

Significance
Diverse microbial communities colonize plant roots. They feed on carbon-rich root exudates which contain a diverse mix of chemicals including primary and specialized metabolites. Here, we show that specialized metabolites act as selective antibiotics to shape the root bacterial communities. By growing single isolates of maize root bacteria in the presence of benzoxazinoids (BXs) in vitro, we find that the strains differ greatly in their tolerance to BXs. Their different levels of tolerance largely explained their abundance on BX-exuding roots. Our work shows how plant specialized metabolites act to shape the maize root microbial community and thus deepens our mechanistic understanding of how plants shape their microbiome.
Keywords: plant specialized metabolites, benzoxazinoids, maize, root microbiome, in vitro phenotyping
Abstract
Plants exude specialized metabolites from their roots, and these compounds are known to structure the root microbiome. However, the underlying mechanisms are poorly understood. We established a representative collection of maize root bacteria and tested their tolerance against benzoxazinoids (BXs), the dominant specialized and bioactive metabolites in the root exudates of maize plants. In vitro experiments revealed that BXs inhibited bacterial growth in a strain- and compound-dependent manner. Tolerance against these selective antimicrobial compounds depended on bacterial cell wall structure. Further, we found that native root bacteria isolated from maize tolerated the BXs better compared to nonhost Arabidopsis bacteria. This finding suggests the adaptation of the root bacteria to the specialized metabolites of their host plant. Bacterial tolerance to 6-methoxy-benzoxazolin-2-one (MBOA), the most abundant and selective antimicrobial metabolite in the maize rhizosphere, correlated significantly with the abundance of these bacteria on BX-exuding maize roots. Thus, strain-dependent tolerance to BXs largely explained the abundance pattern of bacteria on maize roots. Abundant bacteria generally tolerated MBOA, while low abundant root microbiome members were sensitive to this compound. Our findings reveal that tolerance to plant specialized metabolites is an important competence determinant for root colonization. We propose that bacterial tolerance to root-derived antimicrobial compounds is an underlying mechanism determining the structure of host-specific microbial communities.
Diverse communities of microbes including bacteria, fungi, oomycetes, and protists colonize plant roots (1–3). Collectively functioning as a microbiome, these microbes provide several benefits to their host. They can improve plant growth through the production of plant hormones (4, 5), improve plant nutrient uptake (6–8), and protect plants against pathogens (9–11). The root microbiome is mainly recruited from soil, and thus, its composition resembles the surrounding soil microbiome (1, 12). Plants further shape the composition of their root microbiome by root morphological traits (13, 14), immune responses (15, 16), and secretion of diverse root exudates (17–19). Through root exudation, plants release up to 25% of their assimilated carbon to the surrounding soil (20, 21), which attracts and nourishes soil microbes (22, 23). Root exudates contain sugars, amino acids, or organic acids but importantly also contain plant specialized metabolites (17, 18, 24).
Plant specialized metabolites govern the interactions of plants with the environment (25). Among numerous functions, they were shown to shape the root and rhizosphere microbiomes (17, 18, 26). Of note, microbiome structure is not solely defined by plant but also by microbe-derived specialized exometabolites (27). Studies on plant metabolites often compare microbiome composition on roots or rhizospheres of wild-type plants relative to biosynthesis mutants defective in the production of a specialized metabolite. Glucosinolates (28), camalexin (29), triterpenes (22), and coumarins (30–32) were shown to structure the root microbiome of the model plant Arabidopsis thaliana. Sorgoleone exuded by sorghum (Sorghum bicolor) (33), gramine produced by barley (Hordeum vulgare) (34), and benzoxazinoids (BXs) (35–38), diterpenoids (39), zealexins (40), and flavonoids (14) released by maize (Zea mays) shape the root microbial communities of their respective hosts. While it is well documented that plant specialized metabolites present drivers for root microbiome assembly, the underlying mechanisms that explain community structure remain largely unknown.
BXs are specialized compounds produced by sweet grasses (Poaceae), which include important crops such as maize, wheat, and rye (41). These indole-derived alkaloids are especially abundant in young maize seedlings and actively growing tissues, accounting for up to 1% of plant dry weight (42). Besides structuring the root and rhizosphere microbiomes (35–38), BXs defend against insect pests and pathogens and play a role in defense signaling (43). Further, BXs function as phytosiderophores by chelating iron and thus improving plant nutrition (44). The major BX exuded by maize to the surrounding rhizosphere is DIMBOA-Glc (37). In SI Appendix, Table S1, we document all abbreviated BX compounds (like DIMBOA, AMPO,...) with their full names and structures. In the rhizosphere, it is deglycosylated to DIMBOA and rapidly converted to MBOA (6-methoxy-benzoxazolin-2-one), where it is stable for days to weeks (45). Through microbial activity, MBOA is further converted to AMPO, which then accumulates and remains detectable in soil for months to years (45–47). AMPO, an aminophenoxazinone, has allelopathic function by suppressing the growth of neighboring plants (45, 48–50). While maize primarily synthesizes methoxylated BXs, rye or barley mainly produce nonmethoxylated analogs following the same chemical conversion pathway [DIBOA-Glc > DIBOA > BOA > APO; SI Appendix, Table S1; (41, 48, 51)].
BXs are known to affect microbes. Diverse responses, mainly of individual bacterial strains, have been reported for various BX compounds. On one end, BXs are toxic as demonstrated with pure compounds such as MBOA that inhibited the growth of bacteria like Streptococcus aureus and Escherichia coli (52). On the other end, DIMBOA-derived compounds affect bacterial behavior. They were shown to reduce bacterial motility and biofilm formation of the bacterial pathogen Ralstonia solanacearum (53). DIMBOA was shown to act as a chemoattractant for host localization by the beneficial rhizobacterium Pseudomonas putida (54). HDMBOA, a dimethoxylated form of DIMBOA, was found to reduce the virulence of the pathogen Agrobacterium tumefaciens (55). A few studies have addressed how bacteria cope with BX compounds. For instance, strains of Pantoea and Bacillus species can degrade BXs and convert them into various metabolites (51, 56). Some other bacteria were found to tolerate BXs, such as e.g. Photorhabdus, an endosymbiotic bacterium of nematodes, that tolerates high levels of MBOA (57). Mechanistically, this tolerance to MBOA was based on an aquaporin-like membrane channel aqpZ. Finally, the antimicrobial activity of BXs strongly differs among taxonomically diverse bacteria, as shown for Arabidopsis root strains when testing their growth in the presence of the nonmethoxylated BX compounds BOA and APO (58). While this study convincingly revealed strain-level antimicrobial activity of BXs, it remains unclear whether bacterial tolerance to plant specialized metabolites could explain the structure of root microbiomes.
Given that BXs generally structure the microbiome of maize roots (35–38), that exudation of BX leads to accumulation of MBOA in the rhizosphere (37), and that bacteria widely differ in their tolerance to BXs (58), we hypothesized that bacterial tolerance to BXs, in particular to MBOA, explains the BX-dependent community structure of the maize root microbiome. To test this hypothesis, we established a strain collection from roots of BX-exuding maize plants, similar to culture collections of other plant species including Arabidopsis (59), clover (60), rice (61), lotus (62), and maize (63, 64). Although strain collections only represent the culturable fraction of microbiomes, they allow to bridge cultivation with culture-independent methodologies and thereby present powerful resources for the in-depth studying of the molecular mechanisms in microbiomes (65). In a second step, we screened the maize root bacteria (MRB) for their tolerance against pure BXs and aminophenoxazinones present in the maize rhizosphere. Among the 52 isolates tested, we found a broad spectrum of strain-level tolerances to MBOA without a phylogenetic clustering. Mapping these strains to root microbiome profiles revealed that their levels of tolerance to MBOA largely explained their BX-dependent abundance on maize roots. Overall, our findings reveal that a bacterium’s tolerance to the root-derived antimicrobials is a key trait for defining its abundance in the root microbiome.
Results
BX Exudation Does Not Alter Bacterial Community Size on Maize Roots.
Microbial communities can vary in size (microbiome members collectively increase or decrease in abundance without changing composition) and/or composition (some microbiome members increase, while others decrease without affecting community size). BXs shape community composition as shown by classical amplicon sequencing, which measures relative abundances (35–38). However, BX effects on community size remained unknown, but given their antimicrobial activities (58), we hypothesized that BXs may also reduce the overall community size. Thus, we quantified bacterial community size which represents the total number of microbial cells i) by plating root extracts and scoring colony-forming units and ii) by quantitative PCR measurements of bacterial DNA relative to plant root DNA. We compared roots of BX-exuding (wild-type) and BX-deficient (bx1 mutant; see Materials and Methods) maize in two greenhouse experiments with field soil. Additionally, we quantified bacterial community size using DNA extracts of earlier field experiments with wild-type and bx1 plants (35). For the greenhouse experiments, the cultivation-dependent quantifications of bacterial cell numbers and the load of bacterial DNA on roots did not differ between the two genotypes—findings that were further confirmed by the field samples (SI Appendix, Fig. S1). Thus, we concluded that BX exudation, at least under the conditions tested, does not affect community size of the bacterial maize root microbiome.
Culture Collection Covers Abundant Bacteria of the Maize Root Microbiome.
We built a culture collection of MRB (hereafter, MRB collection) isolated from roots of wild-type B73 maize grown in natural soil in the greenhouse. We used the same soil from the Changins site (CH) where the structuring of the maize root microbiome by BXs was first observed (37). We determined the taxonomy of each isolate by sequencing parts of the 16S rRNA gene using Sanger technology. The MRB collection consists of 151 bacterial isolates representing 17 taxonomic families across the five major phyla Pseudomonadota (n = 69), Actinobacteriota (n = 56), Bacillota (n = 23), Bacteroidota (n = 2), and Deinococcota (n = 1; Fig. 1). Among those were typical root-colonizing families such as Pseudomonadaceae, Microbacteriaceae, Rhizobiaceae, and Sphingomonadaceae (1). To investigate how the MRB strains corresponded to community members (amplicon sequence variants, ASV or operational taxonomic units, OTU) in profiles of maize roots, we mapped the 16S rRNA Sanger sequences to microbiome profiles of roots, from which they were isolated (37). This analysis revealed that MRB strains mapped to members that accounted for 24% of total microbiome abundance at the sequence level (SI Appendix, Fig. S2A) with 112 isolates being abundant (>0.1% abundance, respectively) and 34 low abundant (<0.1%) community members (Fig. 1, inner ring).
Fig. 1.
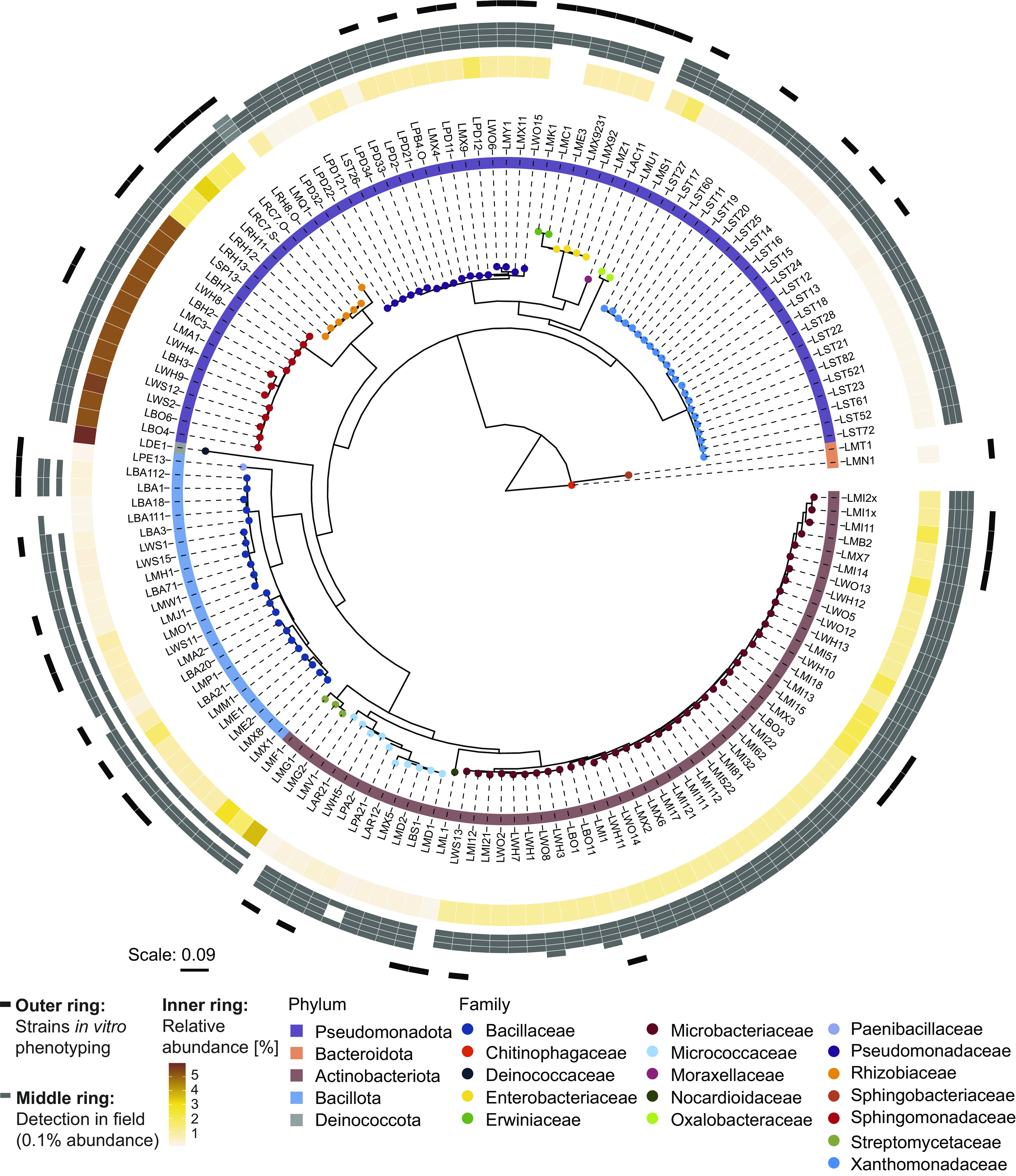
MRB collection. Maximum likelihood phylogeny, constructed from the alignment of 16S rRNA gene sequences. Leaf nodes are colored by family taxonomy, and the ring next to the strain IDs reports phylum taxonomy. The rings on the outside of the tree show the abundance of the strains in different microbiome datasets: The inner ring is colored according to the relative abundance (%) of the corresponding sequence in the microbiome profile of the roots, from which the isolates were isolated from (origin of isolation). Gray boxes in the middle ring mark when a strain was detected (>0.1% abundance) in root microbiome datasets of maize grown in greenhouse and field experiments (Changins field data from ref. (37); Reckenholz and Aurora data from ref. (35)) or a greenhouse experiment with field soil (Sheffield data from ref. (36)). The order of datasets (references) in the ring with gray boxes: inside to outside. The outer ring denotes in black the representative strains used for tolerance assays.
Next, we assessed whether the MRB isolates could be detected in root microbiomes of maize grown in the field and other soils. We mapped the 16S rRNA sequences of the MRB isolates to the following microbiome datasets: field-grown maize in Changins (26), Reckenholz (both CH), and Aurora in the United States (35), and a pot experiment with field soil from a location in Sheffield, United Kingdom (36) (Fig. 1, middle ring). Consistent with the pot experiments for isolation, the majority of MRB isolates (139/151) also mapped to abundant (>0.1%) root microbiome members of maize grown in the field in Changins from where the soil was used for the isolation experiments (SI Appendix, Fig. S2B). Similarly, most MRB isolates were also detected as abundant members in maize root microbiomes in other field soils from Switzerland (117/151), the United States (140/151), and the United Kingdom (84/151). Isolates belonging to Pseudomonadaceae, Microbacteriaceae, and Oxalobacteraceae were detected in all four soils as abundant members. Taken together, these results show that the MRB collection taxonomically covers the major bacterial families and largely represents the abundant members of maize root microbiomes in different field soils.
MBOA Is the Most Selective Exudate Compound Inhibiting MRB In Vitro.
To investigate the tolerance of MRB to BXs, we exposed MRB strains to varying compound concentrations and measured bacterial growth based on optical density in liquid cultures over time. We calculated the area under the bacterial growth curve [area under the curve (AUC)], normalized it to the growth in the control treatment, and defined a tolerance index (TI) for each strain across all tested concentrations (See Materials and Methods and SI Appendix, Fig. S3 A–C). We screened a representative set of 52 isolates (Fig. 1, outer ring) with this in vitro growth assay testing purified DIMBOA-Glc as well as synthetic MBOA and AMPO. We did not include DIMBOA, the bioactive aglycon of DIMBOA-Glc (37), as it was fully converted to MBOA in the absence of bacteria during an assay (SI Appendix, Fig. S3D). In contrast to defining medium inhibitory concentrations (IC50), the TI method allows tolerance comparisons among strains including ones that are not inhibited at the highest tested concentration. The maximal TI value 1 indicates that a strain is not inhibited. We defined bacteria with TI values 0.75 to 1 as tolerant, with values 0.5 to 0.75 as strains of intermediate tolerance, and values <0.5 classify susceptible strains. SI Appendix, Fig. S3C exemplifies the approach with the MBOA-tolerant Pseudomonas LPD2 (inhibited only at the highest concentrations, TI = 0.88) and the MBOA-susceptible Rhizobium LRC7.O (inhibited already at the lowest concentration, TI = 0.25).
Fig. 2 reports the tolerance indices of all tested MRB strains to DIMBOA-Glc, MBOA, and AMPO (underlying growth data and statistical analysis in concomitant SI Appendix, Supplementary Figures). For DIMBOA-Glc, the main compound secreted by maize roots (SI Appendix, Table S1), we could only test two concentrations due to the limited availability (500 and 2,500 µM). The majority of MRB strains was tolerant to DIMBOA-Glc (TI > 0.75, 45/52 strains), half of the strains benefiting with improved growth (TI > 1, 26/52; Fig. 2A and SI Appendix, Fig. S4). Only six strains showed intermediate tolerance to DIMBOA-Glc predominantly belonging to the family of Bacillaceae, while only one strain was strongly inhibited by DIMBOA-Glc.
Fig. 2.

Taxa- and compound-specific tolerance of MRB to BXs and aminophenoxazinones. TIs of individual MRB strains to (A) DIMBOA-Glc, (B) MBOA, and (C) AMPO; their chemical structures are shown in (D). Bargraphs report the mean TIs ±SE of n = 3 (DIMBOA-Glc) or n = 6 (MBOA and AMPO) replicates. See concomitant SI Appendix, Supplementary Figures for the underlying growth data and the pairwise statistical analyses. Colors by family taxonomy.
In contrast to DIMBOA-Glc, MBOA, which is the major compound detected in the rhizosphere (SI Appendix, Table S1) and known for its antimicrobial activity (52, 54, 57), strongly inhibited more than a third of the strains (22/52 strains) and moderately affected another third of the MRB strains (18/52 strains; Fig. 2B and SI Appendix, Fig. S5). The most susceptible strains belonged to the Rhizobiaceae and Moraxellaceae families. Only 12 strains were tolerant to MBOA (TI > 0.75), belonging to Pseudomonadaceae, Bacillaceae, and Microbacteriaceae. Strains belonging to the same family typically showed a similar tolerance level to MBOA (Fisher exact test: tolerance group ~ family, P < 0.001). Among Microbacteriaceae, we found the strongest phenotypic heterogeneity, ranging from the most tolerant strain LMI1x to the second most susceptible strain LMI1 and with many strains of intermediate tolerance.
The broad range of tested MBOA concentrations permitted to validate the robustness of the TI approach. We tested whether TI-based findings were i) affected by the number of concentrations used for calculation and ii) whether the TI is primarily driven by the high concentrations. For this, we recalculated bacteria’s tolerances either including only 3 concentrations (defined as TI3conc) or excluding data of the two highest concentrations (defined as TIlow). Both rankings of the strains by TI3conc (SI Appendix, Fig. S6A) and TIlow (SI Appendix, Fig. S6B) resembled the findings based on the original TI calculated on all concentrations (Fig. 2B). Direct comparisons revealed that both TI3conc and TIlow correlated strongly with the original TI (SI Appendix, Fig. S6 C and D). Hence, the TI is also robust with only three concentrations (as in the case of DIMBOA-Glc), and it is not specifically biased by testing also very high concentrations. Further, this analysis indicates that future screenings can be done with fewer and lower concentrations of MBOA.
Finally, we tested the aminophenoxazinone AMPO, the direct microbial metabolization product of MBOA that accumulates at low levels in the rhizosphere (SI Appendix, Table S1). Because AMPO is insoluble at concentrations >50 µM, it was not tested up to the same concentrations as DIMBOA-Glc and MBOA. Most MRB strains were tolerant (43/52 strains) or only moderately affected (6/52 strains), and only 3 strains were susceptible to AMPO (TI < 0.5; Fig. 2C and SI Appendix, Fig. S7 A and B). The affected strains belonged to Bacillaceae and Micrococcaceae (Fisher exact test: tolerance group ~ family, P < 0.05). Comparing bacterial tolerances of AMPO with MBOA revealed a weak, yet significant negative correlation, meaning that AMPO-tolerant bacteria were not necessarily also MBOA-tolerant (SI Appendix, Fig. S7C). We grew these bacteria in a separate experiment in equimolar amounts of MBOA or AMPO for directly comparing the toxicities of both compounds (SI Appendix, Fig. S8). While only one strain (LWO6) was affected in its growth (AUC > 0.75) by 50 µM MBOA, twenty strains were impacted in their growth by the same concentration of AMPO, indicating that AMPO is more toxic than MBOA. However, AMPO only accumulates at very low levels in the rhizosphere (SI Appendix, Table S1), and we only find a small fraction of the MRB to be susceptible (TI < 0.5) to this compound (Fig. 2C). This, together with the 14× higher amounts of MBOA in the rhizosphere (SI Appendix, Table S1) and the main finding that MRB exhibited the broadest range of tolerances to MBOA (Fig. 2B), lead us to conclude that MBOA is the most selective BX compound in the rhizosphere.
Compound-Specific Growth Inhibition of MRB In Vitro.
We also examined bacterial tolerance to nonmethoxylated compound analogs of MBOA and AMPO: BOA and APO (SI Appendix, Table S1). Given the fact that methoxy groups have been reported to increase the reactivity of BXs (66), we hypothesized that the nonmethoxylated compounds BOA and APO would exert weaker antimicrobial activity on the MRB than their methoxylated relatives MBOA and AMPO. For BOA, most strains were tolerant (16/52 strains) or moderately tolerant (30/52 strains), while only 6 strains were susceptible (SI Appendix, Fig. S9 A and B). Hence, bacterial tolerance was generally higher to BOA compared to MBOA (Fig. 2B). Similar taxonomic groups were susceptible (Rhizobiaceae) or tolerant (Bacillaceae) to both BOA (Fisher exact test: tolerance group ~ family, P < 0.001) and MBOA. This finding was further supported by a significant positive correlation of the TIs of all MRB isolates to these two related compounds (SI Appendix, Fig. S9C).
The spectrum of bacterial tolerance to APO included a large proportion of tolerant MRB strains (37/52 strains), a few moderately affected (7/52 strains), and a few susceptible isolates (8/52; SI Appendix, Fig. S10 A and B). Overall, bacterial tolerance was lower to APO than AMPO (Fig. 2C), indicating that APO is more toxic than its methoxylated relative AMPO. Again, we find similar taxonomic groups to share APO (Fisher exact test: tolerance group ~ family: P < 0.05) and MBOA tolerances, which were significantly correlated (SI Appendix, Fig. S10C). The positive correlations of bacterial tolerances between the non- and methoxylated compound pairs (BOA/MBOA and APO/AMPO) suggest similar modes of toxicity and tolerance. Bacterial tolerances did not correlate between the nonmethoxylated compounds BOA and APO (SI Appendix, Fig. S10D). In summary, we found higher bacterial tolerance to nonmethoxylated BOA (compared to MBOA), while the MRB tolerated the nonmethoxylated APO less than AMPO. Hence, the methoxy group (66) increases the toxicity of MBOA but not the aminophenoxazinones AMPO.
Root Bacteria from Non-BX-Exuding Plants Are Less BX-Tolerant.
Having identified that MRB selectively tolerate MBOA, the most abundant compound accumulating in the rhizosphere (Fig. 2B and SI Appendix, Table S1), we hypothesized that bacteria from a non-BX-exuding plant should be less tolerant to BXs than bacteria isolated from a BX-exuding host. To test this hypothesis, we mapped the 16S rRNA sequences from MRB to the strain collection of Arabidopsis bacteria [AtSphere collection (59)] and selected the most similar bacteria and tested them for tolerance to MBOA. We found that most strains (33/53) were clearly susceptible to MBOA and that 16/53 were moderately affected (Fig. 3A and SI Appendix, Fig. S11). Only 4/53 AtSphere strains were tolerant to MBOA compared to 12/52 tolerant MRB strains (Fig. 2B). Comparing the taxonomic groups of both collections, we found the AtSphere strains of the families Enterobacteriaceae, Microbacteriaceae, Pseudomonadaceae, and Sphingomonadaceae to be significantly less tolerant to MBOA than the corresponding MRB strains (Fig. 3B). In the MRB collection, strains of these families showed higher tolerance to MBOA. For the other families, we found no differences in tolerance between the two collections, presumably because these groups are per se tolerant (Bacillaceae) or susceptible (Micrococcaceae and Rhizobiaceae) to MBOA. These results show that bacteria isolated from a BX-exuding host are generally more tolerant to MBOA than bacteria isolated from a non-BX-exuding plant, which is suggestive of adaptation to host-specific exudate metabolites.
Fig. 3.
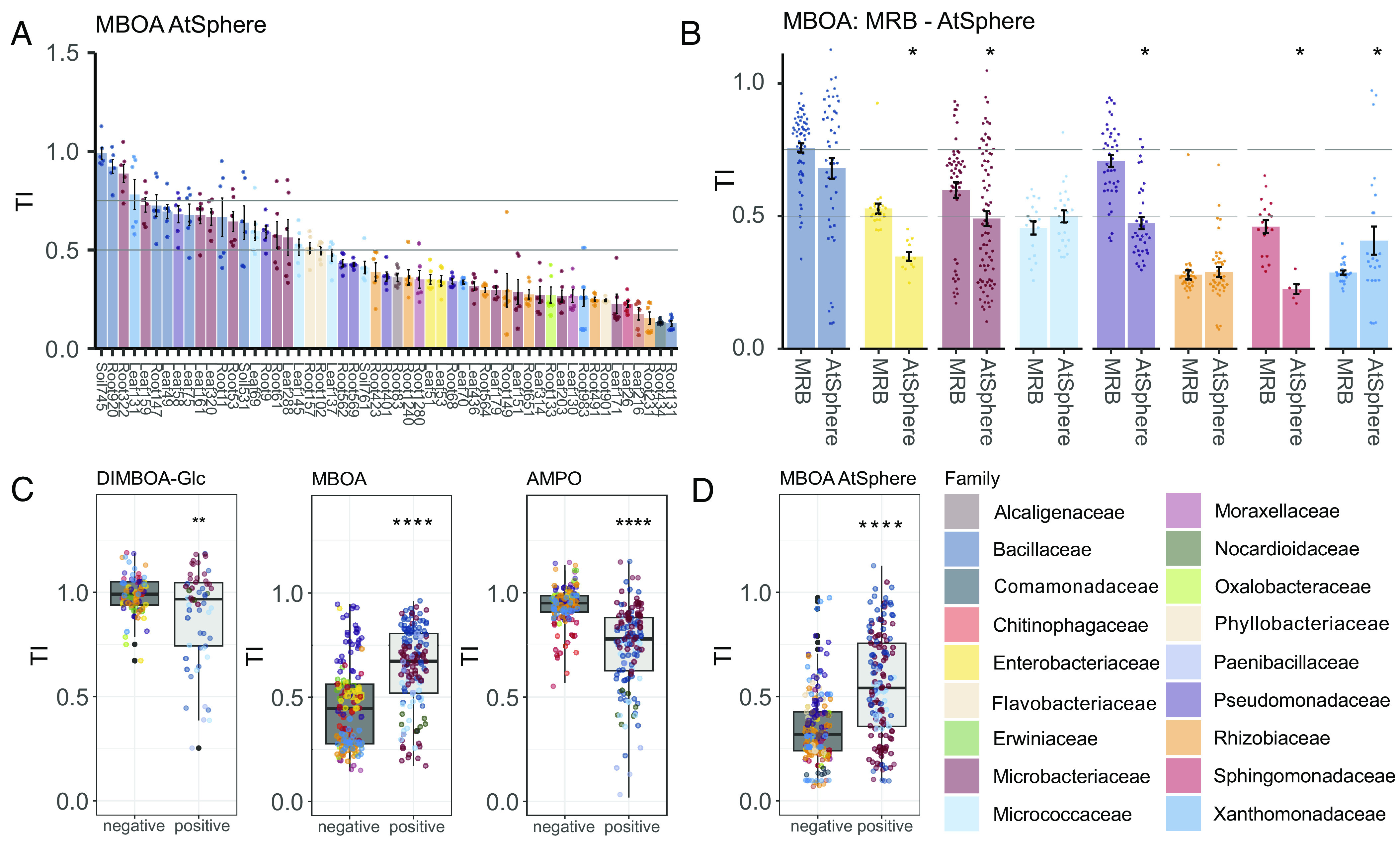
Host-specific tolerance of plant bacteria and cell wall structure defining tolerance. (A) TIs of Arabidopsis bacteria of the AtSphere collection to MBOA. Bargraph reports the mean TIs ±SE of 6 replicates. See SI Appendix, Fig. S12 for the underlying growth data and the pairwise statistical analyses. (B) Comparison of TIs to MBOA of MRB and AtSphere strains, summarized by taxonomic families. Strains are represented based on 6 replicate measurements, statistically compared using t tests (asterisks denote significance: P < 0.05*). (C) TIs to DIMBOA-Glc, MBOA, and AMPO summarized for gram-negative and gram-positive MRB strains and (D) AtSphere strains. Graphs report replicate measurements per strain (n = 3 for DIMBOA-Glc, n = 6 for MBOA and AMPO) and statistical analysis (t test, asterisks denote significance: P < 0.01**, P < 0.0001****). Colors by family taxonomy.
Bacterial Tolerance to BXs Is Related to Their Cell Wall Structure.
Since we found taxonomically related strains to have similar tolerance levels to different BX compounds (Fig. 3B), we tested whether tolerance depended on phylogenetically distinctive features such as cell wall structure. Cell walls of gram-positive bacteria are characterized by a thick peptidoglycan layer, while gram-negative bacteria have a thin peptidoglycan layer located between an inner and an outer membrane. Gram-positive MRB isolates were significantly more tolerant to MBOA (Fig. 3C) and BOA (SI Appendix, Fig. S12A) compared to the gram-negative ones. We found the opposite for AMPO (Fig. 3C) and APO (SI Appendix, Fig. S12B), as well as for DIMBOA-Glc (Fig. 3C) with gram-negative bacteria being more tolerant. Consistently, gram-positive AtSphere strains were also more tolerant to MBOA than gram-negatives (Fig. 3D). Together, this suggests that cell wall structure can partially explain the tolerance patterns of the different bacteria to BXs and aminophenoxazinones.
Bacterial Tolerance to MBOA Explains BX-Dependent Abundance on Maize Roots.
To test the hypothesis that bacterial tolerance to BXs explains the BX-dependent structuring of maize root microbiomes, we squared our in vitro BX tolerance data with microbiome profiles of BX-exuding wild-type and BX-deficient bx1 maize lines. These microbiome profiles were from the same plants from which most of the MRB strains were isolated from (26). After mapping the 16S rRNA sequences of the MRB strains to the OTUs of the sequencing data, we first tested for differences in their mean abundances between wild-type and bx1 roots and rhizospheres (SI Appendix, Fig. S13). OTUs of the Bacillaceae and Microbacteriaceae were generally enriched on wild-type plants, while Xanthomonadaceae and Rhizobiaceae OTUs were depleted—a finding, reminiscent of the in vitro tests where these families contained BX-tolerant or nontolerant strains, respectively (Fig. 2). Next, we correlated the TIs of the MRB strains to the different BX compounds with the abundance changes of their corresponding OTUs on wild-type vs. bx1 microbiomes. Our specific focus lay in determining which BX compounds would best explain the differential abundances of the OTUs. Bacterial tolerance to MBOA correlated best and significantly positively with BX-dependent abundance in root and rhizosphere microbiomes (Fig. 4 A and B). This means that MBOA-tolerant strains mapped to abundant OTUs on BX-exuding roots and their rhizospheres while susceptible strains correspond to low abundant OTUs. Fig. 4C summarizes the comparisons for the other tested compounds. Also, a positive correlation was found for bacterial tolerance to BOA (SI Appendix, Fig. S14A), which was weaker than MBOA. In contrast, bacterial tolerance to AMPO correlated negatively with BX-dependent abundance in root and rhizosphere microbiomes, while tolerance to APO did not correlate (SI Appendix, Fig. S14 B and C). Finally, bacterial tolerance to DIMBOA-Glc did not correlate with BX-dependent abundance (SI Appendix, Fig. S14D), allowing to conclude that DIMBOA-Glc, despite being the most abundant compound in maize root exudates (SI Appendix, Table S1), is not explaining BX-dependent structuring of maize microbiomes. It is noteworthy that the strongest positive correlation was found for MBOA, which is both the most abundant compound accumulating in the rhizosphere (SI Appendix, Table S1) and the compound to which we found the broadest range of bacterial tolerances (Fig. 2B). We conclude that tolerance to MBOA explained best the bacterial abundance on BX-exuding vs. BX-deficient roots and that MBOA—with its antibiotic activity—acts as a key driving factor to structure the maize root microbiome.
Fig. 4.
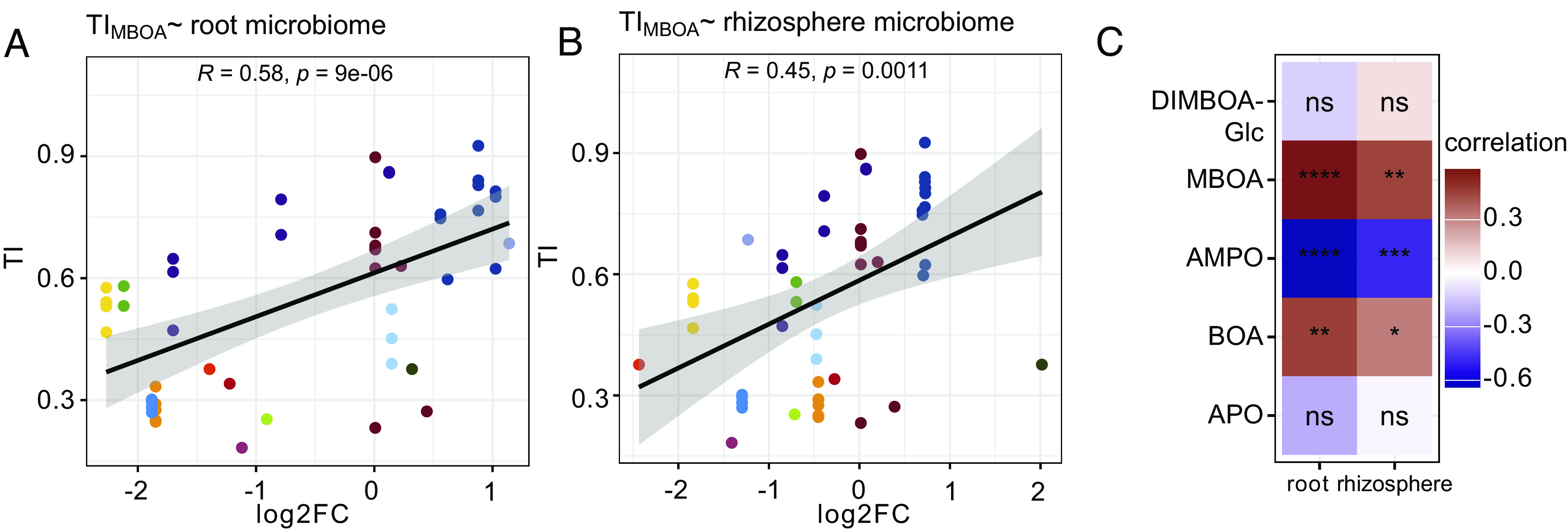
Correlations between in vitro tolerance and BX-dependent abundance in root and rhizosphere microbiomes. Correlations between TIs of all MRB strains to MBOA with the log2 fold changes (log2FC; comparing wild-type vs. bx1 plants) of their corresponding OTUs in (A) root and (B) in rhizosphere microbiome profiles. The correlation coefficient R and the P-value of the Pearson’s product-moment correlation test are reported inside the panels. Each data point reports the mean TI of a strain (means based on n = 6 replicates), colors by family taxonomy. The heatmap in (C) summarizes this analysis for all tested compounds (see SI Appendix, Fig. S14 for additional correlation analyses with DIMBOA-Glc, AMPO, BOA, and APO). Colors refer to the correlation coefficient R, and the statistical significance is indicated by asterisks (levels: ns = not significant, P < 0.05*, P < 0.01**, P < 0.001***, and P < 0.001****).
Discussion
Maize plants shape the composition of their root microbiome through root BX exudation (35–38). To investigate the underlying mechanisms, we established a strain collection of MRB and tested their tolerance to BXs using in vitro growth assays. We show that BXs selectively act as antimicrobials on members of the maize root microbiome. Further, we find that bacterial tolerance to the major BX in the rhizosphere (MBOA) correlates with their abundance on BX-exuding maize roots. Below, we discuss the underlying mechanisms and biological implications of our findings.
BXs Act as Selective Antimicrobials and Are Better Tolerated by Bacteria Isolated from a BX-Exuding Host Plant.
Various plant specialized metabolites have been studied for their selective antimicrobial activities against root microbes. However, such metabolite–microbe interactions are rarely interpreted for native vs. nonhost contexts. Nonhost refers to context where the root microbes and root metabolites do not belong to the same host. Nonhost examples include that coumarins inhibit the growth of the bacterial pathogen R. solanacearum (isolated from tobacco leaves) (67, 68) or that beneficial Pseudomonas simiae (isolated from wheat rhizosphere) and Pseudomonas capeferrum (isolated from potato rhizosphere) tolerate high levels of coumarins, compounds that are not produced by tobacco, wheat or potato (31). Also, the metabolite–microbe interaction of BXs and Arabidopsis bacteria (58) is a nonhost example because Arabidopsis does not produce BXs. Here, the nonmethoxylated BXs compounds BOA and APO, both specialized metabolites of wheat and rye, were found to selectively inhibit the growth of nonhost Arabidopsis bacteria in a strain- and compound-specific manner. Fewer studies considered native host context where root metabolites and root microbes belong to the same plant species. A native host example is the study of Arabidopsis root bacteria for their tolerance to coumarins (30). The highly antimicrobial coumarin fraxetin inhibited the growth of bacterial strains belonging to the Burkholderiaceae, while this was not the case for the less toxic scopoletin. We are not aware of studies that specifically compared native vs. nonhost contexts.
For our study, testing maize and Arabidopsis bacteria for their tolerance against the specialized metabolites of maize, we considered both native and nonhost contexts. The screening in native host context revealed selective growth inhibition of MRB by BXs in an isolate- and compound-dependent manner (Fig. 2). We found highly selective antimicrobial activities of the different BX compounds tested, even with strains that are tolerant to high concentrations of the most abundant and selective BX in the rhizosphere (MBOA). Tolerant strains mostly belonged to the gram-positive Bacillaceae, while gram-negative Xanthomonadaceae and Rhizobiaceae were generally susceptible to MBOA (Fig. 3C). This general pattern of tolerant gram-positive and susceptible gram-negative bacteria was consistent with our “nonhost” screening of Arabidopsis bacteria for MBOA tolerance (Fig. 3D). Also, the general conclusion of selective growth inhibition in an isolate-dependent manner applied to the Arabidopsis bacteria. Importantly, the direct comparison by host context revealed that the “nonhost” bacteria from Arabidopsis were generally less tolerant to the specialized compounds of maize than their “native host” counterparts isolated from maize (Fig. 3B). This finding suggests that members of the root microbiome have adapted to the antimicrobial root exudates of their host plant. Further research is required to test whether our results can be generalized, for instance, by screening the AtSphere (59) and MRB (this study) collections for their tolerance against specialized Arabidopsis compounds like coumarins. If adaptation to host antimicrobials were broadly applicable, this would imply that host-specific mechanisms have evolved in the genomes that enable root bacteria to tolerate host-specific substances. Therefore, genomic studies, e.g., comparing the genomes of Arabidopsis and MRB, should allow the identification of the underlying genetic components for tolerance to host-specific antimicrobial compounds.
Cell Wall Structure Defines BX Tolerance.
We found, both in native host and nonhost contexts, that gram-positive bacteria were more tolerant to MBOA than gram-negative bacteria (Fig. 3 C and D), revealing that cell wall properties are important for bacterial tolerance to BXs. For the tolerant gram-positive bacteria, this suggests that the thick peptidoglycan layer of the cell wall presents a first level of protection, possibly by preventing the entry of MBOA into the cell. Seemingly consistent, the cell wall of gram-negative bacteria consists of a thin peptidoglycan layer. This thin peptidoglycan layer is located between an inner and an outer membrane and possibly the exposure of the membranes explains the susceptibility of gram-negative bacteria to BXs. Little is known about the mode of action of BXs against microorganisms (66, 69). Suggested mechanisms of BX toxicity include intercalation with DNA, chelation, or active import into microbial cells due to their siderophore-like function. For the overall activity, the lipophilicity of BXs, which influences diffusion across cellular membranes, is important. Our work supports this with the finding of the opposed tolerances of bacteria to the less lipophilic (M)BOA vs. the more lipophilic A(M)PO (Fig. 3C and SI Appendix, Fig. S9F). Hence, we infer that bacterial membranes are key factors for BX toxicity.
Mechanisms of BX Tolerance.
There is a key mechanistic question emerging from the finding of opposed tolerances of gram-negative vs. gram-positive bacteria: How do generally susceptible gram-negative bacteria gain tolerance to BXs? Options for increased tolerance are related to the transport of the compounds across the membranes, either by blocking their import (if an active process) or by exporting them out of the cell. The recent study by ref. (57) provided the first mechanistic insights from studying how the gram-negative Photorhabdus bacterium tolerates MBOA (57). Analysis of isolates with increased MBOA tolerance, which were selected after experimental evolution in the presence of MBOA, revealed multiple membrane-related mechanisms for BX tolerance. For instance, a mutation in the aquaporin-like membrane channel aqpZ conferred MBOA tolerance, which suggests a mechanism related to preventing BX import. Alternative to transport, a third possible mechanism for increased tolerance would be to metabolize the compounds. More in-depth studies are needed to uncover the underlying mechanisms of bacterial tolerance to BXs. Again, strain collections like the AtSphere (59) and MRB (this study), will be powerful resources to investigate, e.g., the BX metabolizing traits root bacteria using phenotypic and genomic strain comparisons.
Tolerance to Specialized Compounds Explains Community Structure.
Root microbiome structure is thought to arise from plant and microbial processes, both involving small metabolites. Microbes, for instance, contribute to community structure by competing with their microbiome peers based on specialized exometabolites. Such antimicrobial compounds were recently shown to act as competence determinants for Pseudomonas bacteria in the root microbiome (27). Bulgarelli et al. (1) proposed a two-step selection model for the plant processes with the central idea that root exudates “fuel an initial substrate-driven community shift in the rhizosphere, which converges with host genotype–dependent fine-tuning” of the root microbiome (1). The first step is relatively well understood, where the microbial recruitment from the surrounding soil, also known as the “rhizosphere effect”, is mostly fueled by primary metabolites that function as carbon substrates for microbial growth. For step two, plant specialized metabolites were repeatedly identified as key factors for microbiome assembly among several other drivers (15, 16). Typically, these conclusions stem from shifts in microbiome profiles of plant mutants that lack certain specialized metabolites compared to wild-type plants (22, 31, 32, 35–38). However, the mechanisms by which plant specialized metabolites shape microbiome composition and the processes that regulate microbial community structure remain poorly understood. Here, we provide evidence that bacterial tolerance to plant specialized metabolites explains characteristic microbiome composition. We systematically determined the tolerances of MRB to BXs (Fig. 2 and SI Appendix, Fig. S9) and found that bacterial tolerance to MBOA, the most abundant antimicrobial metabolite in exudates of their host plant, correlated significantly with their abundance on BX-exuding roots (Fig. 4). This was not the case for DIMBOA-Glc (SI Appendix, Fig. S14D), despite being the most abundant compound in maize root exudates (SI Appendix, Table S1). Thus, our results indicate that MBOA-tolerance is the important trait for abundant colonization of maize roots, even if further experiments would be required to validate this finding. This could include the use of synthetic communities composed of strains with different BX tolerance levels and exposing them to different BXs or inoculate axenic roots of wild-type and mutant maize with BX tolerant and susceptible strains and measure the BX-dependent colonization.
Linking Bacterial Tolerance to the Establishment of a Healthy Root Microbiome.
Many specialized metabolites of plants have dual functions, both suppressing pathogens and recruiting beneficials. Thus, their exudation is an important tool for plants to steer the establishment of a healthy root microbiome (70). Such dual functions are also known for coumarins and BXs. Scopoletin, the dominant coumarin in the rhizosphere of Arabidopsis, inhibits the soil-borne fungal pathogens Fusarium oxysporum and Verticillium dahlia while promoting P. simiae and P. capeferrum, both beneficial rhizobacteria (31). Similarly, BXs inhibit fungal pathogens Setosphaeria turtica, Exserohilum turcicum, and Fusarium spp. (43) and reduce the virulence of the phytopathogenic bacterium A. tumefaciens (55). At the same time, BXs enrich beneficial bacteria (14, 31, 54, 70, 71) by acting as chemoattractant for the beneficial rhizobacterium P. putida to maize roots (54). Although most of these examples are from nonhost contexts, they suggest that plants assemble a health-promoting root microbiome with the exudation of their specialized compounds. The established MRB collection allowed us to study this also in a native host context. Here, the screening of the MRB strains for BX tolerance revealed indications toward the establishment of a healthy root microbiome with “pathogen suppression” and “recruitment of beneficials”. Suppression of pathogen implies their susceptibility to BXs, which is what we have observed for all Agrobacterium strains in the collection (Fig. 2). Analogously, recruitment of beneficial implies that they tolerate the BXs, which is what we see for isolates of Pseudomonas and Bacillus (Fig. 2), both families with known beneficial plant bacteria. Now, functional work, e.g., exploring the functional plant phenotypes of the MRB strains, will corroborate the links between susceptibility and tolerance to BXs with pathogen suppression and recruitment of beneficials, respectively.
In conclusion, based on our results and the general facts that many specialized metabolites of plants have an antimicrobial function (22, 30–32, 66) and that many of them are key factors shaping community structure (22, 31, 32, 35–38), we propose that bacterial tolerance to root-derived antimicrobials is a mechanism that determines host-specific microbial community composition.
Materials and Methods
Quantification of Root Bacterial Community Size.
We quantified the size of the bacterial community on roots of B73 and bx1 mutant plants from two independent experiments (SI Appendix, Table S2) by plating the cultivable bacteria and by quantitative PCR. See SI Appendix, Supplementary Methods and Table S2 for details. The mutant bx1 is defective in BX1, a homologue of the tryptophan synthase alpha that catalyzes the formation of indole as the precursor for the BX biosynthesis pathway. BX1 commits the first step of BX biosynthesis and is considered the branchpoint from primary metabolism, and therefore, the mutant bx1 is ideal to interrupt the pathway (41, 72). The bx1 mutant was repeatedly shown to be defective in BXs (37) accumulating >10% of wild-type levels (residual BXs are due to alternative synthesis of indole; see ref. (73)).
Establishment of MRB Culture Collection.
The culture collection of MRB (Dataset S1) was built with strains isolated in five independent experiments (SI Appendix, Table S2). All strains were isolated from greenhouse pot experiments with Changins soil, i.e., batches of the same soil where we first demonstrated the microbiome structuring activity of BXs (37). Most strains originate from wild-type maize plants (inbred line B73), and a small subset of strains was isolated from BX-deficient bx1(B73) plants. Most strains were isolated from “dirty roots” including the root and the rhizosphere fraction (marked as “RoRh”, Dataset S1), and a few strains were isolated from washed roots, rhizosphere, or soil extracts (described in SI Appendix, Supplementary Methods). The extracts were diluted from 1:10–3 to 1:10–6 in 10 mM MgCl2 for plating so that spreading of 50 μL extract with a delta cell spreader (Sigma-Aldrich, St. Louis, USA) resulted in a density of 100 to 300 colony forming units on square plates (12 × 12 cm, Greiner bio-one, Kremsmünster, Austria). The media used for isolation are listed in SI Appendix, Table S4. The plates were incubated at room temperature (23 °C) for 5 to 10 d; single colonies were picked and re-streaked on full-strength tryptic soy broth (TSB; Sigma-Aldrich) or Luria-Bertani medium (LB; Carl Roth, Karlsruhe, Germany) until the isolates were visibly pure colonies. Bacterial strains were routinely sub-cultured at 25 °C to 28 °C in TSB or LB liquid or solid medium amended with 15 g/l agar (Sigma-Aldrich).
For cryopreservation, single colonies of pure strains were inoculated in full-strength liquid TSB or LB medium, grown for 2 d at 28 °C with 180 rotations per minute shaking, and then mixed with the same volume of 40% sterile glycerol (Sigma-Aldrich) in single screw cap microtubes (Sarstedt, Nürnbrecht, Germany). The resulting 20% glycerol stocks were slowly frozen down and stored at –80 °C. The same liquid cultures, of which the glycerol stocks were prepared, were used for Sanger sequencing-based isolate identification using the 16S rRNA gene as described in SI Appendix, Supplementary Methods and Table S3. All sequences together with taxonomies and metadata of the MRB are listed in Dataset S1.
We identified the MRB strains among OTUs or ASVs of published 16S rRNA gene profiles of maize root communities (35–37). See SI Appendix, Supplementary Methods for a detailed description of the mapping method. The sequence similarities of the strains to the microbiome members (ASVs) are listed in Dataset S2.
Phylogenetic analysis was performed with the Sanger-based 16S rRNA gene sequences. They were first concatenated, then aligned using MAFFT v. 7.475 (74) with default options, and analyzed with RAxML v. 8.2.12 (75). The multithreaded version “raxmlHPC-PTHREADS” was used with the options “-f a -p 12345 -x 12345 -T 23 -m GTRCAT” with 1,000 bootstrap replicates. The phylogenetic tree was visualized (Fig. 1) and annotated in R (package ggtree, 102).
To make the MRB collection a useful resource for future studies, we selected a taxonomically representative subset of 54 isolates and sequenced, assembled, and annotated their genomes. Detailed assembly statistics including genome size, N50 (sequence length of the shortest contig at 50% of the total assembly length), number of genes, number of scaffolds, and GC content are listed in Dataset S1. We generated the genomes of the MRB strains in four efforts (Dataset S1) using PacBio and Illumina as detailed in SI Appendix, Supplementary Methods. To assemble the genomes, we used similar pipelines as described in detail in SI Appendix, Supplementary Methods. The raw sequencing data have been deposited in the European Nucleotide Archive (http://www.ebi.ac.uk/ena) with the study accession PRJEB65362 (sample IDs ERS16291034 to ERS16291087) and the genome assemblies at NCBI (http://www.ncbi.nlm.nih.gov/) under the BioProject ID PRJNA1009252 (Dataset S1).
The MRB strain collection is maintained in the laboratory of the authors, and we share it including upgrades (e.g., more strains or genomes) with the research community upon request.
High-Throughput Growth Phenotyping of MRB Strains.
To screen MRB strains for their tolerance against various BXs and degradation compounds, we developed a custom, high-throughput, in vitro, liquid culture–based growth system (76). In brief, many bacteria strains are cultured in parallel and in a replicated manner in many 96-well plates, which are handled with a stacker (BioStack 4, Agilent Technologies, Santa Clara, United States), so that the connected plate reader (Synergy H1, Agilent Technologies) records bacterial growth via optical density (OD600, absorbance at 600 nm) over time. In 50% TSB (SI Appendix, Table S4), we tested the following compounds and concentrations: i) DIMBOA-Glc (500 and 2,500 µM), purified from maize seedlings (purification method is detailed in SI Appendix, Supplementary Methods); ii) synthetic MBOA and BOA (both Sigma-Aldrich) at 250, 500, 625, 1,250, 2,500, and 5,000 μM; iii) AMPO at 10, 25, and 50 μM, synthesized in our lab following a published procedure (77) and iv) synthetic APO (Sigma-Aldrich) at 10, 25, 50, and 100 μM (SI Appendix, Table S5). We included controls with the solvent dimethyl sulfoxide (Sigma-Aldrich), of which the concentration was kept constant in all treatments. BX quantities were validated following an established analytical protocol (78).
We set up separate runs for the different compounds and in one run; we always tested all concentrations of a compound against 52 MRB strains. Every compound was repeated in at least 2 runs. The assay setup is further detailed in SI Appendix, Supplementary Methods. The bacterial growth data were analyzed in R (version 4.0, R core Team, 2016). For growth, we calculated the AUC [function auc() of package MESS (79)] and normalized growth in a treatment relative to the control. Such normalized bacterial growth data of a given concentration was statistically assessed (compound vs control) using one-sample t tests and P-values adjusted for multiple hypothesis testing. As a measure of tolerance of a given strain to a given compound, a specific TI was calculated from the normalized AUC values across all tested concentrations of that compound. This calculation uses again the auc() function taking the AUC across the normalized AUC values across all tested concentrations. Including the 0 µM controls, the TIs of DIMBOA-Glc was calculated based on 3 concentrations, TIs MBOA and BOA included 7 concentrations and TIs of AMPO and APO included 4 concentrations. For validations of the TI approach, we calculated TIs to MBOA i) including only 3 concentrations (0, 500, and 2,500 µM, defined as TI3conc) and ii) excluding data of the two highest concentrations (0 to 1,250 µM MBOA, defined as TIlow). In contrast to defining medium inhibitory concentrations (IC50), the TI method is more broadly applicable as it allows tolerance comparisons including strains that are not inhibited at the highest tested concentration. The TI ranges from 1 (full tolerance, no growth inhibition at highest tested concentration) to 0 (full susceptible, no growth at lowest tested concentration), and we classified strains as tolerant (TI ≥ 0.75), intermediate (0.75 > TI ≥ 0.5), or susceptible (TI < 0.5). TI variation was assessed across strains using ANOVA (TI ~ Strain) of which we report the false discovery rate corrected P values of pair-wise strain-to-strain comparisons (Tukey HSD test). TI variation was also assessed across tolerance classes testing for a taxonomic signal using a Fisher’s exact test (TI class ~ family). TIs were compared between different compounds using Pearson correlation. To test whether bacterial tolerance to BXs explains the BX-dependent structuring of maize root microbiomes (Fig. 3), we analyzed the TI data relative to the microbiome data of wild-type and bx1 mutant maize (37). After mapping the MRB strains to the sequencing data (SI Appendix, Supplementary Methods), we determined differential colonization of the corresponding OTUs on wild-type vs. bx1 roots and rhizospheres. Next, we correlated strain TI with differential colonization (log2FC) of corresponding OTUs using Pearson and its product-moment test.
All code used for statistical analysis and graphing is available from https://github.com/PMI-Basel/Thoenen_et_al_BX_tolerance. The following further R packages were used: Tidyverse (80), Broom (81), DECIPHER (82), DESeq2 (83), emmeans (84), ggthemes (85), multcomp (86), phyloseq (87), phytools (88), and vegan (89) in combination with some custom functions.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Acknowledgments
We thank Prof. Dr. Julia Vorholt (ETH Zurich) and Prof. Dr. Paul Schulze-Lefert (Max Planck Institute for Plant Breeding Research Cologne) for sharing the bacterial strains from the AtSphere collection. Big thanks go to Niklas Schandry (Gregor Mendel Institute Vienna, later Ludwig-Maximilians-Universität Munich) for the introduction to high-throughput chemical phenotyping and the sharing of code for the analysis of the bacterial growth curves. Further, we acknowledge Dr. Pamela Nicholson (next-generation sequencing platform University of Bern) for technical support with sequencing and Kerstin Schneeberger (Agroscope Zurich) for the help with genome assemblies. We thank Selma Cadot for the preparation of the root DNA samples used for quantitative PCR, Corinne Suter for the technical assistance with DNA extraction and culturing of bacteria, Sandro Rechsteiner for isolation of first MRB strains, and Florian Enz for plant maintenance. This work was supported by the Interfaculty Research Collaboration “One Health” of the University of Bern (www.onehealth.unibe.ch). This work was also supported by MINECO, Spain, RYC-2015–19154 and grant SEV‐2015‐0533 funded by MCIN/AEI/10.13039/501100011033 and by the CERCA Programme/Generalitat de Catalunya to I.R.-S.
Author contributions
L.T., M.E., and K.S. designed research; L.T., C.G., V.G., and G.D.-H. performed research; P.M., C.A.M.R., C.H.A., I.R.-S., and R.B. contributed new reagents/analytic tools; L.T., M.K., and J.W. analyzed data; and L.T., M.E., and K.S. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
Raw data and analysis code data have been deposited in GitHub (https://github.com/PMI-Basel/Thoenen_et_al_BX_tolerance) (90). Some study data available [The MRB strain collection is maintained in the laboratory of the authors, and we share it including upgrades (e.g., more strains or genomes) with the research community upon request].
Supporting Information
References
- 1.Bulgarelli D., Schlaeppi K., Spaepen S., van Themaat E. V. L., Schulze-Lefert P., Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 64, 807–838 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Lundberg D. S., et al. , Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulgarelli D., et al. , Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488, 91–95 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Lugtenberg B. J. J., Kamilova F., Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 63, 541–556 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Eichmann R., Eichmann R., Richards L., Schäfer P., Hormones as go-betweens in plant microbiome assembly. Plant J. 105, 518–541 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fabiańska I., Sosa-Lopez E., Bucher M., The role of nutrient balance in shaping plant root-fungal interactions: Facts and speculation. Curr. Opin. Microbiol. 49, 90–96 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Tao K., Tao K., Tao K., Kelly S., Radutoiu S., Microbial associations enabling nitrogen acquisition in plants. Curr. Opin. Microbiol. 49, 83–89 (2019). [DOI] [PubMed] [Google Scholar]
- 8.van der Heijden M. G. A., Martin F., Selosse M.-A., Sanders I. R., Mycorrhizal ecology and evolution: The past, the present, and the future. New Phytolog. 205, 1406–1423 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Durán P., et al. , Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 175, 973–983 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carrión V. J., et al. , Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 366, 606–612 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Pascale A., Proietti S., Pantelides I. S., Stringlis I. A., Modulation of the root microbiome by plant molecules: The basis for targeted disease suppression and plant growth promotion. Front. Plant Sci. 10, 1741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hacquard S., et al. , Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microbe 17, 603–616 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez P. A., et al. , Systems biology of plant-microbiome interactions. Mol. Plant 12, 804–821 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Yu P., et al. , Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation. Nat. Plants 7, 481–499 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Teixeira P. J. P., Colaianni N. R., Fitzpatrick C. R., Dangl J. L., Beyond pathogens: Microbiota interactions with the plant immune system. Curr. Opin. Microbiol. 49, 7–17 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Lebeis S. L., et al. , Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 349, 860–864 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Sasse J., Martinoia E., Northen T. R., Feed your friends: Do plant exudates shape the root microbiome? Trend Plant Sci. 23, 25–41 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Jacoby R. P., Koprivova A., Kopriva S., Pinpointing secondary metabolites that shape the composition and function of the plant microbiome. J. Exp. Bot. 72, 57–69 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pang Z., et al. , Linking plant secondary metabolites and plant microbiomes: A review. Front. Plant Sci. 12, 621276–621276 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Massalha H., Korenblum E., Korenblum E., Tholl D., Aharoni A., Small molecules below-ground: The role of specialized metabolites in the rhizosphere. Plant J. 90, 788–807 (2017). [DOI] [PubMed] [Google Scholar]
- 21.van Dam N. M., Bouwmeester H. J., Metabolomics in the rhizosphere: Tapping into belowground chemical communication. Trend Plant Sci. 21, 256–265 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Huang A. C., et al. , A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 364, eaau6389 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Zhalnina K., et al. , Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 3, 470–480 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Canarini A., et al. , Root exudation of primary metabolites: Mechanisms and their roles in plant responses to environmental stimuli. Front. Plant Sci. 10, 157–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erb M., Kliebenstein D. J., Plant secondary metabolites as defenses, regulators, and primary metabolites: The blurred functional trichotomy. Plant Physiol. 184, 39–52 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lareen A., Burton F., Schäfer P., Plant root-microbe communication in shaping root microbiomes. Plant Mol. Biol. 90, 575–587 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Getzke F., et al. , Cofunctioning of bacterial exometabolites drives root microbiota establishment. Proc. Natl. Acad. Sci. U.S.A. 120, e2221508120 (2023). 10.1073/pnas.2221508120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kudjordjie E. N., Sapkota R., Nicolaisen M., Arabidopsis assemble distinct root-associated microbiomes through the synthesis of an array of defense metabolites. PLOS One 16, e0259171 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koprivova A., et al. , Root-specific camalexin biosynthesis controls the plant growth-promoting effects of multiple bacterial strains. Proc. Natl. Acad. Sci. U.S.A. 116, 15735–15744 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harbort C. J., et al. , Root-secreted coumarins and the microbiota interact to improve iron nutrition in Arabidopsis. Cell Host Microbe 28, 825–837 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stringlis I. A., et al. , MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl. Acad. Sci. U.S.A. 115, 5213–5222 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Voges M. J. E. E. E., et al. , Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc. Natl. Acad. Sci. U.S.A. 116, 12558–12565 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang P., Chai Y. N., Roston R., Dayan F. E., Schachtman D. P., The Sorghum bicolor root exudate sorgoleone shapes bacterial communities and delays network formation. mSystems 6, e00749-20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maver M., et al. , The indole-alkaloid gramine shapes the bacterial communities thriving at the barley root-soil interface. bioRxiv [Preprint] (2021), 10.1101/2020.12.07.414870 (Accessed 3 January 2022). [DOI]
- 35.Cadot S., et al. , Specific and conserved patterns of microbiota-structuring by maize benzoxazinoids in the field. Microbiome 9, 103–103 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cotton T. E. A., et al. , Metabolic regulation of the maize rhizobiome by benzoxazinoids. ISME J. 13, 1647–1658 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu L., et al. , Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 9, 2738–2738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kudjordjie E. N., Sapkota R., Steffensen S. K., Fomsgaard I. S., Nicolaisen M., Maize synthesized benzoxazinoids affect the host associated microbiome. Microbiome 7, 59–59 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy K. M., et al. , Bioactive diterpenoids impact the composition of the root-associated microbiome in maize (Zea mays). Sci. Rep. 11, 333 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding Y., et al. , Genetic elucidation of interconnected antibiotic pathways mediating maize innate immunity. Nat. Plants 6, 1375–1388 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Frey M., et al. , Benzoxazinoid biosynthesis, a model for evolution of secondary metabolic pathways in plants. Phytochemistry 70, 1645–1651 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Zhou S., Richter A., Jander G., Beyond defense: Multiple functions of benzoxazinoids in maize metabolism. Plant Cell Physiol. 59, 1528–1537 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Robert C. A. M., Mateo P., The chemical ecology of benzoxazinoids. Chimia 76, 928 (2022). 10.2533/chimia.2022.928. [DOI] [PubMed] [Google Scholar]
- 44.Hu L., et al. , Plant iron acquisition strategy exploited by an insect herbivore. Science 361, 694–697 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Macías F. A., et al. , Degradation studies on benzoxazinoids. Soil degradation dynamics of 2,4-Dihydroxy-7-methoxy-(2H)-1,4-benzoxazin-3(4H)-one (DIMBOA) and its degradation products, phytotoxic allelochemicals from gramineae. J. Agric. Food Chem. 52, 6402–6413 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Kumar P., Kumar P., Gagliardo R. W., Chilton W. S., Soil transformation of wheat and corn metabolites mboa and DIM2BOA into aminophenoxazinones. J. Chem. Ecol. 19, 2453–2461 (1993). [DOI] [PubMed] [Google Scholar]
- 47.Gfeller V. J., et al. , Plant secondary metabolite-dependent plant-soil feedbacks can improve crop yield in the field. Elife 12, e84988 (2022). 10.1101/2022.11.09.515047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niemeyer H. M., Hydroxamic acids derived from 2-hydroxy-2H-1,4-benzoxazin-3(4H)-one: Key defense chemicals of cereals. J. Agric. Food Chem. 57, 1677–1696 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Schulz M., Marocco A., Tabaglio V., Macías F. A., Molinillo J. M. G., Benzoxazinoids in rye allelopathy–From discovery to application in sustainable weed control and organic farming. J. Chem. Ecol. 39, 154–174 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Venturelli S., et al. , Plants release precursors of histone deacetylase inhibitors to suppress growth of competitors. The Plant Cell 27, 3175–3189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulz M., et al. , Pantoea ananatis converts MBOA to 6-Methoxy-4-nitro-benzoxazolin-2(3H)-one (NMBOA) for Cooperative degradation with its native root colonizing microbial consortium. Nat. Product Commun. 13, 1275–1278 (2018). [Google Scholar]
- 52.Bravo H. R., Copaja S. V., Lazo W., Antimicrobial activity of natural 2-benzoxazolinones and related derivatives. J. Agric. Food Chem. 45, 3255–3257 (1997). [Google Scholar]
- 53.Guo B., et al. , Extract from Maize (Zea mays L.): Antibacterial activity of DIMBOA and its derivatives against Ralstonia Solanacearum. Molecules 21, 1397 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neal A. L., Ahmad S., Gordon-Weeks R., Ton J., Benzoxazinoids in root exudates of maize attract Pseudomonas putida to the rhizosphere. PLOS One 7, e35498 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maresh J. J., Zhang J., Lynn D. G., The innate immunity of maize and the dynamic chemical strategies regulating two-component signal transduction in Agrobacterium tumefaciens. ACS Chem. Biol. 1, 165–175 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Schütz V., et al. , Conversions of benzoxazinoids and downstream metabolites by soil microorganisms. Front. Ecol. Evol. 7, 238 (2019). [Google Scholar]
- 57.Machado R. A. R., et al. , Engineering bacterial symbionts of nematodes improves their biocontrol potential to counter the western corn rootworm. Nat. Biotechnol. 38, 600–608 (2020). [DOI] [PubMed] [Google Scholar]
- 58.Schandry N., et al. , Plant-derived benzoxazinoids act as antibiotics and shape bacterial communities. bioRxiv [Preprint] (2021). 10.1101/2021.01.12.425818 (Accessed 30 April 2022). [DOI]
- 59.Bai Y., et al. , Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528, 364–369 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Hartman K., van der Heijden M. G. A., Roussely-Provent V., Walser J.-C., Schlaeppi K., Deciphering composition and function of the root microbiome of a legume plant. Microbiome 5, 2–2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J., et al. , NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 37, 676–684 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Wippel K., et al. , Host preference and invasiveness of commensal bacteria in the Lotus and Arabidopsis root microbiota. Nat. Microbiol. 6, 1150–1162 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beirinckx S., et al. , Tapping into the maize root microbiome to identify bacteria that promote growth under chilling conditions. Microbiome 8, 1–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Niu B., et al. , Simplified and representative bacterial community of maize roots. Proc. Natl. Acad. Sci. U.S.A. 114, 201616148 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vorholt J. A., Vogel C., Carlström C. I., Müller D. B., Establishing causality: Opportunities of synthetic communities for plant microbiome research. Cell Host Microbe 22, 142–155 (2017). [DOI] [PubMed] [Google Scholar]
- 66.de Bruijn W. J. C., Gruppen H., Vincken J.-P., Structure and biosynthesis of benzoxazinoids: Plant defence metabolites with potential as antimicrobial scaffolds. Phytochemistry 155, 233–243 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Yang L., et al. , New Insights into the Antibacterial Activity of Hydroxycoumarins against Ralstonia solanacearum. Molecules 21, 468–468 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu D., Ding W., Zhang Y., Liu X., Yang L., Oleanolic acid induces the type III secretion system of Ralstonia solanacearum. Front. Microbiol. 6, 1466–1466 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wouters F. C., Blanchette B., Gershenzon J., Vassão D. G., Plant defense and herbivore counter-defense: Benzoxazinoids and insect herbivores. Phytochem. Rev. 15, 1127–1151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong Y., Zhou Q., Hao Y., Huang A. C., Crafting the plant root metabolome for improved microbe-assisted stress resilience. New Phytolog. 234, 1945–1950 (2021). 10.1111/nph.17908. [DOI] [PubMed] [Google Scholar]
- 71.Nakayasu M., et al. , Tomato roots secrete tomatine to modulate the bacterial assemblage of the rhizosphere. Plant Physiol. 186, 270–284 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maag D., et al. , Highly localized and persistent induction of Bx1-dependent herbivore resistance factors in maize. Plant J. 88, 976–991 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Richter A., et al. , Indole-3-glycerolphosphate synthase, a branchpoint for the biosynthesis of tryptophan, indole, and benzoxazinoids in maize. Plant J. 106, 245–257 (2021). [DOI] [PubMed] [Google Scholar]
- 74.Katoh K., Misawa K., Kuma K., Miyata T., MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stamatakis A., RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thoenen L., et al. , Customisablehigh-throughput chemical phenotyping of root bacteria. in press.
- 77.Macías F. A., et al. , Isolation and synthesis of allelochemicals from gramineae: Benzoxazinones and related compounds. J. Agric. Food Chem. 54, 991–1000 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Gfeller V., et al. , Plant secondary metabolite-dependent plant-soil feedbacks can improve crop yield in the field. eLife 12, e84988 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ekstrøm C., MESS: Miscellaneous Esoteric Statistical Scripts (R package, Version 0.5.12, 2016). http://CRAN.R-project.org/package=MESS.
- 80.Wickham H., et al. , Welcome to the tidyverse. J. Open Source Softw. 4, 1686 (2019). [Google Scholar]
- 81.Robinson D., broom: An R package for converting statistical analysis objects into tidy data frames. arXiv: Computation [Preprint] (2014). 10.48550/arXiv.1412.3565 (Accessed 30 June 2022). [DOI]
- 82.Wright E. S., Using DECIPHER v2.0 to analyze big biological sequence data in R. R J. 8, 352–359 (2016). [Google Scholar]
- 83.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lenth R., Sigmann H., Love J., Buerkner P., Herve M., emmeans: Estimated Marginal Means, Aka Least-Squares Means (R package, Version 1.8.8, 2019). https://cran.r-project.org/web/packages/emmeans/index.html.
- 85.Arnold J. B., Extra themes, scales and geoms for “ggplot2” (Version 4.2.4, R package ggthemes, 2019). https://cran.r-project.org/web/packages/ggthemes/index.html. [Google Scholar]
- 86.Hothorn T., Bretz F., Westfall P. H., Simultaneous inference in general parametric models. Biometrical J. 50, 346–363 (2008). [DOI] [PubMed] [Google Scholar]
- 87.McMurdie P. J., Holmes S., phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLOS One 8, e61217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Revell L. J., phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223 (2012). [Google Scholar]
- 89.Oksanen J., et al. , vegan: Community Ecology Package (R package, Version 2.6.4, 2019). https://cran.r-project.org/web/packages/vegan/index.html.
- 90.Thoenen L., Kreuzer M., Waelchli J., Schlaeppi K., Thoenen_et_al_BX_tolerance. GitHub. https://github.com/PMI-Basel/Thoenen_et_al_BX_tolerance. Deposited 11 September 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Data Availability Statement
Raw data and analysis code data have been deposited in GitHub (https://github.com/PMI-Basel/Thoenen_et_al_BX_tolerance) (90). Some study data available [The MRB strain collection is maintained in the laboratory of the authors, and we share it including upgrades (e.g., more strains or genomes) with the research community upon request].