

ABSTRACT
COL6 (collagen type VI)-related myopathies (COL6-RM) are a distinct group of inherited muscle disorders caused by mutations of COL6 genes and characterized by early-onset muscle weakness, for which no cure is available yet. Key pathophysiological features of COL6-deficient muscles involve impaired macroautophagy/autophagy, mitochondrial dysfunction, neuromuscular junction fragmentation and myofiber apoptosis. Targeting autophagy by dietary means elicited beneficial effects in both col6a1 null (col6a1–/–) mice and COL6-RM patients. We previously demonstrated that one-month per os administration of the nutraceutical spermidine reactivates autophagy and ameliorates myofiber defects in col6a1–/– mice but does not elicit functional improvement. Here we show that a 100-day-long spermidine regimen is able to rescue muscle strength in col6a1–/– mice, with also a beneficial impact on mitochondria and neuromuscular junction integrity, without any noticeable side effects. Altogether, these data provide a rationale for the application of spermidine in prospective clinical trials for COL6-RM.
Abbreviations: AChR: acetylcholine receptor; BTX: bungarotoxin; CNF: centrally nucleated fibers; Colch: colchicine; COL6: collagen type VI; COL6-RM: COL6-related myopathies; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; NMJ: neuromuscular junction; Spd: spermidine; SQSTM1/p62: sequestosome 1; TA: tibialis anterior; TOMM20: translocase of outer mitochondrial membrane 20; TUNEL: terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling.
KEYWORDS: Autophagy, collagen VI, nutraceutical, skeletal muscle, spermidine
Introduction
Macroautophagy (hereafter referred to as autophagy) is a tunable catabolic process that ensures the lysosomal-mediated degradation of organelles and cytoplasmic cargoes and proteins, thus facilitating their recycling and the subsequent release of fundamental biomolecules [1,2]. Skeletal muscle tissue, being constantly subjected to mechanical and metabolic stresses during contractile activity, requires a precise regulation of the autophagy process to cope with the ensuing cellular damages that otherwise would progressively accumulate over time within myofibers [3,4].
About one decade ago, a tight association between autophagic flux impairment and myopathic pathology was demonstrated in COL6-RM, a distinctive and heterogeneous class of congenital muscular dystrophies characterized by progressive muscle wasting and weakness and caused by mutations of COL6 (collagen type VI) genes [5–7]. Work on the most-characterized animal models for COL6-RM, the col6a1 null (col6a1–/–) mouse, together with studies in patients’ muscle biopsies, allowed to throw light on the mechanisms underlying the myopathic defects, showing that the pathogenesis of COL6-RM involves autophagy failure, mitochondrial dysfunction and spontaneous apoptosis of myofibers, together with neuromuscular junction (NMJ) defects ([8–11] for a review see [12]).
Since autophagy is a very dynamic process which can be finely modulated at multiple levels by a wide range of endogenous and exogenous stimuli, a number of studies in the past years were aimed at the normalization of the autophagic process in COL6-deficient muscles. Indeed, reactivation of autophagy in col6a1–/– mice was achieved by genetic, pharmacological and dietary strategies [9,10,13], also leading to a successful pilot clinical trial in COL6-RM patients [13]. However, none of these interventions can be adopted as optimal therapies for patients, since they present several potential issues and drawbacks for long-term regimens.
A different and more attractive strategy for achieving autophagy reactivation in COL6-deficient muscles is based on the administration of nutraceutical compounds, such as the polyamine spermidine (Spd) and the stilbenoid pterostilbene [10,14]. Spd is a cationic polyamine ubiquitously present in living organisms and with several beneficial activities in cells, ranging from promotion of protein translation to protection from oxidative damage [15–17]. Interestingly, Spd acts as an autophagy-inducer and anti-aging compound, being therefore considered a caloric-restriction mimetic [18,19]. Moreover, dietary or externally supplied Spd appeared to be safe and it has well-documented beneficial effects on health and life span promotion in diverse animal models, as well as in humans [18–24].
In our previous work, we showed that Spd administration to col6a1–/– mice elicits beneficial effects in several myopathic hallmarks [10]. In particular, i.p. injection of 50 mg/kg Spd for 10 consecutive days and per os administration of 30 mM Spd in drinking water for 30 days led to an amelioration of mitochondria ultrastructural defects and a significantly lower incidence of apoptotic myonuclei, with decreased myofiber degeneration. However, none of the Spd regimens investigated in that study was able to induce a significant amelioration of contractile force in col6a1–/– mice [10], leaving open the field to further studies aimed at addressing the question whether in the long-term Spd is able to improve muscle strength besides its benefits on muscle morphology.
Here we aimed at addressing such aspect, which is of major relevance for the translation of Spd in the clinical setting and its prospective therapeutic use in clinical trials for COL6-RM. Towards this aim, we focused on oral administration of Spd in drinking water, being the most feasible route for prospective clinical applications. Our data show that long-term (100-day-long) per os administration of 30 mM Spd is successful in rescuing muscle strength in col6a1–/– mice by inducing both autophagic and mitophagic fluxes, also leading to a marked amelioration of mitochondria ultrastructure, NMJ integrity and myofiber degeneration. Altogether, these findings therefore point at Spd treatment as a promising and feasible strategy for therapeutic interventions aimed at the treatment of COL6-RM, also suggesting that it may be beneficial in other muscle disorders whose pathomolecular features involve defective autophagy.
Results and discussion
Long-term Spd treatment rescues muscle strength in COL6-deficient mice
In our previous work, we showed that one-month per os administration of 30 mM Spd to col6a1–/– mice was able to reactivate autophagy, with beneficial effects on some structural features of COL6-deficient muscles, such as mitochondria abnormalities and myofiber degeneration [10]. However, that approach did not allow to obtain a functional recovery of the myopathic phenotype, as it was unable to induce a significant amelioration of muscle strength.
To assess whether longer Spd administration regimens lead to a more robust impact on muscle function, we treated wild-type and col6a1–/– mice for 60 and 100 days with 30 mM Spd, added to their drinking water. At the end of the treatments (6 months of age for all groups of mice), we assessed muscle strength by quantification of the in vivo contractile force of the plantar flexor muscles (mainly represented by the gastrocnemius muscle). The shorter Spd regimen (60 days) led to significantly increased absolute force production in col6a1–/– mice (Figure 1A), which however became only a positive trend when normalized for muscle mass (Figure 1B), thus pointing at a moderately positive effect with this regimen. On the other hand, 100-day-long Spd treatment achieved a more robust force amelioration in col6a1–/– mice, with a significant improvement of both absolute (Figure 1C) and normalized (Figure 1D) force. Of note, 30 mM Spd administration for 100 days led to a complete recovery of muscle strength, as both absolute and normalized forces of col6a1–/– mice were not significantly different from those of untreated wild-type mice. To further evaluate functional amelioration, we performed the four-limb hanging test, assessing the ability of the mice to hang on a reversed grid over time. Untreated col6a1–/– animals displayed a significantly lower holding impulse compared to wild-type animals, and both 60-day and 100-day Spd treatments significantly improved the hanging performance of col6a1–/– mice (Figure 1E). Furthermore, and at difference from the previous findings with high-dose Spd i.p. injection for 10 days [10] per os administration of Spd at 30 mM for either 60 days or 100 days did not negatively impinge on contractile strength and hanging performance of wild-type animals (Figure 1A–E).
Figure 1.
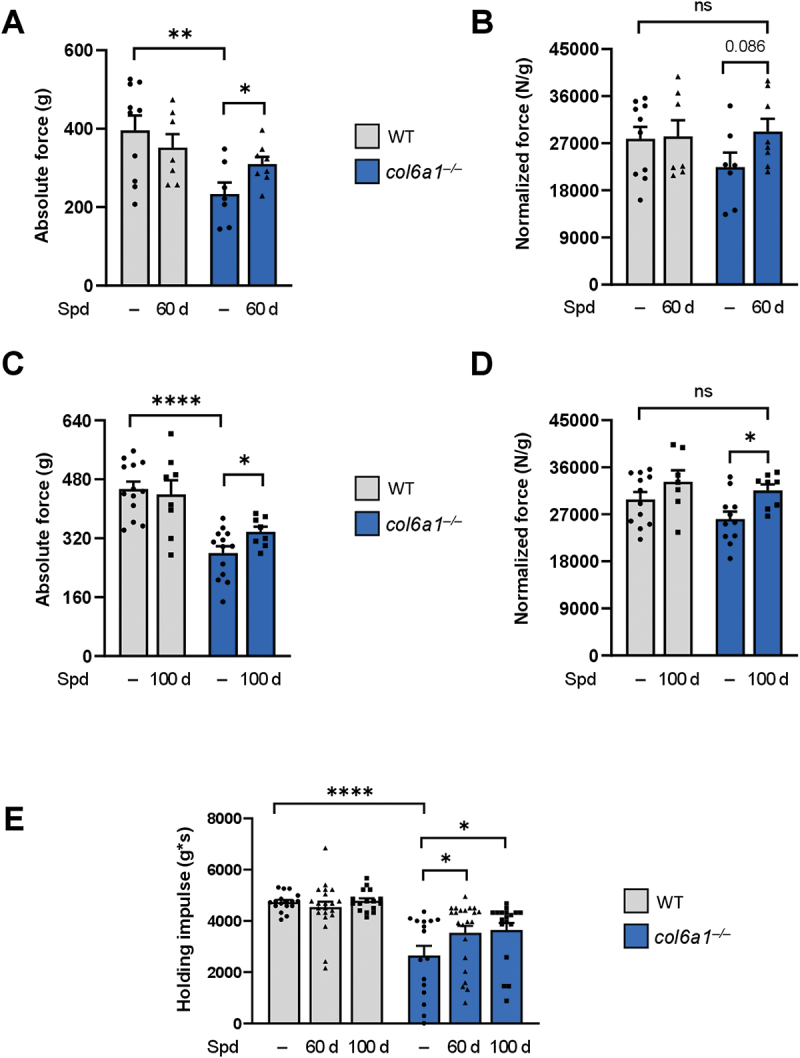
Long-term Spd treatment rescues muscle strength in col6a1–/– mice. (A-D) In vivo quantification of the absolute tetanic force (panels A and C) and tetanic force normalized on muscle weight (panels B and D) of plantar flexor muscles, at 100 Hz frequency of stimulation, in wild-type and col6a1–/– mice under untreated conditions (–) and after 60 days (60 d, panels A and B) or 100 days (100 d, panels C and D) per os administration of 30 mM Spd. Data are provided as mean±s.e.m. (n = 8–12 muscles, each group; ****, P < 0.0001; **, P < 0.01; *, P < 0.05; ns, not significant; Student’s t-test). (E) Bar plot displaying the hanging impulse, calculated as the recorded hanging time performance normalized per body weight with the four-limb hanging test, in wild-type and col6a1–/– mice under untreated conditions (–) and after 60 days (60 d) or 100 days (100 d) per os administration of 30 mM Spd. Data are provided as mean±s.e.m. (n = 16–22 mice, each group; ****, P < 0.0001; *, P < 0.05; two-way ANOVA test with Holm-Šídák’s post hoc test for multiple comparison). WT, wild type.
These findings, together with the evidence that water and Spd consumption did not differ between the two genotypes and that body and muscle weights were not negatively affected by Spd treatment (Figure S1), confirmed the safety of long-term per os Spd administration and indicated that it is able to improve muscle function in the myopathic COL6-deficient mouse model.
Long-term Spd treatment ameliorates the major histopathological defects of COL6-deficient muscles
Myofiber degeneration is a well-known myopathic sign in both col6a1–/– mice and patients affected by COL6-RM [5,7,8,25,26]. To assess whether, as for high-dose Spd treatments in our previous work [10], prolonged oral Spd administrations were able to prevent the myofiber apoptosis and muscle degeneration of col6a1–/– mice, we evaluated the amount of centrally nucleated myofibers (CNF) and apoptotic nuclei in tibialis anterior (TA) muscles of col6a1–/– mice treated or not with Spd. Oral Spd supplementation for 100 days significantly decreased the percentage of CNF in col6a1–/– mice, pointing to a reduction of degeneration/regeneration events (Figure 2A,B). In addition, TUNEL analysis revealed a significant decrease in the incidence of apoptotic myonuclei in TA of col6a1–/– muscle upon both 60- and 100-day Spd treatments (Figure 2C). A more general morphometric analysis of TA muscle from Spd-treated mice highlighted no detrimental macroscopic effects in the myofiber cross-sectional area and in fibrosis content (data not shown), again pointing at the high tolerability of Spd and the lack of major adverse effects at the used doses and regimens.
Figure 2.
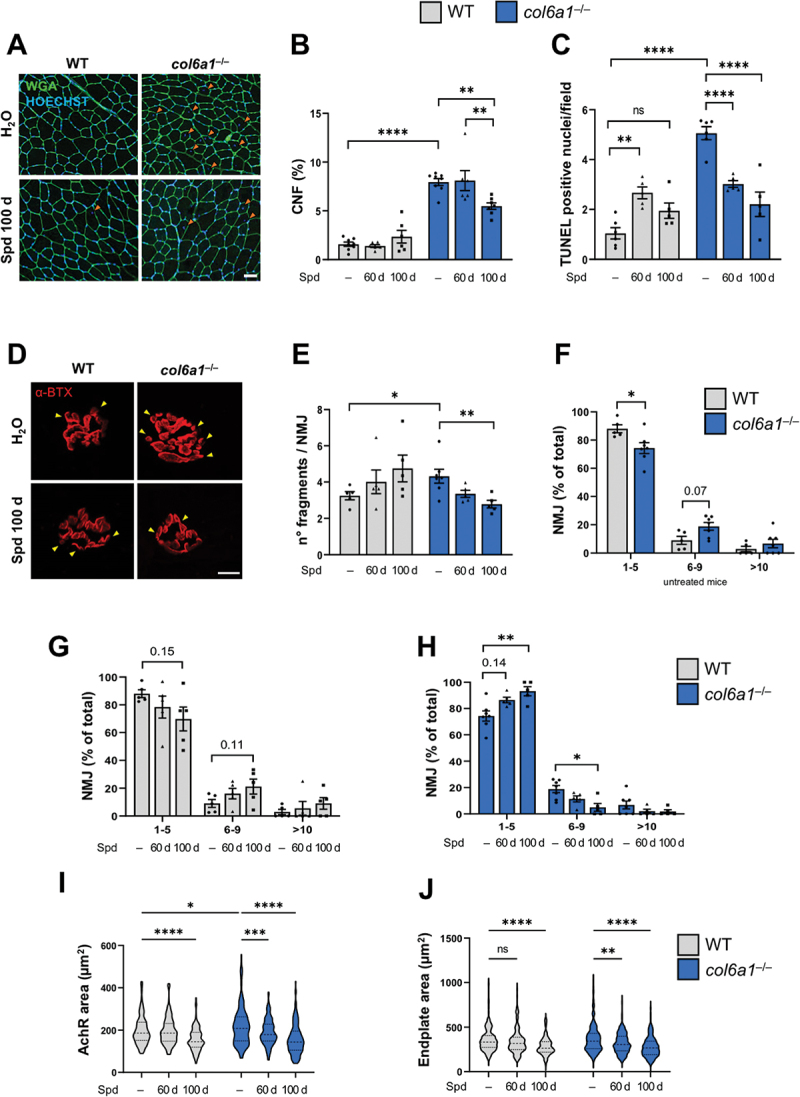
Long-term Spd treatment ameliorates the histopathological defects of col6a1–/– muscles. (A) Representative images of TA cross-sections from wild-type and col6a1–/– mice treated (Spd 100 d) or not (H2O) with 30 mM Spd administered per os for 100 days. Sections were labeled with wheat germ agglutinin (green) and Hoechst (blue). Orange arrowheads point at centrally nucleated myofibers. Scale bar: 50 µm. (B) Bar plot of the percentage of centrally nucleated myofibers (% CNF), quantified from images as in (A), in wild-type and col6a1–/– mice under untreated conditions (–) and after 60 days (60 d) or 100 days (100 d) per os administration of 30 mM Spd. Data are provided as mean±s.e.m. (n = 6–8, each group; ****, P < 0.00001; **, P < 0.01; two-way ANOVA test with Holm-Šídák’s post hoc test for multiple comparison). (C) Bar plot of the number of TUNEL-positive nuclei in TA cross sections from wild-type and col6a1–/– mice under untreated conditions (–) and after 60 days (60 d) or 100 days (100 d) per os administration of 30 mM Spd. Data are provided as mean±s.e.m. (n = 5–6, each group; ****, P < 0.0001; **, P < 0.01; ns, not significant; two-way ANOVA test with Holm-Šídák’s post hoc test for multiple comparison). (D) Representative confocal max-stack immunofluorescent images of NMJ in diaphragm whole-mount preparations from wild-type and col6a1–/– mice treated (Spd 100 d) or not (H2O) with 30 mM Spd administered per os for 100 days. Sections were labeled with α-bungarotoxin (α-BTX, in red). Yellow arrowheads point at NMJ fragments. Scale bar: 10 µm. (E) Bar plot of the number of AChR fragments per NMJ, quantified from 3D reconstruction of images as in (A), in wild-type and col6a1–/– mice under untreated conditions (–) and after 60 days (60 d) or 100 days (100 d) per os administration of 30 mM Spd. At least 35 NMJ were analyzed for each muscle. Data are provided as mean±s.e.m. (n = 5–7 mice, each group; *, P < 0.05; **, P < 0.01; Mann-Whitney test and Kruskal-Wallis test with Dunn’s post hoc test for multiple comparison). (F-H) Bar plot of NMJ distribution over three separate classes based on the number of AChR fragments (e.g., 1 to 5, 6 to 9, more than 10), quantified from 3D reconstruction of images as in (A), in untreated wild-type and col6a1–/– mice (F), and in wild-type mice (G) and col6a1–/– mice (H) under untreated conditions (–) and after 60 days (60 d) or 100 days (100 d) per os administration of 30 mM Spd treatment. At least 35 NMJ were analyzed for each muscle. Data are provided as mean±s.e.m. (n = 5–7 mice, each group; *, P < 0.05; **, P < 0.01; Mann-Whitney test and Kruskal-Wallis test with Dunn’s post hoc test for multiple comparison). (I-J) Violin plot of AChR area (in µm2, panel I) and of endplate area (in µm2, panel J) of NMJ in wild-type and col6a1–/– mice under untreated conditions (–) and after 60 days (60 d) or 100 days (100 d) per os administration of 30 mM Spd (n = 137–218 NMJ sampled from 5–7 mice, each group; ****, P < 0.0001; ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, not significant; two-way ANOVA test with Holm-Šídák’s post hoc test for multiple comparison). WT, wild type.
We recently showed that COL6 is a component of the synaptic cleft, and its deficiency in col6a1–/– mice leads to increased NMJ fragmentation and increased acetylcholine receptor (AChR) area [11]. To assess whether NMJ morphology of COL6-deficient muscles is modulated by supplementation of nutraceutical autophagy-inducing compounds, we quantified the number of AChR fragments per NMJ in diaphragm of control and Spd-treated wild-type and col6a1–/– mice. Whole-mount staining of AChR with fluorophore-conjugated α-bungarotoxin confirmed that the NMJ of untreated col6a1–/– mice were more fragmented when compared to wild-type samples, as expected (Figure 2D–F). Interestingly, Spd treatments led to a progressive NMJ remodeling and a significant amelioration of AChR fragmentation in col6a1–/– samples (Figure 2D,E), with an almost negligible effect in wild-type NMJs (Figure 2D,E–G). This improvement was also reflected by NMJ redistribution toward lower fragment classes in col6a1–/– diaphragm upon Spd administration (Figure 2H). In agreement with these data, the postsynaptic area, which is known to be enlarged in col6a1–/– NMJ [11], was progressively reduced in both wild-type and col6a1–/– mice upon Spd treatments, as displayed by AChR area (Figure 2I) and endplate area (Figure 2J).
Taken together, these data reveal that oral Spd administration induces muscle remodeling in a dose-dependent manner in col6a1–/– mice, ameliorating several histopathological hallmarks of COL6-deficient myopathic muscles, such as the incidence of CNF, apoptotic nuclei and fragmented NMJ.
Muscle autophagic and mitophagic fluxes are reactivated by long-term Spd administration in COL6-deficient mice
A key pathophysiological mechanism causing accretion of dysfunctional mitochondria, myofiber degeneration and muscle weakness in COL6-deficient mice is the defective autophagy [9]. Our previous work showed that 30-day oral Spd administration is able to moderately promote autophagy in muscles of col6a1–/– mice, an effect that becomes evident in 24-h fasted animals [10]. Therefore, we aimed at assessing whether oral Spd administration for 100 days elicited more robust effects in reactivating the autophagic flux of COL6-deficient mice. Toward this aim, we performed autophagic flux analysis by monitoring the lipidation of MAP1LC3B (hereafter referred as LC3) upon Spd administration and after colchicine (Colch) injection, to inhibit the late stages of autophagosome maturation to autolysosome. Western blotting on protein extracts from whole TA muscle lysates showed that untreated col6a1–/– mice displayed a significantly lower accumulation of LC3-II upon Colch injection when compared to the corresponding wild-type samples (Figure 3A, B), as expected and in agreement with the previously described autophagy impairment [10,13]. Interestingly, muscle extracts of col6a1–/– mice treated for 100 days with Spd displayed a significantly higher Colch-dependent LC3-II accumulation when compared to the corresponding samples of untreated col6a1–/– animals (Figure 3A,B), pointing at robust autophagy reactivation even in the absence of fasting.
Figure 3.
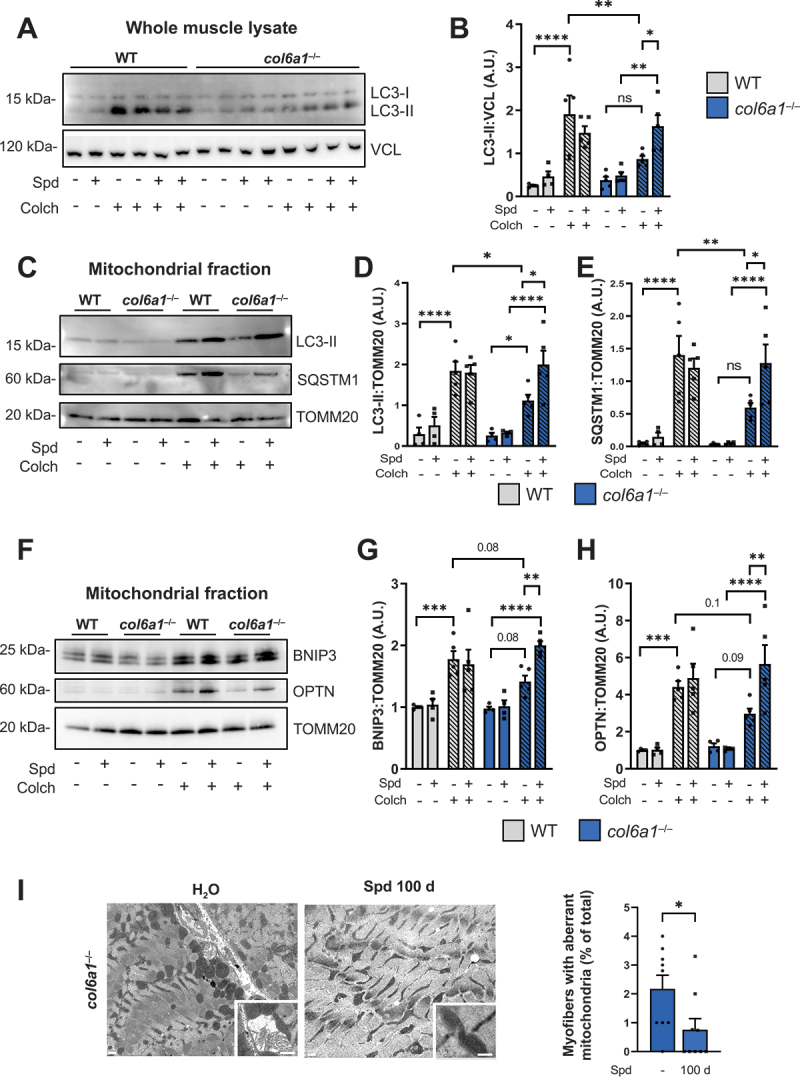
Autophagic and mitophagic fluxes of col6a1–/– muscles are reactivated by long-term Spd administration. (A, B) Western blotting (panel A) and densitometric quantification (determined from three independent western blot experiments, panel B) of the non lipidated (LC3-I) and lipidated (LC3-II) forms of LC3B protein in whole muscle protein extracts of wild-type and col6a1–/– mice treated (Spd +) or not (Spd –) with 30 mM Spd administered per os for 100 days, and injected (Colch +) or not (Colch –) with 0.4 mg/kg colchicine. VCL (vinculin) was used as loading control. Data are provided as mean±s.e.m. (n = 4–5 mice, each group; ****, P < 0.0001; **, P < 0.01; *, P < 0.05; ns, not significant; two-way ANOVA test with Holm-Šídák’s post hoc test for multiple comparison). (C-E) Western blotting (panel C) and densitometric quantification (determined from three independent western blot experiments) of the non lipidated (LC3-I) and lipidated (LC3-II) forms of LC3B (panel D) and of SQSTM1/p62 (panel E) in mitochondrial fraction of muscle extracts of wild-type and col6a1–/– mice treated (Spd +) or not (Spd -) with 30 mM Spd administered per os for 100 days, and injected (Colch +) or not (Colch –) with 0.4 mg/kg colchicine. TOMM20 was used as loading control. Data are provided as mean±s.e.m. (n = 4–5 mice, each group; ****, P < 0.0001; **, P < 0.01; *, P < 0.05; two-way ANOVA test with Holm-Šídák’s post hoc test for multiple comparison). (F-H) Western blotting (panel F) and densitometric quantification (determined from three independent western blot experiments) of BNIP3 (panel G) and OPTN1 (panel H) in mitochondrial fraction of muscle extracts of wild-type and col6a1–/– mice treated (Spd +) or not (Spd -) with 30 mM Spd administered per os for 100 days, and injected (Colch +) or not (Colch –) with 0.4 mg/kg colchicine. TOMM20 was used as loading control. Data are provided as mean±s.e.m. (n = 4–5 mice, each group; ****, P < 0.0001; ***, P < 0.001; **, P < 0.01; two-way ANOVA test with Holm-Šídák’s post hoc test for multiple comparison). (I) Representative transmission electron images of diaphragm sections from col6a1–/– mice treated (Spd 100 d) or not (H2O) with 30 mM Spd 30 mM administered per os for 100 days. Bottom squares for each image show higher magnification details of mitochondria. Scale bars: 400 nm. The bar plot on the right shows the quantification of the percentage of myofibers with aberrant mitochondria in col6a1–/– mice in untreated conditions (Spd –) and after 100 days per os administration of 30 mM Spd (100 d). Data are provided as mean±s.e.m. (n = 9, each group; *, P < 0.05; Mann-Whitney test). A.U., arbitrary unit; WT, wild type.
Besides bulk autophagy, we investigated the selective removal of mitochondria through mitophagy, by performing autophagic flux studies in mitochondria-enriched protein extracts of quadriceps muscles from control and 100-day Spd-treated wild-type and col6a1–/– mice subjected to Colch injection. These experiments suggested that the mitophagic flux is also impaired in COL6-deficient muscles, as displayed by the lower Colch-dependent accumulation of LC3-II (Figure 3C,D) and of the autophagic cargo receptor SQSTM1/p62 (Figure 3C,E) in mitochondria from untreated col6a1–/– mice compared to untreated wild-type animals. Noteworthy, the selective removal of mitochondria by mitophagy was also reactivated upon 100-day Spd oral administration, as shown by the significant Colch-dependent accumulation of LC3-II and SQSTM1 in mitochondria-enriched muscle extracts from Spd-treated col6a1–/– mice when compared to the corresponding samples from untreated col6a1–/– animals (Figure 3C–E). To verify that the detection of this two autophagic markers in the mitochondrial-enriched fraction reflected the mitophagic flux, beyond the presence of any possible contamination of other subcellular components, we evaluated the abundance of two of the most specific mitophagic receptors, BNIP3 and OPTN, in mitochondria-enriched muscle extracts of wild-type and col6a1–/– animals subjected to Colch injection. In agreement with the concept of an impaired mitophagic flux in COL6-deficient muscles, both BNIP3 and OPTN displayed a decreasing trend in their protein amounts in col6a1–/– mice compared to wild-type mice, as well as a significantly higher Colch-dependent accumulation in col6a1–/– mice upon 100-day Spd oral administration (Figure 3F–H). These findings were further corroborated by immunofluorescence microscopy on TA cross-sections followed by quantitative colocalization of LC3B and the mitochondrial marker ATP5A (Figure S2A), as well as of the mitochondrial marker TOMM20 and the endo-lysosomal marker LAMP1 (Figure S2B). These experiments revealed that 100-day-long Spd treatment leads to increase colocalization of mitochondrial markers with LC3B (Figure S2A) and LAMP1 (Figure S2B) in col6a1–/– animals, confirming a major impact of Spd in reactivating mitophagy, thus mediating mitochondria turnover.
Aberrant mitochondria with altered morphology of cristae represent a well-known feature of COL6-deficient mice and patients affected by COL6-RM [8,25]. Transmission electron microscopy on diaphragm ultrathin sections, coupled with mitochondria morphometric analysis, revealed a significantly decreased incidence of myofibers with mitochondrial abnormalities in col6a1–/– animals treated for 100 days with Spd, together with a structural improvement of the mitochondrial cristae (Figure 3I). Based on these results, and despite some limitations for mitophagy assessment in skeletal muscle tissue in vivo [27,28], we can speculate that the beneficial Spd-dependent amelioration of mitochondrial morphology in col6a1–/– mice is relying at least in part on the improved mitochondria quality control elicited by mitophagy reactivation, which assures the removal of dysfunctional mitochondria and prevents the release of pro-apoptotic factors.
Conclusions
Prolonged oral administration of 30 mM Spd to col6a1–/– mice, provided in drinking water for 100 days, is safe and beneficial, being able to recover functional and structural muscle defects. Our results indicate that Spd, by reactivating both bulk autophagy and mitophagy, leads to amelioration of mitochondria ultrastructure, decreased myofiber degeneration and improvement of NMJ morphology, promoting a marked recovery of muscle force in col6a1–/– mice.
Altogether, these findings support the efficacy of Spd as an autophagy-inducing nutraceutical for skeletal muscle, which may be of benefit for the prospective treatment of patients affected by COL6-RM. The safety and feasibility of Spd administration, used as a food or dietary supplement, may allow higher patient compliance without the risk of major side effects. Further clinical studies in the near future will allow to establish the basis for Spd-based trials in COL6-RM and other diseases whose underlying mechanisms involve impaired autophagy in skeletal muscles.
Materials and methods
Mouse maintenance and treatments
Age-matched C57BL/6N wild type and col6a1–/– female mice [11] were housed in a controlled environment with 12 h light/12 h dark cycle, a temperature of 23°C and with free access standard chow (Mucedola Srl, 4RF25). Spd (Sigma-Aldrich, S4139) was administered per os in drinking water at a final concentration of 30 mM. Control mice drank water without the addition of Spd. To avoid excessive deamination of the active compound, water with Spd was replaced three times a week [18]. The volume of drunk water was measured every day for each cage and then divided by the number of animals in the cage to assess the amount of Spd taken by mice daily. For autophagic flux analysis, mice were i.p. injected twice with 0.4 mg/kg of colchicine (Sigma-Aldrich, C9754), respectively 24 h and 12 h before sacrifice [29]. Animals were sacrificed by cervical dislocation at 6 months of age, and the tissues of interest were immediately dissected and fixed or frozen. To avoid undesired effects due to circadian variations in the basal autophagic flux, mice were sacrificed between 7.30 and 10 a.m. The adopted procedures were approved by the Ethics Committee of the University of Padova and carried out according to all pertinent Italian laws (OPBA, n. 100/2020-PR).
In vivo force measurements
Plantar flexor (gastrocnemius, plantaris and soleus) muscle strength was measured in living mice, as previously described [30]. Briefly, animals were anesthetized, and muscle contractile performance was measured in vivo using a 305B muscle lever system (Aurora Scientific Inc.). The tetanic contraction was reached for stimulations equal to 100 Hz, and force values obtained in these settings were compared between the different conditions, as they represent the maximum force the plantar flexors can produce. Animals were then sacrificed by cervical dislocation and muscles were collected, weighted and frozen. Force was normalized to the muscle mass as an estimate of specific force.
Four-limb hanging test
Mice were placed on a grid, which was kept upside down above an empty cage filled with bedding. The hanging time, i.e., the time until the animal fall, was recorded. A fixed limit of 180 seconds was set, and each mouse was tested twice on two consecutive days. Holding impulse was obtained by multiplying the hanging time for the respective mouse body weight. The maximum holding time was used for the analysis according to TREAT-NMD standard operating procedures (DMD_M.2.1.005; https://treat-nmd.org).
Histology and immunofluorescence
Cross sections (10-μm-thick) of TA muscle were incubated with wheat germ agglutinin (WGA) conjugated with Alexa Fluor 488 (1.0 μg/mL; Thermo Fisher Scientific, W11261) and Hoechst 33,258 (2.5 μg/mL; Thermo Fisher Scientific, H3570) in phosphate-buffered saline (PBS; Thermo Fisher Scientific, 18912014), for 20 min at room temperature. Slides were washed twice in PBS for 5 min under gentle shaking and mounted in 80% glycerol in PBS. For each sample, the entire stained TA section was acquired with a Stellaris SP8 confocal microscope (Leica Microsystems). Centrally nucleated myofibers were counted manually from the same muscle images. For immunofluorescence analysis, slides were incubated in cold 1:1 methanol-acetone solution for 10 min at − 20°C, washed twice in PBS for 15 min under gentle shaking and saturated with 10% goat serum in PBS for 30 min at room temperature. The following primary antibodies were incubated overnight: rabbit anti-LC3B (1:100; Thermo Fisher Scientific, PA1–16930), rat anti-LAMP1 (1:100; Developmental Studies Hybridoma Bank/DSHB, 1D4B), rabbit anti-TOMM20 (1:100; Santa Cruz Biotechnology [SCBT], sc -11,415), mouse anti-ATP5F1A/ATP5A (1:200; Abcam, Ab14748). Slides were washed in PBS and incubated for 1 h at room temperature with the appropriate secondary antibodies: goat anti-rabbit IgG (1:800; Jackson ImmunoResearch, 111-165-144), donkey anti-rat IgG (1:500; Jackson ImmunoResearch, 712-225-153) and goat anti-mouse IgG2b (1:500, Thermo Fisher Scientific, A-21141). Random fields were acquired with a Stellaris SP8 confocal microscope. Quantitative colocalization analysis was performed with Squassh software [31].
TUNEL assay
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) assay was performed with the In Situ Cell Death Detection Kit, TMR red (Roche, 12156792910), according to the manufacturer’s instruction. Briefly, transversal cryosections (10-μm-thick) of TA muscle were fixed for 10 min in methanol-acetone 1:1 solution. After three washes in PBS, slices were permeabilized for 5 min with 0.1% Triton X-100 (Sigma-Aldrich, T9284) and 0.1% sodium citrate, washed in PBS and then incubated for 1 h at 37°C in a moist chamber with the labeling solution. Nuclei were counterstained with Hoechst 33,258 (2.5 μg/mL). Finally, slides were washed in PBS and mounted in 80% glycerol in PBS. For each sample, 25–35 randomly chosen images were acquired with a Stellaris SP8 confocal microscope (Leica Microsystems) at 40× magnification. TUNEL-positive nuclei were counted manually on maximum projection images obtained with Fiji software.
NMJ morphometry
Diaphragm muscle was dissected and immediately fixed in 4% paraformaldehyde for 30 min at room temperature, then stored at 4°C in TBS buffer (66 mM Tris, pH 7.4, 41 mM NaCl) until use. To assess NMJ morphology, whole-mount immunofluorescence was performed on diaphragm samples as previously described [11]. Briefly, the diaphragm was simultaneously permeabilized and saturated in permeabilization buffer (4% IgG-free bovine serum albumin [Sigma-Aldrich, A7030], 10% goat serum [Sigma-Aldrich, S26-M], 0.5% Triton X-100 in TBS) for 10 h at room temperature under gentle shaking. Samples were incubated overnight with Alexa Fluor 555-conjugated α-bungarotoxin (α-BTX; Thermo Scientific, B35451), to label post-synaptic motor endplates. After extensive washings in TBS, samples were mounted in an adapted microscope slide with 80% glycerol in PBS. For each sample, at least 30–40 randomly chosen NMJ were acquired using a Stellaris SP8 confocal microscope (Leica Microsystems) at 63× magnification with a 2.5× zoom factor and with a z-step size of 1 μm. AChR-positive areas (corresponding to α-BTX-positive areas) and endplate areas (whole area of the post-synaptic button, within α-BTX-positive boundaries) were semi-automatically measured with the aNMJ-morph ImageJ macro [32]. The number of NMJ fragments was manually counted from 3D reconstruction obtained with Fiji software.
Western blotting
Frozen TA samples were ground in liquid nitrogen and lysed in SDS extraction buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 10% glycerol, 0.5 mM dithiothreitol, 2% SDS, 1% Triton X-100) supplemented with protease (Sigma-Aldrich, 04693132001) and phosphatase (Sigma-Aldrich, P5726) inhibitors. For subcellular fractionation of mitochondria (Figure S3), freshly collected quadriceps were processed as previously described [29] and lysates stored at −80°C until use. Proteins were quantified by BCA Protein Assay Kit (Thermo-Fisher, 23225). SDS-PAGE of protein lysates (20 μg) was carried out in 12% polyacrylamide Novex NuPAGE Bis-Tris gels (Invitrogen, NP0342BOX) and electrotransferred onto PVDF membrane (Thermo Fisher Scientific, 88518). Membranes were saturated for 1 h in 5% nonfat milk in TBS containing 0.1% Tween 20 (TBS-T; Sigma-Aldrich, P7949) and incubated overnight at 4°C with primary antibodies diluted in 2.5% milk in TBS-T. The following primary antibodies were used: rabbit anti-LC3B (1:1,000; Thermo Fisher Scientific, PA1–16930), guinea pig anti-SQSTM1/p62 (1:800; Progen, GP62-C), mouse anti-VCL/vinculin (1:1,000; Sigma-Aldrich, clone VIN-11-5), rabbit anti-TOMM20 (1:800; SCBT, sc -11,415), rabbit anti-OPTN (1:1500; Proteintech, 10837–1-AP), rabbit anti-BNIP3 (1:1000; Cell Signaling Technology, 3769), mouse anti-EEA1 (1:500; SCBT, sc -137,130), mouse anti-RAB5 (1:500; SCBT, sc -46,692), mouse anti-GAPDH (1:10000; Millipore, MAB374); rat anti-LAMP1 (1:1000; DSHB, 1D4B) and rabbit anti-CALR/calreticulin (1:1000; GeneTex, GTX111627). Horseradish peroxidase-conjugated secondary antibodies (1:2000; Bethyl Laboratories, A120101P, A90-116P) were used, and the signal detected with WesternBright ECL (Advansta, K-12045-D50). Densitometric quantification was carried out by FIJI software.
Transmission electron microscopy
Diaphragm muscles were fixed overnight at 4°C with 2.5% glutaraldehyde in 0.1 M cacodylate buffer, washed with 0.1 M cacodylate buffer, post-fixed for 2 h with 1% osmium tetroxide, and embedded in Epon812 (Electron Microscopy Sciences, E14900). Ultrathin sections were stained with uranyl acetate and lead citrate and observed with a Philips EM400 electron microscope operating at 100 kV.
Statistical analysis
Statistical tests [unpaired two-tailed Student’s t-test, unpaired two-tailed Mann-Whitney test, Kruskal-Wallis test and two-way analysis of variants (ANOVA)] were used as described in the figure legends (GraphPad). Statistical significance was set at P < 0.05. All histograms display individual values, and the number of biological replicates (always greater than three) is indicated in the figures’ captions.
Supplementary Material
Funding Statement
This work was supported by Italian Ministry of University and Research (2015FBNB5Y and 201742SBXA), Telethon Foundation (GGP19229), and University of Padova.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2241125
References
- [1].Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873. PubMed PMID: 18006683. doi: 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- [2].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. PubMed PMID: 29618831. doi: 10.1038/s41580-018-0003-4 [DOI] [PubMed] [Google Scholar]
- [3].Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584(7):1411–1416. PubMed PMID: 20132819. doi: 10.1016/j.febslet.2010.01.056 [DOI] [PubMed] [Google Scholar]
- [4].Drake J, Yan Z. Mitophagy in maintaining skeletal muscle mitochondrial proteostasis and metabolic health with ageing. J Physiol. 2017;595(20):6391–6399. PubMed PMID: 28795394. doi: 10.1113/JP274337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bönnemann CG. The collagen VI-related myopathies: Ullrich congenital muscular dystrophy and Bethlem myopathy. Handb Clin Neurol. 2011;101:81–96. PubMed PMID: 21496625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cescon M, Gattazzo F, Chen P, et al. Collagen VI at a glance. J Cell Sci [PubMed PMID: 26377767]. 2015;128:3525–3531. doi: 10.1242/jcs.169748 [DOI] [PubMed] [Google Scholar]
- [7].Lamandé SR, Bateman JF. Collagen VI disorders: Insights on form and function in the extracellular matrix and beyond. Matrix Biol. 2018;71–72:348–367. PubMed PMID: 29277723. doi: 10.1016/j.matbio.2017.12.008 [DOI] [PubMed] [Google Scholar]
- [8].Irwin W, Bergamin N, Sabatelli P, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet. 2003;35(4):367–371. PubMed PMID: 14625552. doi: 10.1038/ng1270 [DOI] [PubMed] [Google Scholar]
- [9].Grumati P, Coletto L, Sabatelli P, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2010;16(11):1313–1320. PubMed PMID: 21037586. doi: 10.1038/nm.2247 [DOI] [PubMed] [Google Scholar]
- [10].Chrisam M, Pirozzi M, Castagnaro S, et al. Reactivation of autophagy by spermidine ameliorates the myopathic defects of collagen VI-null mice. Autophagy. 2015;11(12):2142–2152. PubMed PMID: 26565691. doi: 10.1080/15548627.2015.1108508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cescon M, Gregorio I, Eiber N, et al. Collagen VI is required for the structural and functional integrity of the neuromuscular junction. Acta Neuropathol. 2018;136(3):483–499. PubMed PMID: 29752552. doi: 10.1007/s00401-018-1860-9 [DOI] [PubMed] [Google Scholar]
- [12].Castagnaro S, Gambarotto L, Cescon M, et al. Autophagy in the mesh of collagen VI. Matrix Biol [PubMed PMID: 33373668]. 2021;100–101:162–172. doi: 10.1016/j.matbio.2020.12.004 [DOI] [PubMed] [Google Scholar]
- [13].Castagnaro S, Pellegrini C, Pellegrini M, et al. Autophagy activation in COL6 myopathic patients by a low-protein-diet pilot trial. Autophagy. 2016;12(12):2484–2495. PubMed PMID: 27656840. doi: 10.1080/15548627.2016.1231279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Metti S, Gambarotto L, Chrisam M, et al. The polyphenol pterostilbene ameliorates the myopathic phenotype of Collagen VI deficient mice via autophagy induction. Front Cell Dev Biol [PubMed PMID: 33134297]. 2020;8:997. doi: 10.3389/fcell.2020.580933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pegg AE. Functions of polyamines in mammals. J Biol Chem. 2016;291(29):14904–14912. PubMed PMID: 27268251. doi: 10.1074/jbc.R116.731661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Igarashi K, Kashiwagi K. Modulation of cellular function by polyamines. Int J Biochem Cell Biol. 2010;42(1):39–51. PubMed PMID: 19643201. doi: 10.1016/j.biocel.2009.07.009 [DOI] [PubMed] [Google Scholar]
- [17].Madeo F, Hofer SJ, Pendl T, et al. Nutritional aspects of spermidine. Annu Rev Nutr. 2020;40(1):135–159. PubMed PMID: 32634331. doi: 10.1146/annurev-nutr-120419-015419 [DOI] [PubMed] [Google Scholar]
- [18].Eisenberg T, Knauer H, Schauer A, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11(11):1305–1314. PubMed PMID: 19801973. doi: 10.1038/ncb1975 [DOI] [PubMed] [Google Scholar]
- [19].Madeo F, Eisenberg T, Pietrocola F, et al. Spermidine in health and disease. Science. 2018;359(6374):PubMed PMID: 29371440. doi: 10.1126/science.aan2788. [DOI] [PubMed] [Google Scholar]
- [20].Schroeder S, Hofer SJ, Zimmermann A, et al. Dietary spermidine improves cognitive function. Cell Rep. 2021;35(2):108985. PubMed PMID: 33852843. doi: 10.1016/j.celrep.2021.108985 [DOI] [PubMed] [Google Scholar]
- [21].Pekar T, Bruckner K, Pauschenwein-Frantsich S, et al. The positive effect of spermidine in older adults suffering from dementia: First results of a 3-month trial. Wien Klin Wochenschr. 2021;133(9–10):484–491. PubMed PMID: 33211152. doi: 10.1007/s00508-020-01758-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Schwarz C, Stekovic S, Wirth M, et al. Safety and tolerability of spermidine supplementation in mice and older adults with subjective cognitive decline. Aging. 2018;10(1):19. PubMed PMID: 29315079. doi: 10.18632/aging.101354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wirth M, Benson G, Schwarz C, et al. The effect of spermidine on memory performance in older adults at risk for dementia: A randomized controlled trial. Cortex [PubMed PMID: 30388439]. 2018;109:181–188. doi: 10.1016/j.cortex.2018.09.014 [DOI] [PubMed] [Google Scholar]
- [24].Eisenberg T, Abdellatif M, Schroeder S, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22(12):1428. PubMed PMID: 27841876. doi: 10.1038/nm.4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Angelin A, Tiepolo T, Sabatelli P, et al. Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc Natl Acad Sci U S A. 2007;104(3):991–996. PubMed PMID: 17215366. doi: 10.1073/pnas.0610270104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bonaldo P, Braghetta P, Zanetti M, et al. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet [PubMed PMID: 9817932]. 1998;7:2135–2140. doi: 10.1093/hmg/7.13.2135 [DOI] [PubMed] [Google Scholar]
- [27].Gottlieb RA, Carreira RS. Autophagy in health and disease. 5. Mitophagy as a way of life. Am J Physiol Cell Physiol. 2010;299:C203–210. PubMed PMID: 20357180. doi: 10.1152/ajpcell.00097.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy [PubMed PMID: 33634751]. 2021;17:1–382. doi: 10.1080/15548627.2020.1797280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gambarotto L, Metti S, Chrisam M, et al. Ambra1 deficiency impairs mitophagy in skeletal muscle. J Cachexia Sarcopenia Muscle. 2022;13(4):2211–2224. PubMed PMID: 35593053. doi: 10.1002/jcsm.13010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Baraldo M, Zorzato S, Dondjang AHT, et al. Inducible deletion of Raptor and mTOR from adult skeletal muscle impairs muscle contractility and relaxation. J Physiol. 2022;600(23):5055–5075. PubMed PMID: 36255030. doi: 10.1113/JP283686 [DOI] [PubMed] [Google Scholar]
- [31].Rizk A, Paul G, Incardona P, et al. Segmentation and quantification of subcellular structures in fluorescence microscopy images using Squassh. Nat Protoc. 2014;9(3):586–596. PubMed PMID: 24525752. doi: 10.1038/nprot.2014.037 [DOI] [PubMed] [Google Scholar]
- [32].Minty G, Hoppen A, Boehm I, et al. Anmj-morph: a simple macro for rapid analysis of neuromuscular junction morphology. R Soc Open Sci. 2020;7(4):200128. PubMed PMID: 32431902. doi: 10.1098/rsos.200128 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.