

ABSTRACT
STING1 (stimulator of interferon response cGAMP interactor 1) plays an essential role in immune responses for virus inhibition via inducing the production of type I interferon, inflammatory factors and macroautophagy/autophagy. In this study, we found that STING1 activation could induce not only canonical autophagy but also non-canonical autophagy (NCA) which is independent of the ULK1 or BECN1 complexes to form MAP1LC3/LC3-positive structures. Whether STING1-induced NCA has similar characters and physiological functions to canonical autophagy is totally unknown. Different from canonical autophagy, NCA could increase single-membrane structures and failed to degrade long-lived proteins, and could be strongly suppressed by interrupting vacuolar-type H+-translocating ATPase (V-ATPase) activity. Importantly, STING1-induced NCA could effectively inhibit DNA virus HSV-1 in cell model. Moreover, STING1 [1–340], a STING1 mutant lacking immunity and inflammatory response due to deletion of the tail end of STING1, could degrade virus through NCA alone, suggesting that the antiviral effect of activated STING1 could be separately mediated by inherent immunity, canonical autophagy, and NCA. In addition, the translocation and dimerization of STING1 do not rely on its immunity function and autophagy pathway. Similar to canonical autophagy, LC3-positive structures of NCA induced by STING1 could finally fuse with lysosomes, and the degradation of HSV-1 could be reverted by inhibition of lysosome function, suggesting that the elimination of DNA virus via NCA still requires the lysosome pathway. Collectively, we proved that besides its classical immunity function and canonical autophagy pathway, STING1-induced NCA is also an efficient antiviral pathway for the host cell.
Abbreviations: ATG: autophagy related; Baf: bafilomycin A1; CASM: conjugation of LC3 to a single membrane; CGAS: cyclic GMP-AMP synthase; cGAMP: cyclic GMP-AMP; CQ: chloroquine; CTD: C-terminal domain; CTT: C-terminal tail; ER: endoplasmic reticulum; ERGIC: ER-Golgi intermediate compartment; HSV-1: herpes simplex virus 1; IRF3: interferon regulatory factor 3; IFNs: interferons; LAMP1: lysosomal associated membrane protein 1; LAP: LC3-associated phagocytosis; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MOI: multiplicity of infection; RB1CC1/FIP200: RB1 inducible coiled-coil 1; STING1: stimulator of interferon response cGAMP interactor 1; TBK1: TANK binding kinase 1; TGOLN2/TGN46: trans-golgi network protein 2; ULK1: unc-51 like autophagy activating kinase 1; V-ATPase: vacuolar-type H+-translocating ATPase; VSV: vesicular stomatitis virus
KEYWORDS: autophagy, HSV-1, inherent immunity, lysosome degradation, noncanonical autophagy, STING1
Introduction
Innate immunity constitutes the first line for defending host cells against invading microbial pathogens, which can play a wide range of antiviral responses and release pro-inflammatory signals [1,2]. Through pattern recognition receptors, innate immunity can quickly perceive the danger signal from pathogenic microorganisms, activate adaptive immunity, and start the immune response for host organism. STING1 (stimulator of interferon response cGAMP interactor 1) can initiate the recognition of exogenous DNA introduced by pathogen infection or endogenous DNA abnormally released by host via upstream kinase CGAS (cyclic GMP-AMP synthase) [3,4]. After activation, the conformation of STING1 changes and transports from the endoplasmic reticulum (ER) to endoplasmic reticulum-Golgi intermediate compartment (ERGIC) and Golgi membrane [5]. In this process, STING1 recruits and phosphorylates TBK1 (TANK binding kinase 1), resulting in the phosphorylation of downstream IRF3 (interferon regulatory factor 3), and then the phosphorylated IRF3 will be dimerized and translocate into the nucleus, inducing the production of type I interferon, and finally activate the innate immune response to resist pathogen invasion [6,7]. So the innate immunity mediated by STING1 constitutes the body’s external defense against the threat of pathogens.
STING1 protein is composed of 379 amino acids, mainly composed of four transmembrane domains (STING1 [1-149]) that enable STING1 to locate on ER or mitochondria, and cytoplasmic oriented C-terminal functional domain (STING1[CTD]) that mediates homodimerization for STING1 activation via binding to the cGAMP [8]. Cytoplasmic C-terminal tail (STING1[CTT]/STING1[340–379]) of STING1[CTD] binds and combines with downstream TBK1 and IRF3 to ensure the immune efficacy and releases type I interferon and inflammatory factors, thus effectively inhibits pathogens [9,10]. Besides the above antiviral response pathway, more and more studies have shown that STING1 could also effectively inhibit DNA viral infection through autophagy which is independent of immunity and inflammatory function [11,12]. In fact, HSV-1 has evolved different mechanisms to evade autophagy, but how STING1 activation initiates autophagy and whether STING1-induced autophagy or autophagy-related process contributes to HSV-1 inhibition has not been fully elaborated.
Macroautophagy/autophagy is an evolutionally conserved catabolic process of cells, and is an important homeostatic mechanism involving the degradation of long-lived proteins and the formation of microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3) positive double membrane vesicles called autophagosomes, which sequester damaged organelles, protein aggregates, and invading intracellular pathogens for degradation [13]. Conserved from yeast to human, canonical autophagy takes place through a series of steps including vesicle initiation which mainly depends on the ULK1 (unc-51 like autophagy activating kinase 1) complex, nucleation which depends on the BECN1 (beclin 1) complex, ATG9 and WIPIs, expansion which mainly depends on ATG12–ATG5 and LC3–PE/LC3-II conjugation systems, closure which mainly depends on RB1CC1/FIP200 (RB1 inducible coiled-coil 1) and the endosomal sorting complex required for transport (ESCRT), followed by autophagosomes fusion with lysosomes for cargo degradation [14]. Canonical autophagy induction via STING1 is a primordial function of the CGAS pathway which could effectively restrain HSV-1 virus [11,12]. However, the specific mechanism of STING1 mediated autophagy in cell internal defense is still very limited.
Recently, noncanonical autophagy (NCA) has also been reported when STING1 is activated [12,15]. NCA usually does not require the participation of some ATG genes, but can still normally recruit LC3 molecules to various vacuoles [16], such as LC3-associated phagocytosis (LAP) [17], LC3-associated endocytosis (LANDO) [18], and entosis [19]. In the process of NCA, a parallel LC3 lipid pathway involves the conjugation of LC3 to a single membrane (CASM) in the lysosome chamber. Therefore, NCA is also defined as a CASM process [20]. Despite evidence showing that xenophagy and LAP, two special forms of autophagy, could effectively phagocytize invading pathogens and degrade them by fusing with lysosomes in an LC3-dependent manner [17,21]. Whether STING1-induced NCA can resist HSV-1 virus as canonical autophagy is totally unknown, and the physiological function of STING1-induced NCA requires further exploration.
It is unclear whether STING1-induced NCA has the similar function like canonical autophagy which requires the formation of double-layer autophagosomes and fusion with lysosomes to realize the degradation and recycling of long-lived proteins and intracellular organelles [22]. In addition, we and other groups have reported that vacuolar-type H+-translocating ATPase (V-ATPase) inhibitor bafilomycin A1 (Baf), which originally blocks canonical autophagy and increase the level of LC3-II due to lysosome dysfunction, could significantly reduce the level of LC3-II in NCA induced by chemical AMDE-1 (autophagy modulator with dual effects) and osmotic stress agent chloroquine (CQ) [23,24]. Moreover, Xu, et al. found that ADP ribosylation modification of V-ATPase with SopF could effectively inhibit NCA as well [25,26]. Although V-ATPase is a universal regulator of LAP and NCA [11,15,27], whether V-ATPase is also crucial for STING1-induced NCA remains a question.
In this study, we found that LC3 lipidation could be induced by STING1 or STING1 truncation without innate immune region STING1 [1-340] in ULK1 complex and BECN1 complex deficient cells. In addition, STING1-induced NCA did not affect the translocation and dimerization of STING1, but lost the capability to degrade the long-lived proteins and the formation of double-membrane structures. More importantly, STING1 could effectively suppress HSV-1 virus through NCA alone which was independent of its immunity and inflammatory functions. Such antiviral response through NCA alone also relied on lysosome pathway for virus degradation. This study explored a novel physiological function and laid a foundation for the subsequent antiviral mechanism of NCA induced by STING1 activation.
Results
Activation of STING1 can induce autophagy
In both HeLa and HEK-293A cells, the index LC3-II of autophagy level was significantly increased after transient transfection of HA-STING1 plasmids (Figure 1a and S1A). The level of LC3-II accumulated by the lysosomal inhibitor CQ was further increased after the combination of STING1 overexpression or a specific agonist of STING1 cGAMP (Figure 1b and S1B). At the same time, the increase level of p-TBK1 indicated that STING1 was really activated (Figure 1b and S1B). Moreover, autophagy flux assay using GFP-LC3 HeLa or MEFs also confirmed that overexpression of STING1 could induce autophagy (Figure 1c and S1C). Besides, plasmids harboring full-length STING1 (STING1[FL]), N-terminal domain 1–340 of STING1 lacking immunity function segment (STING1 [1-340]), C-terminal domain segments (149–379) of STING1 facing cytoplasmic functional regions (STING1[CTD]), and four transmembrane domains of STING1 (STING1 [1-149]) were constructed. We found that the activation of STING1 by cGAMP or DNA virus HSV-1 could further induce LC3-II of DLD1 cells which could express the C-terminal domain fragment of STING1, indicating that autophagy could be effectively induced once CTD functional domain was contained (Figure 1d,e). Similarly, GFP-RFP-LC3 was applied and found that like canonical autophagy inducer torin1 instead of STING1 [1-149], STING1[FL], STING1 [1-340], and STING1[CTD] showed significant autophagy induction activity (Figure 1f).
Figure 1.
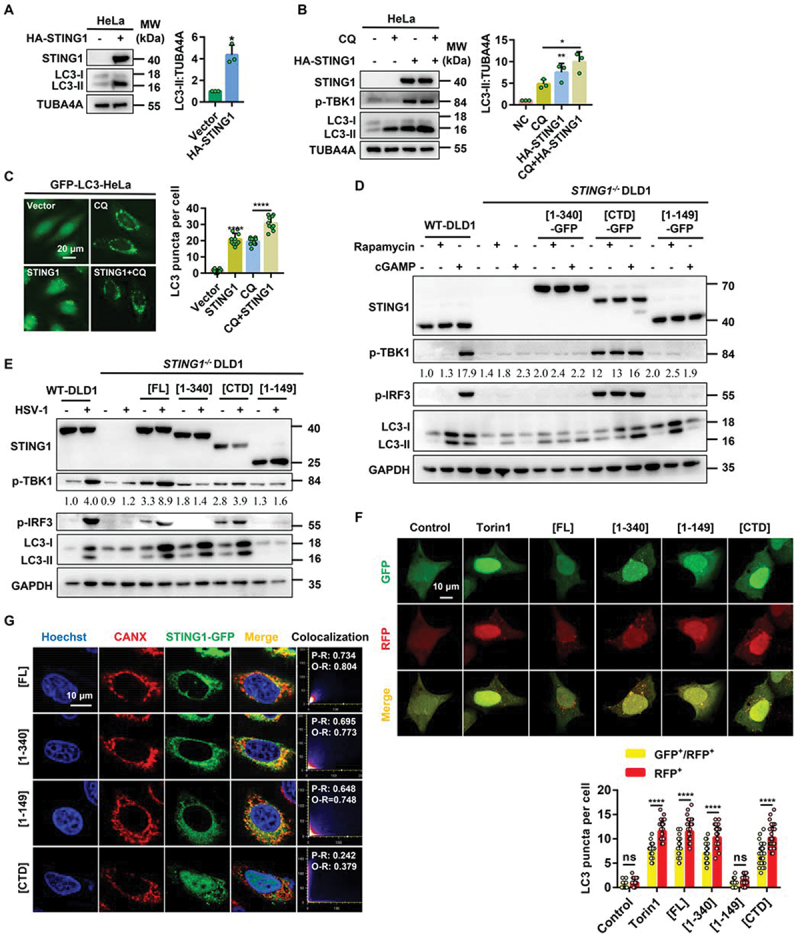
STING1 overexpression and cGAMP induce autophagy. (a) Immunoblot analysis of LC3-ΙΙ levels in HeLa cells that were transfected with vector or HA-STING1 (0.8 µg/mL) for 24 h. (b) Immunoblot analysis of LC3-ΙΙ levels in HeLa cells that were transfected with vector or HA-STING1 with or without CQ treatment at a final concentration of 40 µM. (c) GFP-LC3 puncta formation was analyzed in HeLa cells stably expressing GFP-LC3 that transfected with empty vector or STING1 plasmid with or without CQ (40 µM) treated for 6 h. (d) Western blotting of cell lysates from wild type and STING1-deficient DLD1 cells were transfected with full-length STING1 (STING1[FL]-GFP) or the indicated truncated plasmids labeled with GFP-tag and treated with rapamycin (10 µM) or cGAMP (1 µM) for 6 h. The ratio of p-TBK1:GAPDH was calculated and labeled. (e) Immunoblot analysis of LC3-ΙΙ levels in WT and STING1−/− DLD1 cells that transfected with STING1 or its truncated mutants (STING1 [1-340], STING1[CTD], STING1 [1-149]) with or without HSV-1-GFP virus infected at a multiplicity of infection (MOI) of 5 for 6 h. The ratio of p-TBK1:GAPDH was calculated and labeled. (f) HEK-293A cells stably expressing GFP-RFP-LC3 were treated with 2 μM of torin1 for 6 h or transiently transfected with STING1[FL] or the indicated truncated plasmids. After 24 h expression, cells were harvested and re-seeded on confocal culture dish. The colocalization of GFP and RFP puncta was examined and quantified. Bar: 10 μm. At least 50 cells were counted from each group. (g) WT HeLa cells stably expressing GFP-tagged STING1 or its truncated mutants (STING1 [1-340], STING1[CTD], and STING1 [1-149]) were fixed and stained with marker antibody for ER (CANX [calnexin]) and images were then captured by confocal microscopy. Scale bar: 10 µm. The degree of colocalization for STING1 and CANX was quantified by Pearson’s and overlap coefficient by ImageJ.
*P<0.05, **P<0.01, ***P<0.001, ns, not significant.
When STING1 is activated by cGAMP, it will transfer to the Golgi apparatus from the ER, and finally reach the peripheral body of the nucleus to play inherent function [7]. STING1[FL], STING1 [1-340], and STING1 [1-149] could locate on the ER and mitochondria because they all contained four transmembrane domains, while STING1[CTD] without the four transmembrane domain dispersed in the cytoplasm (Figure 1g and S1D). These data demonstrated that activation of STING1 could induce autophagy, but the four-layer transmembrane domains of STING1 [1-149] failed to induce autophagy.
Activation of STING1 can also induce NCA
As a canonical autophagy inducer, torin1 failed to induce autophagy in RB1CC1/FIP200-, ATG13-, and ATG9A-deficient cells. Similar to AMDE-1, a well-known chemical inducer for NCA [23], activation of STING1 by gene overexpression could increase the level of LC3-II in the above cells (Figure 2a–d and S2A-B). Although CQ is usually used as a lysosome inhibitor, higher dose with longer treatment of CQ could also induce NCA due to osmotic imbalance [24]. So the increase of LC3-II induced by STING1 could be further accumulated by CQ. Interestingly, SQSTM1/p62, the substrate of canonical autophagy [28], did not degrade during the NCA process induced by STING1[FL] and STING1 [1-340] (Figure 2b,c). Different from LAP, which is highly dependent on BECN1 [17], STING1-induced NCA did not require BECN1 (Figure 2d). In addition, STING1 [1-340] without immune function could significantly induce the production of NCA as well, indicating that although STING1 is an immune molecule, STING1-induced NCA did not belong to LAP (Figure 2d). On the contrary, STING1 failed to induce LC3-II in atg5−/− MEF cells (Figure 2e). We then found that cGAMP, a specific agonist of STING1, could also significantly activate endogenous STING1 and led to the degradation of STING1, subsequently induced NCA in ulk1-/- and rb1cc1-/- MEF cells (Figure 2f), suggesting that activation of STING1 by protein overexpression or chemical agonists could both induce NCA which was independent of ULK1 complex or BECN1 complex but was dependent of ATG5.
Figure 2.
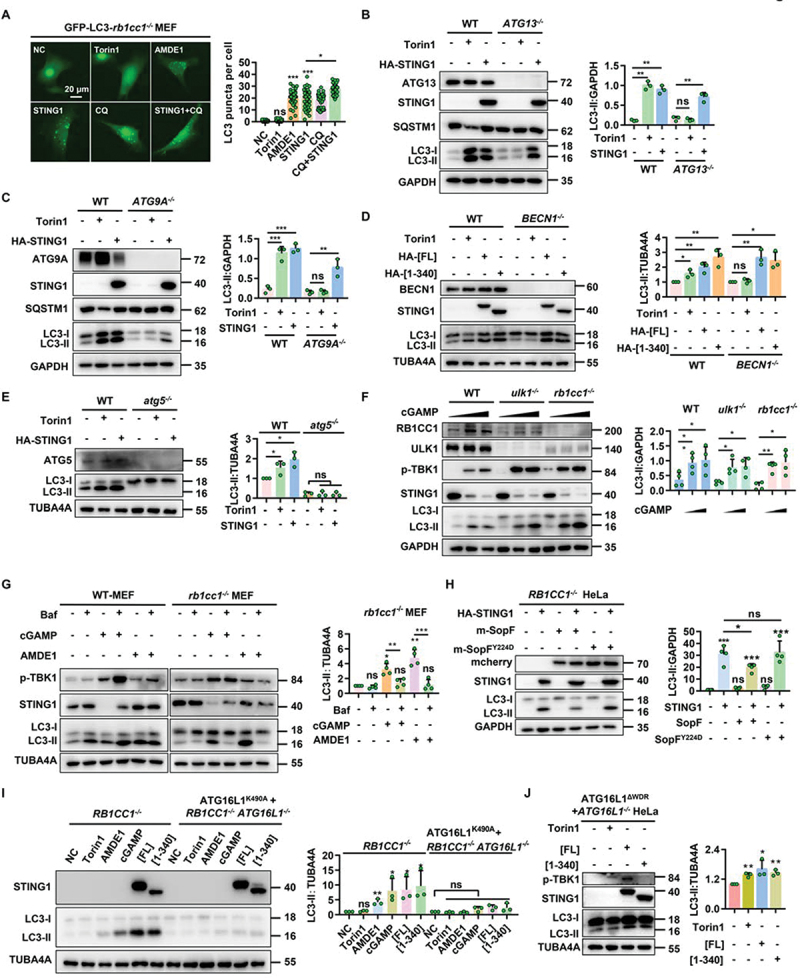
STING1 can significantly induce non canonical autophagy in ULK1- and BECN1-deficient cells. (a) rb1cc1−/− MEFs stably expressing GFP-LC3 with or without transfected of HA-STING1 plasmid were treated with indicated chemicals (AMDE-1, 10 μM; CQ, 20 μM; torin1, 2 μM) for 6 h. Quantification of LC3 puncta per cell was shown on the right side. 50 randomly-selected cells per condition were counted. (b-c) Western blotting of cell lysates from WT-HeLa, ATG13-/- HeLa, and ATG9A-/- HeLa cells were treated with torin1 (2 μM) for 6 h or transfected with HA-STING1 plasmid for 24 h. (d) WT and BECN1−/− HEK-293T cells were treated with torin1 (2 μM) for 6 h or transfected with HA-STING1[FL] or the C-terminal truncation (STING1 [1-340]) plasmids for 24 h. Cell lysates were subjected to western blotting analysis with the indicated antibodies. (e) WT and atg5-/- MEF cells were treated with torin1 (2 μM) for 6 h or transfected with HA-STING1 for 24 h. Cell lysates were subjected to western blotting analysis with the indicated antibodies. (f) WT-MEFs, rb1cc1-/- MEFs and ulk1-/- MEFs were treated with increasing amounts of cGAMP (0 µM, 1 µM and 2 µM respectively) for 3 h, followed by western blotting assays of LC3-ΙΙ conversion. (g) WT and rb1cc1-/- MEF cells were treated with indicated chemicals (AMDE-1, 10 μM; bafilomycin A1 (Baf), 0.5 μM; cGAMP, 1 μM) for 6 h. Immunoblotting was then performed to analyze the LC3-ΙΙ conversion. (h) Western blotting of cell lysates from RB1CC1−/− HeLa cells lacking endogenous STING1 expressing were transfected with HA-STING1 or mcherry-SopF and its mutant mcherry-SopFY224D plasmids for 24 h, and then assessed for LC3-ΙΙ conversion in the right panel. (i) RB1CC1−/− HeLa cells and RB1CC1 and ATG16L1 double knockout cells stably expressing ATG16L1K490A were treated with indicated chemicals (AMDE-1, 10 μM; cGAMP, 1 μM; Torin1, 2 μM) for 6 h or transiently transfected with HA-STING1[FL] or the C-terminal truncation (STING1 [1-340]) plasmids for 24 h, and then assessed for LC3-ΙΙ conversion in the right panel. (J) Restoring the ATG16L1ΔWDR domain by stably expressing on ATG16L1−/− HeLa cells treated with torin1 (2 µM) for 6 h or transiently transfected with HA-STING1[FL] or the C-terminal truncation (STING1 [1-340]) plasmids for 24 h. Cell lysates were subjected to western blotting analysis with the indicated antibodies.
*P<0.05, **P<0.01, ***P<0.001, ns, not significant.
Chemical-induced NCA by CQ or AMDE-1 could be suppressed by V-ATPase inhibitor Baf [23,24,29]. So we wondered whether Baf had the same effect on STING1-induced NCA. As shown in Figure 2g, although Baf led to a significant accumulation of LC3-II in WT MEF cells as a lysosome inhibitor, it strongly inhibited the NCA induced by STING1 and AMDE-1 in rb1cc1-/- MEF cells (Figure 2g). Furthermore, SopF, a unique protein of Salmonella which makes V-ATPase lose the ability to recruit ATG16L1 by ADP ribose modification of V-ATPase complex [25,26], could significantly inhibit STING1 or cGAMP-induced NCA. While SopFY224D, which does not bind to V-ATPase, failed to downregulate the level of LC3-II (Figure 2h and S2C), suggesting STING1-induced NCA required functional V-ATPase.
Moreover, ATG16L1K490A is a recently found allele with a mutation in the C-terminal WD40 repeats of ATG16L1 required for conjugation of LC3 to single membranes but not autophagosome formation [30]. We then used RB1CC1 knockout and RB1CC1 and ATG16L1 double-knockout cells to rescue ATG16L1K490A HeLa cells to see their contribution on NCA. As shown in Figure 2i, the rescue of ATG16L1 instead of ATG16L1K490A have much more LC3-II after the treatment of cGAMP, AMDE-1, or overexpression of STING1. Since the WD40 repeats (WDR) domain of ATG16L1 is required for its NCA role in LC3 lipidation at single membranes [15], rescuing ATG16L1ΔWDR in ATG16L1-/- HeLa cells will only mediate the canonical autophagy process. Similar to torin1 stimulation, overexpression of STING1 could induce LC3-II accumulation in NCA-deficient ATG16L1ΔWDR HeLa cells (Figure 2j). These data indicated that, besides canonical autophagy, activation of STING1 by protein expression or cGAMP could also induce NCA. Such NCA mediated by the WDR domain of ATG16L1 could be blocked by V-ATPase inhibitor.
STING1 activation promotes HSV-1 clearance in cells lacking canonical autophagy
The antiviral effect of STING1 and the infection ability of DNA virus HSV-1 were firstly verified in THP-1 cells, which have higher expression level of endogenous STING1 [31]. Activation of STING1 by the specific agonist cGAMP could significantly reduce the infection efficiency of HSV-1 virus. Nevertheless, the infection rate of HSV-1 increased significantly in THP-1 cells after knocking down STING1 (Fig. S3A-B). These results indicated that STING1, as a DNA recognition molecule, could resist DNA virus under the activation of cGAMP. We then focused on the role of autophagy proteins on STING1-mediated HSV-1 clearance. Using flow cytometry to detect the virus infection rate, we found that both cGAMP stimulation and STING1 overexpression could effectively inhibit the infection rate of HSV-1 in WT and rb1cc1-/- MEF cells, which have higher basal level of STING1 [11]. Infection rate could be further downregulated by the combination of cGAMP and STING1 overexpression (Figure 3a,b). Furthermore, cGAMP stimulation alone could no longer reduce the virus infection rate in ATG13-/- and ATG9A-/- HeLa cells, due to the lower basal level of STING1 [12] (Figure 3c,d). However, transfection of STING1 could still effectively inhibit the virus infection rate in those HeLa cells and could be further reduced by cGAMP (Figure 3c,d). Although cGAMP itself could induce NCA in ATG13-/- HeLa cells independent of endogenous STING1 (Fig. S2B), its antiviral activity also depended on the expression level of STING1. These data suggested that the deletion of upstream gene of canonical autophagy does not inhibit the antiviral function of STING1.
Figure 3.
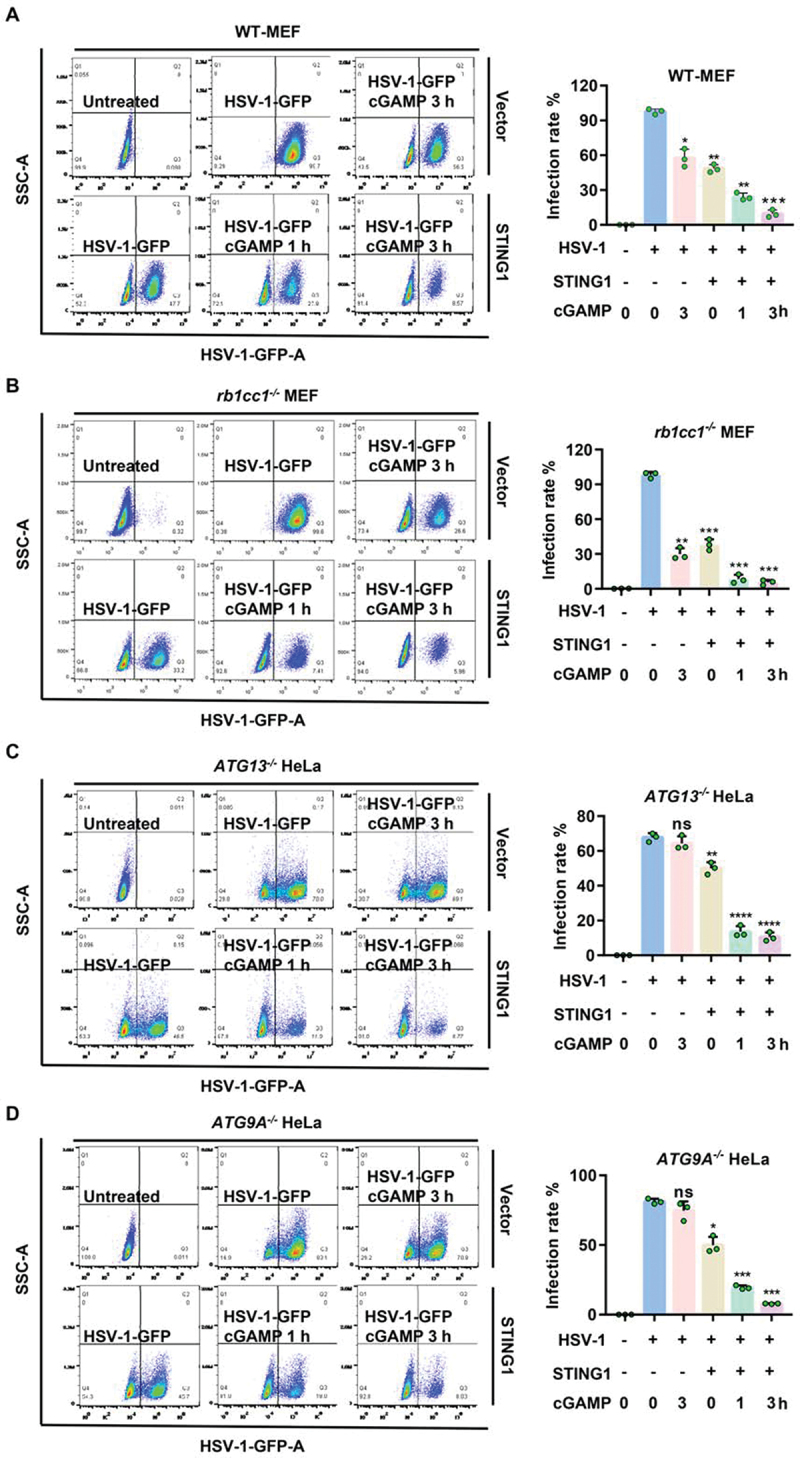
STING1 show obvious resistance to HSV-1 virus in both WT and autophagy gene deficient cells. (A-D) WT (A) or rb1cc1−/− MEF (B) cells containing endogenous STING1 expressing [11] and ATG13−/− HeLa (C) and ATG9A−/− HeLa (D) cells lacking endogenous STING1 expression were transfected with HA-STING plasmids. After 24 h incubation, cells were then infected with HSV-1-GFP virus at a multiplicity of infection (MOI) of 5 for 6 h, with or without 1 μM of cGAMP stimulated for the indicated time. GFP-positive cells were analyzed by flow cytometry. Quantification charts of HSV-1-GFP virus infection rate in (A-D) were shown in the right panels. Data analysis was performed using FlowJo software and presented as mean±SEM from 3 individual experiments.
*P<0.05, **P<0.01, ***P<0.001 vs with positive control based on HSV-1-GFP infection alone, ns, not significant.
STING1 can inhibit HSV-1 through noncanonical autophagy pathway alone, canonical autophagy pathway alone, or immunity function alone
In order to clarify how STING1 exert its antiviral ability, we compared different STING1 mutants with or without immunity function in cells with or without canonical autophagy and NCA function. The C-terminal tail of STING1 was removed to block the ability of downstream TBK1 and IRF3 to bind with STING1, which made STING1 lose its original innate immunity function, but retain the function of autophagy [9]. Comparing with the control HeLa cells without STING1 expression [11,12], unlike MEF cells, cGAMP alone could not change the infection rate of HSV-1 of HeLa cells due to the lack of endogenous STING1 in HeLa cells (Figure 4A). On the contrary, transfection of full-length STING1 inhibit the infection rate. Such inhibition could be found when STING1 [1-340] was transfected (Figure 4A), suggesting HeLa cell could still inhibit HSV-1 without immunity function. We then used the cells mentioned in Figure 2J and found that ATG16L1-/- HeLa cells expressing ATG16L1ΔWDR and transfected with STING1 [1-340] could significantly inhibit HSV-1 (Figure 4B), suggesting STING1-induced canonical autophagy alone could reduce the infection rate. Moreover, transfection of STING1 [1-340] could still effectively inhibit HSV-1 virus replication, and the virus infection rate was further decreased under the treatment of cGAMP in ATG13-/- and ATG9A-/- HeLa cells (Figure 4C and S4A), suggesting that STING1 activation could inhibit virus through only NCA pathway which is independent of its immunity domain and canonical autophagy function. As we known, ATG16L1 is required for both canonical and NCA, so deletion of ATG16L1 gene will lead to the failure of LC3 lipidation [32], which is a key process for different types of autophagy (Fig. S4B). Different from ATG13-/- and ATG9A-/- HeLa cells, STING1 [1-340] could no longer suppress virus replication in ATG16L1-/- HeLa cells (Fig. S4C), indicating that lacking canonical autophagy and NCA pathways and deficient of immunity domain of STING1 simultaneously, cells could strikingly lost the capability to inhibit HSV-1 infection. Similarly, using cells mentioned in Figure 2I we found that without canonical autophagy and NCA pathways, cells transfected with STING1[FL] alone instead of STING1 [1-340] could still reduce the infection rate (Fig. S4D). Importantly, such antiviral effects in figure S5C could be rescued by overexpressing full-length STING1 in ATG16L1−/− HeLa cells (Figure 4D). Although different types of autophagy were all blocked in ATG16L1-/- cells, STING1[FL] could still effectively exert its antiviral effect via its original innate immune function. These results implied that besides immunity function and canonical autophagy pathway, novel NCA pathway induced by STING1 could also be an efficient way to inhibit virus infection.
Figure 4.
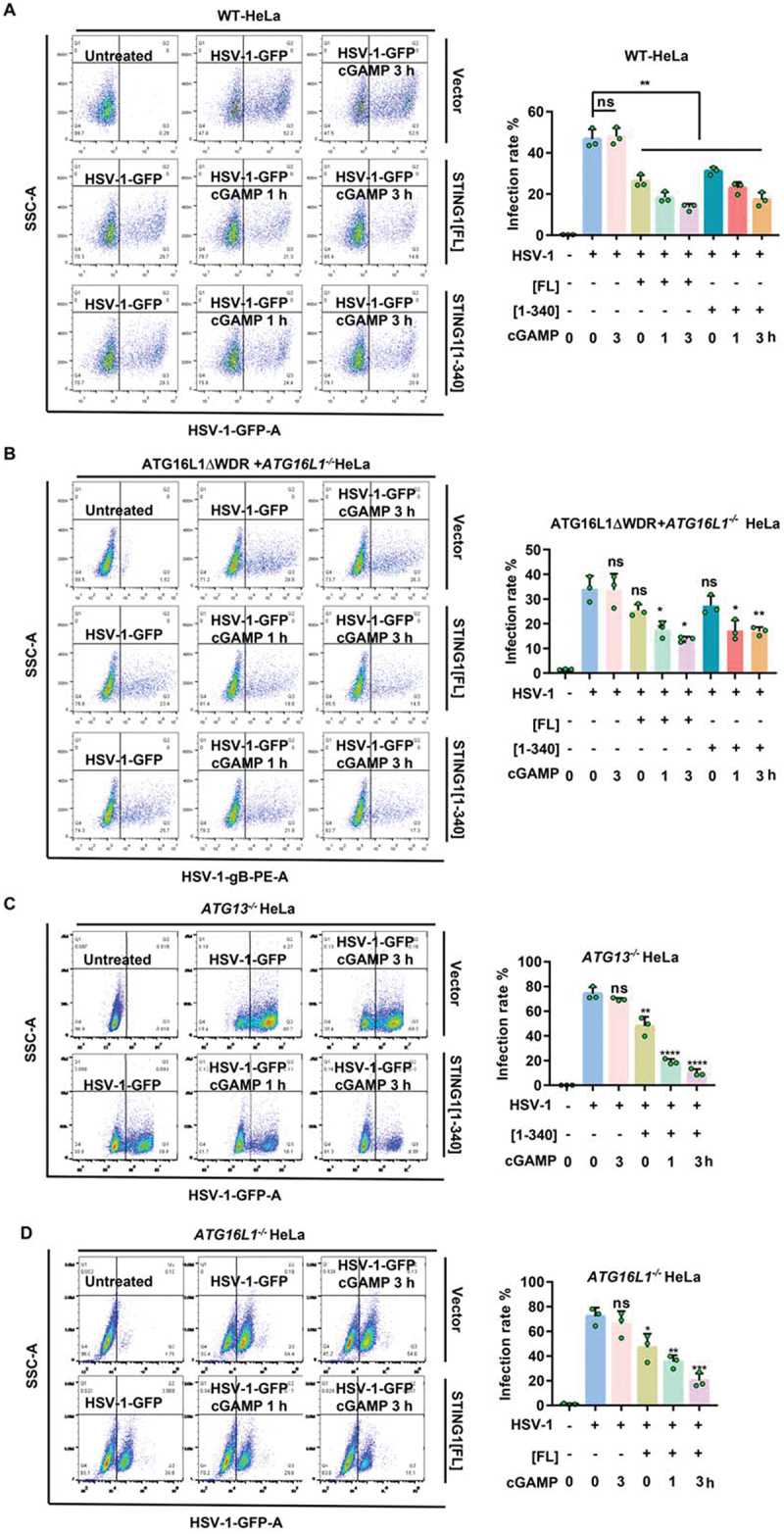
STING1 can inhibit HSV-1 through noncanonical autophagy pathway alone, canonical autophagy pathway alone, or immunity function alone. (A) WT HeLa cells which lacking endogenous STING1 expressing were transfected with STING1 truncated mutants STING1 [1-340] plasmids or full-length STING1[FL] plasmid. After 24 h expression, cells were infected with HSV-1-GFP virus at a MOI of 2.5 for 6 h, with or without 1 μM of cGAMP stimulated for the indicated time. GFP-positive cells were analysis by flow cytometry. Quantification of HSV-1-GFP virus infection rate is shown in the right panel. (B) After STING1[FL] or STING1 [1-340] plasmids were transfected into NCA-deficient ATG16L1ΔWDR-GFP HeLa cells for 24 h, HSV-1-GFP virus were then infected for 6 h, with or without 1 μM of cGAMP stimulated for the indicated time. PE flow detection channel was used to detect HSV-1-GFP virus coat gB protein to avoid the interference of GFP in the basal of the ATG16L1ΔWDR-GFP ATG16L1-/-HeLa cells. PE positive cells were analysis by flow cytometry. (C-D) ATG13-/- HeLa cells which have canonical autophagy dysfunction (C) and ATG16L1-/- HeLa cells which have autophagy dysfunction (D) were transfected with STING1 truncated mutants STING1 [1-340] (C) or full length STING1 (D). After 24 h expression, cells were then infected with HSV-1-GFP virus at a MOI of 5 for 6 h, with or without 1 μM of cGAMP stimulated for the indicated time. GFP-positive cells were analyzed by flow cytometry. Quantification charts of HSV-1-GFP virus infection rate were shown in the right panels. Data analysis was performed using FlowJo software and presented as mean±SEM from 3 individual experiments.
*P<0.05, **P<0.01, ***P<0.001 vs with positive control based on HSV-1-GFP infection alone, ns, not significant.
cGAMP and HSV-1 cannot stimulate immune response and inflammatory factors in STING1 [1-340] cells
Previously, a truncated plasmid lacking immune function, STING1 [1-340], was used to show that STING1 can inhibit the virus via NCA pathway. Recent reports have proved that STING1 can still inhibit pathogen infection through inflammatory response even independent of its original innate immune activity [33,34]. However, we are not clear whether such antiviral effect of STING1 [1-340] in cells lacking canonical autophagy is still caused by any inflammatory factors or not. Therefore, the level of inflammatory factors was detected in different cells under the action of cGAMP and HSV-1. The deletion of canonical autophagy gene Rb1cc1 did not affect the NCA and immunity level, so in both WT and rb1cc1-/- MEF cells, the levels of p-TBK1 and LC3-II increased under cGAMP or HSV-1 virus treatment (Figure 5A). In the meantime, cGAMP or HSV-1 could significantly increase the RNA levels of inflammatory cytokines such as Ifnb, Il6, Tnf, and Cxcl10 due to the contribution of endogenous full length STING1 in MEF cells (Figure 5B). However, cGAMP stimulation failed to directly promote the release of inflammatory factors in ATG13-/- HeLa cells due to the lack of endogenous STING1 (Figure 5C). While, after transfection of STING1[FL], the level of inflammatory factors increased, and further enhanced when combined with cGAMP. More importantly, transfection of STING1 [1-340] could not upregulate the levels of the response of inflammatory factors as STING1[FL] did (Figure 5C). Such pattern was also confirmed when HSV-1 infection was applied to ATG13−/− or BECN1−/− HeLa cells (Figure 5D,E). Taken together, the antiviral effect of STING1 [1-340] which failed to induce immunity response in cells lacking canonical autophagy gene is mainly contributed to NCA activity instead of the production of inflammatory factors.
Figure 5.
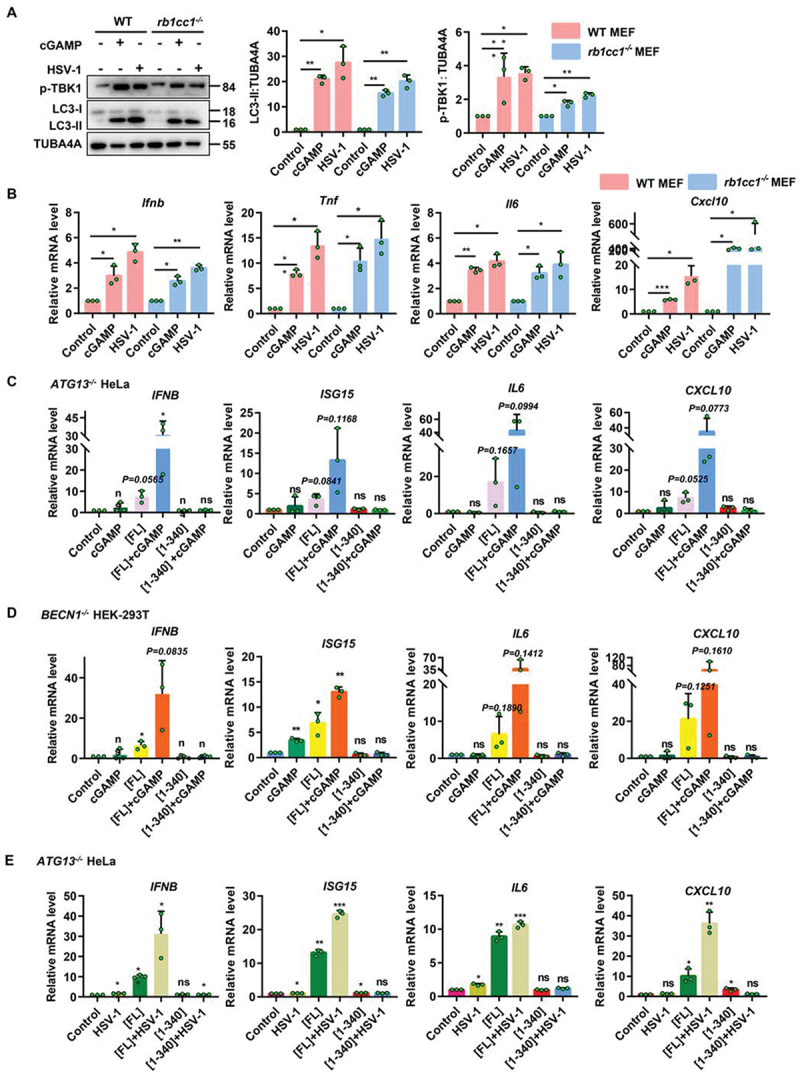
Both cGAMP and HSV-1 can stimulate STING1 to produce immune response and inflammatory factors, but not STING1 [1-340]. (A-B) WT and rb1cc1-/- MEF cells containing endogenous STING1 expressing were treated with 1 μM of cGAMP for 6 h or infected with HSV-1-GFP virus at a MOI of 5 for 6 h. Cell lysates were subjected to immunoblot analysis of LC3-ΙΙ conversion and phosphorylation degree of TBK1 (A). And mRNA was extracted for RT–Qpcr analysis of expression of the genes for Ifnb, Tnf, Il6, and Cxcl10 (B). (C-D) ATG13-/- HeLa (C) and BECN1-/- HEK-293T (D) cells lacking endogenous STING1 expressing were transfected with control plasmid (pcDNA3.1) or STING1 plasmids (STING1[FL] and STING1 [1-340]). After 24 h expression, cells were treated with or without 1 μM of cGAMP for 3 h. Total RNA was isolated to measure the expression of indicated genes by reverse transcription qPCR. (E) ATG13-/- HeLa cells lacking endogenous STING1 expressing were transfected with control plasmid (pcDNA3.1) or STING1 (STING1[FL] and STING1 [1-340]) for 24 h before infected with or without HSV-1-GFP virus at a MOI of 5 for 6 h. Total RNA was isolated to measure the expression of indicated genes by reverse transcription qPCR. Representative data were shown from three independent experiments; n = 3.
*P<0.05, **P<0.01, ***P<0.001, ns, not significant.
Deletion of CTT does not change the translocation and dimerization properties of STING1
STING1 activation by cGAMP requires its translocation from the ER to the Golgi apparatus and subsequent polymerization to recruit and activate the kinase TBK1 and, subsequently, the transcription factor IRF3, ultimately leading to the production of various cytokines, including type I type I interferon (IFNs) [7,10]. We found both STING1 activators cGAMP and HSV-1 could promote the dimerization of endogenous STING1 in MEF cells (Figure 6A). It remains unknown that after removing the original innate immune and inflammatory response ability of STING1, whether STING1 [1-340] could still have the similar characteristics of translocation and dimerization of STING1. Therefore, STING1-/- HeLa cells were used for transient transfection of STING1[FL] and STING1 [1-340]. Both constructs demonstrated increased dimerization after cGAMP treatment in non-reduced electrophoresis (Figure 6B and S5A). In addition, in RB1CC1-/- HeLa and BECN1-/- HEK-293T cells in which NCA could be induced by cGAMP or HSV-1, STING1[FL] and STING1 [1-340] could still dimerize (Figure 6C and S5B). To rule out the possible effect of autophagy pathway on dimerization, ATG7-/- HeLa cells without any autophagy activity were analyzed and found that STING1 [1-340] could still undergo obvious dimerization, suggesting the dimerization of STING1 was irrelevant to autophagy function (Figure 6D). To differentiate whether the multi-bands of STING1 were specific or not, anti-Flag and anti-GFP antibodies were additionally applied to confirm that those bands are specific STING1-related proteins (Figure 6B–D). Moreover, the enhanced level of p-TBK1 in STING1[FL] group was largely downregulated in STING1 [1-340] group even after cGAMP or HSV-1 treatment, which is consistent with the finding that no inflammatory factors could be induced in STING1 [1-340] HeLa cells due to the lack of C-terminal tail 340–379 of STING1.
Figure 6.
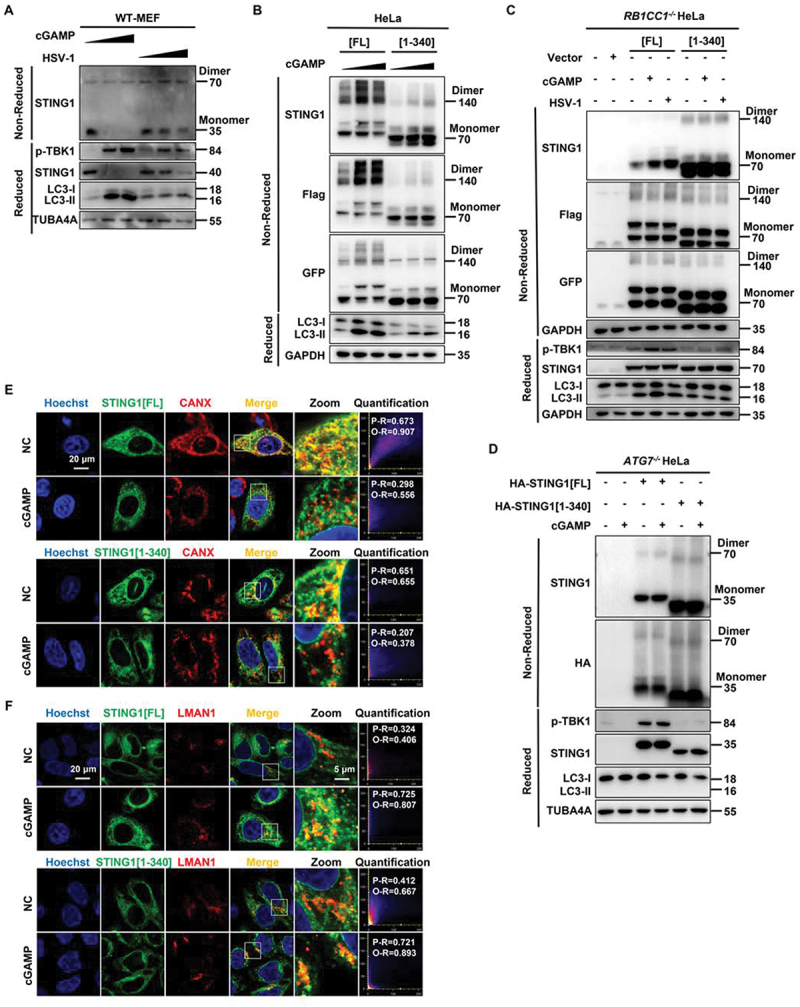
Translocation and dimerization of STING1 is unchanged in STING1 [1-340] which merely affected innate immune function. (A) WT MEF cells were treated with increasing amounts of cGAMP (0 µM, 1 µM, and 2 µM respectively) for 3 h or infected with increasing MOI of HSV-1-GFP virus (0, 2 and 5 respectively) for 6 h. Cell lysates were analyzed by non-reduced gel (for endogenous STING1 dimerization) or reduced gel followed by immunoblotting with the indicated antibodies. (B) WT HeLa cells stably expressing Flag- and GFP-tagged STING1 or the C-terminal truncation (STING1 [1-340]) were treated with increasing amounts of cGAMP (0 µM, 1 µM and 2 µM respectively) for 3 h. Cell lysates were analyzed by non-reduced gel (for recombinant Flag-STING1-GFP dimerization) or reduced gel followed by immunoblotting with the indicated antibodies. (C) RB1CC1-/- HeLa cells which lacking endogenous STING1 expressing were transfected with control plasmid (pcDNA3.1) or STING1 plasmids (STING1[FL] or STING1 [1-340]) for 24 h before treated with cGAMP (1 μM) for 3 h or infected with HSV-1-GFP virus at a MOI of 5 for 6 h. Cell lysates were analyzed by non-reduced gel (for recombinant Flag-STING1-GFP dimerization) or reduced gel followed by immunoblotting with the indicated antibodies. (D) ATG7-/- HeLa cells which autophagy dysfunction and lacking endogenous STING1 expressing were transfected with HA-tagged STING1 or the C-terminal truncation (STING1 [1-340]) for 24 h, cells were then treated with cGAMP (1 μM) for 3 h. Cell lysates were analyzed by non-reduced gel (for recombinant HA-STING1 dimerization) or reduced gel followed by immunoblotting with the indicated antibodies. (E-F) HeLa cells stably expressing GFP-tagged STING1 or the C-terminal truncation (STING1 [1-340]) were fixed and stained with marker antibody for ER (CANX) and the ER-Golgi intermediate compartment (LMAN1/ERGIC-53) after treated with or without 1 μM of cGAMP for 3 h, and images were then captured by confocal microscopy. All images are representative of at least three independent experiments in which > 95% of the cells displayed similar staining. Scale bar: 20 µm. The degree of colocalization for STING1 and CANX or LMAN1 was quantified by Pearson’s and overlap coefficient by Image J.
Furthermore, GFP-labeled STING1[FL] and STING1 [1-340] were stained with different organelle markers such as CANX (calnexin), LMAN1/ERGIC-53, and TGOLN2/TGN46. After cGAMP treatment, both STING1[FL] and STING1 [1-340] could translocate from ER to ERGIC or trans-Golgi membrane (Figure 6E,F and S5C). These results suggested that the deletion of C-terminal tail does not change the translocation and dimerization properties of STING1.
STING1-induced noncanonical autophagy degrades the virus through lysosomes
Although STING1 could induce NCA, we are not clear whether such NCA process could still have the similar hallmarks of canonical autophagy such as long-lived protein degradation function and double membrane structures formation [35]. We firstly compared the wild type and rb1cc1−/− MEF cells with or without cGAMP treatment to measure the long-lived proteins degradation activity using non-radioactive methods [36]. We found that, different from the context of canonical autophagy in which cGAMP could increase the degradation in WT MEFs, STING1-induced NCA mediated by cGAMP could not degrade long-lived proteins in rb1cc1-/- MEFs (Figure 7A,B). We then detected the cellular structures by transmission electron microscope of rb1cc1−/− MEFs before and after cGAMP treatment. As shown in Figure 7C, double membrane structures were not increased but more single membrane vesicles accumulated, suggesting that unlike canonical autophagy, STING1-induced NCA mainly induced single membrane structures.
Figure 7.
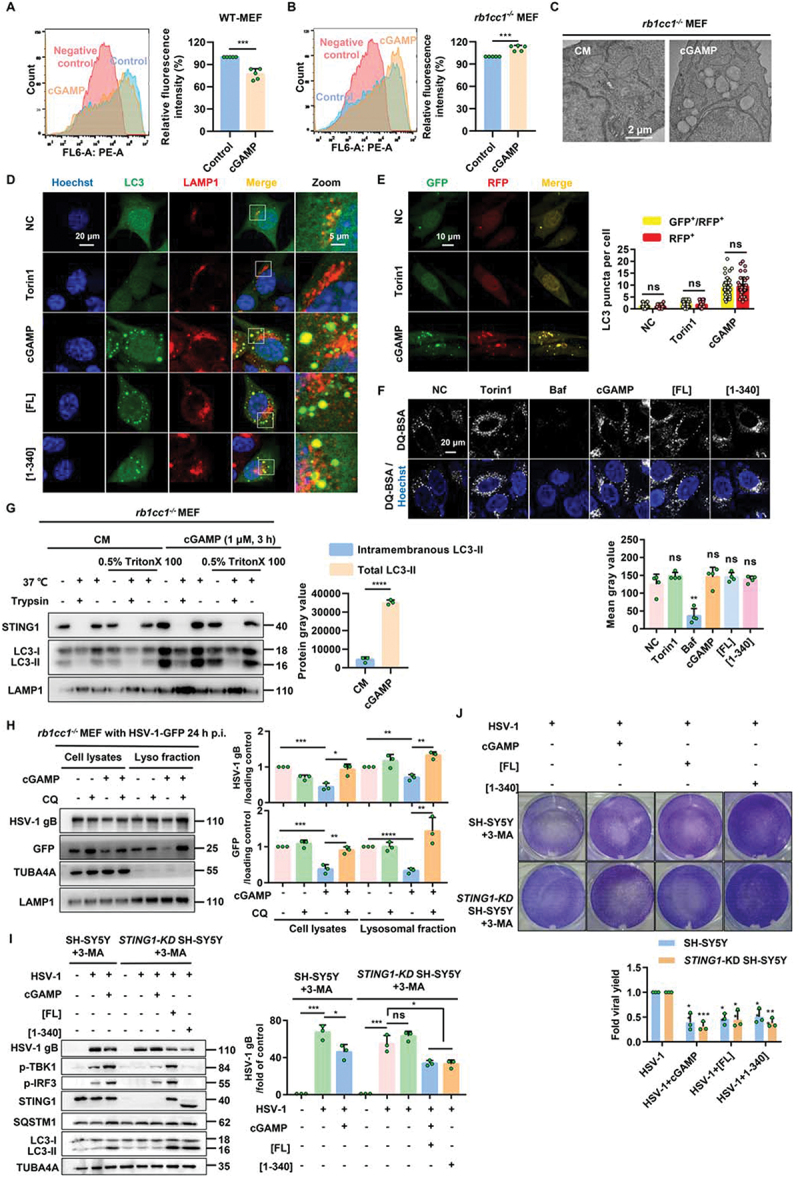
Non canonical autophagy induced by STING1 can degrade the virus through lysosomes.(A-B) Flow cytometry was performed to analyze the fluorescence intensity within the WT MEF (A) and rb1cc1-/- MEF (B) cells, and the histograms represent the relative average fluorescence intensity of 10,000 cells from different samples. Negative control: cells without AHA labeling but were subjected to click reaction. Data for the relative signal intensity were expressed as the ratio of 1 μM of cGAMP treated for 6 h cells to the untreated control cells. (C) Representative images of transmission electronic microscope of rb1cc1-/- MEF cells stimulated with 1 μM of cGAMP for 6 h. (D) Colocalization of NCA autophagosomes and lysosomes induced by STING1. rb1cc1-/- MEF cells stably expressing GFP-LC3 were treated with indicated chemicals (torin1, 2 μM; cGAMP, 1 μM) for 6 h, or transfected with STING1 plasmids (STING1[FL] or STING1 [1-340]) for 24 h, followed by immunostaining of LAMP1 (red). Scale bar: 20 µm. (E) rb1cc1-/- MEF cells which containing endogenous STING1 expressing were transiently transfected with RFP-GFP-LC3 plasmid. After 24 h expression, cells were then harvested and re-seeded on confocal culture dish. Cells were treated with indicated chemicals (torin1, 2 μM; cGAMP, 1 μM) for 6 h, followed by fixation. The colocalization of GFP and RFP puncta was examined and quantified. Scale bar: 10 µm. At least 50 cells were counted from each group. (F) Representative single-plane confocal micrographs of WT HeLa cells incubated with DQ-Red BSA were starved for 2 h by EBSS in the absence or presence of 500 nM Baf, 2 μM torin1, 1 μM cGAMP or transfected with STING1 plasmids (STING1[FL] or STING1 [1-340]) for 24 h. The cell nucleus is stained using DAPI (blue). Scale bar: 20 µm. And then assessed for average fluorescence value of DQ-BSA in the right panel. At least 50 cells were counted from each group. (G) Protease protection assay of homogenates from rb1cc1−/− MEF cells treated with 1 μM of cGAMP for 3 h. Diagrams show subcellular location of LC3-II and endogenous STING1 detected by western blotting. (H) Detect the virus content in lysosomes. Rb1cc1-/- MEF cells which contain endogenous STING1 were infected with HSV-1 virus at MOI = 5. After 24-h infection, cells were harvested and re-seeded on culture dish. Cells were then treated with indicated chemicals (CQ, 40 μM; cGAMP, 1 μM) for 6 h. Lysosomal fraction was collected, followed by immunoblotting with indicated antibodies. Representative data were shown from three independent experiments; n = 3.
*P<0.05, **P<0.01, ***P<0.001, ns, not significant.
Lysosomes are major organelles for degradation of damaged proteins or pathogens after fusion with autophagosomes [37], so we analyzed the colocalization of LC3 and LAMP1 (lysosomal associated membrane protein 1) to see whether STING1-induced NCA could finally fuse with lysosome. After activating STING1 by cGAMP or overexpression of STING1[FL] or STING1 [1-340], the increased LC3-positive structures could significantly overlap with lysosomes (Figure 7D). Tandem LC3 assay also indicated that cGAMP could induce higher ratio of RFP+ GFP+ dots (Figure 7E). To rule out the possibility that lysosome function was suppressed by cGAMP or STING1 overexpression which could cause the LC3 accumulation on the lysosome or the failure of quenching GFP to form RFP+ GFP+ dots, we measured the lysosome function by DQ-BSA assay [38]. As shown in Figure 7F, except V-ATPase inhibitor Baf which impaired the pH of lysosome, torin1, cGAMP, and the overexpression of STING1[FL] and STING1 [1-340] did not suppress lysosome degradation function. So we speculated that the accumulated LC3-positive structures induced by STING1 activation could fuse with lysosomes without disturbing the degradation function of lysosomes. We then performed a protease protection assay to see LC3-II was located inside or outside of the membrane of subcellular organelles. We could see that, after trypsin digestion most lipidated LC3 was digested even without membrane permeabilization by Triton X-100, suggesting that LC3-II proteins are mainly located on the outside of subcellular organelle membranes such as lysosome in rb1cc1−/− MEFs after cGAMP stimulation (Figure 7G).
Since HSV-1 could be inhibited by cGAMP through NCA, and LC3-positive structures could finally fuse with lysosome in rb1cc1−/− MEF cells, we were eager to know whether HSV-1 could be degraded by NCA through lysosomes. When HSV-1 degradation was blocked by lysosome inhibitor CQ, more HSV-1 virus would be detected by GFP or HSV-1 gB in lysosomal fraction (Figure 7H), suggesting HSV-1 could be degraded by STING1 activation through lysosome pathway after NCA induction.
Those results indicated that STING1-induced NCA can impair long-lived proteins degradation and double membrane formation, but has no effect on the fusion of LC3-positive structures with lysosomes, and can degrade HSV-1 through lysosome pathway.
Discussion
STING1 was originally reported to exert its antiviral function by releasing type I interferon and inflammatory factors [3,33]. In addition, STING1 was reported to effectively mediate antiviral response and tumor immune escape in the form of its interferon independent pattern [39,40]. Yamashiro et al. and Wu et al. generated the STING1S365A/S365A mutant mouse that precisely ablate IFN-dependent activities while preserving IFN-independent activities of STING1. STING1S365A/S365A mice are able to protect against HSV-1 infection, despite lacking the STING1-mediated IFN response. This challenges the prevailing view and suggests that STING1 could control HSV-1 infection through IFN-independent activities [39,40]. Indeed, STING1S365A is still able to induce autophagy-like formation of LC3 puncta. Although the mechanism that mediates protection to HSV-1 downstream of the STING1[CTT] and TBK1 remains unclear, we propose this might be through an autophagy or autophagy-like process. Moreover, canonical autophagy, as another important function of STING1, can also effectively resist DNA virus [12]. Recently, we and other groups found that STING1 could also induce NCA. However, whether STING1-induced NCA have similar character to canonical autophagy and could contribute to antiviral response are not clear at all. In this work, we compared the hallmarks of autophagy and found STING1-induced NCA is significantly different from canonical autophagy. More importantly, we found that STING1 could effectively inhibit the virus in NCA cell models lacking the innate immunity, inflammatory response, and canonical autophagy functions of STING1. Our data suggests NCA might be a novel mechanism to mediate the antiviral response of STING1.
Compared to canonical autophagy, which required nearly 40 ATG proteins to form double membrane autophagosomes to degrade long-lived proteins or pathogens through lysosomes [22], NCA has only limited studies for its functions and biological processes. As STING1 activation could strongly induce both canonical autophagy and NCA, we thought it would be important to differentiate these two processes for this vital intrinsic immune molecule. Although other types of NCA or CASM like LAP, LANDO, and chemical-induced NCA were reported, not too much protein degradation function was measured for those NCA [17–19,23]. Here we firstly demonstrated that different from canonical autophagy, STING1-induced NCA could not degrade long-lived proteins. In addition, the membrane structure of NCA induced by STING1 is still a controversial issue until now. According to previous reports [12], STING1 induced obvious double-layer membrane autophagy structure in ULK1- and BECN1-deficient cells under cGAMP stimulation. Nevertheless, Fischer et al. found that the NCA induced by STING1 is an obvious monolayer membrane structure through correlative light electron microscope [15]. In the evidence we provided by transmission electron microscope, we proved that cGAMP could significantly induce NCA in rb1cc1−/− MEF cells with increasing monolayer vesicles. Based on these two hallmarks of autophagy, we demonstrated that STING1-induced NCA has distinct structural and functional properties compared to canonical autophagy.
As a well-known form of NCA, LAP can effectively phagocytize and eliminate invasive pathogens with the participation of BECN1 complex [17]. While, NCA induced by STING1 is largely different from LAP that both ULK1 complex and BECN1 complex are not required. Interestingly, we have ever reported that chemical-induced NCA by AMDE-1 and niclosamide was also independent of ULK1 and BECN1 complex [23,41], but ATG16L1 is indispensable, suggesting different types of NCA may require different ATG proteins. In addition, we and other groups found that chemical-induced NCA could also be strongly inhibited by V-ATPase inhibitor Baf [29]. So we thought V-ATPase and ATG16L1 might play key roles in the process of STING1-induced NCA as well. Interestingly, during the time we carried out this work, more and more studies indicated that WD40 repeats domain of ATG16L1 is required for LC3 lipidation at single membranes, and V-ATPase might be a main binding target for the WD40 repeats domain of ATG16L1 in STING1-induced LC3 lipidation process and xenophagy model [15,25,30]. Similar to our hypothesis, STING1-induced NCA could be strongly blocked by Baf and bacterial protein SopF which can inactivate V0 complex of the V-ATPase. Moreover, both the deletion of WD40 repeats domain of ATG16L1 and mutation of the K490 site on ATG16L1 could greatly block the binding of ATG16L1 to V-ATPase and obviously inhibit NCA. So, this study confirmed that STING1-induced NCA might be mediated by V-ATPase-ATG16L1 axis. While how STING1 initiates such axis remains unclear.
STING1 proteins need to undergo dimerization, translocation from the ER to the Golgi apparatus to exert immune effects by recruiting TBK1 and IRF3 [6,7]. In this work, although the deletion of CTT domain (STING1 [1-340]) failed to increase the phosphorylation level of TBK1 and IRF3 and lost immune and inflammatory response of STING1, it did not affect the translocation and dimerization of STING1 in NCA cells or ATG7-deficient cells without any autophagy function, suggesting the dimerization is separated from autophagy processes. Previous studies have shown that STING1 could resist the virus through the release of type I interferon and the NFKB/NF-κB inflammatory pathway [3,33]. But interferon-independent antiviral effect of STING1 suggests that there might exist other new pathways involved in the antiviral response of STING1 [39,40]. By combination of different mutant of STING1 with or without immune function and different cell lines with or without autophagy to measure the antiviral ability, we finally demonstrated that STING1-induced NCA alone could efficiently downregulate the virus infection rate. Interestingly, cGAMP could also induce NCA in cells without STING1, but such NCA could hardly inhibit virus infection, indicating that STING1 is crucial in the antiviral process.
Since NCA could accumulate LC3-positive vesicles, we tested the role of lysosome which might be necessary for vesicle fusion and virus degradation. NCA induced by STING1 activation does not affect the degradation function of lysosomes by DQ-BSA assay. Whereas, in rb1cc1−/− MEF cells, we found that most of the GFP-LC3 positive structures induced by STING1 were colocalized with lysosomes, suggesting STING1-induced GFP-LC3 puncta could fuse with lysosome but could not be quenched. One possibility is that the lipidated LC3 protein of NCA was mainly fixed outside the lysosomal membrane instead of facing the lysosomal acidic environment for degradation or quenching. This hypothesis was verified by protease protection experiment, which proved that LC3-II proteins were mainly distributed outside the subcellular organelle membrane. In view of the diffused distribution of HSV-1 virus in cells after infection, it is hard to detect the colocalization of lysosome and virus by immunofluorescence tests. On this basis, we further designed the lysosome extraction experiment to explore whether the NCA induced by STING1 can really degrade HSV-1 virus through lysosome after adding the lysosome inhibitor CQ. Although higher dose and longer duration of CQ treatment might also induce NCA due to osmotic imbalance [24], the results showed that CQ alone could not inhibit HSV-1 virus. The detection of viral coat protein gB and GFP tag carried by the HSV-1 showed that STING1 effectively induced NCA to inhibit the HSV-1, and then degraded the virus through lysosomal pathway. The above data clearly showed that HSV-1 virus could be apparently degraded through lysosome during NCA induced by STING1, which is different from traditional interferon-mediated virus inhibition.
Besides the induction mechanism of innate immunity mediated by pattern recognition receptors [42], autophagy has evolved into a host defense mechanism against immune and pathogenic stress through canonical and noncanonical pathways, and mediates the immune self-tolerance of cells to internal and external threats [43,44]. Although its deployment speed is not as fast as that of innate immunity, it can lead to a broader antiviral response and induce a pro-inflammatory signal cascade. Those different self-defense mechanisms of the host cell constitute its internal defense and innate immunity.
To sum up, we uncovered a new mechanism that NCA induced by STING1 activation is important for STING1 to resist DNA virus. Our study also shows a physiological significance for NCA, and provides a certain reference for the antiviral research of NCA in the future.
Materials and methods
Materials
2’3’-cGAMP was from InvivoGen (tlrl-nacga23), and delivered into cells by permeabilization with 10 μM of digitonin (Solarbio, ID0410). The concentration of cGAMP used in stimulating MEF cells was 1 μM unless indicated otherwise. AMDE-1 (4N–049) was from Key Organics. Torin1 (HY-13003) was from MedChemExpress. Chloroquine (CQ, A506569) was from Sangon Biotech. Bafilomycin A1 (B-1080) was from LC Laboratories. Dequenched-BSA (DQ-BSA) Red (D12051) were from Life Technologies.
Antibodies
Antibodies against human STING1 (D2P2F, 13647), p-TBK1 (D52C2), p-IRF3 (Ser396, 4D4G), LAMP1 (D2D11), ATG13 (E1Y9V), ATG9A (D409D), BECN1 (3738S), CANX (2679P), HA (2367S), and ATG4B (D1G2R) were from Cell Signaling Technology; antibodies against LC3B (L7543), ATG5 (A0731), ULK1 (A7481), and TUBA4A/α-tubulin (T6074) were from Sigma-Aldrich; antibodies to LC3B (PM036), ATG5 (M153), SQSTM1/p62 (PM045), and ATG16L1 (M150) were from MBL International; antibodies against LMAN1/ERGIC53 (sc -398,777), GFP (sc-9996), and TOMM20 (FL-145) were from Santa Cruz Biotechnology; antibody against TGOLN2/TGN46 (AHP500GT) was from Bio-Rad. Secondary antibodies conjugated with Alex Fluor 488, Alex Fluor 594, or horseradish peroxidase were from Invitrogen and Jackson Immuno Research Laboratories, respectively.
Cell culture and DNA transfection
WT HeLa cells, ATG16L1−/−, ATG4B−/− HeLa cells, WT mouse embryonic fibroblasts (MEFs), rb1cc1/fip200−/−, ulk1−/−, and atg5−/− MEFs, have been described previously [23]. THP-1 and DLD1 cells were from National Collection of Authenticated Cell Cultures. ATG13−/− Hela cells were provided by Lei Liu (Institute of zoology, Chinese Academy of Sciences, Beijing). ATG9A−/− Hela cells were provided by Dr. Yueguang Rong (Tongji Medical College of HUST, Wuhan). BECN1−/− HEK-293T cells were provided by Dr. Jun Cui (Sun Yat-sen University, Guangzhou). THP-1 and DLD1 cells were cultured in RPMI 1640. Others cells were cultured in DMEM (Thermo Scientific, SH3024301) supplemented with 10% (v:v) fetal bovine serum (Gibco, A31608) and standard supplements in a 37°C, 5% (v:v) CO2 incubator. For starvation, Earle’s balanced salt solution (EBSS, Sigma, E2888) was used.
For transient transfection, human cDNAs encoding N-terminal HA-tagged STING1 and its mutants were cloned into pcDNA3.1(+) or pEGFP-N3. For stable cell line preparation, human cDNAs encoding N-terminal Flag-tagged STING1 and its mutants were cloned into a pLVX-AcGFP-N1 lentiviral vector. These lentiviruses were packaged in HEK-293T cells and transduced into target cells. Plasmids for the expression of WT and Y224D-SopF were provided by Dr. Feng Shao and Dr. Yue Xu (NIBS, Beijing). Plasmids were transfected into cells using Lipofectamine 2000 (Invitrogen, 11668019).
Immunoblotting
Cell lysates were prepared with RIPA buffer (Beyotime Biotechnology, P0013B) with a protease and phosphatase inhibitor cocktail (Thermo Scientific, 78440). The proteins in the cell lysates were subjected to SDS-PAGE using a 12% (w:v) gel. The separated proteins were transferred to a PVDF membrane (Millipore, Billerica, MA). The transferred membrane was blocked with 5% (w:v) skim milk dissolved in TBST buffer (20 mM Tris-HCl, pH 8.0, 137 mM NaCl, and 0.1% Tween 20 [Mikxlife, TC279]). The membrane was washed three times with TBST buffer and then incubated with primary antibody (1:1000–2000) overnight at 4°C. The membrane was subsequently visualized by image analyzer (Tanon, 5200) followed by incubation with the secondary antibody. The density of LC3-II was first normalized to the loading control. Then, the ratio of LC3-II:loading control was quantified.
Immunostaining
Cells were cultured on glass-bottom culture dishes (Nest Scientific, 801002). After treatment, cells were fixed with 4% paraformaldehyde for 20 min, and subsequently permeabilized with 0.01% Triton X-100 (BBI, A600198) for 30 min at room temperature. Cells were blocked in fetal goat serum (Boster Biological Technology, AR1009) after washing three times with phosphate-buffered saline (PBS, Hyclone, SH302561.01). Then cells were incubated with primary antibodies (1:100–150) prepared with fetal goat serum overnight at 4°C. Cells were washed with PBS for three times and incubated with fluorescence-labeled secondary antibody Alexa Fluor 488 or DyLight 594 secondary antibodies (ThermoFisher Scientific, 710369, 35560). Images were observed using laser confocal microscope (Olympus, FV3000) or EVOS FL Auto (Life Technologies, Bothell, WA). For manual quantification of the puncta formation, at least three optical fields with at least 50 cells per experimental condition were analyzed. Data from repeated experiments were subjected to statistical analysis.
Reverse transcription quantitative PCR (RT-Qpcr)
Total RNA was extracted from cultured cells with Trizol reagent (Invitrogen, 15596026) and then was reversely transcripted to cDNA using One-step RT Kit (Takara Biotechnology, Japan). The mRNA levels of target genes were detected using SYBR-Green Quantitative PCR kit (Takara Biotechnology, RR067A) by iCycler iQ system (Bio-Rad, USA). Amplification conditions were 15 min at 95°C, followed by 40 cycles of 30 s at 95°C, 1 min at 55°C, and 30 s at 72°C. All PCR reactions were done in triplicate. Mouse or human-specific primers were synthesized by Invitrogen (Table S1). GAPDH or β-Actin were served as endogenous control.
Virus propagation
HSV-1-GFP strain was obtained from MOE Key Laboratory of Gene Function and Regulation (Guangzhou, China). HSV-1-GFP strain was propagated and titer measured by plague assays on Vero cells and used at the indicated multiplicity of infection in MEF or HeLa cells. To prepare viral stock, adsorption of HSV-1-GFP on Vero cells at 37°C for 1 h, unbound viruses were removed by washing three times with phosphate-buffered saline (PBS, Hyclone, SH302561.01). Coated cells with DMEM containing 2% FBS and incubated at 37°C for 10 h in an atmosphere of 5% CO2. Viral stock was clarified by centrifugation at 1000×g for 10 min. Supernatants containing the viruses were collected, filtered and concentrated by PEG8000 precipitation.
Electron microscopy
Electron microscopy was performed by Japan Electron Optics Laboratory Co., Ltd., JEM-1400 PLUS based on a chemical-fixation method. Chemical-fixation solution containing 2.5% glutaraldehyde was diluted in 0.1 M phosphate buffer, pH 7.5, and rb1cc1−/− MEF cells were fixed for more than 2 h after centrifugation at 800×g for 3 min. These samples were washed with 0.1 M phosphate buffer three times for at least 30 min, followed by further fixation with 1% osmic acid (TED PELLA, 18456) diluted in 0.1 M phosphate buffer for 1 h at 4°C and the same washing process as above. Then the samples were dehydrated using different concentrations of ethanol (30%, 50%, 70%, 90%, 100%) for 5 min each, a mixture of anhydrous acetone and anhydrous ethanol (1:1) for 10 min, and anhydrous acetone two times for 10 min each. After dehydration, these samples were infiltrated with a 1:1, 1:2 and 0:1 mixture of anhydrous acetone and Epon812 resin (TED PELLA, GP18010) at 38°C for 4 h, followed by embedding in Epon812 resin for overnight. Polymerization was performed at 37°C, 45°C, and 60°C for 12 h, 12 h, and 48 h respectively, and then the resin was ultra-thin sectioned using a 70 nm-thick ultramicrotome (UC-7). After that, the sections were stained with 2% uranyl acetate (EMS, 22400) diluted in 70% ethanol for 30 min and lead citrate (TED PELLA, 19314) diluted in double distilled water for 15 min. After washed by double distilled water and dried under the baking lamp, samples were observed through a transmission electron microscope (JEM-1400 PLUS) at a voltage of 100 kV and images were acquired using a CCD camera (EMSIS, MORADA).
Flow cytometry
MEF or HeLa cells were treated with or without 1 μM of cGAMP for indicated time, or transfected with STING1 plasmids or not for 24 h, then subjected to HSV-1 virus with GFP-labeled infection at a multiplicity of infection (MOI) of 5. At 12 h p.i., cells were washed twice with fluorescence activated cell sorting (FACS) buffer (20 mM PBS, pH 7.5, 1% BSA), fixed with 2% paraformaldehyde, and analyzed on BD FACS Calibur (BD Biosciences) in FL1 channel (Ex 488 nm, Em 525 nm) for EGFP signal detection. Data analysis was performed using FlowJo software.
DQ-BSA Red assay
Cells were incubated with 10 µg/mL of DQ-BSA Red (Thermofisher, D12051) in EBSS medium at 37°C for 2 h, and then new medium with or without the indicated compounds were added for another 4 or 6 h, and then fluorescence microscopy was used to detect the intensity of red fluorescent.
Long-lived protein degradation assay
Long-lived protein degradation assay was performed in accordance with the protocol reported before [36]. Cells to be tested were incubated with L-methionine-free DMEM supplemented with 4 mM of glutamine (BBI Life Sciences, GB0224), 0.2 mM of L-cystine (Solarbio, C7480), 1 mM of sodium pyruvate (Macklin, S817535) and 50 μM of AHA (MCE, HY-140346A) with 10% dialyzed FBS (Biological Industries, 2148391). After incubation for 18 h, returned to normal medium and incubated for 2 h. Cells were subjected to 1 μM of cGAMP in the presence of 10×L-methionine for 12 h. Then cells were collected, fixed, washed, permeabilized before incubated in the click reaction master mix (50 μM TAMRA alkyne [AAT Bioquest, AAT-487], 1 mM TCEP [Calbiochem, 580560], 100 μM TBTA [GLPBIO, GC45003], 1 mM CuSO4 [Sangon Biotech, C3008] in PBS) in the dark for 2 h. After click reaction, resuspended in 500 μL PBS and analyzed by flow cytometry.
Protease-protection assay
The protease-protection assay was performed as previously described. Cells were treated with cGAMP for 24 h prior to collection by trypsinization (Sango biotech, A610629–0050) at 37°C. Cells were next centrifuged and resuspended in 0.7 ml of ice-cold homogenization buffer (10 mM HEPES-KOH, 0.07 M sucrose, 0.22 M D-mannitol, 1 mM EDTA, 1 mM DTT, pH 7.5). All following steps were performed on ice or at 4°C. The proteins were extracted by rupturing the cells using 27 G needles with a 1 ml syringe for 10 times. Then, cells were centrifuged twice at 300×g to discard the nuclear pellet. Each sample was divided into 2 tubes for subsequent permeabilization and counterpart non-permeabilization with 5% Triton X-100 in homogenization buffer to a working concentration of 0.5%. Each 2 samples above were divided into 3 parts, one was treated with 1 mg/ml of trypsin to a final concentration of 100 μg/ml at 37°C for 30 min. The rest two parts were added with an equivalent volume of homogenization buffer at 37°C or ice for 30 min. All samples were prepared for further Western blot analysis.
Extraction of lysosomal fraction
Lysosomal protein fraction was extracted using a lysosomal protein extraction kit (Bestbio, BB-31,452–2) according to the manufacturer’s instructions. In brief, collected cells were suspended with lysosome extraction reagent A and oscillated on ice for 10 min. Then the cells were homogenized 35 times in a Dounce homogenizer (Dongguan Kemao Biological Technology, KM-10-024-2) followed by centrifugation at 1,000×g for 5 min. Supernatant fractions were centrifuged at 30,000×g for 30 min and the supernatant was collected and labeled as the cytosolic fraction. The resulting precipitates were suspended with lysosome extraction reagent B and further centrifuged at 30,000×g for 30 min. The precipitates were suspended with lysosome extraction reagent C and oscillated for 20 min followed by centrifugation at 14,000×g for 15 min. The supernatant was taken as the lysosomal protein fraction.
Statistics
The results shown are representative of at least three independent experiments. The data are presented as the mean±standard error of mean. The statistical significance of data was verified by the Student’s t-test wherever required. P-value <0.05 was considered as being significant.
Supplementary Material
Acknowledgements
Dedicated to the 20th anniversary of School of Pharmaceutical Sciences, Sun Yat-sen University. We thank Jun Cui (Sun Yat-sen University, Guangzhou, China) for HSV-1-GFP virus and BECN1-/- HEK-293T cells, Du Feng (Guangzhou Medical University, Guangzhou, China) for WT and atg5−/− MEFs, Yueguang Rong (Tongji Medical College of HUST, Wuhan) for ATG9A-/- HeLa cells, Lei Liu (Chinese Academy of Sciences, Beijing, China) for ATG13-/- HeLa cells, and Feng Shao & Yue Xu (NIBS, Beijing, China) for SopF and SopFY224D plasmids. This work was supported by the National Natural Science Foundation of China (31970699, 31671437), the Guangdong Basic and Applied Basic Research Foundation (2021A1515010766, 2019A1515011030), the Key-Area Research and Development Program of Guangdong Province (2020B1111110003), the Guangdong Provincial Key Laboratory of Construction Foundation (2019B030301005), and the key Research and Development Plan of Guangzhou City (202206080007).
Funding Statement
This work was supported by the National Natural Science Foundation of China (31970699, 31671437), the Guangdong Basic and Applied Basic Research Foundation (2021A1515010766, 2019A1515011030), the Key-Area Research and Development Program of Guangdong Province (2020B1111110003), the Guangdong Provincial Key Laboratory of Construction Foundation (2019B030301005), the key Research and Development Plan of Guangzhou City (202206080007), and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y093).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2237794
References
- [1].Takeuchi O, Akira S.. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. PMID: 20303872. doi: 10.1016/j.cell.2010.01.022 [DOI] [PubMed] [Google Scholar]
- [2].Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–678. PMID: 18724357. doi: 10.1038/nature07317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788–792. PMID: 19776740. doi: 10.1038/nature08476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen Q, Sun L, Chen ZJ. Regulation and function of the Cgas–STING pathway of cytosolic DNA sensing. Nat Immunology. 2016;17(10):1142–1149. PMID: 27648547. doi: 10.1038/ni.3558 [DOI] [PubMed] [Google Scholar]
- [5].Zhang BC, Nandakumar R, Reinert LS, et al. STEEP mediates STING ER exit and activation of signaling. Nat Immunology. 2020;21:868–879. PMID: 32690950. doi: 10.1038/s41590-020-0730-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of Cgas-STING signalling. Nat Rev Mol Cell Biol. 2020;21:501–521. PMID: 32424334. doi: 10.1038/s41580-020-0244-x [DOI] [PubMed] [Google Scholar]
- [7].Dobbs N, Burnaevskiy N, Chen D, et al. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe. 2015;18(2):157–168. PMID: 26235147. doi: 10.1016/j.chom.2015.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang X, Shi H, Wu J, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Molecular Cell. 2013;51(2):226–235. PMID: 23747010. doi: 10.1016/j.molcel.2013.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shang G, Zhu D, Li N, et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat Struct Mol Biol. 2012;19(7):725–727. PMID: 22728660. doi: 10.1038/nsmb.2332 [DOI] [PubMed] [Google Scholar]
- [10].Ergun SL, Fernandez D, Weiss TM, et al. STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell. 2019;178(2):290–301.e10. PMID: 31230712. doi: 10.1016/j.cell.2019.05.036 [DOI] [PubMed] [Google Scholar]
- [11].Liu D, Wu H, Wang C, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Diff. 2019;26(9):1735–1749. PMID: 30568238. doi: 10.1038/s41418-018-0251-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gui X, Yang H, Li T, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567(7747):262–266. PMID: 30842662. doi: 10.1038/s41586-019-1006-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. PMID: 21248839. doi: 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rodgers MA, Bowman JW, Liang Q, et al. Regulation where autophagy intersects the inflammasome. Antioxid Redox Signaling. 2014;20(3):495–506. PMID: 23642014. doi: 10.1089/ars.2013.5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fischer TD, Wang C, Padman BS, et al. STING induces LC3B lipidation onto single-membrane vesicles via the V-ATPase and ATG16L1-WD40 domain. J Cell Bio. 2020;219(12):PMID: 33201170. doi: 10.1083/jcb.202009128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2011;13:7–12. PMID: 22166994. doi: 10.1038/nrm3249 [DOI] [PubMed] [Google Scholar]
- [17].Chamilos G, Akoumianaki T, Kyrmizi I, et al. Melanin targets LC3-associated phagocytosis (LAP): A novel pathogenetic mechanism in fungal disease. Autophagy. 2016;12(5):888–889. PMID: 27028978. doi: 10.1080/15548627.2016.1157242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Heckmann BL, Teubner BJW, Tummers B, et al. LC3-associated endocytosis facilitates β-amyloid clearance and mitigates neurodegeneration in murine alzheimer’s disease. Cell. 2019;178(3):536–551.e14. PMID: 31257024. doi: 10.1016/j.cell.2019.05.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Overholtzer M, Mailleux AA, Mouneimne G, et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131(5):966–979. PMID: WOS:000251800900018. doi: 10.1016/j.cell.2007.10.040 [DOI] [PubMed] [Google Scholar]
- [20].Durgan J, Lystad AH, Sloan K, et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Molecular Cell. 2021;81(9):2031–2040.e8. PMID: 33909989. doi: 10.1016/j.molcel.2021.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sui X, Liang X, Chen L, et al. Bacterial xenophagy and its possible role in cancer: A potential antimicrobial strategy for cancer prevention and treatment. Autophagy. 2017;13(2):237–247. PMID: 27924676. doi: 10.1080/15548627.2016.1252890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dupont N, Leroy C, Hamaï A, et al. Long-lived protein degradation during autophagy. Methods Enzymol. 2017;588:31–40. PMID: 28237108. doi: 10.1016/bs.mie.2016.09.074 [DOI] [PubMed] [Google Scholar]
- [23].Gao Y, Liu Y, Hong L, et al. Golgi-associated LC3 lipidation requires V-ATPase in noncanonical autophagy. Cell Death Dis. 2016;7(8):e2330. PMID: 27512951. doi: 10.1038/cddis.2016.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Florey O, Gammoh N, Kim SE, et al. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy. 2015;11(1):88–99. PMID: 25484071. doi: 10.4161/15548627.2014.984277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xu Y, Zhou P, Cheng S, et al. A bacterial effector reveals the V-ATPase-ATG16L1 axis that initiates xenophagy. Cell. 2019;178:552–566 e20. doi: 10.1016/j.cell.2019.06.007 PMID: 31327526. [DOI] [PubMed] [Google Scholar]
- [26].Xu Y, Cheng S, Zeng H, et al. ARF GTPases activate Salmonella effector SopF to ADP-ribosylate host V-ATPase and inhibit endomembrane damage-induced autophagy. Nat Struct Mol Biol. 2022;29(1):67–77. PMID: 35046574. doi: 10.1038/s41594-021-00710-6 [DOI] [PubMed] [Google Scholar]
- [27].Hooper KM, Jacquin E, Li T, et al. V-ATPase is a universal regulator of LC3-associated phagocytosis and non-canonical autophagy. J Cell Bio. 2022;221. doi: 10.1083/jcb.202105112. PMID: 35511089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zellner S, Behrends C. Autophagosome content profiling reveals receptor-specific cargo candidates. Autophagy. 2021;17(5):1281–1283. PMID: 33779494. doi: 10.1080/15548627.2021.1909410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang R, Wang J, Hassan A, et al. Molecular basis of V-ATPase inhibition by bafilomycin A1. Nat Commun. 2021;12(1):1782. PMID: 33741963. doi: 10.1038/s41467-021-22111-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fletcher K, Ulferts R, Jacquin E, et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. Embo J. 2018;37(4):PMID: 29317426. doi: 10.15252/embj.201797840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jia M, Qin D, Zhao C, et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat Immunology. 2020;21:727–735. PMID: 32541831. doi: 10.1038/s41590-020-0699-0 [DOI] [PubMed] [Google Scholar]
- [32].Gammoh N. The multifaceted functions of ATG16L1 in autophagy and related processes. J Cell Sci. 2020;133(20): PMID: 33127840. doi: 10.1242/jcs.249227 [DOI] [PubMed] [Google Scholar]
- [33].Wang W, Hu D, Wu C, et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLOS Pathogens. 2020;16(3):e1008335. PMID: 32187211. doi: 10.1371/journal.ppat.1008335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Abdul-Sater AA, Tattoli I, Jin L, et al. Cyclic-di-GMP and cyclic-di-AMP activate the NLRP3 inflammasome. EMBO Rep. 2013;14(10):900–906. PMID: 24008845. doi: 10.1038/embor.2013.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14(2):207–215. PMID: 28933638. doi: 10.1080/15548627.2017.1378838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang J, Zhang J, Lee YM, et al. Nonradioactive quantification of autophagic protein degradation with L-azidohomoalanine labeling. Nat Protoc. 2017;12(2):279–288. PMID: 28079880. doi: 10.1038/nprot.2016.160 [DOI] [PubMed] [Google Scholar]
- [37].Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151(6):1256–1269. PMID: 23217709. doi: 10.1016/j.cell.2012.11.001 [DOI] [PubMed] [Google Scholar]
- [38].Marwaha R, Sharma M. DQ-Red BSA trafficking assay in cultured cells to assess cargo delivery to lysosomes. Bio-protocol. 2017;7(19): PMID: 29082291. doi: 10.21769/BioProtoc.2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yamashiro LH, Wilson SC, Morrison HM, et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat Commun. 2020;11(1):3382. PMID: 32636381. doi: 10.1038/s41467-020-17156-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wu J, Dobbs N, Yang K, et al. Interferon-independent activities of mammalian STING mediate antiviral response and tumor immune evasion. Immunity. 2020;53(1):115–126.e5. PMID: 32640258. doi: 10.1016/j.immuni.2020.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu Y, Luo X, Shan H, et al. Niclosamide triggers non-canonical LC3 lipidation. Cells. 2019;8(3):248. PMID: 30875964. doi: 10.3390/cells8030248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. 2020;180(6):1044–1066. PMID: 32164908. doi: 10.1016/j.cell.2020.02.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Silwal P, Kim IS, Jo EK. Autophagy and host defense in nontuberculous mycobacterial infection. Front Immunol. 2021;12:728742. PMID: 34552591. doi: 10.3389/fimmu.2021.728742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Rey-Jurado E, Riedel CA, González PA, et al. Contribution of autophagy to antiviral immunity. FEBS Lett. 2015;589(22):3461–3470. PMID: 26297829. doi: 10.1016/j.febslet.2015.07.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.