

Abstract
Intracellular free Ca2+ ([Ca2+]i) dysregulation occurs in coronary smooth muscle (CSM) in atherosclerotic coronary artery disease (CAD) of metabolic syndrome (MetS) swine. Our goal was to determine how CAD severity, arterial structure, and MetS risk factors associate with [Ca2+]i dysregulation in human CAD compared to changes in Ossabaw miniature swine. CSM cells were dispersed from coronary arteries of explanted hearts from transplant recipients and from lean and MetS swine with CAD. CSM [Ca2+]i elicited by Ca2+ influx and sarcoplasmic reticulum (SR) Ca2+ release and sequestration was measured with fura-2. Increased [Ca2+]i signaling was associated with advanced age and a greater media area in human CAD. Decreased [Ca2+]i signaling was associated with a greater number of risk factors and a higher plaque burden in human and swine CAD. Similar [Ca2+]i dysregulation exhibited in human and Ossabaw swine CSM provides strong evidence for the translational relevance of this large animal model.
Keywords: Atherosclerosis, obesity, metabolic syndrome, histology, sarco-endoplasmic reticulum Ca2+ ATPase, Ca2+ influx, Ca2+ release, animal model, risk factors
Graphical Abstract
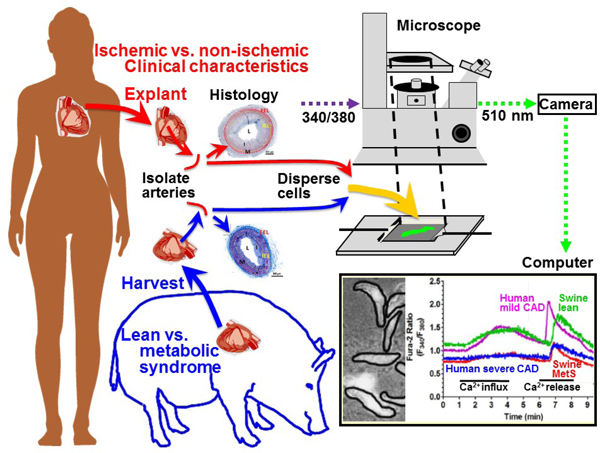
1. Introduction
Metabolic syndrome (MetS) is characterized by a clustering of three or more of the following five conditions: central obesity, hypertension, dyslipidemia, insulin resistance, and glucose intolerance [1]. MetS affects one-third of all adults in the United States and increases the risk of developing coronary artery disease (CAD), which continues to be the primary cause of mortality worldwide and accounts for 1 in 7 deaths in the United States [1]. CAD-induced ischemic cardiomyopathy is the leading cause of heart failure, followed by dilated non-ischemic cardiomyopathy, hypertension, and valvular heart disease [2].
CAD is a progressive disease where initial endothelial damage leads to lipid and inflammatory cell infiltration of the arterial wall, causing medial thickening and neointima formation [3]. This arterial restructuring is exacerbated by the proliferation and recruitment of coronary smooth muscle (CSM) cells to the plaque, which is accomplished by the phenotypic switching of CSM from a differentiated, contractile phenotype to a proliferative, migratory phenotype [4]. These migratory CSM cells secrete and deposit extracellular matrix like collagen, elastin, and fibrin into the thickening artery wall. While the presence of CSM inside the plaque contributes to plaque stability, over time the accumulation of lipid and cellular debris results in a necrotic core and plaque destabilization that often leads to plaque rupture and myocardial infarction. In one study, patients hospitalized for ST segment elevation myocardial infarction who had multivessel CAD had an 8-year mortality from heart failure rate of 11%, as opposed to only a 1% mortality rate for individuals similarly hospitalized without multivessel CAD [5]. Further, CSM dedifferentiation into an osteogenic phenotype is associated with vascular calcification, which is directly related to increased mortality and cardiac events [6], plaque instability, and rupture [7].
Ca2+ is a vital secondary messenger involved in the regulation of several key smooth muscle cell functions, such as transcription [8, 9], migration [10, 11], proliferation [4, 12–14], and contraction [4, 15]. Previous research (reviewed in [16]) has shown that CAD is accompanied by alterations in many CSM Ca2+ transporters, including voltage-gated Ca2+ channels [17, 18], transient receptor potential channels [19], sarco-endoplasmic reticulum Ca2+ ATPases [17, 19–21], plasma membrane Ca2+ ATPases [17], and Na+/Ca2+ exchangers [17]. Our lab recently showed that intracellular Ca2+ ([Ca2+]i) handling alterations that accompany CAD occur in a biphasic manner in Ossabaw miniature swine, with enhanced Ca2+ signaling in early, mild disease and decreased Ca2+ signaling in late, severe disease [22]. Furthermore, when the plaque was separated from the arterial wall in diseased coronary arteries, CSM isolated exclusively from the plaque region exhibited decreased SR Ca2+ store and SR Ca2+ pump activity, while CSM isolated from the arterial wall exhibited increased SR Ca2+ store SR Ca2+ pump activity [22].
There is difficulty in finding an animal model for CAD, as there are many risk factors and uncontrollable variables in the human population. Our lab has characterized the Ossabaw miniature swine model of MetS and CAD [23]. Due to their “thrifty genotype,” Ossabaw swine have a propensity to develop all characteristics of MetS when fed an atherogenic diet [24, 25]. While Ossabaw swine develop diffuse, human-like coronary plaques [21], CSM [Ca2+]i handling patterns have never been compared to freshly isolated CSM from human patients. Therefore, the aims of the current study are: 1) to determine how disease severity, arterial restructuring, and MetS risk factors are associated with [Ca2+]i dysfunction in fresh, non-cultured human CSM and 2) to determine whether CSM [Ca2+]i dysregulation in Ossabaw miniature swine is similar to CSM [Ca2+]i dysregulation in human CAD patients. These results will aid in characterizing the association between pathological arterial remodeling and dysfunctional CSM [Ca2+]i handling in human heart failure patients and will strengthen the Ossabaw miniature swine as a translational model for CAD pathology and at the [Ca2+]i signaling level.
2. Materials and Methods
2.1. Collection of human tissue.
Explanted human hearts were collected from 24 patients (15 male, 9 female; aged 51.0 ± 2.5 years) undergoing heart transplantation surgery at Methodist Hospital in Indianapolis, IN between 2015 and 2018. Epicardial coronary arteries were dissected from the heart at the time of removal and stored for no longer than 24 hours in a physiological salt solution, which preserves intracellular Ca2+ regulatory function (e.g. [22, 24, 25]). Intracellular Ca2+ measures were successfully conducted on cells from arteries of all 24 hearts. Patients were defined by histology of the coronary segments as non-ischemic or ischemic for plaque burden up to 75% and >75%, respectively.
2.2. Animals.
All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee at Indiana University School of Medicine with the recommendations outlined by the National Research Council and the American Veterinary Medical Association Panel on Euthanasia [26, 27]. Ossabaw miniature swine were fed either standard chow diet (5L80; Purina Test Diet, Richmond, IN) or a hypercaloric, atherogenic diet (n = 7 for both groups) [22, 24, 25]. Pigs were euthanized via cardiectomy and coronary arteries were removed for further analysis.
2.3. Swine metabolic phenotyping.
Blood was collected at time of euthanasia for analysis (ANTECH Diagnostics, Fishers, IN).
2.4. Histology.
Segments from proximal epicardial arteries (2–4 mm in length) were placed in 10% phosphate-buffered formalin for 24–48 h, then transferred to 70% ethanol. The proximal epicardial arteries used for histology and intracellular Ca2+ measures were not bypassed previously. We assume that blood flow never was zero even in the ischemic arteries defined by the >75% plaque burden. Histology of arterial cross-sections was performed in the Department of Anatomy and Cell Biology at Indiana University School of Medicine. Verhoeff-Van Gieson elastin stain was used to determine media area and plaque burden, which we define as the percentage of the original lumen that is occupied by atherosclerotic plaque. Von Kossa stains calcified areas black to determine vascular calcification. Masson’s Trichrome stain was used to visualize collagen (blue) and cellular composition (red). Images were captured with a Leica DM3000 microscope connected to Leica Application Suites V4.1 software (Leica Microsystems GmbH, Wetzlar, Germany) and analyzed using Adobe Photoshop® CS6.
2.5. Assessment of [Ca2+]i regulation.
Whole-cell [Ca2+]i levels were measured at room temperature (22–25°C) by using the fluorescent Ca2+ indicator fura-2 AM (InCa++ Ca2+ Imaging System, Intracellular Imaging, Cincinnati, OH) as previously described [19, 21, 24, 25] and following the standards set in the field [28, 29]. Briefly, freshly dispersed smooth muscle cells from the proximal 45 mm of the left anterior descending artery were incubated with 3.0 μM fura-2 AM (Molecular Probes, Eugene, OR) to load the cells with fura-2. An aliquot of cells loaded with fura-2 was placed on a coverslip contained within a constant-flow superfusion chamber that was mounted on the microscope (model TMS-F, Nikon, Melville, NY), with flow maintained at a constant rate of 1–2 mL/min to change solutions. Basal Ca2+ levels were measured in physiologic salt solution. Calcium influx and maximal sarcoplasmic reticulum (SR) Ca2+ loading was accomplished by depolarization with high (80 mM) K+ solution to activate voltage-gated Ca2+ channels. SR Ca2+ stores were released with 5 mM caffeine in Ca2+-free solution to activate SR ryanodine receptors. A caffeine wash-out phase was used to determine sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) function via the undershoot below baseline during this recovery period [30–32]. Fura-2 in CSM was excited by light from a 300 W xenon arc lamp that was passed through a computer-controlled filter changer containing 340 nm and 380 nm bandpass filters. Whole-cell fura-2 fluorescence was expressed as the 340 nm/380 nm ratio of fura-2 emission at 510 nm.
2.6. Statistics.
Statistical analysis was performed using GraphPad Prism 5.0 (San Diego, CA). Unpaired student’s t test was performed for comparisons in swine and one-way analysis of variance with Newman–Keuls post hoc analysis for comparison of human groups. Data are presented as mean ± standard error. Correlations were determined using Pearson’s product-moment coefficient of correlation. Simple regression analyses were performed to determine statistical significance of the correlations. Statistical significance was set at p<0.05.
3. Results
3.1. Structure of human and swine coronary arteries.
Representative histological stains for humans (Fig. 1a-f) and swine (Fig. 1g-l) are shown. Human arteries show great diversity in disease state and structure and have been grouped based on percent plaque burden (Fig. 1m-o). Fig. 1m (blue symbols) illustrates the average plaque burden and variability in the three human groups. While calcification was similar between human groups due to high within-group variability (Fig. 1n), fibrosis as measured by percent collagen was increased in the ischemic >75% plaque burden group compared to the non-ischemic <50% plaque burden group (Fig. 1o). Similar to the human groups, swine with MetS-induced CAD exhibited increased plaque burden (Fig. 1m), similar vascular calcification (Fig. 1n), and greater collagen content (Fig. 1o) compared to their lean counterparts. The two bars on the x-axes of the graphs signify that statistics were done only between experimental groups of the same species.
Fig. 1. Histological staining reveals similar pathological remodeling in both humans and swine.
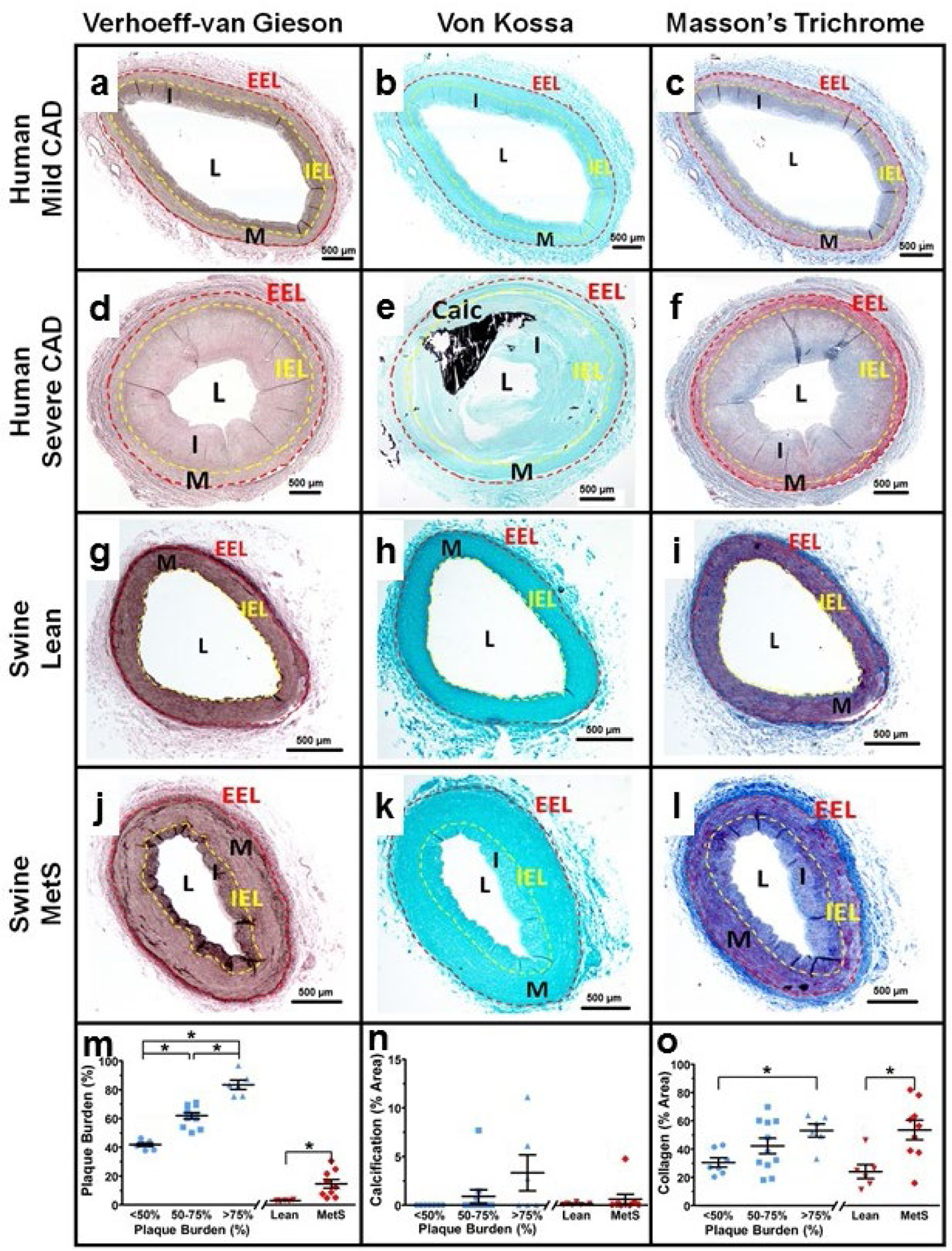
Coronary artery histological sections from a human with non-ischemic <50% plaque burden (a-c), a human with an ischemic >75% plaque burden (d-f), a lean swine (g-i), and a MetS swine (j-l). Verhoeff-Van Gieson staining was used to determine plaque burden and the areas of the tunica media and intima using the visible external elastic lamina (EEL, red dashed line) and internal elastic lamina (IEL, yellow dashed line) (a, d, g, j). Von Kossa staining was used to determine percent of vascular calcification (black) (b, e, h, k). Trichrome staining was used to determine the percent collagen (blue) (c, f, i, l). Humans (bars and blue symbols) were binned based on percent plaque burden and swine (bars and red symbols) showed significantly increased plaque burden in the MetS group compared to the lean group (m). Percent vascular calcification was not significantly different between human or swine groups (n). The percent collagen increased with disease in both the human and swine groups (o). EEL, external elastic lamina; IEL, internal elastic lamina; M, tunica media; I, tunica intima; L, lumen.
3.2. Clinical characteristics are similar in humans with different CAD severities.
Clinical characteristics of patients and swine are in Table 1. Heart function parameters were similar between the groups, including ejection fraction, left ventricular end diastolic pressure, and coronary output, but only qualitatively recorded in patient records. The quantified systolic and diastolic blood pressures were not different (Table 1), which is consistent with qualitatively described similar heart function between the clinical groups. There were no sex-specific differences in any of the measured parameters. Overall, while these groups differ in CAD severity, they exhibit comparable metabolic and functional disease parameters, i.e. similar increased cardiometabolic risk above healthy controls. Swine on an atherogenic diet developed significantly higher weight, systolic blood pressure, fasting blood glucose, total cholesterol, and triglyceride levels compared to lean, healthy controls, indicative of MetS.
Table 1.
Clinical characteristics of human subjects and swine
| Non-ischemic | Ischemic | |||
|---|---|---|---|---|
| Humans | <50% PB | 50–75% PB | >75% PB | p |
| N | 7 | 11 | 6 | - |
| Anthropometric Data | ||||
| Height (cm) | 170 ± 4 | 176 ± 3 | 176 ± 4 | N.S. |
| Weight (kg) | 81 ± 6 | 86 ± 5 | 86 ± 10 | N.S. |
| BMI (kg/m2) | 28 ± 2 | 27 ± 1 | 27 ± 2 | N.S. |
| Clinical Data | ||||
| Age (years) | 52 ± 5 | 51 ± 4 | 49 ± 5 | N.S. |
| Male/Female | 3/4 | 7/4 | 5/1 | N.S. |
| SBP (mmHg) | 107 ± 5 | 110 ± 5 | 104 ± 4 | N.S. |
| DBP (mmHg) | 67 ± 5 | 70 ± 5 | 69 ± 4 | N.S. |
| Ex-smoker | 2 (29%) | 3 (27%) | 4 (67%) | N.S. |
| LVAD | 3 (43%) | 7 (64%) | 4 (67%) | N.S. |
| Presence of MetS | 3 (43%) | 4 (36%) | 3 (50%) | N.S. |
| No. MetS risk factors | 1.9 ± 0.7 | 2.1 ± 0.4 | 2.5 ± 0.6 | N.S. |
| Biochemistry Data | ||||
| Fasting bG (mg/dL) | 128 ± 18 | 106 ± 15 | 106 ± 10 | N.S. |
| HbA1c (%) | 5.6 ± 0.2 | 5.5 ± 0.1 | 5.0 ± 0.3 | N.S. |
| Total cholesterol (mg/dL) | 139 ± 7 | 161 ± 20 | 136 ± 18 | N.S. |
| LDL (mg/dL) | 75 ± 7 | 97 ± 17 | 81 ± 18 | N.S. |
| HDL (mg/dL) | 40 ± 4 | 38 ± 4 | 34 ± 2 | N.S. |
| LDL/HDL Ratio | 2.0 ± 0.2 | 2.7 ± 0.4 | 2.3 ± 0.5 | N.S. |
| Triglycerides (mg/dL) | 117 ± 19 | 128 ± 14 | 108 ± 13 | N.S. |
| Comorbidities | ||||
| Atrial Fibrillation | 2 (29%) | 5 (45%) | 3 (50%) | N.S. |
| Diabetes mellitus | 2 (29%) | 2 (18%) | 0 (0%) | N.S. |
| Kidney Disease | 3 (43%) | 3 (27%) | 1 (17%) | N.S. |
| Cancer | 1 (14%) | 1 (9%) | 0 (0%) | N.S. |
| Clinical Depression | 3 (43%) | 3 (27%) | 4 (67%) | N.S. |
| Treatments | ||||
| Aspirin | 4 (57%) | 10 (91%) | 3 (50%) | N.S. |
| ACEI/ARB | 2 (29%) | 4 (36%) | 2 (33%) | N.S. |
| β-blocker | 3 (43%) | 8 (73%) | 2 (33%) | N.S. |
| Ca-blocker | 1 (14%) | 1 (9%) | 0 (0%) | N.S. |
| Diuretics | 4 (57%) | 9 (82%) | 4 (67%) | N.S. |
| Lipid-lowering drugs | 2 (29%) | 4 (36%) | 2 (33%) | N.S. |
| Anti-diabetic drugs | 2 (29%) | 2 (18%) | 0 (0%) | N.S. |
| Anti-arrhythmic drugs | 4 (57%) | 8 (73%) | 4 (67%) | N.S. |
| Antidepressants | 3 (43%) | 3 (27%) | 4 (67%) | N.S. |
| Swine | Lean | MetS | p | |
| N | 7 | 7 | - | |
| Weight (kg) | 77 ± 4 | 110 ± 3 | <0.05 | |
| Age (years) | 2.6 ± 0.1 | 2.8 ± 0.2 | N.S. | |
| Male/Female | 4/3 | 1/6 | N.S. | |
| SBP (mmHg) | 82 ± 3 | 90 ± 5 | <0.05 | |
| DBP (mmHg) | 59 ± 3 | 60 ± 5 | N.S. | |
| Fasting bG (mg/dL) | 69 ± 3 | 80 ± 3 | <0.05 | |
| Total cholesterol (mg/dL) | 80 ± 6 | 391 ± 108 | <0.05 | |
| Triglycerides (mg/dL) | 52 ± 6 | 67 ± 4 | <0.05 | |
Data are presented as number (%) or mean ± SEM. PB, plaque burden; SBP, systolic blood pressure; DBP, diastolic blood pressure (humans conscious; swine under anesthesia); LVAD, left ventricular assistance device; bG, blood glucose; HbA1c, glycated hemoglobin; HDL, high-density lipoprotein; ACEI, angiotensin converting enzyme inhibitors; ARB, angiotensin II receptor blockers; Ca, calcium. MetS was defined by a blood pressure above 130/85 mm Hg, a fasting blood glucose above 110 mg/dL, an HDL-C level below 40 mg/dL for men or below 50 mg/dL for women, a triglyceride level above 150 mg/dL, and a BMI above 30.0 kg/m2. N.S. = not significant
3.3. Disease severity, arterial structure, and metabolic parameters are correlated to changes in [Ca2+]i handling.
Fig. 2 shows a sample [Ca2+]i tracing from a representative human CSM cell. Table 2 shows greater CAD severity, as measured by the intima/media ratio and percent collagen [22], was significantly correlated to decreased Ca2+ influx through voltage-gated Ca2+ channels and a decreased SR Ca2+ store release. In contrast to those inverse correlations, an increased media area was positively correlated to increased Ca2+ influx and SR Ca2+ store release. Increased vascular calcification was correlated to a decreased Ca2+ influx. Body mass index (BMI) and the number of MetS risk factors present in the patient were correlated to a decreased Ca2+ influx. Dyslipidemia, including increased LDL and total cholesterol levels, was correlated to decreased Ca2+ influx. Age was significantly correlated to an increased SR Ca2+ store release. Correlation graphs are presented in Figs. 3 and 4. Similar correlations on the Ossabaw miniature swine model of MetS and CAD are published [22, 24, 25].
Fig. 2. Sample Ca2+ tracing showing the change in the F340/F380 excitation fluorescence emission ratio from a human CSM cell.
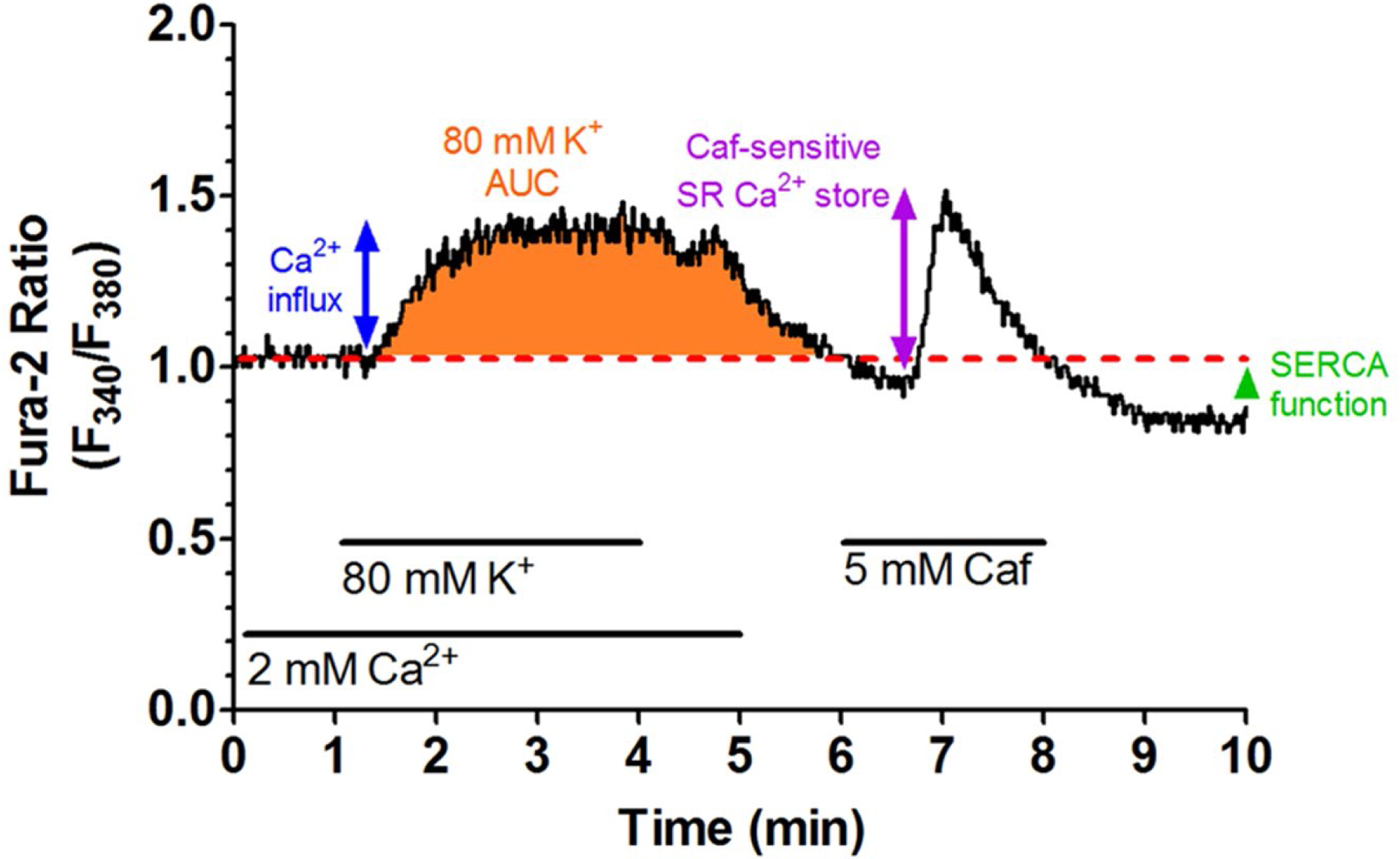
Treatments and duration are indicated by solid lines. Baseline Ca2+ values were established during the first minute in a physiological salt solution (red dashed line). Cell depolarization with an 80 mM K+ solution initiated Ca2+ influx via voltage-gated Ca2+ channels. The height of the Ca2+ influx peak (blue arrow) and the area under the curve (AUC, orange area) were calculated to quantify Ca2+ influx activity. SR Ca2+ store was released by activating ryanodine receptors with 5 mM caffeine (Caf) and was measured by the height of the caffeine-induced peak (purple arrow). The undershoot from baseline was used to measure SERCA activity (green caret)
Table 2.
Linear regression analyses for CSM [Ca2+]i handling measures versus histology measurements and patient parameters
| Humans | Swine | |||
|---|---|---|---|---|
| p | R | p | R | |
| Structural (Histology) Parameters | ||||
| Percent Media vs. | ||||
| Ca2+ Influx | 0.01 | 0.51 | 0.48 | 0.22 |
| 80K AUC | 0.03 | 0.44 | 0.45 | 0.24 |
| SR Store Release | 0.08 | 0.38 | 0.41 | 0.26 |
| Intima/Media Ratio vs. | ||||
| Ca2+ Influx | 0.02 | −0.50 | 0.04 | −0.59 |
| 80K AUC | 0.03 | −0.43 | 0.05 | −0.58 |
| Percent Total Collagen vs. | ||||
| Ca2+ Influx | 0.01 | −0.51 | 0.04 | −0.60 |
| 80K AUC | <0.01 | −0.60 | 0.12 | −0.47 |
| SR Store Release | 0.02 | −0.49 | 0.33 | −0.31 |
| Percent Calcification vs. | ||||
| Ca2+ Influx | 0.02 | −0.48 | 0.96 | −0.01 |
| Patient Clinical Parameters | ||||
| BMI vs. | ||||
| Ca2+ Influx | 0.03 | −0.42 | - | - |
| 80K AUC | 0.01 | −0.52 | - | - |
| Age vs. | ||||
| SR Store Release | 0.02 | 0.47 | 0.76 | 0.10 |
| Number of MetS Risk Factors vs. | ||||
| 80K AUC | 0.01 | −0.51 | - | - |
| LDL Cholesterol vs. | ||||
| Ca2+ Influx | 0.01 | −0.48 | - | - |
| Total Cholesterol vs. | ||||
| Ca2+ Influx | 0.04 | −0.41 | 0.09 | −0.51 |
Fig. 3. Significant correlation of human coronary smooth muscle Ca2+ signaling to histological measures.
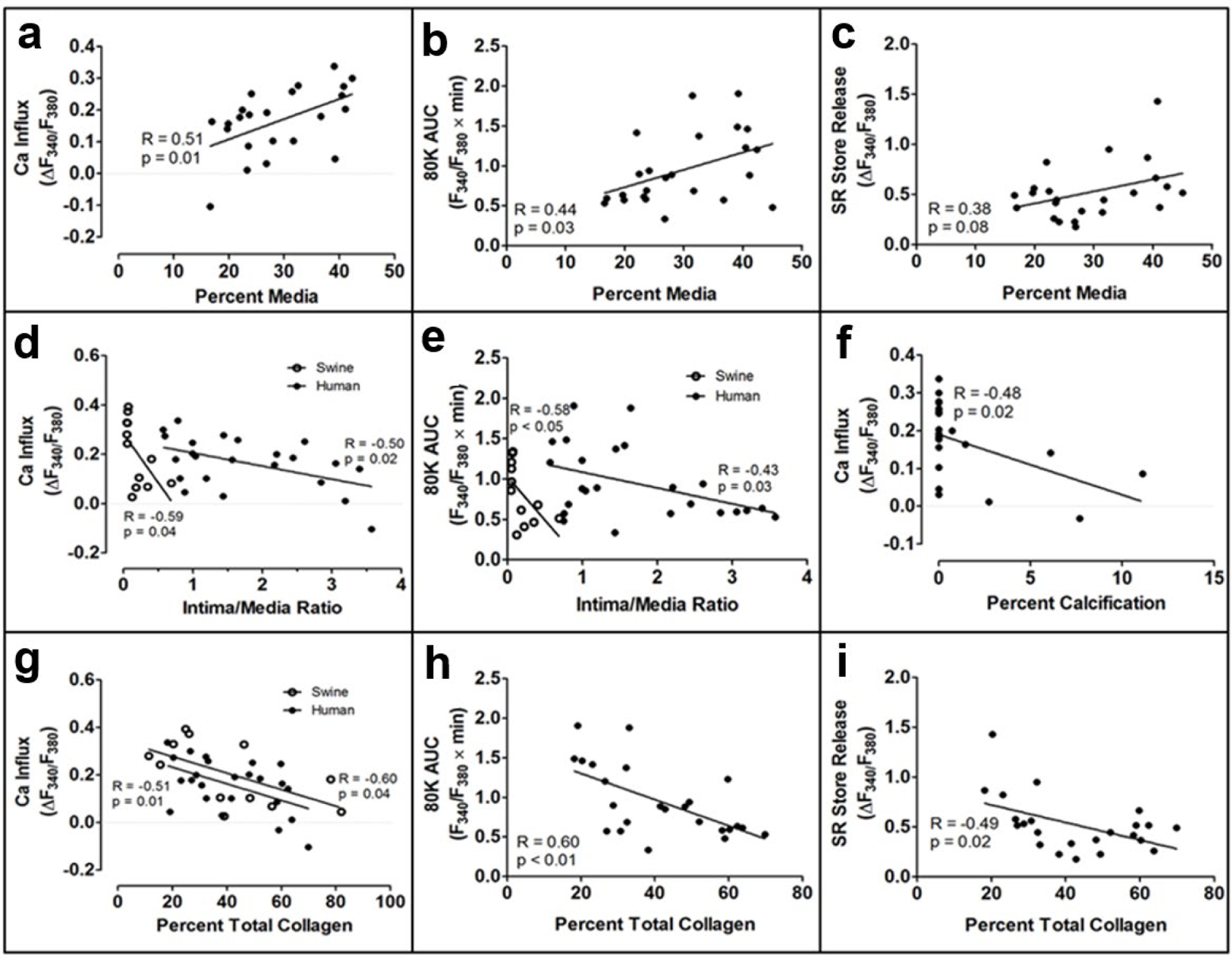
The percent media (a-c), intima/media ratio (d-e), percent calcification (f), and percent collagen (g-i) were significantly correlated to Ca2+ influx (a, d, f, g), area under the 80 mM K+ curve (b, e, h), and SR store release (c and i). Data points for swine (open circles) are included when the correlation was significant, as well (d, e, and g)
Fig. 4. Significant correlation of human coronary smooth muscle Ca2+ signaling to cardiometabolic patient data.
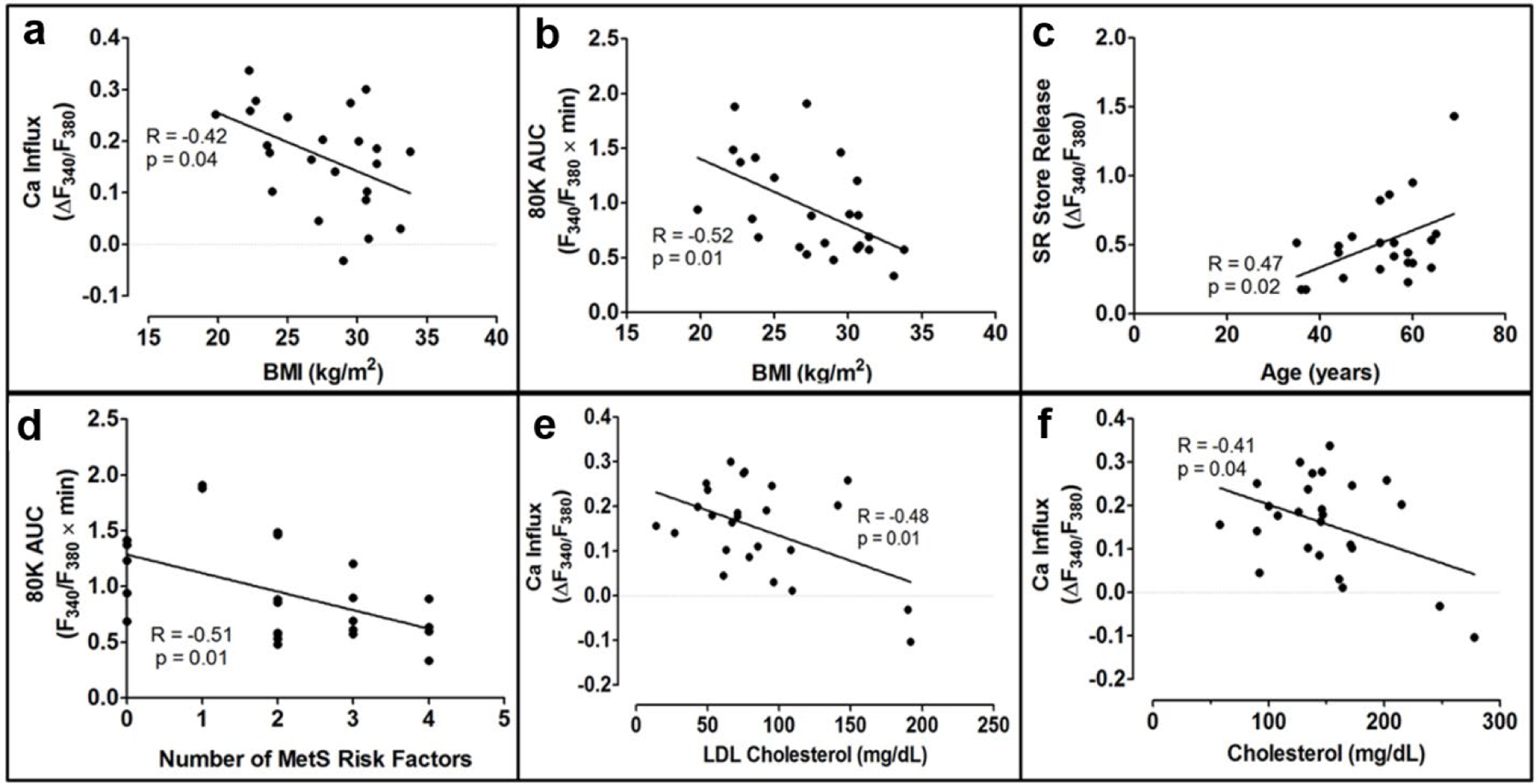
Patient BMI (a and b), age (c), the number of MetS risk factors (d), and dyslipidemia (e-f) were correlated to Ca2+ influx (a, d, and e), area under the 80 mM K+ curve (b and d), and SR store release (c)
3.4. [Ca2+]i handling in Ossabaw swine recapitulates [Ca2+]i handling in human CSM in both health and disease.
Representative single cell tracings of [Ca2+]i are shown a lean swine, MetS swine, human with mild CAD, and human with severe CAD (Fig. 5a). The [Ca2+]i tracing from the human with mild CAD closely resembles the [Ca2+]i from the lean swine, while the [Ca2+]i tracing from the human with severe CAD closely resembles the [Ca2+]i from the MetS swine. Humans with less than 50% and 50–75% plaque burden did not exhibit any differences in [Ca2+]i handling, while humans with greater than 75% plaque burden had a significantly lower Ca2+ influx (Fig. 5b), SR Ca2+ store release (Fig. 5c), and undershoot (Fig. 5d). Swine with MetS-induced CAD showed remarkably similar directional changes, with a lower Ca2+ influx (Fig. 5b), SR Ca2+ store release (Fig. 5c), and undershoot (Fig. 5d) in the MetS group compared to the lean group. This clearly illustrates that similar [Ca2+]i dysregulation is present in CAD in humans and Ossabaw miniature swine.
Fig. 5. Intracellular Ca2+ measures in CSM cells.
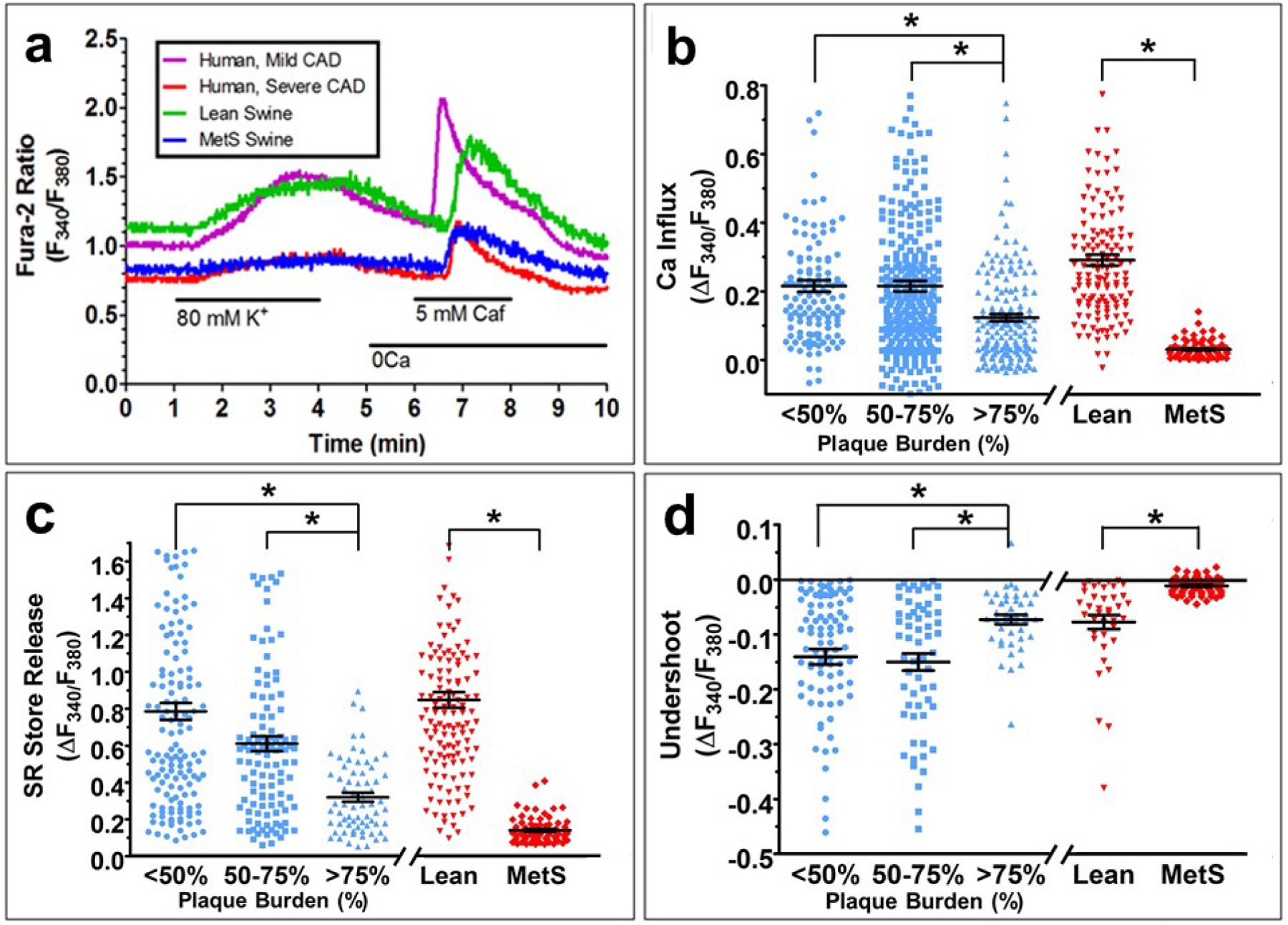
Typical Ca2+ tracings show the change in the F340/F380 excitation fluorescence emission ratio from human and swine CSM cells. There are similarities between humans with mild, non-ischemic CAD and lean swine and between humans with severe, ischemic CAD and MetS swine (a). Mild, non-ischemic CAD was defined by a plaque burden below 50%, while severe, ischemic CAD was defined by a plaque burden of over 75%. Both humans and swine with CAD exhibit decreased Ca2+ influx (b), SR Ca2+ store capacity (c), and SERCA function (d). Group data are plotted as mean ± SE with black lines and bars; individual cells are show in blue symbols for humans and red symbols for swine.
4. Discussion
This study provides insight into [Ca2+]i handling in freshly isolated CSM from explanted hearts of human cardiomyopathy patients. We found that a thickened media layer, which is associated with mild CAD and aging in pigs [24], was correlated to enhanced [Ca2+]i signaling. Advanced CAD progression, as measured by plaque burden and percent collagen, and the number of MetS/cardiometabolic risk factors, BMI, and LDL and total cholesterol levels, are correlated to decreased [Ca2+]i signaling. Increased vascular calcification was accompanied by a decrease in Ca2+ influx. These trends exemplify that [Ca2+]i regulation is compromised in patients with severe, more occlusive CAD and in patients with certain MetS risk factors. By measuring [Ca2+]i from freshly dispersed, non-cultured CSM from explanted hearts of human patients, we provide novel insight into the intricacies of [Ca2+]i dysregulation in diseased human CSM ex vivo and how CAD severity and certain metabolic risk factors correlate with this dysregulation. The [Ca2+]i dysregulation patterns seen in Ossabaw miniature swine with MetS, CAD, and advanced age are consistent with the human data [22, 24, 25], supporting the strong clinical relevance of this large animal model on the cellular Ca2+ signaling level.
Recently, our lab has clarified that CSM Ca2+ dysregulation occurs in a biphasic manner during CAD progression, with increased [Ca2+]i handling in early CAD and decreased [Ca2+]i handling in late CAD [22]. Dysregulation of Ca2+ signaling pathways are associated with CSM dedifferentiation into a synthetic or osteogenic phenotype, which is followed by proliferation, migration to the growing neo-intima, and deposition of hydroxyapatite crystals in the extracellular matrix leading to vascular calcification [33]. Often, this change in phenotype occurs due to CSM adaptations to changes in the extracellular environment, such as increased reactive oxygen species and dyslipidemia [34–36].
Heart failure is a complex, heterogeneous disease with many different etiologies, risk factors, and pathophysiologies. CAD is the leading cause of heart failure and progression of CAD is related to progression of left ventricular dysfunction, a common characteristic of heart failure [37]. In the current study we used histology obtained from the proximal segment of an epicardial coronary artery from patients with cardiomyopathy to classify their coronary disease state. The argument could be made that this stratification reduces the differences between groups. However, while the clinical diagnosis of ischemic cardiomyopathy is an important predictor of 5-year mortality, the extent of CAD is a much better predictor of survival in heart failure patients [38]. Histology can be considered a “snapshot” of one specific cross-section of the artery, not precisely indicative of total artery health. However, as both humans and Ossabaw swine with metabolic syndrome develop diffuse coronary plaque throughout the proximal, middle, and distal sections of the artery [21], the histology might be considered a representative snapshot of overall coronary health and plaque development. Therefore, basing human cohorts on coronary plaque burden seems to be a reasonable stratification strategy and the relative benefits and weaknesses of using clinical diagnosis versus coronary plaque burden should be explored further to identify the more sound stratification strategy.
Patients in this study had different cardiac and CAD severities and similar clinical characteristics. It is important to note, however, that the patients have similarly increased cardiometabolic risk above healthy subjects. There is enough variability within all the groups that enabled excellent regression analysis. This allowed us to test for associations between cardiometabolic risk factors and [Ca2+]i regulation. Additionally, we show that the number of MetS risk factors, as opposed to the diagnosis of MetS itself, affects [Ca2+]i regulation. This is consistent with several studies on MetS and CAD [39–41]. One study found that Japanese patients with either dilated non-ischemic cardiomyopathy or ischemic cardiomyopathy have an incidence of MetS twice as frequently as the general population and have comparable metabolic components, indicating that the risk factors associated with MetS influence the etiology of both ischemic and non-ischemic cardiomyopathy [42].
It is important to note that over half of the patients in this study had left ventricular assist device (LVAD) support. LVADs deliver blood continuously from the left ventricle to the aorta, mechanically unloading the left ventricle and restoring total systemic blood pressure [43]. The continuous blood flow produced by LVAD support (as opposed to pulsatile flow accomplished with a native heartbeat) has several implications for regional flow dynamics, including coronary flow. In fact, implantation of a continuous flow LVAD is associated with a decrease in total coronary blood flow in a swine model [44]. While it appears that coronary artery endothelial function is not impaired by long-term LVAD support [45], the coronary arteries of human patients develop remodeling with increased fibrosis [46]. Intracellular Ca2+ changes in coronary smooth muscle usually accompany structural changes [16]. While studies have shown that LVAD support can increase Ca2+ cycling in cardiomyocytes [47], there have been no studies investigating coronary smooth muscle [Ca2+]i handling in LVAD patients. Our study was not statistically powered to resolve a difference between patients with or without LVAD support. Therefore, it is difficult to determine exactly how LVAD support would affect [Ca2+]i in coronary smooth muscle cells. This would be a promising research direction for future investigations.
Although humans and swine cannot and should not be directly compared, it is apparent that, while humans in general have a greater plaque burden than swine (Fig. 1m), swine exhibit more highly altered [Ca2+]i handling in all the measured parameters (Fig. 5b-d). This may be due to the duration of the disease and severity of the risk factors. Atherosclerosis is a chronic disease occurring over several decades in the human population. Conversely, the Ossabaw swine with MetS-induced CAD are on a diet specifically designed to expedite plaque development over a time span of only 11 months, which would only be 7–10 years of a human lifespan. This could lead to more rapid changes in the cellular milieu, potentially causing more extreme adaptations in CSM leading to more severe [Ca2+]i dysregulation, as compared to the slower, more chronic condition in humans. This also supports the concept that [Ca2+]i dysregulation occurs before and perhaps triggers the structural changes in the artery.
There are limitations of this study. First, hearts from healthy humans without heart failure were not included due to scarcity of tissue. Therefore, this study indirectly compares arteries from pathological human hearts to physiologically healthy, lean swine. Our coronary plaque-based stratification of the severity of disease in the humans is not ideal. However, as we are concerned with relationships in [Ca2+]i handling as a function of cardiometabolic risk and are not comparing these two species directly, we can still extrapolate from the data that the directional changes in [Ca2+]i dysregulation patterns from a state of mild or no disease to a state of greater disease is maintained in both species. Finally, the major weakness of this study is the relatively small human sample set, which affects the generalizability of these results. Future studies should expand on these findings by including a greater number of patients.
An adequate animal model for macrovascular coronary artery disease and subsequent heart failure is of utmost importance, as a better understanding of the pathophysiology of cardiomyopathies could lead to the development of more effective heart failure therapeutics. This report is the first characterization of [Ca2+]i dysregulation in freshly harvested CSM from explanted human hearts. The data strongly support the clinical relevance of the Ossabaw miniature swine model of MetS and CAD. A reliable, clinically relevant animal model that recapitulates human disease on a cellular level provides far more confidence of the translatability of the data.
Acknowledgements
The authors acknowledge the Indiana University School of Medicine Histology Core and Dr. Keith Condon for processing the histology and use of their equipment. Jill K. Badin’s Ph.D. thesis dated August 2019 contained some of the data in this manuscript and can be found at: https://scholarworks.iupui.edu/bitstream/handle/1805/20549/Badin_iupui_0104D_10379.pdf?isAllowed=y&sequence=1
Funding:
This research was funded by National Institutes of Health HL125385, P30 DK097512, the Joshua Diabetes Research Fund, and the Indiana University School of Medicine Center of Excellence in Cardiovascular Research.
Abbreviations
- BMI
Body mass index
- [Ca2+]i
Intracellular free calcium
- CSM
Coronary smooth muscle
- CAD
Coronary artery disease
- LDL
Low-density lipoprotein
- LVAD
Left ventricular assist device
- MetS
Metabolic syndrome
- SERCA
Sarco-endoplasmic reticulum Ca2+ ATPase
- SR
Sarcoplasmic reticulum
Footnotes
This is the author's manuscript of the article published in final edited form as:
Badin, J. K., Eggenberger, C., Rodenbeck, S. D., Hashmi, Z. A., Wang, I.-W., Garcia, J. P., Alloosh, M., & Sturek, M. (2022). Intracellular Ca2+ Dysregulation in Coronary Smooth Muscle Is Similar in Coronary Disease of Humans and Ossabaw Miniature Swine. Journal of Cardiovascular Translational Research, 15(1), 167–178. https://doi.org/10.1007/s12265-021-10153-5
Conflict of interest: The authors declared that they do not have anything to disclose regarding conflict of interest with respect to this manuscript.
Compliance with Ethical Standards
Ethical approval for use of animals: All institutional and national guidelines for the care and use of laboratory animals were followed and approved by the Institutional Animal Care and Use Committee at the Indiana University School of Medicine..
Ethical approval for human subjects: This article does not contain any studies with human participants performed by any of the authors, as approved by exemption, per the use of discarded human tissue, by the Indiana University Institutional Review Board.
References
- 1.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. (2018). Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation, 137(12), e67–e492. doi 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 2.Wexler RK, Elton T, Pleister A, Feldman D (2009). Cardiomyopathy: an overview. American Family Physician, 79(9), 778–784. doi [PMC free article] [PubMed] [Google Scholar]
- 3.Libby P, Ridker PM, Hansson GK (2011). Progress and challenges in translating the biology of atherosclerosis. Nature, 473(7347), 317–325. doi 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 4.Owens GK, Kumar MS, Wamhoff BR (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiological Reviews, 84(3), 767–801. doi 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 5.van der Schaaf RJ, Timmer JR, Ottervanger JP, Hoorntje JC, de Boer MJ, Suryapranata H, et al. (2006). Long-term impact of multivessel disease on cause-specific mortality after ST elevation myocardial infarction treated with reperfusion therapy. Heart, 92(12), 1760–1763. doi 10.1136/hrt.2005.086058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. (2016). Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation, 133(4), e38–60. doi 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 7.Karwowski W, Naumnik B, Szczepanski M, Mysliwiec M (2012). The mechanism of vascular calcification - a systematic review. Medical Science Monitor, 18(1), RA1–11. doi [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hill-Eubanks DC, Werner ME, Heppner TJ, Nelson MT (2011). Calcium signaling in smooth muscle. Cold Spring Harbor Perspectives in Biology, 3(9), a004549. doi 10.1101/cshperspect.a004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wamhoff BR, Bowles DK, McDonald OG, Sinha S, Somlyo AP, Somlyo AV, et al. (2004). L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a rho kinase/myocardin/SRF-dependent mechanism. Circulation Research, 95(4), 406–414. doi 10.1161/01.RES.0000138582.36921.9e. [DOI] [PubMed] [Google Scholar]
- 10.Lundberg MS, Curto KA, Bilato C, Monticone RE, Crow MT (1998). Regulation of vascular smooth muscle migration by mitogen-activated protein kinase and calcium/calmodulin-dependent protein kinase II signaling pathways. J Mol Cell Cardiol, 30(11), 2377–2389. doi 10.1006/jmcc.1998.0795. [DOI] [PubMed] [Google Scholar]
- 11.Pauly RR, Bilato C, Sollott SJ, Monticone R, Kelly PT, Lakatta EG, et al. (1995). Role of calcium/calmodulin-dependent protein kinase II in the regulation of vascular smooth muscle cell migration. Circulation, 91(4), 1107–1115. doi 10.1161/01.cir.91.4.1107. [DOI] [PubMed] [Google Scholar]
- 12.House SJ, Potier M, Bisaillon J, Singer HA, Trebak M (2008). The non-excitable smooth muscle: calcium signaling and phenotypic switching during vascular disease. Pflugers Arch, 456(5), 769–785. doi 10.1007/s00424-008-0491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kruse HJ, Bauriedel G, Heimerl J, Hofling B, Weber PC (1994). Role of L-type calcium channels on stimulated calcium influx and on proliferative activity of human coronary smooth muscle cells. Journal of Cardiovascular Pharmacology, 24(2), 328–335. doi [PubMed] [Google Scholar]
- 14.Nilsson J, Sjolund M, Palmberg L, Von Euler AM, Jonzon B, Thyberg J (1985). The calcium antagonist nifedipine inhibits arterial smooth muscle cell proliferation. Atherosclerosis, 58(1–3), 109–122. doi 10.1016/0021-9150(85)90059-0. [DOI] [PubMed] [Google Scholar]
- 15.Jiang H, Stephens NL (1994). Calcium and smooth muscle contraction. Molecular and Cellular Biochemistry, 135(1), 1–9. doi 10.1007/BF00925956. [DOI] [PubMed] [Google Scholar]
- 16.Sturek M (2011). Ca2+ regulatory mechanisms of exercise protection against coronary artery disease in metabolic syndrome and diabetes. Journal of Applied Physiology, 111(2), 573–586. doi 10.1152/japplphysiol.00373.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witczak CA, Wamhoff BR, Sturek M (2006). Exercise training prevents Ca2+ dysregulation in coronary smooth muscle from diabetic dyslipidemic Yucatan swine. Journal of Applied Physiology, 101(3), 752–762. doi 10.1152/japplphysiol.00235.2006. [DOI] [PubMed] [Google Scholar]
- 18.Berwick ZC, Dick GM, OĽeary HA, Bender SB, Goodwill AG, Moberly SP, et al. (2013). Contribution of electromechanical coupling between Kv and Ca v1.2 channels to coronary dysfunction in obesity. Basic Res Cardiol, 108(5), 370. doi 10.1007/s00395-013-0370-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards JM, Neeb ZP, Alloosh MA, Long X, Bratz IN, Peller CR, et al. (2010). Exercise training decreases store-operated Ca2+ entry associated with metabolic syndrome and coronary atherosclerosis. Cardiovasc Res, 85(3), 631–640. doi 10.1093/cvr/cvp308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill BJ, Price EM, Dixon JL, Sturek M (2003). Increased calcium buffering in coronary smooth muscle cells from diabetic dyslipidemic pigs. Atherosclerosis, 167(1), 15–23. doi 10.1016/s0021-9150(02)00381-7. [DOI] [PubMed] [Google Scholar]
- 21.Neeb ZP, Edwards JM, Alloosh M, Long X, Mokelke EA, Sturek M (2010). Metabolic syndrome and coronary artery disease in Ossabaw compared with Yucatan swine. Comparative Medicine, 60(4), 300–315. doi [PMC free article] [PubMed] [Google Scholar]
- 22.McKenney-Drake ML, Rodenbeck SD, Owen MK, Schultz KA, Alloosh M, Tune JD, et al. (2016). Biphasic alterations in coronary smooth muscle Ca2+ regulation in a repeat cross-sectional study of coronary artery disease severity in metabolic syndrome. Atherosclerosis, 249(1–9. doi 10.1016/j.atherosclerosis.2016.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sturek M, Alloosh M, Sellke FW (2020). Swine disease models for optimal vascular engineering. Annu Rev Biomed Eng, 22(25–49. doi 10.1146/annurev-bioeng-082919-053009. [DOI] [PubMed] [Google Scholar]
- 24.Badin JK, Bruning RS, Sturek M (2018). Effect of metabolic syndrome and aging on Ca2+ dysfunction in coronary smooth muscle and coronary artery disease severity in Ossabaw miniature swine. Experimental Gerontology, 108(247–255. doi 10.1016/j.exger.2018.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Badin JK, Kole A, Stivers B, Progar V, Pareddy A, Alloosh M, et al. (2018). Alloxan-induced diabetes exacerbates coronary atherosclerosis and calcification in Ossabaw miniature swine with metabolic syndrome. Journal of Translational Medicine, 16(1), 58. doi 10.1186/s12967-018-1431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Institute for Laboratory Animal Research. 2010. Guide for the care and use of laboratory animals Washington, D.C.: National Academy Press [Google Scholar]
- 27.AVMA Panel on Euthanasia.American Veterinary Medical Association. (2001). 2000 Report of the AVMA panel on euthanasia. JAVMA, 218(669–696. doi [DOI] [PubMed] [Google Scholar]
- 28.Grynkiewicz G, Poenie M, Tsien RY (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J.Biol.Chem, 260(3440–3450. doi [PubMed] [Google Scholar]
- 29.Oliver AE, Baker GA, Fugate RD, Tablin F, Crowe JH (2000). Effects of temperature on calcium-sensitive fluorescent probes. Biophysical Journal, 78(4), 2116–2126. doi 10.1016/S0006-3495(00)76758-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dineen SL, McKenney ML, Bell LN, Fullenkamp AM, Schultz KA, Alloosh M, et al. (2015). Metabolic Syndrome Abolishes Glucagon-Like Peptide 1 Receptor Agonist Stimulation of SERCA in Coronary Smooth Muscle. Diabetes, 64(9), 3321–3327. doi 10.2337/db14-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKenney-Drake ML, Territo PR, Salavati A, Houshmand S, Persohn S, Liang Y, et al. (2016). 18F-NaF PET imaging of early coronary artery calcification. JACC: Cardiovascular Imaging, 9(627–628. doi 10.1016/j.jcmg.2015.02.026. [DOI] [PubMed] [Google Scholar]
- 32.Rodenbeck SD, Zarse CA, McKenney-Drake ML, Bruning RS, Sturek M, Chen NX, et al. (2017). Intracellular calcium increases in vascular smooth muscle cells with progression of chronic kidney disease in a rat model. Nephrology, Dialysis, Transplantation, 32(3), 450–458. doi 10.1093/ndt/gfw274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bobe R, Hadri L, Lopez JJ, Sassi Y, Atassi F, Karakikes I, et al. (2011). SERCA2a controls the mode of agonist-induced intracellular Ca2+ signal, transcription factor NFAT and proliferation in human vascular smooth muscle cells. J Mol Cell Cardiol, 50(4), 621–633. doi 10.1016/j.yjmcc.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karagiannis GS, Weile J, Bader GD, Minta J (2013). Integrative pathway dissection of molecular mechanisms of moxLDL-induced vascular smooth muscle phenotype transformation. BMC Cardiovascular Disorders, 13(4. doi 10.1186/1471-2261-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spillmann F, Miteva K, Pieske B, Tschope C, Van Linthout S (2015). High-density lipoproteins reduce endothelial-to-mesenchymal transition. Arteriosclerosis, Thrombosis, and Vascular Biology, 35(8), 1774–1777. doi 10.1161/ATVBAHA.115.305887. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Ji L, Jiang R, Zheng L, Liu D (2014). Oxidized high-density lipoprotein induces the proliferation and migration of vascular smooth muscle cells by promoting the production of ROS. J Atheroscler Thromb, 21(3), 204–216. doi 10.5551/jat.19448. [DOI] [PubMed] [Google Scholar]
- 37.Gheorghiade M, Bonow RO (1998). Chronic heart failure in the United States: a manifestation of coronary artery disease. Circulation, 97(3), 282–289. doi 10.1161/01.cir.97.3.282. [DOI] [PubMed] [Google Scholar]
- 38.Bart BA, Shaw LK, McCants CB Jr., Fortin DF, Lee KL, Califf RM, et al. (1997). Clinical determinants of mortality in patients with angiographically diagnosed ischemic or nonischemic cardiomyopathy. Journal of the American College of Cardiology, 30(4), 1002–1008. doi 10.1016/s0735-1097(97)00235-0. [DOI] [PubMed] [Google Scholar]
- 39.Kim JY, Mun HS, Lee BK, Yoon SB, Choi EY, Min PK, et al. (2010). Impact of metabolic syndrome and its individual components on the presence and severity of angiographic coronary artery disease. Yonsei Medical Journal, 51(5), 676–682. doi 10.3349/ymj.2010.51.5.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gui MH, Ling Y, Liu L, Jiang JJ, Li XY, Gao X (2017). Effect of Metabolic Syndrome Score, Metabolic Syndrome, and Its Individual Components on the Prevalence and Severity of Angiographic Coronary Artery Disease. Chinese Medical Journal (Engl.), 130(6), 669–677. doi 10.4103/0366-6999.201611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahmadi A, Leipsic J, Feuchtner G, Gransar H, Kalra D, Heo R, et al. (2015). Is metabolic syndrome predictive of prevalence, extent, and risk of coronary artery disease beyond its components? Results from the multinational coronary CT angiography evaluation for clinical outcome: an international multicenter registry (CONFIRM). PLoS One, 10(3), e0118998. doi 10.1371/journal.pone.0118998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miura Y, Fukumoto Y, Shiba N, Miura T, Shimada K, Iwama Y, et al. (2010). Prevalence and clinical implication of metabolic syndrome in chronic heart failure. Circulation Journal, 74(12), 2612–2621. doi 10.1253/circj.cj-10-0677. [DOI] [PubMed] [Google Scholar]
- 43.Klotz S, Jan Danser AH, Burkhoff D (2008). Impact of left ventricular assist device (LVAD) support on the cardiac reverse remodeling process. Progress in Biophysics and Molecular Biology, 97(2–3), 479–496. doi 10.1016/j.pbiomolbio.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 44.Ootaki Y, Kamohara K, Akiyama M, Zahr F, Kopcak MW Jr., Dessoffy R, et al. (2005). Phasic coronary blood flow pattern during a continuous flow left ventricular assist support. European Journal of Cardio-Thoracic Surgery, 28(5), 711–716. doi 10.1016/j.ejcts.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Symons JD, Deeter L, Deeter N, Bonn T, Cho JM, Ferrin P, et al. (2019). Effect of Continuous-Flow Left Ventricular Assist Device Support on Coronary Artery Endothelial Function in Ischemic and Nonischemic Cardiomyopathy. Circulation: Heart Failure, 12(8), e006085. doi 10.1161/CIRCHEARTFAILURE.119.006085. [DOI] [PubMed] [Google Scholar]
- 46.Ambardekar AV, Weiser-Evans MCM, Li M, Purohit SN, Aftab M, Reece TB, et al. (2018). Coronary Artery Remodeling and Fibrosis With Continuous-Flow Left Ventricular Assist Device Support. Circulation: Heart Failure, 11(5), e004491. doi 10.1161/CIRCHEARTFAILURE.117.004491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei X, Li T, Hagen B, Zhang P, Sanchez PG, Williams K, et al. (2013). Short-term mechanical unloading with left ventricular assist devices after acute myocardial infarction conserves calcium cycling and improves heart function. JACC: Cardiovascular Interventions, 6(4), 406–415. doi 10.1016/j.jcin.2012.12.122. [DOI] [PMC free article] [PubMed] [Google Scholar]