

Abstract
Transforming growth factor β (TGF-β) is cytostatic towards damage-induced compensatory hepatocyte proliferation. This function is frequently lost during hepatocarcinogenesis, thereby switching the TGF-β role from tumour suppressor to tumour promoter. In the present study, we investigate Smad7 overexpression as a pathophysiological mechanism for cytostatic TGF-β inhibition in liver damage and hepatocellular carcinoma (HCC). Transgenic hepatocyte-specific Smad7 overexpression in damaged liver of fumarylacetoacetate hydrolase (FAH)-deficient mice increased compensatory proliferation of hepatocytes. Similarly, modulation of Smad7 expression changed the sensitivity of Huh7, FLC-4, HLE and HLF HCC cell lines for cytostatic TGF-β effects. In our cohort of 140 HCC patients, Smad7 transcripts were elevated in 41.4% of HCC samples as compared with adjacent tissue, with significant positive correlation to tumour size, whereas low Smad7 expression levels were significantly associated with worse clinical outcome. Univariate and multivariate analyses indicate Smad7 levels as an independent predictor for overall (P < 0.001) and disease-free survival (P = 0.0123). Delineating a mechanism for Smad7 transcriptional regulation in HCC, we identified cold-shock Y-box protein-1 (YB-1), a multifunctional transcription factor. YB-1 RNAi reduced TGF-β-induced and endogenous Smad7 expression in Huh7 and FLC-4 cells respectively. YB-1 and Smad7 mRNA expression levels correlated positively (P < 0.0001). Furthermore, nuclear co-localization of Smad7 and YB-1 proteins was present in cancer cells of those patients. In summary, the present study provides a YB-1/Smad7-mediated mechanism that interferes with anti-proliferative/tumour-suppressive TGF-β actions in a subgroup of HCC cells that may facilitate aspects of tumour progression.
Keywords: hepatocellular carcinoma (HCC), human tissue, liver cancer, transforming growth factor β (TGF-β), Y-box protein-1 (YB-1)
INTRODUCTION
Hepatocellular carcinoma (HCC) is the second highest cause of cancer-related mortality worldwide [1]. Its aetiology varies, being continent- and country-dependent, with hepatitis C virus (HCV) infection, hepatitis B virus (HBV) infection and alcohol intoxication as major causes. Much information on molecular mechanisms of HCC development is available; however, a breakthrough in designing targeted molecular therapies has not been achieved. One central player in the underlying paracrine and autocrine cytokine network acting on any liver cell type is transforming growth factor β (TGF-β).
Upon liver damage, TGF-β is rapidly induced and its levels rise with increasing disease severity, thus orchestrating multiple processes, including cell proliferation, differentiation, apoptosis, inflammation, angiogenesis, epithelial–mesenchymal transition (EMT), migration and invasion [2]. TGF-β drives stellate cell activation and myofibroblast transdifferentiation. Furthermore, TGF-β acts cytostatically during liver regeneration and compensatory hepatocyte proliferation [3]. Loss of these cytostatic TGF-β effects is critical for malignant conversion of stressed hepatocytes in progressed stages of liver disease [4].
The canonical TGF-β signalling pathway initiates with ligand binding to the type II receptor, which is subsequently activated by autophosphorylation and binds and phosphorylates the type I receptor. The active ligand–receptor complex recruits and phosphorylates intracellular signalling mediators, receptor (R)-Smads 2/3. Subsequent complex formation with Smad4 increases the retention time of Smad complexes in the nucleus, thereby facilitating transcriptional target gene regulation. Inhibitory Smad6 is specific for interference with R-Smad1/5/8 [downstream mediators of bone morphogenetic proteins (BMPs)], whereas Smad7 generally blunts R-Smad-dependent TGF-β and BMP signalling [5]. Loss of tumour-suppressive TGF-β action (i) by mutation of pathway core components or (ii) by selective inhibition of the cytostatic downstream signalling branch is a critical early step of epithelial cell carcinogenesis. The latter possibility reflects the situation in HCC, where fully available TGF-β signalling is redirected towards tumour progression and metastasis [6]. Several molecular mechanisms of such interferences are known in HCC, including loss of Smad3 adaptor β-spectrin expression [7] or mitogen-activated protein kinase (MAPK)-directed Smad3 linker phosphorylation [8]. Another mechanism to deter TGF-β signalling is up-regulation of inhibitory Smad7. We have previously shown that overexpression of Smad7 in liver inhibits TGF-β-mediated R-Smad phosphorylation and interferes with hepatocellular damage and fibrogenesis in bile duct ligation and CCl4-treated rodent models [9,10]. Smad7 overexpression was also associated with aggressiveness of squamous cell carcinoma, cholangiocarcinoma, breast and gastric cancer [11–14]. Smad7 immunoreactivity was found in tumour cells in 61% of 41 advanced HCCs, whereas dysplastic nodules and early HCCs were negative, indicating a potential role in late-stage hepatocellular carcinoma [15]. Contradictory results showing loss of Smad7 expression were recently reported for another HCC patients collective [16,17]. Taken together, data on the role of Smad7 in human HCC are conflicting in regard to its tumour-suppressive or oncogenic potential. So far, conclusions on the correlation of Smad7 expression to patient survival are based on relatively small sample cohorts.
In the present study, we analysed Smad7 effects on damage-dependent compensatory hepatocyte proliferation in the Fah−/− mouse model and on cytostatic TGF-β signalling in HCC cell lines. In contrast with former studies, we investigated Smad7 expression in a large cohort of matched tumour/surrounding tissue samples (n = 140) linked to a broad variety of clinicopathological parameters.
METHODS
Patient samples
Matched pairs of HCC and adjacent non-tumour tissues from 140 patients were included in the present study (for further details, see the Supplementary Online Data). Tumour characteristics were confirmed histologically excluding mixed-type HCC. Informed consent was obtained from all patients and tissue procurement was approved by the local Medical Ethics Committees.
Mouse strains and animal husbandry
Fah−/−mx.creAlbSmad7 mice were generated from B6,129-Fahtm1Mgo (Fah−/−) [18], Smad7Gt(Alb-loxpLacZloxp-Smad7)1Ty1 and Tg(Mx1-Cre)1Cgn [19] mice. Animals were kept under standard conditions with 12 h day/12 h night cycle and access to food and water ad libitum. To keep Fah−/− mice and derivatives healthy, they were treated with 7.5 mg/l 2-(2-nitro-4-fluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC; Swedish Orphan Biovitrum). The animal experiments were approved by the district government of Niedersachsen, Germany.
To generate hepatocyte-specific Smad7 transgenic mice (Smad7Gt(Alb-loxpLacZloxp-Smad7)1Ty1), four identical 800 bp insulator elements of the human β-globin cluster [20,21] were cloned into the pBR322*3 vector [22]. The albumin enhancer/promoter region [23] was ligated into an expression vector containing a rabbit β-globin intron and a human growth hormone (hGH) polyA site [24]. The fragment comprising promotor, intron and polyA site was purified and inserted between two pairs of insulators into the previously generated pBR322*3 derivative. A lacZ gene with a nuclear localization signal followed by a hGH polyA site and flanked by two loxP sites [25] was inserted between the β-globin intron and polyA site of the expression vector. cDNA for murine Smad7 was derived from total RNA of FVB/NHSD mice by RT-PCR (reverse transcription polymerase chain reaction) and inserted between 3′-loxP and 3′-polyA sites of the expression vector. Transgenic mice were generated on FVB/NHSD genetic background with isolated and purified Smad7-expression vector DNA according to standard techniques [26]. Offspring were analysed for presence of the Smad7 transgene by transgene-specific RT-PCR (S7-tg-for: 5′-ACAGCTCAATTGGGACAACA-3′, S7-tg-rev: 5′-ATGCATGCCTGGAATCCC-3′; product size 830 bp). Smad7 transgenic mice were backcrossed on to a C57Bl/6J genetic background and bred with Alb-Cre transgenic mice to generate hepatocyte-specific Smad7 transgenic mice. Recombination efficacy of the Smad7 transgene construct was assessed by lacZ staining and PCR (polymerase chain reaction) using primers selectively detecting Smad7 (RT-S7-for: 5′-GTGTTGCTGTGAATCTTACGG-3′, RT-S7-rev: 5′-GATGAAGATGGGGTAACTGCT-3′; product size: 476 bp) or the Smad7 transgene (RT-S7-tg-for: 5′-TCACCTTTCCTATCAACCCC-3′, RT-S7-tg-rev: 5′-CGCTCCTTGAGTTTCTTGAG-3′; product size: 560 bp).
Primary cells
Non-neoplastic tissue samples from liver resections were obtained from patients undergoing partial hepatectomy for meta-static liver tumours of colorectal cancer. Primary human hepatocytes (PHHs) were isolated and cultivated as described in [27] according to the guidelines of the charitable state-controlled foundation HTCR (Human Tissue and Cell Research, Regensburg, Germany) with informed patient consent and approved by the local ethical committee of the University of Regensburg. All experiments involving human tissues and cells have been carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Cell culture and treatment
Huh7, HepG2, Hep3B, PLC/PRF/5, HLE, HLF, Huh6 and FLC-4 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Lonza Group) supplemented with 10% FBS (Invitrogen), 4 mM l-glutamine (Lonza) and 100 units/ml penicillin/streptomycin (Biochrom).
DNA methylation analysis
Bisulfite treatment of genomic DNA was performed using the EZ DNA methylation Kit (Zymo Research). Using 25 ng of bisulfite-treated DNA as template, a 320 bp fragment containing 45 CpG dinucleotides was amplified with primer Smad7-2i5′ (5′-TTTATGTAGGAAGTYGAGGTTGG-3′) and Smad7-2i3′ (5′-CAACAACAACAACAAAAACCC-3′) (reaction conditions: 95 °C for 5 min; 40 cycles of 95 °C for 30 s, 60 °C of 45 s, 72 °C for 30 s; final incubation 72 °C for 5 min). The PCR product was analysed by direct Sanger sequencing using Smad7-2i3′ as sequencing primer.
Since it was not possible to amplify the Smad7 promoter region [28] after bisulfite modification, methylation of this region was analysed by cutting unmodified DNA with a methylation-sensitive restriction enzyme and subsequent amplification with flanking primers (qPCR (quantitative real-time polymerase chain reaction) with SYBR Green for online PCR product detection). Incubation with restriction enzymes HpaII and Bst1236I, control digests (with HindIII) and subsequent qPCR was performed as described previously [29]. The sequences of flanking primers were: Smad-7-F1, 5′-AAAGCGACAGGGTGTCTAGACG-3′; Smad-7-R1, 5′-TCTGCTCGGCTGGTTCCA-3′; Smad-7-F2, 5′-CACGGAAGATCCTGTCCCC-3′; and Smad-7-R2, 5′-GCTCTGTCTCGGGCAGCTC-3′. Nine CpG dinucleotides within the Smad7 promoter region and the beginning of exon 1 were analysed.
Immunoblot analysis
RIPA-buffer-lysed cells were separated by SDS/PAGE and transferred to nitrocellulose membranes (Pierce). For blocking the membranes, 5% non-fat dried skimmed milk powder in TBST was used. Primary antibodies used were: anti-Smad7 (MAB2029, R&D Systems), anti-[Y-box protein-1 (YB-1)] (ABIN353739, antibodies-online), anti-p21 (SAB4500065, Sigma–Aldrich), anti-pSmad2 (Ser465/Ser467), anti-Smad2, anti-pSmad3, anti-actin, anti-Mcl1, anti-Bcl-xL, anti-c-Myc and anti-tubulin (3010, 5339, 9520, 4961, 4572, 2762, 9402 and 2146 respectively, Cell Signaling Technology). Secondary antibodies used were: horseradish-peroxidase-linked anti-rabbit and anti-mouse antibodies (sc-2004 and sc-2005 respectively, Santa Cruz Biotechnology). The membrane was developed with Supersignal Substrate (34077, Pierce).
Immunohistochemical/immunofluorescent staining
Tissues were fixed in 4% buffered formalin solution and embedded in paraffin. Tissue sections (4 μm thick) were generated and used within 1 week. Antigen retrieval was performed by microwave treatment in EDTA buffer (1 mM, pH 8.0). Slides were blocked with peroxidase-blocking reagent (S2003, Dako) for 30 min and 10% H2O2 for 15 min at room temperature. For immunohistochemistry, slides were washed with PBS and incubated with the following antibodies: anti-Ki67 (1:500, ab-15580, Abcam), anti-p21 (1:50 dilution, M7202, DAKO), anti-Smad7 (1:100 dilution, ZB-8, Santa Cruz), anti-pSmad2 (1:100 dilution, 3010, Cell Signaling Technology) at 4 °C overnight. Slides were washed with PBS, incubated with streptavidin-conjugated anti-horseradish peroxidase antibody for 30 min and developed with diaminobenzidine (D5905, Sigma–Aldrich). Slides were counterstained with haematoxylin. Immunoreactivity was examined under a light microscope and quantification was performed as described in [30]. For YB-1/Smad7 co-staining, slides were washed with PBS and incubated with the anti-YB-1 antibody (1:100 dilution) at 4 °C overnight. Then slides were washed with PBS and incubated with the anti-Smad7 antibody (1:100; ZB-8, Santa Cruz Biotechnology) at 4 °C overnight. Then, Alexa Fluor® 488-conjugated donkey anti-rabbit IgG and Alexa Fluor® 633-conjugated rabbit anti-mouse IgG (A-21206 and A-21063 respectively, Life Technologies) were applied for 30 min at room temperature. After washing, samples were mounted with Cytomation Fluorescent Mounting Medium (S3023, Dako) and examined with a confocal microscope (DM IRE2, Leica). Sections treated only with secondary antibodies were used as negative controls.
Overexpression and RNAi
YB-1 expression plasmid (pDream2.1/YFP-YB-1-CFP) was previously described [31]. YB-1 knockdown oligonucleotides were from Qiagen (GS4904). Smad7 knockdown oligonucleotides were from Life Technologies (s69506). Plasmid transfection was performed using Fugene HD (04709705001, Roche). siRNA transfection was performed with RNAiMAX (13778, Invitrogen). Overexpression and knockdown efficiency was verified by qPCR and/or immunoblotting.
MTT assay
After TGF-β or compound treatment, cells were incubated with MTT reagent (M5655, Sigma–Aldrich) for 4 h, according to the manufacturer’s protocol. Absorbance was measured at 570 nm (reference 630 nm).
RNA isolation and qPCR
RNA from frozen tissue samples was extracted using the standard TRIzol® protocol (15596, Life Technologies). Total RNA was reverse-transcribed using Omnitect Reverse Transcription Kit (205313, Qiagen). Expression analysis for Smad7 and 18S rRNA was done with Taqman probes (Hs00178696_m1 and Hs03003631_g1, Applied Biosystems) as follows: 95 °C for 10 min; 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Expression analysis for YB-1 was performed with SYBR Green (4367659, Life Technologies). YB-1 primers: forward 5′-GGGTGCAGGAGAACAAGGTA-3′; reverse 5′-TCTTCATTGCCGTCCTCTCT-3′. 18S rRNA primers: forward 5′-AAACGGCTACCACATCCAAG-3′; reverse 5′-CCTCCAATGGATCCTCGTTA-3′. For data analysis, fold change in gene expression was normalized to 18S rRNA.
Statistics
Smad7 transcripts were quantified by qPCR. For normalization, 18S rRNA was used as a reference. Smad7 expression in cancer tissue was calculated by relating to Smad7 transcripts of corresponding adjacent non-tumour liver tissue. Smad7 and YB-1 mRNA expression differences between tumours and paired non-tumour tissues were analysed by Student’s paired two-tailed t test. Correlation between Smad7 expression and clinicopathological features were analysed by Fisher’s exact test. X-tile software was used to search for critical cut-off point (between ΔΔCT = 0.000225549 and ΔΔCT = 0.000227791) linking Smad7 expression level and patient survival time [32]. The Kaplan–Meier method (log-rank test) was used for survival rates. Statistical analyses were conducted using GraphPad Prism 5.0. Univariate analysis was calculated by Kaplan–Meier method (log-rank test). Multivariate analysis with 95% confidence intervals was done using Cox proportional hazard regression model with stepwise selection (forward, likelihood ratio). Correlation of YB-1 and Smad7 was done by Spearman rank-order coefficient method. Statistical analyses were conducted using SPSS (version 16.0). For each analysis, P < 0.05 indicated statistical significance. *p < 0.05, **p < 0.01, ***p < 0.001. Results are given as means +/− S.D.
RESULTS
Smad7 expression in Fah−/− mice
To analyse the impact of TGF-β signalling inhibition on compensatory hepatocyte proliferation following liver damage, we selected the fumarylacetoacetate hydrolase Fah−/− mouse model. It reflects the human disease hereditary tyrosinaemia type 1 (HT1) and is characterized by fast and severe liver damage and extremely high susceptibility to liver cancer. Treatment with NTBC, which is also used for HT1 patients, keeps Fah−/− mice alive. Upon NTBC withdrawal, the mice exhibit all of the phenotypic and biochemical manifestations of the human disease on an accelerated time scale, progressing to end-stage liver disease including HCC [18]. Fah−/−-MxCre mice were cross-bred with AlbSmad7 mice. Hepatocyte-specific Smad7 expression was induced with poly(I):poly(C) injections 2 weeks before the experiment (Figure 1a). Mice were killed after 2 weeks of treatment/withdrawal of NTBC. Smad7 overexpression in hepatocytes of Fah−/−S7+ mice was demonstrated by immunohistochemistry (Figure 1b). Consequently, down-regulation of p-Smad2 and p-Smad3 could be detected in these animals (Figure 1b and Supplementary Figure S1i). Liver damage upon NTBC withdrawal was obvious from haematoxylin/eosin (H/E) staining (Figure 1c), increased serum levels of glutamate pyruvate transaminase (GPT), glutamic oxaloacetic transaminase (GOT) and bilirubin, decreased glucose values and increased liver to body weight ratios (Supplementary Figures S1a–S1e). Upon liver damage, animals with induced Smad7 expression displayed significantly higher proliferation rates than Fah−/−S7–animals indicated by decreased p21 and increased Ki67 immunostaining (Figures 1d and 1e) accompanied by decreased TGF-β expression (Supplementary Figure S1f). Smad7 overexpression further reduced cleaved caspase 3 protein expression in NTCB-treated and untreated mice (Supplementary Figure S1g), indicating inhibition of TGF-β-dependent apoptosis in these animals. Accordingly, Supplementary Figure S1h shows p21 mRNA down-regulation and BcL-xL mRNA up-regulation (not significant) upon Smad7 overexpression. Additonally, c-Myc and Mcl-1 up-regulation as well as p21 down-regulation were shown by immunoblotting (Supplementary Figure S1i). Taken together, up-regulated Smad7 decreases the cytostatic effect of TGF-β in a liver damage model in vivo, thus enhancing compensatory proliferation. We hypothesize that hepatocyte-specific Smad7 up-regulation is a potential mechanism for hyperproliferation of hepatocytes during malignant progression upon chronic liver damage.
Figure 1. Ectopic expression of Smad7 in Fah−/− mice.
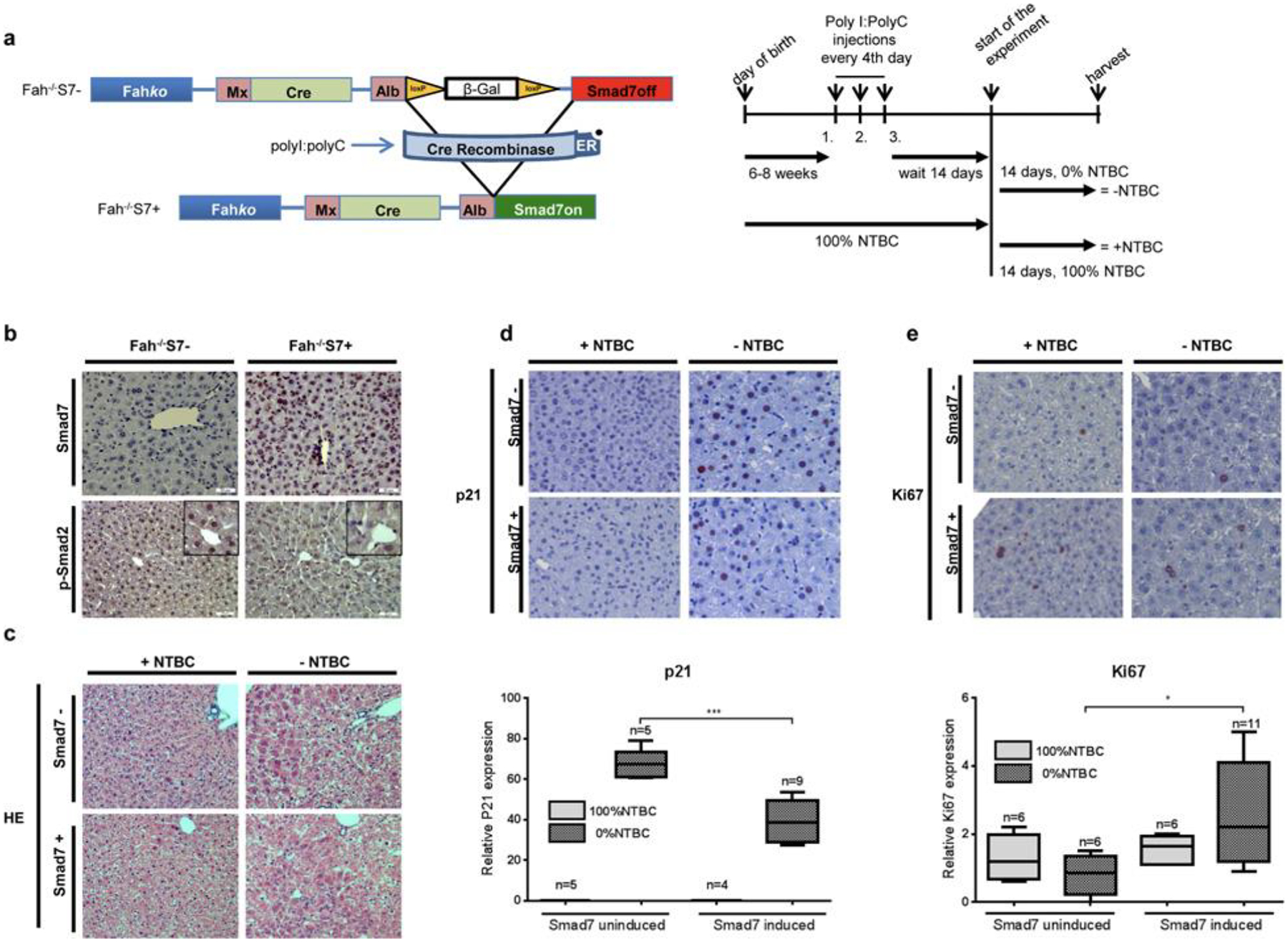
(a) In Fah−/−MxCreAlbSmad7 mice, liver damage was induced by NTBC withdrawal and Cre recombinase was activated by poly(I):poly(C) treatment for hepatocyte-specific Smad7 expression. Animals were killed after 2 weeks. (b) Induced Smad7 and decreased pSmad2 expression was detected by immunohistochemistry in Fah−/−/S7+ animals. (c) H/E staining and (d) immunohistochemical examination of p21; quantification of p21-positive hepatocyte nuclei is shown. (e) Smad7-dependent decrease in antiproliferative p21 expression is reflected in significantly increased proliferative activity, as shown by quantified Ki67-positive nuclear staining.
Smad7 expression in HCC cell lines
To investigate this further, we compared intrinsic Smad7 expression in eight HCC cell lines and cultured normal PHHs from eight preparations. We found high Smad7 levels in FLC-4 and Huh6 cells, low levels in PLC and HepG2 cells, and intermediate levels in Hep3B, Huh7, HLE and HLF cells (Figure 2a). All of the cell lines exhibited higher Smad7 expression levels than normal PHHs. On the basis of our cell line clustering [33], Huh7 and FLC-4 cells were chosen as model systems for responsiveness or insensitivity towards TGF-β-dependent proliferation control respectively (Figure 2b).
Figure 2. Smad7 inhibits TGF-β-mediated cytostasis in HCC cell lines.
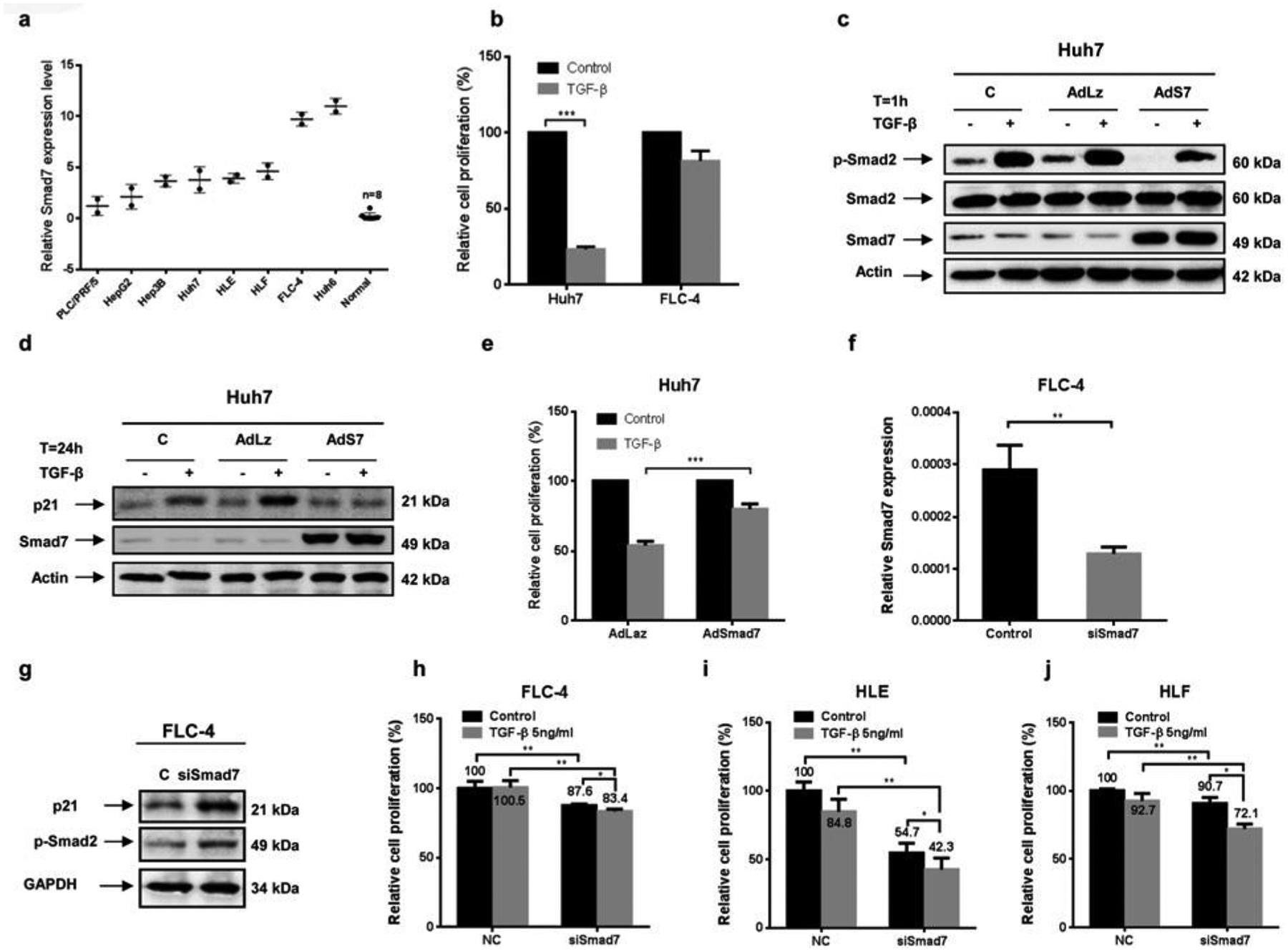
(a) Endogenous expression levels of Smad7 mRNA in normal primary human hepatocytes and human HCC cell lines measured by qPCR. (b) TGF-β-mediated proliferation inhibition was found in Huh7 cells with intermediate Smad7 expression, whereas FLC-4 cells with high Smad7 expression were not responsive, as measured by MTT assay. (c–e) Adenovirus-mediated ectopic Smad7 expression inhibited (c) endogenous and TGF-β-induced Smad2 phosphorylation, (d) p21 protein expression and (e) decreased the proliferation-antagonizing effect of TGF-β in Huh7 cells. (f) Knocking down Smad7 expression in FLC-4 (g) induced p21 protein expression and Smad2 phosphorylation and sensitized (h) FLC-4, (i) HLE and (j) HLF cells for TGF-β-mediated proliferation inhibition.
Huh7 cells with intermediate Smad7 expression levels demonstrated reduced proliferation rates and induction of Smad2 phosphorylation and p21 expression upon TGF-β incubation (Figures 2b, 2c and 2d). Ectopic overexpression of Smad7 in Huh7 cells decreased basal and TGF-β-dependent Smad2 phosphorylation, abrogated TGF-β-induced p21 expression and impaired its inhibitory effect on proliferation (from 54.1% to 80.2%; Figures 2c, 2d and 2e). Conversely, knockdown of Smad7 in FLC4 cells with high intrinsic Smad7 levels using RNAi (Figure 2f) induced p21 expression and phosphorylation of Smad2 (Figure 2g), thus partially restoring the cytostatic response to TGF-β (Figure 2h). To strengthen our findings, two other mesenchymal HCC cell lines, HLE and HLF, both show significant induction of proliferation inhibition after Smad7 knockdown (Figures 2i and 2j). Interestingly, the effect of Smad7 knockdown on proliferation inhibition, even by autocrine TGF-β signalling (comparing black bars) is prominent in HLE cells.
These data suggest that intrinsic up-regulation of Smad7 serves as a countermechanism to modulate TGF-β-dependent cytostasis in HCC cell lines.
High relative Smad7 expression levels in human HCC is associated with survival advantage
We next analysed Smad7 mRNA expression levels in 140 tumour and tumour-surrounding tissue samples from human HCC patients with different aetiologies (Table 1) by qPCR (Figure 3a). Clinical data for most patients enrolled were available, subclassifying the cohort according to age, gender, tumour number, tumour size, degree of differentiation, vascular invasion, Union for International Cancer Control (UICC) stages, HBV serology, cirrhosis, AFP (α-fetoprotein) and AST (aspartate aminotransferase) levels.
Table 1.
Aetiology of HCC development in the study population
| Alcohol | NASH | HCV+alcohol | HCV | HBV+alcohol | HBV | N/A | Total | |
|---|---|---|---|---|---|---|---|---|
| Number of patients | 8 | 16 | 1 | 3 | 4 | 78 | 30 | 140 |
N/A, not available.
Figure 3. Smad7 mRNA expression in 140 HCC samples relative to 18S ribosomal RNA expression.
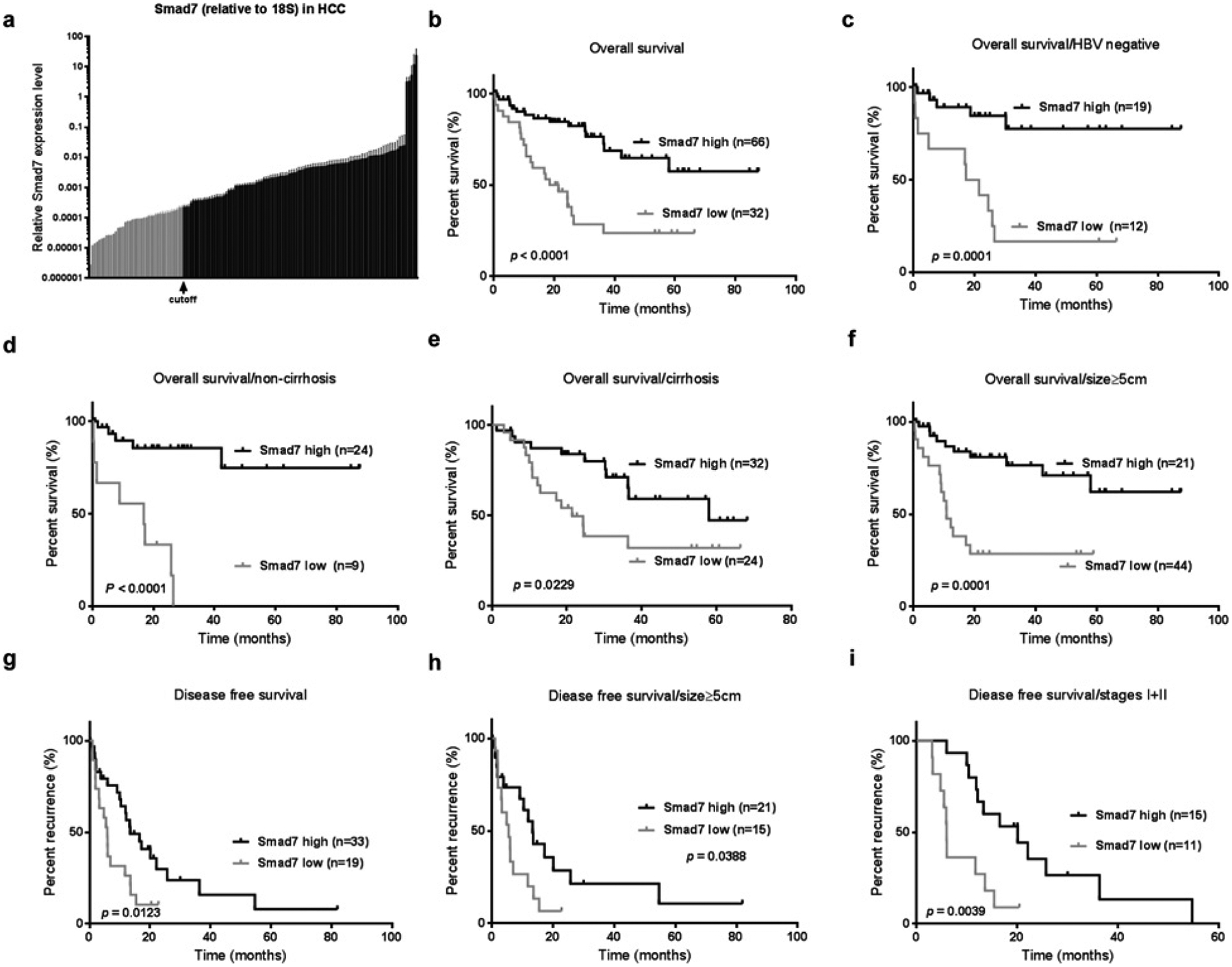
(a) Smad7 expression levels in HCC samples were determined by qPCR. The X-tile algorithm was applied to determine critical cut-off with respect to patient survival data. Kaplan–Meier analysis of OS for (b) high-compared with low-Smad7-expressing patients (P < 0.0001) subclassified according to (c) HBV infection status (P = 0.0001), (d and e) cirrhosis status (P < 0.0001 and P = 0.02) and (f) tumour size > 5 cm (P = 0.0001). Kaplan–Meier analysis of DFS (g) for high-compared with low-Smad7-expressing patients (P = 0.0123) and subclassified for (h) tumour size (P = 0.0388) and (i) UICC stage I and II (P = 0.039).
Univariate analysis defined Smad7 mRNA levels as significantly associated with overall survival (OS) (P < 0.001) and disease-free survival (DFS) (P = 0.012). Multivariate analysis suggested that Smad7 mRNA expression levels were an independent predictor of OS [hazard ratio (HR) = 0.275, 95% confidence interval (CI) = 0.144–0.527, P < 0.001] and DFS (HR = 0.432, 95% CI = 0.220–0.849, P = 0.015]. Multivariate analysis also revealed association of tumour number, vascular invasion and stages with OS and tumour stages with DFS (Table 2).
Table 2.
Univariate and multivariate analysis of factors associated with OS and DFS of 140 HCC patients
| OS | DFS | |||||||
|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | |||||
| Variable | P value | P value | HR | 95% CI | P value | P value | HR | 95% CI |
| Age (<54 versus ⩾54) | 0.193 | 0.143 | ||||||
| Gender (female versus male) | 0.561 | 0.112 | ||||||
| Tumour number (solitary versus multiple) | 0.012 | 0.017 | 2.811 | 1.213–6.844 | 0.054 | |||
| Tumour size (<5 versus ⩾5) | 0.584 | 0.206 | ||||||
| Vascular invasion (no versus yes) | 0.002 | 0.003 | 2.6 | 1.373–4.925 | 0.443 | |||
| UICC stages (I+II versus III+IV) | <0.001 | 0.001 | 3.735 | 1.762–7.916 | 0.001 | 0.001 | 3.204 | 1.600–6.417 |
| Anti-HBV (positive versus negative) | 0.848 | 0.546 | ||||||
| Cirrhosis (no versus yes) | 0.863 | 0.688 | ||||||
| AFP (ng/ml) (<20 versus ⩾20) | 0.208 | 0.308 | ||||||
| Smad7 (low versus high) | <0.001 | <0.001 | 0.275 | 0.144–0.527 | 0.012 | 0.015 | 0.432 | 0.220–0.849 |
Factors associated with OS display significant P values for vascular invasion, tumour stages and Smad7 mRNA expression levels, whereas for DFS, tumour stages and Smad7 mRNA were significant.
The critical cut-off linking Smad7 expression with patient survival was determined by X-tile analysis [32] classifying 99 patients as high-Smad7-expressing and 41 patients as low-Smad7-expressing (Table 3). Kaplan–Meier analysis revealed that lower Smad7 levels were correlated with worse clinical outcome. The median OS was significantly shortened in patients with low Smad7 (20 months compared with undefined, P < 0.0001; Figure 3b). Median DFS was also significantly shortened in patients with low Smad7 (5.87 months compared with 13.43, P = 0.0123, Figure 3g). Furthermore, Smad7 displayed prognostic relevance in distinct HCC subgroups classified by the following clinical features, i.e. (i) regarding OS: HBV-negative (P = 0.0001, Figure 3c), non-cirrhosis (P < 0.0001, Figure 3d), cirrhosis (P = 0.02, Figure 3e) and size ⩾5 cm (P = 0.0001, Figure 3f); and (2) regarding DFS: size ⩾5 cm (P = 0.0388, Figure 3h) and stages (P = 0.0039, Figure 3i).
Table 3.
Correlation between Smad7 expression and clinicopathological characteristics of 140 HCC patients
| Low (n = 41) | High (n = 99) | P value | |
|---|---|---|---|
| Age (years, mean ± S.D.) | 52.61 ± 13.63 (n = 33) | 55.43 ± 13.18 (n = 83) | 0.31 |
| Gender | 0.20 | ||
| Male | 29 | 61 | |
| Female | 4 | 20 | |
| Number of tumours | 0.52 | ||
| Solitary | 15 | 33 | |
| Multiple | 6 | 8 | |
| Tumour size (cm) | |||
| <5 | 9 | 21 | 0.82 |
| ⩾5 | 24 | 48 | |
| Degree of differentiation | 0.06 | ||
| Poor | 19 | 30 | |
| Moderate | 14 | 45 | |
| Good | 1 | 11 | |
| Vascular invasion | 1 | ||
| (+) | 14 | 28 | |
| (−) | 16 | 31 | |
| UICC stages | 0.64 | ||
| I+II | 14 | 26 | |
| III+IV | 13 | 31 | |
| Anti-HBV | 0.65 | ||
| (+) | 23 | 43 | |
| (−) | 12 | 17 | |
| Cirrhosis | 0.01 | ||
| (+) | 28 | 43 | |
| (−) | 11 | 49 | |
| AFP (ng/ml) | 1 | ||
| <20 | 11 | 24 | |
| ⩾20 | 19 | 40 | |
| AST | 60.69 ± 37.76 | 137.84 ± 385.59 | 0.14 |
Smad7 mRNA expression was determined in 140 patients. Using the X-tile algorithm, the critical cut-off linking Smad7 expression levels with patient survival was determined. Significant correlations were observed for cirrhosis by Fisher’s exact test (P = 0.01). No relationship with any other clinicopathological features was found.
Interestingly, Smad7 expression levels were found to be significantly correlated with patients’ cirrhosis status (P = 0.0122), but not with other clinical features (Table 3). Additionally, correlation of Smad7 expression levels in surrounding tissue with survival, but not other clinical parameters, could be demonstrated (Supplementary Figure S2 and Supplementary Table S2). The critical cut-off was determined by the X-tile algorithm. These data indicate a substantial role for TGF-β signalling regulation in surrounding areas for tumour aggressiveness.
In summary, these findings indicate that high Smad7 expression in hepatocellular liver tumours is common, and suggests a beneficial outcome regarding OS and DFS, as well as in the following subgroups: non-HBV, cirrhosis, non-cirrhosis, tumour size and tumour stages.
Smad7 expression in human HCC patients compared with adjacent tissue of the same patient
Commonly, marker protein expression in cancerous tissue is compared with surrounding tissue of the same patient. In our cohort, Smad7 mRNA expression in HCC as compared with surrounding tissue was >2-fold up-regulated in 41.4% of the patients, whereas, in 14.3% of HCC samples, Smad7 expression was <0.5-fold compared with the corresponding surrounding tissue (Figure 4a). Interestingly, 11.4% of HCC samples demonstrated a more than 10-fold up-regulation of Smad7 expression (Figure 4a). On average in the complete cohort, Smad7 transcripts were 4.16-fold more abundant in HCC as compared with matched non-tumour tissue (Figure 4b). Performing aetiology subclassification, Smad7 was significantly more frequently up-regulated in the HCC compared with the surrounding tissue in the HBV collective as compared with alcoholic liver disease (P < 0.001) or non-alcoholic steatohepatitis (P < 0.001) patients (Figure 4c). However, as only limited sample numbers of aetiologies other than HBV were included in our cohort, these correlations need to be verified in larger collectives.
Figure 4. Fold change of Smad7 expression in tumour relative to non-tumour tissue in a cohort of 140 HCC patients of different aetiologies.
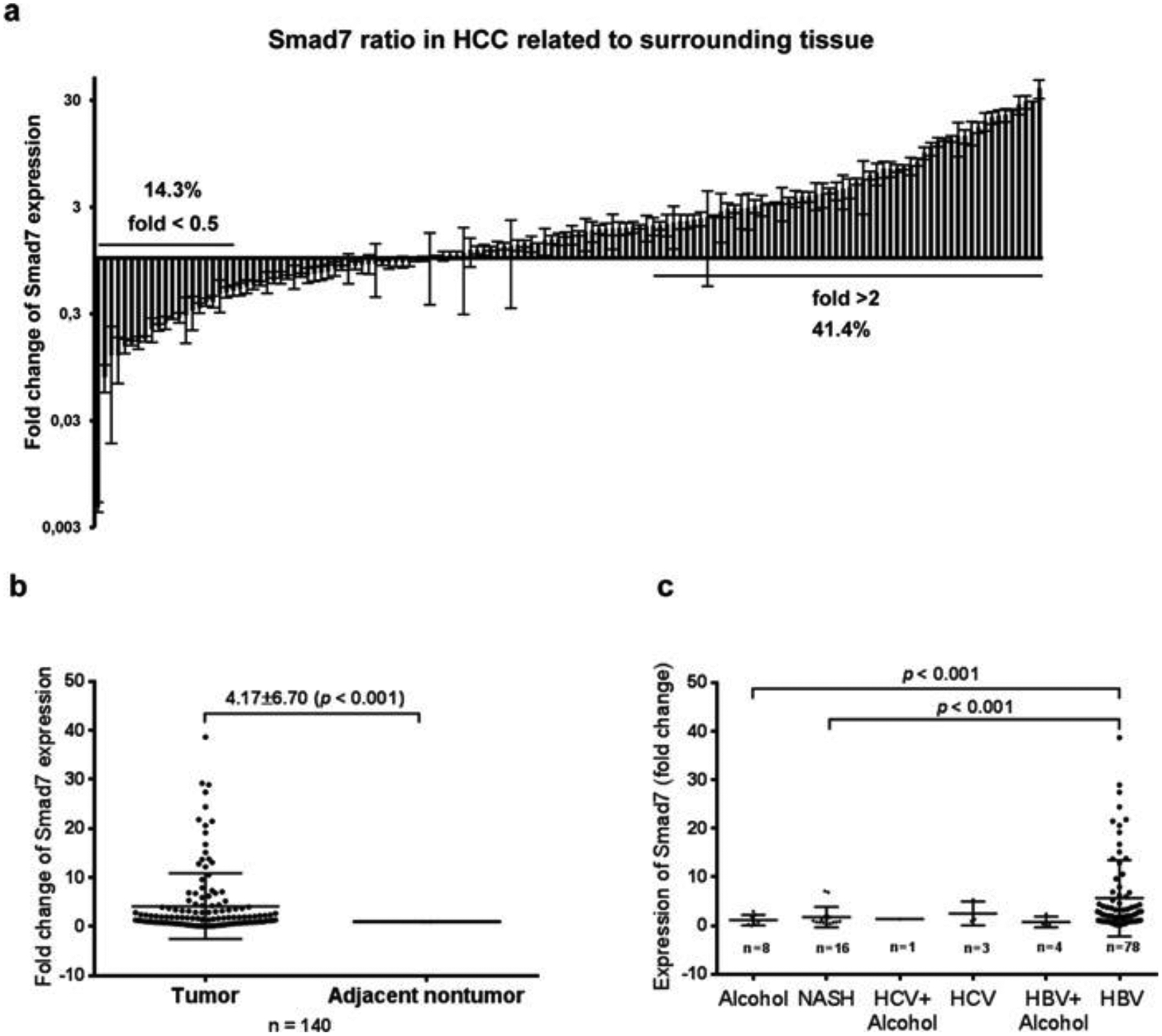
(a) Approximately 14.3% of patients displayed repressed (<0.5-fold) and 41.4% increased (>2-fold) Smad7 expression. 11.4% of patients showed more than 10-fold increased expression. (b) Medium fold change of Smad7 expression in HCC as compared with surrounding tissue in the entire cohort was 4.165 ± 0.567 (P < 0.001); (c) Subgrouping of HCC patients based on variant aetiologies of the underlying disease related to fold change of Smad7 expression in HCC compared with non-tumour tissue; significant differences were observed between HBV patients and alcoholic liver disease patients (P < 0.001), and between HBV and non-alcoholic steatohepatitis (NASH) patients (P < 0.001); n of other aetiologies was not sufficient for correlation analysis.
Details of the correlation analysis for Smad7 expression in HCC compared with surrounding tissue of the same patient regarding clinicopathological features are listed in Table 4. Significant correlation was only found between up-regulated Smad7 transcripts in HCC compared with surrounding tissue and tumour size >5 cm (P = 0.03). For other clinicopathological features, including patient survival data (results not shown), no significant correlations were found. As Smad7 is a very sensitive regulator of TGF-β signalling, we also wanted to include slightly changed Smad7 expression levels into the correlation analysis. Applying ratio thresholds of < 1 or ⩾1 for HCC compared with surrounding tissue to our analysis, we could confirm correlation of up-regulated Smad7 to tumour size and found an additional correlation with UICC stages III and IV (Supplementary Table S1). Using the X-tile algorithm, no critical cut-off showing correlation of Smad7 expression ratio (HCC/surrounding tissue) with survival could be determined.
Table 4.
Correlation between Smad7 expression and clinicopathological characteristics of 140 HCCs
| Smad7 expression (mRNA) | |||
|---|---|---|---|
| Clinicopathological variables | Down (<0.5) n = 20 | Up (>2) n = 58 | P value |
| Age (years, mean ± S.D.) | 53.69 ± 9.82 (n = 16) | 54.07 ± 14.09 (n = 44) | 0.91 |
| Gender | |||
| Male | 14 | 36 | 0.72 |
| Female | 2 | 8 | |
| Number of tumours | |||
| Solitary | 8 | 25 | 1.00 |
| Multiple | 1 | 5 | |
| Tumour size (cm) | |||
| <5 | 7 | 5 | 0.03 |
| ⩾5 | 9 | 33 | |
| Degree of differentiation | 0.74 | ||
| Poor | 6 | 24 | |
| Moderate | 9 | 26 | |
| Good | 2 | 4 | |
| Vascular invasion | |||
| (+) | 6 | 13 | 1 |
| (−) | 10 | 22 | |
| UICC stages | |||
| I+II | 8 | 16 | 0.54 |
| III+IV | 7 | 24 | |
| Anti-HBV | |||
| (+) | 8 | 35 | 0.69 |
| (−) | 3 | 9 | |
| Cirrhosis | 1 | ||
| (+) | 11 | 32 | |
| (−) | 8 | 26 | |
| AFP (ng/ml) | 0.72 | ||
| <20 | 4 | 9 | |
| ⩾20 | 9 | 27 | |
| AST (units/litre) | 253.80 ± 174.20 (n = 15) | 56.15 ± 6.20 | 0.28 |
Expression levels of Smad7 mRNA in 140 HCC samples as compared to surrounding tissue was determined. Applying thresholds of <0.5- and >2-fold regulation, significant correlations were observed for tumour size by Fisher’s exact test (P value 0.03). No relationship with any other clinicopathological features was found.
Taken together, our results suggest that up-regulated Smad7 transcription in HCC tissue as compared with surrounding tissue of the same patients, especially in the HBV cohort, positively correlates with tumour size and possibly with advanced stages III/IV.
Cold-shock protein YB-1 is a key regulator of Smad7 expression in HCC cell lines
We were then interested in finding a mechanism for Smad7 expression regulation in HCC as discussed above. As we previously identified a prominent CpG island in the Smad7 promoter, we thought modulated promoter methylation might lead to increased transcription rates [28]. A total of nine out of 41 CpGs were interrogated at distal and proximal promoter regions, but we could not identify any DNA methylations in the eight HCC cell lines described above. This indicates that up-regulated Smad7 expression in HCC cell lines does not relate to differential promoter methylation (for Huh7 cells, see Supplementary Figure S3). Thus we assumed a different mechanism of Smad7 expression regulation.
In hepatic stellate cells, we previously delineated that the ancestral multitasking protein YB-1 can directly associate with the Smad7 promoter and affects transcription rates [34]. To analyse similar events in HCC cell lines, overexpression and knockdown experiments were set up. Ectopic expression of YB-1 in Huh7 cells induced Smad7 expression by 2.6-fold (Figure 5a), whereas RNAi targeting YB-1 expression decreased TGF-β-dependent Smad7 expression in Huh7 cells 1.9-fold (Figure 5b) and intrinsic Smad7 expression in FLC-4 cells 3.5-fold (Figure 5c). Moreover, by qPCR we found a significant correlation (rs = 0.93, P = 0.001) between transcript levels of YB-1 and Smad7 in eight HCC cell lines (Figure 5d). To translate findings into patients, we quantified YB-1 mRNA expression in a random selection of samples from the above cohort of HCC patients. A significant positive correlation between YB-1 and Smad7 mRNA expression levels (HCC normalized to surrounding tissue of the same patients) was calculated (rs = 0.75, P < 0.0001) (Figure 5e). By immunofluorescence, cancer cells displaying nuclear co-localization of YB-1 and Smad7 proteins were seen in HCC tissue (Figure 5f).
Figure 5. YB-1 induces Smad7 expression in HCC cell lines.
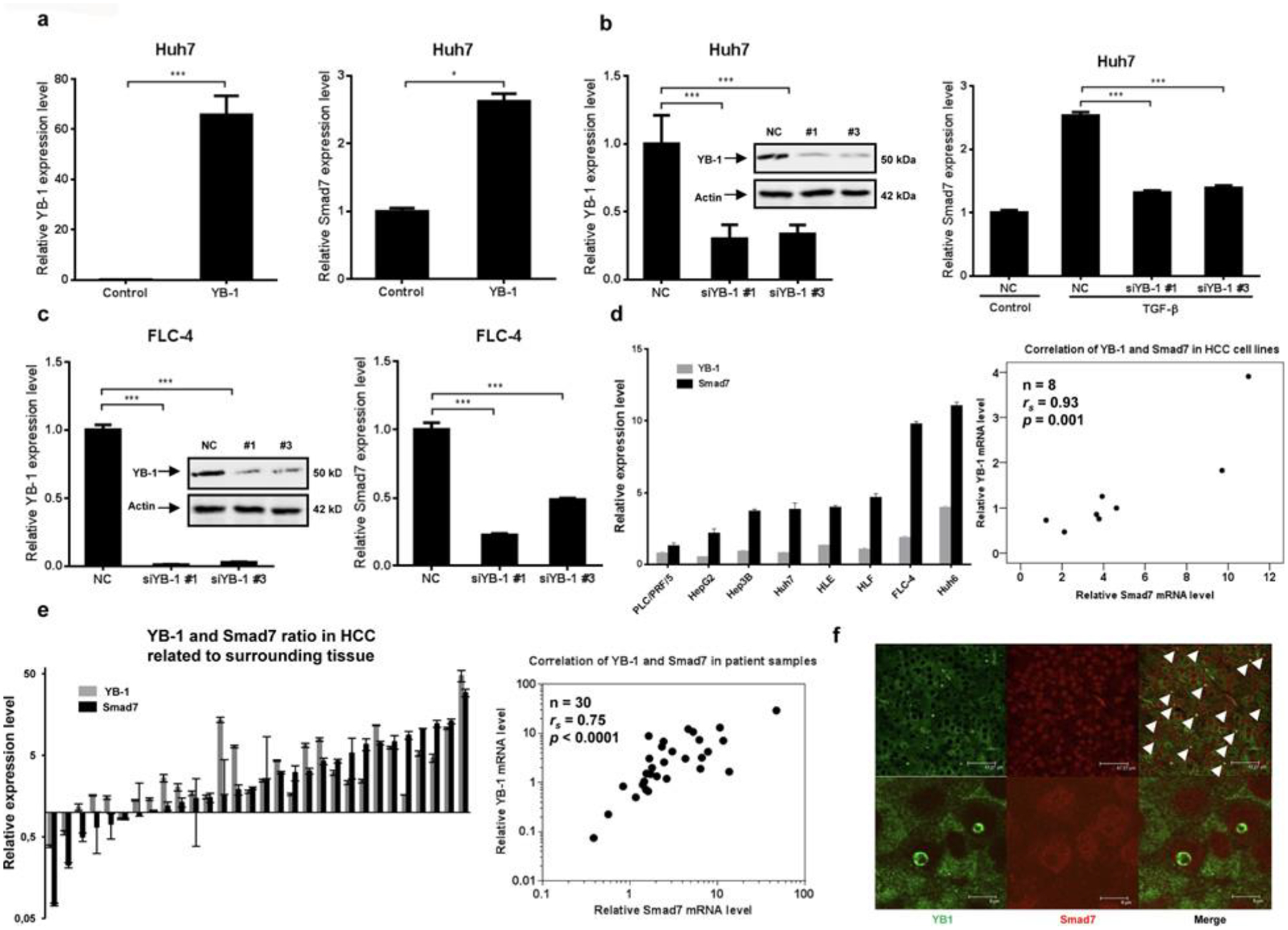
(a) Expression of YB-1 in Huh7 cells upon transient transfection with a corresponding expression plasmid induces Smad7 mRNA expression. (b and c) Knockdown of YB-1 using RNAi modulates TGF-β-induced (b) and respective endogenous (c) Smad7 expression in Huh7 (b) and in FLC-4 (c) cells. (d and e) Co-expression of YB-1 and Smad7 mRNA was analysed by qPCR in (d) eight HCC cell lines and (e) 30 HCC samples. Significant correlation between differential expression levels of both genes when comparing HCC with surrounding tissue was identified (P = 0.001 and P < 0.0001 respectively); rs, Spearman rank order correlation coefficient; (f) nuclear co-staining of YB-1 (green, arrows and inlay) and Smad7 (red) was frequently found in cancer cells of HCC patients. The bar histograms summarize qPCR results for mRNA levels; representative immunoblot analyses showing knockdown are presented.
DISCUSSION
In the present study, we have gained further insight into the role of the TGF-β inhibitor Smad7 in hepatocarcinogenesis. First, we showed that ectopic overexpression of Smad7 in hepatocytes enhances compensatory proliferation upon liver damage in the Fah−/− mouse model, indicating its potential as a negative regulator of cytostatic TGF-β signalling. Secondly, similarly, modulation of Smad7 expression in HCC cell lines with RNAi or Smad7 expression constructs has an impact on sensitivity for cytostatic TGF-β effects. Thirdly, in a cohort of 140 patients, we found up-regulated Smad7 transcripts in 41.4% of HCC tissue samples compared with matched surrounding tissue with significant correlation to tumour size. Furthermore, low relative Smad7 expression levels both in HCC samples as well as in the surrounding tissue were significantly associated with a bad prognosis. Univariate and multivariate analyses indicate Smad7 levels in HCC as an independent predictor for OS and DFS. Finally, we identified up-regulated expression of transcription factor YB-1 as a mechanism for up-regulated Smad7 mRNA expression in HCC cells and human patients’ HCC tissue.
It is widely accepted that one early major step of progression from chronic liver disease to HCC is abrogation of cytostatic TGF-β effects. Besides previously identified mechanisms [7,35], we hypothesized that Smad7 up-regulation in precancerous disease stages may inhibit tumour-suppressive TGF-β signalling, thus facilitating carcinogenesis. Accordingly, proliferative activity in damaged liver tissue is enhanced by hepatocyte-specific ectopic Smad7 expression in the Fah−/− mouse model, at least partially by interfering with TGF-β-induced p21 expression. Furthermore, oncogenic c-Myc, anti-apoptotic Mcl1 (possibly Bcl-xL) and cleaved caspase 3 are up-regulated. In line with the tumour-promoting Smad7 concept, Matsuzaki et al. [36] found chronic autocrine TGF-β stimulation of Huh7 and HepG2 cells leading to continuous cycles of Smad7 induction, which negatively affects Smad2 signalling and TGF-β-mediated cytostatic responses. Data investigating the apoptotic response of TGF-β-treated Hep3B and Huh7 cells are also consistent with such a hypothesis [37,38]. In line with this, the present study showed that Smad7 knockdown in FLC-4 cells with up-regulated Smad7 expression restores the cytostatic TGF-β response, further arguing for the presence of tumour-promoting Smad7 effects in liver cancer cells. Finally, we recently found that Smad7-mediated loss of cytostatic TGF-β function is critical for tumour-initiating cancer cell development in a mouse model of HCV plus alcohol-mediated hepatocarcinogenesis in HCV patients with HCC [4].
Clinically, comparing Smad7 mRNA expression in the tumour area with surrounding non-tumour tissue of the same patient indicates frequent up-regulation in HCC, especially in patients with large tumours, and, if very tight thresholds are applied, additionally in advanced tumour stages. When comparing relative Smad7 mRNA levels in HCC tissue using the X-tile algorithm [32], we found a significant positive correlation between Smad7 expression levels and patients’ OS and DFS, in part linked to other clinicopathological parameters. Interestingly, also Smad7 expression levels in surrounding tissue were positively associated with prognosis. This suggests that not the ratio of Smad7 up-regulation compared with surrounding tissue, but rather absolute Smad7 levels within the tumour as well as in surrounding tissue affect survival. Thus regulation of TGF-β signalling in the tumour environment is probably as important for HCC development as TGF-β signalling regulation in the tumour itself. In other words, our data suggest that high levels of Smad7 in surrounding tissue as well as in tumour tissue are beneficial for the patient. However, that ratio determines whether the tumour is likely to grow. If Smad7 is only up-regulated in the tumour, TGF-β signalling in the surrounding tissue seems to support tumour growth. Furthermore, we want to note that HCC-surrounding tissue rarely represents healthy tissue, but rather exhibits pathological signalling changes itself. This might affect the missing correlation of Smad7 expression in HCC related to surrounding areas to survival, but explain the association to large late stage tumours.
In line with our data, Xia et al. [17] recently found a correlation of low Smad7 expression with poor patient survival. However, in contrast with our patients’ collective, they reported decreased Smad7 expression in HCC as compared with the surrounding tissue in the majority of their samples, which again reflects the heterogeneity of HCC. More specifically, this might be due to different staging and, e.g., the grading of cirrhosis in the reference material, as compared with our cohort.
These clinical data support the hypothesis that increased expression of Smad7 in a subcohort of HCC patients might be involved in the host resistance to late-stage hepatocarcinogenesis by interfering with tumour-promoting activities of TGF-β. Such assumption is supported by data demonstrating enhanced liver carcinogenesis in progressed stages of diethylnitrosamine treatment-induced carcinogenesis in Smad7-deficient mice, as compared with control mice [16]. Accordingly, positive Smad7 immunostaining was found in 61% of advanced human HCCs, whereas dysplastic nodules and early HCCs were negative [15]. In comparison, decreased Smad7 mRNA expression described in three out of ten HCC tissues was correlated with cancer-cell-specific increased Smad2 nuclear staining [39], thus facilitating tumourigenic TGF-β/Smad2 signalling.
On the basis of the above knowledge, we conclude that, similar to TGF-β, Smad7 function is highly context- and disease-stage-dependent, and thus may be either tumour suppressive or oncogenic. In our patients’ cohort, especially in the non-HBV-derived, non-cirrhotic and large tumour HCC subclass, high Smad7 levels would account for tumour suppression.
Part of the present study investigated the mechanism of Smad7 expression regulation. Previously, we have described participation of the cold-shock transcription factor YB-1 in Smad7 transcription in hepatic stellate cells [34]. Knockdown and overexpression experiments with Huh7 and FLC-4 cells identified YB-1 as critical factor for Smad7 expression levels in HCC cells as well. Moreover, YB-1 and Smad7 expression were significantly correlated in cancer tissue of HCC patients including frequent nuclear co-localization. Given heterogeneity of HCC and TGF-β signalling cross-talk with many other pathways [Akt, extracellular-signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK)], this correlation of course is not 100%, and we do not expect YB-1 to be the only Smad7 regulation mechanism in liver/HCC. However, in line with our data, nuclear YB-1 antagonizes TGF-β/Smad3 signalling in fibrogenesis, e.g. in regulating collagen gene expression [40]. Furthermore, adenoviral infection and Col1A2 promoter-driven expression of YB-1 as well as a small compound, HSc025, that promotes nuclear translocation of YB-1, were able to improve liver injury in CCl4-treated mice [41,42]. On the basis of our findings, such intervention may also have therapeutic potential for HCC treatment. Accordingly, DNA-binding protein A (DbpA), another member of the cold-shock protein family, was identified as significant adverse prognostic marker of HCC in 82 analysed patients [43]. However, investigation of the clinical and diagnostic value of YB-1 protein fragment in plasma samples revealed that YB-1/p18 was not a good marker for HCC, but proved powerful in detecting malignancies other than HCC [44].
Supplementary Material
CLINICAL PERSPECTIVES.
As information on Smad7 and its impact on TGF-β signalling regulation in HCC is still limited and often controversial, we aimed to get more specific insight.
Our data suggest that high Smad7 expression in cells with epithelial character is associated with loss of proliferation control and is potentially oncogenic. In contrast, high expression of Smad7 in HCC as well as surrounding tissue might be tumour suppressive as indicated by increased overall survival. Furthermore, elevated YB-1 and Smad7 expression are correlated frequent events in HCC.
We hypothesize that undifferentiated interpretations of Smad7 expression data do not allow predictions on HCC development and TGF-β-directed treatment options, as the outcome remains highly contextual. In contrast, defining HCC subclasses based on clinicopathological features of patients might facilitate diagnostic and predictive conclusions in the future.
ACKNOWLEDGEMENTS
Human HCC samples were provided by Professor Otto Kollmar (Department of General and Visceral Surgery, University Hospital Göttingen, Göttingen, Germany) and the Department of General, Visceral, Vascular and Pediatric Surgery (University of Saarland, Homburg/Saar, Germany), Professor Heike Allgayer (Department of Experimental Surgery and Molecular Oncology of Solid Tumors, Medical Faculty Mannheim, University of Heidelberg and German Cancer Research Center, Heidelberg, Germany), Professor Chun Fang Gao (Department of Laboratory Medicine, Eastern Hepatobiliary Hospital, Second Military Medical University, Shanghai, China) and Professor Thomas Weiss and the tissue bank at the Center for Liver Cell Research (Department of Pediatrics and Juvenile Medicine, University of Regensburg Hospital, Regensburg, Germany). The sample collection provided by Professor Thomas Weiβ was supported by the charitable state controlled foundation HTCR (Human Tissue and Cell Research) supplying human tissue for research purposes. We also thank Britta Bauche for providing preliminary results to start this project.
FUNDING
This study was funded by ESF Baden Württemberg (www.esf-bw.de) [Margarete von Wrangell Stipend], Deutsche Forschungsgemeinschaft (DFG) [grant number SFB-TRR77], the BMBF [Virtual Liver network, VLN] and the EU [IT-Liver consortium, Marie Curie Training Network].
Abbreviations:
- AFP
α-fetoprotein
- AST
aspartate aminotransferase
- BMP
bone morphogenetic protein
- CI
confidence interval
- DFS
disease-free survival
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- H/E
haematoxylin/eosin
- hGH
human growth hormone
- HR
hazard ratio
- HT1
hereditary tyrosinaemia type 1
- N/A
not available
- NTBC
2-(2-nitro-4-fluoromethylbenzoyl)-1,3-cyclohexanedione)
- OS
overall survival
- PCR
polymerase chain reaction
- PHH
primary human hepatocyte
- qPCR
quantitative real-time polymerase chain reaction
- R-Smad
receptor Smad
- RT-PCR
reverse transcription polymerase chain reaction
- TGF-β
transforming growth factor β
- UICC
Union for International Cancer Control
- YB-1
Y-box protein 1
REFERENCES
- 1.Ferlay J, Soerjomataram II, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman DD and Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–E386 [DOI] [PubMed] [Google Scholar]
- 2.Dooley S and ten Dijke P (2012) TGF-beta in progression of liver disease. Cell Tissue Res 347, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karkampouna S, Ten Dijke P, Dooley S and Julio MK (2012) TGFbeta signaling in liver regeneration. Curr. Pharm. Des 18, 4103–4113 [DOI] [PubMed] [Google Scholar]
- 4.Chen CL, Tsukamoto H, Liu JC, Kashiwabara C, Feldman D, Sher L, Dooley S, French SW, Mishra L, Petrovic L et al. (2013) Reciprocal regulation by TLR4 and TGF-beta in tumor-initiating stem-like cells. J. Clin. Invest 123, 2832–2849 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Miyazono K, ten Dijke P and Heldin CH (2000) TGF-beta signaling by Smad proteins. Adv. Immunol 75, 115–157 [DOI] [PubMed] [Google Scholar]
- 6.Reichl P, Haider C, Grubinger M and Mikulits W (2012) TGF-beta in epithelial to mesenchymal transition and metastasis of liver carcinoma. Curr. Pharm. Des 18, 4135–4147 [DOI] [PubMed] [Google Scholar]
- 7.Baek HJ, Pishvaian MJ, Tang Y, Kim TH, Yang S, Zouhairi ME, Mendelson J, Shetty K, Kallakury B, Berry DL et al. (2011) Transforming growth factor-beta adaptor, beta2-spectrin, modulates cyclin dependent kinase 4 to reduce development of hepatocellular cancer. Hepatology 53, 1676–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagata H, Hatano E, Tada M, Murata M, Kitamura K, Asechi H, Narita M, Yanagida A, Tamaki N, Yagi S et al. (2009) Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor-suppression in rat hepatocellular carcinoma. Hepatology 49, 1944–1953 [DOI] [PubMed] [Google Scholar]
- 9.Dooley S, Hamzavi J, Ciuclan L, Godoy P, Ilkavets I, Ehnert S, Ueberham E, Gebhardt R, Kanzler S, Geier A et al. (2008) Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology 135, 642–659 [DOI] [PubMed] [Google Scholar]
- 10.Dooley S, Hamzavi J, Breitkopf K, Wiercinska E, Said HM, Lorenzen J, Ten Dijke P and Gressner AM (2003) Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 125, 178–191 [DOI] [PubMed] [Google Scholar]
- 11.Chen YK, Huang AH, Cheng PH, Yang SH and Lin LM (2013) Overexpression of Smad proteins, especially Smad7, in oral epithelial dysplasias. Clin. Oral Investig 17, 921–932 [DOI] [PubMed] [Google Scholar]
- 12.Osawa H, Nakajima M, Kato H, Fukuchi M and Kuwano H (2004) Prognostic value of the expression of Smad6 and Smad7, as inhibitory Smads of the TGF-beta superfamily, in esophageal squamous cell carcinoma. Anticancer Res 24, 3703–3709 [PubMed] [Google Scholar]
- 13.Theohari I, Giannopoulou I, Magkou C, Nomikos A, Melissaris S and Nakopoulou L (2012) Differential effect of the expression of TGF-beta pathway inhibitors, Smad-7 and Ski, on invasive breast carcinomas: relation to biologic behavior. APMIS 120, 92–100 [DOI] [PubMed] [Google Scholar]
- 14.Huang Q, Liu L, Liu CH, Shao F, Xie F, Zhang CH and Hu SY (2012) Expression of Smad7 in cholangiocarcinoma: prognostic significance and implications for tumor metastasis. Asian Pac. J. Cancer Prev 13, 5161–5165 [DOI] [PubMed] [Google Scholar]
- 15.Park YN, Chae KJ, Oh BK, Choi J, Choi KS and Park C (2004) Expression of Smad7 in hepatocellular carcinoma and dysplastic nodules: resistance mechanism to transforming growth factor-beta. Hepatogastroenterology 51, 396–400 [PubMed] [Google Scholar]
- 16.Wang J, Zhao J, Chu ES, Mok MT, Go MY, Man K, Heuchel R, Lan HY, Chang Z, Sung JJ and Yu J (2013) Inhibitory role of Smad7 in hepatocarcinogenesis in mice and in vitro. J. Pathol 230, 441–452 [DOI] [PubMed] [Google Scholar]
- 17.Xia H, Ooi LL and Hui KM (2013) MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology 58, 629–641 [DOI] [PubMed] [Google Scholar]
- 18.Grompe M, al-Dhalimy M, Finegold M, Ou CN, Burlingame T, Kennaway NG and Soriano P (1993) Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev 7, 2298–2307 [DOI] [PubMed] [Google Scholar]
- 19.Kuhn R, Schwenk F, Aguet M and Rajewsky K (1995) Inducible gene targeting in mice. Science 269, 1427–1429 [DOI] [PubMed] [Google Scholar]
- 20.Fleenor DE and Kaufman RE (1993) Characterization of the DNase I hypersensitive site 3′ of the human beta globin gene domain. Blood 81, 2781–2790 [PubMed] [Google Scholar]
- 21.Li Q and Stamatoyannopoulos G (1994) Hypersensitive site 5 of the human beta locus control region functions as a chromatin insulator. Blood 84, 1399–1401 [PubMed] [Google Scholar]
- 22.Blessing M, Nanney LB, King LE, Jones CM and Hogan BL (1993) Transgenic mice as a model to study the role of TGF-beta-related molecules in hair follicles. Genes Dev 7, 204–215 [DOI] [PubMed] [Google Scholar]
- 23.Gorski K, Carneiro M and Schibler U (1986) Tissue-specific in vitro transcription from the mouse albumin promoter. Cell 47, 767–776 [DOI] [PubMed] [Google Scholar]
- 24.Werner S, Weinberg W, Liao X, Peters KG, Blessing M, Yuspa SH, Weiner RL and Williams LT (1993) Targeted expression of a dominant-negative FGF receptor mutant in the epidermis of transgenic mice reveals a role of FGF in keratinocyte organization and differentiation. EMBO J 12, 2635–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sauer B and Henderson N (1988) Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. U.S.A 85, 5166–5170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pluck A and Klasen C (2009) Generation of chimeras by microinjection. Methods Mol. Biol 561, 199–217 [DOI] [PubMed] [Google Scholar]
- 27.Damm G, Pfeiffer E, Burkhardt B, Vermehren J, Nüssler AK and Weiss TS (2013) Human parenchymal and non-parenchymal liver cell isolation, culture and characterization. Hepatol. Int 7, 951–958 [DOI] [PubMed] [Google Scholar]
- 28.Stopa M, Benes V, Ansorge W, Gressner AM and Dooley S (2000) Genomic locus and promoter region of rat Smad7, an important antagonist of TGFbeta signaling. Mamm. Genome 11, 169–176 [DOI] [PubMed] [Google Scholar]
- 29.Bettstetter M, Dechant S, Ruemmele P, Vogel C, Kurz K, Morak M, Keller G, Holinski-Feder E, Hofstaedter F and Dietmaier W (2008) MethyQESD, a robust and fast method for quantitative methylation analyses in HNPCC diagnostics using formalin-fixed and paraffin-embedded tissue samples. Lab. Invest 88, 1367–1375 [DOI] [PubMed] [Google Scholar]
- 30.Marhenke S, Buitrago-Molina LE, Endig J, Orlik J, Schweitzer N, Klett S, Longerich T, Geffers R, Sanchez Munoz A, Dorrell C et al. (2014) p21 promotes sustained liver regeneration and hepatocarcinogenesis in chronic cholestatic liver injury. Gut 63, 1501–1512 [DOI] [PubMed] [Google Scholar]
- 31.Mertens PR, Martin IV, Frye BC, Rauen T, Strauch S, Pabst M and Geier A (2012) Rat Mrp2 gene expression is regulated by an interleukin-1beta-stimulated biphasic response with enhanced transcription and subcellular shuttling of YB-1. Eur. J. Cell Biol 91, 533–541 [DOI] [PubMed] [Google Scholar]
- 32.Camp RL, Dolled-Filhart M and Rimm DL (2004) X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin. Cancer Res 10, 7252–7259 [DOI] [PubMed] [Google Scholar]
- 33.Dzieran J, Fabian J, Feng T, Coulouarn C, Ilkavets I, Kyselova A, Breuhahn K, Dooley S and Meindl-Beinker NM (2013) Comparative analysis of TGF-beta/Smad signaling dependent cytostasis in human hepatocellular carcinoma cell lines. PLoS ONE 8, e72252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dooley S, Said HM, Gressner AM, Floege J, En-Nia A and Mertens PR (2006) Y-box protein-1 is the crucial mediator of antifibrotic interferon-gamma effects. J. Biol. Chem 281, 1784–1795 [DOI] [PubMed] [Google Scholar]
- 35.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX and Mishra L (2003) Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science 299, 574–577 [DOI] [PubMed] [Google Scholar]
- 36.Matsuzaki K, Date M, Furukawa F, Tahashi Y, Matsushita M, Sugano Y, Yamashiki N, Nakagawa T, Seki T, Nishizawa M et al. (2000) Regulatory mechanisms for transforming growth factor beta as an autocrine inhibitor in human hepatocellular carcinoma: implications for roles of smads in its growth. Hepatology 32, 218–227 [DOI] [PubMed] [Google Scholar]
- 37.Yamamura Y, Hua X, Bergelson S and Lodish HF (2000) Critical role of Smads and AP-1 complex in transforming growth factor-beta-dependent apoptosis. J. Biol. Chem 275, 36295–36302 [DOI] [PubMed] [Google Scholar]
- 38.Zhang H, Ozaki I, Mizuta T, Hamajima H, Yasutake T, Eguchi Y, Ideguchi H, Yamamoto K and Matsuhashi S (2006) Involvement of programmed cell death 4 in transforming growth factor-beta1-induced apoptosis in human hepatocellular carcinoma. Oncogene 25, 6101–6112 [DOI] [PubMed] [Google Scholar]
- 39.Matsuzaki K, Date M, Furukawa F, Tahashi Y, Matsushita M, Sakitani K, Yamashiki N, Seki T, Saito H, Nishizawa M et al. (2000) Autocrine stimulatory mechanism by transforming growth factor beta in human hepatocellular carcinoma. Cancer Res 60, 1394–1402 [PubMed] [Google Scholar]
- 40.Higashi K, Inagaki Y, Suzuki N, Mitsui S, Mauviel A, Kaneko H and Nakatsuka I (2003) Y-box-binding protein YB-1 mediates transcriptional repression of human alpha 2(I) collagen gene expression by interferon-gamma. J. Biol. Chem 278, 5156–5162 [DOI] [PubMed] [Google Scholar]
- 41.Higashi K, Tomigahara Y, Shiraki H, Miyata K, Mikami T, Kimura T, Moro T, Inagaki Y and Kaneko H (2011) A novel small compound that promotes nuclear translocation of YB-1 ameliorates experimental hepatic fibrosis in mice. J. Biol. Chem 286, 4485–4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inagaki Y, Kushida M, Higashi K, Itoh J, Higashiyama R, Hong YY, Kawada N, Namikawa K, Kiyama H, Bou-Gharios G et al. (2005) Cell type-specific intervention of transforming growth factor beta/Smad signaling suppresses collagen gene expression and hepatic fibrosis in mice. Gastroenterology 129, 259–268 [DOI] [PubMed] [Google Scholar]
- 43.Yasen M, Kajino K, Kano S, Tobita H, Yamamoto J, Uchiumi T, Kon S, Maeda M, Obulhasim G, Arii S and Hino O (2005) The up-regulation of Y-box binding proteins (DNA binding protein A and Y-box binding protein-1) as prognostic markers of hepatocellular carcinoma. Clin. Cancer Res 11, 7354–7361 [DOI] [PubMed] [Google Scholar]
- 44.Tacke F, Kanig N, En-Nia A, Kaehne T, Eberhardt CS, Shpacovitch V, Trautwein C and Mertens PR (2011) Y-box protein-1/p18 fragment identifies malignancies in patients with chronic liver disease. BMC Cancer 11, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.