

Abstract
Wallerian degeneration (WD) is a conserved axonal self-destruction program implicated in several neurological diseases. WD is driven by the degradation of the NAD+ synthesizing enzyme NMNAT2, the buildup of its substrate NMN, and the activation of the NAD+ degrading SARM1, eventually leading to axonal fragmentation. The regulation and amenability of these events to therapeutic interventions remain unclear. Here we explored pharmacological strategies that modulate NMN and NAD+ metabolism, namely the inhibition of the NMN-synthesizing enzyme NAMPT, activation of the nicotinic acid riboside (NaR) salvage pathway and inhibition of the NMNAT2-degrading DLK MAPK pathway in an axotomy model in vitro. Results show that NAMPT and DLK inhibition cause a significant but time-dependent delay of WD. These time-dependent effects are related to NMNAT2 degradation and changes in NMN and NAD+ levels. Supplementation of NAMPT inhibition with NaR has an enhanced effect that does not depend on timing of intervention and leads to robust protection up to 4 days. Additional DLK inhibition extends this even further to 6 days. Metabolite analyses reveal complex effects indicating that NAMPT and MAPK inhibition act by reducing NMN levels, ameliorating NAD+ loss and suppressing SARM1 activity. Finally, the axonal NAD+/NMN ratio is highly predictive of cADPR levels, extending previous cell-free evidence on the allosteric regulation of SARM1. Our findings establish a window of axon protection extending several hours following injury. Moreover, we show prolonged protection by mixed treatments combining MAPK and NAMPT inhibition that proceed via complex effects on NAD+ metabolism and inhibition of SARM1.
Keywords: NAD+, Neurodegeneration, Niacin, SARM1, NMNAT2, Neuropathy
1. Introduction
Wallerian degeneration (WD) is a highly conserved program of axonal fragmentation classically encountered in experimental axotomy in the peripheral nervous system (PNS) but also present in traumatic, toxic and degenerative neuropathies and certain non-neuropathic conditions, such as impairments of axonal transport (Coleman and Hoke, 2020; Conforti et al., 2014). WD is closely associated with the regulation of axonal metabolism of nicotinamide adenine dinucleotide (NAD+, Fig. 1) and although WD has several decision points that are not yet fully understood, the interruption of supply of the labile nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) and its further rapid depletion by members of the stress mitogen-activated protein kinase (MAPK) pathway or the MYC binding protein 2 (MYCBP2 or PHR1) ubiquitin ligase complex have been proposed as initiating events (Summers et al., 2018; Loreto et al., 2020). In turn, depletion of NMNAT2 leads to the arrest of the axonal synthesis of NAD+, a promiscuous redox cofactor essential for axonal maintenance, as well as accumulation of the NAD+ precursor nicotinamide mononucleotide (NMN) in the first hours after injury (Summers et al., 2018; Di Stefano et al., 2017; Di Stefano et al., 2015; Gilley and Coleman, 2010; Sasaki et al., 2016; Essuman et al., 2018; Essuman et al., 2017). More recently, the focus has shifted to sterile alpha and toll/interleukin receptor motif-containing protein (SARM1) (Osterloh et al., 2012) that is thought to execute WD via its role as a NADase that leads to further depletion of NAD+ stores (Essuman et al., 2017; Gerdts et al., 2015) or possibly via poorly understood downstream pro-degenerative signals (Li et al., 2022). SARM1 activation is a necessary and sufficient instructive signal for WD in vitro as well as in models of axotomy and peripheral neuropathy (Osterloh et al., 2012; Geisler et al., 2016; Turkiew et al., 2017). Deletion of Sarm1 is also protective of CNS axons in models of traumatic axonal injury (TAI) (Henninger et al., 2016; Marion et al., 2019; Ziogas and Koliatsos, 2018). Dual leucine kinase (DLK, or MAP3K12) and leucine zipper kinase (LZK, or MAP3K13), members of the stress MAPK cascade that are triggered with axonal injury (Fernandes et al., 2012; Miller et al., 2009; Watkins et al., 2013; Welsbie et al., 2017; Welsbie et al., 2013; Welsbie et al., 2019; Yang et al., 2015) may reinforce SARM1 signaling, for example via DLK/LZK-mediated degradation of palmitoylated NMNAT2 (Summers et al., 2018) or via c-Jun N-terminal kinase 1 (JNK1)-mediated phosphorylation of SARM1 (Murata et al., 2018).
Fig. 1.
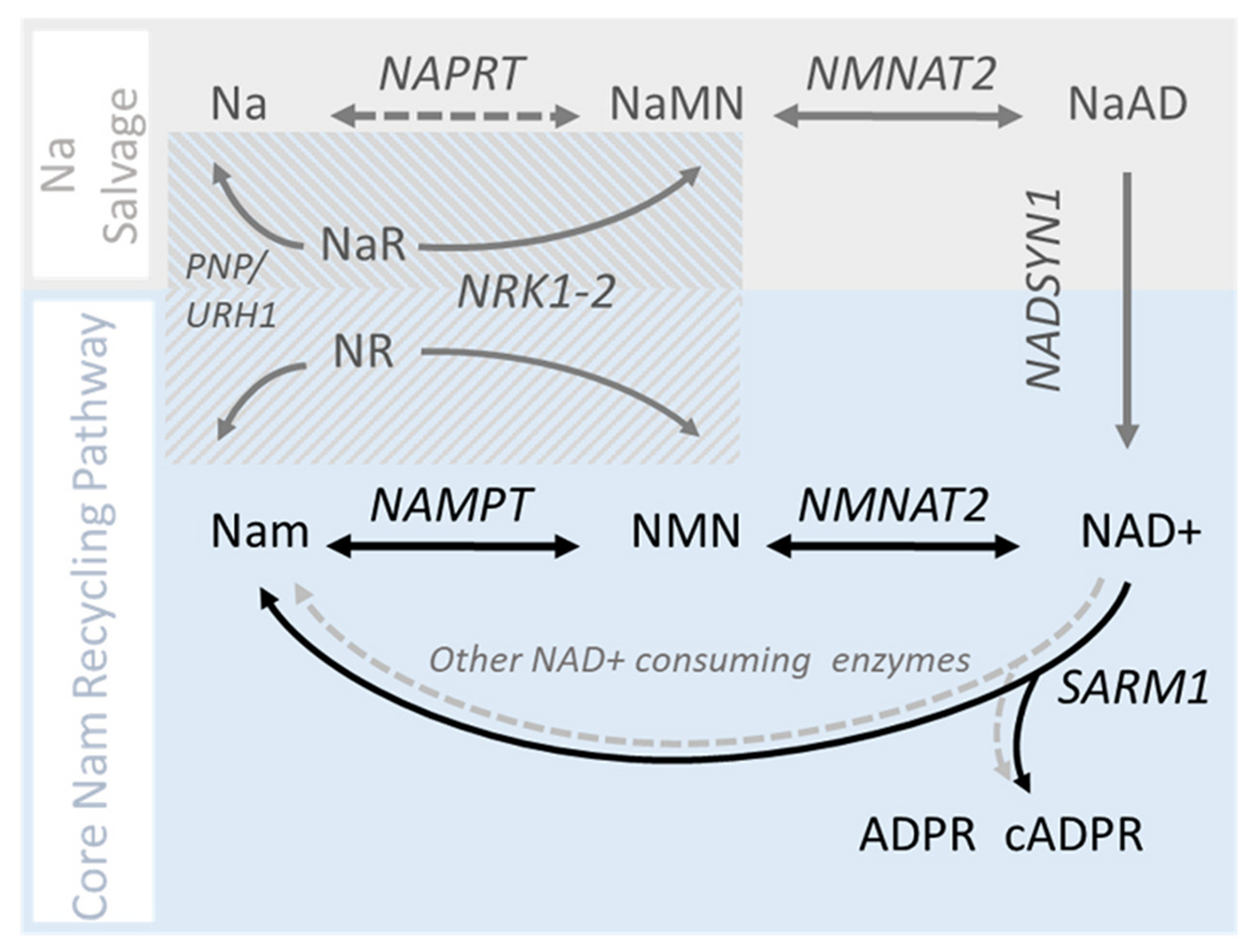
Metabolic pathways of NAD+ synthesis in mammalian axons.
Axonal NAD+ synthesis occurs predominantly via the NAD+ salvage pathway (blue box). NAD+ degradation by SARM1 or other enzymes (not shown) regenerates Nam which is then recycled back to NAD+ by the successive action of NAMPT and NMNAT2. The contribution of the nicotinic acid (Na) salvage pathway (also known as Preiss-Handler pathway; grey box) under basal conditions is thought to be minimal (Liu et al. 2018), whereas the contribution of the accessory nicotinamide riboside kinase pathway (striped box) is not well characterized. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Although the mechanisms of SARM1 activation and SARM1-dependent axon degeneration have not been fully characterized, there is evidence for a role of NMN and NAD+ as positive and negative modulators of SARM1 activity, respectively (Bratkowski et al., 2020; Figley et al., 2021; Jiang et al., 2020; Sporny et al., 2020; Zhao et al., 2019; Hopkins et al., 2021). Based on this work and also the upstream role of MAPK-dependent NMNAT2 degradation as a key driver of WD, here we use inhibitors of Nicotinamide phosphoribosyltransferase (NAMPT) or MAPKs as means to modulate NMN biosynthesis and NMNAT2 levels on injured mouse dorsal root ganglion (DRG) axons. We define the effects of these inhibitors on axonal degeneration, characterize the temporal dynamics and metabolic signatures of these effects, and explore mechanisms of action.
2. Materials and methods
2.1. Primary cell cultures
Dorsal root ganglion cells (DRGs) were collected from E14 mouse embryos (CD1 or SARM1 KO), enzymatically dissociated in dissection buffer (HBSS, 10 mM sodium pyruvate, 20 mM HEPES, 0.25 mM glucose, 1× penicillin/streptomycin) containing 0.1% trypsin for 20 min at 37 °C, and then purified on tissue culture plate for 2–3 h in DRG media at 37 °C. Designated number of cells were plated in DRG media containing Neurobasal (21103–049, Gibco), 1× Glutamax (35050–061, Gibco), 1× penicillin/streptomycin (15140–122, Gibco), 1 μM uridine (U3003, Sigma), 1 μM 5-fluoro-2′-deoxyuridine (F0503, Sigma), 2% B27 (17504–044, Invitrogen) and 50 ng/mL nerve growth factor (NGF, BT5025, Envigo Bioproducts Inc.) on PDL (200 μg/mL)/Laminin (10 μg/mL)-coated plates or microfluidic chambers.
2.2. Degeneration Index experiments
Dorsal root ganglion neurons were plated in the cell compartment of polydimethylsiloxane (PDMS) - based microfluidic champers at a density of 78 K per cm2) that allow separation of axons from cell bodies (Fig. 2) or in a drop culture format in 24-well plates at 25 K per well. Maintenance media was replaced by 50% every 3–4 days.
Fig. 2.
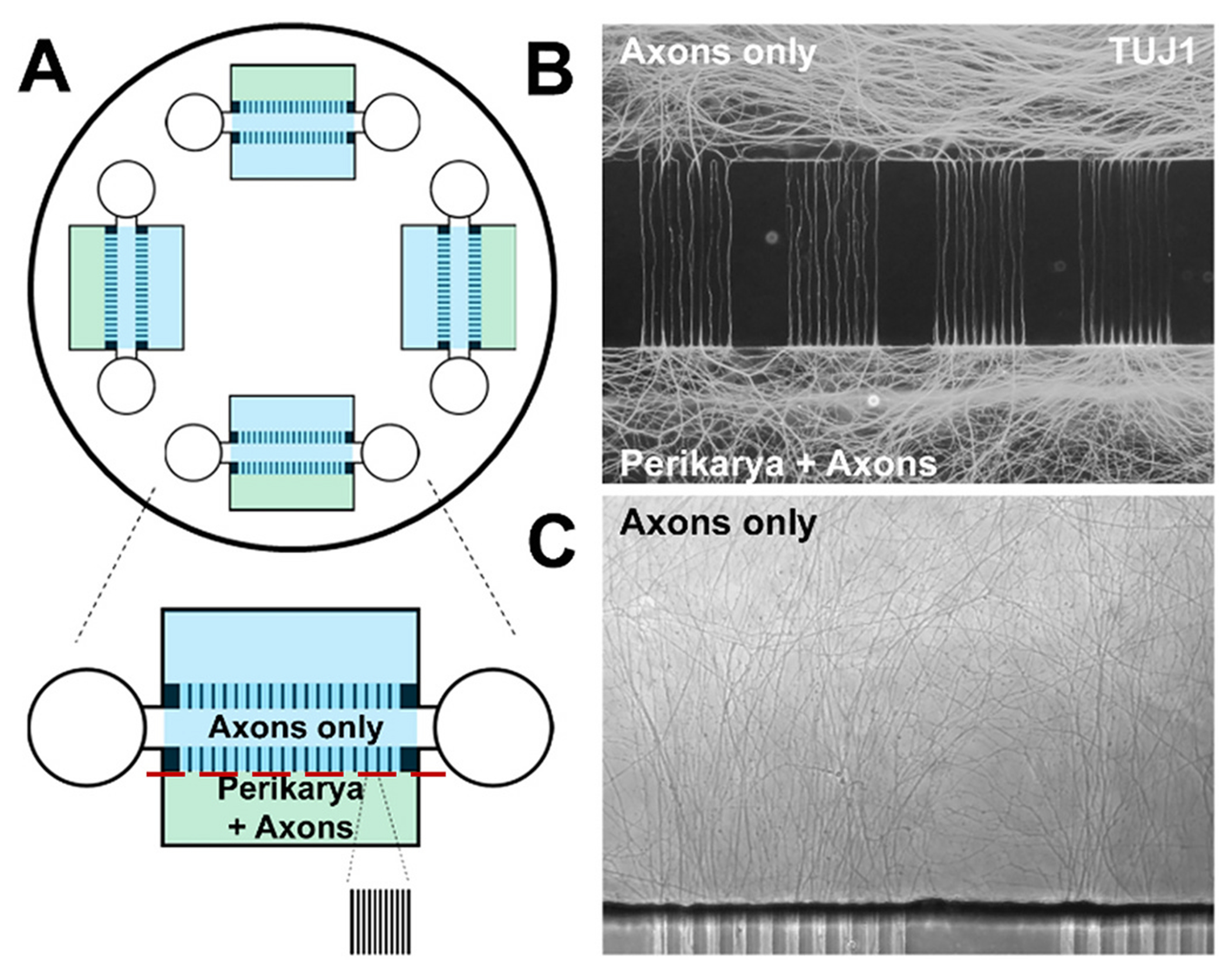
Primary dorsal root ganglion neuron culture in microfluidic devices.
A. Diagram of PDMS-based microfluidic device used in DI experiments. Red line indicates plane of axotomy. B. DRG axons populating the axon compartment (top) and axons/processes in the perikaryal compartment (bottom) visualized with TUJ1 immunohistochemistry. C. Axons visualized with phase contrast microscopy. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Experiments for determination of degeneration index (DI) were performed on the 14th day in vitro (DIV). In our preliminary experiments, we noted that axotomy at earlier DIVs (<10), i.e. when axons are still growing rapidly, would cause rapid fragmentation (data not shown). We selected a more advanced stage of culturing, i.e. DIV 12–14, for axotomy experiments because it offered optimal density for image analysis and a slower fragmentation time curve that allows for better interrogation of time dependent events and which is more proximal to in vivo observations. Pharmacological treatments were performed before or after axotomy with the following compounds and concentrations unless stated otherwise: FK866 (100 nM, 13,287), GNE3511 (500 nM, 13,287), CHS828 (250 nM, 11,021), STF31 (2 μM, 11,173) GPP (1 μM, 13,670) and STF118804 (250 nM, 15,283); all from Cayman Chemical Company (Ann Arbor, MI). Stock compounds were dissolved in DMSO, aliquoted and stored at −20 °C. Nicotinic acid ribose (NaR, 500 μM, AMBH93D590D5) and NMN (500 μM, N3501) were from Sigma, and stock solutions were dissolved in H20 and stored at −20 °C in aliquots for single use.
After axotomy with a razor blade, 5–7 bright field images were taken manually at 20× using Nikon Eclipse TS100 to monitor degeneration/fragmentation of axons. Out of focus images or with significant artifacts that would hinder analysis were excluded before image processing. Images were analyzed with ImageJ to calculate degeneration index (DI) blind to treatment assignment. Script is available upon request. DI quantifications of multiple fields per biological replicate were mean averaged. For each experiment 2–3 biological replicates were used for statistical analysis. Due to some variance in absolute DI outcomes across separate experiments, statistical comparisons are made only within each experimental scenario.
2.3. Metabolite extraction and analysis
Dorsal root ganglion neurons were plated as a drop culture (250 K/20 μL /well) in 6-well plates. At 19 DIV, axotomy and pharmacological treatments were performed as indicated with 3–6 biological replicates per tested condition. A longer culture compared to DI experiments was required to achieve adequate yield. Before metabolic extraction, the plate was cooled on ice, cell bodies including proximal axons were removed, and the remaining axons were washed with 3 mL ice-cold PBS three times. Metabolites were extracted by incubating axons with 1 mL of ice-cold methanol:water (1:0.9), pH 7.0 on ice for 10 min. Axons were detached and the solution with the axons was transferred to 2 mL Eppendorf tubes. Samples were spun down at 1000 g for 5 min. at 4 °C and then the supernatant were transferred to a new 2 mL tube, flash frozen in liquid nitrogen and stored at −80 °C. Axon pellets were extracted in RIPA buffer with protease inhibitors and used for protein determination by BCA (Bio-Rad, Hercules, CA) for the purpose of normalization.
Samples were then divided into 400 μL aliquots and dried down by speed vac (Savant, Thermo Scientific). Before analysis, samples were reconstituted in 100 μL water (optima grade, Fisher Scientific), centrifuged (10,000 g) for 15 min at 4 °C and the supernatants were subjected to high pressure liquid chromatography (HPLC, Shimadzu, Kyoto, Japan). HPLC was comprised of a SIL-30ACMP 6-MTP Autosampler, Nexera LC-30 CE HPLC Pumps and a UPLC C18 column from Agilent (859700–902: Zorbax RRHD SB-C18, 2.1x150mm, 1.8 μm particle size, 80 A pore size). Mobile phase A was water with 0.1% formic acid and mobile phase B was acetonitrile with 0.1% formic acid. Metabolites were eluted with the following gradients: 0% mobile phase B from 0 to 2 min, 40% mobile phase B from 2 to 7 min, 100% mobile phase B from 7 to 8 min, 0% mobile phase.
Targeted assays used a Shimadzu 8060 triple quadrupole (QQQ) mass spectrometer equipped with an electrospray ionization source used in both the positive and negative mode. Resolution was set to 1 Da. Source conditions were set with nebulizing gas flow at 3 L/min, heating gas flow set to 10 L/min, interface temperature set to 300 °C, DL temperature set to 250 °C, heat block temperature set to 400 °C, and drying gas flow set to 10 L/min. Serial dilutions of standards for each metabolite in water (optima grade, Fisher Scientific) were used for calibration. Metabolites were quantified by Labsolutions Insight quantitative analysis tool (Shimadzu, Kyoto, Japan) with standard curves prepared for each metabolite in dilution from 25 to 20,000 fmol on column (R2 > 0.99), and then normalized by total protein estimates. Quality control pools were created from the samples and run before, after, and every ten samples within each batch to control for system performance and instrument drift. A limited number of internal standards (Nicotinic acid-13C6, Nicotinamide-13C3,15 N, L-Tryptophan-13C11,15 N2) were spiked into each sample for sample quality control, which was used as an exclusion criteria for any sample that varied in internal standard beyond 30% of the mean. Limit of quantitation was also calculated as 10× standard deviation of the x-intercept divided by slope. Although concentrations of metabolites were estimated and normalized by total protein per sample, the lack of heavy standards precluded absolute quantitation of each metabolite, therefore metabolite levels and substrate/product ratios for each experiment are expressed in relative terms to designated control, while statistical comparisons are made only within each experimental scenario.
2.4. Western blotting
Dorsal root ganglia were plated as a drop culture (250 K /20 μL) in 6-well plates and experimental treatments were performed as described above for metabolomic experiments. After removal of cell bodies and proximal axons, and washing with ice cold PBS, transected axons were directly lysed with 100 μL of 1× sample buffer (161–0737, Bio-Rad) containing phosphatase inhibitor (04906845001, Roche), protease inhibitor/EDTA (78,430, Thermofisher) and beta-mercaptoethanol. Lysates were transferred to Eppendorf tubes and vortexed for 1 min. Protein determination was performed by the RC-DC method (Bio-Rad).
Samples were denatured at 70 °C for 10 min before gel electrophoresis. Samples and visible/near infrared molecular weight standards (Chameleon 800 nm, Li-Cor, Germany) were electrophoresed on NuPAGE 4–12% Bis-Tris gels (Invitrogen, USA) with SDS NuPAGE MOPS Running Buffer. Proteins were transferred to a PVDF membrane using an iBlot transfer device (Invitrogen, USA). Membranes were air dried, rehydrated and then blocked with 50% Odyssey blocking solution-tris buffered saline (Li-Cor, Germany) and incubated overnight at 4 °C with the target antibodies: mouse alpha-spectrin (Millipore Cat# MAB1622, RRID:AB_11214057) mouse actin (Santa Cruz Biotechnology Cat# sc-47,778 HRP, RRID:AB_2714189), rabbit SCG10 (Proteintech Cat# 10586–1-AP, RRID:AB_2197283), rabbit pThr183/Tyr185-JNK (Cell Signaling Technology Cat# 9251, RRID:AB_33165). Membranes were washed with TBS with 0.1% tween-20 and incubated with secondary antibodies (1:15000; IRDye 800CW or 680RD; Li-Cor, Germany). Signals were read at 700 or 800 nm on an Odyssey CLx infrared scanner (Li-Cor, Germany) and analyzed with Image Studio (vs 5.2, Li-Cor, Germany). Intensities for each target protein were normalized to actin for semi-quantitative assessment of abundancies.
In our preliminary experiments we were not able to detect NMNAT2 in axonal extracts by regular western blot due to its very low expression and antibody specificity. For this reason we used the NaveniFlex MR 100 proximity ligation assay (Navinci Diagnostics, Uppsala, Sweden) to amplify the signal as follows: After transfer, PVDF membranes were blocked in 50% Intercept Blocking Buffer containing 100 μg/mL salmon sperm DNA. Then membranes were incubated in primary antibodies at 4 °C overnight. NMNAT2 was detected by a pair of mouse monoclonal NMNAT2 antibody (1:30, Sigma-Aldrich Cat# WH0023057M1, RRID:AB_1842701) and rabbit polyclonal NMNAT2 antibody (1:100, Santa Cruz Biotechnology Cat# sc-134,935, RRID:AB_10610925) in primary antibody diluent. The following steps were done according to manufacturer’s manual at 37 °C: Probing for mouse and rabbit antibodies, oligonucleotides hybridization, T4 DNA ligation and polymerization and binding to IR700 conjugated detection probe, and all washes in between was done with TBS containing 0.05% Tween 20 at room temperature.
2.5. Human embryonic stem cell-derived neuron culture and DI experiments
Human embryonic stem cells, H9 (WiCell, WI, RRID:CVCL_U176) maintained under feeder-free conditions (StemFlex media, Life technologies) were rapidly differentiated to a cholinergic phenotype with Sendai virus mediated delivery of synthetic neurogenic transcription factor (CH-SeV, Elixirgen, MD), as described (Goparaju et al., 2017) plated in microfluidic devices as above and maintained in cholinergic neuron maintenance media (CH-MM, Elixirgen, MD). On day 5 of differentiation, neurons were transduced with a lentivirus for the expression of hsyn-tdTomato (SL100290, Signagen Laboratories,) and visualization of neurites. Experimental procedures were identical to the ones involving mouse neurons. Images were acquired with a Zeiss Axio Observer Z1 epifluorescence microscope and degeneration index was calculated based on the tdTomato signal.
2.6. Statistical analysis
Sample number was defined as the number of biological samples (e.g. culture wells) that were independently manipulated and measured. Experimental sample numbers were determined based on sample sizes used in the field and our preliminary investigations. Data distribution was assumed to be normal. Data were plotted and statistical analyses were performed with GraphPad Prism (vs 9.1.1, San Diego, CA) as indicated. Statistical significance was determined at p = 0.05. In case of multiple comparisons, adjusted p-values are presented unless otherwise stated.
3. Results
3.1. NAMPT inhibition delays WD in a time-dependent manner
Based on evidence that the rise of NMN levels secondary to NMNAT2 loss may drive, at least in part, WD (Loreto et al., 2020; Di Stefano et al., 2017; Di Stefano et al., 2015), we revisited the issue of NAMPT inhibition as a strategy to lower NMN levels and thus protect axons from degeneration. NAMPT inhibition is a translationally relevant strategy given the existence of clinically available and CNS penetrant inhibitors such as Daporinad (FK866). However, although NAMPT inhibition was shown to be protective in in vitro systems, the reported efficacy was variable, likely depending on time of application (Di Stefano et al., 2017; Di Stefano et al., 2015), and its effect on SARM1 activation was not characterized. To explore the protective role of NAMPT inhibition, we used primary DRG cultures in a microfluidic compartmentalized system (Fig. 2) and quantitated axon fragmentation of transected axons based on DI. Our findings confirm that the effect of NAMPT inhibition with FK866 is time-dependent and is optimal when applied in the first 2 h post injury (See Supplementary Fig. 1). Of note, although axon integrity was protected up to 24 h, the effect of NAMPT inhibition was not as prolonged as that afforded by SARM1 deletion (Fig. 3A-B).
Fig. 3.
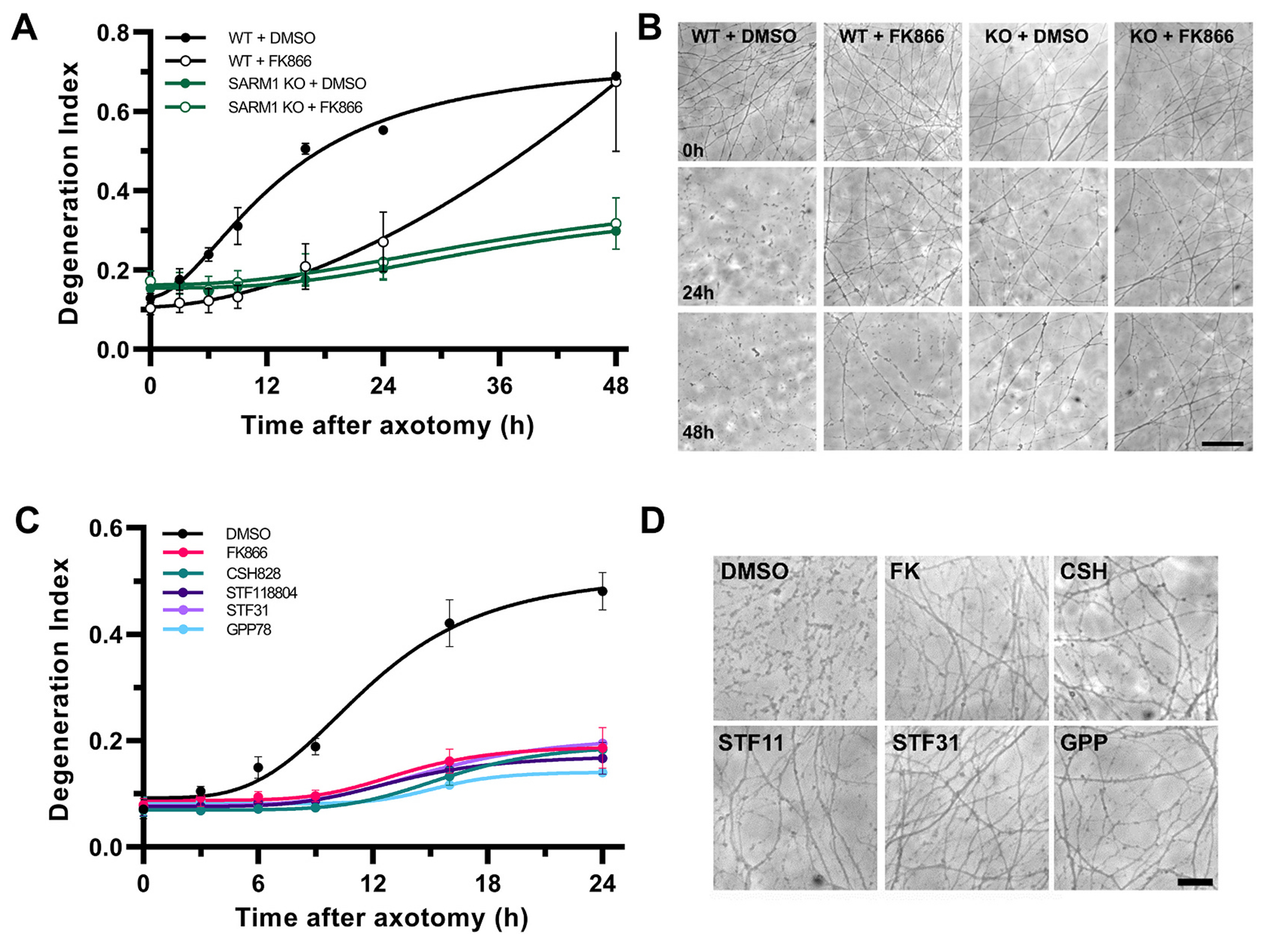
NAMPT inhibition delays axon fragmentation.
A. Degeneration Index analysis comparing NAMPT inhibition at 2 h post axotomy with SARM1 deletion. Two-way ANOVA for the effect of time (F6,36 = 94.40, p < 0.0001), treatment/genotype (F3,6 = 4.14, p = 0.067) and their interaction (F18,36 = 12.85, p < 0.0001), with Holm-ţídák’s multiple comparisons shows that the NAMPT inhibitor FK866 significantly delays axon fragmentation for up to 24 h compared to vehicle treated WT axons (for WT + DMSO vs WT + FK866 t42 = 2.68, p = 0.04; for WT + FK866 vs SARM1 KO + DMSO t42 = 2.28, p = 0.09) whereas SARM1 deletion with or without NAMPT inhibition protects against fragmentation for at least 48 h compared to wt axons (t42 = 5.33–5.83, p < 0.0001). B. Representative phase contrast micrographs from A. Scale bar, 50 μm C. Structurally different NAMPT inhibitors (FK866, CSH828, GPP78, STF31, STF118804) similarly protect against axon fragmentation after axotomy shown by DI analysis. Two-way ANOVA for the effect of time (F5,35 = 68.08, p < 0.0001), treatment (F5,7 = 30.66, p = 0.0001) and their interaction (F25,35 = 11.19, p < 0.0001), with Holm-ţídák’s multiple comparisons, reveals robust protection against axon fragmentation post axotomy up to 24 h for all treated groups versus vehicle treated axons, t42 = 10.64–12.67, p < 0.0001. D. Representative phase contrast micrographs taken at 24 h post axotomy for C. Scale bar, 25 μm. Error bars indicate ±1 SEM.
To further validate that the observed protective effect of FK866 is specifically related to the inhibition of NAMPT, we tested a number of structurally distinct NAMPT inhibitors (FK866, CSH828, STF118804, STF31 and GPP78) given 2 h post-lesion. Robust protection was observed for up to 24 h for all compounds (Fig. 3C-D) and at all dilutions tested (nM - μM range; Supplementary Fig. 2).
We also hypothesized that pre-treatment/early treatment with NAMPT inhibitors might have a parallel “toxic” effect because of a further reduction in NAD+ biosynthesis in the presence of residual axonal NMNAT2 (Sasaki et al., 2016). We therefore explored whether the protective effect of NAMPT inhibition on axonal fragmentation is mediated via reduction in levels of the NAMPT product NMN in a time-dependent manner. To address this question, we added FK866 to DRGs at the time of injury to suppress endogenous NMN synthesis and introduced NMN (500 μM) at the time of injury and 2 or 4 h thereafter. We found that NMN addition at 4 h leads to rapid degeneration of axons, whereas earlier introduction has a less robust effect (Fig. 4).
Fig. 4.
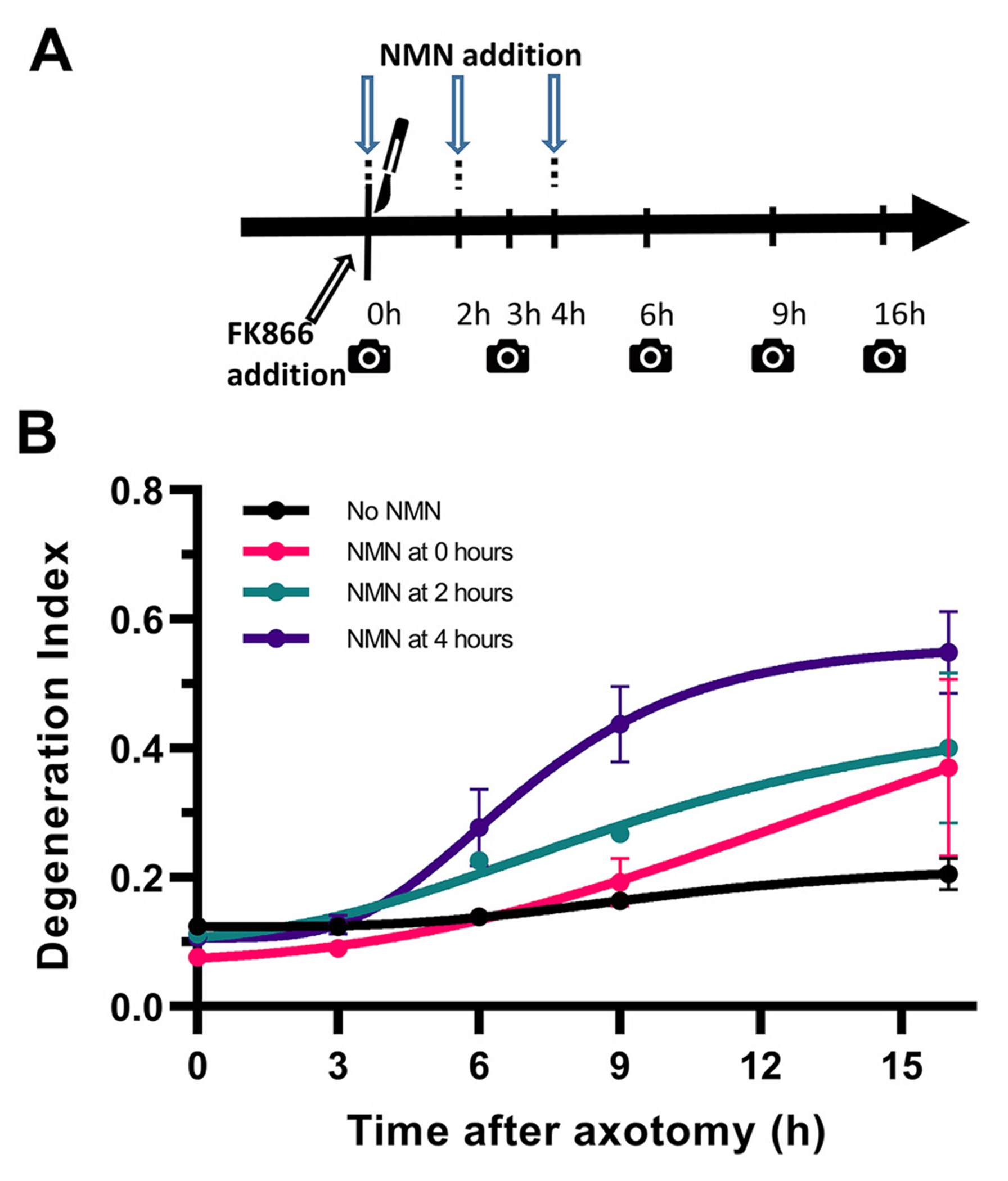
Time-dependent effects of NMN in initiating axon fragmentation.
A. The NAMPT inhibitor FK866 was added at the time of injury to suppress endogenous NMN synthesis and NMN was added at the time of injury, or 2 and 4 h later (arrows). B. Degeneration index analysis indicates that introduction of NMN initiates axon fragmentation in a time-dependent manner, with introduction at 4 h leading to a more rapid axon fragmentation, whereas earlier interventions have a slower effect. Holm-ţídák’s multiple comparisons after two-way ANOVA for the effect of time (F4,16 = 28.01,p < 0.0001), treatment (F3,4 = 5.30, p = 0.071) and their interaction (F12,16 = 2.50, p = 0.045) shows that NMN addition at 4 h leads to higher fragmentation index compared to FK866 control at both 9 (t20 = 4.03, p = 0.0039) and 16 h post-axotomy (t20 = 5.05, p = 0.0004), but NMN addition at 2 h leads to significant fragmentation only at 16 h (t20 = 0.047). Error bars indicate ±1 SEM.
Taken together, these findings indicate that the role of NMN (and, respectively, NAMPT inhibition) in injured axons varies based on timing, with an evident vulnerability of the axon to the effect of NMN at 2–4 h post injury.
3.2. NAMPT inhibition causes complex, time- dependent effects on NAD+ and SARM1 metabolomics
To better understand the mechanism of NAMPTi-mediated protection, we analyzed key metabolites in the NAD+ salvage and SARM1 pathways in transected DRG axons (Fig. 5) from drop cultures which prevent contamination from proximal axons or cell bodies. First, we confirmed previous observations (Di Stefano et al., 2017; Di Stefano et al., 2015) that transection of untreated axons leads to a progressive increase in axonal NMN; these levels triple by 8 h following injury, before any visible fragmentation of the axon. Conversely, NAD+ levels decrease by 40% at 2 h, with a further decline to 80% by 6 h post injury. Cyclic ADPR (cADPR) levels, recently proposed as a marker of SARM1 activity (Sasaki et al., 2020), begin to increase 2 h after injury and peak at 8 h, at which point they are 4-fold higher compared to levels in unlesioned axons (Fig. 5A). While the measurement of static metabolite levels can be informative, changes in product/substrate ratios specific to an enzyme are a relative indicator of the conversion of a substrate to product and may serve as a strong, albeit indirect, marker of enzymatic activity. On this foundation, we explored the time course of relative changes in product/substrate ratios for NAMPT, NMNAT2 and SARM1 (see Fig. 1 for corresponding metabolites). We found a progressive decrease in the NAD+/NMN ratio leading to 20-fold drop by 10 h following injury that may represent a substantial reduction of NMNAT2 activity, and a corresponding 20- to 30-fold increase in the cADPR/NAD+ ratio, consistent with increased SARM1 activity.
Fig. 5.
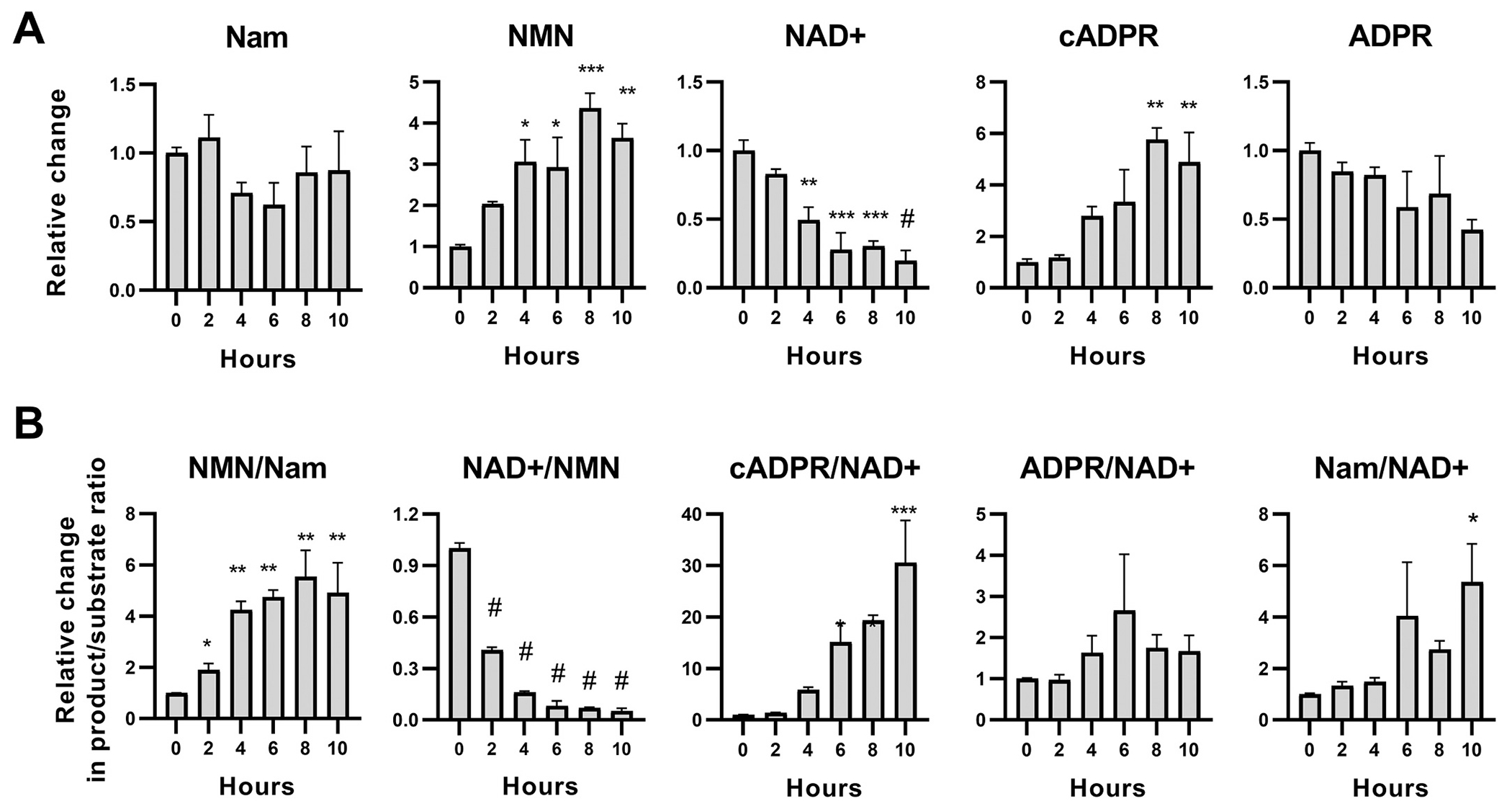
Changes in core Nam salvage pathway metabolites in transected axons.
A. Relative changes in protein-normalized levels of members of the core Nam salvage pathway in distal axons following transection. Injury leads to significant decreases of NAD+ levels, F1,15 = 50.18; p < 0.0001; and significant increases in NMN, F1,15 = 20.56; p = 0.0004) and the SARM1-related NAD+degradation product cADPR (F1,15 = 24.97; p = 0.0002). There is also a trend for decreasing ADPR levels, F1,15 = 7.003; p = 0.018, with no change in Nam levels. B. Analysis of product/substrate ratios (from A) as surrogates for the activity of key enzymes in the core Nam recycling pathway. Axonal injury leads to progressive increases in the NMN synthetic activity (NMN/Nam ratio, F1,15 = 20.28; p = 0.0004), loss of NAD+ synthetic activity (NAD+/NMN ratio; F1,15 = 31.07; p < 0.0001) and significant NAD+ degrading activity (cADPR/NAD+ ratio, F1,15 = 44.95; p < 0.0001 but not for ADPR/NAD+; F1,15 = 1.34; p = 0.27). These are consistent with loss of NMNAT2 function and increased activity of SARM1. Time-course changes are analyzed with Deming linear regression (Fdf) but differences from baseline (0 h) are also indicated with t-tests, as * p < 0.05, ** p < 0.01, *** p < 0.001, # p < 0.0001 (unadjusted). Error bars indicate ±1 SEM.
Based on the axon-protective effect of NAMPT inhibition, the great similarity in the time course of NMN and cADPR changes post injury, and previous suggestive evidence (Di Stefano et al., 2017; Di Stefano et al., 2015; Zhao et al., 2019), we then asked whether NMN may play a role in activating SARM1, and whether lowering NMN via NAMPT inhibition would block SARM1 activation. Cultures were treated with FK866 at a time optimized for best axonal protection (2 h post axotomy) or vehicle (DMSO) and metabolites were extracted from axonal samples at 2 and 6 h post treatment (Fig. 6). SARM1 activation was indirectly assessed via measuring levels of its product cADPR (Essuman et al., 2017; Sasaki et al., 2020) and the cADPR/NAD+ index. In uninjured axons, NAMPT inhibition for 6 h results only in modest, non-significant reductions of NMN, NAD+ and cADPR, and no changes in indices of enzymatic activity (Fig. 6A). In transected axons, the effects of NAMPT inhibition are more complex. At 2 h post-treatment, FK866 is not only ineffective in preventing the injury-induced increase in NMN, but in fact increases NMN by 48% compared to vehicle and also increases NAD+ by 34% with no effect on cADPR levels (Fig. 6B). These unexpected observations are at odds with the predicted suppression of NMN and NAD+ levels seen in perikarya of the same axons (Supplementary Fig. 3).
Fig. 6.
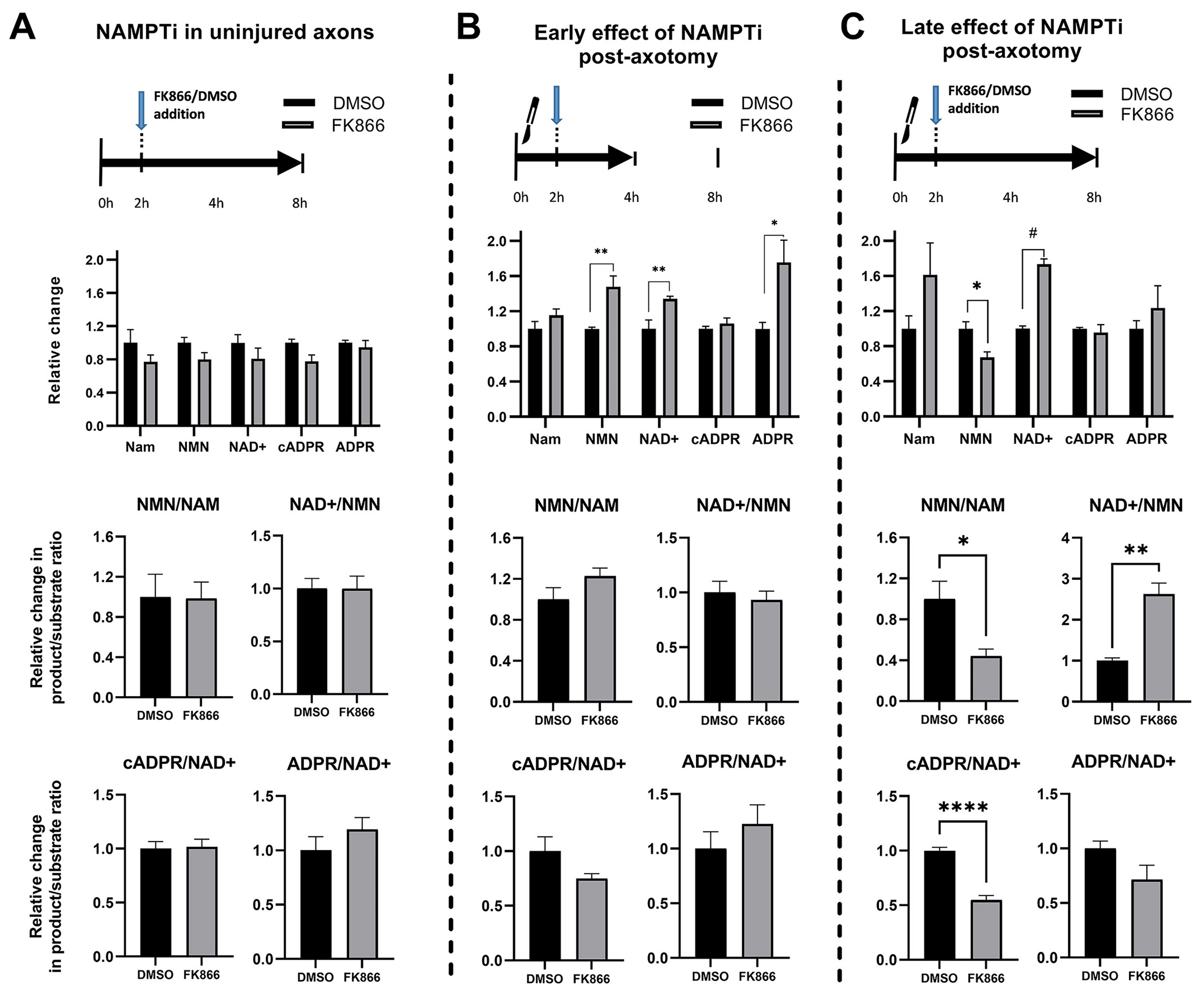
Effects of NAMPT inhibition on core Nam salvage metabolites in naïve and injured axons.
Relative changes in protein-normalized metabolite concentrations (top) and in enzymatic product/substrate ratios (middle and bottom) shown after delayed treatment (2 h post axotomy) with the NAMPT inhibitor FK866 or vehicle (DMSO). A. In uninjured axons (6 h post-treatment), NAMPT inhibition does not alter significantly enzymatic activity or levels of metabolites. B. NAMPT inhibition in transected axons leads to significant increases in both NMN (t10 = 3.80; p = 0.003) and NAD+ levels (t10 = 3.28; p = 0.008) early after treatment (2 h post-treatment); and no significant changes in enzymatic activities. C. Prolonged treatment of transected axons (6 h post-treatment) is associated with reduced NMN (t7 = 3.31; p = 0.013) but increased NAD+ levels (t7 = 10.0; p = 0.00002) compared to vehicle-treated axons. There is also reduced NMN synthetic activity (NMN/Nam ratio, t7 = 3.29, p = 0.013), increased NAD+ synthetic activity (NAD+/NMN ratio, t7 = 5.24, p = 0.001) and reduced NAD+ degrading activity (cADPR/NAD+ ratio, t7 = 8.41, p < 0.0001). Differences with vehicle (DMSO)-treated axons were analyzed with Student’s t-test. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001. Error bars indicate ±1 SEM.
With continued (6-h) treatment, however, NAMPT inhibition decreases NMN by 33% and also increases NAD+ by 70%, effectively increasing the NAD+/NMN ratio 2.6 times compared to vehicle. Although NAMPT inhibition does not change absolute cADPR levels on either time point, it does cause a significant 50% decrease in cADPR/NAD+ at 6 h, an effect consistent with reduced SARM1 activity (Fig. 6C).
These data suggest that NAMPT inhibition in transected axons has complex, time-dependent effects on NMN and NAD+ levels. Instead of a generic suppression of NAD+ as might have been expected, NAMPT inhibition profoundly modulates the NAD+ and SARM1 metabolome by protecting NAD+ levels and suppressing SARM1 activity.
3.3. The engagement of the NMN-independent NaR salvage pathway maximizes the protective effect of NAMPT inhibitors
Although neuronal NAD+ biosynthesis involves mainly the core nicotinamide (Nam) recycling pathway that proceeds through NMN, the nucleoside nicotinic acid riboside (NaR) can also be utilized by neurons for NAD+ synthesis in the nicotinic acid (Na) salvage (Preiss-Handler) pathway that does not require NMN as intermediary (Fig. 1) (Liu et al., 2018; Yang and Sauve, 2016). For example, it was previously shown that boosting this pathway increases NAD+ synthesis in the absence of the precursor NMN and protects axons from vincristine- mediated neurotoxicity (Liu et al., 2018). We therefore asked whether NAMPT inhibition supplemented with NaR may also offer superior protection in our axotomy model.
As illustrated in Fig. 7, combining FK866 with NaR leads to a robust protection against axonal fragmentation regardless of the time of NAMPT inhibition, while NaR offers no protection by itself (Fig. 7A). Given its predicted effect in increasing NAD+ synthesis, we anticipated that the efficacy of NaR supplementation would decrease in a time-dependent fashion commensurate with the progressive loss of the NAD-synthesizing enzyme NMNAT2 (Summers et al., 2018; Loreto et al., 2020). We therefore treated transected axons with the NAMPT inhibitor FK866, and then added NaR at different time points up to 24 h post injury. We found that NaR supplementation is superior to NAMPT inhibition alone, and that the addition of NaR can be delayed up to 9 h with no loss of efficacy, resulting in robust protection for several days (Fig. 7B); that level of protection is similar to the one afforded by SARM1 deletion. This finding indicates that augmentation of NAD+ synthesis may not be the main mechanism of added protection, an idea also consistent with the lack of efficacy of NaR alone. Therefore, although our findings using the axotomy model are consistent with the basic findings of Liu et al. (2018) in the vincristine model and show that the pharmacological efficacy of the FK866 + NaR combination is similar to that of gene deletion, they cast doubt on the role of NAD+ increase as a protective mechanism in this setting.
Fig. 7.
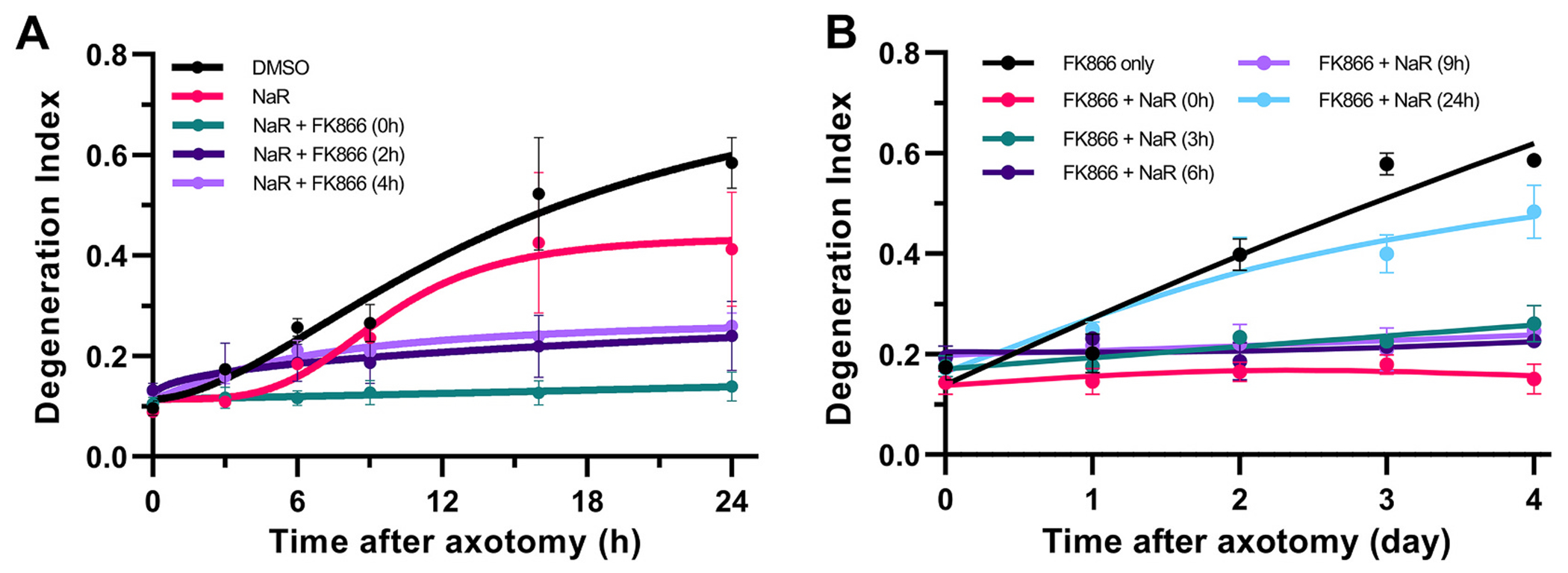
Prolonged protection of transected axons by supplementation of NAMPT inhibition with NaR.
A. The protective effect of NAMPT inhibition is augmented in the presence of NaR. Transected axons were treated with NaR at the time of injury and with the NAMPT inhibitor FK-866 at the time of injury or 2 and 4 h later; or NaR alone. Statistical analysis of DI was performed by two-way ANOVA for the effect of time (F5,25 = 23.48, p < 0.0001), treatment (F4,5 = 5.37, p = 0.047) and their interactions (F20,25 = 4.08, p = 0.0006) with Holm-ţídák’s multiple comparisons. In the presence of NaR, FK-866 addition up to 4 h post-axotomy is equally effective compared to vehicle treated axons at preventing axon degeneration at 24 h with t30 = 4.8–6.58 p < 0.0003). NaR has no protective effect by itself. B. The supplementation of NaR can be delayed up to 9 h with equally protective effect on axon fragmentation. Transected axons were treated with the NAMPT inhibitor FK866 after injury and NaR was added concurrently or at 3, 6, 9, or 24 h post axotomy. Statistical analysis of DI was performed by two-way ANOVA for the effect of time (F4,32 = 62.77, p < 0.0001), treatment (F5,8 = 17.05, p = 0.0004) and their interactions (F20,32 = 16.82, p < 0.0001) with Holm-ţídák’s multiple comparisons. Supplementation of NaR 3, 6 or 9 h post-axotomy in the presence of FK866, significantly protects axons compared to FK866 only treatment (t40 = 8.0–11.85, p < 0.0001) up to 4 days post-injury. Supplementation of NaR at 24 h has no additive effect on FK866 treatment alone. Error bars indicate ±1 SEM.
3.4. Supplementation of NAMPT inhibitors with NaR further decreases NMN and suppresses SARM1 activation in injured axons
Results presented in the previous section cast doubt on the role of NAD+ augmentation as a mechanism of the enhanced protective effect of the FK866-NaR combination. To explore the metabolic basis of the robust and lasting protection of this regimen which is similar to that of SARM1 deletion, we treated axons at the time of injury with FK866 alone, FK866 plus NaR, or vehicle (DMSO), and extracted axonal metabolites 8 h later (Fig. 8). As predicted, the FK866 + NaR combination does not significantly increase NAD+ above levels achieved with FK866 treatment alone (Fig. 8A). However, it does cause a further reduction in levels of NMN. As a result, the combination of FK866 with NaR leads to an even greater increase in the NAD+/NMN ratio compared to FK866 alone, generating levels similar to those of uninjured axons (Fig. 8B). In addition, the combination further suppresses SARM1 activity, as revealed by a significant 21% decrease in absolute cADPR levels and a greater decrease in the cADPR/NAD+ and ADPR/NAD+ ratios compared to FK866 alone (Fig. 8A-B). Of note, although metabolites of the Na salvage pathway are very low or undetectable in naïve or injured axons, stimulation of this pathway with NaR in the combined FK866 + NaR treatment leads to predictable increases in deamido-NMN (NaMN) and deamido-NAD (NaAD) (Supplementary Fig. 4). These data indicate that NaR synergizes with NAMPT inhibition in an unexpected way, i.e. by further suppressing NMN synthesis and SARM1 activity while preserving NAD+ gains achieved with NAMPT inhibition alone.
Fig. 8.
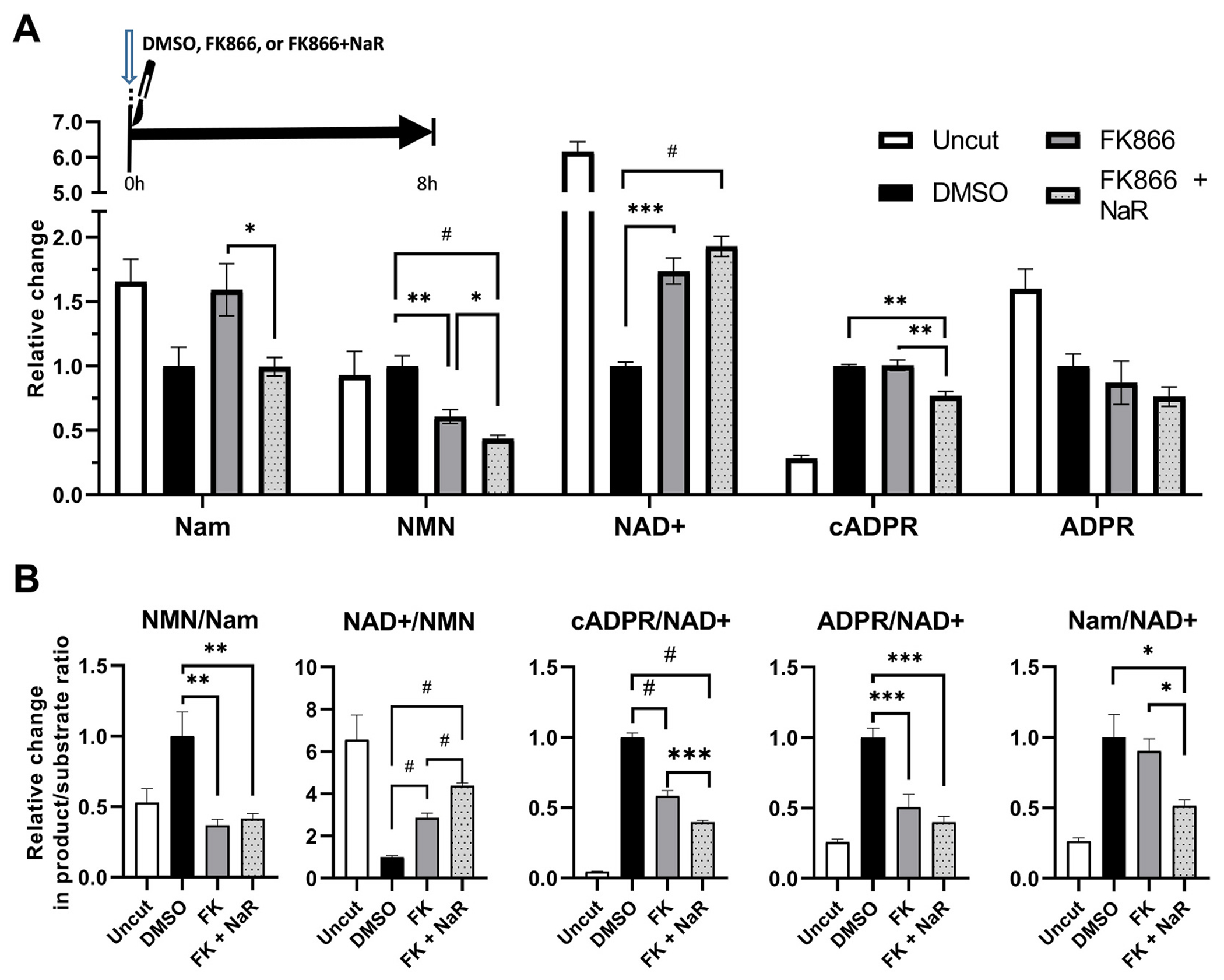
Supplementation of NAMPT inhibition with NaR favorably modulates the metabolic profile of transected axons.
A. Relative changes in protein-normalized levels of metabolites in distal axons at 8 h, following transection and treatment at the time of injury with NAMPT inhibitor FK866 alone or in combination with NaR, or vehicle (DMSO). NAMPT inhibition alone or in combination with NaR suppresses NMN (t10 = 5.03, p = 0.001 and t10 = 7.61, p < 0.001) and protects NAD+ levels (t10 = 6.46, p = 0.0001 and t10 = 8.60, p < 0.0001) compared to vehicle treated transected axons. Compared to NAMPT inhibition alone, NaR supplementation leads to further reduction of NMN levels (t10 = 2.31, p = 0.044) and only trend increase in NAD+ levels (t10 = 1.8, p = 0.11); while it also suppresses cADPR levels (t10 = 5.13, p = 0.001). B. Product/substrate ratios of key enzymatic steps. NAMPT inhibition alone or in combination with NaR, leads to reductions in the NMN/Nam (t10 = 4.44, p = 0.004 and t10 = 4.34, p = 0.004), increases in the NAD+/NMN (t10 = 8.26, p < 0.0001 and t10 = 15.84, p < 0.0001), and reductions in the cADPR/NAD+ ratios, consistent with reduced NAMPT, increased NMNAT2, and reduced NAD+ degrading activities respectively. The combination of FK866 + NaR is superior to FK866 alone with regards the effects on NAD+/NMN (t10 = 10.09, p < 0.0001) and cADPR/NAD+ ratios (t10 = 4.96,p < 0.0006). * = p < 0.05, ** = p < 0.01, *** = p < 0.001, # = p < 0.0001 after Holm-šídák’s multiple comparisons. Error bars indicate ±1 SEM.
3.5. Inhibition of the DLK/LZK MAPK pathway extends and enhances the protective effects of NAMPT inhibitors on transected axons
It has been proposed that the loss of NAD+ and the accumulation of NMN after axonal injury are driven by the rapid reduction in levels of NMNAT2 (Loreto et al., 2020). Basal levels of NMNAT2 are regulated by two distinct pathways based on palmitoylation status: an atypical Skp1-cullin 1-F-box E3 ligase complex degrading the non-palmitoylated fraction; and the stress-induced DLK/LZK MAPK cascade, a highly conserved retrograde signal of axonal injury (Welsbie et al., 2017; Welsbie et al., 2013; Welsbie et al., 2019; Yang et al., 2015; Shin et al., 2012) targeting the predominant palmitoylated fraction (Summers et al., 2018). Here we explored whether inhibition of the latter pathway post-injury prevents the loss of axonal NMNAT2 and, as a result, interferes with WD. We blocked the stress MAPK cascade at the DLK/LZK level with the selective DLK inhibitor GNE3511 (Patel et al., 2015).
The suppression of JNK phosphorylation and preservation of levels of the microtubule-binding protein superior cervical ganglion 10 (SCG10), an axon-maintenance factor that is rapidly degraded after axonal injury in a JNK-dependent manner (Shin et al., 2012), show that our GNE3511 treatment protocol was effective in inhibiting DLK (Fig. 9A-B). As predicted, DLK inhibition by GNE3511 was effective in preventing the loss of NMNAT2 caused by injury (Fig. 9A-B).
Fig. 9.
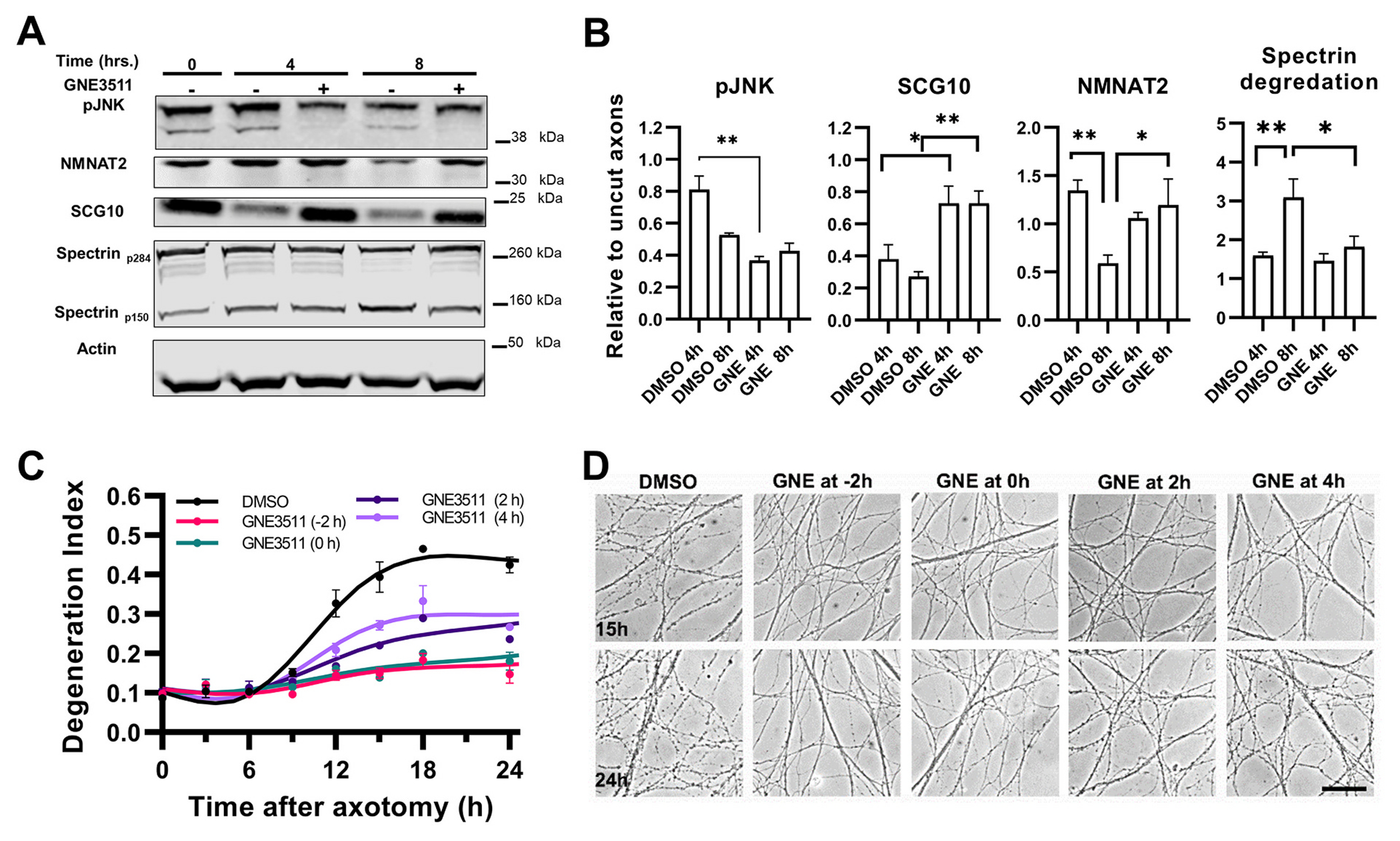
Western blot and Degeneration Index analysis reveal protective effects of DLK inhibition in transected axons.
A-B. Semi-quantitative Western blot analysis of distal axons at 4 and 8 h after transection and treatment with the DLK inhibitor GNE3511 or vehicle (DMSO). A shows representative immunoblot and B quantification of signals normalized to actin and with reference to uninjured/untreated axon samples. GNE3511 suppresses phosphorylation of JNK at 4 h, t5 = 6.17, p = 0.0005. NMNAT2 is significantly degraded between 4 and 8 h post-axotomy (t5 = 3.5, p = 0.016) in vehicle treated axons but not in the presence of GNE (t5 = 2.8, p = 0.046). Similarly, GNE suppresses the injury associated degradation of SCG10 at both time points (t5 = 3.06, p = 0.031; and t5 = 4.02, p = 0.008 respectively), and the injury associated degradation of spectrin at 8 h (calculated as the index of the p150 fragment to total spectrin, p284; t5 = 3.08, p = 0.03). Signals were analyzed by 2-way ANOVAs with Holm-šídák’s multiple comparisons. C. Axons were treated with GNE 2 h before or at the time of transection, or 2 and 4 h later, or with vehicle (DMSO) at the time of injury. Statistical analysis of DI was performed by two-way ANOVA for the effect of time (F7,35 = 164.7, p < 0.0001), treatment (F4,5 = 47.78, p = 0.0004) and their interactions (F28,35 = 16.22, p < 0.0001) with Holm-šídák’s multiple comparisons. GNE treatment at any time point offers protection against fragmentation early after axotomy (12h) compared to vehicle treated axons (t40 = 5.7–8.8, p < 0.0001). However, pre-treatment or treatment at the time of injury shows significantly less degeneration compared to delayed treatment at 24 h (t40 = 2.71–5.8, p < 0.03–0.0001). D. Representative photomicrographs from C. Scale bar, 50 μm. Error bars indicate ±1 SEM.
After validating that our GNE3511 treatment effectively engages the MAPK targets, we assessed the effect of MAPK inhibition on axon fragmentation. We found that treatment with GNE3511 before or at the time of injury causes a significant delay in WD, whereas later treatment is not as effective (Fig. 9C-D). Because NAMPT inhibition and DLK/LZK MAPK inhibition work via different mechanisms, the former by modulating NMN levels and the latter by preserving levels of NMNAT2, we explored whether the combined inhibition might have an additive effect. In this experiment, instead of purely using a NAMPT inhibitor like FK866, we used FK866 together with NaR as an “FK866 plus” strategy, based on the fact that supplementation the NAMPT inhibitor with NaR offers superior protection that does not depend on time of treatment. Transected axons were treated with either GNE-3511 or DMSO at the time of injury and with FK866 plus NaR at 0 h or 8 h hours after injury. Our results show that treatment with FK866 plus NaR at 0 h or 8 h after axotomy offers robust protection against degeneration for a period of 4 and 3 days, respectively. In the presence of GNE3511, protection is further extended to 6 and 5 days, whereas GNE3511 alone delays axon degeneration by only several hours (Fig. 10). The combination of optimized FK866 + NaR treatment with GNE3511 is also effective in human embryonic stem cell-derived neurons subjected to axotomy, with significant delay in axon fragmentation for up to 48 h (Fig. 11).
Fig. 10.
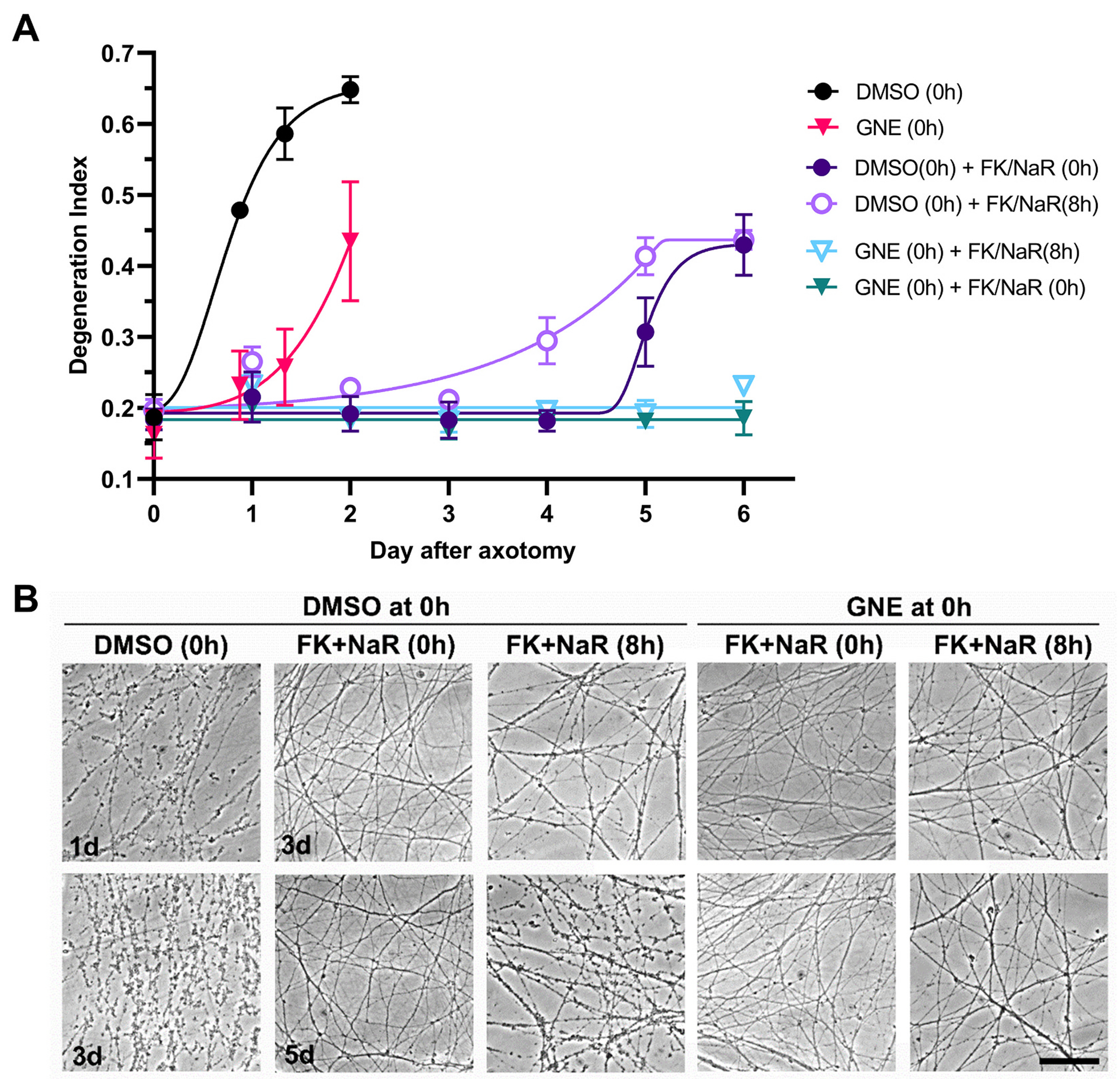
MAPK inhibition further enhances the protective effect of NAMPT inhibition-NaR combination. A. Transected axons were treated at the time of injury with the DLK inhibitor GNE-3511 or vehicle (DMSO) and with the combination of FK866/NaR either at the time of injury or with an 8 h delay. Statistical analysis of degeneration index was performed by two-way ANOVA for the effect of time (F2.2,17.2 = 49.43, p < 0.0001), treatment (F3,8 = 9.58, p = 0.005) and their interactions (F18,48 = 13.99, p < 0.0001) with Holm-šídák’s multiple comparisons. There are no significant differences between any of the FK + NaR groups up to day 4. However the combined treatment of GNE with FK + NaR (whether early or delayed) affords better protection against fragmentation compared to FK + NaR treatment alone at day 6 (t56 = 6.8–8.4, p < 0.0001). Error bars indicate ±1 SEM. B. Representative photomicrographs used in counts. Scale bar, 50 μm.
Fig. 11.
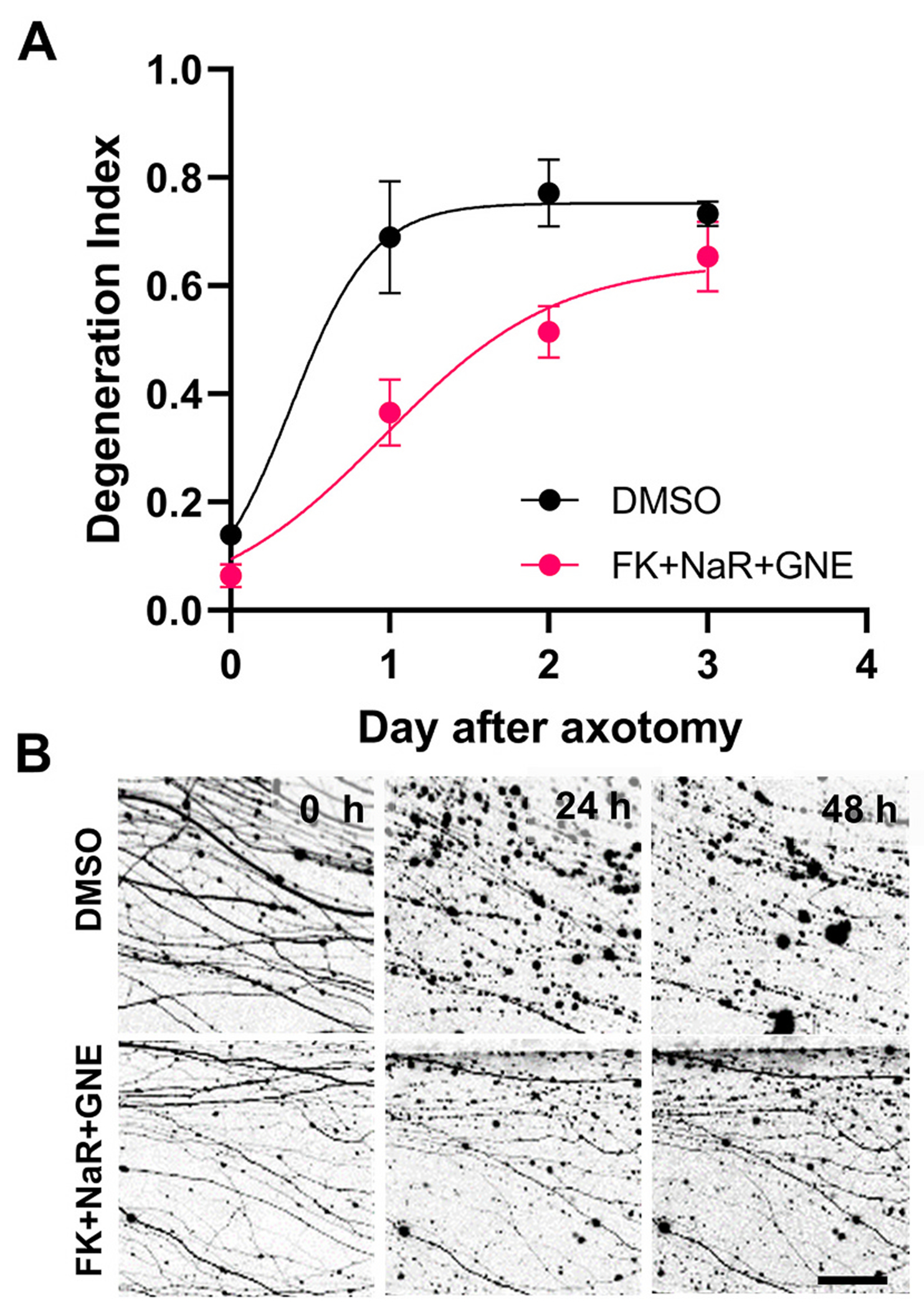
Combinatorial pharmacological treatment delays fragmentation of human axons after transection.
Human embryonic stem cell-derived cholinergic neurons expressing tdTomato were subjected to axotomy and treated with FK866 + NaR + GNE3511 or vehicle. A. Degeneration index analysis reveals significant protection of the combinatorial treatment regimen at 24 (t16 = 4.04, p = 0.004; and 48 h (t16 = 3.21, p = 0.016). B. Representative epifluorescent images from A showing expression of tdTomato. Scale bar, 25 μm.
To further explore the metabolic profile of MAPK inhibition, we treated transected axons with GNE3511, FK866 plus NaR, or their combination, at the time of injury, and analyzed NAD+ and SARM1 metabolites 8 h later (Fig. 12). The comparison of the three treatment groups revealed significant differences in NAD+ and NMN, but not in cADPR levels. As expected by the relative protection of NMNAT2 levels by DLK inhibition, we found that NAD+ levels are significantly higher with GNE3511 than FK866 + NaR treatment, while the combined three-way treatment achieved NAD+ levels in-between those seen with GNE3511 and FK866 + NaR. In the case of NMN, FK866 + NaR treatment was associated with the lowest levels, while the addition of GNE, surprisingly, led to a modest increase (Fig. 12A). When we analyzed product/substrate ratios among treatments, we found significant differences in NAD+/NMN and cADPR/NAD+ (serving as indices of NMNAT2 and SARM1 activity, respectively). Treatment with GNE3511 alone significantly increased the NAD+/NMN ratio compared to both FK866 + NaR alone and FK866 + NaR plus GNE3511. Similarly, GNE3511 alone or in combination suppressed the cADPR/NAD+ ratio by 3.5 and 2 times compared to FK866 + NaR alone (Fig. 12B). The metabolic effects of DLK inhibition were also reflected by homologous effects in the Na salvage pathway (Fig. 1) in axons that were treated with FK866 + NaR: GNE3511 co-treatment led to doubling of NaAD levels and of the NaMN/NaAD ratio (Supplementary Fig. 5), an action consistent with increased NMNAT2 activity. These highly favorable effects of MAPK inhibition are consistent with the protection of NMNAT2 levels seen at the same time frame (Fig. 9A-B).
Fig. 12.
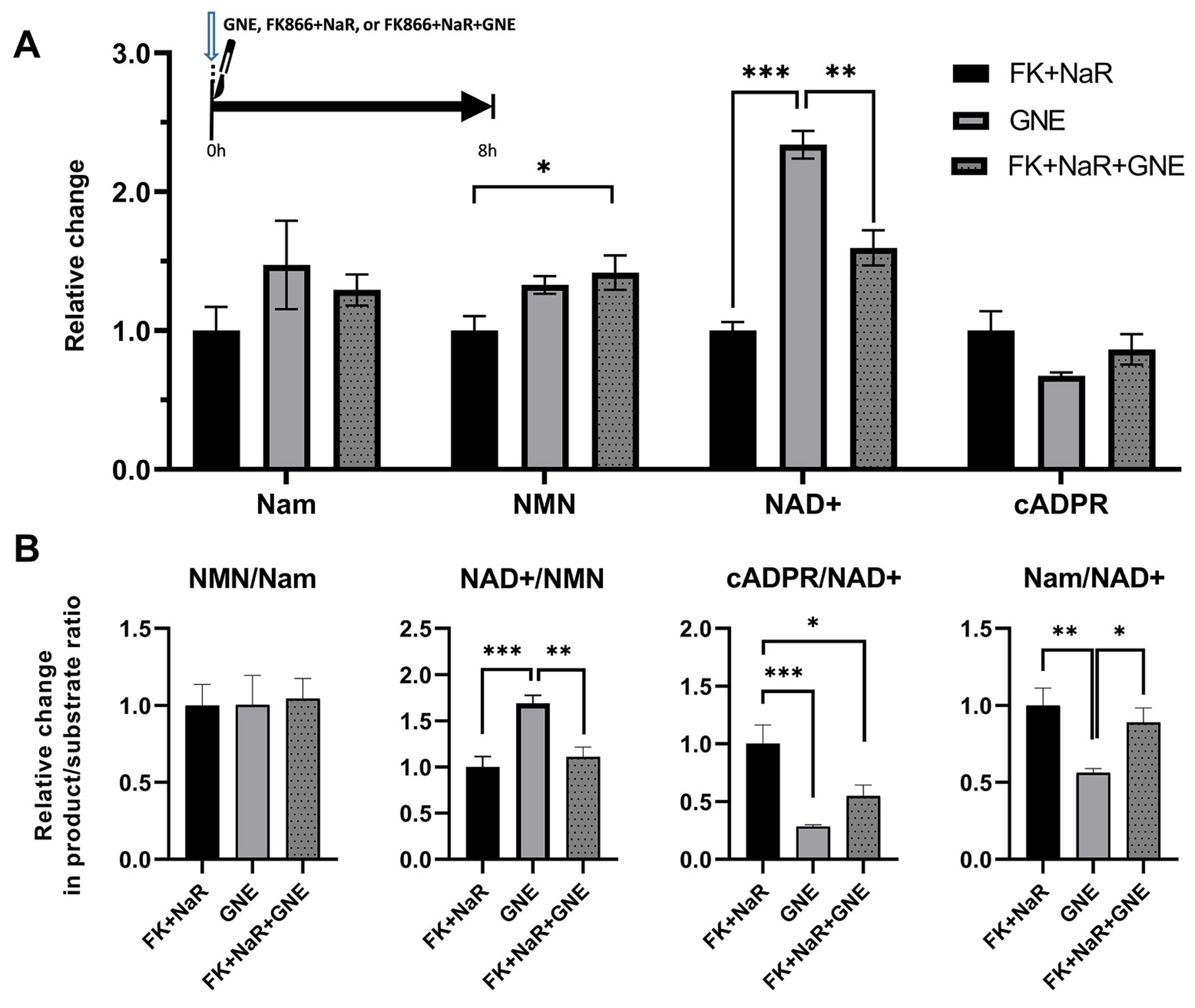
Metabolic effects of MAPK inhibition with and without NAMPT inhibition+NaR in transected axons.
A. Relative changes in protein-normalized levels of metabolites in distal axons at 8 h, following transection and treatment at the time of injury with the MAPK inhibitor GNE-3511, the NAMPT inhibitor FK866 in combination with NaR, or with the three compounds together. One-way ANOVA analysis with Holm-šídák’s multiple comparisons for effects between the three treatment groups revealed significant changes in NAD+ (F2,15 = 44.87, p < 0.0001) and NMN levels (F2,15 = 4.79, p = 0.023) but not in other metabolites. Treatment with GNE resulted in greater protection of NAD+ levels compared to both FK + NaR (t15 = 9.45, p < 0.001) or triple combination (t15 = 5.25, p = 0.002). The triple combination had slightly higher NMN levels compared to FK + NaR alone (t15 = 2.93, p = 0.03). B. Relative changes in product/substrate ratios of key enzymatic steps. GNE treatment alone leads to a greater increase in the NAD+/NMN ratio than FK + NaR (t15 = 4.77, p = 0.0007) or FK + NaR + GNE (t15 = 4.01, p = 0.002); while GNE alone or in combination better reduces the cADPR/NAD+ ratio (t15 = 4.66, p = 0.0009; and t15 = 1.73, p = 0.02) compared to FK + NaR. * = p < 0.05, ** = p < 0.01, *** = p < 0.001. Error bars indicate ±1 SEM. ADPR levels were below the lower limit of quantification and are not shown.
3.6. SARM1 activation negatively correlates with the NAD+/NMN ratio in the context of axotomy and NAMPT inhibition
Changes in NMN and NAD+ levels have been implicated in WD, not only due to the nature of these metabolites as substrate and product of NMNAT2, respectively, but also because of their proposed roles as allosteric activator (NMN) and inhibitor (NAD+) of SARM1 (Bratkowski et al., 2020; Figley et al., 2021; Jiang et al., 2020; Sporny et al., 2020). In our material, axotomy leads to a progressive increase in NMN and a progressive loss of NAD+, generating an up to 10-fold decrease in the NAD+/NMN ratio at 8 h post axotomy (Fig. 6B). In all FK866 treatment scenarios laid out in Section 3, regardless of variable and often unexpected changes in NMN and NAD+, there is a significant increase in the NAD+/NMN ratio, in a way mimicking the effect of NMNAT activity: NAMPT inhibition ultimately leads to 3-fold increase in the NAD+/NMN ratio at 8 h post axotomy, whereas the combination of FK866 with NaR brings the NAD/NMN+ ratio close to that of uninjured controls (Fig. 8), an effect also replicated by DLK inhibition (Fig. 11).
In order to explore the relationship between NAD+ or NMN levels and SARM1 activity, we explored correlations between NAD+, NMN or the NAD+/NMN ratio with cADPR levels in our biological samples. We first analyzed individual replicates from a heterogeneous set of samples including naïve, treated, and untreated injured axons (from experiments illustrated in Figs. 8 and S3). We found that NAD+ negatively correlates with cADPR levels (Spearman’s ρ = −0.61, p = 0.0015), probably because of the great difference in NAD+ levels between injured and uninjured samples but NAD+ levels cannot explain the cADPR variance within injured samples (ρ = −0.12, ns). Conversely, NMN levels correlate with cADPR levels in injured axon samples (ρ = 0.73, p = 0.0006) but not when naïve axons are also included in the analysis (ρ = 0.10, ns). On the other hand, the NAD+/NMN ratio is highly predictive of cADPR levels in either scenario (ρtotal = −0.72, p < 0.0001; ρtransected = −0.63, p < 0.0047; Fig. 13). We did not find correlations with other products of SARM1, i.e. ADPR (ρ = 0.23, ns) and Nam (ρ = 0.16, ns); or with metabolites of the Na salvage pathway. Finally, we confirmed these correlations in another independent set of samples (from experiments illustrated in Fig. 12). Again cADPR levels are negatively correlated with the NAD+/NMN ratio (ρ = −0.85, p < 0.0001) but have no significant correlation with either NAD+ (ρ = −0.43, ns) or NMN (ρ = 0.40, ns) (Fig. 13).
Fig. 13.
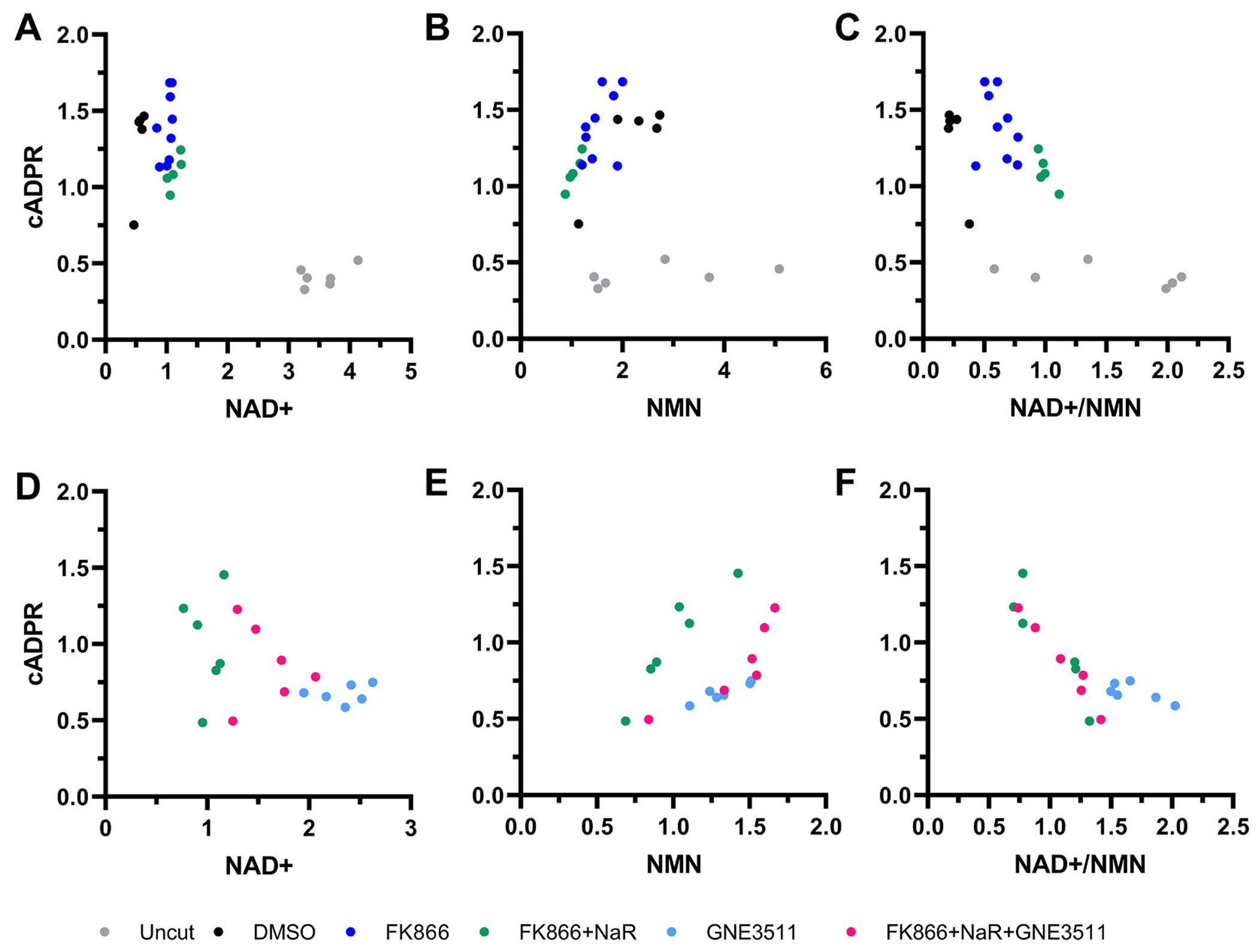
The NAD+/NMN ratio better predicts cADPR levels than NAD+ and NMN levels.
Scatter plots that show relationships of relative cADPR levels with NAD+ (A, D), NMN (B, E) or NAD+/NMN ratios (C, F) within individual axon samples. Values are from replicates of experiments described in Fig. 7 (A-C) and Fig. 12 (D-F). Values are expressed relative to the average values of the FK866 + NaR group. See text for details.
These findings are consistent with the hypothesis that the NAD+/NMN ratio, rather than the absolute levels of each metabolite relates to SARM1 activity (Bratkowski et al., 2020; Figley et al., 2021; Jiang et al., 2020; Sporny et al., 2020). The association of the NAD+/NMN ratio with cADPR in biological samples confirms and expands previous observations in cell-free assays (Figley et al., 2021) and indicates the central role of NAD+ and NMN metabolism in the regulation of SARM1 activity.
4. Discussion
In the present study we used pharmacological agents that modulate pathways of NAD+ metabolism i.e. the Nam recycling pathway, the Na salvage pathway, and the DLK/LZK MAPK cascade, and established their effects in preventing axon fragmentation after axotomy. We find that combinatorial pharmacological strategies that better preserve NAD+ and the NAD+/NMN balance, and suppress SARM1 activity are associated with better outcomes. Inhibition of the NMN-synthesizing enzyme NAMPT is sufficient to delay WD in a time-dependent manner, whereas supplementation with NaR leads to complete suppression of axon fragmentation up to 4 days post injury. The decision point of axon fragmentation occurs within a specified time-window and is related to inverse changes in NMN and NAD+ levels, probably driven by a MAPK-dependent degradation of NMNAT2. As in the case of NAMPT inhibition, blockade of the injury-induced MAPK activity is also sufficient to delay axon fragmentation and supplements the protective effect of the former. Although metabolite analyses reveal complex and time-dependent effects, they indicate that even partial suppression of SARM1 activity by the modulation of NAD+ metabolic pathways is sufficient to suppress axon fragmentation. Finally, our results are consistent with the notion that the ratio of NAD+ to NMN is one of the drivers of SARM1 activity (Bratkowski et al., 2020; Figley et al., 2021; Jiang et al., 2020; Sporny et al., 2020).
Wallerian degeneration (WD) has been implicated in a spectrum of neurological disorders from mechanical axonal injury and peripheral neuropathies to neurodegenerative diseases (Coleman and Hoke, 2020; Conforti et al., 2014; Koliatsos and Alexandris, 2019; Figley and DiAntonio, 2020). Since the discovery of the axon-protective protein Wlds, a degradation-resistant fusion protein of NMNAT1, and the identification of SARM1 as the executioner of WD, axonal NAD+ metabolism has been placed at the center of WD (Conforti et al., 2014; Figley and DiAntonio, 2020; Coleman and Freeman, 2010). In several key genetic experiments, increases in baseline levels of NAD+ delayed axon fragmentation, whereas prevention of injury-associated rise of NMN was robustly protective (Sasaki et al., 2016). Although such genetic manipulations led to the identification of several key steps, including the importance of NMN and NAD+ in driving or inhibiting WD, respectively (Di Stefano et al., 2017; Sasaki et al., 2016), they also establish supranormal baseline levels of target enzymes and metabolites (up to 20-fold) that may not be relevant to the native state. Moreover, important questions remain in the understanding of decision points and regulatory events. The present study, in which we took a targeted pharmacological approach to protect wt axons, allows for a more physiological exploration of the role of NMN and NAD+ regulation in WD and offers new ideas on axon rescue.
4.1. Delayed degeneration and favorable modulation of axonal NAD+ metabolism by NAMPT inhibition
Our finding that NAMPT inhibition delays WD is consistent with previous observations (Loreto et al., 2020; Di Stefano et al., 2015; Sasaki et al., 2016; Loreto et al., 2015). Additionally, we have shown that the efficacy of NAMPT inhibition is time-dependent, with maximum protection when treatment is initiated within 2 h post injury. This is consistent with previous observations on markedly reduced efficacy of treatment prior to injury (Di Stefano et al., 2015; Sasaki et al., 2016). However, our experiments revealed surprising effects of NAMPT inhibition beyond the reduction in NMN as originally hypothesized (Loreto et al., 2020; Di Stefano et al., 2015; Sasaki et al., 2016). Although NAMPT inhibition leads to predictable reductions in both NMN and NAD+ in cell bodies, it has a biphasic effect on NMN in injured axons, i.e. a paradoxical increase in the early hours post injury followed by partial suppression later. Partial inhibition of NAMPT is an unlikely explanation, because doses used are at saturating levels (>250 times the Kd) and FK866 is a non-competitive inhibitor. Furthermore, our initial dose titration experiments did not reveal dose-response effects on axon protection in the range of doses used. The persistence of NMN in axons in the presence of a non-competitive NAMPT inhibitor, suggests alternative sources of NMN that deserve further exploration.
An important novel finding is that NAMPT inhibition (within 2 h of injury) leads to a relative protection of axonal NAD+ levels at least up to 8 h post axotomy. It remains to be seen whether the relative protection of NAD+ levels can be fully explained by reduced degradation due to decreased SARM1 enzymatic activity or other mechanisms involving NMNAT2-independent pathways.
4.2. NaR synergizes with NAMPT inhibition for the modulation of NMN levels and offers robust protection against axotomy
While NAMPT inhibition delays axon fragmentation, its combination with NaR leads to robust protection for several days, similar to that previously seen in the vincristine model (Liu et al., 2018). However, again, the protective mechanism of NaR supplementation is not the expected one. NaR supplementation does not significantly increase NAD+ levels compared to NAMPT inhibition alone, but instead leads to further NMN reductions. More importantly, unlike NAMPT inhibition alone, NaR supplementation leads to a significant reduction in levels of cADPR, a putative biomarker of SARM1 (Sasaki et al., 2020), and further suppresses SARM1 enzymatic activity (as indicated by the cADPR/NAD+ ratio). As argued in the previous section, injured axons may use another source of NMN that is NAMPT-independent. Given the structural similarities of NaR with the NMN precursor nicotinamide riboside (NR) and the similarity in other substrates, it is plausible that excess NaR may be a competitor of NR in pathways affecting NMN synthesis. This interpretation is consistent with the fact that NaR supplementation is highly efficacious without increasing NAD+ levels above levels seen with NAMPT inhibition alone, and is still robustly effective even at late time-points post injury, i.e. after NMNAT2 is largely degraded. Although we cannot exclude residual NMNAT2 activity at later time points or the involvement of its mitochondrial isoform NMNAT3, these findings indicate that the protective effect of NaR supplementation may not be necessarily due to an increase in NAD+ synthesis as originally thought (Liu et al., 2018) but may include allostatic changes in NMN synthesis through other mechanisms.
It is also possible that, in parallel to reducing NMN, NaR and/or its derivatives (NaMN, NaAD) may act as allosteric inhibitors of SARM1. Indeed, it was recently shown that micromolar concentrations of NaR can substantially inhibit the enzymatic activity of purified human SARM1 (Angeletti et al., 2022). However, despite the high doses used in the present study (500 μM), NaR on its own had no detectable effect on axon fragmentation, making this explanation less likely. It was also recently shown by Sasaki and colleagues that NaMN may compete with NMN and act as an allosteric inhibitor of purified SARM1 in the micromolar range (Sasaki et al., 2021). In our samples we found that supplementation with NaR in the presence of a NAMPT inhibitor leads to both a substantial increase in levels of NaMN and to an inversion of the NMN/NaMN ratio, although we didn’t detect a consistent correlation of either with cADPR levels. The above findings suggest that the protective effect of the combination of NAMPT inhibition with NaR may involve multiple parallel actions on SARM1 and NAD+/NMN metabolism.
4.3. NMNAT2, the NAD+/NMN ratio and SARM1 activation
Our data indicate that SARM1 activity is closely related to the NAD+/NMN ratio, in view of the negative correlation of the latter with cADPR levels. Conversely, interventions that either preserve or mimic NMNAT activity seem to have an inhibitory effect on SARM1 and thus suppress WD. This notion is consistent with recent evidence on cell-free assays suggesting that NMN and NAD+ compete for the same allosteric site and regulate recombinant SARM1 activity in opposite directions (Bratkowski et al., 2020; Figley et al., 2021; Jiang et al., 2020; Sporny et al., 2020). Our results thus confirm, for the first time, that the NAD+/NMN ratio is an important driver of SARM1 activity in biological systems as well.
Our finding that the pharmacological inhibition of DLK post injury suppresses NMNAT2 degradation and delays WD, extends previous findings on the role of DLK in regulating baseline levels of NMNAT2 (Summers et al., 2018). This is relevant because the activity of the DLK/LZK pathway is increased in both the distal axon and perikarya after injury (Welsbie et al., 2013; Welsbie et al., 2019; Yang et al., 2015). Moreover, MAPK inhibition is associated with the preservation of NMNAT2 activity as indicated by greater protection of NAD+ levels compared to NAMPT inhibition and a further improvement of the NAD+/NMN ratio. Although these effects can be attributed to the protection of NMNAT2, it is also possible that MAPK activity may regulate SARM1 directly via phosphorylation (Murata et al., 2018) or through other mechanisms. This may also explain why the addition of a DLK inhibitor to FK866 + NaR-treated axons results in further reduction in SARM1 activity without, additional improvement of the NAD+/NMN ratio. Despite the strong and sometimes superior metabolic effects of DLK inhibition in the first hours after axonal injury, its protective effect against axon fragmentation is not as long lasting, a finding that may relate to a passive component of NMNAT2 depletion in the absence of its transport or local synthesis. Moreover, the efficacy of DLK inhibition is time dependent, with a short time window of few hours post-axotomy, which is consistent with the time course of NMNAT2 degradation.
4.4. Translational potential
Our findings indicate robust effects of NAMPT inhibitors on axonal viability that become substantially enhanced and resemble the effect of SARM1 deletion with the addition of a DLK inhibitor and the NAD+ precursor NaR. Such protective effects may extend to axonal physiology and may also apply to in vivo models. The first prediction is based on electrophysiological studies on transected axons from SARM1 KO or Wlds mice (Fernandes et al., 2018). The second is supported by the highly conserved nature of WD and the already established effects of Wlds and SARM1 KO in several simple and complex models of disease (Coleman and Hoke, 2020).
Some observations from rodent DRG neurons may also apply to human embryonic stem cell-derived neurons, as indicated by a substantial delay in axon fragmentation after combinatorial treatment with FK866, NaR and GNE3511. While these data support the idea that the roles of NAD and NMN in axonal viability are conserved in human neurons and are therapeutically relevant, the shorter duration of the effect of treatment may suggest the involvement of additional pathways contributing to SARM1 activation or possible differences between species that are yet to be identified.
Suppression of NMN synthesis by NAMPT inhibitors, particularly in combination with NaR, represents a promising therapeutic target given the availability of CNS penetrant, clinically tested and safe inhibitors like FK866 (Daporinad). The translational potential of this approach is further supported by the existence of a therapeutic window extending several hours after injury. NAMPT inhibition may be also promising because of its independent role in inhibiting neuroinflammatory responses and ameliorating secondary injury in multiple models of injury (Esposito et al., 2012; Galli et al., 2020; Tan et al., 2020; Wu et al., 2017; Zhang et al., 2015; Zou et al., 2020). Similarly, the role of the MAPK pathway is broader than simply regulating WD. Dual leucine zipper kinase signaling is associated with retrograde neuronal death from axonal injury or trophic factor deprivation and its suppression has significant rescue effects (Watkins et al., 2013; Welsbie et al., 2017; Welsbie et al., 2013; Welsbie et al., 2019; Fernandes et al., 2014; Ghosh et al., 2011). Therefore, DLK inhibition might have a dually protective role in attenuating both axonal and perikaryal degeneration. As in the case of NAMPT inhibition, small molecules that block DLK/LZK e.g. sunitinib are CNS penetrant, have been through clinical trials, and are approved as drugs for certain non-neurological diseases (Welsbie et al., 2017; Gore et al., 2015; Tang et al., 2012). Potential therapeutic applications of small-molecule combinations tested here are broad, from chemotherapy-induced peripheral neuropathy and glaucoma to CNS neurodegenerative disease, especially because altered MAPK signaling and reduction in NMNAT2 levels may play a role in several of these disorders (Ali et al., 2016; Le Pichon et al., 2017; Ljungberg et al., 2012).
Supplementary Material
Funding
This work was supported by grants to V.E.K. from the National Eye Institute (RO1EY028039), the National Institute of Neurological Disorders and Stroke (R01NS114397), the TEDCO Maryland Stem Cell Research Fund (2020-MSCRFD-5402) and from generous gifts from the Kate Sidran Family and the Giller Family foundations.
Abbreviations:
- ADPR
adenosine diphosphate ribose
- cADPR
cyclic adenosine diphosphate ribose
- DI
degeneration index
- DIV
day in vitro
- DLK
dual leucine kinase (also known as MAP3K12)
- JNK
c-Jun N-terminal kinase
- LZK
leucine zipper kinase (also known as MAP3K13)
- MAPK
mitogen-activated protein kinase
- Na
nicotinic acid
- NaAD
nicotinic acid adenine dinucleotide or deamido-NAD
- NAD+
nicotinamide adenine dinucleotide
- Nam
nicotinamide
- NaMN
nicotinic acid mononucleotide or deamido-NMN
- NAMPT
nicotinamide phosphoribosyltransferase
- NaR
nicotinic acid ribose
- NMN
nicotinamide mononucleotide
- NMNAT2
nicotinamide mononucleotide adenylyltransferase 2
- NR
nicotinamide riboside
- NRK
nicotinamide riboside kinase
- SARM1
sterile alpha and toll/interleukin receptor motif-containing protein 1
- WD
Wallerian degeneration
Footnotes
Ethics approval
All procedures were performed according to the National Institute of Health Guide for the Care and Use of Experimental Animals and were approved by the Johns Hopkins University Animal Care and Use Committee.
CRediT authorship contribution statement
Athanasios S. Alexandris: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing. Jiwon Ryu: Data curation, Formal analysis, Investigation, Methodology, Visualization. Labchan Rajbhandari: Methodology, Resources. Robert Harlan: Methodology, Investigation. James McKenney: Investigation. Yiqing Wang: Investigation. Susan Aja: Project administration. David Graham: Supervision. Arun Venkatesan: Supervision. Vassilis E. Koliatsos: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2022.105808.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- Ali YO, Allen HM, Yu L, Li-Kroeger D, Bakhshizadehmahmoudi D, Hatcher A, et al. , 2016. NMNAT2:HSP90 complex mediates Proteostasis in Proteinopathies. PLoS Biol. 14 (6), e1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeletti C, Amici A, Gilley J, Loreto A, Trapanotto AG, Antoniou C, et al. , 2022. SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. iScience 25 (2), 103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratkowski M, Xie T, Thayer DA, Lad S, Mathur P, Yang YS, et al. , 2020. Structural and mechanistic regulation of the pro-degenerative NAD hydrolase SARM1. Cell Rep. 32 (5), 107999. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR, 2010. Wallerian degeneration, wld(s), and nmnat. Annu. Rev. Neurosci 33, 245–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MP, Hoke A, 2020. Programmed axon degeneration: from mouse to mechanism to medicine. Nat. Rev. Neurosci 21 (4), 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Gilley J, Coleman MP, 2014. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci 15 (6), 394–409. [DOI] [PubMed] [Google Scholar]
- Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, et al. , 2015. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 22 (5), 731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano M, Loreto A, Orsomando G, Mori V, Zamporlini F, Hulse RP, et al. , 2017. NMN Deamidase delays Wallerian degeneration and rescues axonal defects caused by NMNAT2 deficiency in vivo. Curr. Biol 27 (6), 784–794. [DOI] [PubMed] [Google Scholar]
- Esposito E, Impellizzeri D, Mazzon E, Fakhfouri G, Rahimian R, Travelli C, et al. , 2012. The NAMPT inhibitor FK866 reverts the damage in spinal cord injury. J. Neuroinflammation 9, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J, 2017. The SARM1 toll/Interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron. 93 (6), 1334–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman K, Summers DW, Sasaki Y, Mao X, Yim AKY, DiAntonio A, et al. , 2018. TIR domain proteins are an ancient family of NAD(+)-consuming enzymes. Curr. Biol 28 (3), 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes KA, Harder JM, Fornarola LB, Freeman RS, Clark AF, Pang IH, et al. , 2012. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol. Dis 46 (2), 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes KA, Harder JM, John SW, Shrager P, Libby RT, 2014. DLK-dependent signaling is important for somal but not axonal degeneration of retinal ganglion cells following axonal injury. Neurobiol. Dis 69, 108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes KA, Mitchell KL, Patel A, Marola OJ, Shrager P, Zack DJ, et al. , 2018. Role of SARM1 and DR6 in retinal ganglion cell axonal and somal degeneration following axonal injury. Exp. Eye Res 171, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figley MD, DiAntonio A, 2020. The SARM1 axon degeneration pathway: control of the NAD(+) metabolome regulates axon survival in health and disease. Curr. Opin. Neurobiol 63, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figley MD, Gu W, Nanson JD, Shi Y, Sasaki Y, Cunnea K, et al. , 2021. SARM1 is a metabolic sensor activated by an increased NMN/NAD(+) ratio to trigger axon degeneration. Neuron. 109 (7), 1118–36 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli U, Colombo G, Travelli C, Tron GC, Genazzani AA, Grolla AA, 2020. Recent advances in NAMPT inhibitors: a novel immunotherapic strategy. Front. Pharmacol 11, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Doan RA, Strickland A, Huang X, Milbrandt J, DiAntonio A, 2016. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain. 139 (Pt 12), 3092–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J, 2015. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science (New York, N Y.) 348 (6233), 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW, 2011. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J. Cell Biol 194 (5), 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coleman MP, 2010. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 8 (1), e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goparaju SK, Kohda K, Ibata K, Soma A, Nakatake Y, Akiyama T, et al. , 2017. Rapid differentiation of human pluripotent stem cells into functional neurons by mRNAs encoding transcription factors. Sci. Rep 7, 42367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore ME, Szczylik C, Porta C, Bracarda S, Bjarnason GA, Oudard S, et al. , 2015. Final results from the large sunitinib global expanded-access trial in metastatic renal cell carcinoma. Br. J. Cancer 113 (1), 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henninger N, Bouley J, Sikoglu EM, An J, Moore CM, King JA, et al. , 2016. Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1. Brain. 139 (Pt 4), 1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins EL, Gu W, Kobe B, Coleman MP, 2021. A novel NAD signaling mechanism in axon degeneration and its relationship to innate immunity. Front. Mol. Biosci 8, 703532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Liu T, Lee CH, Chang Q, Yang J, Zhang Z, 2020. The NAD(+)-mediated self-inhibition mechanism of pro-neurodegenerative SARM1. Nature. 588 (7839), 658–663. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Alexandris AS, 2019. Wallerian degeneration as a therapeutic target in traumatic brain injury. Curr. Opin. Neurol 32 (6), 786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pichon CE, Meilandt WJ, Dominguez S, Solanoy H, Lin H, Ngu H, et al. , 2017. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med 9 (403). [DOI] [PubMed] [Google Scholar]
- Li Y, Pazyra-Murphy MF, Avizonis D, de Sa Tavares Russo M, Tang S, Bergholz JS, et al. , 2022. Activation of Sarm1 produces cADPR to increase intra-axonal calcium and promote axon degeneration in CIPN. J Cell Biol 221 (2), e202106080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HW, Smith CB, Schmidt MS, Cambronne XA, Cohen MS, Migaud ME, et al. , 2018. Pharmacological bypass of NAD(+) salvage pathway protects neurons from chemotherapy-induced degeneration. Proc. Natl. Acad. Sci. U. S. A 115 (42), 10654–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungberg MC, Ali YO, Zhu J, Wu CS, Oka K, Zhai RG, et al. , 2012. CREB-activity and nmnat2 transcription are down-regulated prior to neurodegeneration, while NMNAT2 over-expression is neuroprotective, in a mouse model of human tauopathy. Hum. Mol. Genet 21 (2), 251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loreto A, Di Stefano M, Gering M, Conforti L, 2015. Wallerian degeneration is executed by an NMN-SARM1-dependent late ca(2+) influx but only modestly influenced by mitochondria. Cell Rep. 13 (11), 2539–2552. [DOI] [PubMed] [Google Scholar]
- Loreto A, Hill CS, Hewitt VL, Orsomando G, Angeletti C, Gilley J, et al. , 2020. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol. Dis 134, 104678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion CM, McDaniel DP, Armstrong RC, 2019. Sarm1 deletion reduces axon damage, demyelination, and white matter atrophy after experimental traumatic brain injury. Exp. Neurol 321, 113040. [DOI] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A, 2009. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci 12 (4), 387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata H, Khine CC, Nishikawa A, Yamamoto KI, Kinoshita R, Sakaguchi M, 2018. C-Jun N-terminal kinase (JNK)-mediated phosphorylation of SARM1 regulates NAD(+) cleavage activity to inhibit mitochondrial respiration. J. Biol. Chem 293 (49), 18933–18943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, et al. , 2012. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 337 (6093), 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Cohen F, Dean BJ, De La Torre K, Deshmukh G, Estrada AA, et al. , 2015. Discovery of dual leucine zipper kinase (DLK, MAP3K12) inhibitors with activity in neurodegeneration models. J. Med. Chem 58 (1), 401–418. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Nakagawa T, Mao X, DiAntonio A, Milbrandt J, 2016. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD(+) depletion. Elife. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Engber TM, Hughes RO, Figley MD, Wu T, Bosanac T, et al. , 2020. cADPR is a gene dosage-sensitive biomarker of SARM1 activity in healthy, compromised, and degenerating axons. Exp. Neurol 329, 113252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Zhu J, Shi Y, Gu W, Kobe B, Ve T, et al. , 2021. Nicotinic acid mononucleotide is an allosteric SARM1 inhibitor promoting axonal protection. Exp. Neurol 345, 113842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, DiAntonio A, 2012. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron. 74 (6), 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporny M, Guez-Haddad J, Khazma T, Yaron A, Dessau M, Shkolnisky Y, et al. , 2020. Structural basis for SARM1 inhibition and activation under energetic stress. Elife. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers DW, Milbrandt J, DiAntonio A, 2018. Palmitoylation enables MAPK-dependent proteostasis of axon survival factors. Proc. Natl. Acad. Sci. U. S. A 115 (37), E8746–E8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z, Chen L, Ren Y, Jiang X, Gao W, 2020. Neuroprotective effects of FK866 against traumatic brain injury: involvement of p38/ERK pathway. Ann. Clin. Transl. Neurol 7 (5), 742–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SC, Lagas JS, Lankheet NA, Poller B, Hillebrand MJ, Rosing H, et al. , 2012. Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int. J. Cancer 130 (1), 223–233. [DOI] [PubMed] [Google Scholar]
- Turkiew E, Falconer D, Reed N, Hoke A, 2017. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst 22 (3), 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, et al. , 2013. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci. U. S. A 110 (10), 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, et al. , 2013. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc. Natl. Acad. Sci. U. S. A 110 (10), 4045–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Mitchell KL, Jaskula-Ranga V, Sluch VM, Yang Z, Kim J, et al. , 2017. Enhanced functional genomic screening identifies novel mediators of dual leucine zipper kinase-dependent injury signaling in neurons. Neuron. 94 (6), 1142–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Ziogas NK, Xu L, Kim BJ, Ge Y, Patel AK, et al. , 2019. Targeted disruption of dual leucine zipper kinase and leucine zipper kinase promotes neuronal survival in a model of diffuse traumatic brain injury. Mol. Neurodegener 14 (1), 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GC, Liao WI, Wu SY, Pao HP, Tang SE, Li MH, et al. , 2017. Targeting of nicotinamide phosphoribosyltransferase enzymatic activity ameliorates lung damage induced by ischemia/reperfusion in rats. Respir. Res 18 (1), 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Sauve AA, 2016. NAD(+) metabolism: bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 1864 (12), 1787–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS, et al. , 2015. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell. 160 (1–2), 161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XQ, Lu JT, Jiang WX, Lu YB, Wu M, Wei EQ, et al. , 2015. NAMPT inhibitor and metabolite protect mouse brain from cryoinjury through distinct mechanisms. Neuroscience. 291, 230–240. [DOI] [PubMed] [Google Scholar]
- Zhao ZY, Xie XJ, Li WH, Liu J, Chen Z, Zhang B, et al. , 2019. A cell-permeant mimetic of NMN activates SARM1 to produce cyclic ADP-ribose and induce non-apoptotic cell death. iScience. 15, 452–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziogas NK, Koliatsos VE, 2018. Primary traumatic Axonopathy in mice subjected to impact acceleration: a reappraisal of pathology and mechanisms with high-resolution anatomical methods. J. Neurosci 38 (16), 4031–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Xie L, Wang W, Zhao G, Tian X, Chen M, 2020. FK866 alleviates cerebral pyroptosis and inflammation mediated by Drp1 in a rat cardiopulmonary resuscitation model. Int. Immunopharmacol 89 (Pt A), 107032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.