

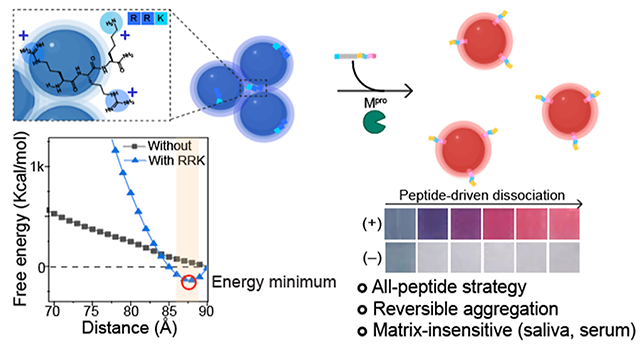
Abstract
Colorimetric biosensors based on gold nanoparticle (AuNP) aggregation are often challenged by matrix interference in biofluids, poor specificity, and limited utility with clinical samples. Here, we propose a peptide-driven nanoscale disassembly approach, where AuNP aggregates induced by electrostatic attractions are dissociated in response to proteolytic cleavage. Initially, citrate-coated AuNPs were assembled via a short cationic peptide (RRK) and characterized by experiments and simulations. The dissociation peptides were then used to reversibly dissociate the AuNP aggregates as a function of target protease detection, i.e., main protease (Mpro), a biomarker for severe acute respiratory syndrome coronavirus 2. The dissociation propensity depends on peptide length, hydrophilicity, charge, and ligand architecture. Finally, our dissociation strategy provides a rapid and distinct optical signal through Mpro cleavage with a detection limit of 12.3 nM in saliva. Our dissociation peptide effectively dissociates plasmonic assemblies in diverse matrices including 100% human saliva, urine, plasma, and seawater, as well as other types of plasmonic nanoparticles such as silver. Our peptide-enabled dissociation platform provides a simple, matrix-insensitive, and versatile method for protease sensing.
Keywords: colorimetric biosensor, DLVO theory, reversible aggregation, dissociation peptide, matrix-insensitive, SARS-CoV-19
Graphical Abstract
1. INTRODUCTION
Colorimetric biosensors using gold nanoparticles (AuNPs) are a popular tool for in vitro detection of various disease biomarkers (e.g., proteins, enzymes, nucleic acids, and metal ions).1–4 Colorimetry is easy, simple, affordable, and does not require a skilled operator.5 A simple spectrophotometer and/or naked eye can be used for detection, and a relatively small amount of AuNPs can provide sufficient visible color changes due to their high molar absorption coefficients.6
Most research on this topic has focused on aggregation-based color changes facilitated by electric dipole–dipole interactions between proximal AuNPs leading to plasmonic coupling and a bathochromic shift (red to blue color shift). This aggregation is governed by DLVO theory.7 This theory suggests that AuNPs remain colloidally stable because the attractive Van der Waals (VdW) forces are balanced by the repulsive steric forces and electrostatic forces. When used for sensors, hydrogen bonding,8 DNA pairings,1,4 antibody–antigen,9 electrostatic,6 and hydrophobic2 interactions can change the electrostatic repulsion, steric repulsion, or VdW attraction to induce such aggregation and thus a color change. This aggregation is nearly always irreversible because the VdW forces scale to the sixth power of separation distance. Thus, the VdW forces become insurmountably large, and the particles fall into an energy well—they are permanently aggregated.
Sensors based on this mechanism often have poor specificity and limited utility with clinical samples for several reasons. First, citrate-stabilized AuNPs are very sensitive to pH, solvent polarity, and ionic strength—all of these can cause undesired aggregation and thus require surface modifications of the particles (e.g., PEGylation, BSPP).10–12 Second, biofluids such as plasma, serum, saliva, sweat, or urine can lead to a protein corona, thus changing the surface charge and preventing electrostatic-based plasmonic coupling.13 Third, AuNP colloidal stability is notoriously dependent on ionic strength—physiological salt concentrations can lead to charge screening of the electrostatic repulsions thus allowing attractive VdW to predominate. Although a few studies have reported aggregation-based colorimetric assays in biofluids, they often require sample dilution or removal of the analyte from the matrix.6,14,15 Finally, AuNP aggregation can amplify eventually leading to aggregates larger than 1 μm that lose colloidal stability and become colorless.3
To address these issues, we recently proposed colorimetric assays based on dissociation.14 Here, particle aggregation was first triggered by a short cationic peptide containing arginine and lysine that induced plasmonic coupling of anionic AuNPs coated with citrate. Most importantly, this aggregation was reversible upon addition of steric stabilizers such as thiol-PEG molecules (HS-PEGs).16 We then made a PEG-peptide conjugate containing a cleavage sequence specific to trypsin.14 The first value of this approach was a remarkable insensitivity to the matrix—dispersion could even be done in seawater and bile. Second, the aggregated AuNPs were stable for months and could even be dried to completeness but could still be redissolved and used to detect proteases.
The work reported here satisfied three goals inspired by this prior work. First, we used computational methods to better understand the mechanism of this reversible aggregation. We hypothesized that the short cationic peptide has a steric bulk that maintains some separation distance between the AuNPs thus preventing runaway attractive VdW attraction. Second, we used an all-peptide strategy that is simpler and requires no PEG–peptide couplings. We demonstrated that charge, hydrophilicity, peptide length, and ligand architecture can impact on the dissociation efficiency. Finally, we construct a practical sensor that is made of the optimized dissociation domain with a biomolecular recognition element of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease, i.e., Mpro.17,18 After protease cleavage, released peptides successfully provided a rapid color readout of Mpro with a limit of detection (LoD) of 12.3 nM in saliva. Furthermore, our dissociation peptide can dissociate AuNP aggregates in various matrixes including 100% human urine, plasma, and seawater and can be applied to other types of plasmonic nanoparticles (e.g., silver).
2. RESULTS AND DISCUSSION
2.1. Short Cationic Peptides for Reversible Aggregation.
To induce reversible aggregation of the AuNPs, we used positively charged Arg- and Lys-based peptide residues (i.e., RRK). The RRK peptide could induce plasmonic coupling by electrostatic attractions between negatively charged citrate molecules and guanidine and amine groups in RRK (Figure 1a).7,19 The size of AuNPs increased upon addition of RRK peptides, as confirmed by transmission electron microscopy (TEM) and dynamic light scattering (DLS) (Figures 1b and S1). UV–vis spectroscopy showed that the plasmonic resonance peak (520 nm) of AuNPs redshifted to 648 nm; visually, the sample changed color from red to blue (Figure 1c). Raman spectroscopy show C–C stretching at 984 cm−1 and C–N stretching at 1443 cm−1 from Arg residues in RRK (Figure 1d).20 Lastly, R, RRK, and RRKRRK peptides (from 0.5 to 30 μM) were used to induce AuNP aggregates to study the impact of charge number on particle aggregation. The results showed that RRKRRK and RRK peptides effectively induced particle aggregation, whereas AuNPs did not aggregate with just one R even at 8-fold higher concentration than RRK (Figure 1e). A higher positive charge number can decrease the critical coagulation concentration (CCC).21 The RRK peptide was chosen in this study because particle aggregation induced by a strong cationic peptide (i.e., RRKRRK) can result in irreversible aggregation.
Figure 1.
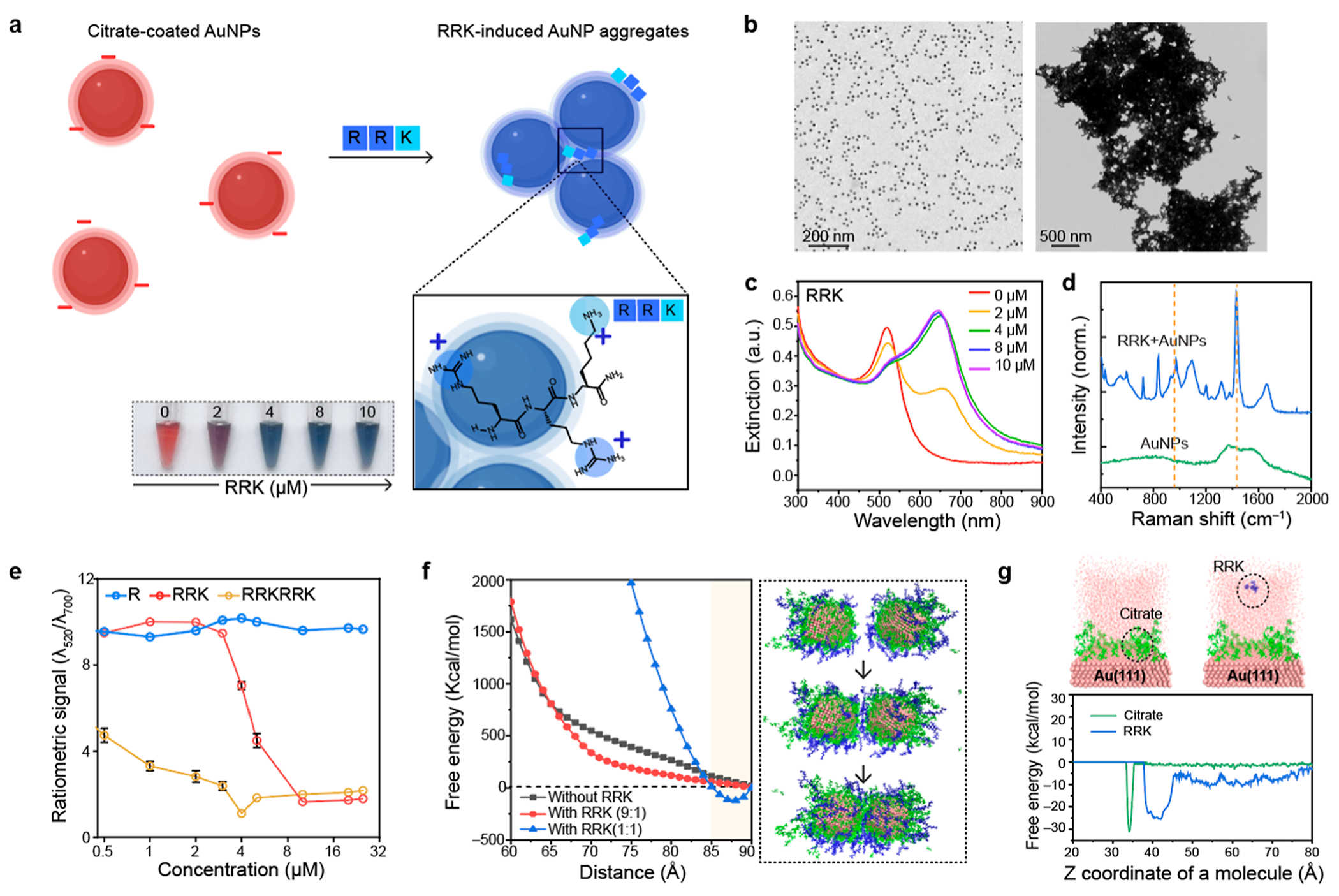
Short cationic peptides for reversible aggregation. (a) Schematic illustration of RRK-based particle aggregation. AuNPs were aggregated by electrostatic attractions between negatively charged citrate on AuNPs and positively charged RRK peptides. The inset photograph shows color change from red to blue as a function of the RRK peptide (2–10 μM). (b) TEM images of citrate-coated AuNPs (left) and RRK-induced AuNP aggregates (right). (c) UV–vis spectrum of RRK-induced AuNP aggregates. The plasmonic resonance peak of AuNPs was redshifted due to the plasmonic coupling. (d) Raman shifts before and after adding RRK peptides into citrate-coated AuNPs. The Raman peak at 1443 cm−1 was attributed to the C–N stretching in the Arg residue.20 (e) Ratiometric signal (λ520/λ700) of AuNPs after adding R, RRK, and RRKRRK peptides with different concentrations 0.5–32 μM, respectively. The error bars represent the standard deviation of three independent samples. (f) SMD simulations for free energy investigation as a function of AuNP distance at 298 K and 1 atm. Energy minimum point was observed after adding RRK peptides. Black, red, and blue lines indicate citrate to RRK molar ratios of 1:0, 9:1, and 1:1, respectively. The inset images indicate the simulation stages along a trajectory of the citrate-coated AuNPs with the RRK (9:1) system. (g) MTD free energy investigation for a system with 1 RRK on the Au(111) surface (right) and a system with 1 citrate on the Au(111) surface (left). The Z coordinate value was calculated based on the center mass of RRK and citrate molecule as shown in the inset images. The upper surface of the Au(111) slab was located at 31 Å. The MTD results observed no surface ligand exchange on Au(111) during the electrostatic interactions between RRK and citrate molecules.
To further study RRK-based plasmonic coupling, we adopted both quantum mechanics (QM) computation and molecular dynamics (MD) simulation. MD simulation revealed that the surface environment of AuNP changed as a function of RRK molecules interacting with citrate-coated AuNPs (Figure S2). Furthermore, we computed free energies before and after adding RRK peptides into the citrate-coated AuNPs via steered MD (SMD) simulation. Initially, the citrate-coated AuNPs were favorably dispersed due to strong electrostatic repulsions.
After adding enough RRK peptides, a free energy minimum point at 87 Å was observed, which suggests a separation distance where nanoparticles were reversibly plasmonically coupled (Figures 1f and S3). Lastly, metadynamic (MTD) simulations evaluated the binding mechanism between the Au (111) surface and citrate or RRK molecule, respectively (Figure 1g). The Z coordinates of citrate and RRK molecule are 34 and 39 Å, thus indicating that RRK peptides are favored to bind on the top of a citrate-coated Au (111) surface rather than replacing citrate molecules.
2.2. Peptide-Enabled Dissociation of AuNP Aggregates.
Peptide-based ligands for AuNPs are of particular interest because of their structural- and chemical-versatilities that can provide high colloidal stability, functionalization, and prevention of protein adsorptions.22–25 Peptides which could provide electrostatic repulsion, steric distance, and hydrophilicity can attenuate electrostatic attractions induced by RRK, leading to reversible aggregation. We designed our dissociation peptides (i.e., A1 peptide) to have three major components: charge, spacer, and anchoring groups (Figure 2a). First, we used Glu (E) and Lys (K) amino acids to provide steric distance and strong hydration layer.23 Negatively charged EE residues provide electrostatic repulsion, and positively charged KK residues in the vicinity of the thiol could increase the grafting kinetics of thiols onto the citrated-coated AuNPs.26 Second, Pro-based linker residues provide a space between charge and anchoring group, further increasing the stability of the AuNPs.27 Lastly, Cys (C) amino acid contains a thiol side chain that binds to the surface of AuNPs via strong Au–S bonds.28
Figure 2.
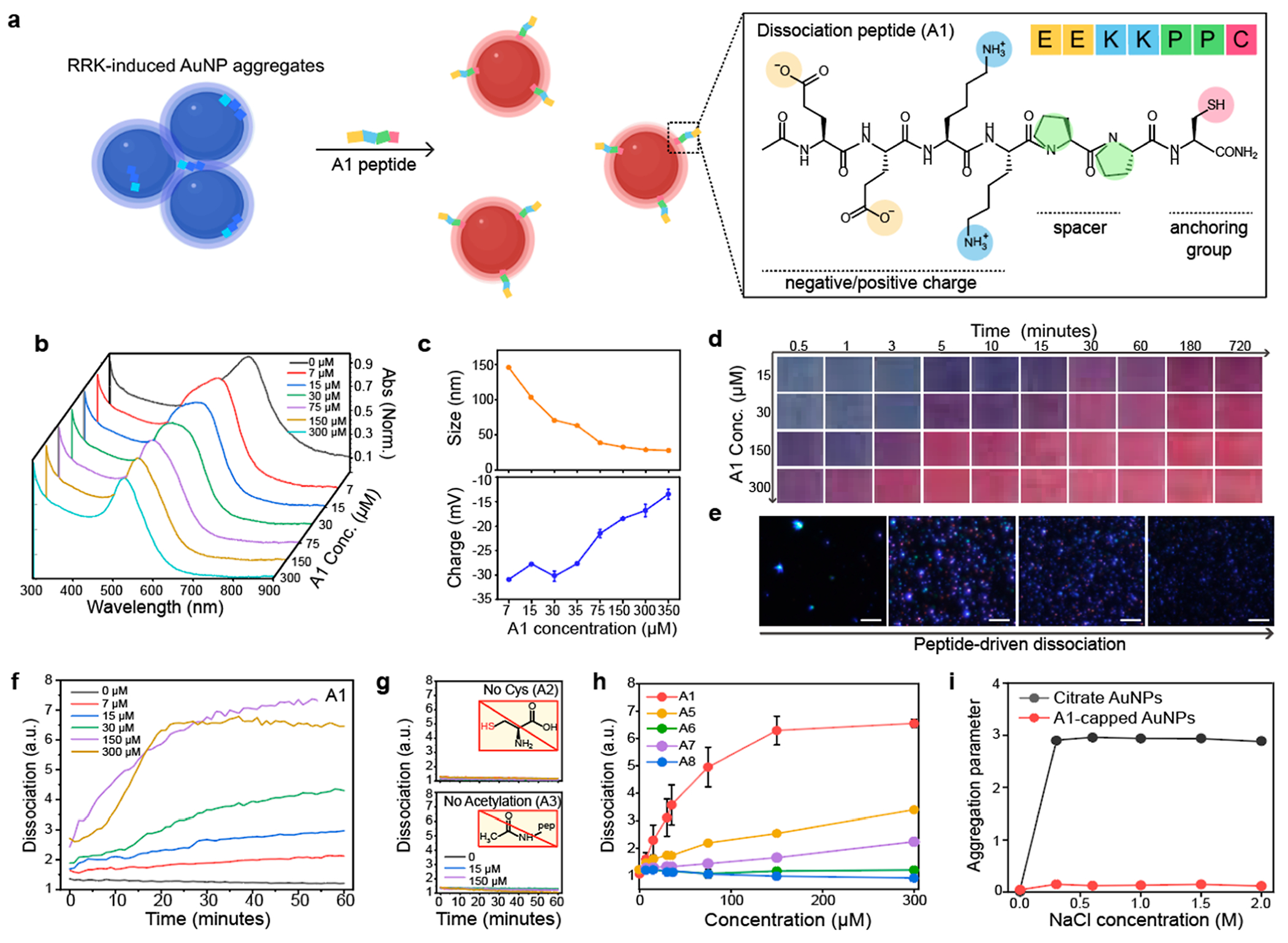
Peptide-enabled dissociation of AuNP aggregates. (a) Schematic illustration of peptide-based particle dissociations. AuNP aggregates induced by RRK peptides were reversibly dissociated by the A1 peptides. The structural component of the A1 peptide contains charge, spacer, and anchoring group. (b) UV–vis spectrum shows that the plasmonic resonance peak of AuNP aggregates blueshifted upon addition of the A1 peptide (7–300 μM). (c) Hydrodynamic diameter and the surface charge after adding the A1 peptide. (d) Time-dependent photographs show 150 μM of the A1 peptide required to dissociate AuNP aggregates. x and y axis indicate time and the A1 concentration, respectively. (e) Darkfield images of AuNP aggregates (left) and the dissociated AuNPs (right). The scale bar indicates 10 μm. Blue dots represent actual AuNPs dissociated by the A1 peptide. (f) Time-dependent particle dissociation driven by the A1 peptide. The ratiometric signal (λ520/λ700) was referred to as dissociation (y axis). (g) Particle dissociation was quenched without Cys (A2) and acetylation (A3). (h) Dissociation capacity of the A1, A5, A6, A7, and A8 peptides. (i) Dissociated AuNPs by the A1 (red) showed higher colloidal stability than citrate-coated AuNPs (black). Panel (c,h,i) repeated three independent times and showed similar results.
After inducing AuNP aggregates by the RRK peptides, the A1 peptides with different concentrations from 7 to 300 μM were used to dissociate AuNP aggregates. The plasmonic resonance peak of AuNP aggregates blueshifted to 520 nm after particle dissociation (Figure 2b). The hydrodynamic diameter of AuNP aggregates (>1 μm) was reduced to 27 ± 0.07 nm, and the surface charge of AuNPs was—13.4 ± 1.03 mV because of surface modification with neutral charged A1 peptides (150 μM) and residual citrates (Figure 2c). Particle dissociation induced color change from blue to red and a decrease in size as confirmed by time-dependent photographs, multi-laser wavelength nanoparticle tracking analysis (MNTA), and TEM (Figures 2d,e and S4). The ratiometric signal (λ520/λ700) indicated that A1 peptide (>150 μM) rapidly dissociated AuNP aggregates in 20 min (Figure 2f).
To further study the role of each amino acid in the A1 peptide, we synthesized eight peptide sequences (from A2 to A8) for control experiments (Figure S5). Table 1 includes peptide sequence, net charge, critical dissociation concentrations (CDC), and design rationale. First, we replaced the Cys residue with Gly (i.e., A2) to confirm the role of the anchoring group. Not surprisingly, the A2 peptide failed to dissociate AuNP aggregates due to the absence of the Au–S binding (Figures 2g and S6). Second, we hypothesized that negatively charged EE residues were required for particle dissociation because they provide electrostatic repulsions for citrate-coated AuNPs. To prove this, the A1 peptide without acetylation (i.e., A3) and one Lys residue (i.e., A4) were synthesized. The A3 and A4 peptides showed no particle dissociations, confirming that positively charged peptides could not dissociate AuNP aggregates likely due to lack of electrostatic repulsions (Figures 2g and S7).
Table 1.
A1–A8 Peptides to Study Role of Structural Components in the Dissociation Peptide
| peptide name | peptide sequence Ace: acetylation, Am: amide |
net charge | M.W. (g mol–1) | CDC (μM) | description |
|---|---|---|---|---|---|
| A1 | Ace–EEKKPPC–Am | 0 | 870.43 | 150 | dissociation peptide |
| A2 | Ace–EEKKPPG–Am | 0 | 824.44 | X | remove Cys |
| A3 | NH2-EEKKPPC–Am | +1 | 828.42 | X | no acetylation |
| A4 | Ace–KEEKKPPC–Am | +1 | 998.52 | X | single Lys |
| A5 | Ace–EEPPKKC–Am | 0 | 870.43 | >300 | change the position of P spacer |
| A6 | Ace–KKEEPPC–Am | 0 | 870.43 | X | switch position of E and K |
| A7 | Ace–EEPPC–Am | −2 | 614.24 | >300 | Glu only |
| A8 | Ace–KKPPC–Am | +2 | 612.34 | X | Lys only |
We further studied the role of Glu, Lys, and Pro amino acids in A1 peptides (Figure 2h). For example, a Pro-based spacer can provide a rigid and self-assembling monolayer (SAM) that increases the colloidal stability of AuNPs.29 When the spacer was moved to the site between Glu and Lys residues (i.e., A5), the dissociation capacity was 50% lower than the A1 peptide. In addition, when the position of Glu was switched with Lys (i.e., A6), particle dissociation was quenched, meaning that the position of positively charged residue is important to maintain the negatively charged electrical double layers. A1 peptide without Lys (i.e., A7) decreased to 30% of the dissociation capacity likely due to the decrease in grafting kinetics of thiol–Au bonds and hydration layers.23,26 Lastly, the dissociated AuNPs were incubated in different concentrations (0.5 to 2 M) of NaCl to examine the colloidal stability of the dissociated AuNPs (Figure 2i). The aggregation parameter defines the variations of absorbance ratio between 520 and 600 nm at the initial and final conditions (see Supporting Information).6,22 The results showed that the A1 peptide-capped AuNPs showed higher colloidal stability than citrate-coated AuNPs due to its longer peptide length, rigid SAM, and improved hydrophilicity.22
2.3. Impact of Hydrophilicity and Steric Bulk on Particle Dissociation.
Pro-, Ala-, and Gly-based spacers have different hydrophobic and hydrophilic natures, rigidity, and flexibility.23,30 For example, Pro residue is more hydrophobic and rigid (i.e., low mobility), while Gly residue is more hydrophilic and flexible. To investigate the impact of the rigidity and hydrophilicity of the peptides on the particle dissociation, we synthesized A1, A10, and A11 peptides that have different spacers: PP, AA, and GG (Figures 3a,b, and S8). We also synthesized the peptide without a spacer (i.e., A9) to confirm the impact of the spacer on particle dissociation. The results showed that the A1, A9, A10, and A11 peptides dissociated AuNP aggregates, changing color from blue to red within 10 min, respectively (Figures 3c,d, and S9). The A11 peptide showed the highest dissociation capacity compared to other spacers (Figure 3e). For example, the CDC of A11, A1, and A10 peptides were 16, 40, and 150 μM, respectively, indicating that higher hydrophilic and flexible spacer could enhance the dissociation process. We further compared colloidal stability of the dissociated AuNPs by A1, A9, A10, and A11 peptides. The dissociated AuNPs were incubated in NaCl (from 0.5 to 2 M) for 1 h and measured variations of absorbance between 520 and 600 nm. The A9-capped AuNPs (i.e., without spacer) showed relatively low colloidal stability compared to Pro-, Ala-, and Gly-capped AuNPs (Figure 3f). Pro-capped AuNPs showed a two-fold lower aggregation parameter than Gly- or Ala-capped AuNPs possibly due to their rigid structure and SAM.23 Lastly, Fourier-transform infrared spectroscopy (FTIR) data confirmed peaks at 1600 and 1400 cm−1 attributed to carboxyl groups in Glu amino acid (Figure 3g).
Figure 3.
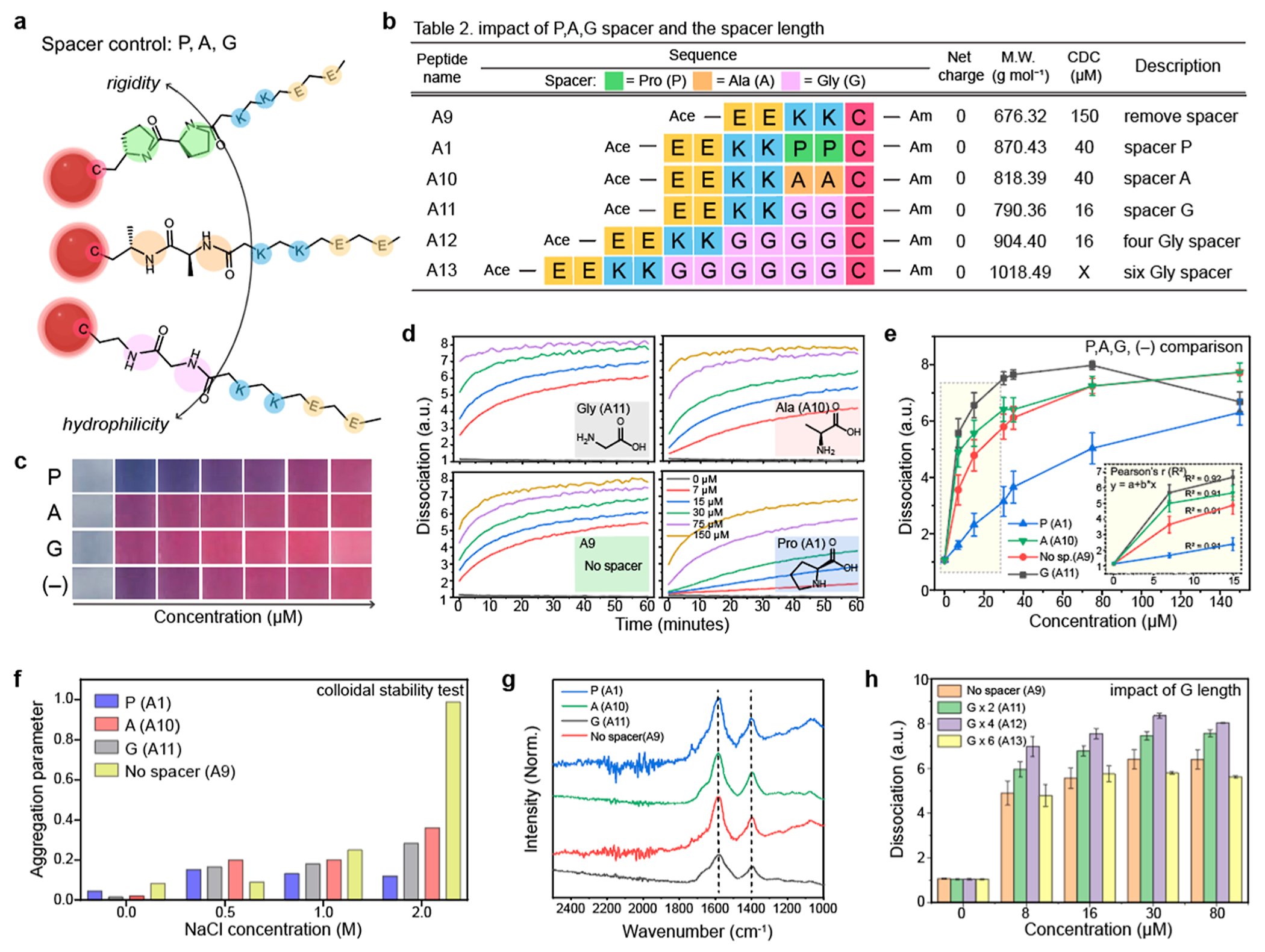
Impact of hydrophilicity and steric bulk on particle dissociation. (a) Different Pro-, Ala-, and Gly-spacers have different nature of rigidity and hydrophilicity which can impact on the dissociation capacity. Table 2 in (b) describes peptide sequences that are designed to investigate the impact of spacers. (c) Photographs of the dissociated AuNPs by the PP, AA, and GG spacers and without spacer (−) as a negative control. (d) Time-dependent particle dissociations driven by the A1, A9, A10, and A11 peptides, respectively. (e) Gly spacer showed a higher dissociation capacity than the Pro- and Ala- spacers. (f) Aggregation parameter of the dissociated AuNPs driven by the A1, A9, A10, and A11 peptides. The results showed that the peptide with spacer can provide higher colloidal stability for AuNPs than the peptide without spacer. (g) FTIR data of the dissociated AuNPs by the A1, A9, A10, and A11 peptides. The peaks at 1400 and 1600 cm−1 were attributed to the carboxyl group in the Glu amino acid. (h) Impact of the spacer length on the particle dissociation. Increasing the length of the spacer (from two to four) improved dissociation capacity, while the spacer with six Glu (i.e., A13) showed lower dissociation capacity than the A12 peptide. The panel (e,f,h) repeated three independent times and showed similar results.
Next, we examined the impact of the peptide length on the particle dissociation. Gly spacer was selected because it showed the highest dissociation capacity compared to the Pro- and Ala-spacers. The A11, A12, and A13 peptides which contain two, four, and six repeated Gly amino acids were synthesized for the test (Figure 3b). The average peptide length of four repeated Gly (GGGG) is known around 18–20 Å.23 The particle dissociation was improved as a function of the increased number of Gly spacer (from two to four) (Figures 3h and S10). The A11 and A12 peptides dissociate AuNP aggregates with the concentration of 16 μM, respectively. However, the A13 peptide showed 25% lower dissociation capacity than the A12 peptides at the same peptide concentration (30 μM). These results revealed that the particle dissociation relies on the peptide length: the spacer length until 20 Å improved particle dissociation while over 20 Å could inversely impact the dissociation capacity likely due to steric hindrance caused by the large size of the spacer.14
2.4. Protease Detection with Dissociation Peptide.
We then applied our dissociation strategy for Mpro detection.17,18 We used the A18 peptide which contains three major structural components: dissociation domain (CGGKKEE at the N terminus), cleavage site (AVLQ↓ SGF), and one Arg at the C terminus for dissociation shielding according to the A4 peptides (Figure 4a). The A18 fragments released by Mpro in PB buffer dissociated AuNP aggregates, changing color from blue to red, while the A18 peptides without Mpro showed no color changes and became transparent due to the colloidal settlement (Figure 4b). Matrix assisted laser absorption ionization time of flight mass spectrometry (MALDI-TOF MS) data confirmed the mass peaks of the A18 fragments (CGGKKEEAVLQ: 1203.84) which is a result of the Mpro proteolysis (Figure 4c). After Mpro incubation (200 nM) for 1 h, the A18 fragments with different concentrations from 8 to 80 μM were used for the dissociation. The result showed that at least 40 μM of the A18 peptide (CDC) was required to dissociate AuNP aggregates, and the dissociation quickly occurred within 10 min (Figure 4d). The plasmonic resonance peak of AuNP aggregates blueshifted to 520 nm which was an absorption peak of pristine AuNPs (Figure 4e). The size of AuNP aggregates was reduced to 26 ± 0.30 nm, and the surface charge was −27 ± 0.95 mV as a function of the released A18 fragments by Mpro cleavage (Figure 4f).
Figure 4.
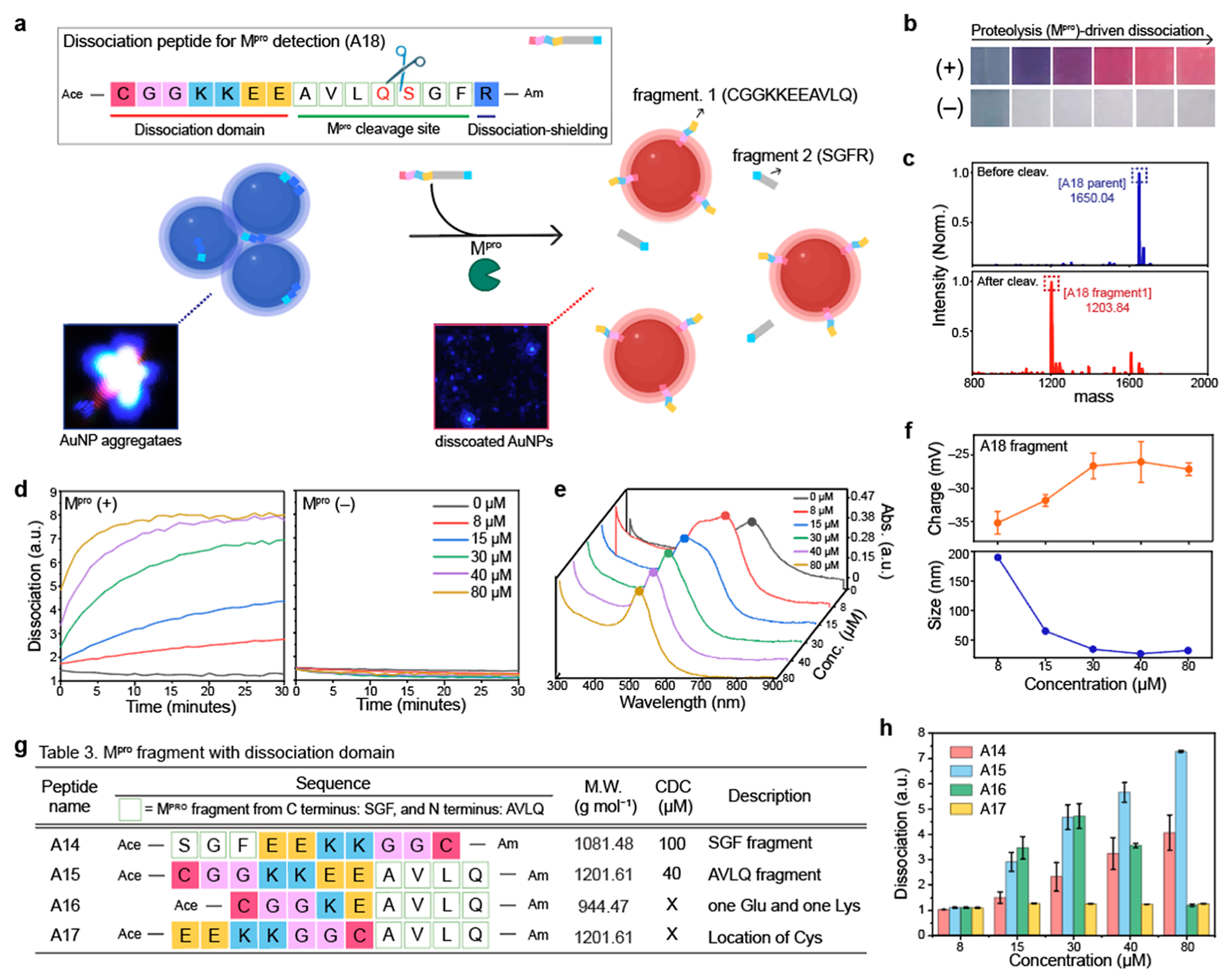
Mpro detection using dissociation strategy. (a) Schematic illustrates that Mpro cleavage releases dissociation domains, changing the color from blue to red. Our dissociation peptide (i.e., A18) consists of three parts: dissociation domain (CGGKKEE), cleavage site (AVLQ↓.SGF), and dissociation shielding site (R). The inset images are before and after particle dissociation obtained by darkfield microscopy. Blue dots indicate actual AuNP aggregates (left) and the dissociated AuNPs (right). (b) Color changes with (+) and without (−) Mpro in PB buffer. The released A18 fragment (CGGKKEEAVLQ) dissociated AuNP aggregates, changing the color from blue to red. (c) MALDI-TOF MS data before and after Mpro cleavage, confirming the mass peaks of the A18 parent and its fragment. (d) Time-dependent particle dissociation by the A18 fragments. The results showed that at least 40 μM of the A18 fragment was required for particle dissociations. (e) UV–vis spectrum before and after particle dissociation by the A18 fragments with different concentrations (8–80 μM). (f) Changes in the size and surface charge after the dissociation induced by Mpro cleavage. Table 3 in (g) describes peptide sequences that are designed to confirm the best location and order of the dissociation domain for Mpro detection. (h) Particle dissociations driven by the A14, A15, A16, and A17 peptides. The results show that the dissociation domain located at C-terminus showed the highest dissociation capacity. In addition, the thiol group at the tail showed higher dissociation affinity than the thiol group in the middle. The panel (f,h) repeated three independent times and showed similar results.
Next, we synthesized four different peptide sequences to study the impact of the fragments (e.g., SGF or AVLQ), charge density, and the location of the Cys residue on the particle dissociation (Figures 4g and S11). Mpro cleavage requires an AVLQ↓SGF site (Mpro cleaves after Arg), and the two different fragments (e.g., AVLQ or SGF) could impact the dissociation capacity of the peptide. To decide the location of the dissociation domain, we synthesized a dissociation domain with AVLQ (i.e., A15) and SGF (i.e., A14), respectively. The A15 peptide showed higher dissociation capacity than the A14 peptide, indicating that adding the dissociation domain at the N-terminus is a better approach than adding it at the C-terminus (Figures 4h and S12). In addition, the A17 peptide which has lower charge components decreased the dissociation capacity, and the location of the Cys amino acid can impact particle dissociation. For example, the Cys at the N terminus showed a 5-fold higher dissociation capacity than the peptide with Cys in the middle as Cys in the middle gives less passivation of AuNPs, thus preventing particle dissociation.23 In conclusion, we placed our dissociation domain (i.e., CGGKKEE) at the N terminus to efficiently activate particle dissociation by Mpro proteolysis.
2.5. Matrix-Insensitive Mpro Detection.
Our dissociation strategy offers matrix-insensitive target detection because the dissociation mechanism is less interrupted by operating mediums (e.g., proteins, ions) compared to aggregation-based biosensors.14 Since SARS-CoV-2-infected patients might release viral proteases to respiratory fluids,31 our A18 peptides were examined to dissociate AuNP aggregates in saliva or EBC (Figure 5a). We performed a stepwise assay by first incubating the A18 peptide with different Mpro concentrations (0.3 to 47 nM) for 30 min at 37 °C. Then, AuNP aggregates were added as a readout for 1 h. Figure 5b showed that the A18 fragments cleaved by Mpro in saliva or EBC dissociated AuNP aggregates within 10 min. Figure 5c plots the ratiometric signal (λ520/λ700) against different Mpro concentrations (from 0.6 to 150 nM), showing that higher Mpro concentrations quickly activated particle dissociation than at lower concentrations. The LoD for Mpro was determined to be 12.3 nM in saliva, 16.7 nM in the EBC, and 16.4 nM in PB buffer, respectively. The CDC of the A18 fragment was 40 μM. When the concentration of the A18 fragment was above CDC, the color turned to red. Otherwise, the color became transparent due to colloidal settlement in saliva or EBC (Figure S13).
Figure 5.
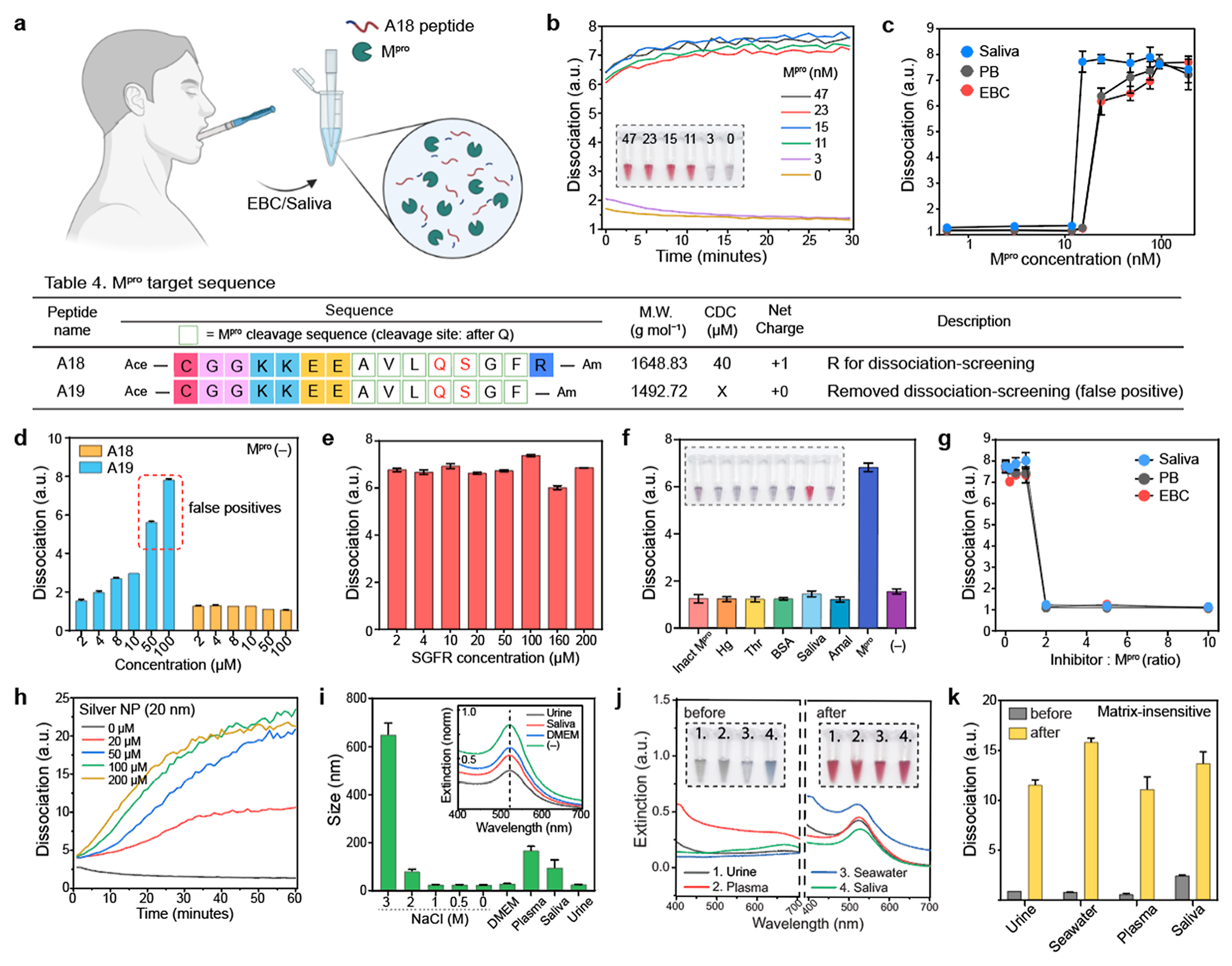
Matrix-insensitive Mpro detection (a) Schematic illustration of Mpro detection in EBC or saliva. The released A18 fragment by Mpro cleavage was used for colorimetric biosensing in saliva or EBC. (b) Time-dependent Mpro detection from 0 to 47 nM in saliva. The inset photograph shows that our dissociation strategy can provide a clear readout of the positive Mpro sample above 11 nM in saliva. (c) Detection limits of Mpro in saliva, EBC, and PB buffer, respectively. Table 4 describes peptide sequences that are designed to verify the role of the dissociation screening domain. (d) One Arg at the C-terminus can prevent false positives. False positives occurred in A19 when the peptide concentration was over 50 μM, while A18 showed no false positive in the absence of Mpro. (e) A18 fragment from C terminus (i.e., SGFR) had negligible impact on the dissociation process. (f) Specificity test using different biological proteins [e.g., inactivated Mpro (inact Mpro), hemoglobin (Hg), thrombin (Thr), BSA, saliva, and amalyase (Amal)]. (g) GC376 inhibitor assay test in saliva, EBC, and PB buffer, respectively. (h) The released A18 fragments by Mpro cleavage can dissociate other types of plasmonic assemblies such as AgNP aggregates. (i) After particle dissociation, the A18-capping AuNPs maintained high colloidal stability in different biological media (e.g., urine, saliva, plasma, DMEM) and extreme conditions (e.g., 2 M NaCl). (j) Plasmonic resonance peaks of the AuNP aggregates blueshifted after particle dissociation in 100% of (1) human urine, (2) plasma, (3) seawater, and (4) saliva. The inset photographs show before (left) and after (right) adding dissociation peptides. (k) Ratiometric signal (λ520/λ700) of the dissociated AuNPs in diverse matrixes, indicating that our dissociation strategy is less affected by the sample matrix. The panel (c–g,i–k) repeated three independent times and showed similar results.
To prevent non-desired particle dissociation, a positively charged domain was placed at the C terminus for dissociation screening: one R in A18 and no R in A19 as a negative control (Table 4 in Figure 5 and Figure S14). In the absence of Mpro, the A19 peptide over 50 μM can cause non-desired particle dissociation, while the A18 peptide showed no false positives (Figures 5d and S15). We further confirmed that the release of SGFR fragments during Mpro proteolysis had negligible impact on the dissociation process (Figures 5e and S16). We conducted a specificity test for Mpro using several related proteins such as hemoglobin (hg), inactivated Mpro (incubated at 60 °C for 3 h), thrombin (thr), bovine serum albumin (BSA), human saliva, and α-amylase (amal). Figure 5f shows that only the positive control (i.e., 200 nM Mpro) produced a prominent optical signal due to the release of the dissociation peptide by proteolytic cleavage (Figure S17). To define the enzymatic role in the protease in the colorimetric assays, a competitive inhibitor (GC376) for Mpro was used for the test. Mpro (200 nM) was incubated with increasing molarity of GC376 (i.e., 0–2 μM) in different operating mediums such as saliva, EBC, and PB buffer for 10 min prior to adding the A18 substrates. Figure 5g indicates that particle dissociation was prevented by the addition of inhibitors (from 400 nM to 2 μM) due to the formation of Mpro–GC376 complexes (Figures S18 and S19).
Next, we applied our dissociation strategy on silver nanoparticles (AgNPs, 20 nm in size) because AgNPs offer a higher order of extinction coefficient compared to AuNPs.32,33 As expected, the release of the A18 fragments by Mpro cleavage could dissociate AgNP aggregates, changing color from blue to yellow (Figures 5h and S20). Other plasmonic nanostructures including gold nanorods or bimetallic nanoparticles might also be able to be dissociated. Notably, the A18 peptide was designed for gold or silver nanoparticles with a size of 20 nm and negatively charged citrate ligands. Different nanostructures might require a different peptide sequence to provide sufficient electrical repulsion, hydrophilicity, and steric hindrance for dissociation. Our dissociation strategy can also improve the colloidal stability of plasmonic nanoparticles. After the particle dissociation, the A18-capped AuNPs maintained high colloidal stability in extreme conditions such as Dulbecco’s modified eagle medium (DMEM), human plasma, saliva, human urine, and NaCl (from 0.5 to 2 M) (Figure 5i). Lastly, our dissociation strategy is less affected by matrix interference. We replaced the sample matrix with 100% human plasma, urine, saliva, and seawater after RRK-based AuNPs aggregation. After adding dissociation peptides, reversible aggregation still occurred, leading to a blueshift of the plasmonic resonance peak; this in turn changes color from blue to red in 100% human plasma, urine, saliva, and seawater (Figures 5j,k and S21). These results emphasize that our peptide-based dissociation strategy provides a simple and versatile approach for reversible aggregation of plasmonic assemblies, offering a new mechanism for designing a matrix-insensitive plasmonic biosensor.
3. CONCLUSIONS
In summary, we developed peptide-driven dissociation of plasmonic assemblies as a response to Mpro detection of SARS-CoV-2. This strategy eliminated the need for surface modifications of AuNPs and complex couplings (e.g., PEG–peptide) for protease sensing. Both computational and experimental methods were used to understand reversible aggregation by a short cationic RRK peptide. Using 19 different peptide sequences, we verified that the dissociation capacity relies on hydrophilicity, charge density, ligand architecture, and steric distance. After incorporating the dissociation domain with an Mpro cleavage site, a colorimetric assay using UV–vis spectroscopy was tested to confirm the reproducibility and applicability of our platform for Mpro detection.
With an optimized peptide sequence, our dissociation strategy successfully produced a distinct optical signal as a function of the released peptides by Mpro cleavage with a detection limit of 12.3 nM in saliva. The dissociation-screening site at the C terminus enhances proteolytic cleavage (around 3-fold)34 and prevents false positives. Inhibitor assay and specificity test further confirmed the critical role of Mpro in the dissociation process, showing no non-specific activation. Our dissociation peptides successfully dissociated silver clusters, demonstrating the capability of our dissociation platform for potential future studies involving a variety of plasmonic nanomaterials including anisotropic or multi-component nanostructures35 that can induce distinct changes in extinction spectra through the dissociation process. Lastly, we demonstrated that our dissociation strategy can be less interrupted by matrixes such as human plasma, urine, and seawater, and the dissociated AuNPs maintained high colloidal stability. This peptide-driven dissociation strategy holds significant promise in various fields such as colloidal science, biochemistry, and plasmonic biosensors with diverse applications ranging from disease diagnosis and drug detection to environmental monitoring.
4. METHODS
4.1. Experimental Details.
4.1.1. Preparation of AuNPs and AuNP Aggregates.
Citrate-stabilized AuNPs with a size of 13 nm were synthesized using the Turkevich method.36 Briefly, 45 mg of HAuCl4·3H2O was dissolved in 300 mL of MQ water under generous stirring (600 rpm) and boiling condition at 120 °C. Then, 150 mg sodium citrate (dissolved in 5 mL of MQ water) was rapidly injected, and the reaction was left under boiling conditions for 20 min. The color of the solution changed from purple to gray and dark reddish. The resulting product was cooled down and was stored at room temperature for future use. The optical density of the final product was 1.45 (concentration ~3.6 nM, ε520 = 4.0 × 108 M−1 cm−1). Notably, 500 mL of round flask was cleaned with Aqua regia and distilled water (three times) before the synthesis.
Briefly, 10 μM of RRK peptide was used to aggregate 100 μL of AuNPs (conc ~3.6 nM). The particle aggregation rapidly occurred (less than 10 s), changing color from red to blue. Then the desired dissociation peptides were used to dissociate the AuNP aggregates. It is notable that our AuNPs are stabilized by citrate. Different surface ligands (e.g., BSPP) and different sizes (40 or 60 nm) of AuNPs require different amounts of the RRK peptide for particle aggregation. It is notable that surface ligands with large molecular weight (e.g., PEG1k or PVP55k) are difficult to aggregate using RRK peptides.
4.1.2. Dissociation of AuNP Aggregates Using Proteolysis of Peptides.
Briefly, a dried peptide powder was dissolved in phosphate buffer (20 mM, pH 8.0) and incubated with the Mpro at a molar ratio of 3000:1 (substrate/enzyme ratio) for 0.5 h at 37 °C. To confirm the Mpro cleavage site, the sample was purified using a C18 column (5 μm, 9.4 × 250 mm) and eluted with a flow rate of 3 mL/min over 30 min with a linear gradient from 10 to 95% to ACN in H2O. After the purification, the molecular weight of a fragment peptide was confirmed by using ESI-MS (positive or negative mode) or/and matrix (e.g., HCCA) assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS, Bruker Autoflex Max) in the Molecular Mass Spectrometry Facility at UC San Diego.
To test particle dissociation in saliva and EBC condition, the desired amounts (conc, 30 μM) of the dissociation peptides (A18, Ace–CGGKKEEAVLQSGFR–Am) were incubated with Mpro in 100% of saliva or EBC for 0.5 h at 37 °C. Then, the 40 μL of A18 fragment peptides in saliva or EBC were mixed with 100 μL of AuNP aggregates for colorimetric sensing. The mass peaks of the A18 peptide and its fragment were confirmed by MALDI-TOF MS examination, respectively.
4.1.3. Dissociation Strategy in Diverse Matrixes.
Briefly, RRK peptides (8–10 μM) were first used to trigger AuNP aggregation in distilled water. The sample was centrifuged at 1 g for 5 min to remove the supernatant. Then, the pellet was re-dispersed in 100% human saliva, plasma, urine, and seawater. After re-dispersion, the desired amounts of the dissociation peptide were used to dissociate AuNPs aggregates in different matrixes. Both A11 and A12 peptides successfully dissociated the aggregated AuNPs in diverse matrixes. The experiment was performed with three replicates, and the microplate reader was used to measure spectral scanning from 300 to 900 nm before and after dissociation. The ratiometric signal (λ520/λ700) was referred as dissociation. The data was blanked to remove the background signal.
4.2. Computational Details.
4.2.1. Investigation of RRK Interaction on a Citrate-Coated AuNP Using MD Simulations.
The forcefields applied in MD simulations were based on AMBER37 forcefield (RRK, citrate, Na, Cl), TIP3P38 forcefield (water), and EAM/Fs potential (Au) from Ackland et al.39 The pair interactions were determined by general mixing rule except the RRK|Au and citrate|Au interactions, which were constructed based on the parameterization of QM interaction energies. In MD simulations, a long-range particle–particle particle-mesh solver, Van der Waals cutoff 10 Å, timesteps 1.0 fs, and SHAKE algorithm40 for water molecules and hydrogen atoms were adopted.
To investigate the binding phenomenon for RRK molecules toward a citrate-coated AuNP system, we performed MD simulations using LAMMPS engine.41 The initial structure contained a 5 nm-diameter Au nanoparticle, 80 citrate molecules, 240 Na ions, and 4670 water molecules. MD simulation was initiated with 500 conjugated gradient steps, followed by the canonical ensemble (NVT) to heat up a system into 298 K. Afterwards, the isobaric/isothermal ensemble (NPT) was proceeded to optimize systemic density at 298 K/1 atm and NVT ensemble was further applied to equilibrate a system. Based on the equilibrated citrate-coated Au nanoparticle structure, furthermore, we constructed a citrate|RRK Au nanoparticle system by randomly placing 80 RRK molecules and 240 Cl ions around the citrate-coated Au nanoparticle and embedding it into water solvents, thereby a structure with AuNP|80 citrate||240 Na|80 RRK|240 Cl|33622 water was constructed. With the same procedure as mentioned in this section, an equilibrated AuNP|80 citrate|240 Na|80 RRK|240 Cl|33622 water system was obtained, demonstrating the RRK binding phenomenon.
4.2.2. Determining Free Energies as a Function of Nanoparticle Distance Using Steered Molecular Dynamics Simulations.
SMD simulations were performed to investigate free energy values as two Au nanoparticles approached each other. In this study, we adopted the same forcefield parameters, long-range solver, cutoff point, and SHAKE algorithm as mentioned in MD simulation (Supporting Information Section 2.2) section. There were two systems performed: a system without RRK molecules and a system with RRK molecules, where the first system contained two 5 nm-diameter Au nanoparticles, 870 citrate, 2160 Na, and 29238 water, and the second system contained two 5 nm-diameter Au nanoparticles, 870 citrate, 2160 Na, 95 RRK, 285 Cl, and 45542 water. Each model was initially equilibrated using the same procedure as MD simulation and further performed a 3.7 ns SMD simulation to investigate free energies as a function of Au nanoparticle distance. In SMD simulation, we adopted a harmonic restraint with a force constant of 100 kcal/mol Å2, where the equilibrium value of the harmonic restraint was gradually changed from 91 Å into 55 Å, and saved free energy values as two Au nanoparticles approached each other, thereby the free energy values at different Au nanoparticle distance were determined.
4.2.3. Free Energy Investigation Using Metadynamics Approach.
To explore the molecular behavior when a molecule approaches to an Au(111) surface, we constructed systems with periodic boundaries in x, y coordinates and a finite boundary in the z coordinate and explored free energy values using the MTD approach.42 Two systems were constructed: (1) a single citrate molecule on an Au(111) slab, representing as the procedure to form a citrate-coated Au surface, and (2) a single RRK molecule on a citrate-coated Au surface, representing as the procedure for adding RRK molecules into a citrate-coated Au system. The corresponding number of Na+(Cl−) ions were added to form a charge neutral system, and the system cell size was (57.48778, 49.78588, 150 Å) in (x, y, z). The Au(111) slab position was fixed at 19 Å/31 Å (bottom/top), and the slab–slab interactions were turned off via inserting empty volume in z with a factor 2.0.
Each system was initialized using 500 steps CG minimization and further heated up into 298 K using the Nose–Hoover thermostat (NVT ensemble). Afterward, 1 ns NVT ensemble was adopted to equilibrate a system. In MTD section, the 50 ns trajectory was proceeded, and the z coordinate was measured as the center of mass of the citrate molecule in the (1) system and the center of mass of the RRK molecule in the (2) system, where Gaussian functions with a weight of 1.0 kcal/mol and a width 1.25 Å were deposited every 0.2 ps into each system. As shown in the results (Figure S3), a weaker peak with a relatively far distance from an Au (111) surface was found in the (2) system, implying RRK molecule interacted with citrates and bound on a citrate-coated Au surface, which reduced the charge effect on Au surfaces and further resulted in the NP aggregation phenomenon.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of Health (#1R21AI157957 and #1R01DE031114). This work was performed in part at San Diego Nanotechnology Infrastructure (SDNI) of University of California San Diego supported by (NSF ECCS-1542148), and by the Cellular and Molecular Medicine Electron Microscopy core facility (NIH S10 OD 023527). We further thank the Cellular and Molecular Medicine Electron Microscopy Core via UCSD-CMM-EM Core, RRID: SCR_022039. This work was performed in part at the San Diego Nanotechnology Infrastructure (SDNI) of UCSD which is supported by the National Science Foundation (grant ECCS-2025752). The author acknowledges funds from Sloan Research Fellowship and UCSD ENLACE program supported by Dr. Olivia Graeve. The author also acknowledges Biorender for supporting artwork design.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.3c09627.
Material information and experimental designs (including peptide synthesis, specificity test, inhibitor test, stability test, simulation method), description of DLVO theory, TEM image of RRK-induced Au clusters, ESI–MS data of all the peptide sequence, photographs of dissociated AuNPs, time-dependent ratiometric signal, full UV–vis spectrum of specificity test, inhibition test, SGFR fragment test, AgNP test, and matrix-insensitive test (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsami.3c09627
The authors declare no competing financial interest.
Contributor Information
Wonjun Yim, Materials Science and Engineering Program, University of California San Diego, La Jolla, California 92093, United States.
Maurice Retout, Department of Nano and Chemical Engineering, University of California San Diego, La Jolla, California 92093, United States.
Amanda A. Chen, Department of Nano and Chemical Engineering, University of California San Diego, La Jolla, California 92093, United States
Chuxuan Ling, Department of Nano and Chemical Engineering, University of California San Diego, La Jolla, California 92093, United States.
Lubna Amer, Materials Science and Engineering Program, University of California San Diego, La Jolla, California 92093, United States.
Zhicheng Jin, Department of Nano and Chemical Engineering, University of California San Diego, La Jolla, California 92093, United States.
Yu-Ci Chang, Materials Science and Engineering Program, University of California San Diego, La Jolla, California 92093, United States.
Saul Chavez, Department of Nano and Chemical Engineering, University of California San Diego, La Jolla, California 92093, United States.
Karen Barrios, Department of Nano and Chemical Engineering, University of California San Diego, La Jolla, California 92093, United States.
Benjamin Lam, Department of Nano and Chemical Engineering, University of California San Diego, La Jolla, California 92093, United States.
Zhi Li, Shu Chien-Gene Lay Department of Bioengineering, University of California San Diego, La Jolla, California 92093, United States.
Jiajing Zhou, Department of Nano and Chemical Engineering, University of California San Diego, La Jolla, California 92093, United States.
Lingyan Shi, Shu Chien-Gene Lay Department of Bioengineering, University of California San Diego, La Jolla, California 92093, United States.
Tod A. Pascal, Department of Nano and Chemical Engineering, University of California San Diego, La Jolla, California 92093, United States
Jesse V. Jokerst, Materials Science and Engineering Program, University of California San Diego, La Jolla, California 92093, United States; Department of Nano and Chemical Engineering and Department of Radiology, University of California San Diego, La Jolla, California 92093, United States
REFERENCES
- (1).Liu J; Lu Y A Colorimetric Lead Biosensor Using DNAzyme-Directed Assembly of Gold Nanoparticles. J. Am. Chem. Soc 2003, 125, 6642–6643. [DOI] [PubMed] [Google Scholar]
- (2).Jin Z; Li Y; Li K; Zhou J; Yeung J; Ling C; Yim W; He T; Cheng Y; Xu M; et al. Peptide Amphiphile Mediated Co-assembly for Nanoplasmonic Sensing. Angew. Chem., Int. Ed 2023, 62, No. e202214394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Guo L; Xu Y; Ferhan AR; Chen G; Kim D-H Oriented Gold Nanoparticle Aggregation for Colorimetric Sensors with Surprisingly High Analytical Figures of Merit. J. Am. Chem. Soc. 2013, 135, 12338–12345. [DOI] [PubMed] [Google Scholar]
- (4).Liu J; Lu Y Accelerated Color Change of Gold Nanoparticles Assembled by DNAzymes for Simple and Fast Colorimetric Pb2+ Detection. J. Am. Chem. Soc 2004, 126, 12298–12305. [DOI] [PubMed] [Google Scholar]
- (5).Ghosh SK; Pal T Interparticle Coupling Effect on the Surface Plasmon Resonance of Gold Nanoparticles: from Theory to Applications. Chem. Rev 2007, 107, 4797–4862. [DOI] [PubMed] [Google Scholar]
- (6).Jin Z; Mantri Y; Retout M; Cheng Y; Zhou J; Jorns A; Fajtova P; Yim W; Moore C; Xu M; et al. A Charge-Switchable Zwitterionic Peptide for Rapid Detection of SARS-CoV-2 Main Protease. Angew. Chem., Int. Ed 2022, 61, No. e202112995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kim T; Lee K; Gong M.-s.; Joo S-W Control of Gold Nanoparticle Aggregates by Manipulation of Interparticle Interaction. Langmuir 2005, 21, 9524–9528. [DOI] [PubMed] [Google Scholar]
- (8).Lin SY; Wu SH; Chen C. h. A Simple Strategy for Prompt Visual Sensing by Gold Nanoparticles: General Applications of Interparticle Hydrogen Bonds. Angew. Chem., Int. Ed 2006, 45, 4948–4951. [DOI] [PubMed] [Google Scholar]
- (9).Zhang L; Mazouzi Y; Salmain M; Liedberg B; Boujday S Antibody-Gold Nanoparticle Bioconjugates for Biosensors: Synthesis, Characterization and Selected Applications. Biosens. Bioelectron 2020, 165, 112370. [DOI] [PubMed] [Google Scholar]
- (10).Wang Y; Quinsaat JEQ; Ono T; Maeki M; Tokeshi M; Isono T; Tajima K; Satoh T; Sato S.-i.; Miura Y; et al. Enhanced Dispersion Stability of Gold Nanoparticles by the Physisorption of Cyclic Poly (Ethylene Glycol). Nat. Commun 2020, 11, 6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yim W; Takemura K; Zhou J; Zhou J; Jin Z; Borum RM; Xu M; Cheng Y; He T; Penny W; et al. Enhanced Photoacoustic Detection of Heparin in Whole Blood via Melanin Nanocapsules Carrying Molecular Agents. ACS Nano 2021, 16, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jin Z; Yeung J; Zhou J; Retout M; Yim W; Fajtová P; Gosselin B; Jabin I; Bruylants G; Mattoussi H; et al. Empirical Optimization of Peptide Sequence and Nanoparticle Colloidal Stability: The Impact of Surface Ligands and Implications for Colorimetric Sensing. ACS Appl. Mater. Interfaces 2023, 15, 20483–20494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cedervall T; Lynch I; Lindman S; Berggård T; Thulin E; Nilsson H; Dawson KA; Linse S Understanding the Nanoparticle–Protein Corona Using Methods to Quantify Exchange Rates and Affinities of Proteins for Nanoparticles. Proc. Natl. Acad. Sci 2007, 104, 2050–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Retout M; Jin Z; Tsujimoto J; Mantri Y; Borum R; Creyer MN; Yim W; He T; Chang Y-C; Jokerst JV Di-Arginine Additives for Dissociation of Gold Nanoparticle Aggregates: A Matrix-Insensitive Approach with Applications in Protease Detection. ACS Appl. Mater. Interfaces 2022, 14, 52553–52565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Liang X; Wei H; Cui Z; Deng J; Zhang Z; You X; Zhang X-E Colorimetric Detection of Melamine in Complex Matrices based on Cysteamine-Modified Gold Nanoparticles. Analyst 2011, 136, 179–183. [DOI] [PubMed] [Google Scholar]
- (16).Jokerst JV; Lobovkina T; Zare RN; Gambhir SS Nanoparticle PEGylation for Imaging and Therapy. Nanomed 2011, 6, 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; et al. Structure of Mpro from SARS-CoV-2 and Discovery of its Inhibitors. Nature 2020, 582, 289–293. [DOI] [PubMed] [Google Scholar]
- (18).Owen DR; Allerton CM; Anderson AS; Aschenbrenner L; Avery M; Berritt S; Boras B; Cardin RD; Carlo A; Coffman KJ; et al. An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374, 1586–1593. [DOI] [PubMed] [Google Scholar]
- (19).Yim W; Zhou J; Sasi L; Zhao J; Yeung J; Cheng Y; Jin Z; Johnson W; Xu M; Palma-Chavez J; et al. 3D-Bioprinted Phantom with Human Skin Phototypes for Biomedical Optics. Adv. Mater 2023, 35, 2206385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhu G; Zhu X; Fan Q; Wan X Raman Spectra of Amino Acids and Their Aqueous Solutions. Spectrochim. Acta, Part A 2011, 78, 1187–1195. [DOI] [PubMed] [Google Scholar]
- (21).Chang Y-C; Jin Z; Li K; Zhou J; Yim W; Yeung J; Cheng Y; Retout M; Creyer MN; Fajtová P; et al. Peptide Valence-Induced Breaks in Plasmonic Coupling. Chem. Sci 2023, 14, 2659–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lévy R; Thanh NT; Doty RC; Hussain I; Nichols RJ; Schiffrin DJ; Brust M; Fernig DG. Rational and Combinatorial Design of Peptide Capping Ligands for Gold Nanoparticles. J. Am. Chem. Soc 2004, 126, 10076–10084. [DOI] [PubMed] [Google Scholar]
- (23).Nowinski AK; Sun F; White AD; Keefe AJ; Jiang S Sequence, Structure, and Function of Peptide Self-Assembled Monolayers. J. Am. Chem. Soc 2012, 134, 6000–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mokashi-Punekar S; Walsh TR; Rosi NL Tuning the Structure and Chiroptical Properties of Gold Nanoparticle Single Helices via Peptide Sequence Variation. J. Am. Chem. Soc 2019, 141, 15710–15716. [DOI] [PubMed] [Google Scholar]
- (25).Liu X; Zhang Q; Knoll W; Liedberg B; Wang Y Rational Design of Functional Peptide–Gold Hybrid Nanomaterials for Molecular Interactions. Adv. Mater 2020, 32, 2000866. [DOI] [PubMed] [Google Scholar]
- (26).Olmedo I; Araya E; Sanz F; Medina E; Arbiol J; Toledo P; Alvarez-Lueje A; Giralt E; Kogan MJ How Changes in the Sequence of the Peptide CLPFFD-NH2 Can Modify the Conjugation and Stability of Gold Nanoparticles and Their Affinity for β-Amyloid Fibrils. Bioconjugate Chem. 2008, 19, 1154–1163. [DOI] [PubMed] [Google Scholar]
- (27).Jin Z; Ling C; Li Y; Zhou J; Li K; Yim W; Yeung J; Chang Y-C; He T; Cheng Y; et al. Spacer Matters: All-Peptide-Based Ligand for Promoting Interfacial Proteolysis and Plasmonic Coupling. Nano Lett. 2022, 22, 8932–8940. [DOI] [PubMed] [Google Scholar]
- (28).Yim W; Zhou J; Mantri Y; Creyer MN; Moore CA; Jokerst JV Gold Nanorod–Melanin Hybrids for Enhanced and Prolonged Photoacoustic Imaging in the Near-Infrared-II Window. ACS Appl. Mater. Interfaces 2021, 13, 14974–14984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chen S; Cao Z; Jiang S Ultra-Low Fouling Peptide Surfaces Derived from Natural Amino Acids. Biomaterials 2009, 30, 5892–5896. [DOI] [PubMed] [Google Scholar]
- (30).YináYi M An AM1 and PM3 Molecular Orbital and Self-Consistent Reaction-Field Study of the Aqueous Solvation of Glycine, Alanine and Proline in Their Neutral and Zwitterionic Forms. J. Chem. Soc., Perkin Trans 1991, 2 (4), 531–537. [Google Scholar]
- (31).Huang N; Pérez P; Kato T; Mikami Y; Okuda K; Gilmore RC; Conde CD; Gasmi B; Stein S; Beach M; et al. SARS-CoV-2 Infection of the Oral Cavity and Saliva. Nat. Med 2021, 27, 892–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Retout M; Gosselin B; Mattiuzzi A; Ternad I; Jabin I; Bruylants G Peptide-Conjugated Silver Nanoparticles for the Colorimetric Detection of the Oncoprotein Mdm2 in Human Serum. ChemPlusChem 2022, 87, No. e202100450. [DOI] [PubMed] [Google Scholar]
- (33).Retout M; Mantri Y; Jin Z; Zhou J; Noël G; Donovan B; Yim W; Jokerst JV Peptide-Induced Fractal Assembly of Silver Nanoparticles for Visual Detection of Disease Biomarkers. ACS Nano 2022, 16, 6165–6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Shan YF; Xu GJ Study on Substrate Specificity at Subsites for Severe Acute Respiratory Syndrome Coronavirus 3CL Protease. Acta Biochim. Biophys. Sin 2005, 37, 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ivanchenko M; Jing H Smart Design of Noble Metal–Copper Chalcogenide Dual Plasmonic Heteronanoarchitectures for Emerging Applications: Progress and Prospects. Chem. Mater 2023, 35, 4598–4620. [Google Scholar]
- (36).Kimling J; Maier M; Okenve B; Kotaidis V; Ballot H; Plech A Turkevich Method for Gold Nanoparticle Synthesis Revisited. J. Phys. Chem. B 2006, 110, 15700–15707. [DOI] [PubMed] [Google Scholar]
- (37).Salomon-Ferrer R; Case DA; Walker RC An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev.: Comput Mol. Sci 2013, 3, 198–210. [Google Scholar]
- (38).Mark P; Nilsson L Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar]
- (39).Ackland GJ; Tichy G; Vitek V; Finnis MW Simple N-body potentials for the noble metals and nickel. Philos. Mag. A 1987, 56, 735–756. [Google Scholar]
- (40).Ryckaert J-P; Ciccotti G; Berendsen HJ Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- (41).Plimpton S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys 1995, 117, 1–19. [Google Scholar]
- (42).Mahinthichaichan P; Vo QN; Ellis CR; Shen J Kinetics and Mechanism of Fentanyl Dissociation from the μ-Opioid Receptor. JACS Au 2021, 1, 2208–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.