer patients treated with combination bevacizumab therapy, including 10 responders and 10 nonresponders, indicated that ECM deposition in liver metastases and a highly activated fatty acid oxidation (FAO) pathway were elevated in nonresponders after antiangiogenic therapy compared with responders. In mouse models of liver metastatic colorectal cancer (mCRC), anti-VEGF increased ECM deposition and FAO in colorectal cancer cells, and treatment with the FAO inhibitor etomoxir enhanced the efficacy of antiangiogenic therapy. Hepatic stellate cells (HSC) were essential for matrix stiffness–mediated FAO in colon cancer cells. Matrix stiffness activated lipolysis in HSCs via the focal adhesion kinase (FAK)/yes-associated protein (YAP) pathway, and free fatty acids secreted by HSCs were absorbed as metabolic substrates and activated FAO in colon cancer cells. Suppressing HSC lipolysis using FAK and YAP inhibition enhanced the efficacy of anti-VEGF therapy. Together, these results indicate that bevacizumab-induced ECM remodeling triggers lipid metabolic cross-talk between colon cancer cells and HSCs. This metabolic mechanism of bevacizumab resistance mediated by the physical tumor microenvironment represents a potential therapeutic target for reversing drug resistance.
Significance:
Extracellular matrix stiffening drives bevacizumab resistance by stimulating hepatic stellate cells to provide fuel for mCRC cells in the liver, indicating a potential metabolism-based therapeutic strategy for overcoming resistance.
Graphical Abstract
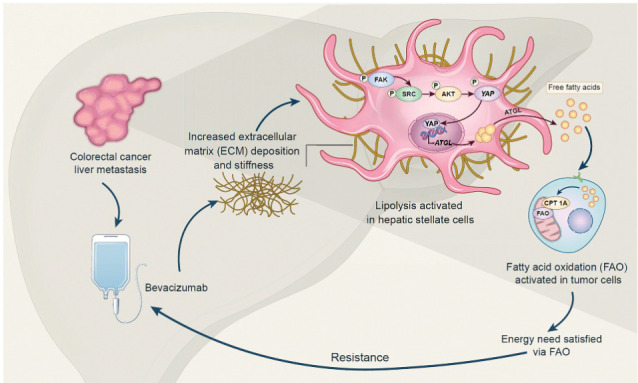
Introduction
The average 5-year overall survival (OS) rate for colorectal cancer is 60%, making it the third most prevalent cancer in the world (1). Primary colorectal cancer can be treated surgically; however, patients with metastatic colorectal cancer (mCRC) have a considerably lower chance of surviving. Synchronous liver metastasis is present in 20–30% of patients at the time of diagnosis, and 50 to 70% of patients with colorectal cancer eventually develop hepatic metastases, which are the major cause of mortality (2).
The anti-VEGF antibody bevacizumab, combined with chemotherapy is the current standard treatment for mCRC based on superior OS improvement (3). However, survival benefits are limited and the disease eventually progresses. Previous studies have shown that the dosage and timings of antiangiogenic treatment have a significant impact on its toxicity characteristics (4, 5), and VEGF withdrawal itself is associated with increased extracellular matrix (ECM) deposition in malignant tumors (6, 7). The pathophysiologic mechanisms of acquired resistance to antiangiogenic therapy are still unknown, partly because of the poor understanding of the impact of antiangiogenic therapy on the tumor microenvironment.
The tumor microenvironment is characterized by both physical and biological abnormalities (8). Disease progression can be driven by changes in the mechanical properties of tissues (9). Previous studies have found that ECM stiffness and collagen cross-linking are associated with disease development in various solid tumors (10, 11). It has been reported that tissue stiffness is higher in liver metastases than in primary colorectal tumors, which is caused by highly activated metastasis-associated fibroblasts (MAF), resulting in enhanced angiogenesis and antiangiogenic therapy resistance (12). Tumors also flexibly cooperate with aberrant metabolic networks through a number of distinct and common pathways to sustain proliferation, even in the nutritionally deficient tumor microenvironment (13). Mounting evidence points to a crucial relationship between mechanotransduction and cellular metabolism (14, 15). However, boosting glycolysis alone is inadequate to satisfy all the metabolic demands of proliferating cells. Tumors tend to grow or metastasize to host environments rich in adipocytes. In breast cancer, melanoma, ovarian cancer peritoneal metastasis, and prostate cancer bone metastasis, stromal adipocytes act as lipid reservoirs and hydrolyze lipids to produce free fatty acids (FFA), which can be absorbed and metabolized by tumor cells (16–18). This adipocyte-cancer cell cross-talk is an adaptive metabolic process established by cancer cells to take full advantage of lipid storage in the tumor microenvironment. Hepatic stellate cells (HSC) are known to account for 15% of the total number of hepatic stromal cells, and quiescent HSCs store large amounts of retinol and are also known as lipid storage cells in the liver. During activation, HSCs are transformed into myofibroblasts, resulting in the loss of lipid droplets and ECM production. Lipid droplet hydrolysis, which contains retinoids and triglycerides, and the release of FFAs are hallmarks of activated HSCs (19, 20).
In this study, we delineated a metabolic reprogramming adaptation to therapeutic-stress-controlling tumor development from the perspective of the tumor's physical microenvironment. We suggested that mechanically dismantling the metabolic cross-talk within the tumor may provide potential combinatorial approaches for antiangiogenic therapy in liver mCRC.
Materials and Methods
Patient samples
All patients were histologically diagnosed with colorectal cancer at Nanfang Hospital, Southern Medical University (Guangzhou, China) and received palliative treatment with mFOLFOX6 [oxaliplatin, leucovorin, and 5-fluorouracil (5-FU)] or FOLFIRI (irinotecan, leucovorin, and 5-FU) chemotherapy with bevacizumab. Pre- and posttreatment paired samples obtained from 20 patients with colorectal cancer liver metastases between 2018 and 2021 were analyzed, including 10 responders and 10 nonresponders, according to the RECIST version 1.1, all of which had experienced the best objective responses of partial response (PR) initially. Responders ended up with stable disease or PR at the time when posttreatment samples were collected; while nonresponders ended up with progressive disease. None of these 20 patients had liver cirrhosis before treatment. Pre- and posttreatment paired samples from another 3 patients with colorectal cancer liver metastases, who had been diagnosed with liver cirrhosis by CT before receiving palliative treatment, were analyzed. All patients provided written informed consent, agreed to a protocol that was authorized by the institutional review board (Nanfang Hospital Ethics Review Board, Southern Medical University; permit number: NFEC‐2017‐206) and allowed for thorough tumor examination after providing written informed permission. The Declaration of Helsinki and the International Ethical Guidelines for Biomedical Research Involving Human Subjects were both followed in this work. Detailed clinical information is described in Supplementary Table S1.
Cell lines and culture
The human colon cancer cell lines- DLD1 (RRID: CVCL_0248) and HCT116 (RRID: CVCL_0291) and, human umbilical vein endothelial cells (HUVEC; RRID: CVCL_2959) were purchased from ATCC. MC38 (RRID: CVCL_B288) murine colon cancer cells were obtained from Jennio Biotech Co., Ltd. The immortalized human HSC cell line, LX-2 (RRID: CVCL_5792) was purchased from Procell Biotech Co., Ltd. The murine HSC cell line, JS-1 (RRID: CVCL_B6RW) was obtained from Huifeng Biotech Co., Ltd. DLD1 and HCT116 cells were cultured in RPMI1640 (catalog no. SH30809.01, HyClone) supplemented with 10% FBS (catalog no. SV30160.03, HyClone). MC38 cells and HUVECs were cultured in DMEM (catalog no. SH30022.01, HyClone) supplemented with 10% FBS. LX-2 and JS-1 cells were cultured in Stellate Cell Medium (catalog no. 5301, ScienCell) supplemented with 2% FBS and 1% stellate cell growth supplement (catalog no. 5352, ScienCell).
Mycoplasma testing was routinely performed for all cell lines. Human cell lines were authenticated by short tandem repeat sequencing.
Animals
C57BL/6 mice (female, 4–5 weeks old) were obtained from the Animal Center of the Southern Medical University. All animal experimental procedures adhered to specific pathogen-free standards and the guidelines of the Laboratory Animal Center of Southern Medical University (permit number: NFYY-2018–0639). The Animal Care and Use Committee of Nanfang Hospital, Southern Medical University, approved all animal experiments.
Liver metastasis model
C57BL/6 mice (female, 4–5 weeks old) were anaesthetized through intraperitoneal injection of 1% sodium pentobarbital (0.15 mL/kg). After administering anesthesia, the animals were placed in a supine position, and the right subcostal region was shaved and disinfected. Afterwards, a 1-cm subcostal incision approximately 0.5 cm below the ribcage was made. After reaching the peritoneum the median liver lobe was gently exposed by using the rear end of the microsurgical forceps. Approximately 1 × 105 of MC38-Luc cells (a MC38 cell line that stably expresses firefly luciferase) were collected and suspended in 25-μL sterile PBS. 25 μL of the suspension was slowly injected under the liver capsule. The presence of white protrusions suggested the effective implantation of a tumor. After returning the liver body its original position, the abdomen was closed. Aseptic technique was used throughout the procedure. Treatments were initiated 7 days after injection.
HSC/tumor coinjection subcutaneous tumor model
Murine HSCs were isolated from C57BL/6 mice (female, 4–5 weeks old), as described previously (21). MC38 cells (0.5 × 106) were mixed with or without HSCs (0.5 × 106) and coinjected into the left thighs of C57BL/6 mice after dilution with PBS to 100 μL. Treatments were initiated 7 days after HSC/tumor injection. The volume of tumor nodules was measured every 3 days using the following formula: tumor volume = (width)2 × length/2, until day 27. After euthanasia on day 28, tumors were collected.
Polyacrylamide hydrogels
Polyacrylamide gels were prepared as described previously (22). Briefly, acrylamide and bis-acrylamide in variable ratios were mixed with tetramethylethylenediamine and ammonium persulfate for polymerization; the formula used is presented in Supplementary Table S2. The mixtures were degassed for 15 minutes to eliminate oxygen from the solution. Gel surfaces were activated to facilitate cell attachment by exposure to the heterobifunctional crosslinker Sulfo-SANPAH (catalog no. ab145610, Abcam) at 0.5 mg/mL in 50 nmol/L HEPES under UV light. Following activation, the gels were washed with PBS to remove excess crosslinkers, then incubated with collagen I (catalog no. C8062, Solarbio) solution (0.05 mg/mL in PBS, pH 7.4) for 30 minutes. Following a PBS rinse and sterilization by microwave heating, the polyacrylamide gels were placed on a 6-well cell culture plate for cell seeding and further studies.
Statistical analysis
Each experiment was performed in at least three replicates. Data that met the assumptions of the test are shown as the mean ± SD and were analyzed with Prism Version 7.0 (GraphPad Software). Statistical analyses including Student t test, one-way ANOVA, two-way ANOVA, or Kruskal–Wallis test, were considered to be significant when P values were below 0.05. The variance was statistically similar between the groups.
Additional experimental procedures are provided in Supplementary Methods. Detailed information on reagents and antibodies in this work are provided in Supplementary Table S3. Primer sequences are listed in Supplementary Table S4.
Data availability statement
The data generated in this study are available within the article and its Supplementary Data files. All other raw data are available upon request from the corresponding author. The mass spec lipidomics data was uploaded to MetaboLights (MTBLS8402).
Results
Bevacizumab increases ECM deposition in liver metastases in a dose-dependent manner
To explore the correlation between ECM deposition and bevacizumab resistance, we first performed morphology analyses of puncture biopsy specimens from patients with mCRC who responded (10 cases) or did not respond (10 cases) to chemotherapy with bevacizumab (Supplementary Fig. S1A). Although there was no difference in ECM deposition between responders before or after bevacizumab treatment, we found that nonresponders had significantly higher ECM deposition in liver metastases after treatment compared with that before treatment (Fig. 1A). Previous studies have found a therapeutic window for antiangiogenic therapy; overdosing can reduce efficacy (5). We investigated whether low-dose VEGF inhibition in mouse models might limit ECM deposition and improve the efficacy of antiangiogenic therapy. We used mouse models of liver mCRC to evaluate tumor growth in response to 5-FU alone or combined with various degrees of VEGF inhibition. The VEGF antibody B20.4–1.1 (a VEGF-blocking antibody abbreviated as B20) was administered at 1 mg/kg, 2.5 mg/kg, or 5 mg/kg (Fig. 1B). Even at low doses, B20 could suppress tumor growth by 50% compared with immunoglobulin G (IgG)-treated controls, whereas tumor growth was not significantly affected by the varying doses of B20 (Fig. 1C). Moreover, high doses of B20 failed to reduce tumor vessels ulteriorly, compared with low doses (Supplementary Fig. S1B), consistent with previous studies (6, 23). To evaluate changes in the mechanical characteristics of liver mCRC after anti-VEGF therapy, we analyzed the morphology of metastases harvested from mice treated with B20. These findings demonstrated a dose-dependent rise in ECM deposition in the MC38 liver mCRC model (Fig. 1D).
Figure 1.

Bevacizumab increases ECM deposition in colorectal cancer liver metastases in a dose-dependent manner. A, Representative hematoxylin and eosin (H&E), Masson and picrosirius red (left) on serial sections showing ECM deposition change in liver metastases from patients with colorectal cancer before and after receiving chemotherapy and bevacizumab treatment. Quantification of intraindividual comparisons of collagen volume fraction (right) in paired samples (two-tailed paired t test; n = 10 per group). B, Schematic representation of the liver metastasis models used in C and D. Mice were given treatment with control IgG, 5-FU+IgG, 5-FU+1 mg/kg B20, 5-FU+2.5 mg/kg B20 or 5-FU+5 mg/kg B20 (5-FU 50 mg/kg; twice a week, 3 weeks). C, Representative in vivo bioluminescent images (left) and quantification of bioluminescent signals (right) of liver metastases in mice with indicated treatment (one-way ANOVA; n = 5 per group). D, Representative hematoxylin and eosin, Masson, picrosirius red images (left) on serial sections and quantification of collagen volume fraction (right) showing ECM deposition in liver metastases in mice with indicated treatment (one-way ANOVA; n = 5 per group). E, Schematic representation of the liver metastasis models used in F and G. Mice were administered 5-FU+B20 or 5-FU+B20+BAPN (5-FU, 50 mg/kg; B20, 5 mg/kg; BAPN, 100 mg/kg; twice a week, 3 weeks). F, Representative in vivo bioluminescent images (left) and quantification of bioluminescent signals (right) of liver metastases in mice with indicated treatment (two-tailed unpaired t test; n = 5 per group). G, Representative hematoxylin and eosin, Masson, picrosirius red images (left), and quantification of collagen volume fraction (right) showing ECM deposition in liver metastases in mice with indicated treatment (two-tailed unpaired t test; n = 5 per group). Data are graphed as the mean ± SD. Scale bar, 100 μm. ns, not significant; P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B and E, Created with BioRender.com.)
To further validate this possible impact of ECM deposition after VEGF-targeted therapy, we combined 5-FU and B20 with a known pharmacologic inhibitor β-aminopro-pionitril (BAPN) of lysyl oxidase to decrease ECM stiffening in the mouse models of liver mCRC (Fig. 1E). Inhibition of mechanotransduction by BAPN prevented tumor progression (Fig. 1F), decreased ECM deposition (Fig. 1G), and further improved the efficacy of antiangiogenic therapy (Supplementary Fig. S1C).
To investigate whether bevacizumab would also change the mechanical characteristics of paracancerous tissues, we performed morphology analyses of pre- and posttreatment paired normal liver tissues from patients with mCRC mentioned above and another 3 patients with mCRC with liver cirrhosis before treatment. We observed that bevacizumab treatment did not induce ECM deposition in paracancerous tissues (Supplementary Fig. S1D).
Anti-VEGF therapy-induced ECM deposition in liver metastases is related to activation of the fatty acid oxidation pathway
It is almost certain that restriction of angiogenesis induces significant metabolic alterations in tumors, despite these alterations being largely uncharacterized. Antiangiogenic drugs have been shown to trigger lipid-dependent metabolic reprogramming as an alternative mechanism of drug resistance (24). To determine whether these metabolic adaptations are connected with abnormal ECM deposition, we performed an IHC analysis of specimens from patients with mCRC. Posttreatment specimens from nonresponders had a higher expression of the rate-limiting fatty acid oxidation (FAO) enzyme carnitine palmitoyltransferase 1A (CPT1A), which transports fatty acids into mitochondria for further oxidation by converting acyl-CoAs into acylcarnitines (25), similar to ECM deposition. However, there was no significant difference in the expression of CPT1A between the pre- and posttreatment specimens from responders (Fig. 2A).
Figure 2.

Anti-VEGF therapy-induced ECM deposition in liver metastases is associated with activation of FAO metabolism. A, Left, representative IHC images showing CPT1A expression in liver metastases from colorectal cancer patients before and after receiving chemotherapy and bevacizumab treatment. Right, IHC scores of CPT1A of intraindividual comparisons in paired samples (two-tailed paired t test; n = 10 per group). B, Representative IHC images on serial sections of MC38 liver metastases of mice with indicated treatment. C, Representative IHC images on serial sections of MC38 liver metastases of mice with indicated treatment. D, Schematic representation of the liver metastasis models used in E and F. Mice were given treatment with IgG, etomoxir (ETX) +IgG, 5-FU+B20 or 5-FU+B20+ETX (5-FU 50 mg/kg, B20 5 mg/kg; etomoxir 15 mg/kg; twice a week, 3 weeks). E, Representative in vivo bioluminescent images (left) and quantification of bioluminescent signals (right) of liver metastases in mice treated with indicated treatment (one-way ANOVA; n = 5 per group). F, Representative hematoxylin and eosin (H&E), Masson, picrosirius red images (left) on serial sections and quantification of collagen volume fraction (right) showing ECM deposition in liver metastases in mice with indicated treatment (one-way ANOVA; n = 5 per group). G, FAO rate of DLD1 and HCT116 cells cultured on 1 kPa, 10 kPa, and 25 kPa polyacrylamide hydrogels (one-way ANOVA; n = 5 independent experiments). Data are graphed as the mean ± SD. Scale bar, 100 μm. ns, not significant; P > 0.05; *, P < 0.05; ***, P < 0.001. (D, Created with BioRender.com.)
We performed a series of animal studies to further confirm the links between ECM deposition, FAO metabolism, and bevacizumab. IHC staining revealed that FAO metabolic targets were significantly upregulated in B20-treated liver metastases in a dose-dependent manner (Fig. 2B), consistent with ECM deposition. In addition, pharmacologic inhibition of lysyl oxidase decreased ECM deposition induced by antiangiogenic therapy; and decreased FAO metabolic targets expression (Fig. 2C). Notably, patients with liver cirrhosis before treatment tended to show poor respond to bevacizumab (Supplementary Fig. S1E; Supplementary Table S1). Moreover, pre- and posttreatment samples from these patients showed a high CPT1A expression. There was no difference in either ECM deposition or CPT1A expression between pre- and posttreatment specimens of patients with liver cirrhosis (Supplementary Fig. S1F). These findings suggest that the expression of Cpt1a is positively correlated with ECM deposition in liver metastases. Next, we combined chemotherapy and B20 with a known pharmacologic inhibitor of CPT1, etomoxir, to understand the effects of FAO metabolism on anti-VEGF therapy (Fig. 2D). We observed that treatment with etomoxir enhanced the efficacy of antiangiogenic therapy in mouse models of liver mCRC (Fig. 2E; Supplementary Fig. S1G), indicating that activation of FAO metabolism might be related to bevacizumab resistance. Meanwhile, inhibition of CPT1 had no impact on ECM deposition in liver metastases (Fig. 2F), suggesting that ECM deposition might interfere with FAO metabolism. These findings support the role of ECM deposition in activating the FAO pathway in liver metastases and suggest that these metabolic alterations contribute to bevacizumab resistance.
Stiffness is modulated by the cytoskeleton and the ECM at the tissue level (8). To determine whether mechanical signals transmitted by ECM stiffness regulate FAO metabolism, we established polyacrylamide hydrogels of precisely defined stiffnesses [1 kilopascal (kPa), 10 kPa, and 25 kPa] as supports for in vitro culturing of the human colon cancer cell lines DLD1 and HCT116 and MC38 murine colon cancer cells. However, matrix stiffness did not directly impact the FAO rate of colon cancer cells (Fig. 2G).
HSCs serve as essential intermediaries for matrix stiffness–mediated FAO activation in colon cancer cells
The above data indicated that there might be an intermediary in the tumor microenvironment. Activated HSCs act as fibroblasts, produce ECM proteins, and play key roles in liver fibrosis (26). We hypothesized that HSCs are associated with the mechanical cues that induce metabolic reprogramming. To test this hypothesis, we performed a mixed-culture experiment on HSCs and colon cancer cells. Human HSCs LX-2 and murine HSCs JS-1 were labeled with carboxyfluorescein succinimidyl ester and cocultured with their respective species colon cancer cells on polyacrylamide hydrogels for 48 hours and then isolated by fluorescence activated cell sorting (Fig. 3A; Supplementary Fig. S2A; ref. 27). In all three cocultured colon cancer cell lines, we found that the FAO rate and the expression of key genes in the FAO pathway increased proportionally with stiffness, along with decreased reactive oxygen species (ROS) levels (Fig. 3B and C; Supplementary Fig. S2B–S2E). We reasoned that, in the presence of HSCs, colon cancer cells would reprogram FAO metabolism upon exposure to a stiff matrix. To further confirm this finding, we mixed MC38 cells with or without primary murine HSCs and subcutaneously coinjected them into C57BL/6 mice (28). Four groups of subcutaneous tumor models were treated with control IgG, 5-FU + IgG, 5-FU + B20, or 5-FU + B20 +BAPN (Fig. 3D). The results showed that in the presence of HSCs, the combined treatment of BAPN with 5-FU + B20 significantly inhibited the proliferation of subcutaneous tumors compared with the other three groups. Although chemotherapy and antiangiogenic therapy were less effective in suppressing tumors arising from MC38/HSCs coinjections than those arising from MC38 mono-injections, this difference could be eliminated by the combination of BAPN (Fig. 3E and F). ECM remodeling and the expression of key genes in the FAO pathway were significantly increased in tumors arising from MC38/HSCs coinjections treated with chemotherapy and antiangiogenic therapy, compared with the other groups. These upregulations could be inhibited by cotreatment with BAPN. In the absence of HSCs, antiangiogenic therapy did not induce ECM deposition or activation of the FAO pathway in subcutaneous tumors (Fig. 3G). Consistent with our in vitro model, matrix stiffness requires HSCs as essential intermediaries to activate FAO metabolism in colon cancer cells. With the presence of HSCs, tumors showed stronger resistance to antiangiogenic therapy. Still, cotreatment with BAPN could restore the efficacy of antiangiogenic therapy in tumors arising from MC38/HSCs coinjections (Fig. 3H).
Figure 3.

HSCs are essential for matrix stiffness–mediated FAO metabolic reprogramming in colon cancer cells. A, Schematic representation of the HSCs-colon cancer cells coculture system. HSCs were labeled with CFSE before coculture with colon cancer cells for 48 hours on polyacrylamide hydrogels. B and C, FAO rate (B) and Western blot analysis (C) of DLD1 and HCT116 cells cocultured with LX-2 cells on 1 kPa, 10 kPa, and 25 kPa polyacrylamide hydrogels (one-way ANOVA; n = 5 independent experiments). D, Schematic representation of HSC/MC38 coinjection subcutaneous tumor models used in E–G. MC38 cells (0.5×106) were subcutaneously injected with or without murine primary HSCs (0.5×106) into C57BL/6 mice. After 7 days, mice were treated with IgG, 5-FU+IgG, 5-FU+B20, or 5-FU+B20+BAPN (5-FU 50 mg/kg, B20 5 mg/kg, BAPN 100 mg/kg; twice a week, 3 weeks). E, Representative images of subcutaneous tumors with indicated treatment. F, Relative tumor growth curves (top) and relative tumor volume (bottom) of subcutaneous tumors with indicated treatment (one-way ANOVA; n = 5 per group). G, Left, representative hematoxylin and eosin (H&E), Masson, picrosirius red images, and IHC images on serial sections of subcutaneous tumors with indicated treatment. Right, quantification of collagen volume fraction in subcutaneous tumors with indicated treatment. Scale bar, 100 μm (one-way ANOVA; n = 5 per group). H, Representative immunofluorescence images (left) showing tumor vessels (stained for CD31) and quantification of vessel density (right) in subcutaneous tumors with indicated treatment. Scale bar, 50 μm (one-way ANOVA; n = 5 per group). Data are graphed as the mean ± SD. ns, not significant; P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001. (A, Created with BioRender.com.)
Stiffness-induced HSCs promote FAO metabolism in colon cancer cells by secreting FFAs
To determine whether direct contact between colon cancer cells and HSCs was necessary for metabolic reprogramming, we cultured LX-2 cells and JS-1 cells on polyacrylamide gels of different stiffnesses for 48 hours and harvested the supernatant as conditioned medium (CM) for further experiments. We prepared 1 kPa_CM, 10 kPa_CM and 25 kPa_CM from LX-2 and JS-1 cells. The FAO rate assay showed that more active FAO metabolism was observed in colon cancer cells after treatment with CM from HSCs grown on the stiff gels (Fig. 4A; Supplementary Fig. S3A). ROS levels were significantly decreased in 25 kPa_CM treated colon cancer cells, and ATP production was increased (Fig. 4B and C; Supplementary Fig. S3B and S3C). We also investigated the oxygen consumption rate, which is a measure of mitochondrial activity. Compared with those treated with 1 kPa_CM and 10 kPa_CM, cells treated with 25 kPa_CM displayed higher basal and maximal respiration rates and enhanced intracellular ATP levels, suggesting that mitochondrial activity was significantly increased by CM from HSCs on stiff substrates (Fig. 4D; Supplementary Fig. S3D). CD36 is a fatty acid translocase and transporter of FFAs, and its expression is positively correlated with the uptake of extracellular FFAs (29). The expression of key genes in the FAO pathway was also found upregulated proportionally to stiffness in colon cancer cells, so as the expression of CD36 (Fig. 4E and F; Supplementary Fig. S3E and S3F). These results suggest that FAO metabolism in colon cancer cells may be promoted by increasing the uptake of FFAs.
Figure 4.

CM derived from HSCs cultured on the stiff substrates upregulates FAO metabolism in colon cancer cells. A–C, FAO rate (A), ROS content (B), and ATP level (C) of DLD1 or HCT116 cells treated with CM from LX-2 cells cultured on 1 kPa, 10 kPa, and 25 kPa polyacrylamide hydrogels for 48 hours (one-way ANOVA; n = 5 independent experiments). D, The basal respiration, maximal respiration, spare respiration capacity, and ATP production of DLD1 cells with indicated treatment were measured by OCR measurements (one-way ANOVA; n = 5 independent experiments). E and F, Relative expression of FAO-related genes in DLD1 and HCT116 cells treated with indicated CM by qPCR analysis (E) and Western blot analysis (one-way ANOVA; n = 5 independent experiments; F). Data are graphed as the mean ± SD. ns, not significant; P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Previous studies have reported that liver stiffness can activate the differentiation of HSCs into myofibroblasts (21). HSC activation is characterized by the loss of lipid droplets (30). We hypothesized that matrix stiffness could induce the secretion of fatty acids by the HSCs. To test this hypothesis, we examined the FFA content in the supernatant from HSCs cultured on polyacrylamide gels with different stiffness and found that FFAs secreted by HSCs were increased on stiff substrates (Supplementary Fig. S4A). To directly assess the fatty acids species secreted by HSCs, we performed targeted lipidomic profiling of supernatant from HSCs cultured on polyacrylamide gels with different stiffnesses in vitro. Mass spectrometry–based lipid profiling of 35 individual medium- and long-chain fatty acid species was performed on these samples. We calculated the z-score for each species and identified the fatty acids species that were increased across the supernatant samples. Overall, the most significantly increased fatty acids were monounsaturated fatty acids (e.g., oleic acid) and polyunsaturated fatty acids (e.g., docosahexaenoic acid; Fig. 5A; Supplementary Fig. S4B and S4C). To confirm the physiologic relevance of fatty acids, we determined whether oleic acid was sufficient to promote FAO metabolism in colon cancer cells in a serum-free medium and the absence of HSCs. We treated DLD1, HCT116, and MC38 cells with oleic acid and found that treatment promoted the FAO metabolism in all cell lines (Fig. 5B–D; Supplementary Fig. S4D–S4G).
Figure 5.

High stiffness-induced HSCs promote FAO metabolism in colon cancer cells through secreting FFAs. A, Heat map of targeted lipidomic profiling of medium- and-long-chain fatty acids species with significant differential expression in supernatant derived from LX-2 cells cultured on 1 kPa, 10 kPa, and 25 kPa (Kruskal–Wallis test; n = 6 per group). B–D, FAO rate (B), ATP level (C), and Western blot analysis (D) of DLD1 and HCT116 cells treated with BSA or oleic acid (OA, 100 μmol/L; two-tailed unpaired t test; n = 5 independent experiments). E, Schematic representation of the lipid transfer experiments procedures. HSCs were cultured on polyacrylamide hydrogels with defined stiffnesses. After lipid droplets labeled with BODIPY, HSCs were cocultured with colon cancer cells using a Transwell system. F, Representative images (left) and quantification (right) of labeled lipids within DLD1 and HCT116 cells, after coculturing for 48 hours with LX-2 cells that were prestimulated on 1 kPa, 10 kPa, and 25 kPa polyacrylamide hydrogels. Scale bar, 5 μm (one-way ANOVA; n = 5 independent experiments). Data are graphed as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (E, Created with BioRender.com.)
The above data indicate that FFAs secreted by HSCs on stiff substrates promote FAO metabolism in colon cancer cells, but it is unclear whether this is a result of direct FFAs transfer or an indirect signal that triggers FAO for intracellular lipid depletion in tumor cells. We tested whether they were direct donors to colon cancer cells using a lipid “pulse-chase” approach. LX-2 and JS-1 cells were seeded on polyacrylamide gels with different stiffnesses and loaded with boron-dipyrromethene (BODIPY)-labeled fatty acids. Labeled HSCs were washed to eliminate extracellular lipids and cocultured with colon cancer cells on top using a Transwell system (Fig. 5E). Within 24 hours, we observed BODIPY-labeled fatty acids within the cytoplasm of colon cancer cells, suggesting that lipid species derived from HSCs can be transferred to colon cancer cells. Moreover, matrix stiffness drove the transfer of more fatty acids from the HSCs (Fig. 5F; Supplementary Fig. S4H). These data provide evidence of intratumoral metabolic cross-talk between HSCs and tumor cells, which are activated by ECM stiffening.
Matrix stiffness activates lipolysis in HSCs via the focal adhesion kinase—yes-associated protein pathway
Given the changes in tumor cell metabolism observed in the HSCs cocultured with colon cancer cells and the parallel increases in FFAs content of HSCs supernatant, we wanted to determine whether matrix stiffness affected lipid metabolism in HSCs. Lipid droplet staining showed that ECM stiffening decreased lipid droplet accumulation in the HSCs (Fig. 6A; Supplementary Fig. S5A). The breakdown of lipid droplets is best characterized in adipocytes, in which key roles are assigned to adipose triglyceride lipase (ATGL), its coactivator abhydrolase domain containing 5 (ABHD5), hormone-sensitive lipase (LIPE), and monoacylglycerol lipase (MGLL). In this process, ATGL first hydrolyses triglycerides into diacylglycerol, which is then decomposed into monoacylglycerol by LIPE. Ultimately, monoacylglycerol is catalytically hydrolyzed into glycerol by MGLL and fatty acids are produced at each hydrolytic step (31). We examined the mRNA and protein expression levels of lipolysis-related genes in HSCs. Consistent with the decreased lipid droplet accumulation on stiff substrates, we observed an increase in lipolysis-related gene expression in HSCs, including the total expression of ATGL, ABHD5, MGLL, and the phosphorylation level of LIPE (Fig. 6B; Supplementary Fig. S5B and S5C). We used atglistatin, an ATGL-specific inhibitor, to suppress the ATGL activity upregulated by mechanical cues in HSCs (Fig. 5D) and found stiffness-mediated upregulation of FFA secretion and lipid droplet depletion could be restored by atglistatin treatment (Fig. 6C; Supplementary Fig. S5E and S5F). These data raise the possibility that matrix stiffness triggers lipolysis in HSCs and provides FFAs as an energy source for activating FAO metabolism in tumor cells. Thus, we treated HSCs with atglistatin before performing HSCs–colon cancer cell lipid transfer experiments. As expected, pharmacologic inhibition of ATGL also reduced the transfer of BODIPY-labeled fatty acids derived from HSCs on stiff substrates into colon cancer cells (Fig. 6D; Supplementary Fig. S5G). Furthermore, CM from atglistatin-treated HSCs on stiff substrates blunted the upregulation of both FAO rate and protein expression of key genes in the FAO pathway in tumor cells, compared with that in the respective control groups (Fig. 6E and F; Supplementary Fig. S5H and S5I). In addition, HSCs on stiff substrates had higher expression levels of activation markers and showed well-spread fibrous morphology compared with those on soft substrates (Supplementary Fig. S6A–S6C).
Figure 6.

Matrix stiffness activates lipolysis in HSCs. A, Representative images (left) and quantification (right) of lipid level in LX-2 cells culture on 1 kPa, 10 kPa, and 25 kPa polyacrylamide hydrogels (one-way ANOVA; n = 5 independent experiments). B, Western blot analysis of lipolytic genes in LX-2 cells cultured on 1 kPa, 10 kPa, and 25 kPa polyacrylamide hydrogels. C, Representative images (top) and quantification (bottom) of lipid level in LX-2 cells cultured on indicated stiffness after treatment with DMSO or 10 μmol/L atglistatin (ATGLi) for 24 hours (one-way ANOVA; n = 5 independent experiments). D, Representative images (top) and quantification (bottom) of labeled lipids within DLD1 and HCT116 cells, after coculturing for 48 hours with LX-2 cells that were prestimulated with indicated treatment (one-way ANOVA; n = 5 independent experiments). E and F, FAO rate (E) and Western blot analysis (F) of DLD1 and HCT116 cells treated with indicated CM (one-way ANOVA; n = 5 independent experiments). Data are graphed as the mean ± SD. Scale bar, 5 μm. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Next, we explored the molecular mechanisms by which matrix stiffness mediates lipolysis in HSCs. Mechanical cues regulate the Hippo pathway effectors yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), which govern cellular responses to ECM stiffness (32). Increased interaction of ECM proteins in tumor microenvironments results in integrin-mediated outside-in signaling, which activates focal adhesion kinase (FAK) and Src (33). Active Src is capable of directly phosphorylating and activating YAP in breast carcinoma cells (34). YAP/TAZ target gene connective tissue growth factor (CTGF) was found significant upregulated in both LX-2 and JS-1 cells on stiff substrates (Supplementary Fig. S6A and S6B), suggesting a high activation status of the Hippo pathway. Thus, we hypothesized that FAK-Src signaling might contribute to stiffness-induced YAP activation in HSCs. To test this hypothesis, we examined the protein levels of these signaling cascades in both LX-2 and JS-1 cells cultured on polyacrylamide hydrogels with different stiffnesses. We found that matrix stiffness significantly elevated the levels of FAK, Src, and AKT phosphorylation, along with increased levels of YAP/TAZ and significantly decreased levels of YAP phosphorylation (Fig. 7A; Supplementary Fig. S7A). To further assess whether the FAK–YAP axis is involved in stiffness-enhanced HSCs lipolysis, we performed quantitative polymerase chain reaction (qPCR) and Western blotting. The mRNA and protein levels of key enzymes ATGL in lipolysis were significantly decreased by both pharmacologic inhibition of FAK (PF573228) and pharmacologic inhibition of YAP (verteporfin), but not the other key enzymes (Fig. 7B; Supplementary Fig. S7B and S7C), suggesting that matrix stiffness regulates HSCs lipolysis at least partly through the FAK–YAP–dependent upgradation pathway. We found that PF573228 and verteporfin could restore lipid droplet deletion and suppress FFAs secretion in HSCs on stiff substrates (Fig. 7C; Supplementary Fig. S7D and S7E). We prestimulated HSCs with FAK and YAP inhibitors for 24 hours and washed them with PBS to remove the remaining inhibitors. Pretreated HSCs were re-cultured in basal medium for another 24 hours, before performing lipid transfer experiments or collecting the supernatant as CM. The results show that inhibiting FAK and YAP blunted mechanical force-induced lipid cross-talk between HSCs and tumor cells (Fig. 7D; Supplementary Fig. S7F). Consistent with these results, FAO metabolism upregulation in tumor cells induced by CM from HSCs on stiff substrates was blunted with FAK and YAP inhibitors (Fig. 7E and F; Supplementary Fig. S7G and S7H). These findings demonstrate that the FAK–YAP–dependent mechanotransduction pathway is essential for stiffness-induced lipid cross-talk in the tumor niche.
Figure 7.

Matrix stiffness induces lipolysis in HSCs via activating the FAK–YAP signaling pathway. A, Western blot analysis of LX-2 cells cultured on 1 kPa, 10 kPa, and 25 kPa polyacrylamide hydrogels. B, Western blot analysis of LX-2 cells cultured on 25 kPa treated with DMSO, 10 μmol/L FAK inhibitor PF-573228 (FAKi), or 0.1 μmol/L YAP inhibitor verteporfin (YAPi) for 24 hours. C, Representative images (left) and quantification (right) of lipid level in LX-2 cells with indicated treatment (one-way ANOVA; n = 5 independent experiments). D, Representative images (left) and quantification (right) of labeled lipids within DLD1 and HCT116 cells after coculturing for 48 hours with LX-2 cells treated as indicated. Scale bar, 5 μm (one-way ANOVA; n = 5 independent experiments). E and F, FAO rate (E) and Western blot analysis (F) of DLD1 and HCT116 cells treated with indicated CM (one-way ANOVA; n = 5 independent experiments). Data are graphed as the mean ± SD. Scale bar, 5 μm. ns, not significant; P > 0.05; **, P < 0.01; ***, P < 0.001.
FAK and YAP inhibition enhances the efficacy of anti-VEGF therapy
To test whether metabolic cross-talk is involved in bevacizumab resistance and can maintain protumoral behavior induced by matrix stiffening, we assessed alterations in cell proliferation. Increased proliferation was observed in colon cancer cells when treated with CM from HSCs on stiff substrates (Supplementary Fig. S8A). We then examined whether treated colon cells could affect angiogenesis using an in vitro HUVEC model. CM from HSCs grown on stiff substrates enhanced the ability of tumor cells to induce tube formation and bevacizumab resistance of HUVECs (Supplementary Fig. S8B). Chicken chorioallantoic membrane assay also confirmed that colon cancer cells treated with CM from HSCs on stiff substrates had a significantly stronger capacity for angiogenesis (Supplementary Fig. S8C). Leveraging this metabolic mechanism, we sought to determine whether repressing FAO activation could overcome bevacizumab resistance in vitro. We treated colon cancer cells with etomoxir and CM from HSCs, and collected the supernatant of cancer cells after switching back to basal medium for 24 hours. The results revealed that repressing FAO activation significantly weakened the capacity for angiogenesis of cancer cells (Supplementary Fig. S8D and S8E). These data indicate that bevacizumab resistance in liver metastases is associated with matrix stiffness-coordinated cell-to-cell metabolic communication.
Finally, we tested whether inhibiting of FAK or YAP activity could potentially enhance the antiangiogenic efficacy in vivo (Fig. 8A). The combination of FAK or YAP inhibitors with 5-FU and B20 resulted in significant suppression of tumor proliferation (Fig. 8B and C). Consistent with in vitro results, FAO metabolic target expression in tumors arising from MC38/HSCs coinjections was also decreased by combined inhibition treatment compared with those treated with 5-FU+IgG or 5-FU+B20. Only in tumors arising from MC38/HSCs coinjections did both combination therapies show significantly synergistic powers in tumor angiogenesis suppression compared with the other subgroups. On the contrary, inhibition of FAK or YAP activity did not impact the expression of FAO metabolic targets or vessel density in tumors arising from MC38 mono-injections (Fig. 8D and E). In summary, our study provides evidence that an ECM stiffening-trigged intratumoral metabolic cross-talk, regulated by FAK–YAP signaling, mediates bevacizumab resistance through activating FAO metabolism in mCRC. Understanding the metabolic adaption in tumors under therapeutic stress and elucidating the underlying mechanism may contribute to more efficient multimodality treatments for patients with mCRC.
Figure 8.

FAK and YAP inhibition enhances the efficacy of anti-VEGF therapy. A, Schematic representation of HSC/MC38 coinjection subcutaneous tumor models used in B–E. MC38 cells (0.5×106) were subcutaneously injected with or without primary murine HSCs (0.5×106) into C57BL/6 mice. After 7 days, mice were treated with 5-FU+IgG, 5-FU+B20, 5-FU+B20+FAKi, or 5-FU+B20+YAPi (5-FU, 50 mg/kg; B20, 5 mg/kg; FAKi, 5 mg/kg; YAPi, 10 mg/kg; twice a week, 3 weeks). B, Representative images of subcutaneous tumors of mice treated with indicated treatment. C, Relative tumor growth curves (top) and relative tumor volume (bottom) of subcutaneous tumors with indicated treatment (one-way ANOVA; n = 5 per group). D, Representative IHC images of serial sections of subcutaneous tumors of mice with indicated treatment. Scale bar, 100 μm. E, Representative immunofluorescence images (left) showing tumor vessels (stained for CD31) and quantification of vessel density (right) in subcutaneous tumors with indicated treatment. Scale bar, 50 μm (one-way ANOVA; n = 5 per group). Data are graphed as the mean ± SD. ns, not significant; P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001. (A, Created with BioRender.com.)
Discussion
Bevacizumab has been used in patients with advanced colorectal tumors based on OS improvement. However, the survival benefit of anti-VEGF therapy in patients with mCRC is limited to a few months because of acquired resistance, which remains a major challenge in mCRC treatment (35). The mechanisms driving bevacizumab resistance require further investigation. Here, we report the mechanism of acquired resistance to anti-VEGF therapy in liver mCRC. In this study, we revealed that stromal matrix stiffening induced by antiangiogenic therapy leads to lipid metabolic cross-talk between HSCs and tumor cells, which plays a vital role in treatment resistance in liver mCRC.
Accumulating evidence has shown that under therapeutic stress, tumor cells can exhibit plasticity to adapt to a drug-treated environment, whereas the tumor microenvironment evolves in response to therapeutic stress (36). A comprehensive understanding of the evolution of tumor cells and the microenvironment during treatment is essential to uncover the mechanisms of resistance and to explore strategies that improve outcomes. To better understand the evolution of the tumor microenvironment under treatment stress, our use of pretreatment and posttreatment dynamic specimens from the same patients with liver mCRC for pairing analysis strengthens the clinical relevance of these findings. Anti-VEGF treatment has been reported to lead to abnormal deposition of ECM components (6). Notably, we found ECM deposition in patients who did not respond to bevacizumab. Consistent with the results of orthotopic mouse models of liver mCRC treated with different doses of B20 (7), these data support the concept of a therapeutic window, beyond which inappropriate VEGF inhibition will lead to inadequate perfusion and rebound angiogenesis. This observation emphasizes the practical applicability of our preclinical data and motivates further exploration into the potential benefit of judicious dosage of VEGF-targeting drugs in order to promote individualized therapy.
Shen and colleagues showed that increased metastases stiffness plays an important role in mCRC and influences the effect of antiangiogenic therapy on intra-tumoral blood vessel reduction. MAFs in liver metastases support sprouting angiogenesis by cytokines with concomitant local ECM remodeling (12). Although activated-HSC/MAFs are a major contributor to ECM remodeling of liver metastases, it is unknown if tumor stiffness can in return regulate the biology of the activated-HSC/MAFs and their tumor-promoting effects. In this study, we show that tumor-associated stiffness, built by activated HSCs, in return, activates metabolic reprogramming in HSCs and strengthens tumor's capacity to overcome therapeutic stress so as to form “an amplification loop” for metastatic growth in the liver.
Targeting the tumor stroma for therapeutic purposes is a novel concept that has gained attention in recent years (37). It remains to be seen whether alternative targets within the ECM, such as the direct targeting of HSCs or MAFs, could offer a therapeutic strategy and enhance the efficacy of systemic treatment in these patients. However, the therapeutic use of stroma inhibitors may have unanticipated outcomes. Deletion of a subset of stromal cells that express fibroblast activation proteins can limit tumor progression by enhancing antitumor immunity in lung carcinoma (38). Berna and colleagues reported that the depletion of carcinoma-associated fibroblasts leads to immunosuppression and accelerates pancreatic cancer (39). These contradictory results suggest that targeting the tumor stroma as a therapeutic approach might require consideration of the heterogeneous nature of the tumor microenvironment.
While prior context-specific studies have demonstrated that mechanical signatures of the tumor microenvironment continuously modulate cell functions, such as growth, survival, apoptosis, morphogenesis and metabolic activity (40), we focused exclusively on lipid metabolites and metabolic pathways. The full scope of the effect of the stiffness of the tumor microenvironment on metabolic reprogramming may be substantially greater. The “seed and soil” concept postulates that tumor cells develop best when their environment enhances their capacity to multiply and prevent cell death. In vivo, the "soil" is the microenvironment of the tumor, which is currently believed to have a crucial role in metastasis and medication resistance (41). While we identified lipid cross-talk between HSCs and tumor cells as a major mechanism for drug resistance in response to matrix stiffness, it is vital to determine whether comparable metabolic interaction occurs among other cells within the liver metastases microenvironment. The resolution of spatial omics and the development of single-cell metabolomics will be crucial for deciphering the metabolic architecture of tumors.
In summary, using clinical specimens and a number of in vitro and in vivo models, we identified matrix stiffness-mediated lipid communication as a potential therapeutic target for treating liver mCRC. These results have far-reaching implications for understanding how cells regulate metabolic adaptations based on extracellular mechanical cues. Our study also revealed that matrix stiffness-mediated HSCs-tumor cells lipid cross-talk contributes to the development of resistance to anti-VEGF therapy. We further showed that FAK and YAP inhibitors significantly improved the efficacy of bevacizumab to provide new strategies for reversing drug resistance.
Supplementary Material
Supplementary Methods
Figure. S1 Increased ECM deposition and active FAO metabolism induce Bevacizumab resistance in liver metastases.
Figure.S2 HSCs are essential for matrix stiffness-mediated FAO metabolic reprogramming in colon cancer cells.
Figure. S3 Conditioned medium derived from HSCs cultured on the stiff substrates upregulates FAO metabolism in colon cancer cells.
Figure. S4 High stiffness-induced HSCs promote FAO metabolism in colon cancer cells through secreting FFAs.
Figure. S5 Matrix stiffness activates lipolysis in HSCs.
Figure. S6 Matrix stiffness induces HSC activation.
Figure. S7 Matrix stiffness induces lipolysis in HSCs via activating the FAK-YAP signaling pathway.
Figure. S8 Matrix stiffness induced lipid crosstalk between HSCs and colon cancer cells promote tumor proliferation and angiogenesis.
Supplementary Table S1. Patient Information
Supplementary Table S2. The composition of polyacrylamide gels
Supplementary Table S3. Reagents or Resources
Supplementary Table S4. Primer sequences
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 82073303 to W. Liao, No. 82272635 to W. Liao, and No. 82102731 to R. Zhou), the President Fund of Nanfang Hospital (No. 2019Z025 to W. Liao), and the Natural Science Foundation of Guangdong Province of China (2022A1515012418 to R. Zhou). Schematic representations were created with BioRender.com.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
No disclosures were reported.
Authors' Contributions
Y. Zheng: Resources, data curation, visualization, methodology, writing–original draft. R. Zhou: Conceptualization, resources, visualization. J. Cai: Data curation, validation. N. Yang: Data curation, writing–review and editing. Z. Wen: Data curation, writing–review and editing. Z. Zhang: Data curation. H. Sun: Data curation. G. Huang: Data curation. Y. Guan: Data curation. N. Huang: Writing–review and editing. M. Shi: Writing–review and editing. Y. Liao: Writing–review and editing. J. Bin: Writing–review and editing. W. Liao: Conceptualization, resources, supervision, project administration.
References
- 1. Weitz J, Koch M, Debus J, Höhler T, Galle PR, Büchler MW. Colorectal cancer. Lancet 2005;365:153–65. [DOI] [PubMed] [Google Scholar]
- 2. Barkhatov L, Aghayan DL, Scuderi V, Cipriani F, Fretland AA, Kazaryan AM, et al. Long-term oncological outcomes after laparoscopic parenchyma-sparing redo liver resections for patients with metastatic colorectal cancer: a European multicenter study. Surg Endosc 2022;36:3374–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42. [DOI] [PubMed] [Google Scholar]
- 4. Lee SJ, Lee SY, Lee WS, Yoo JS, Sun JM, Lee J, et al. Phase I trial and pharmacokinetic study of tanibirumab, a fully human monoclonal antibody to vascular endothelial growth factor receptor 2, in patients with refractory solid tumors. Invest New Drugs 2017;35:782–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Q, Wang Y, Jia W, Deng H, Li G, Deng W, et al. Low-dose antiangiogenic therapy sensitizes breast cancer to PD-1 blockade. Clin Cancer Res 2020;26:1712–24. [DOI] [PubMed] [Google Scholar]
- 6. Rahbari NN, Kedrin D, Incio J, Liu H, Ho WW, Nia HT, et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci Transl Med 2016;8:360ra135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schiffmann LM, Brunold M, Liwschitz M, Goede V, Loges S, Wroblewski M, et al. A combination of low-dose bevacizumab and imatinib enhances vascular normalization without inducing extracellular matrix deposition. Br J Cancer 2017;116:600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nia HT, Munn LL, Jain RK. Physical traits of cancer. Science 2020;370:eaaz0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kai F, Laklai H, Weaver VM. Force matters: biomechanical regulation of cell invasion and migration in disease. Trends Cell Biol 2016;26:486–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quaas A, Walsh S, et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011;53:1192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Joyce MH, Lu C, James ER, Hegab R, Allen SC, Suggs LJ, et al. Phenotypic basis for matrix stiffness-dependent chemoresistance of breast cancer cells to doxorubicin. Front Oncol 2018;8:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shen Y, Wang X, Lu J, Salfenmoser M, Wirsik NM, Schleussner N, et al. Reduction of liver metastasis stiffness improves response to bevacizumab in metastatic colorectal cancer. Cancer Cell 2020;37:800–17. [DOI] [PubMed] [Google Scholar]
- 13. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell 2017;168:657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salvi AM, DeMali KA. Mechanisms linking mechanotransduction and cell metabolism. Curr Opin Cell Biol 2018;54:114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park JS, Burckhardt CJ, Lazcano R, Solis LM, Isogai T, Li L, et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020;578:621–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Choi J, Cha YJ, Koo JS. Adipocyte biology in breast cancer: from silent bystander to active facilitator. Prog Lipid Res 2018;69:11–20. [DOI] [PubMed] [Google Scholar]
- 17. Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov 2018;8:1006–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sawyer BT, Qamar L, Yamamoto TM, McMellen A, Watson ZL, Richer JK, et al. Targeting fatty acid oxidation to promote anoikis and inhibit ovarian cancer progression. Mol Cancer Res 2020;18:1088–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017;14:397–411. [DOI] [PubMed] [Google Scholar]
- 20. Shmarakov IO, Jiang H, Liu J, Fernandez EJ, Blaner WS. Hepatic stellate cell activation: a source for bioactive lipids. Biochim Biophys Acta Mol Cell Biol Lipids 2019;1864:629–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dou C, Liu Z, Tu K, Zhang H, Chen C, Yaqoob U, et al. P300 acetyltransferase mediates stiffness-induced activation of hepatic stellate cells into tumor-promoting myofibroblasts. Gastroenterology 2018;154:2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Syed S, Karadaghy A, Zustiak S. Simple polyacrylamide-based multiwell stiffness assay for the study of stiffness-dependent cell responses. J Vis Exp 2015; 97:52643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aguilera KY, Rivera LB, Hur H, Carbon JG, Toombs JE, Goldstein CD, et al. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res 2014;74:1032–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iwamoto H, Abe M, Yang Y, Cui D, Seki T, Nakamura M, et al. Cancer lipid metabolism confers antiangiogenic drug resistance. Cell Metab 2018;28:104–17. [DOI] [PubMed] [Google Scholar]
- 25. Setoyama D, Fujimura Y, Miura D. Metabolomics reveals that carnitine palmitoyltransferase-1 is a novel target for oxidative inactivation in human cells. Genes Cells 2013;18:1107–19. [DOI] [PubMed] [Google Scholar]
- 26. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008;88:125–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Myojin Y, Hikita H, Sugiyama M, Sasaki Y, Fukumoto K, Sakane S, et al. Hepatic stellate cells in hepatocellular carcinoma promote tumor growth via growth differentiation factor 15 production. Gastroenterology 2021;160:1741–54. [DOI] [PubMed] [Google Scholar]
- 28. Tu K, Li J, Verma VK, Liu C, Billadeau DD, Lamprecht G, et al. Vasodilator-stimulated phosphoprotein promotes activation of hepatic stellate cells by regulating Rab11-dependent plasma membrane targeting of transforming growth factor beta receptors. Hepatology 2015;61:361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, Wells WA, et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol Cancer Ther 2011;10:427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khomich O, Ivanov AV, Bartosch B. Metabolic hallmarks of hepatic stellate cells in liver fibrosis. Cells 2019;9:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang Y, Chen H, Yang P, Bai X, Shi Y, Vistro WA, et al. Hepatic lipid droplet breakdown through lipolysis during hibernation in Chinese soft-shelled turtle (Pelodiscus sinensis). Aging 2019;11:1990–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. LaQuaglia MJ, Grijalva JL, Mueller KA, Perez-Atayde AR, Kim HB, Sadri-Vakili G, et al. YAP subcellular localization and hippo pathway transcriptome analysis in pediatric hepatocellular carcinoma. Sci Rep 2016;6:30238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma H, Wang J, Zhao X, Wu T, Huang Z, Chen D, et al. Periostin promotes colorectal tumorigenesis through integrin-FAK-Src pathway-mediated YAP/TAZ activation. Cell Rep 2020;30:793–806. [DOI] [PubMed] [Google Scholar]
- 34. Sorrentino G, Ruggeri N, Zannini A, Ingallina E, Bertolio R, Marotta C, et al. Glucocorticoid receptor signaling activates YAP in breast cancer. Nat Commun 2017;8:14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011;473:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cathcart AM, Smith H, Labrie M, Mills GB. Characterization of anticancer drug resistance by reverse-phase protein array: new targets and strategies. Expert Rev Proteomics 2022;19:115–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lampi MC, Reinhart-King CA. Targeting extracellular matrix stiffness to attenuate disease: from molecular mechanisms to clinical trials. Sci Transl Med 2018;10:eaao0475. [DOI] [PubMed] [Google Scholar]
- 38. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer 2016;16:582–98. [DOI] [PubMed] [Google Scholar]
- 39. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25:719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep 2014;15:1243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Langley RR, Fidler IJ. The seed and soil hypothesis revisited–the role of tumor–stroma interactions in metastasis to different organs. Int J Cancer 2011;128:2527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods
Figure. S1 Increased ECM deposition and active FAO metabolism induce Bevacizumab resistance in liver metastases.
Figure.S2 HSCs are essential for matrix stiffness-mediated FAO metabolic reprogramming in colon cancer cells.
Figure. S3 Conditioned medium derived from HSCs cultured on the stiff substrates upregulates FAO metabolism in colon cancer cells.
Figure. S4 High stiffness-induced HSCs promote FAO metabolism in colon cancer cells through secreting FFAs.
Figure. S5 Matrix stiffness activates lipolysis in HSCs.
Figure. S6 Matrix stiffness induces HSC activation.
Figure. S7 Matrix stiffness induces lipolysis in HSCs via activating the FAK-YAP signaling pathway.
Figure. S8 Matrix stiffness induced lipid crosstalk between HSCs and colon cancer cells promote tumor proliferation and angiogenesis.
Supplementary Table S1. Patient Information
Supplementary Table S2. The composition of polyacrylamide gels
Supplementary Table S3. Reagents or Resources
Supplementary Table S4. Primer sequences
Data Availability Statement
The data generated in this study are available within the article and its Supplementary Data files. All other raw data are available upon request from the corresponding author. The mass spec lipidomics data was uploaded to MetaboLights (MTBLS8402).