

ABSTRACT
Colorectal cancer (CRC) is one of the most common malignancies worldwide and remains a major clinical challenge. Periplocin, a major bioactive component of the traditional Chinese herb Cortex periplocae, has recently been reported to be a potential anticancer drug. However, the mechanism of action is poorly understood. Here, we show that periplocin exhibits promising anticancer activity against CRC both in vitro and in vivo. Mechanistically, periplocin promotes lysosomal damage and induces apoptosis in CRC cells. Notably, periplocin upregulates LGALS3 (galectin 3) by binding and preventing LGALS3 from Lys210 ubiquitination-mediated proteasomal degradation, leading to the induction of excessive lysophagy and resultant exacerbation of lysosomal damage. Inhibition of LGALS3-mediated lysophagy attenuates periplocin-induced lysosomal damage and growth inhibition in CRC cells, suggesting a critical role of lysophagy in the anticancer effects of periplocin. Taken together, our results reveal a novel link between periplocin and the lysophagy machinery, and indicate periplocin as a potential therapeutic option for the treatment of CRC.
Abbreviations: 3-MA: 3-methyladenine; ACACA/ACC1: acetyl-CoA carboxylase alpha; AMPK: adenosine monophosphate-activated protein kinase; AO: Acridine orange; ATG5: autophagy related 5; ATG7: autophagy related 7; CALM: calmodulin; CHX: cycloheximide; CRC: colorectal cancer; CQ: chloroquine; CTSB: cathepsin B; CTSD: cathepsin D; ESCRT: endosomal sorting complex required for transport; LAMP1: lysosomal associated membrane protein 1; LMP: lysosomal membrane permeabilization; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MCOLN1/TRPML1: mucolipin TRP cation channel 1; MKI67/Ki-67: marker of proliferation Ki-67; MTOR: mechanistic target of rapamycin kinase; P2RX4/P2X4: purinergic receptor P2X 4; PARP1/PARP: poly(ADP-ribose) polymerase 1; PRKAA/AMPKα: protein kinase AMP-activated catalytic subunit alpha; SQSTM1/p62: sequestosome 1; TFEB: transcription factor EB; TRIM16: tripartite motif containing 16.
KEYWORDS: Autophagic flux, colorectal cancer, LGALS3 (galectin 3), lysophagy, lysosomal damage, periplocin
Introduction
Colorectal cancer (CRC) ranks as the third most commonly diagnosed malignancy and the second leading cause of cancer-related death worldwide, with an incidence of over 1.8 million and mortality of over 0.8 million in 2018 [1]. Despite recent advances in CRC treatment attributable to the improved early detection combining with surgical excision, chemotherapy or radiotherapy, the prognosis for CRC patients still remains poor [2,3]. Adjuvant chemotherapy using fluoropyrimidine (fluorouracil or capecitabine) and oxaliplatin may confer a survival advantage, however, not all patients benefit from these adjuvant regimens [4]. It is therefore imperative to develop novel potential therapeutic agents for more effective treatment of CRC patients.
Macroautophagy/autophagy is a multistep lysosome-mediated intracellular bulk degradation system to digest excess or defective cellular constituents or organelles [5]. The important role of autophagy in cellular stress response as well as in cancer development and progression underscores the excellent potential of using autophagy-targeting agents for cancer treatment [6,7]. Recent findings from our and other groups have demonstrated that autophagy induction may compromise, or contrarily, potentiate the tumor suppressive effects of anticancer drugs, suggesting a context-dependent role of autophagy in cancer treatment [8–13]. In terms of the pro-survival role of autophagy, the selective degradation of dysfunctional organelles (such as mitochondria, lysosomes, endoplasmic reticulum, etc.) has been shown to enable the quality control and turnover of organelles to maintain cell survival in response to cellular stress, including drug treatment [14]. Intriguingly, we, and others, have also observed a lethal role of mitophagy involving the removal of mitochondria with normal function [15–17]. However, whether other types of selective autophagy, particularly lysophagy, can provoke cell death remains largely unknown.
Periplocin is the most cytotoxic compound isolated from Cortex periplocae (a well-known traditional Chinese herb), and is traditionally used as an antirheumatic, diuretic and cardiotonic agent [18]. Importantly, as a cardiac glycoside, periplocin has been characterized as an effective constituent of qili qiangxin capsules, a traditional Chinese medicine prescription approved for the treatment of chronic heart failure with favorable efficacy and safety [18,19]. Recently, growing evidence suggests periplocin also possesses promising anticancer activity in colorectal cancer, liver cancer, gastric cancer, myxofibrosarcoma and pancreatic cancer [20–25]. The anticancer mechanism of periplocin in these reports are limited to apoptosis induction and cell cycle arrest. Whether other cell death mechanism is involved in the anticancer effects of periplocin remains poorly understood.
In this study, we show that periplocin inhibits the growth of CRC cells by inducing lysosomal damage and stimulating excessive lysophagy. Periplocin inhibits the ubiquitin-mediated degradation of LGALS3 (galectin 3), which leads to the massive redirection of LGALS3 to the damaged lysosomes, thereby promoting excessive lysophagy and exacerbating lysosomal damage in CRC cells. These findings demonstrate a novel link between periplocin and lysophagy, which provides preclinical proof of concept for the use of periplocin in CRC treatment.
Results
Periplocin exhibits anticancer effect against CRC in vitro and in vivo
To investigate whether periplocin exhibits anticancer activity in CRC cells, an MTT assay was conducted to assess the growth of various human CRC cell lines (DLD-1, SW480, HCT116, SW620, HT29, LOVO, and RKO) and a human colon mucosal epithelial cell line (NCM460) following 24-h treatment of periplocin with different concentrations. Periplocin treatment markedly inhibited the growth of all tested CRC cell lines in a dose-dependent manner, with IC50 values ranging from 0.12 μM to 0.82 mΜ (Figure 1A,B). Notably, NCM460 cells showed higher tolerance to periplocin treatment, with an IC50 value of 44.95 ± 3.59 μM (Figure 1A,B). The growth inhibition effect of periplocin was also observed in pancreatic cancer and hepatocellular carcinoma cells (Figure S1A,B). In addition, the proliferation of CRC cells was significantly suppressed in response to periplocin treatment, as indicated by reduced numbers of clones (Figure 1C,D) and EdU incorporation (Figure 1E). By contrast, no obvious anti-proliferative effect was observed for periplocin in NCM460 cells (Figure 1C,D). Moreover, we observed an obvious increase in the proportion of cells in the G2/M phase (Figure 1F), upregulation of CDKN1A/p21 level, and downregulation of CCNB1 (cyclin B1) and CDK1 levels (Figure 1G) following periplocin treatment, indicating that periplocin caused cell cycle arrest in CRC cells. Interestingly, periplocin did not significantly affect cell cycle in NCM460 cells (Figure 1F,G). Together, these results suggest that periplocin markedly inhibits CRC cell growth and proliferation in vitro.
Figure 1.
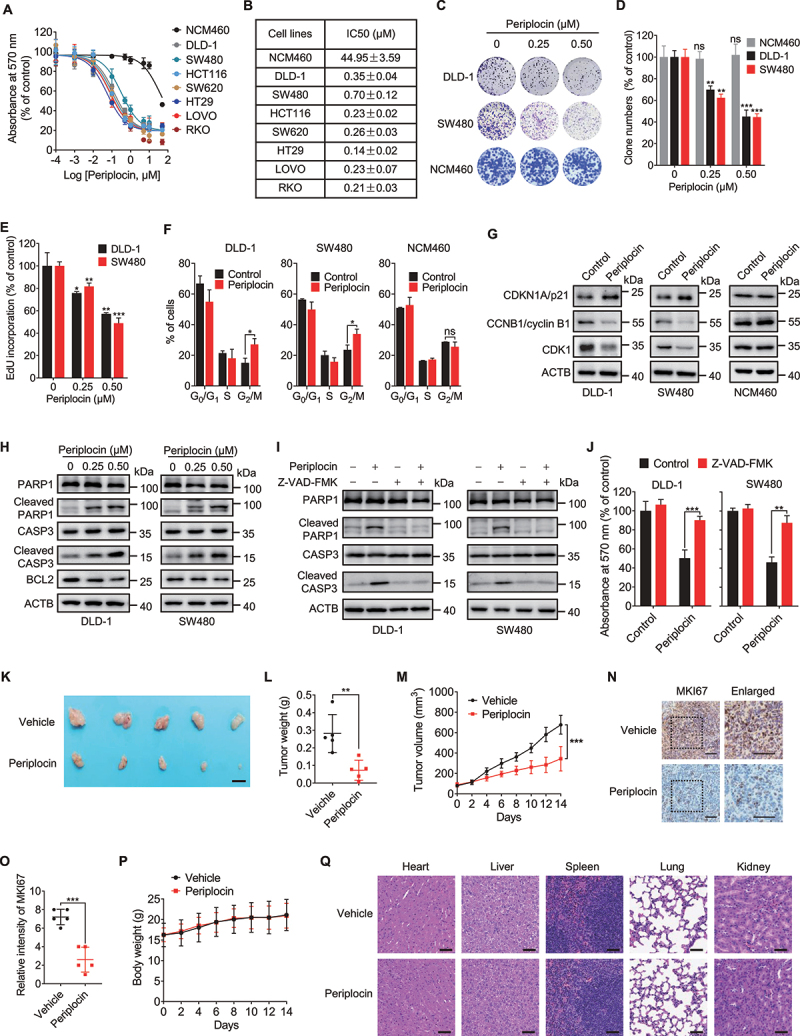
Periplocin exhibits anticancer effect against CRC in vitro and in vivo. (A) Cell viability of human CRC cell lines and colon mucosal epithelial cell line NCM460 treated with periplocin for 24 h at the indicated concentrations. (B) IC50 values of periplocin in CRC cells and NCM460 cells as treated in (A). (C-E) Cells were subjected to periplocin treatment at the indicated concentrations for 24 h. Cell proliferation was determined by colony formation (C and D) and EdU incorporation (E). (F) Cell cycle distribution of DLD-1, SW480, and NCM460 cells treated with 0.50 μM periplocin for 24 h determined by flow cytometry. (G) Immunoblotting analysis of CDKN1A/p21, CCNB1/cyclin B1, and CDK1 in DLD-1, SW480, and NCM460 cells treated with 0.50 μM periplocin for 24 h. (H) Immunoblotting analysis of PARP1, cleaved PARP1, CASP3, cleaved CASP3, and BCL2 in DLD-1 and SW480 cells treated with periplocin for 24 h at the indicated concentrations. (I and J) DLD-1 and SW480 cells were treated with 0.50 μM periplocin for 24 h in the presence or absence of Z-VAD-FMK. Immunoblotting analysis was performed to detect the protein levels of PARP1, cleaved PARP1, CASP3, and cleaved CASP3 (I), and MTT assay was conducted to examine cell growth (J). (K-M) SW480 cells were subcutaneously inoculated into nude mice. Mice were injected with vehicle or periplocin (15 mg/kg/day) for two weeks. Image (K), weight (L), and volume (M) of tumor xenografts were shown. Scale bar: 1 cm. (N and O) Representative images (N) and quantitative analysis (O) of immunohistochemical staining for MKI67 in tumor xenografts. Scale bar: 50 μm. (P) Body weight of tumor-bearing mice was monitored at the indicated periods. (Q) H&E staining of the heart, liver, spleen, lung, and kidney from tumor-bearing mice. Scale bar: 100 μm. Results in (A-J) are representative of three independent experiments. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. ns, non-significant.
It has been previously shown that apoptosis induction is involved in the tumor suppressive effect of periplocin [22,25], inspiring us to ask whether periplocin inhibits the growth of CRC cells by inducing apoptosis. The apoptotic rate was determined by flow cytometric analysis. As shown in Figure S1C,D, periplocin treatment for 24 h revealed obvious apoptosis induction in DLD-1 and SW480 cells. This observation was further confirmed by increased levels of cleaved PARP1/PARP and cleaved CASP3 (caspase 3), as well as decreased level of BCL2 in periplocin-treated CRC cells (Figure 1H). The apoptosis induction caused by periplocin in CRC cells was both dose- and time-dependent (Figure 1H, Figure S1E). The caspase inhibitor Z-VAD-FMK profoundly suppressed periplocin-induced apoptosis (Figure 1I), and rescued periplocin-mediated growth inhibition of CRC cells (Figure 1J). Of Note, no obvious apoptosis was observed in human colon mucosal epithelial NCM460 cells (Figure S1F). Overall, these data suggest that periplocin inhibits the growth of CRC cells, at least partially, by inducing apoptosis.
To confirm the growth inhibition effect of periplocin against CRC in vivo, a mouse xenograft model was generated by subcutaneously inoculating SW480 cells into nude mice. As shown in Figure 1K–M, periplocin-treated mice showed a marked reduction in the size, weight, and growth rate of CRC xenografts compared with the vehicle-treated group. In addition, periplocin treatment resulted in weaker MKI67/Ki-67 staining compared with the controls (Figure 1N,O). Moreover, we found an obvious reduction of tumor-associated macrophages (AIF1/IBA1+), decrease of PECAM1/CD31+ vasculatures, and repression of fibrotic components (PDGFRA+ or ACTA2/α-SMA+) in CRC xenografts obtained from periplocin-treated mice, suggesting that periplocin treatment also changed the microenvironment (Figure S1G,H). Importantly, periplocin treatment had no significant effect on the mice body weight (Figure 1P), the pathological features of major organs of mice (Figure 1Q), or blood biochemical indicators (Figure S1I-Q), suggesting that the therapeutic dose of periplocin had no obvious toxic or adverse effects in mice. Collectively, these results demonstrate that periplocin exhibits potent anti-CRC effects both in vitro and in vivo with minimal toxicity.
Periplocin induces lysosomal damage in CRC cells
To further determine the mechanism underlying the growth inhibition effect of periplocin in CRC cells, we profiled the global protein expression in response to periplocin treatment using TMT-based quantitative proteomics analysis (Table S1). The differentially expressed proteins in periplocin-treated cells compared with controls were then subjected to gene set enrichment analysis (GSEA). Interestingly, the lysosome pathway was significantly enriched (Figure 2A). In detail, periplocin treatment led to a marked decrease in the levels of lysosome-associated proteins, including LAMP1 (lysosomal associated membrane protein 1), LAMP2, CTSB (cathepsin B) and CTSD (cathepsin D) (Figure 2B, Figure S2A), implying the induction of lysosomal damage in periplocin-treated cells. Emerging evidence has revealed that lysosomal damage causes cell death [26], which leads us to hypothesize that lysosomal damage might be involved in periplocin’s anticancer activity in CRC cells.
Figure 2.
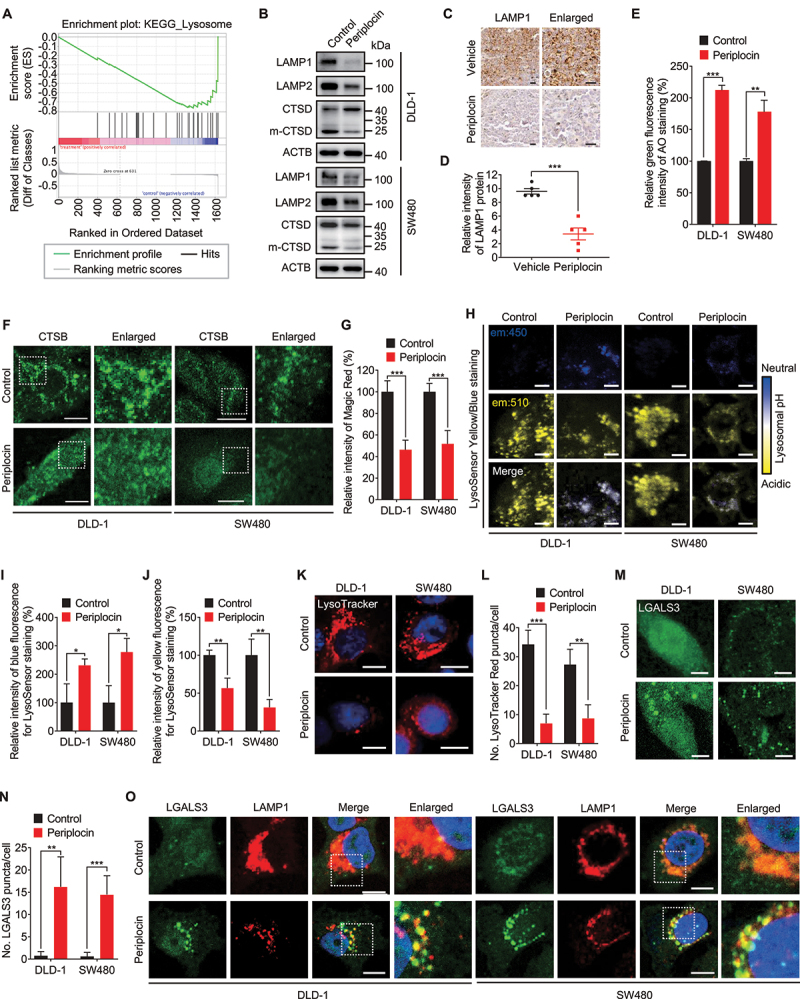
Periplocin induces lysosomal damage in CRC cells. (A) TMT-based quantitative proteomics analysis was used to profile the global protein expression in SW480 cells treated with or without 0.50 μM periplocin for 24 h. The differentially expressed lysosome-associated proteins were enriched by gene set enrichment analysis (GSEA). (B) Immunoblotting analysis of LAMP1, LAMP2 and CTSD under 0.50 μM periplocin treatment for 24 h. m-CTSD, mature CTSD. (C and D) Representative images (C) and quantitative analysis (D) of immunohistochemical staining for LAMP1 in SW480 ×enografts from vehicle- or periplocin-treated mice. Scale bar: 50 μm. (E) Acridine orange (AO, 5 μM) was used to label the lysosomes of cells treated with or without 0.50 μM periplocin for 24 h. Green fluorescence indicating the leakage of lysosomal contents was detected by flow cytometry. (F) Immunofluorescent analysis of CTSB in cells treated with or without 0.50 μM periplocin for 24 h. Scale bar: 10 μm. (G) Quantitation of the relative fluorescent intensity of Magic Red staining in cells treated with or without 0.50 μM periplocin for 24 h. (H-J) Representative images (H) and quantitative analysis (I and J) of LysoSensor Yellow/Blue DND-160 staining in cells treated with or without 0.50 μM periplocin for 24 h. Scale bar: 10 μm. (K and L) Representative images (K) and quantitative analysis (L) of LysoTracker Red DND-99 staining in cells treated with or without 0.50 μM periplocin for 24 h. Scale bar: 10 μm. (M and N), Representative images (M) and quantitative analysis (N) for immunofluorescent staining of LGALS3 in cells treated with or without 0.50 μM periplocin for 24 h. Scale bar: 10 μm. (O) Immunofluorescent analysis of the colocalization of LGALS3 with LAMP1 in cells treated with or without 0.50 μM periplocin for 24 h. Scale bar: 10 μm. Results are representative of three independent experiments. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
To ascertain whether periplocin induces lysosomal damage, we examined the levels of LAMP1 and LAMP2, two integral lysosomal membrane proteins that are essential for the function of lysosomes [27]. In agreement with the proteomics data, periplocin treatment significantly decreased the protein levels of LAMP1 and LAMP2 in both DLD-1 and SW480 cells (Figure 2B). Reduced LAMP1 expression was also observed in tumor xenografts of periplocin-treated mice (Figure 2C,D). Other lysosomal proteins, such as ATP6V1B and ATP6V1H, were also downregulated following periplocin treatment (Figure S2B). The decrease of these lysosomal protein levels in periplocin-treated cells was due to neither proteasomal degradation (as evidence by failed rescue of their reduced levels by MG132 treatment; Figure S2B), nor increased secretion of lysosome proteins (Figure S2C). It has been shown previously that the downregulation of lysosomal membrane proteins renders lysosomes more sensitive to lysosomal membrane permeabilization (LMP) [26,28]. We therefore investigated whether periplocin promotes LMP in CRC cells by detecting lysosomal leakage. Acridine orange (AO) is a metachromatic lysosomotropic dye that exhibits red fluorescence when highly accumulated in the acidic lysosome environment and green fluorescence when dissipated throughout the cytosol. Therefore, it can be used as an indicator for the leakage of lysosomal contents into the cytosol [26]. We stained acidic lysosomes with AO, and observed an obvious increase of AO green fluorescence in periplocin-treated cells, suggesting the cytosolic leakage of AO from the lysosomes (Figure 2E). To further confirm the leakage of lysosomal contents, we performed immunofluorescent staining of CSTB, a classic lysosomal acid hydrolase. Compared with the punctate pattern of intense fluorescence in the control group, periplocin treatment led to a diffuse fluorescence pattern of CTSB throughout the cytosol (Figure 2F). This observation was confirmed by decreased Magic Red fluorescent signaling following periplocin treatment, which revealed a reduction of CTSB activity caused by periplocin (Figure 2G). In line with these observations, periplocin treatment also suppressed the activity of CTSD as evidenced by decreased ratio of the mature CTSD to its precursor (Figure 2B). Consistently, the LysoSensor Yellow/Blue probe staining showed that periplocin-treated cells exhibited weaker yellow fluorescence and stronger blue fluorescence, indicating that periplocin inhibited the acidification of the lysosomes (Figure 2H-J). The damage and deacidification of lysosomes caused by periplocin were further evidenced by decreased staining of the acidotropic LysoTracker Red (Figure 2K,L). It has been reported that the elevated lysosomal pH was reported to be associated with homotypic lysosome fusion mediated by P2RX4/P2X4-CALM (calmodulin) signaling, while decreased lysosomal pH stimulates lysosome fission by activating MCOLN1/TRPML1-CALM axis [29–31]. Although no obvious changes in the protein levels of P2RX4 and CALM were observed (Figure S2D), periplocin treatment caused a significant increase in the interaction between P2RX4 and CALM (Figure S2E,F). Moreover, periplocin-treated cells exhibited increased colocalization of P2RX4 with LAMP1 (Figure S2G). However, the interaction of MCOLN1 with CALM was not affected following periplocin treatment (Figure S2E). These data suggest that periplocin promotes homotypic lysosome fusion, but not lysosome fission.
We next determined whether the lysosomal leakage is associated with LMP by detecting the lysosomal redirection of LGALS3, which senses and binds to the lysosomal β-galactosides exposed on the cytosolic side of the lysosomal membrane when LMP occurs [26,32]. As shown in Figure 2M,N, there was a remarkable accumulation of LGALS3 puncta in periplocin-treated cells compared with control cells. A similar accumulation of LGALS1 (galectin 1) puncta was also observed in periplocin-treated CRC cells (Figure S2H,I). More importantly, although periplocin reduced the protein level of LAMP1 in vitro and in vivo (Figure 2B-D, Figure S2A), the co-occurrence of LGALS3 puncta with LAMP1 was markedly increased in periplocin-treated cells (Figure 2O), indicating LGALS3 was accumulated on the membrane of the lysosomes. Interestingly, an increased colocalization of LGALS3 with P2RX4 was observed in response to periplocin treatment (Figure S2J), suggesting that the injured lysosomes caused by periplocin was also involved in lysosome fusion. Overall, these results illustrate that periplocin induces lysosomal damage and impairs lysosomal function in CRC cells.
Periplocin stimulates lysophagy in CRC cells
In response to lysosomal damage, lysophagy is often activated to eliminate damaged lysosomes or lysosomal debris to maintain lysosomal quality [33]. In addition, it has been previously reported that some cardiac glycosides induce autophagy in lung cancer cells, and increasing evidence has highlighted the pharmacological modulation of autophagy in anticancer therapies [34,35]. Therefore, we postulated that periplocin might induce autophagy in CRC cells. As shown in Figure 3A and Figure S3A, periplocin treatment resulted in obvious conversion of MAP1LC3B/LC3B-I to lipidated LC3B-II, an autophagosome marker, in a dose-dependent manner in various CRC cell lines. However, no apparent LC3B turnover was observed in periplocin-treated NCM460 cells (Figure S3B). Consistently, periplocin treatment promoted the accumulation of both endogenous LC3B (Figure 3B,C) and exogenous GFP-LC3B (Figure S3C,D) puncta in CRC cells. Moreover, CRC xenografts in periplocin-treated mice displayed stronger LC3B staining compared with the vehicle-treated group (Figure S3E,F). These results suggest that periplocin promotes the accumulation of autophagosomes in CRC cells both in vitro and in vivo.
Figure 3.
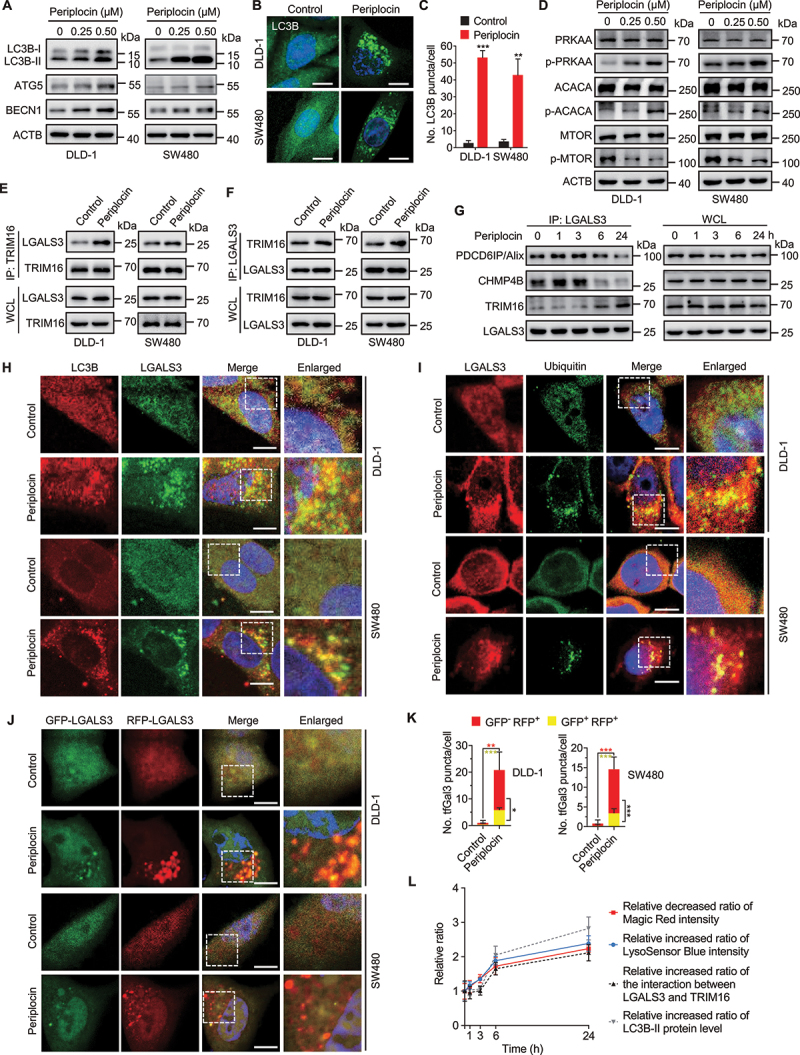
Periplocin stimulates lysophagy in CRC cells. (A) Immunoblotting analysis of LC3B, ATG5, and BECN1 in cells treated with periplocin for 24 h at the indicated concentrations. (B and C) Representative images (B) and quantitative analysis (C) for immunofluorescent staining of endogenous LC3B puncta in cells treated with or without 0.50 μM periplocin for 24 h. (D) Immunoblotting analysis of PRKAA, p-PRKAA (Thr172), ACACA, p-ACACA (Ser79), MTOR, and p-MTOR (Ser2448) in cells treated with periplocin for 24 h at the indicated concentrations. (E and F) Reciprocal co-immunoprecipitation analysis of the interaction between endogenous LGALS3 and TRIM16 in cells treated with or without 0.50 μM periplocin for 24 h. (G) Co-immunoprecipitation analysis of the interaction between endogenous LGALS3 with PDCD6IP/Alix, CHMP4B, and TRIM16 in DLD-1 cells treated with 0.50 μM periplocin at different time periods. (H) Immunofluorescent analysis of the colocalization of LC3B with LGALS3 in CRC cells treated with or without 0.50 μM periplocin for 24 h. Scale bar: 10 μm. (I) Immunofluorescent analysis of the colocalization of LGALS3 with ubiquitin in CRC cells treated with or without 0.50 μM periplocin for 24 h. Scale bar: 10 μm. (J) Representative fluorescent images of CRC cells transiently expressing Mrfp-GFP-tandem fluorescent-tagged LGALS3 (tfGal3) followed by 0.50 μM periplocin treatment for 24 h. Scale bar: 10 μm. (K) Quantitative analysis of the GFP+ RFP+ or GFP− RFP+ LGALS3 puncta in (J). (L) the relative decreased ratio of Magic Red intensity, relative increased ratio of LysoSensor Blue intensity, relative increased ratio of the interaction between LGALS3 and TRIM16, and relative increased ratio of LC3B-II protein level in DLD-1 cells following 0.50 μM periplocin treatment at different time periods. Results are representative of three independent experiments. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
The elevated LC3-II levels and increased autophagosomes are consequences of either activated autophagy initiation or the blocked autophagic flux [36]. To determine whether periplocin initiates autophagy in CRC cells, we examined the expression of BECN1 (beclin 1) and ATG5, which play important roles in autophagy initiation and autophagosome formation [37]. As expected, periplocin treatment dose-dependently elevated the protein levels of BECN1 and ATG5 (Figure 3A). In addition, inhibition of the class III phosphatidylinositol 3-kinase/PtdIns3K using 3-methyladenine (3-MA), or knockdown of either ATG5 or BECN1, significantly attenuated the elevated LC3B-II levels (Figure S3H-K) and accumulated LC3 puncta (Figure S3L-O) in periplocin-treated CRC cells. These results suggest that periplocin stimulates the initiation of autophagy in CRC cells. We then investigated whether periplocin induces complete autophagic flux in CRC cells. As shown in Figure S4A-C, co-administration of chloroquine (CQ, an autolysosomal inhibitor) with periplocin resulted in a further increase in LC3B-II turnover and LC3B puncta accumulation. In addition, using a tandem monomeric RFP-GFP-tagged LC3B construct, we found periplocin treatment resulted in increased formation of RFP+ GFP− fluorescent autolysosomes in CRC cells (Figure S4D,E). Taken together, these data indicate that periplocin promotes autophagic flux in CRC cells.
Next, we investigated whether periplocin triggers lysophagy in CRC cells. It has been reported that, in response to lysosomal damage, AMP-activated protein kinase (AMPK) is activated and MTOR (mechanistic target of rapamycin kinase) is inactivated to initiate lysophagy for the clearance of injured lysosomes [38,39]. We found that the phosphorylation levels of PRKAA/AMPKα (Thr172) and ACACA/ACC1 (acetyl-CoA carboxylase alpha; Tyr79) were elevated by periplocin treatment in a dose-dependent manner, whereas the phosphorylation level of MTOR (Thr2448) was suppressed (Figure 3D), suggesting that periplocin activates AMPK and inactivates MTOR signaling. Previous studies have demonstrated that the recruitment of TRIM16 to LGALS3 on the lysosomes is a key event in initiating the lysophagic machinery [33,40]. Using reciprocal immunoprecipitation, we found periplocin treatment led to a marked increase in the interaction between TRIM16 and LGALS3 (Figure 3E,F). In addition to TRIM16, it has been reported that the endosomal sorting complex required for transport (ESCRT) components could also be recruited to the lysosome membranes by LGALS3 at earlier time points for lysosome membrane repair in response to lysosomal damage [41–43]. Indeed, periplocin treatment increased the interaction of LGALS3 with two ESCRT components, PDCD6IP/Alix and CHMP4B, both of which peaked at 3 h and decreased at 6 h of periplocin treatment (Figure 3G). Interestingly, the binding of CHMP4B with TRIM16 was only enhanced at 6 h of periplocin treatment (Figure 3G). These results suggest that periplocin promotes ESCRT-mediated lysosomal repair at earlier time points and sequentially initiates lysophagy machinery at later time points, and both processes are coordinated by LGALS3.
To further determine whether the lysosomes are engulfed by the autophagosomes, we performed immunofluorescent staining to assess the colocalization of LC3B (an autophagosome marker) and LGALS3 (a marker of damaged lysosomes). The results showed that periplocin significantly promoted the overlap between LC3B and LGALS3, indicating a colocalization of the injured lysosomes with autophagosomes (Figure 3H). During lysophagy, damaged lysosomes are often ubiquitinated to enable selective autophagy of lysosomes [39,44,45]. As shown in Figure 3I and Figure S4F, we observed increased colocalization of ubiquitin with both LAMP1 and LGALS3 in periplocin-treated cells, suggesting that the damaged lysosomes caused by periplocin can be labeled by ubiquitin signal for the initiation of lysophagy. Furthermore, a mRFP-eGFP-tandem fluorescent-tagged LGALS3 (tfGal3) was used to ascertain whether the engulfed lysosomes were subjected to autophagic degradation. The GFP-LGALS3 fluorescence is acid-sensitive and will be quenched in autolysosomes with acidic environment, whereas the monomeric RFP-LGALS3 protein is resistant to autolysosomal degradation [44]. As shown in Figure 3J,K, we observed increased RFP+ GFP− LGALS3 puncta in periplocin-treated CRC cells, suggesting the enveloping of damaged lysosomes by functional autolysosomes. Collectively, these results suggest that periplocin promotes lysophagy in CRC cells. Of note, the interaction of LGALS3 with TRIM16 and LC3B-II level were both increased only at 6 h of periplocin treatment, whereas the decrease in Magic Red intensity and increase in LysoSensor Blue intensity both occurred at 1 h of periplocin treatment and sustained till 24 h treatment (Figure 3L), suggesting that lysophagy is the response to lysosomal damage during periplocin treatment.
Periplocin-induced lysophagy potentiates lysosomal damage in CRC cells
Given the engulfment of damaged lysosomes in the autolysosomes with complete autophagic flux, we next asked whether periplocin-induced lysosphagy mediates the clearance of damaged lysosomes. Blocking lysophagy using ATG5 shRNA markedly rescued the reduced protein levels of LAMP1, ATP6V1B and ATP6V1H in periplocin-treated CRC cells (Figure 4A, Figure S5A,B). The reduced LAMP1 protein level following periplocin treatment was also restored by ATG7 knockdown (Figure 4B). In addition, ATG5 knockdown partially rescued the diffuse fluorescent pattern of CTSB in response to periplocin treatment (Figure 4C, Figure S5C). This observation was further confirmed by the ATG5 or ATG7 knockdown-mediated recovery of decreased Magic Red fluorescent signaling in periplocin-treated CRC cells (Figure 4D,E). Consistently, the impairment of lysophagy by shATG5 or shATG7 knockdown significantly countered periplocin-induced increase of LGALS3 puncta (Figure 4F-I, Figure S5D,E). These data suggest that periplocin-induced lysophagy potentiates, rather than prevents, lysosomal damage and dysfunction in CRC cells.
Figure 4.
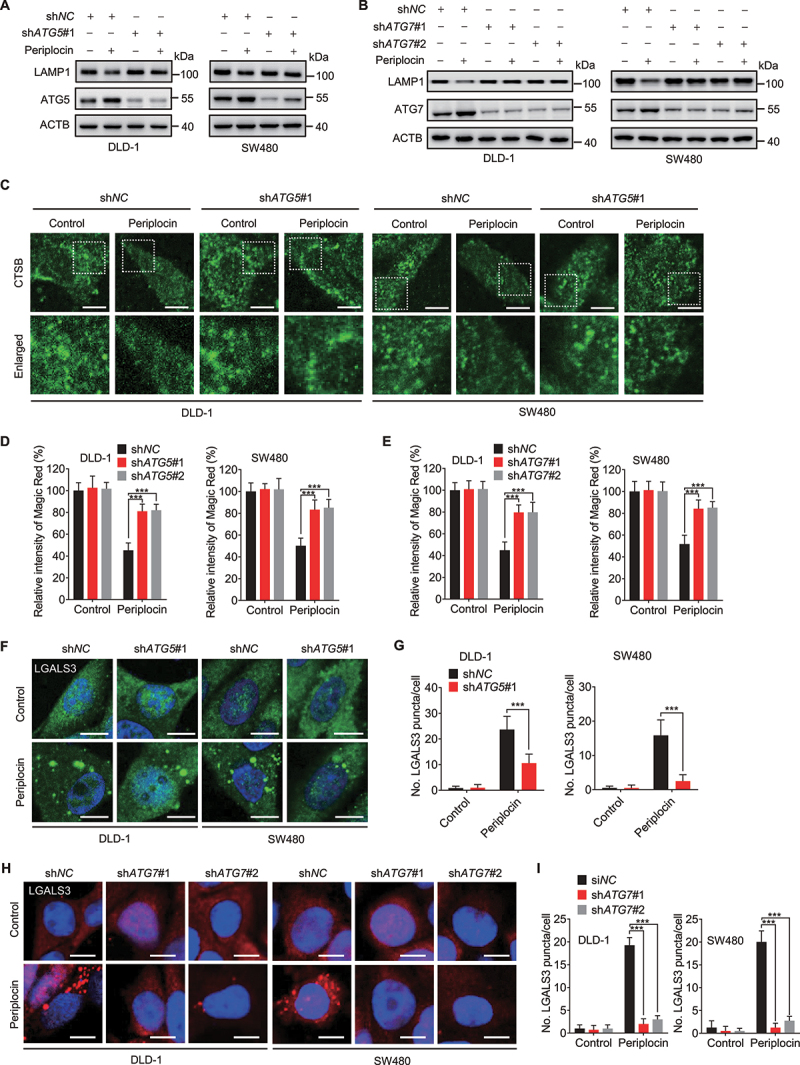
Periplocin-induced lysophagy potentiates lysosomal damage in CRC cells. (A and B) Immunoblotting analysis of LAMP1 in cells with ATG5 (A, shATG5#1) or ATG7 (B, shATG7#1 and shATG7#2) knockdown followed by 0.50 μM periplocin treatment for 24 h. (C) Immunofluorescent analysis of CTSB in cells with or without ATG5 knockdown (shATG5#1) followed by 0.50 μM periplocin treatment for 24 h. Scale bar: 10 μm. (D and E) Quantitation of the relative fluorescent intensity of Magic Red staining in cells with ATG5 (D) or ATG7 (E) knockdown followed by 0.50 μM periplocin treatment for 24 h. (F and G) Representative images (F) and quantitative analysis (G) for immunofluorescent staining of LGALS3 in cells with or without ATG5 knockdown (shATG5#1) followed by 0.50 μM periplocin treatment for 24 h. Scale bar: 10 μm. (H and I) Representative images (H) and quantitative analysis (I) for immunofluorescent staining of LGALS3 in cells with or without ATG7 knockdown followed by 0.50 μM periplocin treatment for 24 h. Scale bar: 10 μm. Results are representative of three independent experiments. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Periplocin-induced lysophagy inhibits CRC cell growth
The resultant increase of lysosomal damage suggests that periplocin-induced lysophagy may inhibit CRC cell growth by disturbing lysosomal homeostasis. To test this hypothesis, we treated CRC cells with 3-MA, wortmannin (a class III phosphatidylinositol 3-kinase inhibitor) or CQ in combination with periplocin, and examined cell growth. As shown in Figure 5A-C, treatment with 3-MA, wortmannin or CQ significantly rescued the growth inhibition of CRC cells in response to periplocin treatment. Similar alleviation of periplocin-mediated proliferation inhibition was also observed by CQ or 3-MA treatment, as evidenced by colony formation (Figure 5D,E) and EdU incorporation (Figure 5F). These findings were further strengthened by the observation that inhibiting lysophagy through ATG5 or ATG7 knockdown resulted in reduced sensitivity of CRC cell growth to periplocin treatment (Figure 5G-K). In addition, the increased levels of cleaved PARP1 and cleaved CASP3 in CRC cells were markedly suppressed by ATG5 or ATG7 knockdown (Figure 5L,M), suggesting that lysophagy inhibition attenuates periplocin-induced apoptosis in CRC cells. Overall, these results indicate that lysophagy contributes to the anticancer effect of periplocin in CRC cells.
Figure 5.
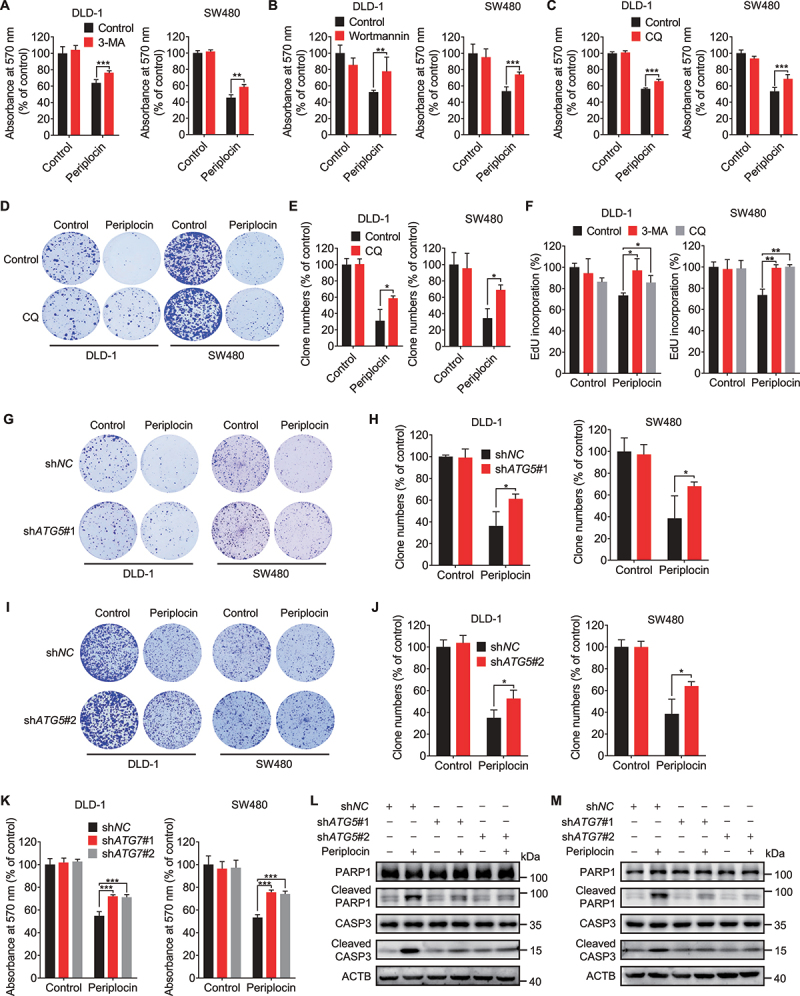
Periplocin-induced lysophagy potentiates the growth inhibition of CRC cells. (A and B) MTT assay of CRC cells treated with 0.50 μM periplocin for 24 h in combination with 5 mM 3-MA (A) or 200 nM wortmannin (B). (C and D) MTT (C) and colony formation (D) assay of cells treated with 0.50 μM periplocin for 24 h in combination with or without chloroquine (CQ, 10 μM). (E) Quantification of clone numbers in (D). (F) CRC cells were treated with 3-MA (5 mM) or CQ (10 μM) in the presence or absence of 0.50 μM periplocin for 24 h. Cell proliferation was measured by EdU incorporation. (G-J) Colony formation assay of cells with or without ATG5 knockdown (G, shATG5#1; I, shATG5#2) followed by 0.50 μM periplocin treatment for 24 h. Quantification of clone numbers was shown (H, shATG5#1; J, shATG5#2). (K) MTT assay of CRC cells with or without ATG7 knockdown followed by 0.50 μM periplocin treatment for 24 h. (L and M) Immunoblotting analysis of PARP1, cleaved PARP1, CASP3, and cleaved CASP3 in DLD-1 cells with ATG5 (L) or ATG7 (M) knockdown followed by 0.50 μM periplocin treatment for 24 h. Results are representative of three independent experiments. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Periplocin binds and prevents ubiquitin-mediated degradation of LGALS3 in CRC cells
We next investigated the mechanism underlying periplocin-mediated lysophagy. The expression of LGALS3, a key lysophagy marker [46], was found to be upregulated following periplocin treatment as evidenced by immunofluorescent staining (Figure 2M). To this end, we presumed that periplocin-induced lysophagy might be attributable to the upregulation of LGALS3. Indeed, periplocin treatment prominently elevated the protein level of LGALS3 as evidence by immunoblotting analysis (Figure 6A). The increased protein level of LGALS3 was further confirmed in tumor xenografts from periplocin-treated mice (Figure 6B,C). However, no obvious change was observed on the mRNA level of LGALS3 in periplocin-treated cells compared with controls (Figure S6A), suggesting that transcriptional regulation was not involved in increased LGALS3 expression following periplocin treatment. Therefore, we postulated that periplocin might elevate LGALS3 by regulating protein stability. Of note, cycloheximide (CHX, a translational inhibitor) treatment led to an obvious decrease in LGALS3 protein level in control cells, but had no obvious effect on the upregulated protein level of LGALS3 in periplocin-treated cells, suggesting periplocin maintains LGALS3 stability (Figure 6D,E). In support of this, treatment with MG132 (a proteasome inhibitor) failed to further enhance the protein level of LGALS3 in response to periplocin treatment (Figure 6F,G). These data suggest that periplocin may prevent proteasomal degradation of LGALS3.
Figure 6.
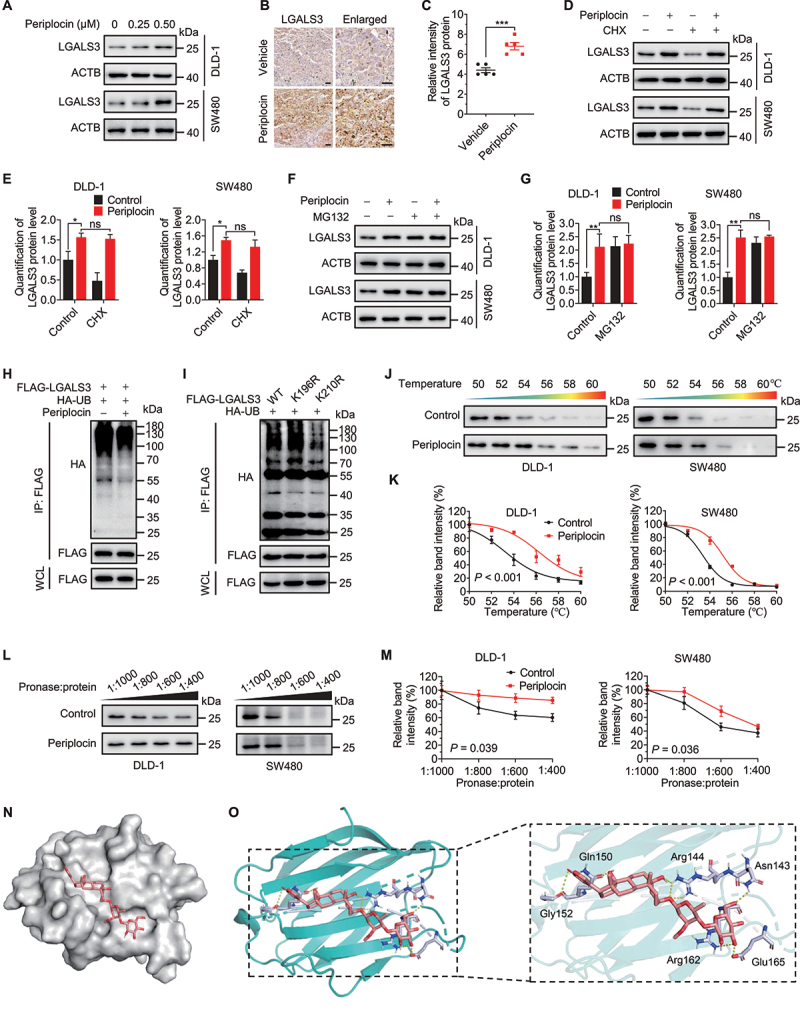
Periplocin binds and prevents ubiquitin-mediated degradation of LGALS3 in CRC cells. (A) Immunoblotting analysis of LGALS3 in cells treated with periplocin for 24 h at the indicated concentrations. (B and C) Representative images (B) and quantitative analysis (C) of immunohistochemical staining for LGALS3 in SW480 ×enografts from vehicle- or periplocin-treated mice. Scale bar: 50 μm. (D) Immunoblotting analysis of LGALS3 in cells treated with 0.50 μM periplocin for 24 h in the presence or absence of cycloheximide (CHX, 50 μg/mL). (E) Quantitation of LGALS3 protein level in (D). (F) Immunoblotting analysis of LGALS3 in cells treated with 0.50 μM periplocin for 24 h in the presence or absence of MG132 (25 μM, 6 h). (G) Quantitation of LGALS3 protein level in (F). (H) FLAG-LGALS3 was co-expressed with HA-tagged ubiquitin (HA-UB) in HEK293T cells, followed by treatment with or without 0.50 μM periplocin for 24 h in the presence of MG132 (25 μM, 6 h). Immunoprecipitation was performed with FLAG beads, followed by immunoblotting with the indicated antibodies. (I) FLAG-LGALS3 WT, FLAG-LGALS3K196R, or FLAG-LGALS3K210R was co-expressed with HA-UB followed by MG132 treatment (25 μM, 6 h). Immunoprecipitation was performed with FLAG beads, followed by immunoblotting with the indicated antibodies. (J and K) Cellular thermal shift assay (CESTA) showing target engagement of LGALS3 by periplocin in CRC cells. (L and M) Drug affinity responsive target stability (DARTS) analysis of periplocin binding with LGALS3. (N and O) Electrostatic surface representation (N) and cartoon representation (O) for the docking model of periplocin binding to H. sapiens LGALS3 protein. Results are representative of three independent experiments. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. ns, non-significant.
We therefore measured the effect of periplocin on LGALS3 ubiquitination. As shown in Figure 6H, periplocin markedly decreased the ubiquitin-conjugated level of LGALS3. In addition, periplocin did not affect the ubiquitination of other proteins, such as PHGDH (phosphoglycerate dehydrogenase) and PRMT1 (protein arginine methyltransferase 1) (Figure S6B,C), and had no obvious effect on the expression of several essential proteasome components (Figure S6D,E). These data suggest that periplocin specifically prevents LGALS3 from ubiquitin-mediated degradation, regardless of the general ubiquitylation status of the proteasome. A previous study of systematic ubiquitination profiling identified Lys196 (K196) and Lys210 (K210) as the potential ubiquitination sites of LGALS3 [47]. To determine the ubiquitination site required for LGALS3 degradation, we generated lysine to arginine mutants of LGALS3 at K196 or Lys210 (LGALS3K196R or LGALS3K210R). As shown in Figure 6I, the ubiquitin-conjugated level of LGALS3K196R mutant was comparable to the wild type (WT), whereas K210 mutation significantly decreased LGALS3 ubiquitination. These results suggest that periplocin elevates LGALS3 level by preventing K210 ubiquitination and proteasomal degradation. In addition, periplocin was found to stabilize the protein level of LGALS3 against thermal changes using a cellular thermal shift assay (CETSA) (Figure 6J,K), implying the binding of periplocin with LGALS3. The interaction between periplocin and LGALS3 was further confirmed by drug affinity responsive target stability (DARTS) analysis, as evidenced by a more stable property of LGALS3 protein against pronase digestion in response to periplocin treatment (Figure 6L,M). Moreover, using semi-flexible docking analysis, we found that LGALS3 showed a good binding activity for periplocin, with a binding energy of −6.689 kcal/mol. Glu165, Arg162, Gly152, Gln150, Arg144, and Asn143 of LGALS3 were identified as possible sites for periplocin binding (Figure 6N,O), which required further experimental investigation. Together, these data suggest that periplocin binds and prevents ubiquitin-mediated degradation of LGALS3 in CRC cells.
Periplocin induces lethal lysophagy by upregulating LGALS3 in CRC cells
To determine whether LGALS3 upregulation is involved in periplocin-mediated lysophagy induction, we examined the turnover of LC3B in LGALS3 knockdown or knockout CRC cells treated with or without periplocin. We found that siRNA-mediated LGALS3 knockdown (siLGALS3#1 and siLGALS3#2, Figure 7A,B) or CRISPR-Cas9-mediated LGALS3 knockout (lgals3 KO#1 and lgals3 KO#2, Figure 7C,D) significantly suppressed periplocin-induced LC3B turnover. Consistently, LGALS3 knockdown countered the periplocin-induced increase of LC3B puncta (Figure 7E,F). Moreover, periplocin treatment caused a marked decrease in SQSTM1/p62 protein level, which could be rescued by LGALS3 knockdown (Figure S6F,G) or knockout (Figure S6H,I). These data suggest that LGALS3 upregulation is required for periplocin-induced lysophagy in CRC cells.
Figure 7.
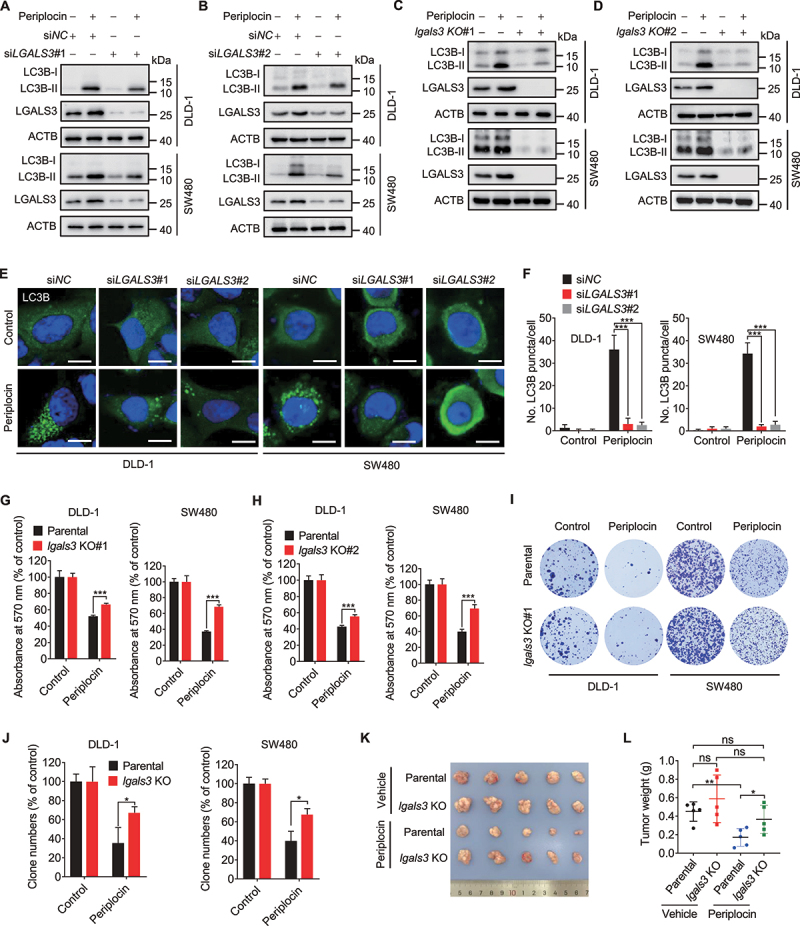
Periplocin induces lethal lysophagy by upregulating LGALS3 in CRC cells. (A and B) Immunoblotting analysis of LC3B turnover in cells transfected with siNC or siLGALS3 followed by 0.50 μM periplocin treatment for 24 h. (C and D) Immunoblotting analysis of LC3B turnover in parental or lgals3 KO cells followed by 0.50 μM periplocin treatment for 24 h. (E and F) Representative images (E) and quantitative analysis (F) for immunofluorescent staining of endogenous LC3B puncta in cells transfected with siNC or siLGALS3 followed by 0.50 μM periplocin treatment for 24 h. Scale bar: 10 μm. (G and H) MTT assay of CRC cells with or without LGALS3 knockout (G, lgals3 KO#1; H, lgals3 KO#2) in response to 0.50 μM periplocin treatment for 24 h. (I) Colony formation assay of parental or lgals3 KO cells treated with or without 0.50 μM periplocin for 24 h. (J) Quantification of clone numbers in (I). (K and L) DLD-1 parental or lgals3 KO cells were subcutaneously inoculated into nude mice. Mice were injected with vehicle or periplocin (15 mg/kg/day) for two weeks. Image (K) and weight (L) of tumor xenografts were shown. Results in A-J are representative of three independent experiments. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. ns, non-significant.
We next investigated whether LGALS3 is required for periplocin’s anti-CRC effect. As shown in Figure 7G,H and Figure S6J, periplocin treatment inhibited the growth of DLD-1 and SW480 cells, which was markedly rescued by LGALS3 knockout (Figure 7G,H) or siRNA-mediated LGALS3 knockdown (Figure S6J). A similar increase in clone numbers was also observed in lgals3 KO cells following periplocin treatment (Figure 7I,J). In addition, LGALS3 knockout significantly alleviated the periplocin-induced G2/M phase arrest (Figure S6K,L). To further support our observations, we subcutaneously inoculated DLD-1 parental or lgals3 KO cells into nude mice. The results showed that LGALS3 knockout significantly rescued the decrease of xenograft tumor growth in periplocin-treated mice (Figure 7K,L). Collectively, these data indicate that LGALS3 is required for the anti-CRC effect of periplocin.
Discussion
In this study, we have investigated the anticancer effect of periplocin in CRC cells and the involved mechanisms. Our results suggest that periplocin inhibits the growth and proliferation of CRC cells by inducing lysosomal damage and exacerbating lysophagy. We found that periplocin binds to LGALS3 and inhibits its proteasomal degradation, leading to LGALS3 upregulation and lysosomal redirection, thereby inducing excessive lysophagy-mediated lysosomal damage and cell death. Therefore, our findings provide a novel insight into periplocin’s anticancer mechanism in CRC cells, and underpin a cytotoxic role of lysophagy induced by periplocin.
There has been a long history of the use of natural product-derived traditional medicines, especially in China, India, Greece and Rome [48]. Many clinical anticancer drugs approved by the FDA are natural products or their derivates (such as paclitaxel, vinblastine, adriamycin), highlighting an indispensable role of natural products in cancer drug discovery [49–51]. Our group has previously reported the anticancer activities of several natural products, including quercetin, ivermectin and 3′-epi-12β-hydroxyfroside [8,10,52]. In this study, we found periplocin, another nature product derived from the traditional Chinese herb Cortex periplocae, showed promising anticancer effects against CRC. In addition, our preclinical data revealed that periplocin had no obvious toxicity in mice. Indeed, as an effective constituent of qili qiangxin capsules, periplocin has been used for the clinical treatment of chronic heart failure [18,19], implying acceptable safety for the clinical use of periplocin. Therefore, periplocin holds great promise to be repurposed as a novel potential anticancer drug for CRC treatment.
Previous studies have demonstrated that periplocin exhibits cytotoxicity in other cancers, such as liver cancer, gastric cancer, myxofibrosarcoma and pancreatic cancer, However, the involved mechanism is either unclear or limited to apoptosis induction [20,22,23,25]. Our data demonstrate that the anticancer activity of periplocin in CRC cells is due to lysosomal damage and excessive lysophagy, which lead to apoptosis. This lysosomal damage induced by periplocin involves stimulation of LMP. Given the basic property of periplocin (pKa = 12.89 ± 0.70, calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02), we propose that periplocin can act like a lysosomotropic agent to be trapped within lysosomes to induce LMP and cause lysosomal damage. It has been reported that LMP can lead to mitochondrial membrane permeabilization (MMP)-mediated caspase-dependent apoptosis, caspase-independent apoptosis-like cell death or necrosis, which may provide a rational explanation for the apoptosis-dependent cell death caused by periplocin [26,53,54]. Similar to other types of organelle-selective autophagy (e.g. mitophagy), lysophagy is generally characterized as a pro-survival mechanism under cellular stress by degrading damaged lysosomes [42]. Interestingly, we found that inhibition of autophagy rescued periplocin-induced lysosomal damage and growth inhibition of CRC cells, indicating that periplocin-induced lysophagy plays a lethal role by exacerbating lysosomal damage and cell death. It has been reported that lysosomes are highly dynamic and heterogeneous. Lysosomes can undergo fusion and fission, which are well coordinated to maintain lysosomal homeostasis [31,55]. We found that periplocin treatment promotes P2RX4-mediated lysosome fusion, but has no obvious effect on lysosome fission. Moreover, P2RX4 was found to co-localize with LGALS3 in periplocin-treated cells, suggesting the involvement of damaged lysosomes during lysosome fusion. Therefore, periplocin-induced lysophagy-mediated clearance of damaged lysosomes can nonspecifically engulf vicinal functional lysosomes when lysosome fusion occurs, leading to excessive lysophagy and subsequent cell death. Our findings thus demonstrate a cytotoxic mechanism of lysophagy, and suggest a context-dependent manner for lysophagy-mediated cell fate decision. In addition, we found that periplocin treatment promoted the translocation of TFEB (transcription factor EB) into the nucleus (Figure S7A,B). Interestingly, periplocin treatment led to increased TFEB nuclear translocation and LGALS3 expression both in a time-dependent manner. While the protein level of LGALS3 increased at 1 h of periplocin treatment, the level of nuclear TFEB increased at 3–6 h after periplocin treatment (Figure S7C,D). These data imply that TFEB nuclear translocation and activation in periplocin-treated CRC cells may represent a compensate mechanism for lysosomal damage through promoting the lysosomal biogenesis.
Target identification is the initial and critical step for understanding the mechanism of action and development of novel natural products [50,56]. In this study, we found periplocin physically engaged LGALS3 in living CRC cells, suggesting LGALS3 is a binding target of periplocin. Further studies are needed to confirm the direct binding using recombinant LGALS3 protein. The binding of periplocin inhibited ubiquitin-mediated degradation of LGALS3, leading to elevated protein level of LGALS3. Periplocin binding might affect the conformational change of LGALS3 and decrease its binding with related E3 ligase, thereby preventing ubiquitin-mediated degradation of LGALS3, which also requires further investigation. Upregulated LGALS3 was then recruited to the lysosomes following periplocin treatment to initiate excessive lysophagy machinery. These results suggest that periplocin-mediated LGALS3 accumulation and excessive lysophagy may merit further exploration as a therapeutic strategy for CRC treatment.
LGALS3 is distributed either in the cytosol or nuclei. Cytosolic LGALS3 can bind the exposed intraluminal carbohydrate chains of lysosomal glycoproteins when lysosomal membrane rupture, followed by recruitment of TRIM16 which serves as a platform for autophagic initiation factors and mobilization of LC3-containing autophagic membranes [33,42,57]. Nuclear LGALS3 serves as a co-transcription factor to promote the expression of multiple genes, including MMP1, F2R/PAR1, and FSCN1/Fascin-1, to promote tumor growth and metastasis [58]. LGALS3 was reported to be overexpressed in tumors according to most studies [59,60], which seems to contradict with our finding describing LGALS3 accumulation leads to cancer cell death. However, in our study, periplocin-induced LGALS3 overexpression was observed in the cytosol, but not nuclei. It implies that the oncogenic role of LGALS3 May be largely dependent on its nuclear translocation, and its cytosolic form may represent tumor-suppressive function, although it cannot be excluded that cytosolic LGALS3 was also found to promote tumor growth or metastasis in some studies. In line with our hypothesis, several findings illustrating the down-expression or tumor-suppressive role of LGALS3 have also been reported [61,62]. Thus, the function of LGALS3 is context-dependent and deserves further investigation.
In summary, our study revealed that periplocin inhibits the growth of CRC cells via inducing lysosomal damage and LGALS3-mediated lethal lysophagy. These findings provide novel insights into the mechanism of action of periplocin’s anticancer efficacy, which supports a preclinical rationale for repurposing periplocin as a therapeutic drug for CRC, and highlights the promise of monitoring context-dependent lysophagy for cancer treatment.
Materials and methods
Cell culture
Human CRC cell lines (DLD-1 [CCL-221], SW480 [CCL-228], HCT116 [CCL-247], SW620 [CCL-227], HT29 [HTB-38], LOVO [CCL-229], RKO [CRL-2577]), human pancreatic cancer cell lines (MIA PaCa-2 [CRL-1420], Panc-1 [CRL-1469]), human hepatocellular carcinoma cell lines (Hep3B [HB-8064], SK-Hep-1 [HTB-52]), and HEK293T (CRL-3216) were obtained from the American Type Culture Collection (ATCC). Human colon mucosal epithelial cell line NCM460 was purchased from MingzhouBio (MZ-0658). Cells were cultured as previously described [63,64]. Briefly, all cell lines were cultured in high glucose Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher Scientific, 12800017) containing 100 U/mL penicillin, 100 U/mL streptomycin, and 10% fetal bovine serum (VivaCell Biosciences, C04001) in a humidified chamber at 37°C under 5% (v:v) CO2 atmosphere. All cell lines are mycoplasma-free.
Reagents and antibodies
Periplocin (13137-64-9) was supplied by Chengdu Herbpurify Co., Ltd. Periplocin was dissolved in DMSO to make a stock solution at 1 mM or 100 mM and stored at −80°C. 3-methyladenine (HY-19312), wortmannin (HY-10197), and oxaliplatin (HY-17371) were purchased from MedChemExpress. DAPI (62248), Lipofectamine 3000 (L3000015), LysoTracker Red DND-99 (L7528), and LysoSensor Yellow/Blue DND-160 (L7545) were obtained from ThermoFisher Scientific. Magic Red was purchased from Abcam (ab270772). MG-132 (S2619), CHX (S7418), and Z-VAD-FMK (S7023) were purchased from Selleck. DMSO (D2650), chloroquine (CQ; C6628), and acridine orange (A6014) were purchased from Sigma-Aldrich.
Antibodies used in this study: ATG5 (12994), LAMP1 (9091), CTSB (31718), ACTA2/α-SMA (19245), PECAM1/CD31 (3528), BCL2 (15071), CDKN1A/p21 (2947), and p-PRKAA/AMPKα (Thr172, 2535) were purchased from Cell Signaling Technology. MKI67/Ki-67 (ab16667), LAMP2 (ab25631), PDGFRA (ab203491), and AIF1/IBA1 (ab178846) was purchased from Abcam. LGALS3 (galectin 3; sc -32,790), TRIM16 (sc -398,851), PDCD6IP/Alix (sc -53,540), PSMB4 (sc -390,878), ATP6V1B (sc -55,544), and ATP6V1H (sc -166,227) were purchased from Santa Cruz Biotechnology. MAP1LC3B/LC3B (NB100–2220) was obtained from Novus Biologicals. ACACA/ACC1 (67373–1-Ig), p-ACACA/ACC1 (Ser79; 29119–1-AP), PRKAA/AMPKα (10929–2-AP), MTOR (66888–1-Ig), p-MTOR (Ser2448; 67778–1-Ig), BECN1 (beclin 1; 11306–1-AP), BCL2 (60178–1-Ig), LGALS1 (galectin 1; 60223–1-Ig), CHMP4B (13683–1-AP), P2RX4/P2X4 (13534–1-AP), CALM/calmodulin (10541–1-AP), and MCOLN1/TRPML1 (15291–1-AP) were purchased from Proteintech. PSMB6 (A4053), FLAG (AE063), HA (AE036), PARP1/PARP (A19596), CASP3/caspase 3 (A0214), cleaved CASP3/caspase 3 (A11021), ATG5 (a1967), ATG7 (a19604), and ACTB/β-actin (AC026) were obtained from ABclonal. LGALS3 (MAB1154) was purchased from R&D Systems. SQSTM1/p62 (PM045) was purchased from MBL Life Science. CDK1 (WL02373) and CCNB1/cyclin B1 (WL01760) were purchased from Wanleibio. Goat anti-mouse Alexa Fluor 488 (A32723), goat anti-mouse Alexa Fluor 594 (A32742), goat anti-rabbit Alexa Fluor 488 (A32731), and goat anti-rabbit Alexa Fluor 594 (A32740) were purchased from ThermoFisher Scientific.
shRNA and siRNA sequences are listed as follows:
shATG5#1: 5′-GCAACUCUGGAUGGGAUUG-3′;
shATG5#2: 5′-CCTTTGGCCTAAGAAGAAA-3′;
shATG7#1: 5′-GCCTGCTGAGGAGCTCTCCAT-3′;
shATG7#2: 5′-GCTTTGGGATTTGACACATTT-3′;
shBECN1#1: 5′-GGAGCCAUUUAUUGAAACUTT-3′;
shBECN1#2: 5′-GGGCGGAAGUCUUCAGCUAGC-3′;
siLGALS3 #1: 5’-AUAUGAAGCACUGGUGAGGUCUAUG-3’;
siLGALS3 #2: 5’- GCCACUGAUUGUGCCUUAUTT-3’.
Tumor xenograft model
Six-week-old female nude mice were purchased from HFK Bioscience. The animal study was approved by the Institutional Animal Care and Treatment Committee of Sichuan University. 5 × 106 SW480 or DLD-1 cells were suspended in PBS (Sangon Biotech, B040100–0005) and subcutaneously grafted into mice. When the tumor volumes reached ~100 mm3, mice were randomly divided into two groups intraperitoneally receiving 0.1 mL of vehicle (10% castor oil [MedChemExpress, HY-107799], 5% DMSO, 10% ethanol, 75% physiological saline [0.9% NaCl]) or periplocin (15 mg/kg/day), respectively. The tumor volumes were measured every three days and calculated as (length × width2)/2. Mice were euthanized after two weeks, and tumor tissues were formalin-fixed immediately.
Immunoblotting and immunoprecipitation
Immunoblotting was performed as previously described [65]. Briefly, cells were rinsed with ice-cold PBS and then lysed in RIPA buffer ((50 mM Tris, pH 7.4, 150 mM NaCl, 1.0 mM EDTA, 0.1% SDS [Sigma-Aldrich, L5750], 1% NP-40 [Solarbio Life Sciences, N8030], 1% sodium deoxycholate [Solarbio Life Sciences, D8331]). The lysates were quantified and then separated by SDS-PAGE, followed by transfer to a PVDF membrane (EMD Millipore, ISEQ00010). The membrane was blocked with 5% skimmed milk in TBST (Sangon Biotech, C520009) for 2 h at room temperature, then incubated with primary antibodies at 4°C overnight and secondary antibodies at room temperature for 1.5 h. Finally, Immobilon Western HRP Substrate (EMD Millipore, WBKLS0500) was used to visualize the immunoreactive bands, and the images were obtained using a ChemiScope 6000 Touch (Clinx Science Instruments).
For immunoprecipitation, cells were lysed in IP lysis buffer (20 mM Tris, pH 7.4, 137 mM NaCl, 10% glycerol, 1% NP-40, and 2 mM EDTA). The lysates were then incubated with 1 μg of the indicated antibodies at 4°C overnight, followed by 3 h incubation with protein A/G agarose beads (EMD Millipore, IP10). After centrifugation, washing for 5 times and boiling with loading buffer, the samples were analyzed by immunoblotting with the indicated antibodies.
Measurement of cell growth, proliferation, apoptosis, and cell cycle
The short-term effects of periplocin on cell growth were evaluated by MTT assay. Cells were seeded in 96-well plates at a density of 5000 cells/well and treated for 24 h. Cells were then incubated with 5 mg/mL MTT (Sigma-Aldrich, M2128) for 4 h and dissolved in DMSO. The absorbance was measured at 570 nm with a spectrophotometer.
Colony formation assay was performed to detect the long-term effects of periplocin on cell growth. Cells were seeded in 24-well plates (500 cells/well) and then subjected to drug treatment. After treatment for 24 h, the medium containing drugs was changed with fresh medium. After two weeks, cells were fixed using methanol, stained with crystal violet (Beyotime Biotech, C0121), and then photographed. The clone numbers were counted using Image J software.
5-ethynyl-20-deoxyuridine (EdU) labeling assay was performed to measure cell proliferation using the Cell-Light EdU Apollo488 In Vitro Kit (Ribobio, C10310–3). Cells seeded in 96-well plates (4000 cells/well) were treated with the indicated drugs for 24 h, then labeled with 50 μM EdU for another 2 h. After fixing with 4% paraformaldehyde in PBS, the samples were stained with reaction cocktail, and the nuclei were subsequently stained with DAPI. Cells were imaged using a fluorescence microscope (Axio Observer 7, ZEISS).
For cell cycle analysis, cells were digested by trypsin, stained with a Cell Cycle Kit (KeyGen Biotech, KGA512), and subjected to cell cycle distribution analysis on a flow cytometer (BD FACSCelesta).
The AnnexinV-FITC/propidium iodide (PI) Detection Kit (KeyGen Biotech, KGA108) was used to measure the ratio of apoptotic cells. Cells were cultured in 6-well plates followed by periplocin treatment for 24 h at the indicated concentrations. Cells were washed twice with PBS, resuspended in 500 mL binding buffer, and then incubated with ANXA5/annexin V-FITC and PI. At least 10,000 live cells were taken for flow cytometric analysis on a flow cytometer (BD FACSCelesta). Oxaliplatin was used as a positive control. The apoptotic induction was also determined by immunoblotting analysis of cleaved PARP1, cleaved CASP3, and BCL2 levels.
Acridine orange staining
Cells were treated with periplocin for 24 h, and then incubated with 5 μM acridine orange (Sigma-Aldrich, A6014) for 15 min at 37°C. After washing and resuspension with PBS, samples were analyzed on a flow cytometer (BD FACSCelesta).
Quantitative RT-PCR analysis
Total RNA was extracted using TRIzol (ThermoFisher Scientific, 15596018). The RT-PCR was performed as previously described [66]. The total RNA (1 μg) was reverse transcribed using the PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, RR047A). The mRNA levels of the indicated genes were quantified using the Bio-Rad iTaq Universal SYBR Green Supermix (Bio-Rad, 1725271) in a CFX96 Real Time System (Bio-Rad Laboratories). The primer pairs of LGALS3 were: forward 5“-CCATTTGAAAGTGGGAAACCA-3”, reverse 5“-CATCATTCACTGCAACCTTGAAG-3”.
Immunohistochemistry
Tumor xenografts were collected and further fixed in 4% paraformaldehyde overnight followed by paraffin embedding. Immunohistochemical staining was performed as described previously [67]. Briefly, sections (5 μm) were dewaxed, rehydrated, and quenched. Antigen retrieval was then performed using citrate buffer. After serum blocking with goat serum (Sigma-Aldrich, G9023) at 37°C for 1 h, slides were incubated with the indicated primary antibodies at 4°C overnight and MaxVision HRP-Polymer anti-Mouse/Rabbit IHC Kit (MXB Biotechnologies, Kit-5010). Finally, slides were stained using diaminobenzidine (MXB Biotechnologies, DAB-0031) and counterstained with Mayer’s hematoxylin (MXB Biotechnologies, CTS-1096).
Cellular thermal shift assay
Cells cultured in 100-mm dishes to 80% confluency were treated with or without periplocin (0.50 μM) for 6 h. Cells were harvested by trypsin, resuspended with PBS, and then divided into 6 aliquots, each of which was heated at 50, 52, 54, 56, 58, 60°C for 5 min. Soluble fractions were then extracted by 3 cycles of freeze-thawing with liquid nitrogen, followed by centrifugation at 17,000 g for 10 min. Finally, samples were analyzed by immunoblotting with LGALS3 antibody.
Drug affinity responsive target stability (DARTS)
Cells were treated with or without periplocin for 4 h, and lysed using RIPA buffer. The pronase (Sigma-Aldrich, 0165921001) was added for proteolysis at room temperature for 1 h. The ratio of pronase:total protein was 1:400, 1:600, 1:800 and 1:1000. Subsequently, the samples were then mixed with the loading buffer and analyzed by immunoblotting analysis with LGALS3 antibody.
TMT (tandem mass tags)-labeled quantitative proteomics analysis
TMT-labeled proteomics analysis was performed using TMTsixplex Isobaric Label Reagent Set (ThermoFisher Scientific, 90061) according to the manufacturer’s instructions. Briefly, cells grown in 100-mm dishes to 80% confluency were treated with or without periplocin (0.50 μM) for 24 h, then harvested and lysed. The samples were reduced and alkylated with TCEP (Sigma-Aldrich, 580560) and IAM (Sigma-Aldrich, I1149), respectively, followed by sequencing-grade trypsin (Promega, V5111) digestion. Subsequently, tryptic peptides were labeled with isobaric TMT tags. The samples were then pooled, dried, desalted and analyzed using a Q Exactive Plus Orbitrap LC-MS/MS System (ThermoFisher Scientific). Data analysis was performed using Proteome Discoverer 1.2 software (ThermoFisher Scientific).
Immunofluorescence
Cells were seeded at 30,000–40,000 cells/well on glass coverslips in 24-well plates followed by transfection or treatment as indicated. The cells were washed with PBS and fixed with 4% paraformaldehyde for 30 min at room temperature. After exposure to PBS containing 0.2% Triton X-100 (Solarbio Life Sciences, T8200) and 5% BSA for 1.5 h, the slides were stained with the indicated primary antibodies at 4°C overnight, and subsequently stained with Alexa Fluor secondary antibodies at room temperature for 2 h. DAPI was used to stain nuclei for 10 min at room temperature. Images were visualized using a Zeiss LSM 710 confocal microscope.
Statistical analysis
All statistical analysis was performed using GraphPad Prism 8.0 software. Two-tailed Student’s t-test was used to compare the variables between two groups. Two-way ANOVA was performed for comparing tumor volume. Data were shown as means ± SD. Significance was indicated as follows: *P < 0.05, **P < 0.01; ***P < 0.001, ns, non-significant.
Supplementary Material
Funding Statement
This work was supported by the National Natural Science Foundation of China (81821002, 82130082, 82341004, 82273122, 82073081, 82172588, 82002963), National Key Research and Development Project of China (2020YFA0509400), Guangdong Basic and Applied Basic Research Foundation (2019B030302012), the Science and Technology Department of Sichuan Province (2017SZ0057), the Chengdu Science and Technology Program (2019-YF05-00715-SN), and 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYGD22007).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2239042
References
- [1].Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018. Nov;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- [2].Benson AB, Venook AP, Al-Hawary MM, et al. NCCN guidelines insights: colon cancer, version 2.2018. J Natl Compr Canc Netw. 2018. Apr;16(4):359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest. 2019 Jul 2;129(8):3006–3017. doi: 10.1172/JCI127201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dekker E, Tanis PJ, Vleugels JLA, et al. Colorectal cancer. Lancet. 2019 Oct 19;394(10207):1467–1480. doi: 10.1016/S0140-6736(19)32319-0 [DOI] [PubMed] [Google Scholar]
- [5].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. doi: 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017. Sep;17(9):528–542. doi: 10.1038/nrc.2017.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dong L, He J, Luo L, et al. Targeting the interplay of autophagy and ROS for cancer therapy: an updated overview on phytochemicals. Pharmaceuticals (Basel). 2023 Jan 8;16(1):92. doi: 10.3390/ph16010092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dou Q, Chen HN, Wang K, et al. Ivermectin induces cytostatic autophagy by blocking the PAK1/Akt Axis in breast cancer. Cancer Res. 2016 Aug 1;76(15):4457–4469. doi: 10.1158/0008-5472.CAN-15-2887 [DOI] [PubMed] [Google Scholar]
- [9].Jiang J, Zhang L, Chen H, et al. Regorafenib induces lethal autophagy arrest by stabilizing PSAT1 in glioblastoma. Autophagy. 2020. Jan;16(1):106–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang K, Liu R, Li J, et al. Quercetin induces protective autophagy in gastric cancer cells: involvement of Akt-Mtor- and hypoxia-induced factor 1alpha-mediated signaling. Autophagy. 2011. Sep;7(9):966–978. [DOI] [PubMed] [Google Scholar]
- [11].Xia Y, Xu F, Xiong M, et al. Repurposing of antipsychotic trifluoperazine for treating brain metastasis, lung metastasis and bone metastasis of melanoma by disrupting autophagy flux. Pharmacol Res. 2021. Jan;163:105295. [DOI] [PubMed] [Google Scholar]
- [12].Yan C, Luo L, Guo CY, et al. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017 Mar 1;388:34–42. doi: 10.1016/j.canlet.2016.11.018 [DOI] [PubMed] [Google Scholar]
- [13].Zhang Z, Zhou L, Xie N, et al. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct Target Ther. 2020 Jul 2;5(1):113. doi: 10.1038/s41392-020-00213-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Anding AL, Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell. 2017 Apr 10;41(1):10–22. doi: 10.1016/j.devcel.2017.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen Y, Chen HN, Wang K, et al. Ketoconazole exacerbates mitophagy to induce apoptosis by downregulating cyclooxygenase-2 in hepatocellular carcinoma. J Hepatol. 2019. Jan;70(1):66–77. [DOI] [PubMed] [Google Scholar]
- [16].Dany M, Gencer S, Nganga R, et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood. 2016 Oct 13;128(15):1944–1958. doi: 10.1182/blood-2016-04-708750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012. Oct;8(10):831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yun WJ, Yao ZH, Fan CL, et al. Systematic screening and characterization of Qi-Li-Qiang-Xin capsule-related xenobiotics in rats by ultra-performance liquid chromatography coupled with quadrupole time-of-flight tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2018 Jul 15;1090:56–64. doi: 10.1016/j.jchromb.2018.05.014 [DOI] [PubMed] [Google Scholar]
- [19].Li X, Zhang J, Huang J, et al. A multicenter, randomized, double-blind, parallel-group, placebo-controlled study of the effects of qili qiangxin capsules in patients with chronic heart failure. J Am Coll Cardiol. 2013 Sep 17;62(12):1065–1072. doi: 10.1016/j.jacc.2013.05.035 [DOI] [PubMed] [Google Scholar]
- [20].Cheng CF, Lu IH, Tseng HW, et al. Antitumor effect of periplocin in TRAIL-Resistant human hepatocellular carcinoma cells through downregulation of IAPs. Evid Based Complement Alternat Med. 2013;2013:958025. doi: 10.1155/2013/958025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Du YY, Liu X, Shan BE. Periplocin extracted from cortex periplocae induces apoptosis of SW480 cells through inhibiting the Wnt/beta-catenin signaling pathway. Ai Zheng. 2009. May;28(5):456–460. [PubMed] [Google Scholar]
- [22].Lohberger B, Bernhart E, Stuendl N, et al. Periplocin mediates TRAIL-induced apoptosis and cell cycle arrest in human myxofibrosarcoma cells via the ERK/p38/JNK pathway. Phytomedicine. 2020 Jun 5;76:153262. doi: 10.1016/j.phymed.2020.153262 [DOI] [PubMed] [Google Scholar]
- [23].Xie G, Sun L, Li Y, et al. Periplocin inhibits the growth of pancreatic cancer by inducing apoptosis via AMPK-Mtor signaling. Cancer Med. 2021. Jan;10(1):325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhao L, Shan B, Du Y, et al. Periplocin from Cortex periplocae inhibits cell growth and down-regulates survivin and c-myc expression in colon cancer in vitro and in vivo via beta-catenin/TCF signaling. Oncol Rep. 2010. Aug;24(2):375–383. [PubMed] [Google Scholar]
- [25].Zhao LM, Li L, Huang Y, et al. Antitumor Effect of Periplocin in TRAIL-Resistant gastric cancer cells via upregulation of death receptor through activating ERK1/2-EGR1 pathway. Mol Carcinog. 2019. Jun;58(6):1033–1045. [DOI] [PubMed] [Google Scholar]
- [26].Wang F, Gomez-Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018. Dec;19(12):918–931. doi: 10.1111/tra.12613 [DOI] [PubMed] [Google Scholar]
- [27].Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009. Sep;10(9):623–635. doi: 10.1038/nrm2745 [DOI] [PubMed] [Google Scholar]
- [28].Fehrenbacher N, Bastholm L, Kirkegaard-Sorensen T, et al. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008 Aug 15;68(16):6623–6633. doi: 10.1158/0008-5472.CAN-08-0463 [DOI] [PubMed] [Google Scholar]
- [29].Cao Q, Yang Y, Zhong XZ, et al. The lysosomal Ca(2+) release channel TRPML1 regulates lysosome size by activating calmodulin. J Biol Chem. 2017 May 19;292(20):8424–8435. doi: 10.1074/jbc.M116.772160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cao Q, Zhong XZ, Zou Y, et al. Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J Cell Bio. 2015 Jun 22;209(6):879–894. doi: 10.1083/jcb.201409071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Saffi GT, Botelho RJ. Lysosome fission: planning for an exit. Trends Cell Biol. 2019. Aug;29(8):635–646. doi: 10.1016/j.tcb.2019.05.003 [DOI] [PubMed] [Google Scholar]
- [32].Aits S, Kricker J, Liu B, et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy. 2015;11(8):1408–1424. doi: 10.1080/15548627.2015.1063871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Papadopoulos C, Meyer H. Detection and clearance of damaged lysosomes by the endo-lysosomal damage response and lysophagy. Curr Biol. 2017 Dec 18;27(24):R1330–R1341. doi: 10.1016/j.cub.2017.11.012 [DOI] [PubMed] [Google Scholar]
- [34].Galluzzi L, Bravo-San Pedro JM, Levine B, et al. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017. Jul;16(7):487–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang Y, Qiu Q, Shen JJ, et al. Cardiac glycosides induce autophagy in human non-small cell lung cancer cells through regulation of dual signaling pathways. Int J Biochem Cell Biol. 2012. Nov;44(11):1813–1824. [DOI] [PubMed] [Google Scholar]
- [36].Mizushima N, Murphy LO. Autophagy assays for biological discovery and therapeutic development. Trends Biochem Sci. 2020. Dec;45(12):1080–1093. doi: 10.1016/j.tibs.2020.07.006 [DOI] [PubMed] [Google Scholar]
- [37].Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014 Mar 27;157(1):65–75. doi: 10.1016/j.cell.2014.02.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jia J, Abudu YP, Claude-Taupin A, et al. Galectins control mTOR in response to endomembrane damage. Mol Cell. 2018 Apr 5;70(1):120–135 e8. doi: 10.1016/j.molcel.2018.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jia J, Bissa B, Brecht L, et al. AMPK, a regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin-directed ubiquitin signal transduction system. Mol Cell. 2020 Mar 5;77(5):951–969 e9. doi: 10.1016/j.molcel.2019.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chauhan S, Kumar S, Jain A, et al. Trims and galectins globally cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016 Oct 10;39(1):13–27. doi: 10.1016/j.devcel.2016.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jia J, Claude-Taupin A, Gu Y, et al. Galectin-3 coordinates a cellular system for lysosomal repair and removal. Dev Cell. 2020 Jan 6;52(1):69–87 e8. doi: 10.1016/j.devcel.2019.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Radulovic M, Schink KO, Wenzel EM, et al. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. Embo J. 2018 Nov 2;37(21). doi: 10.15252/embj.201899753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Skowyra ML, Schlesinger PH, Naismith TV, et al. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018 Apr 6;360(6384). doi: 10.1126/science.aar5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Maejima I, Takahashi A, Omori H, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. Embo J. 2013 Aug 28;32(17):2336–2347. doi: 10.1038/emboj.2013.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Papadopoulos C, Kirchner P, Bug M, et al. VCP/P97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. Embo J. 2017 Jan 17;36(2):135–150. doi: 10.15252/embj.201695148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yao RQ, Ren C, Xia ZF, et al. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. 2021. Feb;17(2):385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Elia AE, Boardman AP, Wang DC, et al. Quantitative proteomic atlas of ubiquitination and acetylation in the DNA damage response. Mol Cell. 2015 Sep 3;59(5):867–881. doi: 10.1016/j.molcel.2015.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cragg GM, Grothaus PG, Newman DJ. Impact of natural products on developing new anti-cancer agents. Chem Rev. 2009. Jul;109(7):3012–3043. doi: 10.1021/cr900019j [DOI] [PubMed] [Google Scholar]
- [49].Cao Y. Obesity protects cancer from drugs targeting blood vessels. Cell Metab. 2018 Jun 5;27(6):1163–1165. doi: 10.1016/j.cmet.2018.05.014 [DOI] [PubMed] [Google Scholar]
- [50].Chen X, Wang Y, Ma N, et al. Target identification of natural medicine with chemical proteomics approach: probe synthesis, target fishing and protein identification. Signal Transduct Target Ther. 2020 May 21;5(1):72. doi: 10.1038/s41392-020-0186-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Efferth T, Saeed MEM, Kadioglu O, et al. Collateral sensitivity of natural products in drug-resistant cancer cells. Biotechnol Adv. 2020. Jan - Feb;38:107342. [DOI] [PubMed] [Google Scholar]
- [52].Sun Y, Huang YH, Huang FY, et al. 3’-epi-12beta-hydroxyfroside, a new cardenolide, induces cytoprotective autophagy via blocking the Hsp90/Akt/mTOR axis in lung cancer cells. Theranostics. 2018;8(7):2044–2060. doi: 10.7150/thno.23304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Boya P, Gonzalez-Polo RA, Poncet D, et al. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene. 2003 Jun 19;22(25):3927–3936. doi: 10.1038/sj.onc.1206622 [DOI] [PubMed] [Google Scholar]
- [54].Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005. Nov;5(11):886–897. doi: 10.1038/nrc1738 [DOI] [PubMed] [Google Scholar]
- [55].Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 2020. Feb;21(2):101–118. doi: 10.1038/s41580-019-0185-4 [DOI] [PubMed] [Google Scholar]
- [56].Song Y, Luo L, Wang K. Off-target identification by chemical proteomics for the understanding of drug side effects. Expert Rev Proteomics. 2020. Oct;17(10):695–697. doi: 10.1080/14789450.2020.1873134 [DOI] [PubMed] [Google Scholar]
- [57].Yoshida Y, Yasuda S, Fujita T, et al. Ubiquitination of exposed glycoproteins by SCF(FBXO27) directs damaged lysosomes for autophagy. Proc Natl Acad Sci U S A. 2017 Aug 8;114(32):8574–8579. doi: 10.1073/pnas.1702615114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kim SJ, Chun KH. Non-classical role of Galectin-3 in cancer progression: translocation to nucleus by carbohydrate-recognition independent manner. BMB Rep. 2020. Apr;53(4):173–180. doi: 10.5483/BMBRep.2020.53.4.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kim SJ, Choi IJ, Cheong TC, et al. Galectin-3 increases gastric cancer cell motility by up-regulating fascin-1 expression. Gastroenterology. 2010. Mar;138(3):1035-45 e1–2. [DOI] [PubMed] [Google Scholar]
- [60].Song S, Byrd JC, Mazurek N, et al. Galectin-3 modulates MUC2 mucin expression in human colon cancer cells at the level of transcription via AP-1 activation. Gastroenterology. 2005. Nov;129(5):1581–1591. [DOI] [PubMed] [Google Scholar]
- [61].Castronovo V, Van Den Brule FA, Jackers P, et al. Decreased expression of galectin-3 is associated with progression of human breast cancer. J Pathol. 1996. May;179(1):43–48. [DOI] [PubMed] [Google Scholar]
- [62].Pacis RA, Pilat MJ, Pienta KJ, et al. Decreased galectin-3 expression in prostate cancer. Prostate. 2000 Jul 1;44(2):118–123. doi: [DOI] [PubMed] [Google Scholar]
- [63].He X, Yin X, Wu J, et al. Visualization of human T lymphocyte-mediated eradication of cancer cells in vivo. Proc Natl Acad Sci U S A. 2020 Sep 15;117(37):22910–22919. doi: 10.1073/pnas.2009092117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hosaka K, Yang Y, Seki T, et al. Therapeutic paradigm of dual targeting VEGF and PDGF for effectively treating FGF-2 off-target tumors. Nat Commun. 2020 Jul 24;11(1):3704. doi: 10.1038/s41467-020-17525-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wang K, Luo L, Fu S, et al. PHGDH arginine methylation by PRMT1 promotes serine synthesis and represents a therapeutic vulnerability in hepatocellular carcinoma. Nat Commun. 2023 Feb 23;14(1):1011. doi: 10.1038/s41467-023-36708-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sun X, He X, Zhang Y, et al. Inflammatory cell-derived CXCL3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism. Gut. 2022. Jan;71(1):129–147. [DOI] [PubMed] [Google Scholar]
- [67].Andersson P, Yang Y, Hosaka K, et al. Molecular mechanisms of IL-33-mediated stromal interactions in cancer metastasis. JCI Insight. 2018 Oct 18;3(20). doi: 10.1172/jci.insight.122375 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.