

Abstract
Most enveloped viruses infect cells by binding receptors at the cell surface and undergo trafficking through the endocytic pathway to a compartment with the requisite conditions to trigger fusion with a host endosomal membrane. Broad categories of compartments in the endocytic pathway include early and late endosomes, which can be further categorized into subpopulations with differing rates of maturation and motility characteristics. Endocytic compartments have varying protein and lipid components, luminal ionic conditions and pH that provide uniquely hospitable environments for specific viruses to fuse. In order to characterize compartments that permit fusion, we studied the trafficking and fusion of viral particles pseudotyped with the vesicular stomatitis virus glycoprotein (VSV-G) on their surface and equipped with a novel pH sensor and a fluorescent content marker to measure pH, motion, and fusion at the single particle level in live cells. We found that the VSV-G particles fuse predominantly from more acidic and more motile endosomes, and that a significant fraction of particles is trafficked to more static and less acidic endosomes that do not support their fusion. Moreover, the fusion-supporting endosomes undergo directed motion.
Keywords: endocytosis, endosomes, organelle motility, viral trafficking, virus entry, fluorescent pH sensor, vesicular stomatitis
Graphical Abstract
Introduction
Viruses infect host cells after binding to host receptors at the cell surface. For most enveloped viruses, infection requires internalization and trafficking through the endocytic pathway. Receptor binding occurs via glycoproteins on the viral surface and conformational changes of these proteins mediate fusion between the viral and host membranes, leading to infection. Some viruses fuse early in the endocytic pathway, while others must be trafficked to late endosomes to encounter conditions required to trigger fusion. These may include processing by proteases, binding to endosomal receptors, as well as favorable conditions of pH, lipid composition, and ionic milieu.1 Late endosomes are more acidic than early endosomes, and endo-lysosomes and lysosomes are more acidic still. In addition, each progressively more acidic compartment in the pathway has its own lipid and protein components.2 In many cases endosomal pH is the primary fusion trigger, though in some cases there are other essential triggers.1 Viruses that fuse in late endosomes include influenza virus and lymphocytic choriomeningitis virus1,3. In contrast, Semliki forest virus4 and avian sarcoma and leukosis virus (ASLV) fuse in early endosomes.5
Early endosomes can be further distinguished as subpopulations with differing characteristics6,7 and rates of maturation.8 These subpopulations can be important for cargo sorting and fate. While some cargos preferentially sort into faster-maturing and more motile “dynamic” early endosomes, other cargos are preferentially sorted to more slowly maturing and less motile “static” early endosomes (nomenclature as per8). Influenza virus is preferentially sorted to a subset of motile endosomes.8 In contrast, ASLV non-preferentially sorts to static and dynamic endosomes in proportion to the abundance of those endosomes while reportedly preferentially fusing with less motile, slow-moving compartments in a cell line expressing TVA950, the transmembrane form of the ASLV receptor.5 Hence among viruses studied with respect to trafficking through subpopulations of endosomes, one virus (influenza) has been reported to preferentially sort to dynamic endosomes, while the other (ASLV) has been reported to preferentially fuse in static endosomes. This stark difference led us to ask whether particles bearing the glycoprotein of another virus, i.e., vesicular stomatitis virus (VSV), fuse in dynamic or static endosomes. We addressed this question by single particle fluorescence microscopy correlating fusion with the pH state and motility of endosomes in live cells. We also characterized the fate of non-fusing particles. Although we chose to study trafficking and fusion directed by the well characterized VSV glycoprotein9–11 as a model, the methods developed in this work should be applicable to study trafficking and fusion of other enveloped viruses at the single particle level in live cells.
VSV is an enveloped virus commonly used as a model for the study of the trafficking and fusion of enveloped viruses. It is a member of the family Rhabdoviridae, primarily causes acute illness in hoofed animals, and can cause mild symptoms in humans.12 Cell entry and fusion of VSV is mediated by its envelope glycoprotein G. VSV-G is a class III fusion protein known for undergoing reversible conformational changes.13,14 The primary host receptor for VSV is the low-density lipoprotein receptor (LDL-R),15,16 which has previously been found to sort into dynamic endosomes.8 After binding its receptor, VSV undergoes a mode of clathrin-mediated endocytosis (CME) requiring actin,17,18 is trafficked through the endocytic pathway encountering progressively lower pH, and is triggered to fuse with an endosomal membrane at low pH.19,20
To examine trafficking and fusion mediated by VSV-G and address the question of whether the virus fuses in dynamic or static endosomes, we use murine leukemia virus (MLV) pseudotyped with VSV-G. Our VSV-G pseudotype particles contain gag-mKate2 as a content marker to demonstrate full fusion and a membrane anchored FRET-based ratiometric pH-sensor to monitor changes in acidification as the virus particles are endocytosed and trafficked in live cells. This system allows us not only to correlate fusion with endosomal pH at the single particle level, but also to follow the velocity and directedness of motion of virus-containing endosomes. By using this approach, we describe differing characteristics in the populations of endosomes that support fusion events versus those that do not. Endosomes that are more motile and more readily acidified support VSV-G driven viral fusion events, whereas more static endosomes do not appear to support fusion of VSV-G pseudoviral particles.
Results
Calibration and characterization of a FRET-based pH sensor in viral particles
Previous work used a FRET-based pH sensor consisting of mTFP1 and eYFP as a chimera with the ICAM1 transmembrane domain (ICAM1-TMD) to monitor pH during the trafficking of MLV bearing the envelope protein from ASLV.5 The paper’s authors identified two key limitations of the mTFP1-eYFP pH sensor: the particles were not observable in the perinuclear region due to noise contributed by cellular autofluorescence exceeding the signal of the sensor, and the sensor could not reliably detect pH values below 5.2. To improve on these issues, we designed a new ratiometric pH sensor using mTFP1 and mCitrine as a FRET pair21 attached to ICAM1-TMD (Fig. 1A, B). mCitrine is approximately 1.5-fold brighter than eYFP and has a lower pKa of 5.7 compared to 6.9 of eYFP.22 Both mTFP1 and mCitrine are translated from a single open reading frame with ICAM1-TMD. The fluorescent proteins are thus expressed in a one-to-one ratio permitting the fluorescence intensity ratio of mTFP1 over mCitrine to be used as a pH indicator (Fig. 1B). The use of a ratiometric probe is key for live cell imaging because changes in focal plane affect both emissions wavelengths equally,23 permitting the measurement of pH even when the probe is on a moving object such as a viral particle or endosome. In unfused particles, ICAM1-TMD anchors mTFP1-mCitrine in the membrane of the viral particle topologically oriented toward the extracellular environment or, following endocytosis, into the endosomal lumen. Following fusion, the FRET pair is expected to sense endosomal pH via its luminal orientation with the TMD now embedded in the endosomal membrane.
Figure 1. Design and calibration of a FRET-based pH sensor to measure cellular trafficking and the pH of fusion of viruses in cells.
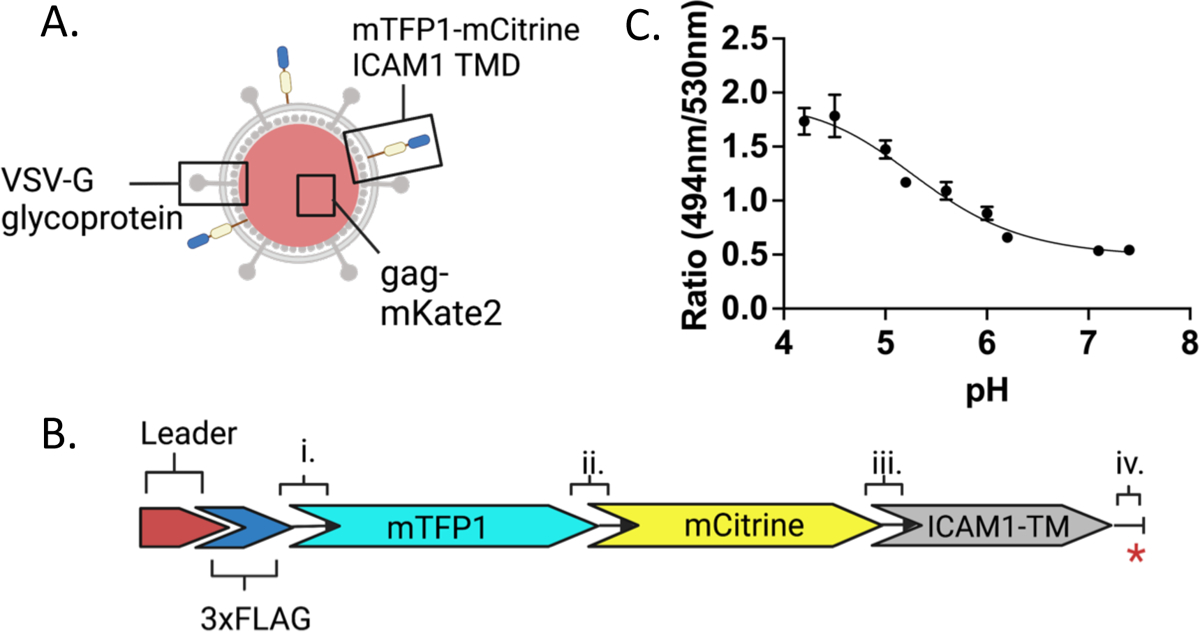
A. Schematic of an MLV particle pseudotyped with VSV-G glycoprotein and bearing a FRET-based pH sensor with mKate2 as a content marker. B. Schematic representation of the pH sensor construct. The pre-protrypsin leader sequence (Leader) is followed by 3xFLAG sequences, followed by the fluorescent protein and ICAM1 transmembrane domain sequences. Linkers between components are i., ii., iii., and iv. where iv. separates the first four amino acids of the ICAM1 cytoplasmic domain and the stop codon indicated by the red asterisk. The sequence of linker ii. between mTFP1 and mCitrine is RSTSLQEFGT. C. The pH sensitive mTFP1-mCitrine FRET pair on the pseudotyped viral particle (as shown in panel A) is calibrated with buffers of known pH on coverslips. The ratio of the fluorescence emission intensities at 494nm/530nm is used as a proxy for FRET efficiency and plotted as a function of pH. The error bars represent the standard errors of the mean (SEM). Numerical values of the mean and SEM and numbers of individual particles evaluated at each pH are listed in Supplemental Figure 1B.
MLV particles pseudotyped with VSV-G and containing the FRET pH-sensor were produced as described in Materials and Methods. The particles were also labelled with a content marker, Gag-mKate2, which is cleaved to Gag and mKate2 during capsid maturation.24 To calibrate the sensor, particles were deposited on a poly-lysine coated glass coverslip and imaged in citrate-phosphate buffers of known pH. Individual, triply labelled particles were selected and the mean emission intensities from 8.9 nm wide spectral bands were extracted from each particle region. Particle regions were background subtracted and the emission intensity ratios of mTFP1 to mCitrine (I494.0–502.9 /I530.0–538.9) were calculated for each pH. The sensor fluorescence ratio undergoes a transition from a high to a low FRET state as the pH is decreased from 7.4 to 4.2 (Fig. 1C). Based on the measured fluorescence, the FRET sensor is well-equipped to distinguish pH in the range of 4.5–6.2 but is less discriminating between pH 6.2 and 7.4. At very low pH (4.2–4.5) there is high particle-particle variability in fluorescence ratio (Supp. Fig. 1A, B). When buffers of decreasing pH were applied sequentially, most (10/13) individual virus particles’ fluorescence ratio responses increased with decreasing pH as expected. However, three of 13 particles were unresponsive or displayed a scattered fluorescence ratio response (Supp. Fig. 2A, B). We do not know why the fluorescence of a few particles displayed these unexpected pH dependencies, but one explanation could be that one or the other fluorescent protein is not properly folded or their folding is differently sensitive to pH in these outlier particles.
To assess the performance of the pH sensor over repeated exposures and the variation of the fluorescence ratio for a single particle on a frame-to-frame basis, virus particles were imaged on a coverslip for 16 minutes in neutral live cell imaging buffer before imaging was paused, a buffer of pH 5.2 was introduced, and imaging was continued (Supp. Fig. 2C). While some particles could be observed for many minutes after the pH change, several particles were visible only briefly after the introduction of low pH buffer or were no longer visible at all. As expected, the fluorescence ratio increased with the addition of low pH buffer for all particles still observed after buffer replacement (Supp. Fig 2C). The mean values (Supp. Fig. 2D) and standard deviations (Supp. Fig. 2E) of the fluorescence ratios over the first six frames (in pH 7.4 buffer) were calculated for each of these particles. The median value of the mean ratio for all particles over the first six frames was 0.58, which corresponds according to the calibration curve of Fig. 1C to pH 6.8. The median value of the standard deviation was 0.045, which is equivalent to a range of pH 6.6–7.2 around the median initial ratio of all particles, slightly below the pH 7.4 buffer surrounding the particles. Once the buffer was shifted to pH 5.2, the median fluorescence ratio value of the mean fluorescence ratio for all particles in the six subsequent frames was 1.38, which corresponds to a pH of 5.1 according to the calibration curve. This is approximately the same as the pH 5.2 buffer surrounding the particles, further verifying the accurate readout of the sensor at low pH.
Visualization, pH dependence and time-course of fusion of VSV-G mediated fusion events
Having established the characteristics of the new FRET-based pH sensor in isolated viral particles, we used this sensor to track the measured pH (as calculated from the calibration curve in Figure 1C) of endosomes harboring VSV-G pseudotyped viruses and their eventual fusion by live cell imaging. Parameters to describe these endosomes included their pH, changes in pH, and their displacement and velocity of motion within the cell. Pseudoviral particles were spun onto A549 cells in the cold, a field of view was selected for imaging, and the cold buffer was replaced with warm (37° C) imaging buffer immediately prior to commencing imaging, which was conducted at 37° C. Fusion events were identified by a sudden drop of the mKate2 signal while the mTFP1 and mCitrine signals persisted, as indicated in the representative example shown in Fig. 2A by the magenta arrow at 28.3 minutes. Fusion events, initially assessed by direct visualization, were confirmed by quantitation of the raw and background subtracted signals of all three channels (Fig. 2B, 2C) as described in the Materials and Methods. The displacement and velocity of the particle shown in panel A were plotted as a function of time in Figs. 2D and 2E, respectively. The displacement and velocity results from this example particle, as well as from many other particles will be described in a later section.
Figure 2. Progression of an individual example virus particle from the start of imaging to fusion and beyond simultaneously observed with three fluorescence channels.
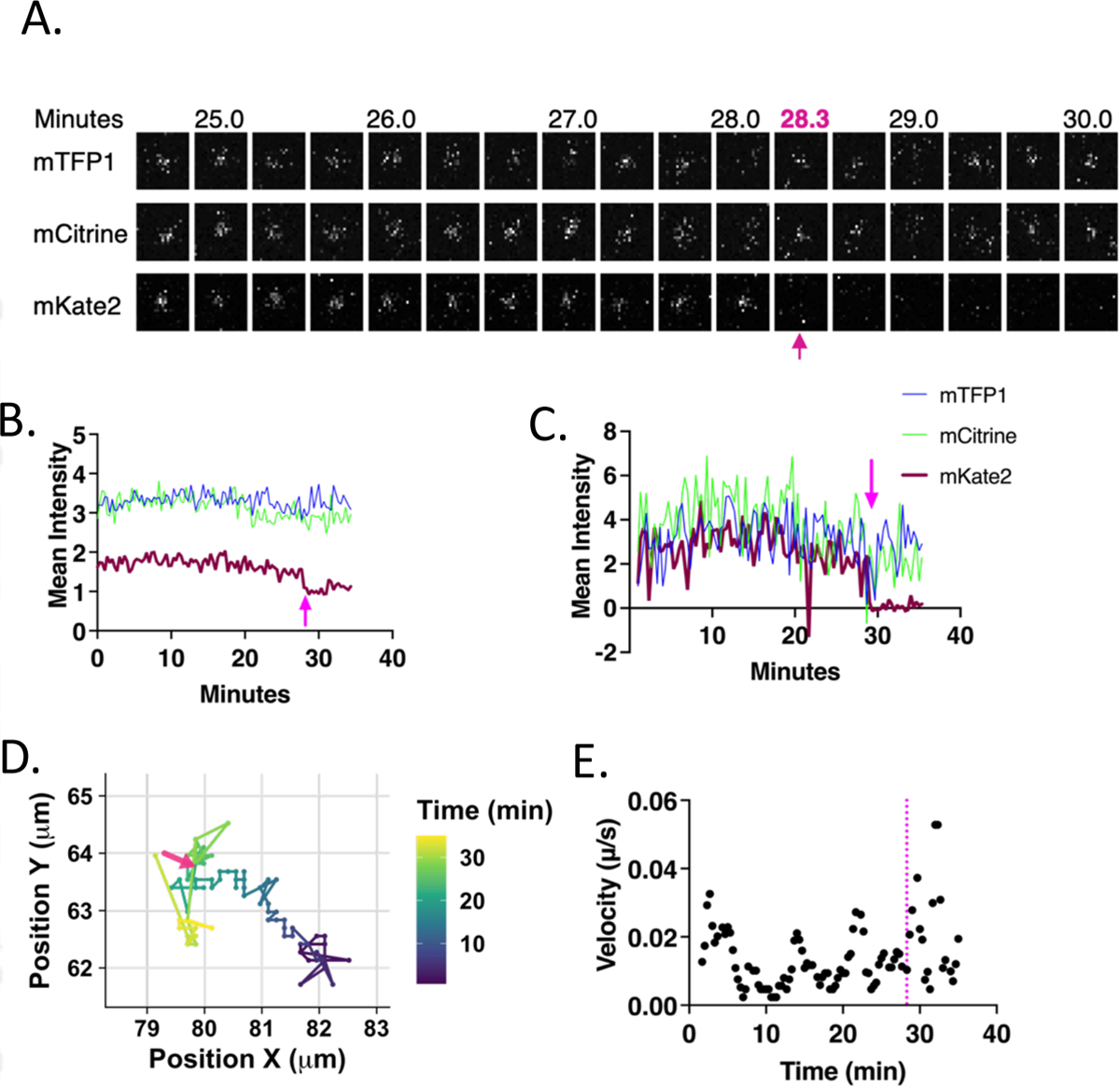
A. Images of the particle in the mTFP1 (494–511nm) (top), mCitrine (521–538nm) (middle), and mKate2 (593–741nm) (bottom) channels. Images are 20s apart and the track is cropped to 10 frames before and 5 frames after fusion. The size of each image is 2.94 × 2.94 μm, centered around the particle centroid determined by tracking. In all images the arrow indicates the time at which fusion ends. B. Raw extracted intensity from a 2.942 μm2 box surrounding the particle centroid and plotted over time. C. Background subtracted intensity using a 1.96 μm diameter inner circle with an outer 0.28 μm diameter annular region from which the mean intensity was calculated and subtracted from the intensity of the inner region. D. Path of the particle over time within a boxed region from the total field of view. The color of the line represents time according to the scale on the right. The arrow indicates the position of the particle at the time that fusion is completed. E. Velocity was calculated by taking the distance between the localization of the particles at adjacent points in time and multiplying by the frame rate (1/20s). A 3-frame (one minute) rolling average of the velocities was taken and plotted as a function of time. The dotted line indicates the time at which fusion is completed.
For each particle undergoing fusion, the ratio of the mTFP1 to mCitrine fluorescence intensity was extracted and plotted at 20 second intervals, the frame rate of imaging, over all frames in which the particle was observed (Fig. 3A). The time of fusion, determined as the time of disappearance of the mKate2 signal (Fig. 2A, 2C), is shown as a vertical dashed line in Fig. 3A. Several interesting phenomena of change in fluorescence ratio were observed immediately prior to or following fusion events. In several examples, the pH sensor signal disappeared or experienced significant frame to frame variation within five minutes following fusion (Fig. 3A: examples ii., iv., v., ix., xii., and xiv.). This is similar to the phenomenon observed when virus particles were rapidly acidified on a coverslip (Supp. Fig. 2C) and may be due to loss of signal from the FRET donor and acceptor, perhaps caused by quenching of the signal due to protonation of the chromophore or irreversible protein unfolding at very low pH (less than pH 4.2). Several fusion events occurred during periods of increasing fluorescence ratio indicating acidification (Fig. 3A: examples iii., vi., vii., and xviii.). Even transient shifts to lower pH may trigger viral content release. A decrease in ratio (alkalization) immediately follows fusion (Fig. 3A: examples vi., vii., x., xi., xiii.) for five of the fusion events. Temporary alkalization of endosomes following fusion could be caused by a leaky fusion event in which there is a loss of contiguity in the endosomal membrane25 allowing the permeation of cytosolic contents into the endosome.
Figure 3. Kinetics and pH characteristics of VSV-G mediated viral particle trafficking and fusion.
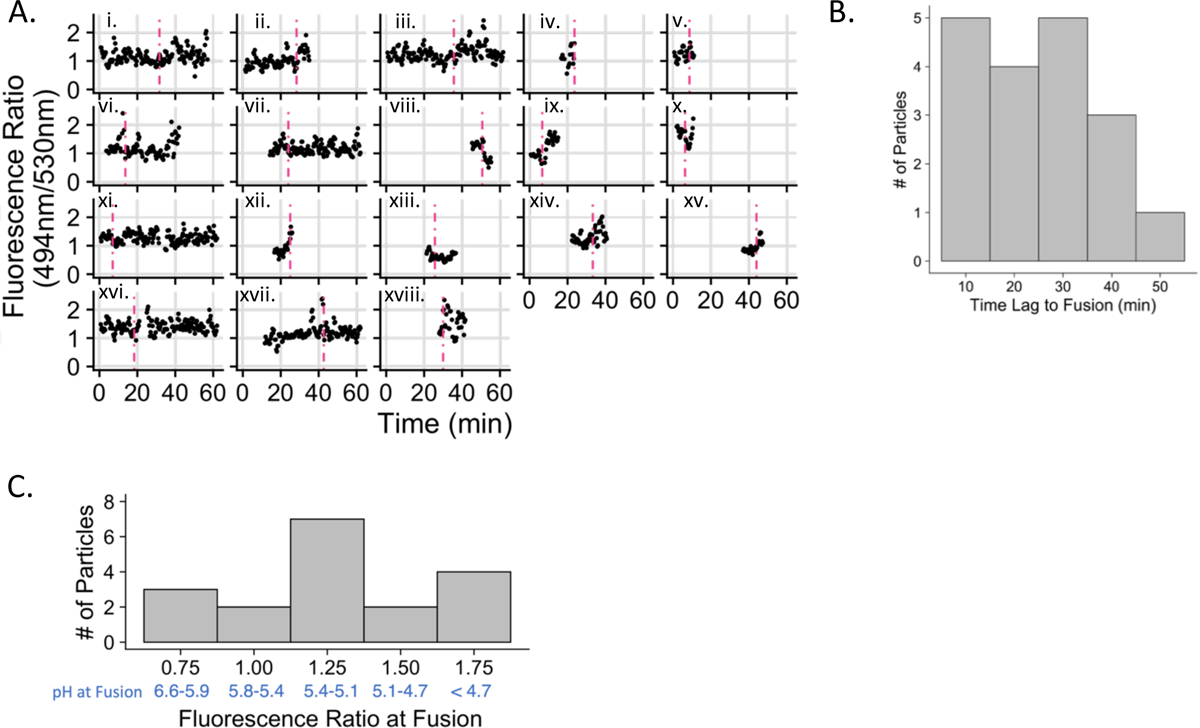
A. Fluorescence emission intensity ratio plotted over time for all particles that fuse. The dashed vertical line represents the time of fusion as determined by the spreading or disappearance of the mKate2 signal. B. Frequency histogram of the time from the beginning of imaging to the time at which fusion occurs. C. Frequency histogram of the 494nm/530nm emission intensity ratios at the time of fusion using a six-frame moving average. The pH range falling within each histogram bin is shown in blue; fluorescence ratios were converted to pH range using the calibration curve (shown in Figure 1C). 18 fusion events were analyzed (out of 88 particles meeting the criteria described in the Materials and Methods) and are represented here (n=18).
The mean time of fusion (content release), counted from the start of imaging and derived from the 18 events shown in Fig. 3A, is distributed primarily in observation times less than 45 minutes with a mean value of 25.2 min (Fig. 3B). The mean fluorescence ratio at fusion was 1.24 +/− 0.08 (SEM), which is the equivalent of pH 5.3 (pH 5.2–5.4). The total range of pH values measured at viral fusion extended from a fluorescence ratio of 0.67 (pH 6.4) to 1.78 (pH 4.2) (Fig. 3C).
For virus particles encountering progressively more acidic environments as they move along the endocytic pathway, the fluorescence ratio is expected to increase. For 8 of the 18 fusing particles, the change in fluorescence ratio from the first time the particle is observed to the time of fusion was positive as expected, with a mean increase of 0.17 +/− 0.77 over all particles observed (Supp. Fig. 3A). Five of the 18 fusing particles had very small fluorescence ratio changes that fell within the expected frame-to-frame variability of the probe (2 SD of frame-to-frame ratio difference for single particles; Supp. Fig. 2E) and thus cannot be interpreted as having a meaningful change in pH during the observed time before they fuse (Supp. Fig. 3A). Five of the 18 fusing particles demonstrated increasing pH between the time the particle was first observed and the time of fusion. This latter result may both reflect physiologic fluctuations in the pH in the endosome and the possibility that observation of some particles may have only commenced soon before fusion (Fig. 3A), when the particle may already have been in a moderately acidic environment.
Rapid fluctuations in pH during the trafficking of fusing virus particles were a particularly interesting phenomenon observed in several cases (Fig. 3A: examples i., iii., xvii.). Similar fluctuations have been observed during trafficking of influenza virus8,26 but were not observed to the same extent during the trafficking of ASLV.5 For trace i. in Fig. 3A, the measured pH decreases from 5.7 to a local minimum of under 4.2 at a rate of at least −0.4 pH units per minute during the four minutes immediately following fusion. The pH is rapidly restored to a calibrated pH of 5.6 at the rate of at least 0.6 pH units per minute during the next two minutes (Supp. Fig. 3B). This pattern of rapid acidification followed by alkalization is also seen in trace xvii. in Fig. 3A and is quantified in Supp. Fig. 3C. Fluctuations in pH over the course of minutes may reflect proton leak variability over time27,28 or other transient changes in endosomes. The very rapid rates of acidification following alkalization are consistent with the previously observed general kinetics of V-ATPase when reacidifying endosomes.28
Comparison of acidification and properties of motion for fusion permissive and non-permissive endosomes
We next compared the features of fusion permissive endosomes and endosomes that did not permit fusion. The FRET-based pH sensor (Fig. 1A, B) is topologically oriented toward the extracellular/luminal space both while embedded in the viral membrane during trafficking and in the endosomal membrane after fusion, permitting measurement of pH during viral trafficking as well as during and following fusion. Fusion events were identified as described in Figure 2 and in the Materials and Methods in 13 separate experiments conducted on seven separate days. All monodisperse, triple labelled virus particles (mTFP1, mCitrine, gag-mKate2) that were observable for at least 20 frames (~7 minutes) were identified in experiments containing at least one fusion event. Of 88 total particles that were detected and tracked, 18 underwent fusion corresponding to a fusion efficiency of 20.4% for qualified triply labelled particles. We empirically observed a number of significant differences between fusion permissive and fusion non-permissive endosomes.
Fusion permissive endosomes display a higher maximum degree of acidification than fusion non-permissive endosomes. Fusion permissive endosomes achieve a median minimum pH of 4.2 compared to a median minimum pH of 5.3 in fusion non-permissive endosomes (Fig. 4A); fluorescence ratios were converted to pH values using the calibration curve of Fig. 1C. The pH sensor can best differentiate between pHs in the range of 4.5–6.2 and the upper plateau of the fit for the calibration curve is at pH 4.2, meaning that values at or below 4.2 represent highly acidic values and cannot be interpreted precisely. Despite the greater acidification of fusion permissive endosomes, both categories of endosomes had similar pH values when they were first observed (pH ~5.5, Fig. 4B), and similar values for the most alkaline pH they ever experienced during the whole observation period (pH ~6, Fig. 4C). Our observation that most virus particles, regardless of their ability to fuse, appear to start in endosomes with calibrated pH values below seven may be indicative of an initial rapid trafficking period into mildly acidic early endosomes during the one to two minutes required to refocus the microscope after the addition of warm buffer. Prior work found that up to 50% of ASLV viral particles reach a compartment of pH 6.2 or lower within two minutes of the initiation of viral trafficking.5
Figure 4. Fusion permissive and fusion non-permissive endosomes exhibit different extents of acidification.
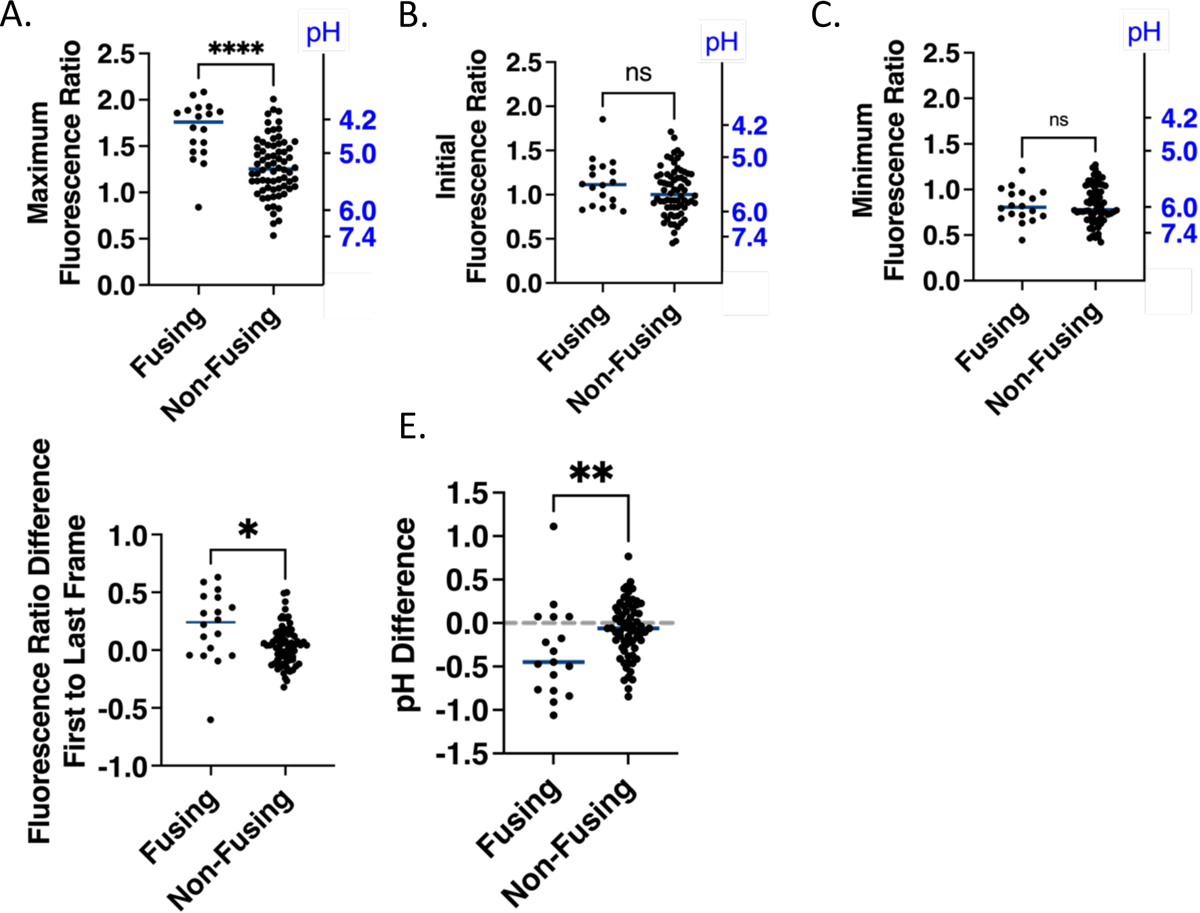
A. Maximum emission intensity ratios (494nm/530nm) achieved in tracks based on a six-frame rolling average for fusion permissive and fusion non-permissive endosomes. The right Y axis displays reference pH values. (Note that the relationship between pH and ratio is not linear; see Figure 1C). All selected particles were non-overlapping with other particles, had apparent diameters less than ~1.5 μm, and were triple labelled. Non-fusing particles are from experiments, in which fusion events were observed in other locations. B. Initial emission intensity ratios over the first 6 observable frames for fusion permissive and fusion non-permissive endosomes. C. Minimum emission intensity ratios in tracks based on a six-frame rolling average for fusion permissive and fusion non-permissive endosomes. D. Difference between emission intensity ratios at the time at which the particle is first observable (start) and the time at which the pH sensor from the particle is last observable (end) for fusion permissive and fusion non-permissive endosomes. E. The initial and final emission intensity ratios from panel D converted to pH using the fit of the calibration curve (Figure 1C) and plotted as pH differences over the observable time for fusion permissive and fusion non-permissive endosomes. No change in pH is indicated with a horizontal dashed line. In all panels, points represent the values for individual particles and horizontal lines represent the median value of each group. Numbers of fusing particles and non-fusing particles are n=18 and n=70, respectively. A Mann-Whitney test (non-parametric assumption) was used to assess the significance of the differences in the distribution of fluorescence ratio values (A-D) or pH (E) (p<0.05 = *, p<0.01 =**, p<0.0001=****, ns, not significant). In E, observations where the initial and final values fell above or below the lower and upper limits of the calibration curve fit were excluded from analysis and the numbers are n=17 for fusing and n=68 for non-fusing particles.
We also calculated the fluorescence ratio difference over the entire time the FRET pair could be observed in a given endosome. According to this analysis, fusion permissive endosomes became more acidified from their starting state than fusion non-permissive endosomes. The median ratio changes were +0.24 and +0.01 units, respectively (Fig. 4D). The difference between the initial and final pH was −0.4 units for fusion permissive and −0.1 units for fusion non-permissive endosomes (Fig. 4E).
Fusion permissive and non-permissive endosomes displayed different displacements and velocities of displacement during the observed trafficking times in the cell. Velocities were measured from particle localizations in consecutive frames and a three-frame (one minute) rolling average was taken for each time point. While the motions of both fusion permissive and non-permissive endosomes were mostly slow and included stationary segments, leading to median velocities of all segments in a track of ~0.01 μm/s (Fig. 5A), the fastest steps in fusion permissive endosomes had higher velocities (~0.05 μm/s) compared to those observed in non-permissive endosomes (~0.01 μm/s) (Fig. 5B). The total displacement, defined by the distance traveled between the coordinate points at which the particle was first and last observed, was determined for both subpopulations of endosomes. The median total displacement of fusion permissive endosomes was higher than that of non-permissive endosomes (Fig. 5C). This result is somewhat surprising in light of the similar median velocities of the two subpopulations of endosomes, but suggests that the high maximum velocities found in fusion permissive endosomes make an outsized contribution to their total displacement. Finally, FRET signals from tracks of fusion permissive and non-permissive endosomes could be observed for a similar length of time (Fig. 5D), demonstrating that the difference in total displacement and maximum velocity did not depend on the total time for which the particle was observed.
Figure 5. Fusion permissive endosomes have higher maximum velocities and travel further than fusion non-permissive endosomes.
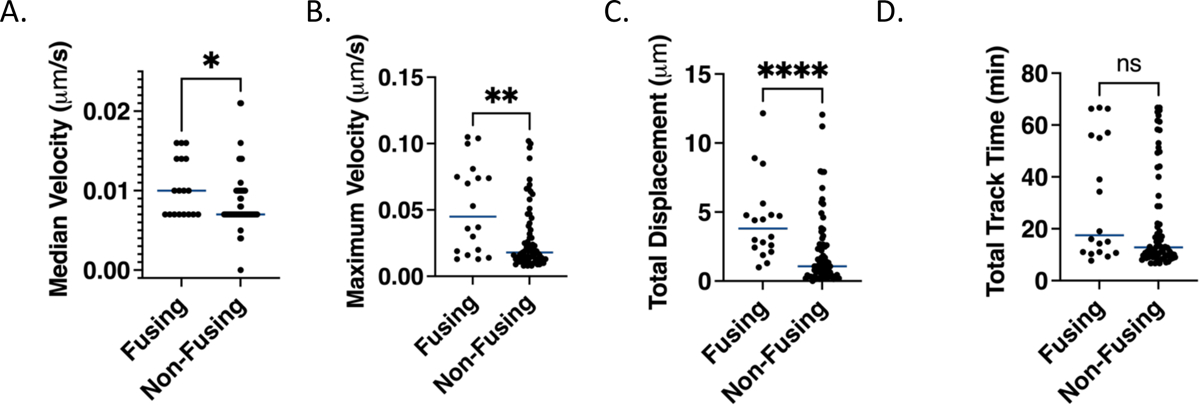
A. Median velocities from all segments in a given track for fusion permissive and fusion non-permissive endosomes. The median value for each endosome is calculated based on a 3-frame (1 minute) rolling average of the velocities of all segments in a given track. B. Maximum velocities determined as the highest velocity of for any segment based on a 3-frame rolling average as in A for fusion permissive and fusion non-permissive endosomes. C. Total displacements calculated from the distance between the coordinate points at which the FRET sensor signal is first and last observed for fusion permissive and fusion non-permissive endosomes. D. Total track lengths, i.e. times that the FRET sensor signal was observable calculated as the time difference between when particles are first and last observed for fusion permissive and fusion non-permissive endosomes. In all panels, points represent the values for individual particles and the horizontal lines represent the medians. A Mann-Whitney test (non-parametric assumption) was used to assess the significance of the differences in the distribution of all values (p<0.05 = *, p<0.01 =**, p<0.0001=****, ns, not significant).
Mean squared displacement and trajectory analysis of fusion permissive and non-permissive endosomes
Given the different maximum velocities and total displacements between fusion permissive and non-permissive endosomes, a mean-squared displacement (MSD) analysis was conducted to determine whether the two subpopulations of endosomes exhibit different modes of motion. We used plots of MSD versus time to distinguish between random (diffusive) and directed motion.29,30 To facilitate this analysis, we took a randomly chosen 18-member subset from the non-fusion permissive endosomes such that equal numbers of the two subpopulations of endosomes were analyzed. The MSD (μm2) at each time step up to 400 seconds (6.7 minutes) was calculated and plotted for the 18 fusion permissive and 18 non-permissive endosomes (Supp. Fig. 4A, B). For better visualization, we also averaged the MSD values for all 18 tracks in each set at each time lag and plotted the averaged MSD at each time lag (Fig. 6A). This averaged MSD plot is dramatically different for fusion permissive and non-permissive endosomes, with fusion permissive-endosomes showing an upwards bending curve indicative of directed motion, while the fusion non-permissive endosomes exhibited a more linear MSD vs. time relationship indicating free (Brownian) diffusion (Fig. 6A). As expected, there is higher variability at longer time steps since these are derived from fewer measurements.29 The same trend observed in the averaged data can be seen in many curves of individual endosomes (Supp. Fig. 4A, B).
Figure 6. Motion analysis reveals behavior consistent with directed motion in fusion-permissive endosomes.
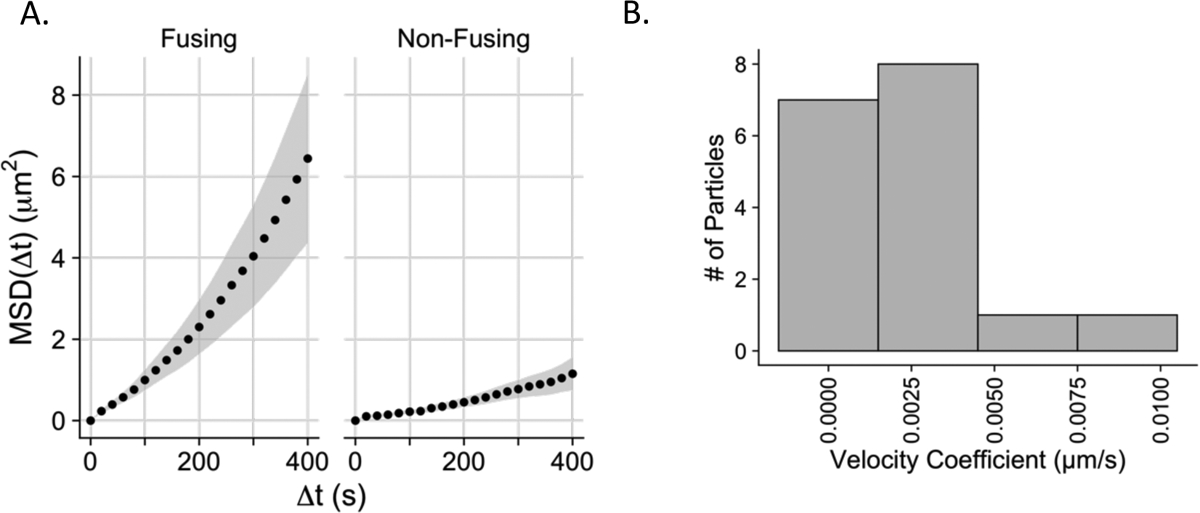
A. Mean square displacements (MSD) (μm2) plotted vs. time step Δt for endosomes harboring fusing and non-fusing virus particles. Each data point represents the mean MSD of 18 endosomes containing fusing or non-fusing particles respectively, at each time step. The grey envelopes represent the standard errors of the mean for all particles in the plotted data set at Δt. B. Distribution of velocities (v) of directed motion of fusing virus particles. To test for a component of directed motion, the MSD of fusing particles was fit with Equation 1 in Materials and Methods. Time steps up to 60% of the total length of the track were fit. 17/18 of the fusing particle MSD curves could be fit with this model; one curve had a decrease in MSD values at higher values of Δt such that it could not be fit with Equation 1.
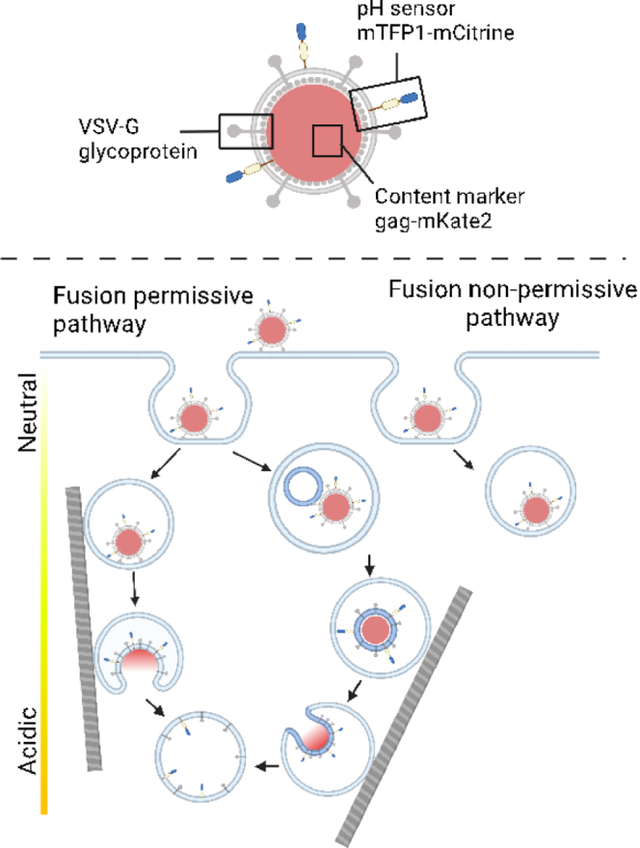
The MSD data from 17 out of 18 fusion tracks could be reasonably fit with a model for directed motion (Equation 1 in Materials and Methods) with the velocity coefficient v as one of the fitting parameters. The particles were binned according to their velocity coefficients and moved with an average velocity coefficient of 0.0027 μm/s (Fig. 6B). The velocity coefficients plotted for the fusion-permissive endosomes are consistent with previously published values for slow directed motion of endosomes containing influenza virus along microtubules or microfilaments.31 The difference in characteristics of motion and acidification between fusion permissive and non-permissive endosomes are indicative of two distinctive populations of endosomes, one of which undergoes acidification and trafficking, and one of which is relatively static on the time-scale of trafficking and fusion of MLV particles bearing VSV-G (Figure 7).
Figure 7. Summary of characteristics for fusion permissive and fusion non-permissive endosomes with a model for viral particle trafficking during cell entry.
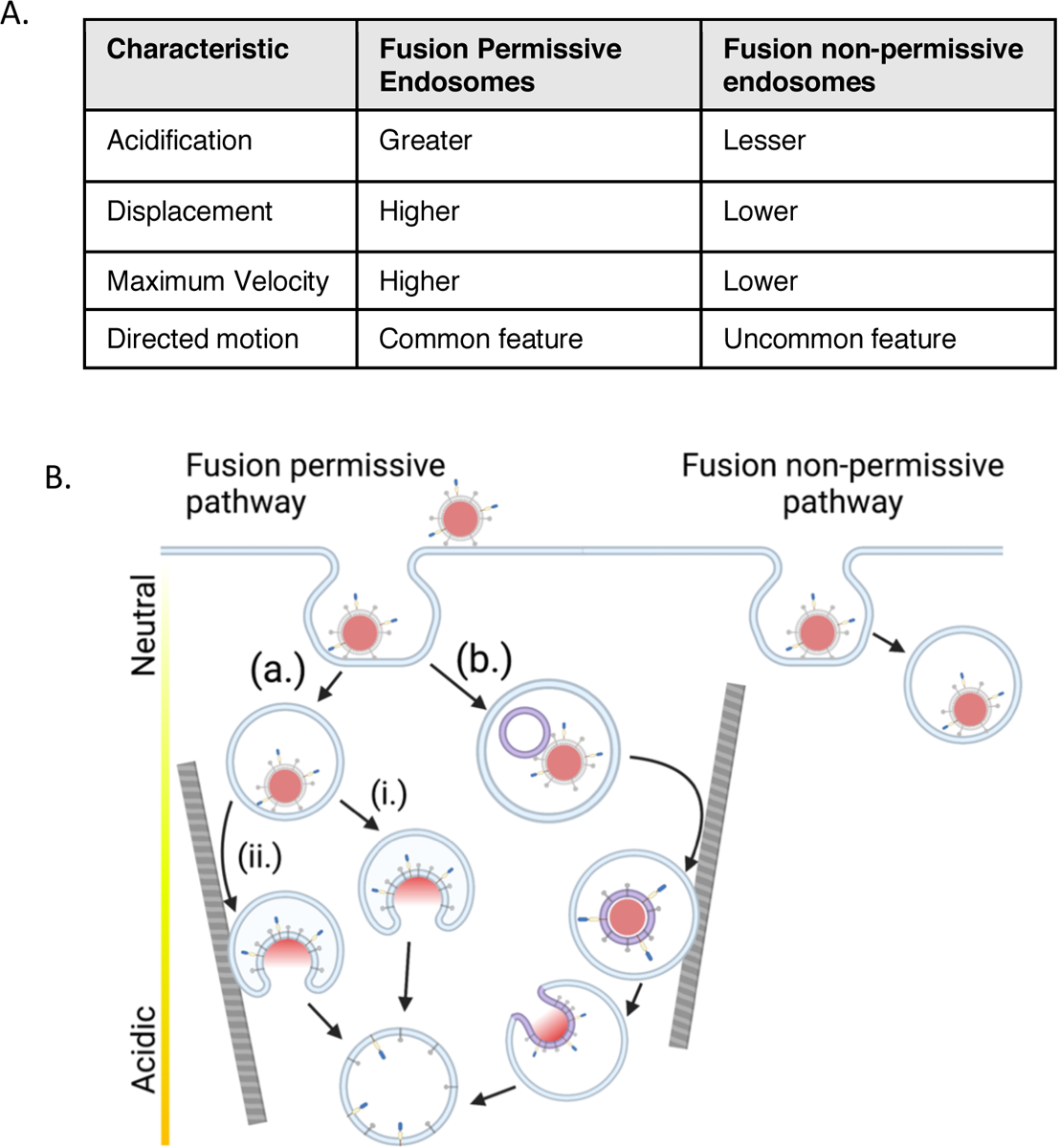
A. Summary of characteristics of fusion permissive and non-permissive endosomes based on the single virus particle tracking and fusion data presented in Figures 4–6. B. Model of trafficking of VSV-G pseudotyped viral particles in fusion permissive and non-permissive endosomes. In this model, fusion is associated with the maturation of the subpopulation of fusion permissive endosomes, many of which also undergo directed motion, likely along elements of the cytoskeleton. The sorting of particles to fusion permissive or non-permissive endosomes does not appear to be preferential (see Discussion). Three fusion scenarios are displayed: In path (a) the virus fuses directly with the limiting endosomal membrane; in path (i) the virus fuses prior to directed motion of the endosome or in an endosome that does not undergo directed motion; in path (ii) the virus fuses after the endosome containing it undergoes directed motion. In path (b) the virus undergoes fusion with an intralumenal vesicle, which then undergoes retrofusion with the limiting membrane of the endosome in a more acidic compartment. The ILV-containing compartment also may or may not undergo directed motion prior to fusion. Components of the viral particle are as represented in Figure 1A.
Discussion
In this study, we have investigated the fusion of single virus particles mediated by the surface glycoprotein VSV-G in whole, live A549 cells and discovered that two subpopulations of endosomes with different characteristics of acidification and intracellular motion play a role in the fate of individual viral particles. Endosomes that did support fusion of internalized viruses were more acidic, more motile and underwent directed motion to a greater extent than endosomes that did not support fusion (Fig. 7A). Based on our findings, we propose a model whereby the sorting of viral particles into more dynamic endosomes disposes particles to undergo fusion; conversely the sorting of virus particles to a more static subpopulation of endosomes traps these particles in locations where they are strongly disinclined to fuse (Fig. 7B).
Distinctive pools of endosomes and viral fusion
The presence of different pools of early endosomes has previously been established,6,7,32 with some less motile early endosomes maturing more slowly, as defined by a longer time lag to the acquisition of Rab7, and some more motile early endosomes maturing more rapidly.8 Maintenance of these separate populations of dynamic and static early endosomes is dependent on intact microtubules and the early endosomal tethering complex CORVET, while sorting of cargo into subpopulations relies on differential clustering of the cargo at the cell surface.6,8 The significance of these separate pools to viral infectivity has only begun to be studied, with differing results as to whether viruses are more likely to be sorted to or fuse from dynamic or static early endosomes.5,8 We found that viral particles pseudotyped with VSV-G had a fusion efficiency (as indicated by content release) of approximately 20%. Fusion permissive endosomes moved a median 3.8 μm during their entire observation times (Fig. 5C) with a mean velocity of 0.16 μm/min (Fig. 6B). They also decreased their pH on average by −0.4 units during the entire observation time (Fig. 4E) and reached an average pH of 5.3 immediately before fusion (Fig. 4C). Most of the remaining 80% of particles, consisting of the non-fusing subset, were contained in endosomes that had a lower motility and lower overall acidification than endosomes bearing particles that fused. For comparison, endosomes bearing virus particles that did not fuse moved on the median only 1.0 μm (Fig. 5C), moved diffusively with no discernible component of directed motion (Fig. 6A), and lowered their pH on average only by −0.1 units during the entire observation time (Fig. 4E). Only 10 of the 70 fusion non-permissive endosomes achieved a change in pH above the median change in pH for fusion-permissive endosomes (Fig. 4E).
The sorting of viruses and physiological cargoes to subpopulations of early endosomes may rely on a number of factors including cargo size and qualities of the receptor. The static subpopulation of endosomes is in the majority (65%) and the minority of endosomes are dynamic (35%) in BSC-1 cells. In the same cells, influenza virus is preferentially sorted to the dynamic endosome subpopulation as compared to static endosomes. When bound with its endogenous ligand, LDL, LDL-R, which also serves as the VSV-G receptor, is preferentially sorted to dynamic endosomes as well.8 In contrast to influenza virus, ASLV pseudotyped viral particles were found to sort predominately (70%) to less motile endosomes in CV-1 cells expressing the transmembrane receptor for ASLV, TVA950.5 The 70% of ASLV sorted to less motile endosomes is consistent with the overall proportion (65%) of static endosomes.8 This suggests that ASLV is sorted to static and dynamic subpopulations in proportion to the abundance of each endosome subpopulation, without preference for either subpopulation. In our current study, VSV-G pseudotyped particles are also sorted without a clear preference for dynamic endosomes.
The indiscriminate sorting of VSV-G pseudotyped particles as compared to the preferential sorting of LDL-bound LDL-R to dynamic endosomes may be due to the smaller size of LDL compared to MLV particles. MLV particles are approximately five times larger in diameter than LDL, the physiological cargo for LDL-R.33,34 The importance of cargo and viral particle size with respect to trafficking has previously been demonstrated. VSV particles required an actin dependent mechanism of clathrin-mediated endocytosis as compared to small, defective viral particles that were otherwise similar.18 The MLV particles bearing VSV-G used in the present study are more spheroid in comparison to the bullet shape of VSV. They are ~25% smaller in length than the maximum dimension of VSV particles, but they are almost twice as wide33,35, suggesting that they, like VSV, are large enough to potentially undergo clathrin-mediated endocytosis in a different manner than relatively small cargo such as LDL. Smaller particle size has also been shown to have important downstream effects in motility; smaller cargo are more likely to undergo fast directed motion.36 Binding avidity for ligands to receptors has also previously been shown to affect cargo fate.37 Therefore, it is possible that the number of glycoproteins on a single viral particle that bind to multiple LDL-Rs in a single clathrin coated pit could also determine to which type of endosome the internalized virus is directed.
In general accordance with our findings that fusion non-permissive endosomes did not achieve the same extent of acidification as fusion permissive endosomes, endosomes containing ASLV particles that failed to fuse showed a pH distribution shifted toward more neutral pH values compared to fusing particles.5 However, while ASLV was observed to undergo an initial period of acidification to a pH of approximately 6.2–6.0 and then stay close to that pH, we observed continued further acidification as well as significant physiological fluctuations in pH in endosomes bearing VSV-G viral particles (Fig. 3A, 4C–E). One limitation of the prior study, as identified by the authors, with respect to measuring pH during trafficking of ASLV particles was difficulty in observing pH values below 6.0 due to cellular autofluorescence and, potentially, the higher pKa of the pH sensor used in that study.5 We did not observe significant cellular autofluorescence and utilized a FRET acceptor fluorescent protein that was 1.5-fold brighter than the acceptor protein used in the previous work. Moreover, mCitrine, used in our work as the acceptor fluorescent protein, has a pKa of 5.7,22 permitting measurements down to pH 4.2 in live cell imaging.
Determining the compartment identity of both fusion permissive and non-permissive populations of endosomes would be fertile ground for future study. While the current experimental system has the advantage of a ratiometric pH probe in combination with a content marker, allowing the measurement of both pH and content release, the combination of fluorophores occupies a broad spectral range (approximately 475–750 nm emission wavelength). Recently developed near infrared fluorescent proteins38 may offer an exciting opportunity to express fluorescent chimeras of compartment markers (such as APPL1, EEA1, Rab5, Rab7 or Rab11) that will not interfere with imaging of the FRET-based pH probe and viral content marker described in this study. One intriguing possibility is that non-fusion permissive endosomes recycle viral particles to the cell surface, a phenomenon recently described for Zika and influenza viruses,39,40 thus providing an additional layer of defense to the host cell.
Characteristics of VSV-G mediated fusion
Viral internalization and trafficking through the endocytic pathway are key requirements for productive fusion and infection by most enveloped viruses.1 We identified individual full fusion events based on content release of mKate2 (Fig. 2A–C, Fig. 3A). By simultaneously tracking the pH sensors on individual particles prior to, during, and following fusion, we were able to quantify pH changes as well as the pH at which full fusion occurred. The mean lag time from initiating trafficking via addition of warm medium to fusion was 25 minutes for the 18 fusing particles observed (Fig. 3B). Although the time course of fusion for viral particles may depend on cell type and viral model, it is interesting that our results are substantially similar to at least three previously published studies, i.e., similar values of approximately 20–35 minutes were obtained in bulk assays for half-maximal infection or fusion of viral particles bearing VSV-G41–43. This indicates that the FRET-based pH sensor does not appear to significantly change the kinetics of trafficking and/or fusion. The heterogeneity of the time course of fusion seen in our single particle experiments likely not only reflects biological variabilities of individual particles, but likely also reflects the heterogeneity of maturation of the endosomes harboring the viral particles, leading to a quite broad range of times to achieve fusion.
To our knowledge, this is the first single-particle measurement of the pH of VSV-G mediated full fusion in intact cells. Our data suggest that most full fusion and content release events occur at a mean pH ranging from 5.2 to 5.4 (Fig. 3C) in whole cells. Interaction of VSV-G with liposomes, reflecting an early stage of fusion, requires a pH trigger reported to be anywhere from approximately 4.0 to 6.011,44 although the biggest structural changes in the VSV-G protein occurs between pH 5.5 and pH 6.5.11 Cell-cell fusion mediated by G protein expressed in the plasma membrane showed half maximal fusion at approximately pH 6.0–6.2,19,20 though the extent of cell-cell fusion has been demonstrated to be greater at pH 5.5.45 The efficiency of VSV fusion (lipid mixing) with a supported lipid bilayer is also greatest at pH 5.5.14 Differences in the target membrane with which VSV-G fuses and ionic conditions in the endosome likely influence the fusion behavior of internalized viruses and environmental conditions in the endosome may differ significantly from these earlier model systems used to study membrane binding and fusion.
Previous findings suggest that VSV-G mediated virus entry includes an intermediate step of fusion whereby viral particles first fuse with intraluminal vesicles (ILVs) within endosomes.43 The endosomes containing these virus-bearing ILVs then mature, acidifying further, and acquiring lysobisphosphatidic acid (LBPA). In the presence of the appropriate lipid composition and pH, ILVs then undergo retrofusion with the limiting membrane enabling the release of viral contents in the cytoplasm43,42 (Fig. 7B). The findings that VSV-G may fuse with intraluminal vesicles is further supported by the fact that anionic lipids, such as LBPA, promote VSV-G content release46,47 and these lipids are potentially enriched in ILVs.48 The lower average pH of content release determined in our experiments compared to the interaction of VSV-G with plasma membranes (e.g., for cell-cell fusion) reported at higher pH could be explained by the proposed mechanism of viral content release requiring ILV retrofusion in the late endosome. Many of the content release events we observe may therefore represent retrofusion events that may require a lower pH than the initial fusion with ILVs and are known to depend on the lipid composition of the late endosomes.43,49,50 The small number (3/18) of content release events we observe at greater than pH 6.0 (Fig. 3C) may represent direct virus fusion with the limiting membrane of the endosome. It is possible content release via retrofusion is the predominate mechanism while fusion with the limiting membrane only predominates when the retrofusion mechanism has been blocked in some way or is not advantageous within a given cell or endosome. This is supported by data showing that content release is markedly less efficient when ILVs cannot be formed or trafficked, but that some content release still occurs under these conditions.3,43
General considerations of FRET-based pH sensors to track viruses and study membrane fusion
The FRET-based pH sensor consisting of mTFP1 and mCitrine enhanced our ability to observe the trafficking of viral particles to low pH compartments and the pH of endosomes following viral fusion. Important was the lower pKa and higher emission intensity of mCitrine compared to FRET donors in previously used sensors. This system in conjunction with a third soluble content probe included in the viral particle to measure content release (full fusion) should be useful to follow the trafficking and fusion of other viruses. Improved far red fluorescence proteins such as mCardinal51 or the recently developed FR-MQV52 could further enhance signal over noise, improving particle tracking and perhaps allowing identification of more fusion events in every field of view. The mTFP1-mCitrine sensor could also be used in future experiments to describe pH and motion of individual physiological cargos during trafficking, thereby solidifying the role of distinctive populations of endosomes on the fate of specific cargoes.
Important to the success of the current study was also the software developed to extract the traces of trafficking particles and their fusion events. Summing the fluorescence from three spectrally well-defined channels and defining appropriate surrounding backgrounds around moving particles in live cells was critical for identifying fusion events (Fig. 2, 3). In addition, this software enabled trajectory analysis to distinguish different modes of motion of organelles or viruses in cells (Fig. 6).
The current study should also serve as a helpful illustration of the virtue and limitations of current pH sensing and tracking methods. As illustrated in many of our figures, the spread of biological variations of individual particles and the uncertainties of the measured parameters can be quite large. Careful calibration of the probes and analytical methods including defining the appropriate statistical descriptions of error is important. Despite these challenges, it is possible to extract meaningful new biological data from single particle tracking and pH measurements in cells as demonstrated in the present work. We expect that the continued development of new technologies in microscopy will further improve signal-to-noise and enable the tracking of many particles in the same field of view at high time resolution, thereby increasing assay throughput.
Materials and Methods
Cell lines
A549 cells came from the lab of Judy White and were verified by ATCC Human Cell Line STR Profiling Service. A549 cells were grown in Ham’s F12 Nutrient Mixture with 10% FBS v/v and 100 units/mL penicillin, 100 units/mL streptomycin, 0.25 micrograms/mL amphotericin B. U2OS and HEK293T17 cells were grown in high glucose DMEM supplemented with 10% FBS v/v, 2mM L-Glutamine, 1 mM sodium pyruvate, 100 units/mL penicillin, 100 units/mL streptomycin, 0.25 micrograms/mL amphotericin B.
Plasmids and cloning
To construct the FRET-based pH sensor, the mTFP1-mCitrine fragment was synthesized by GENEWIZ. The linker between the two constituent fluorescent proteins was RSTSLQEFGT. The gene fragment was then inserted in the place of ecto-pHluorin (EcpH) in EcpH-TM, a gift from Gregory Melikian (Addgene plasmid # 85389; http://n2t.net/addgene:85389; RRID: Addgene_85389).53 The mTFP1-mCitrine gene fragments and EcpH-TM were separately digested using HindIII HF and EcoRI HF (New England Biolabs) according to manufacturer instructions for 1 hr. Digestions were gel purified; then EcPh-TM and the insert were ligated with T4 ligase (NEB). The ligation reaction was heat inactivated and transformed into DH5α competent cells (NEB). Colonies were selected on LB-carbenecillin plates. Plasmids were initially screened by digestion and then the sequence of the open reading frame was verified with primers corresponding to sequences in CMV (forward), TFP (forward), hGH poly(A) (reverse).
Additional plasmids used included Gag-mKate2 (a gift from Gregory Melikian, Emory University), Murine leukemia virus (Friend strain) gag-pol plasmid (a gift from Jean Millet at Cornell University and Jean Dubuisson at the Centre National de la Recherche Scientifique in Lille), pTG-luc (a gift from Gary Whittaker at Cornell University and Jean Dubuisson at the Centre National de la Recherche Scientifique in Lille), VSV-G plasmid (a gift from Michael Whitt at the University of Tennessee).
Preparation of pseudovirus
VSV-G bearing MLV pseudovirus with the FRET-based pH sensor and gag-mKate2 content marker was prepared by transfecting HEK293 T17 cells in Optimem-I using polyethylenimine and a 4:2:0.75:2:3 ratio: pTG-luc:pCMV gag-pol:gag-mKate2:VSV-G:mCitrine-mTFP1-ICAM1. HEK293 T17 cells were seeded at a density of ~5×106 cells/well in a 6 well tissue culture dish 24 hours prior to transfection. Optimem-I medium was replaced after 4–6 hours with high-glucose DMEM supplemented with 10% FBS. Supernatant was collected after 48 hours and purified by low-speed centrifugation, passage through a 0.45 μM filter, and centrifugation at 1.04×105 × g through a 25% sucrose-HM cushion. The supernatant and the cushion were aspirated to the pellet, which was resuspended in HM buffer (130 mM NaCl, 20 mM HEPES, 20 mM MES, pH 7.4) at ~140x concentration relative to the initial volume of viral supernatant. Resuspended pseudovirus was stored in aliquots at −80° C.
The infectivity of generated pseudoviruses was evaluated by luciferase assay as described previously.54 Briefly, A549 target cells were seeded at 2.5×104 cells/well in a 96 well plate and infected by spinning at 4° C. Cells were prepared according to Britelite plus Reporter Gene Assay system (PerkinElmer). The assay was read on a GloMax Explorer (Promega). The pseudovirus preparation used to generate data contained in Figures 1–6 was found to be infectious (Supp. Fig. 5A). Additional viral preparations were found to be similarly infectious (Supp. Fig. 5B) and have comparable calibration curves (Supp. Fig. 5C).
Particle and experimental analysis criteria for calibration and live cell imaging
Stringent quantitative selection criteria were applied to ensure that the pH readout was uniform between calibration and live cell imaging, as well as that tracking would be accurate. Only particles with fluorescence from gag-mKate2 (content marker), mTFP1 (FRET donor) and mCitrine (FRET acceptor) were included in analysis of calibration experiments and live cell imaging. In addition, only particles less than approximately 1.5 μm in apparent diameter were included in the analysis as larger spots were considered to be part of aggregates.
Live cell imaging was performed on 27 plates of cells on 11 separate days. In each plate of cells, a single field of view was imaged, comprising one individual experiment. Of these 27 individual experiments, only 13 individual experiments conducted on 7 separate days had ideal conditions in which the field of view had 5–20 viral particles visible in the collected image series and a viral content release event was observed in the field of view. For four of the individual experiments conducted, more than one fusion event was observed in the field of view. Individual experiments with too many particles in the field of view were excluded because individual particles couldn’t be differentiated and tracked during trafficking.
For the 13 plates retained for analysis, the criteria described above for particle selection was applied (triply labelled, less than 1.5μm in apparent diameter). As an additional measure to avoid including particles where tracking was ambiguous in the analysis, particles were excluded if they could be observed in the same 2.94 μm2 area as another particle at any time while they were tracked. Only particles that could be observed for at least 20 frames (6.67 minutes) were included in the analysis. From a total observation of 5041 particle tracks from the 13 individual experiments, 930 tracks were retained based on the applied criteria of the particle/endosome being tracked for at least 20 frames. After the additional particle inclusion criteria (listed above) were applied, 88 tracks remained. Within these 88 tracks, 18 fusion events occurred. Intensity and trajectory analysis were conducted for all 88 tracks.
Calibration and evaluation of FRET-based pH sensor
6–8 microliters of suspended pseudoviral particles labelled with the FRET-based pH sensor, mCitrine-mTFP1-ICAM1, and the content label, gag-mKate2 were spun for 20 min at 4° C and 200xg onto poly-L-lysine 0.1% V/V (Sigma-Aldrich) coated 35mm imaging dishes with a #1.5 glass insert (Mattek), such that there were ~5–20 particles per field of view. The particles were observed on a laser scanning confocal microscope (LSM880, Carl Zeiss) at 37° C with a 63x (1.4 NA) oil immersion objective. The pH sensor was excited with an Argon laser at 458nm and the content label was excited at 561nm. Emissions intensities were collected in 15 × 8.9nm-width spectral bands between 472nm and 606nm using the GaAsp spectral detector of the microscope. In addition, emissions above 606nm were collected in a photomultiplier tube detector and that intensity was assigned to mKate2.
Mean fluorescence emission intensities at each wavelength were extracted from the region of each triple labelled, detected particle in each field of view and background subtraction was performed.
The donor intensity relative to the acceptor intensity was quantified by measuring the ratio of the intensities from the two respective bands according to I494.0–502.9 /I530.0–538.9. The mean and standard deviations of all evaluated particles for a given prep at a given pH were plotted and fit with a sigmoidal function in GraphPad Prism 9.
Live cell imaging
A549 cells were seeded 24–32 hours prior to imaging at a concentration of approximately 1×105 cells/dish onto a 35mm imaging dish with #1.5 glass slide insert (Mattek) coated with fibronectin. Prior to addition of virus, cells were chilled on ice for 10 minutes, then washed with cold HEPES imaging buffer (Life Technologies) supplemented with 4.5 mg/mL glucose. 8–12 μL of pseudoviral particles were spun onto cells for 20 minutes at 4° C and 200 × g. A single pseudovirus preparation was used to generate the data derived from live cell imaging and displayed in Figures 2–6. Based on the similar infectivity and pH sensor calibration of viral particles from different preparations (Supp. Fig. 5 B,C), we expect that live cell imaging data from additional pseudovirus preparations would produce very similar results to those shown in Figures 2–6. Cells were washed in 2 mL of cold imaging buffer, placed on the microscope with the stage warmed to 37° C and the field-of-view for imaging was selected. After field-of-view and focus were set, viral trafficking was initiated by removal of 4° C imaging buffer and replacement with 37° C imaging buffer. A one to two-minute period of re-focusing followed the addition of 37° C imaging buffer and time zero in all experiments is the initiation of imaging after this initial time lag.
Excitation and detector configurations were the same as described for calibration and evaluation of the FRET-based pH sensor. An additional photomultiplier detector was used to generate a pseudo-DIC image of the cells using the transmitted light from the 458nm excitation. Frames were taken every 20s for at least one hour in a z-stack of 13 planes with 0.535nm between each plane. The image size was (800 × 800 pixel2) with a pixel resolution of 0.141 nm/pixel and an optical zoom of 1.2x. The Definite Focus feature of the microscope was used every 5 frames to avoid vertical drift.
Analysis of live cell imaging
Unless otherwise noted, particles were tracked in 2D using a maximum intensity Z-projection of the integrated intensities from emission bands 494–502 nm, 530–538 nm, and 593–741nm. The cell area was masked for analysis using the pseudo-DIC image to exclude particles directly adhered to the glass insert of the imaging dish. Particles were tracked using the TrackMate.55 Particles were detected using a Laplacian of Gaussian segmentation and an expected minimum particle radius of 700–800 nm and an intensity threshold appropriate to the individual experiment. The simple Linear Assignment Problem (LAP) tracker was used with a maximum search distance of 2 nm for consecutive frames and a search radius of 3 nm for missed frames with a maximum of 2 missed frames permitted. Tracks with less than 4 spots were eliminated. In cases where a particle was visible but below the selected threshold for 2 or more frames, tracks were manually connected. Manual connection of particle localizations was only performed in cases where based on the sparseness of particles we had a high degree of confidence the particle identified by the tracking program at two separated time points was the same.
Tracking results were compiled into tracks and intensities from 494.0–502.9 and 530.0–538.9 were extracted using the MATLAB (R2018a, MathWorks). The mean intensity of the background at all extracted emissions intensities was subtracted from the mean intensity of the particle area, with the background region being a 2 pixel-width annular region around each 14 pixel (1.96μm) diameter circular particle region. This data was used to calculate fluorescence ratios.
An in-house designed MATLAB (R2020b, MathWorks) application extracted and stored image regions of 21×21 pixel dimensions around each tracked particle and from 3 spectral channels. This data was then visualized and analyzed to identify fusion events, displacements and motion by custom built software written in LabView (National Instruments). Emissions collected from wavelengths 494–511 were assigned to channel 1, 521–538 to channel 2, and 593–741nm to channel 3. The extracted image regions corresponding to the trajectory of each file were read into LabView. Fusion was identified as a well-defined decrease of the mKate2 signal over 1 minute or less while signal corresponding to the FRET pair persisted. Only well-centered particles in the extracted region that had an approximate apparent diameter of less than 1.5 μm were considered for fusion analysis and only particles satisfying the same criteria were selected for the comparison set of non-fusing particles.
The velocity coefficient (v), representing the velocity of directed motion apart from drift or diffusion, was obtained using a directed-motion model of mean-squared displacement (MSD, ρ)
| (Equation 1) |
where D is the observed diffusion coefficient and τ is the time lag. Only time lags less than 60% of the total length of the entire tracks were used to fit to equation 1 because of the degree of imprecision in time lags that are long relative to the length of the observation time of the particle.29
pH values were determined from the measured fluorescence emission intensity ratios I494.0–502.9 /I530.0–538.9 using a standard table calculated from the fit in the calibration experiment of Fig. 1C (see Statistical Analysis and Data Fitting).
Statistical analysis and data fitting
Particle localization and emissions intensity data for all experiments was initially aggregated in R Studio (v. 1.1.453, R Studio Team 2020). Velocity, total displacement, fluorescence intensity ratio change, and related values were calculated in R Studio unless otherwise indicated. Statistical analysis was conducted in GraphPad Prism (Version 9.1.2 for Mac) and compared with the distributions of groups without an underlying assumption of parametric distributions (Mann-Whitney test). The two-tailed P-values were calculated and the summary values are reported in the relevant figures.
Supplementary Material
Synopsis:
We used a single particle approach to measure the acidification, motion, and fusion of single viral particles labeled with a fluorescent protein-based pH sensor and a fluorescent protein content marker in live cells. We found that the viral particles are trafficked to different subpopulations of endosomes. Endosomes that support fusion are more motile and become more acidic than endosomes that do not support fusion.
Acknowledgements:
This work was supported by NIH grant R01 AI30557. M.C was partially supported by training grant T32-GM080186 from the NIH. We thank James Casanova, Bettina Winkler and Ryan D’Souza for helpful discussions. For plasmids, we acknowledge Gregory Melikian, Gary Whittaker, Jean Millet, Jean Dubuisson and Michael Whitt. Schematics and models were prepared with BioRender.com.
Footnotes
Conflicts of Interest: The authors declare that they have no conflicts of interest with the contents of this article.
Data availability statement:
The routines to analyze the virus particle tracking data are available from the authors upon request.
Bibliography:
- 1.White JM, Whittaker GR, Author C, White JM. Fusion of Enveloped Viruses in Endosomes. 2016;(19):593–614. doi: 10.1111/tra.12389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30(17):3481–3500. doi: 10.1038/emboj.2011.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pasqual G, Rojek JM, Masin M, Chatton JY, Kunz S. Old World Arenaviruses Enter the Host Cell via the Multivesicular Body and Depend on the Endosomal Sorting Complex Required for Transport. PLOS Pathog. 2011;7(9):e1002232. doi: 10.1371/journal.ppat.1002232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kielian MC, Marsh M, Helenius A. Kinetics of endosome acidification detected by mutant and wild-type Semliki Forest virus. EMBO J. 1986;5(12):3103–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Padilla-Parra S, Matos PM, Kondo N, Marin M, Santos NC, Melikyan GB. Quantitative imaging of endosome acidification and single retrovirus fusion with distinct pools of early endosomes. Proc Natl Acad Sci. 2012;109(43):17627–17632. doi: 10.1073/pnas.1211714109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perini ED, Schaefer R, Stöter M, Kalaidzidis Y, Zerial M. Mammalian CORVET Is Required for Fusion and Conversion of Distinct Early Endosome Subpopulations. Traffic. 2014;15(12):1366–1389. doi: 10.1111/tra.12232 [DOI] [PubMed] [Google Scholar]
- 7.Kalaidzidis I, Miaczynska M, Brewińska-Olchowik M, et al. APPL endosomes are not obligatory endocytic intermediates but act as stable cargo-sorting compartments. J Cell Biol. 2015;211(1):123–144. doi: 10.1083/jcb.201311117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lakadamyali M, Rust MJ, Zhuang X. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell. 2006;124(5):997–1009. doi: 10.1016/j.cell.2005.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roche S, Bressanelli S, Rey FA, Gaudin Y. Crystal Structure of the Low-pH Form of the Vesicular Stomatitis Virus Glycoprotein G. Science. 2006;313(5784):187–191. doi: 10.1126/science.1127683 [DOI] [PubMed] [Google Scholar]
- 10.Roche S, Rey FA, Gaudin Y, Bressanelli S. Structure of the Prefusion Form of the Vesicular Stomatitis Virus Glycoprotein G. Science. 2007;315(5813):843–848. doi: 10.1126/science.1135710 [DOI] [PubMed] [Google Scholar]
- 11.Carneiro FA, Ferradosa AS, Da Poian AT. Low pH-induced conformational changes in vesicular stomatitis virus glycoprotein involve dramatic structure reorganization. J Biol Chem. 2001;276(1):62–67. doi: 10.1074/jbc.M008753200 [DOI] [PubMed] [Google Scholar]
- 12.Liu G, Cao W, Salawudeen A, et al. Vesicular Stomatitis Virus: From Agricultural Pathogen to Vaccine Vector. Pathogens. 2021;10(9):1092. doi: 10.3390/pathogens10091092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albertini AAV, Baquero E, Ferlin A, Gaudin Y. Molecular and cellular aspects of rhabdovirus entry. Viruses. 2012;4(1):117–139. doi: 10.3390/v4010117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim IS, Jenni S, Stanifer ML, et al. Mechanism of membrane fusion induced by vesicular stomatitis virus G protein. Proc Natl Acad Sci. 2016;114(1):E28–E36. doi: 10.1073/pnas.1618883114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci. 2013;110(18):7306–7311. doi: 10.1073/pnas.1214441110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer DG, Tal N, Novick D, Barak S, Rubinstein M. An antiviral soluble form of the LDL receptor induced by interferon. Science. 1993;262(5131):250–253. doi: 10.1126/science.8211145 [DOI] [PubMed] [Google Scholar]
- 17.Sun X, Yau VK, Briggs BJ, Whittaker GR. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology. 2005;338(1):53–60. doi: 10.1016/j.virol.2005.05.006 [DOI] [PubMed] [Google Scholar]
- 18.Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan SPJ. Vesicular Stomatitis Virus Enters Cells through Vesicles Incompletely Coated with Clathrin That Depend upon Actin for Internalization. PLOS Pathog. 2009;5(4):e1000394. doi: 10.1371/journal.ppat.1000394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White J, Matlin K, Helenius A. Cell fusion by Semliki Forest, influenza, and vesicular stomatitis viruses. J Cell Biol. 1981;89(3):674–679. doi: 10.1083/jcb.89.3.674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fredericksen BL, Whitt MA. Vesicular stomatitis virus glycoprotein mutations that affect membrane fusion activity and abolish virus infectivity. J Virol. 1995;69(3):1435–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott BL, Hoppe AD. Optimizing fluorescent protein trios for 3-Way FRET imaging of protein interactions in living cells. Sci Rep. 2015;5:1–13. doi: 10.1038/srep10270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the Environmental Sensitivity of Yellow Fluorescent Protein: MECHANISM AND APPLICATIONS*. J Biol Chem. 2001;276(31):29188–29194. doi: 10.1074/jbc.M102815200 [DOI] [PubMed] [Google Scholar]
- 23.Canton J, Grinstein S. Measuring Lysosomal PH by Fluorescence Microscopy. Vol 126. Elsevier Ltd; 2015. doi: 10.1016/bs.mcb.2014.10.021 [DOI] [PubMed] [Google Scholar]
- 24.Markosyan RM, Cohen FS, Melikyan GB. Time-resolved Imaging of HIV-1 Env-mediated Lipid and Content Mixing between a Single Virion and Cell Membrane. Mol Biol Cell. 2005;16(12):5502–5513. doi: 10.1091/mbc.E05-06-0496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chlanda P, Mekhedov E, Waters H, et al. The hemifusion structure induced by influenza virus haemagglutinin is determined by physical properties of the target membranes. Nat Microbiol. 2016;(April):16050. doi: 10.1038/nmicrobiol.2016.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lakadamyali M, Rust MJ, Babcock HP, Zhuang X. Visualizing infection of individual influenza viruses. Proc Natl Acad Sci. 2003;100(16):9280–9285. doi: 10.1073/pnas.0832269100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grabe M, Oster G. Regulation of Organelle Acidity. J Gen Physiol. 2001;117(4):329–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson DE, Ostrowski P, Jaumouillé V, Grinstein S. The position of lysosomes within the cell determines their luminal pH. J Cell Biol. 2016;212(6):677–692. doi: 10.1083/jcb.201507112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian H, Sheetz MP, Elson EL. Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys J. 1991;60(4):910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saxton MJ, Jacobson K. SINGLE-PARTICLE TRACKING: Applications to Membrane Dynamics. Annu Rev Biophys Biomol Struct. 1997;26(1):373. doi: 10.1146/annurev.biophys.26.1.373 [DOI] [PubMed] [Google Scholar]
- 31.Xia L, Zhang LJ, Tang HW, Pang DW. Revealing Microtubule-Dependent Slow-Directed Motility by Single-Particle Tracking. Anal Chem. 2021;93(12):5211–5217. doi: 10.1021/acs.analchem.0c05377 [DOI] [PubMed] [Google Scholar]
- 32.Miaczynska M, Christoforidis S, Giner A, et al. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004;116(3):445–456. doi: 10.1016/s0092-8674(04)00117-5 [DOI] [PubMed] [Google Scholar]
- 33.Yeager M, Wilson-Kubalek EM, Weiner SG, Brown PO, Rein A. Supramolecular organization of immature and mature murine leukemia virus revealed by electron cryo-microscopy: Implications for retroviral assembly mechanisms. Proc Natl Acad Sci U S A. 1998;95(13):7299–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeon H, Blacklow SC. Structure and physiologic function of the low-density lipoprotein receptor. Annu Rev Biochem. 2005;74:535–562. doi: 10.1146/annurev.biochem.74.082803.133354 [DOI] [PubMed] [Google Scholar]
- 35.Cureton DK, Massol RH, Whelan SPJ, Kirchhausen T. The Length of Vesicular Stomatitis Virus Particles Dictates a Need for Actin Assembly during Clathrin-Dependent Endocytosis. PLOS Pathog. 2010;6(9):e1001127. doi: 10.1371/journal.ppat.1001127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoyama M, Yoshioka Y, Arai Y, et al. Intracellular trafficking of particles inside endosomal vesicles is regulated by particle size. J Controlled Release. 2017;260:183–193. doi: 10.1016/j.jconrel.2017.06.007 [DOI] [PubMed] [Google Scholar]
- 37.Ghaffarian R, Muro S. Distinct Subcellular Trafficking Resulting from Monomeric vs Multimeric Targeting to Endothelial ICAM-1: Implications for Drug Delivery. Mol Pharm. 2014;11(12):4350–4362. doi: 10.1021/mp500409y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matlashov ME, Shcherbakova DM, Alvelid J, et al. A set of monomeric near-infrared fluorescent proteins for multicolor imaging across scales. Nat Commun. 2020;11(1):239. doi: 10.1038/s41467-019-13897-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Owczarek K, Chykunova Y, Jassoy C, Maksym B, Rajfur Z, Pyrc K. Zika virus: mapping and reprogramming the entry. Cell Commun Signal. 2019;17(1):41. doi: 10.1186/s12964-019-0349-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang ZG, Zhao L, Chen LL, et al. Spatiotemporal Quantification of Endosomal Acidification on the Viral Journey. Small.:2104200. doi: 10.1002/smll.202104200 [DOI] [PubMed] [Google Scholar]
- 41.Mingo RM, Simmons J a, Shoemaker CJ , et al. Ebola virus and severe acute respiratory syndrome coronavirus display late cell entry kinetics: evidence that transport to NPC1+ endolysosomes is a rate-defining step. J Virol. 2015;89(5):2931–2943. doi: 10.1128/JVI.03398-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saeed MF, Kolokoltsov AA, Davey RA. Novel, rapid assay for measuring entry of diverse enveloped viruses, including HIV and rabies. J Virol Methods. 2006;135(2):143–150. doi: 10.1016/j.jviromet.2006.02.011 [DOI] [PubMed] [Google Scholar]
- 43.Le Blanc I, Luyet PP, Pons V, et al. Endosome-to-cytosol transport of viral nucleocapsids. Nat Cell Biol. 2005;7(7):653–664. doi: 10.1038/ncb1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eidelman O, Schlegel R, Tralka TS, Blumenthal R. pH-dependent fusion induced by vesicular stomatitis virus glycoprotein reconstituted into phospholipid vesicles. J Biol Chem. 1984;259(7):4622–4628. [PubMed] [Google Scholar]
- 45.Beilstein F, Abou Hamdan A, Raux H, et al. Identification of a pH-Sensitive Switch in VSV-G and a Crystal Structure of the G Pre-fusion State Highlight the VSV-G Structural Transition Pathway. Cell Rep. 2020;32(7):108042. doi: 10.1016/j.celrep.2020.108042 [DOI] [PubMed] [Google Scholar]
- 46.Roth SL, Whittaker GR. Promotion of vesicular stomatitis virus fusion by the endosome-specific phospholipid bis(monoacylglycero)phosphate (BMP). FEBS Lett. 2011;585(6):865–869. doi: 10.1016/j.febslet.2011.02.015 [DOI] [PubMed] [Google Scholar]
- 47.Matos PM, Marin M, Ahn B, Lam W, Santos NC, Melikyan GB. Anionic lipids are required for vesicular stomatitis virus G protein-mediated single particle fusion with supported lipid bilayers. J Biol Chem. 2013;288(18):12416–12425. doi: 10.1074/jbc.M113.462028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, Gruenberg J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature. 1998;392(6672):193–197. doi: 10.1038/32440 [DOI] [PubMed] [Google Scholar]
- 49.Amini-Bavil-Olyaee S, Choi YJ, Lee JH, et al. The Antiviral Effector IFITM3 Disrupts Intracellular Cholesterol Homeostasis to Block Viral Entry. Cell Host Microbe. 2013;13(4):452–464. doi: 10.1016/j.chom.2013.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perrin P, Janssen L, Janssen H, et al. Retrofusion of intralumenal MVB membranes parallels viral infection and coexists with exosome release. Curr Biol. 2021;31(17):3884–3893.e4. doi: 10.1016/j.cub.2021.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chu J, Haynes RD, Corbel SY, et al. Non-invasive intravital imaging of cellular differentiation with a bright red-excitable fluorescent protein. Nat Methods. 2014;11(5):572–578. doi: 10.1038/nmeth.2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mukherjee S, Hung ST, Douglas N, et al. Engineering of a Brighter Variant of the FusionRed Fluorescent Protein Using Lifetime Flow Cytometry and Structure-Guided Mutations. Biochemistry. 2020;59(39):3669–3682. doi: 10.1021/acs.biochem.0c00484 [DOI] [PubMed] [Google Scholar]
- 53.Miyauchi K, Marin M, Melikyan GB. Visualization of retrovirus uptake and delivery into acidic endosomes. Biochem J. 2011;434(3):559–569. doi: 10.1042/BJ20101588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hulseberg CE, Fénéant L, Szymańska-de Wijs KM, et al. Arbidol and Other Low-Molecular-Weight Drugs That Inhibit Lassa and Ebola Viruses. J Virol. 2019;93(8):e02185–18. doi: 10.1128/JVI.02185-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tinevez JY, Perry N, Schindelin J, et al. TrackMate: An open and extensible platform for single-particle tracking. Methods. 2017;115:80–90. doi: 10.1016/j.ymeth.2016.09.016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The routines to analyze the virus particle tracking data are available from the authors upon request.