

Abstract
Gut microbes provide essential services to their host and shifts in their composition can impact host fitness. However, despite advances in our understanding of how microbes are assembled in the gut, we understand little about the stability of these communities within individuals, nor what factors influence its composition over the life of an animal. For this reason, we conducted a longitudinal survey of the gut microbial communities of individual free-ranging woodrats (Neotoma spp.) across a hybrid zone in the Mojave Desert, USA, using amplicon sequencing approaches to characterize gut microbial profiles and diet. We found that gut microbial communities were individualized and experienced compositional restructuring as a result of seasonal transitions and changes in diet. Turnover of gut microbiota was highest amongst bacterial subspecies and was much lower at the rank of Family, suggesting there may be selection for conservation of core microbial functions in the woodrat gut. Lastly, we identified an abundant core gut bacterial community that may aid woodrats in metabolizing a diet of plants and their specialized metabolites. These results demonstrate that the gut microbial communities of woodrats are highly dynamic and experience seasonal restructuring which may facilitate adaptive plasticity in response to changes in diet.
Keywords: herbivore, mark and release, microbiome, neotoma, symbiosis
The gut bacterial communities of herbivorous woodrats were repeatedly sampled for 2 years to identify what factors influence the stability of microbes in the gut of individuals over their natural lifespan.
Introduction
Mammals harbor a complex community of microorganisms along the gastrointestinal tract that provide essential services such as aiding in the breakdown of complex polysaccharides (Flint et al. 2008), synthesizing essential vitamins (LeBlanc et al. 2013), inhibiting colonization by pathogens (Abt and Pamer 2014), modulating host immune responses (Round and Mazmanian 2009), and facilitating tolerance to dietary toxins (Kohl et al. 2014b). The study of gut microbial communities has received exceptional attention over the last decade, largely due to the advances in DNA sequencing technologies that have enabled inventorying the community of microorganisms in the gut. Much of this research effort has employed captive experiments with model animal systems such as laboratory-reared mice (Nguyen et al. 2015, Clavel et al. 2016), or has focused on humans (Thursby and Juge 2017). Such studies provide valuable insights into the factors that influence the composition, structure, and function of the gut microbiome. However, captive experiments are limited to testing only a handful of factors at any one time and are unable to replicate the complexity of interacting factors that shape gut microbial communities of animals in nature. In addition, the gut microbiome of captive animals is often less diverse (Kohl et al. 2014, Schmidt et al. 2019) and has altered composition compared to wild counterparts (McKenzie et al. 2017, Gibson et al. 2019), which limits the generalizability of the results of captive studies to natural populations. For these reasons, there has been a recent call for greater attention to studying the microbiomes of animals under natural conditions (Hird 2017, Cusick et al. 2021).
Over the last decade there has been numerous studies on the gut microbial communities of free-ranging animals that have identified strong predictors of the assemblage of gut microbiota such as diet (Hicks et al. 2018, Murillo et al. 2022), geography (Gomez et al. 2015), and host genetics (Kohl et al. 2018, Weinstein et al. 2021, Nielsen et al. 2022). Additionally, seasonal dynamics may be common in gut microbial communities (Ren et al. 2017, Hicks et al. 2018, Orkin et al. 2018) and these seasonal changes can be highly individualized (Marsh et al. 2022). Despite the growing body of literature on the gut microbiome of free-ranging animals, relatively few have characterized microbial communities within individuals over their natural lifespan, likely due to the difficulties of resampling free-ranging animals. For this reason, we understand little about the temporal dynamics of gut microbial communities within individuals, or how variation in community composition may influence aspects such as host fitness. Therefore, longitudinal surveys of individuals and their gut microbiota are warranted to advance our understanding of the temporal dynamics of such communities at both population and individual scales, and to determine whether shifts in community composition are largely stochastic or are repeatable across timescales.
Here, we report the results of a longitudinal survey of two species of woodrats, the Bryant’s woodrat (Neotoma bryanti), the desert woodrat (N. lepida) and their hybrids from a sympatric population in a desert shrubland in southern, CA, USA. Woodrats are ideal for studying the factors that shape gut microbial communities as they are abundant across most habitats in North America (Goldman 1910), can be resampled with relative ease for longitudinal surveys (Shurtliff et al. 2014) and are amenable to captivity (Egoscue 1957). In particular, studies on woodrats have identified important factors that influence gut microbial community structure including host genetics (Weinstein et al. 2021, Nielsen et al. 2022), diet (Kohl and Dearing 2012, Martínez-Mota et al. 2020, Stapleton et al. 2022), and intestinal parasites (Doolin et al. 2022). Here, we present a survey of individual woodrats in nature to characterize the composition, structure and temporal dynamics of these rodents’ gut microbial communities. From feces, we generated both 16S rRNA gene sequences to profile gut microbial communities and chloroplast trnL (UAA) intron sequences to quantify diet for each individual across multiple captures. Additionally, in our analyses we included available genome-wide ancestry estimates for each individual that were generated as part of a previous survey of this woodrat population (Klure et al. 2023). We leveraged this combination of sequencing approaches to characterize the influence of factors such as identity, ancestry, diet, and season in structuring gut microbial community composition. Using this woodrat system, we aimed to: (i) characterize the stability of gut microbial communities within individuals in nature, (ii) identify factors that drive shifts in microbial community composition, and (iii) determine whether a stable core community of microbiota exists in the gut.
Materials and methods
Sample collection and ancestry determination
Woodrats were repeatedly live-trapped across eight sampling events from January 2019 to November 2020 at a site known as “Whitewater” located in the San Gorgonio Pass, Riverside County, CA, USA within creosote bush (Larrea tridentata) shrublands (latitude/longitude: 33° 55′ N; 116° 38′). This site was selected for this study due to the relative ease of resampling individuals overtime, its high degree of seasonality, and due to the ongoing interspecific hybridization, which allows for the testing of genotypic effects on gut microbial assemblage. Woodrats are a primary prey species and as such have low annual survival rates as compared to larger mammal species and thus, to produce a multiyear cohort of 35 individuals analyzed in this study, it was necessary to sample more than 200 individuals captured more than 400 times (Klure et al. 2023). Sherman traps were baited with oats, set at dusk, and checked at dawn. Woodrats were weighed, sexed and both fresh fecal pellets and a tissue sample from the ear were collected from each individual. Fecal and tissue samples were stored in a liquid nitrogen dewar while in the field and stored at –80°C in the lab prior to processing. Each woodrat received an ear tag with a unique identifier and was released at their site of capture so that individuals could be resampled over time. Trap collected feces were selected for this work as they represent a noninvasive metric for characterizing the gut microbiota of individuals overtime, contain little environmental contamination of microbes (Kohl et al. 2015), and provide census of the microbiota most similar to the community present in the lower gastrointestinal tract, but also to that of the foregut (Kohl et al. 2014a). Due to the ongoing interspecific hybridization present at Whitewater, the ancestry of each individual was previously determined as part of Klure et al. (2023) using a genome-wide single nucleotide polymorphism dataset generated from a reduced representation sequencing approach (Parchman et al. 2012).
Amplicon library preparation and sequencing
We extracted DNA from feces in batches as samples became available once three sampling trips were completed using the Qiagen PowerFecal DNA Isolation Kit (#12830) following the manufacturers protocol. We prioritized individuals for sequencing if they had a high recapture rate (i.e. captured at least three times); however, as parental and backcross N. lepida have low survival rates at Whitewater (Klure et al. 2023), we also included individuals with low recapture rates (i.e. captured only once or twice) to maintain a balanced ancestry representation. In total, we extracted DNA from 129 fecal samples from 35 individuals (∼3.7 samples per individual on average) for the 2019–2020 sampling period representing the following ancestry classes: parental N. bryanti (n = 10), backcross N. bryanti (n = 11), F1/F2 hybrids (n = 5), backcross N. lepida (n = 6), and parental N. lepida (n = 3). We included negative controls for each extraction batch (n = 12) and extracted three preps of a Zymo Microbial Mock Community Standard (ZymoBIOMICS Microbial Community Standard D6300) to assess for any potential environmental or reagent contamination. Library preparation and sequencing of DNA extracts were conducted by the University of Chicago at Urbana-Champaign DNA Sequencing Facility. Individual libraries were prepared using a two-stage PCR protocol as described in Naqib et al. (2018). For each DNA sample, we generated both 16S rRNA gene (V4 subunit) amplicon libraries to estimate microbial composition using the unmodified 515F/806R primer pair (Caporaso et al. 2011, Parada et al. 2016) and chloroplast trnL (UAA) intron amplicon libraries to estimate diet using the unmodified g/h primer pair (Taberlet et al. 2007). Libraries were combined into three separate library pools that each contained ∼50 samples of each library type. These library pools were then sequenced individually across a single lane of an Illumina Miniseq to generate 2 × 150 bp paired-end reads, requiring a total of three sequencing lanes. To test for any potential lane effects, we included controls consisting of the repeated sequencing of a single 16S rRNA library generated from woodrat feces and a 16S rRNA library generated from an artificial microbial mock community (ZymoBIOMICS Microbial Community Standard D6300).
Read processing and generation of final datasets
Paired-end reads were processed in R. v. 4.2.1 (R Core Team 2022) following the dada2 v.1.24.0 (Callahan et al. 2016) workflow. Reads were processed independently based on their respective library type and sequencing lane. First, we removed primer sequences using cutadapt v.3.5 (Martin 2011) and then filtered out reads if they contained ambiguous nucleotides, sites with base-site quality scores less than 10, more than two expected errors as calculated from the nominal definition of the quality score as follows, 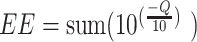 , and if they corresponded to the PhiX loading control. For the 16S rRNA gene reads, we removed 2 bp from the end of the forward and reverse reads due to quality drops at these positions and removed any resulting reads less than 100 bp in length. For the trnL reads, we filtered out any reads less than 14 bp in length, but did not perform length trimming as trnL amplicons vary considerably in length. Sequencing errors were inferred on a per lane basis using 100 000 000 bp from each lane using randomly selected samples as input. Dereplicated reads and their corresponding inferred error rates were then used to identify members of each sequence community as amplicon sequence variants (ASV) at 100% sequence identity (Callahan et al. 2017). We then merged paired-end reads requiring a minimum overlap of 10 bp with no mismatches allowed in the overlap region. We removed chimeric sequences from the resulting sequence tables and then merged each table from each sequencing lane to generate a single data frame containing sequences from all samples for each library type.
, and if they corresponded to the PhiX loading control. For the 16S rRNA gene reads, we removed 2 bp from the end of the forward and reverse reads due to quality drops at these positions and removed any resulting reads less than 100 bp in length. For the trnL reads, we filtered out any reads less than 14 bp in length, but did not perform length trimming as trnL amplicons vary considerably in length. Sequencing errors were inferred on a per lane basis using 100 000 000 bp from each lane using randomly selected samples as input. Dereplicated reads and their corresponding inferred error rates were then used to identify members of each sequence community as amplicon sequence variants (ASV) at 100% sequence identity (Callahan et al. 2017). We then merged paired-end reads requiring a minimum overlap of 10 bp with no mismatches allowed in the overlap region. We removed chimeric sequences from the resulting sequence tables and then merged each table from each sequencing lane to generate a single data frame containing sequences from all samples for each library type.
Microbial taxonomy was assigned to the 16S rRNA gene ASVs using the naïve Bayesian classifier method (Wang et al. 2007) in dada2 with default parameters except that we increased the minimum bootstrap confidence to assign a taxonomic rank from 50% to 70%. Taxonomy assignments were based on the current version at the time of analyses (v.138.1) of the Silva 16S rRNA gene small-subunit reference database (https://www.arb-silva.de/download/arb-files/). We were unable to assign plant taxonomy using dada2 to the trnL ASVs as many of the resulting read lengths were too short for this method (< 50 bp). Instead, we created a custom trnL reference database as described in Weinstein et al. (2021), and aligned the sequences from each sample against this database using BLASTN v.2.9.0+ (https://doi.org/10.1186/1471-2105-10-421). ASVs were assigned taxonomy based on the reference sequence with the lowest E-value, highest % identity, and the closest length to the ASV. We did not assign taxonomy if there was no reference sequence with an E-value < 1, % identity ≥ 90, and length within 10% of that of the ASV sequence. If multiple best hits were identified for an ASV sequence, taxonomy was assigned at each rank if at least 70% of the best hits shared that rank. The script used for the taxonomic classification of the trnL ASVs (classify_sequences.py) and associated documentation is available at https://github.com/robertgreenhalgh/stand/.
We created a phylogenetic tree of the resulting 16S rRNA gene sequences for use in downstream analyses. To do this, we generated a matrix of multiple sequence alignments from the ASV sequences using DECIPHER v.2.24.0 (Wright 2016) with a kmer size of 8. We exported the multiple sequence alignment data in FASTA format using ShortRead v.1.54.0 (Morgan et al. 2009) and generated a maximum-likelihood tree of the 16S rRNA gene sequences using FastTree2 v.2.1.10 (Price et al. 2010) employing the nucleotide and generalized time-reversible model parameters. We then combined the ASV table, taxonomy table, sample metadata and phylogenetic tree stored in Newick format into a single S4 class data frame using phyloseq v.1.40.0 (McMurdie and Holmes 2013).
We performed additional filtering of ASVs in phyloseq for each library type and subsequently rarified the resulting amplicon communities to limit the effects variation in read depth across samples. For the 16S rRNA gene amplicon dataset, we removed singletons, ASVs with taxonomic assignments to Mitochondria or Chloroplast, ASVs with exceptionally low prevalence by removing ASVs not found in at least two individuals, and ASVs that lacked taxonomic assignments at the rank of Family or above. Although the process of rarefying amplicon count data has been a matter of debate due to the perceived loss of data and power for statistical inference (McMurdie and Holmes 2014), we opted to rarefy our libraries as there is strong evidence that that rarefying leads to a reduction in the frequency of type-1 errors with common microbiome analyses (McKnight et al. 2019, Yang and Chen 2022). We generated rarefaction curves with vegan v.2.6–2 (Dixon 2003) and opted to rarefy the 16S rRNA gene libraries to 14687 reads, as this was the lowest read depth achieved by a sample that was still sufficient to capture the diversity of ASVs present in our dataset (Figure S1, Supporting Information). For the trnL dataset, we removed singletons/doubletons and removed ASVs that corresponded to oats (Genus Avena), the bait that was used during trapping. We then removed ASVs that accounted for less than 1% of the relative read abundance in each sample, as these ASVs likely represented environmental contamination or low-frequency dietary items. The resulting trnL libraries were then rarefied to a depth of 4190 reads, the lowest read depth of a sample present in the dataset.
Community diversity and composition analyses
We used vegan to estimate alpha and beta diversity indices for the 16S rRNA and chloroplast trnL libraries. For the trnL libraries, ASVs were binned at the rank of family to avoid overestimating diet richness (i.e. several plant species were represented by multiple ASVs). For estimates of alpha diversity, we estimated observed richness (i.e. observed plant families for the trnL libraries and observed ASVs for the 16S rRNA libraries) and the Shannon–Wiener index (Shannon 1948) as a measurement of both community richness and evenness. We determined significant differences between mean alpha diversity measurements using nonparametric rank sum tests for either population comparisons with the Kruskal–Wallis test (Kruskal and Wallis 1952) or pairwise comparisons of individuals with the Wilcoxon test (Wilcoxon 1945).
For beta diversity comparisons of diet, we created a matrix of pairwise Bray–Curtis distances (Bray and Curtis 1957) and subsequently performed principal coordinate analysis (PCoA) on the resulting trnL distance matrix. We stored the resulting principal coordinate axes in a separate data frame for use in subsequent modeling efforts. For beta diversity analyses of gut microbial communities, we created pairwise distance matrices based on Jaccard (Jaccard 1912), Bray–Curtis, and UniFrac distances (Lozupone and Knight 2005). We then performed PCoA based on the ordination of the above distance matrices. Using the R package vegan, we tested for homogeneity of group dispersions using a multivariate analogue of the Levene’s test (PERMDISP) using the betadisper() function and tested for the significance of our measured factors on structuring microbial community composition using permutational multivariate analysis of variance (PERMANOVA) using the adonis2() function. We included the following variables in the model for each distance metric: ancestry, identity, diet (consisting of the first principal coordinate from the Bray–Curtis diet ordination), season, sex, and reproductive status. We did not include age as a variable in these models as all individuals were adults at the time of their first capture based on body weight (> 100 g). We characterized the precipitative seasonality of Whitewater using climate data from an atmospheric monitoring station located at the Palm Springs International Airport (∼15 km from the site). A total of 140 mm of precipitation occurred between December 2018 and April 2019 and 127 mm between December 2019 and April 2020, accounting for ∼75% and ∼95% of the total precipitation over the following 12-month period, respectively. Thus, we classified these periods as the “wet” season at Whitewater.
Variance partitioning of a 16S rRNA Bray–Curtis distance matrix was performed with vegan using redundancy analysis ordination (RDA) with the following explanatory matrices: identity, diet (consisting of the first four principal components from the diet PCoA analysis as described above), sampling trip, within site capture location (latitude and longitude based on GPS coordinates taken at the site of capture), ancestry (q), and sex. Significance of each of the explanatory matrices was assessed using permutation tests with the anova.cca()function in vegan. The eulerr v.6.1.1 package (https://github.com/jolars/eulerr) was used to plot area-proportional Venn-diagrams of the individual variance and covariance explained by each significant explanatory matrix.
Quantifying microbiome dissimilarity
The R package Microbiome v.1.18 (https://microbiome.github.io/) was used to model microbiome dissimilarity over time within individuals. We used the plasticity() function in an iterative manner to calculate pairwise dissimilarity values at the ASV-level based on Jaccard, Bray–Curtis, unweighted UniFrac, and weighted UniFrac distances for samples from the same individual for all possible comparisons. We repeated this process with Jaccard and Bray–Curtis distances with ASVs binned at the rank of Family. We compared differences in mean dissimilarity values using a pairwise Wilcoxon rank sum test and adjusted resulting P-values for multiple comparisons using the Bonferroni Correction method (Bonferroni 1936). We assessed for a relationship between time (distance in days between samples) on dissimilarity scores using a generalized linear model (GLM) for both Bray–Curtis and weighted UniFrac distances. Lastly, we explored the effects of the % sequence similarity cutoff used to assign sequences to ASV bins on influencing dissimilarity estimates. We used the DECIPHER package to align and cluster the sequences from the original 100% identity ASV sequence table using the following sequence similarity cutoffs for ASV binning: 90% , 92%, 94%, 96%, and 98%. We then recalculated pairwise Bray–Curtis distances between samples from the same individual as described previously and modeled the effect of sequence similarity cutoff on Bray–Curtis distances using linear regression.
Identification of microbes with differential and stable abundances over time
We used the R package NBZIMM v.1.0 (Zhang and Yi 2020) to perform a negative binomial mixed model with zero inflation to identify microbial ASVs with differential abundance based on the variables of time (days since previous sampling) and diet (first principal component from the diet PCoA of Bray–Curtis distances). This type of model is particularly appropriate for modeling longitudinal microbiome data as it can account for within-subject correlation structures and is robust to data sparsity (Zhang and Yi 2020). In our model, we tested for differential abundance of ASVs that had nonzero proportions greater than 0.20 using rarefied counts. We included time and diet as fixed effects, individual as a random effect and fitted the logistic regression with the logarithmic transformation of the initial library size of each sample. We controlled for false discovery rate using the Benjamini–Hochberg procedure and considered ASVs with an adjusted P-value ≤ .001 as taxa with differential abundance (Benjamini and Hochberg 1995). The R package microbiome was used to identify the core taxonomic members of the gut microbiome of wild woodrats. We classified an ASV as belonging to the “core community” if it occurred in every sampling period, was present in ≥ 80% of samples collected at that time, and had a relative abundance of > 0.0001. We assessed whether the structure of the core microbial community was influenced by ancestry, identity, diet, season, sex, and reproductive status using PERMANOVA in the same manner as previously described.
Identifying microbes associated with creosote bush feeding
We investigated the abundance of creosote bush (the most abundant diet item with little seasonality) in the diet of woodrats and its relationship to the abundance of bacterial families present in woodrat feces. We ran a GLM with the base stats package in R for each bacterial family with an offset term consisting of a logarithmic transformation of initial library sizes and used a Quasi-Poisson regression to account for overdispersion. We performed a square root transformation of the relative abundances of bacterial families and creosote bush to improve the fit of the residuals.
Field cafeteria experiment
We conducted a limited free-choice feeding experiment with adult woodrats (n = 7) in March 2019 at Whitewater to assess whether the dietary shifts found in the wet season reflect individual foraging preference or are simply an artifact of plant resource availability. Woodrats were temporarily housed in solid-bottom shoebox cages (48 cm × 27 cm × 20 cm) with pine shavings and a plastic tube for shelter. Each woodrat was provided 10 g each of freshly clipped leaves from creosote bush (L. tridentata), Sahara mustard (Brassica tournefortii) and desert dandelion (Malacothrix glubrata). The leaf bundles were presented at three separate corners of the cage and secured with wire. The two forb species were selected as they were the most abundant ephemeral plants at Whitewater at the time of the experiment and were commonly found cached on middens during trapping. Woodrats were provided water ad libitum and were released at their site of capture after 24 h. After the release of the woodrats, the remaining plant material in each cage was collected, separated by species, and weighed. We calculated foliar intake as follows: foliage consumed (g)/body mass (kg)/day. We compared differences in mean intake using ANOVA with post hoc analysis following the Tukey’s procedure.
Results
Quality control and validation of 16S rRNA libraries
In the initial unfiltered dataset containing both fecal and control samples there were a total of 9102 ASVs, and the 129 fecal samples achieved an average read depth of 41395 ± 12814 (standard deviation) reads. A total of 5899 ASVs were filtered from this dataset due to the following exclusionary criteria: taxonomic classification to Chloroplast or Mitochondria (2409 ASVs), present as a singleton (577 ASVs), did not meet sample prevalence threshold (2765 ASVs), and lacked taxonomic assignment to at least the rank of Family (124 ASVs). In the final filtered dataset, we retained a total of 3227 ASVs and the fecal samples had an average read depth of 33524 ± 14003 prior to rarefying. After rarefying, 127 fecal samples were retained after discarding two samples with read depths less than the rarefied count of 14687. Of the 3227 ASVs retained in this final dataset, only 36.6% were assigned a taxonomic classification at the rank of Genus or below and therefore we used the rank of Family or above for compositional analyses. The 16S rRNA libraries demonstrated consistent results across different sequencing lanes based on the woodrat and mock community control samples (Figure S2A and B, Supporting Information). The negative controls consisting of extraction blanks (n = 12) had an average of 596 ± 890 (∼1.4% of the mean initial reads produced by the fecal samples) and 179 ASVs. Of the 179 ASVs present in the extraction blanks, 68 also occurred in the fecal samples. The read abundances of microbial taxa from blank samples are shown in Figure S3 (Supporting Information) at the rank of Family. The mock community control samples that were extracted alongside the fecal samples and were present in each library pool contained all eight of the expected genera without any evidence of significant contamination (Figure S2A and B, Supporting Information). We repeated a subset of the analyses described in this study using a dataset where we removed the shared ASVs between the negative control and fecal samples and found that this did not influence the overall results of the study (Figure S4, Supporting Information). Based on these results, we determined the 16S rRNA libraries from feces to be of high quality with minimal concerns for significant lane effects, environmental, cross-sample, or reagent contamination and thus, used the original dataset containing all 3277 ASVs for all downstream analyses.
Factors influencing the structure of the gut microbiome of woodrats in nature
Based on the results of the redundancy analysis, we found four factors: identity, sampling period, diet, and capture location explain a significant portion of the variance in gut microbiome assemblage (Fig. 1A; Table S1, Supporting Information). In total, these four factors together explained 30.9% of the overall variance in gut microbial community composition across all samples (Fig. 1A). We also performed redundancy analysis independently for samples from the wet and dry season that revealed identity explained more variance in gut microbiome composition in the dry season (14.03%) and was nonexplanatory in the wet season (Table S1, Supporting Information). Additionally, in the wet season RDA, capture location (1.09%) explained marginally more variance in gut microbiome composition than either the combined or dry season RDAs (Table S1, Supporting Information). We found that season impacted the relative abundances of both dietary items (Fig. 1B) and bacterial families in the gut of woodrats (Fig. 1C). Diet diversity (Shannon’s index) was significantly higher during the wet season (January–April) of each year (Kruskal–Wallis test: χ2 = 5.112, P = .024; Fig. 1D), and gut microbial diversity (Shannon’s index) was significantly lower during the same period (Kruskal–Wallis test: χ2 = 26.564, P < .001; Fig. 1E). Community richness was also impacted by season in similar manner to diversity, where diet richness was higher in the wet season (Kruskal–Wallis test: χ2 = 14.242, P < .001; Figure S5, Supporting Information) and microbial richness was lower in the same period (Kruskal–Wallis test: χ2 = 23.077, P < .001; Figure S5, Supporting Information). Gut microbial community structure was also impacted by season (PERMANOVA: Sum of squares = 1.065, Pseudo-F = 5.054, R2 = 0.030, P = .001) but to a lesser extent than community richness and evenness (Fig. 1F). Using PERMANOVA, we also found that ancestry (q) was a significant factor in explaining variation in microbial composition across all distance metrics assessed; however, its explanatory power was small (R2 < 0.02 for all distance metrics; Table S2, Supporting Information).
Figure 1.

Factors influencing the composition of gut microbiota and diet of woodrats at Whitewater. (A) Euler diagram displaying the significant explanatory factors and their % variance on gut microbiome composition based on redundancy analysis for the entire dataset. (B) Relative abundance of diet items at the family-level averaged by season. (C) Relative abundance of gut bacteria binned by Order by season. (D) Diet diversity by season. Diet was more diverse in the wet season (Kruskal–Wallis test: χ2 = 5.112, P = .024). (E) Gut microbiome diversity by season. Gut microbial communities were more diverse in the dry season (Kruskal–Wallis test: χ2 = 26.564, P < .001). (F) Principal coordinates analysis of the gut microbiome based on Bray–Curtis distances. Season was a significant factor in structuring microbial communities (PERMANOVA: Sum of squares = 1.065, Pseudo-F = 5.054, R2 = 0.030, P = .001). Ellipses represent 95% confidence intervals. Significant differences are denoted by * (* = P < .05; ** P < .01, and *** P < .001).
Stability of individual gut microbial communities
The stability of individual gut microbial communities varied dramatically based on the measure of dissimilarity and the sampling periods being compared. At the ASV-level, we found that nonphylogenetically weighted distance metrics demonstrated substantial ASV turnover within individuals over time (Fig. 2A). In contrast, distance metrics that incorporate phylogenetic relatedness (UniFrac), especially when weighted for the abundance of ASVs, demonstrated substantially less dissimilarity between sampling periods spanning 2–4 months (Fig. 2A). When ASVs were binned to the family-level, there was less dissimilarity within an individual’s gut microbiome over time indicating that the turnover in microbial composition was largely at the level of species and below (Fig. 2B). This same pattern was found when we compared microbiome dissimilarity for each individual with ASVs binned at artificial sequence similarity cutoffs stepwise from 100% to 90% (Figure S6, Supporting Information). Specifically, mean gut microbiome dissimilarity was positively correlated with the level of sequence similarity used to bin ASVs (linear regression: F1:478 = 131.5, R2 = 0.214, P < .001), where gut microbial communities represented by 90% ASV bins had lower dissimilarity estimates compared to higher ASV sequence similarity bins (Figure S6, Supporting Information). We also found that individual gut microbial communities were more dissimilar when compared across seasons for both sampling years (2019: Kruskal–Wallis test: χ2 = 8.08, P = .004; 2020: Kruskal–Wallis test: χ2 = 8.00, P = .005; Fig. 2C). Surprisingly, we found no relationship between gut microbiome dissimilarity and the time in days between samples based on either Bray–Curtis (GLM; P > .05) or weighted UniFrac (GLM; P > .05) distances (Fig. 2D).
Figure 2.

Gut microbiota compositional dissimilarity by time and season at the individual-level. (A) Individual pairwise gut microbiome dissimilarity at the (A) ASV-level and (B) Family-level for each trip comparison and distance metric in chronological order. Lines and filled areas present moving averages and 95% confidence intervals, respectively, and are colored by distance metric. (C) Bray–Curtis dissimilarity from within the same season (i.e. dry to dry, wet to wet) or across seasons (i.e. dry to wet, wet to dry) for each sampling year. Individuals’ gut microbiomes were more dissimilar when sampled across seasons in 2019 (Kruskal–Wallis test: χ2 = 8.08, P = .004) and 2020 (Kruskal–Wallis test: χ2 = 8.00, P = .005). (D) Gut microbiome dissimilarity as a factor of time between sample comparisons. There was no significant relationship between dissimilarity and time between samples for either Bray–Curtis (GLM; P > .05) or weighted UniFrac distances (GLM; P > .05). Significant differences are denoted by * (* = P < .05; ** P < .01, *** P < .001).
Microbes with differential abundance
We found strong individual-specific patterns of gut microbiome stability, where some individuals demonstrated higher stability of their microbial communities than others (e.g. Fig. 3A; summary Figure S7, Supporting Information). We identified 50 ASVs out of the 493 ASVs considered with nonzero proportions > 0.20 (Figure S8, Supporting Information) that had differential abundance either over time (25 ASVs) or with changes in diet (25 ASVs; Fig. 3B). The majority of ASVs (68%) with significant differential abundance over time belonged to three families, Muribaculaceae (10 ASVs), Lachnospiraceae (five ASVs), and Ruminococcaceae (two ASVs). These significant ASVs demonstrated temporal trends in their relative abundance that was consistent across most individuals (Fig. 3C).
Figure 3.

Temporal trends of gut microbial composition in woodrats at Whitewater. (A) Relative abundance of the top eight most abundant bacterial families over time in two individual woodrats. Examples of large shifts (Animal #11) and small shifts (Animal #40) in bacterial family composition over time. (B) Results from a zero-inflated negative binomial mixed model on bacterial ASVs and the factors of time and diet on influencing their abundance. Dashed red line indicates the significance threshold. In total, the abundance of 50 ASVs were significantly correlated with time or diet. (C) Boxplots demonstrating the relative abundance of ASVs that were found to have significant differential abundance over time across all individuals. ASVs were binned at the rank of family.
Stable core microbial community
We identified a stable core microbiome consisting of 26 ASVs belonging to six bacterial families: Muribaculaceae (20 ASVs), Lactobacillaceae (two ASVs), Ruminococcaceae (one ASV), Erysipelotrichaceae (one ASV), Oscillospiraceae (one ASV), and Eggerthellaceae (one ASV). These core ASVs varied little in relative abundance and evenness over time (Fig. 4A) and accounted for an average of 18.20 ± 0.05% of the total read abundance across all samples. Based on PERMANOVA using Bray–Curtis distances, as compared to the gut microbial communities of woodrats with all members present, the core microbial community did not differ in its structure by season (Sum of Squares = 0.174, Pseudo-F = 1.472; R2 = 0.010; P = .145), but did retain a significant signature of host identity (Sum of Squares = 5.978; Pseudo-F = 1.580; R2 = 0.338; P = .001) and diet (Sum of Squares = 0.483; Pseudo-F = 4.080; R2 = 0.027; P = .003). Additionally, as expected, core ASVs demonstrated significantly higher stability across all pairwise comparisons of samples from the same individual (unweighted UniFrac distances; Kruskal–Wallis test: χ2 = 105.37, P < .001; Fig. 4B).
Figure 4.

Core gut bacterial members and their temporal dynamics. (A) Relative abundance of the 26 core ASVs binned at the family-level by sampling date. (B) Core ASVs were significantly more stable than noncore ASVs across all pairwise comparisons of samples from the same individual (Kruskal–Wallis test: χ2 = 105.37, P < .001). Significant differences are denoted by * (* = P < .05; ** P < .01, and *** P < .001).
Bacterial families associated with creosote bush feeding
Creosote bush was the most abundant diet item across seasons (Fig. 1B), consistent with the fact that the majority of woodrats at Whitewater construct their nest in this plant. We identified two microbial families, Eggerthellaceae (GLM: t = 3.727, pseudo-R2 = 0.123, P < .001) and Erysipelotrichaceae (GLM: t = 4.797, pseudo-R2 = 0.188, P < .001) whose relative abundance was positively correlated with the abundance of creosote bush in diet (Figure S9, Supporting Information).
Foraging preferences
The woodrats in the cafeteria trial weighed an average of 180 ± 29 g (standard deviation) and consumed an average of 84 ± 7% of the freshly clipped plant material presented. We found that woodrats had a higher foliar intake of the two ephemeral plant species than creosote bush (ANOVA: F2:15 = 16.24, P < .001; Fig. 5). The ancestry (q) of this woodrat cohort was unknown at the time of the experiment, but later found to consist of a range of ancestry classes consisting of several parental N. bryanti (q = 1.00, 0.97, 0.96), a backcross N. bryanti (q = 0.71), an early generation hybrid (q = 0.53), and a backcross N. lepida (q = 0.38). This ancestry composition is fairly representative of the ancestry distribution of the entire population (Klure et al. 2023). Despite a small sample size and a lack of representation of parental N. lepida, we found that woodrats with more N. lepida ancestry consumed a greater amount of creosote bush during the cafeteria trial (LM: F1:6 = 6.46, R2 = 0.56, P < .05; Figure S10, Supporting Information), consistent with known interspecific differences in their respective tolerance to creosote bush from this site (Dearing et al. 2022).
Figure 5.

Cafeteria trial conducted at Whitewater, CA over 24 h with seven adult woodrats and three plant species. Woodrats preferentially consumed more of the forb species, M. glubrata and B. tournefortii than the shrub, L. tridentata (ANOVA: F2:18 = 16.24, P < .001). Significant differences are denoted by * (* = P < .05; ** P < .01, *** P < .001).
Discussion
We understand little about the temporal dynamics of the gut microbiome of animals in nature, and if present, whether temporal variation in gut microbial community composition is largely stochastic or rather indicative of underlying ecological processes. This gap in knowledge is largely due to the scarcity of studies that survey the gut microbiomes of individuals repeatedly in nature. Therefore, we investigated the stability of the gut microbiome of individual woodrats at a seasonal shrubland in the Mojave Desert. We found exceptionally high turnover of ASVs within the gut microbiome of individuals, but substantially less turnover at higher taxonomic ranks. We also found a strong signal of individuality in gut microbiome composition, but this signal was lost during seasonal restructuring. Additionally, despite a high level of overall turnover in the gut microbiome, we identified a highly stable and abundant core community of microbes that likely aids woodrats in feeding on plants. We discuss these findings and their implications in greater detail below.
Gut microbial community stability is low amongst microbial ASVs and high at the rank of family
At the deepest taxonomic resolution (ASV), we found high turnover of microbial taxa within individuals over time, where most individuals retained less than half of their ASVs across sampling periods spanning as little as 2–4 months. These results are similar to levels of turnover in the gut microbiomes of other small mammals in nature such as red squirrels (T. hudsonicus; Bobbie et al. 2017) and meerkats (S. suricatta; Risely et al. 2022). Additionally, we found no relationship between microbial turnover and time between sampling, which is comparable to that reported from a 13-year longitudinal survey of baboons (Ren et al. 2017). These findings indicate that the gut microbiome is highly dynamic and shifts in taxonomic composition, especially at a species and strain resolution, may be largely stochastic. Possible explanations for this phenomenon may be that ASVs with high turnover contribute little to essential functions of the gut microbiome, or may they be merely “passing through” the gut and are not established residents of the community.
In comparison, when assessed at a higher taxonomic resolution (family), we found substantially lower turnover in gut microbial communities with most turnover occurring in taxa with low abundance. This difference may be the result of selection by the host for the maintenance of core functions in the gut microbiome but not for subspecies diversity. The phylogenetic conservation of function among bacteria is highly variable, dependent on environmental conditions, and influenced by horizonal gene transfer (Louca et al. 2018). Furthermore, the genic content of closely related bacterial strains can vary considerably (Lladó Fernández et al. 2018). However, deeper phylogenetic conservation is known for microbial enzymes that metabolize carbohydrates, fats, and proteins (Koren et al. 2013) and for glycoside hydrolases that breakdown complex polysaccharides such as cellulose and chitin (Berlemont and Martiny 2015). One of the many essential services provided by gut bacteria to mammalian herbivores is the degradation of complex polysaccharides derived from their plant-based diet (Dearing and Kohl 2017). The higher stability of gut microbiota in woodrats at higher taxonomic ranks may indicate selection for the maintenance of essential bacterial functions for digesting plant matter. The most abundant diet item of woodrats at Whitewater is creosote bush, which is both high in fiber and toxic secondary metabolites, primarily phenolics (Meyer and Karasov 1989, Hyder et al. 2002, Arteaga et al. 2005, Dearing et al. 2022). Thus, the successful metabolism of creosote bush foliage likely requires a high diversity of both host and microbial enzymes. Woodrats rely on their gut microbiota to metabolize creosote bush resin and avoid toxicity (Kohl et al. 2014b), further indicating that selection to maintain core functions may be essential in woodrats to meet their nutritional demands. This woodrat–creosote bush system presents a valuable opportunity for additional experimental validation of the functions of core gut microbes as woodrats are amenable to experimental manipulations (Stapleton et al. 2022) and retain a majority of their wild microbiota from nature when in captivity (Martínez-Mota et al. 2020).
Gut microbial communities are individualized and impacted by environmental factors
Host identity played a major role in the composition and stability of the gut microbiome of woodrats in addition to strong effects of season and diet. In longitudinal studies of wood mice (A. sylvaticus) and meerkats (S. suricatta), gut microbiome composition was individualized, especially over short time scales spanning a few months (Marsh et al. 2022, Risely et al. 2022). Additionally, in spotted hyenas (C. crocuta), gut microbial communities progressively individualized throughout the life of an animal and may play a role in facilitating host preferences in diet (Rojas et al. 2022). The formation of an individualized gut microbiome could stem from a variety of factors such as limited transmission of microbiota between hosts, strong host genetic control over the assemblage of gut microbiota and host behavior. Teasing apart the relative contribution of such factors in structuring microbial communities is difficult; however, a few of the measurements taken in this study may provide some insight. For example, we included estimates of genome-wide ancestry in our modeling of microbiome variance, yet found only a modest influence of host genotype on microbiome composition. Thus, as interspecific variation was inadequate to explain variation in gut microbiome composition, it is highly unlikely that there is sufficient interindividual genetic variation to facilitate the individuality we detected in gut microbiome composition. These results are in contrast to another hybrid zone between N. bryanti and N. lepida, Whitney Well, where gut microbial communities are largely species-specific (Nielsen et al. 2022). However, parental individuals at Whitewater are less genetically differentiated, extensively overlap in habitat use and diet, and experience a higher rate of interspecific hybridization than parental individuals at Whitney Well (Klure et al. 2023). These population dynamics may limit the assemblage of species-specific gut microbiota. Conversely, it is possible that the limited sample size of woodrats with N. lepida ancestry in this dataset limited our ability to detect any potential cryptic interspecific differences in gut microbial profiles. Nevertheless, at Whitewater we believe that seasonal changes in interindividual microbial transmission rates among woodrats may be playing a more substantial role in structuring gut microbial communities.
For much of their life, woodrats are solitary and defend their nest sites against congeners (Cameron 1971, Kinsey 1977), which may limit the transmission of microbiota across individuals. Asociality is reduced during the wet season when breeding occurs, approximately between January and April at Whitewater based on the presence of juvenile individuals at the site. During this period, the individualized signature of the gut microbiome disappeared and the capture location of each woodrat explained additional variation in gut microbiome composition. This suggests that the seasonal sociality of woodrats may increase the transmission of gut microbiota across hosts. Additionally, the wet season brings with it a plethora of changes in both abiotic and biotic conditions including drastic changes in diet that likely influence microbial community composition and may induce seasonal restructuring of the gut microbiome.
We found that diet explained a portion of the variation in gut microbiome composition, which has also been documented in several other animal species (Maurice et al. 2015, Grieneisen et al. 2021, Víquez-R et al. 2021, Weinstein et al. 2021). Therefore, it is likely that changes observed in diet across seasons likely contribute to the seasonal restructuring of the gut microbiome of woodrats. In the dry season at Whitewater, woodrats have a less diverse diet dominated primarily by two fibrous and perennial shrubs, creosote bush, and its root hemiparasite white rhatany (Krameria bicolor; Cannon 1911). In comparison, in the wet season, woodrats have a more diverse diet rich in ephemeral plants such as evening primrose (Oenothera spp.), mustards (Brassica spp.), and asters (family Asteraceae). Interestingly, gut microbial diversity followed an inverse pattern of diet diversity, where it was higher in the dry season and lower in the wet season. This negative relationship between microbiome and diet diversity may reflect the season-dependent nutritional quality of plant resources at Whitewater. Ephemeral plants tend to have increased digestibility as they are typically lower in recalcitrant fiber content and less toxic due to less investment in chemical defense compared to perennial shrubs (Meissner and Paulsmeier 1995, Johnstone et al. 2002). In fact, in the cafeteria trial we showed that woodrats at Whitewater preferred herbaceous plants over creosote bush when available, possibly due to an avoidance of creosote bush due to its toxicity (Arteaga et al. 2005), its nutritional quality, or both. Similarly, reduced gut microbiome diversity is seen in several animal species (including humans) feeding on diets low in fiber (Deehan and Walter 2016, Sonnenburg et al. 2016). In contrast, during the dry season, we found that the gut microbial communities of woodrats were more diverse. During this period these microbial communities are exposed to a higher diversity of challenging dietary substrates including complex plant polysaccharides and secondary metabolites that may select for a high diversity of microbial metabolic processes, potentially driving an increase in the overall diversity of the community.
The strong individuality in gut microbiome composition we detected in the dry season also may be influenced by autocoprophagy in woodrats reinforcing individual-specific patterns of gut microbiota assemblage. Coprophagous behavior may be more frequent in woodrats in the dry season as a mechanism to increase nutrient uptake from nutritionally poor sources. Indeed, we have noticed in our laboratory feeding trails that woodrats seem to increase fecal ingestion with increasing levels of creosote bush resin in the diet (Dearing, personal observation). Moreover, this pattern has been demonstrated in the greater cane rat (T. swinderianus), where it increases coprophagic behavior in response to increases in fiber in its diet (Zyi and Delport 2010). Additionally, studies on the role of coprophagy in modulating the gut microbiome in Brandt’s voles (L. brandtii) found that coprophagy stabilizes microbial communities and when prevented, induces dysbiosis (Bo et al. 2020). Therefore, it is probable that seasonal differences in the frequency or type of coprophagy may influence the gut microbiome of woodrats, potentially strengthening the individuality of gut microbial communities during nutrient challenging periods and diminishing its individual signal during the wet season when allocoprophagy may be more common.
Despite turnover, a core microbial community exists in the gut
We identified an abundant core microbial community belonging to a high diversity of bacterial families, the majority of which are rich in fibrolytic functions. These core ASVs were highly abundant and accounted for a disproportionate amount of the total read abundance across samples (∼20%) despite only representing ∼10% of the average diversity within individual gut microbial communities. Approximately two-thirds of the core ASVs belonged to a single family, Muribaculaceae (previously S24-7), a common taxon in the guts of homeothermic animals, and typically the most abundant family in the gut microbiome of woodrats (Kohl et al. 2014b, Weinstein et al. 2021, Stapleton et al. 2022). This family harbors a versatile functional profile with regard to the degradation of complex carbohydrates (Lagkouvardos et al. 2019) and contains tropic guilds with distinct hydrolases that specialize on the degradation of plant glycans (Ormerod et al. 2016). Other core ASVs belonged to additional fibrolytic families such as Lactobacillaceae, Ruminococcaceae, and Erysipelotrichaceae that have rich functional profiles for the degradation of plant glycans (Biddle et al. 2013, Despres et al. 2016, Wu et al. 2021). This function of the microbiome could be critical to herbivorous hosts in accessing the limited dietary protein contained within plant cell walls; therefore, the stability of core ASVs may be due to selection from the host to maintain essential microbial functions in the gut for processing plant foliage, and in particular for processing creosote bush.
Creosote bush is the most abundant item in the diets of woodrats at Whitewater, occurring in animals in every sampling period, and as such may be a key factor in facilitating the stability of core microbes in the guts of woodrats. As previously described, core microbes likely aid woodrats in the processing of complex polysaccharides present in a diet high in creosote bush, but are also likely involved in its detoxification in the gut. Several core microbes belonged to the families Lactobacillaceae and Eggerthellaceae that produce enzymes that degrade plant toxins. In particular, Lactobacillaceae possess a diverse profile of aryl-alcohol dehydrogenases that detoxify phenolics (Kohl et al. 2014b) and Eggerthellaceae produce a diverse set of enzymes that degrade polyphenols (Rodríguez-Daza et al. 2020) including resveratrol (Bode et al. 2013), an inducible metabolite produced by plants in response to herbivory. One of the most compelling examples of the potential for members of Eggerthellaceae to degrade toxic plant metabolites come from studies on Eggerthella lenta. This bacterial species readily degrades cardiac glycosides in the human gut to such an efficiency that it can metabolize digoxin (also commercially known as Digitalis or Lanoxin), a plant-derived medicine used to treat cardiac abnormalities, before its absorption into the blood stream (Haiser et al. 2013). In fact, the abundance of Eggerthellaceae was positively correlated with the abundance of dietary creosote bush, further suggesting it may play a role in its metabolism. In addition to creosote bush and its secondary metabolites, woodrats at Whitewater also consume significant quantities of white rhatany that is chemically defended by a diverse suite of phenolics (Hyder et al. 2002, Jiménez-Estrada et al. 2013). Therefore, in addition to potential fibrolytic services, the core microbial community likely aids woodrats in detoxifying dietary phenols, enabling them to capitalize on toxic diets when less toxic plant species are unavailable.
Conclusion
The gut microbiome of individual woodrats consists of both stable and dynamic microbes. It responds to changes in both environmental abiotic conditions and changes in host behavior. At Whitewater, woodrat gut microbial communities become progressively individualized during highly xeric and nutritional challenging periods, then experience seasonal restructuring during the wet season. This seasonal restructuring may facilitate adaptive plasticity to its host to season-dependent nutritional challenges (Baniel et al. 2021). Woodrats at Whitewater also contain a set of core microbial taxa that may facilitate their ability to capitalize on low digestible and highly toxic diets. This study expands our understanding on the seasonal dynamics of the gut microbiome of wild animals and by incorporating comprehensive measurements of host genetics and behavior, further demonstrates that these seasonal dynamics may be indicative of synchronous ecological processes that may contribute to the adaptive plasticity of the host. Lastly, these results are the product of a limited survey of individual woodrats from a single habitat, and as such, additional longitudinal studies are necessary to identify whether the gut microbial communities of other small mammal species are shaped by similar ecological processes.
Supplementary Material
Acknowledgement
We are grateful to M. Doolin, M. Nelson, T. Stapleton, C. Kohlschein, and C. Hernandez for aid in sample collection. Drs. Talia Karasov, Marjorie Matocq and Danny Nielson provided helpful comments on analyses. We thank the Wildlands Conservancy and preserve manager Kerry Puckett for access to the Mission Creek Preserve. We are grateful to the staff at the University of Illinois at Chicago Sequencing Core for assistance with library preparation and sequencing.
Contributor Information
Dylan M Klure, School of Biological Sciences, University of Utah, 257 S 1400 E rm 201, Salt Lake City, UT, 84112, United States.
M Denise Dearing, School of Biological Sciences, University of Utah, 257 S 1400 E rm 201, Salt Lake City, UT, 84112, United States.
Ethics statement
Woodrat trapping and handling was permitted by the California Department of Fish and Wildlife Scientific Collection Permits SC-008123 and SC-1379S, and University of Utah Institutional Animal Use and Care Committee protocol 19-01005. All animal handling protocols followed the guidelines set by the American Society of Mammologists (Sikes 2016).
Conflict of interest
The authors declare no competing interests.
Funding
This work was supported by the National Science Foundation through DEB award 1342615 and IOS award 1656497 to M.D.D. and the Independent Research/Development Program. D.M.K. was supported by the National Institutes of Health through a T32 Institutional Training Grant (Utah Training Program in Genetics: T32GM141848) and a University of Utah Graduate School Research Fellowship.
Data Availability
The reduced representation sequence data used for genome-wide ancestry analyses is available from the NCBI SRA under BioProject PRJNA767020. The 16S rRNA and chloroplast trnL sequence data with associated metadata is available from the Dryad Data Platform under DOI: 10.5061/dryad.ghx3ffbss.
References
- Abt MC, Pamer EG. Commensal bacteria mediated defenses against pathogens. Curr Opin Immunol. 2014;29:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga S, Andrade-Cetto A, Cárdenas R. Larrea tridentata (Creosote bush), an abundant plant of Mexican and US-American deserts and its metabolite nordihydroguaiaretic acid. J Ethnopharmacol. 2005;98:231–9. [DOI] [PubMed] [Google Scholar]
- Baniel A, Amato KR, Beehner JCet al. Seasonal shifts in the gut microbiome indicate plastic responses to diet in wild geladas. Microbiome. 2021;9:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- Berlemont R, Martiny AC. Genomic potential for polysaccharide deconstruction in bacteria. Appl Environ Microb. 2015;81:1513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddle A, Stewart L, Blanchard Jet al. Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity. 2013;5:627–40. [Google Scholar]
- Bo TB, Zhang XY, Kohl KDet al. Coprophagy prevention alters microbiome, metabolism, neurochemistry, and cognitive behavior in a small mammal. ISME J. 2020;14:2625–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobbie CB, Mykytczuk NCS, Schulte-Hostedde AI. Temporal variation of the microbiome is dependent on body region in a wild mammal (Tamiasciurus hudsonicus). FEMS Microbiol Ecol. 2017;93:81. [DOI] [PubMed] [Google Scholar]
- Bode LM, Bunzel D, Huch Met al. In vivo and in vitro metabolism of trans-resveratrol by human gut microbiota. Am J Clin Nutr. 2013;97:295–309. [DOI] [PubMed] [Google Scholar]
- Bonferroni CE. Teoria Statistica Delle Classi e Calcolo Delle Probabilità. 8:Seeber, 1936. 3–62.Pubbl. R Ist. Sup. Sci. Econ. Commer. Fir. [Google Scholar]
- Bray JR, Curtis JT. An ordination of the Upland Forest communities of Southern Wisconsin. Ecol Monogr. 1957;27:325–49. [Google Scholar]
- Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11:2639–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJet al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron GN. Niche overlap and competition in woodrats. J Mammal. 1971;52:288–96. [PubMed] [Google Scholar]
- Cannon WL. The Root Habits of Desert Plants. Vol. 129. Washington: Carnegie Institution of Washington, 1911. [Google Scholar]
- Caporaso JG, Lauber CL, Walters WAet al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 2011;108:4516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel T, Lagkouvardos I, Blaut Met al. The mouse gut microbiome revisited: from complex diversity to model ecosystems. Int J Med Microbiol. 2016;306:316–27. [DOI] [PubMed] [Google Scholar]
- Cusick JA, Wellman CL, Demas GE. The call of the wild: using non-model systems to investigate microbiome–behaviour relationships. J Exp Biol. 2021;224. 10.1242/JEB.224485/263930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearing MD, Kohl KD. Beyond fermentation: other important services provided to endothermic herbivores by their gut microbiota. Integr Comp Biol. 2017;57:723–31. [DOI] [PubMed] [Google Scholar]
- Dearing MD, Orr TJ, Klure DMet al. Toxin tolerance across landscapes: ecological exposure not a prerequisite. Funct Ecol. 2022;36:2119–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deehan EC, Walter J. The fiber gap and the disappearing gut microbiome: implications for Human nutrition. Trends Endocrinol Metabol. 2016;27:239–42. [DOI] [PubMed] [Google Scholar]
- Despres J, Forano E, Lepercq Pet al. Xylan degradation by the human gut Bacteroides xylanisolvens XB1AT involves two distinct gene clusters that are linked at the transcriptional level. BMC Genomics. 2016;17. 10.1186/S12864-016-2680-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14:927–30. [Google Scholar]
- Doolin ML, Weinstein SB, Dearing MD. Pinworms are associated with taxonomic but not functional differences in the gut microbiome of white-throated woodrats (Neotoma albigula). J Parasitol. 2022;108:408–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egoscue HJ. The desert woodrat: a laboratory colony. J Mammal. 1957;38:472–81. [Google Scholar]
- Flint HJ, Bayer EA, Rincon MTet al. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Micro. 2008;6:121–31. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Nguyen BN, Neumann LMet al. Gut microbiome differences between wild and captive black rhinoceros—implications for rhino health. Sci Rep. 2019;9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman EA. Revision of the wood rats of the genus Neotoma. North Am Fauna. 1910;31:1–124. [Google Scholar]
- Gomez A, Petrzelkova K, Yeoman CJet al. Gut microbiome composition and metabolomic profiles of wild western lowland gorillas (Gorilla gorilla gorilla) reflect host ecology. Mol Ecol. 2015;24:2551–65. [DOI] [PubMed] [Google Scholar]
- Grieneisen L, Dasari M, Gould TJet al. Gut microbiome heritability is nearly universal but environmentally contingent. Science. 2021;373:181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiser HJ, Gootenberg DB, Chatman Ket al. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science. 2013;341:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks AL, Lee KJ, Couto-Rodriguez Met al. Gut microbiomes of wild great apes fluctuate seasonally in response to diet. Nat Commun. 2018;9:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hird SM. Evolutionary biology needs wild microbiomes. Front Microbiol. 2017;8. 10.3389/fmicb.2017.00725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyder PW, Fredrickson EL, Estell REet al. Distribution and concentration of total phenolics, condensed tannins, and nordihydroguaiaretic acid (NDGA) in creosotebush (Larrea tridentata). Biochem Syst Ecol. 2002;30:905–12. [Google Scholar]
- Jaccard P. The distribution of flora in the alpine zone. New Phytol. 1912;11:37–50. [Google Scholar]
- Jiménez-Estrada M, Velázquez-Contreras C, Garibay-Escobar Aet al. In vitro antioxidant and antiproliferative activities of plants of the ethnopharmacopeia from northwest of Mexico. BMC Complem Altern Med. 2013;13. 10.1186/1472-6882-13-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone J, Russell DE, Griffith B. Variations in plant forage quality in the range of the porcupine caribou herd. Ran. 2002;22:83–91. [Google Scholar]
- Kinsey KP. Agonistic behavior and social organization in a reproductive population of Allegheny Woodrats, Neotoma floridana magister. J Mammal. 1977;58:417–9. [Google Scholar]
- Klure DM, Greenhalgh R, Parchman TLet al. Hybridization in the absence of an ecotone favors hybrid success in woodrats (Neotoma spp.). Evolution. 2023;77:959–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl KD, Dearing MD, Bordenstein SR. Microbial communities exhibit host species distinguishability and phylosymbiosis along the length of the gastrointestinal tract. Mol Ecol. 2018;27:1874–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl KD, Dearing MD. Experience matters: prior exposure to plant toxins enhances diversity of gut microbes in herbivores. Ecol Lett. 2012;15:1008–15. [DOI] [PubMed] [Google Scholar]
- Kohl KD, Luong K, Dearing MD. Validating the use of trap-collected feces for studying the gut microbiota of a small mammal (Neotoma lepida). J Mammal. 2015;96:90–3. [Google Scholar]
- Kohl KD, Miller AW, Marvin JEet al. Herbivorous rodents (Neotoma spp.) harbour abundant and active foregut microbiota. Environ Microbiol. 2014;16:2869–78. [DOI] [PubMed] [Google Scholar]
- Kohl KD, Skopec MM, Dearing MD. Captivity results in disparate loss of gut microbial diversity in closely related hosts. Conserv Physiol. 2014;2:cou009–. 10.1093/CONPHYS/COU009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl KD, Weiss RB, Cox Jet al. Gut microbes of mammalian herbivores facilitate intake of plant toxins. Ecol Lett. 2014;17:1238–46. [DOI] [PubMed] [Google Scholar]
- Koren O, Knights D, Gonzalez Aet al. A guide to enterotypes across the human body: meta-analysis of microbial community structures in Human microbiome datasets. PLOS Comput Biol. 2013;9:e1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Statist Assoc. 1952;47:583–621. [Google Scholar]
- Lagkouvardos I, Lesker TR, Hitch TCAet al. Sequence and cultivation study of muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome. 2019;7:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc JG, Milani C, de Giori GSet al. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. 2013;24:160–8. [DOI] [PubMed] [Google Scholar]
- Lladó Fernández S, Větrovský T, Baldrian P. The concept of operational taxonomic units revisited: genomes of bacteria that are regarded as closely related are often highly dissimilar. Folia Microbiol. 2019;64:19–23. [DOI] [PubMed] [Google Scholar]
- Louca S, Polz MF, Mazel Fet al. Function and functional redundancy in microbial systems. Nat Ecol Evol. 2018;2:936–43. [DOI] [PubMed] [Google Scholar]
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microb. 2005;71:8228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie VJ, Song SJ, Delsuc Fet al. The effects of captivity on the mammalian gut microbiome. Integr Comp Biol. 2017;57:690–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight DT, Huerlimann R, Bower DSet al. Methods for normalizing microbiome data: an ecological perspective. Methods Ecol Evol. 2019;10:389–400. [Google Scholar]
- McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217. 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLOS Comput Biol. 2014;10:e1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh KJ, Raulo AM, Brouard Met al. Synchronous seasonality in the gut microbiota of wild mouse populations. Front Microbiol. 2022;13:809735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.j. 2011;17:10. [Google Scholar]
- Martínez-Mota R, Kohl KD, Orr TJet al. Natural diets promote retention of the native gut microbiota in captive rodents. ISME J. 2020;14:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice CF, Knowles S, Ladau Jet al. Marked seasonal variation in the wild mouse gut microbiota. ISME J. 2015;9:2423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner HH, Paulsmeier DV. Plant compositional constituents affecting between-plant and animal species prediction of forage intake. J Anim Sci. 1995;73:2447–57. [DOI] [PubMed] [Google Scholar]
- Meyer MW, Karasov WH. Antiherbivore chemistry of Larrea tridentata: effects on Woodrat (Neotoma Lepida) feeding and nutrition. Ecology. 1989;70:953–61. [Google Scholar]
- Morgan M, Anders S, Lawrence Met al. ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics. 2009;25:2607–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murillo T, Schneider D, Fichtel Cet al. Dietary shifts and social interactions drive temporal fluctuations of the gut microbiome from wild redfronted lemurs. ISME Commun. 2022;2:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqib A, Poggi S, Wang Wet al. Making and sequencing heavily multiplexed, high-throughput 16S ribosomal RNA gene amplicon libraries using a flexible, two-stage PCR protocol. Methods Mol Biol. 2018;1783:149–69. [DOI] [PubMed] [Google Scholar]
- Nguyen TLA, Vieira-Silva S, Liston Aet al. How informative is the mouse for human gut microbiota research?. Dis Model Mech. 2015;8:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DP, Harrison JG, Byer NWet al. The gut microbiome reflects ancestry despite dietary shifts across a hybrid zone. Ecol Lett. 2022;26:1–13. [DOI] [PubMed] [Google Scholar]
- Orkin JD, Campos FA, Myers MSet al. Seasonality of the gut microbiota of free-ranging white-faced capuchins in a tropical dry forest. ISME J. 2019;13:183–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormerod KL, Wood DLA, Lachner Net al. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome. 2016;4:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol. 2016;18:1403–14. [DOI] [PubMed] [Google Scholar]
- Parchman TL, Gompert Z, Mudge Jet al. Genome-wide association genetics of an adaptive trait in lodgepole pine. Mol Ecol. 2012;21:2991–3005. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing, 2022. [Google Scholar]
- Ren T, Boutin S, Humphries MMet al. Seasonal, spatial, and maternal effects on gut microbiome in wild red squirrels. Microbiome. 2017;5:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risely A, Schmid DW, Müller-Klein Net al. Gut microbiota individuality is contingent on temporal scale and age in wild meerkats. Proc R Soc B. 2022;289:20220609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Daza MC, Roquim M, Dudonné Set al. Berry polyphenols and fibers modulate distinct microbial metabolic functions and gut microbiota enterotype-like clustering in obese mice. Front Microbiol. 2020;11:2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas CA, Holekamp KE, Jasso MVet al. Taxonomic, genomic, and functional variation in the gut microbiomes of wild spotted hyenas across 2 decades of study. mSystems. 2022;8. 10.1128/MSYSTEMS.00965-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt E, Mykytczuk N, Schulte-Hostedde AI. Effects of the captive and wild environment on diversity of the gut microbiome of deer mice (Peromyscus maniculatus). ISME J. 2019;13:1293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27:623–56. [Google Scholar]
- Shurtliff QR, Murphy PJ, Matocq MD. Ecological segregation in a small mammal hybrid zone: habitat-specific mating opportunities and selection against hybrids restrict gene flow on a fine spatial scale. Evolution. 2014;68:729–42. [DOI] [PubMed] [Google Scholar]
- Sonnenburg ED, Smits SA, Tikhonov Met al. Diet-induced extinctions in the gut microbiota compound over generations. Nature. 2016;529:212–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton TE, Kohl KD, Denise Dearing M. Plant secondary compound- and antibiotic-induced community disturbances improve the establishment of foreign gut microbiota. FEMS Microbiol Ecol. 2022;98:1–13. [DOI] [PubMed] [Google Scholar]
- Taberlet P, Coissac E, Pompanon Fet al. Power and limitations of the chloroplast trn L (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 2007;35:e14–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J. 2017;474:1823–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Víquez-R L, Speer K, Wilhelm Ket al. A faithful gut: core features of gastrointestinal microbiota of long-distance migratory bats remain stable despite dietary shifts driving differences in specific bacterial taxa. Microbiol Spectr. 2021;9. 10.1128/SPECTRUM.01525-21/SUPPL_FILE/SPECTRUM01525-21_SUPP_1_SEQ12.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JMet al. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microb. 2007;73:5261–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein SB, Martínez-Mota R, Stapleton TEet al. Microbiome stability and structure is governed by host phylogeny over diet and geography in woodrats (Neotoma spp). Proc Natl Acad Sci USA. 2021;118. 10.1073/PNAS.2108787118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcoxon F. Individual comparisons by ranking methods. Biomet Bull. 1945;1:80–3. [Google Scholar]
- Wright ES. Using DECIPHER v2.0 to analyze big biological sequence data in R. R J. 2016;8:352–9. [Google Scholar]
- Wu J, Liu M, Zhou Met al. Isolation and genomic characterization of five novel strains of Erysipelotrichaceae from commercial pigs. BMC Microbiol. 2021;21:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Chen J. A comprehensive evaluation of microbial differential abundance analysis methods: current status and potential solutions. Microbiome. 2022;10:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yi N. NBZIMM: negative binomial and zero-inflated mixed models, with application to microbiome/metagenomics data analysis. BMC Bioinf. 2020;21:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zyi AV, Delport JH. Digestibility of nutrients and aspects of the digestive physiology of the Greater cane rat, Thryonomys swinderianus in two seasons. 2010;45:254–64. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The reduced representation sequence data used for genome-wide ancestry analyses is available from the NCBI SRA under BioProject PRJNA767020. The 16S rRNA and chloroplast trnL sequence data with associated metadata is available from the Dryad Data Platform under DOI: 10.5061/dryad.ghx3ffbss.