

Significance
Amplification of MYC and mutation of p53 are frequently found in human cancers, yet directly targeting it has proven difficult. Thus, exploring alternative methods to hinder their activities would significantly impact cancer treatment. TopBP1 (topoisomerase IIβ-binding protein 1) functions at the convergent point of Rb, PI3K/Akt, and p53 pathways, making it a promising cancer therapeutic target. Our high-throughput screening led to the development of a previously undescribed small-molecular inhibitor targeting the BRCT7/8 domains of TopBP1. In addition to inhibiting mutant p53 function, this inhibitor can block MYC activity by freeing MIZ1, a MYC inhibitor, from TopBP1. It can also inhibit Rad51 foci formation and synergize with inhibitors of PARP1/2 or PARP14, providing a potential synthetic lethal targeted cancer therapy.
Keywords: TopBP1, BRCT domains, small-molecule inhibitors, MYC, p53
Abstract
We have previously identified TopBP1 (topoisomerase IIβ-binding protein 1) as a promising target for cancer therapy, given its role in the convergence of Rb, PI(3)K/Akt, and p53 pathways. Based on this, we conducted a large-scale molecular docking screening to identify a small-molecule inhibitor that specifically targets the BRCT7/8 domains of TopBP1, which we have named 5D4. Our studies show that 5D4 inhibits TopBP1 interactions with E2F1, mutant p53, and Cancerous Inhibitor of Protein Phosphatase 2A. This leads to the activation of E2F1-mediated apoptosis and the inhibition of mutant p53 gain of function. In addition, 5D4 disrupts the interaction of TopBP1 with MIZ1, which in turn allows MIZ1 to bind to its target gene promoters and repress MYC activity. Moreover, 5D4 inhibits the association of the TopBP1-PLK1 complex and prevents the formation of Rad51 foci. When combined with inhibitors of PARP1/2 or PARP14, 5D4 synergizes to effectively block cancer cell proliferation. Our animal studies have demonstrated the antitumor activity of 5D4 in breast and ovarian cancer xenograft models. Moreover, the effectiveness of 5D4 is further enhanced when combined with a PARP1/2 inhibitor talazoparib. Taken together, our findings strongly support the potential use of TopBP1-BRCT7/8 inhibitors as a targeted cancer therapy.
Cancer development involves many steps of genetic alterations and signaling pathway deregulation. Regardless of the events that initiate the development of cancer, during progression most cancers exhibit deregulation of at least one of the common signaling pathways, such as Rb, p53, and PI3(K)/Akt. Previously, we identified topoisomerase IIβ-binding protein 1 (TopBP1) as a therapeutic target that functions at a convergent point of these common oncogenic pathways (1). We then identified calcein as a lead inhibitor that binds the 7th to 8th BRCA1 carboxyl-terminal (BRCT) domains of TopBP1 (TopBP1-BRCT7/8) (1). Because calcein lacks membrane permeability, we treated cancer cells with Calcein AM (acetoxymethyl ester) (CalAM), a cell-permeable derivative that can be rapidly hydrolyzed to form calcein inside the cells, and demonstrated its in vivo anticancer activity (1). This study provides a proof-of-concept evidence for targeting TopBP1-BRCT7/8 in cancer, particularly in tumors that overexpress TopBP1 (1, 2).
TopBP1 contains nine BRCT domains responsible for many protein interactions. Through the diverse protein–protein interactions, TopBP1 is involved in DNA replication, ATR checkpoint activation, DNA repair, mitosis, and transcriptional regulation (3). During G1/S phase entry, E2F1 transcriptionally induces the expression of TopBP1 (4), which in turn interacts with Treslin to promote the initiation of DNA replication (5). In late S and G2 phases, activated Akt phosphorylates TopBP1 (6) and switches the TopBP1-interacting partner from Treslin to E2F1, resulting in the termination of replication initiation and inhibition of E2F1-mediated apoptosis (7). During mitosis, TopBP1 interacts with SLX4, PLK1, MDC1, topoisomerase IIα (8) and Cancerous Inhibitor of Protein Phosphatase 2A (CIP2A) (9) to maintain genome integrity. Upon DNA replication stress, TopBP1 is recruited to stalled replication forks. It then activates ATR (Ataxia telangiectasia mutated and Rad3 related) through a conserved ATR-activating domain (10). In addition to ATR checkpoint activation, TopBP1 plays a crucial role in regulating homologous recombination (HR) DNA repair (11).
Indeed, TopBP1 overexpression is found in most types of cancer (TCGA datasets), and is an independent poor-prognostic factor in breast cancer (12, 13) and ovarian cancer (14). TopBP1 is an E2F target and its expression is constantly induced when Rb/E2F or p53 pathway is deregulated (4). Research from our lab has elucidated several mechanisms by which TopBP1 overexpression promotes cancer development: i) inhibiting E2F1-mediated apoptosis (4, 6, 15), ii) promoting mutant p53 (mutp53) gain of function (GOF) through transcriptional regulation of NF-Y and p63/p73 (13), iii) bypassing the requirement of active Cdk2 to promote DNA replication (7), and iv) paradoxically hindering ATR/Chk1 activation (16).
The activity of TopBP1 in repressing E2F1-mediated apoptosis is regulated by Akt. Akt phosphorylates TopBP1 at Ser1159, which in turn induces its oligomerization through TopBP1-BRCT7/8 domains (6). Oligomerization of TopBP1 promotes its binding to E2F1 and MIZ1 (MYC-interacting zinc finger protein 1), thereby inhibiting E2F1-dependent apoptosis and MIZ1-dependent p21Cip1 expression (6). Thus, compounds that bind and block TopBP1-BRCT7/8 domains, such as CalAM, can reactivate E2F1-dependent apoptosis and induce p21Cip1 expression (1). In addition to mediating TopBP1 binding to wild-type p53 to repress p53 function (12), TopBP1-BRCT7/8 can also bind mutp53 to promote mutp53 GOF and tumor growth by facilitating mutp53 interaction with NF-Y and p63/p73 (13). On the other hand, some p53 contact mutants bind both TopBP1 and Treslin and facilitate their interaction, thereby overriding the requirement of Cdk2 and promoting DNA replication in the late G1 phase (17). Through its BRCT7/8 domains, TopBP1 recruits PLK1 to phosphorylate Rad51 and facilitates the chromatin loading of Rad51 to promote DNA repair. Thus, TopBP1-BRCT7/8 appears to be an attractive target to develop small-molecule inhibitors for cancer therapy. Indeed, we previously demonstrated that the TopBP1-BRCT7/8 inhibitor CalAM could inhibit mutp53 GOF (1). CalAM has also been shown to enhance PARPi (PARP inhibitors) sensitivity (11).
MIZ1 plays a key role in the regulation of MYC activity, and the outcome of the transcriptional response to MYC (activation vs. repression) closely correlates with the ratio of MYC/MIZ1 bound to each promoter (18). Indeed, the continuous degradation of MIZ1 is required for transcriptional activation by MYC in colon cancer cells (19). Thus, enhancement of MIZ1 activity can attenuate the oncogenic activity of MYC and may be exploited as a therapeutic strategy. Since MIZ1 is inhibited by TopBP1 through binding to TopBP1-BRCT7/8 (6, 20), targeting TopBP1-BRCT7/8 may release MIZ1 and result in the anti-MYC effect.
Here, we perform a molecular docking screen, and through hit expansion, we identify several previously undescribed compounds that target TopBP1-BRCT7/8. We show the in vivo anticancer activity of one of the compounds, specifically 5D4, and demonstrate its anti-MYC activity and synergy with PARPi.
Results
A Molecular Docking Screen Identifies a Lead Compound 2H3 that Targets TopBP1-BRCT7/8.
Based on the crystal structure of TopBP1-BRCT7/8, previously we performed a small scale of molecular docking to screen more than 2,000 known compounds and identified CalAM, which could block the interactions of TopBP1-BRCT7/8 with pS1159-TopBP1 phosphopeptide and DNA-binding domain (DBD) of p53 (1). CalAM inhibited tumor growth in breast cancer xenograft models and induced apoptosis in cancer cells highly expressing TopBP1 (1). These data provide proof of concept for targeting TopBP1 in cancer. We then expanded screening to test 200,000 compounds from 3 selected libraries (drug-like, natural product derivatives, and compounds tested in men) and identified compounds with best docking scores (close to or < –8.0). We shortlisted the compounds based on purchasability, predicted cell permeability (polar surface area < 140), and no known targets (in SEA, ZINC, PubChem, and ChemBank databases), and prioritized 99 compounds for testing (Dataset S1). We performed 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay to evaluate the effects of these compounds on cell viability in BT549 and MDA-MB-468 triple-negative breast cancer (TNBC) cell lines and carried out biochemical binding assay to assess their activity in blocking the interaction of TopBP1-BRCT7/8 with pS1159-TopBP1 peptide (1). Among these compounds, Cpd 2H3 showed the most active cytotoxic activity (SI Appendix, Fig. S1). Cpd 2H3, a methylpyrazole-quinuclidin derivative (Fig. 1A), blocked TopBP1-BRCT7/8 binding to the pS1159-TopBP1 phosphopeptide in vitro (IC50 = 20 nM) (Fig. 1B) and attenuated its association with full-length TopBP1 in HEK293T cells (SI Appendix, Fig. S2). The BRCT7 and BRCT8 domains of TopBP1 form a pocket that binds pS1159-TopBP1 phosphopeptide (21) (Fig. 1 C, Top). Molecular docking shows that Cpd 2H3 can be docked well into the pocket (Fig. 1 C, Bottom). Cpd 2H3 exhibited cytotoxic activity in MDAH-2774 and TOV-112D ovarian cancer cells (Fig. 1 D, E, and G) and MDA-MB-468 breast cancer cells (Fig. 1 F and G) but only had a minimal effect in nontransformed MCF10A cells and AML12 mouse hepatocytes (Fig. 1G). Cpd 2H3 also inhibited clonogenic viability of TOV-112D cells (Fig. 1H). It is predicted that Cpd 2H3 has more favorable drug-like properties than CalAM according to Lipinski’s rules (SI Appendix, Table S1).
Fig. 1.
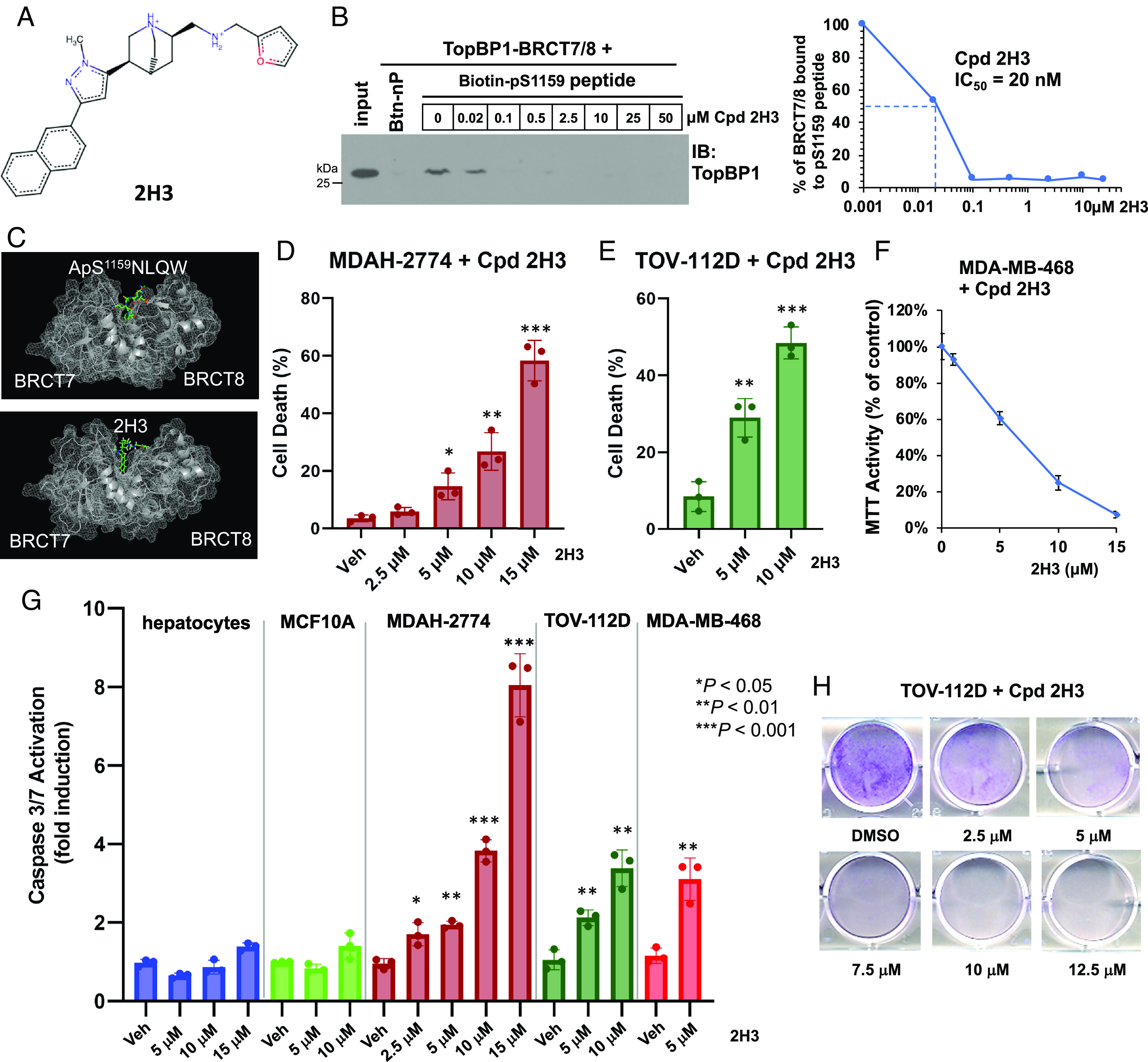
Cpd 2H3 inhibits TopBP1-BRCT7/8 binding to the pS1159 peptide and induces cell death in breast and ovarian cancer cells. (A) Structure of Cpd 2H3. (B) Cpd 2H3 blocks the interaction between TopBP1-BRCT7/8 and pS1159 peptide in vitro. Purified TopBP1-BRCT7/8 was incubated with Cpd 2H3 and biotinylated phospho-S1159 peptide or nonphosphorylated peptide (Btn-nP), followed by Streptavidin Sepharose pulldown. The pulldown of TopBP1-BRCT7/8 was analyzed by immunoblotting (IB) with an anti-TopBP1 antibody recognizing TopBP1-BRCT7/8. (C) Structure of TopBP1-BRCT7/8 complexed with pS1159 peptide or Cpd H3. (Top) pS1159 peptide was overlayed with TopBP1-BRCT7/8 (PDB code: 3AL3), guided by the structure of TopBP1-BRCT7/8 with pBACH peptide, and simulated to a local energy minimum (21). (Bottom) The lowest-energy docking of Cpd 2H3 (ball & stick model) to the pocket of TopBP1-BRCT7/8. (D and E) Cpd 2H3 induces cell death in MDAH-2774 (D) and TOV-112D (E). Cells were treated with dimethyl sulfoxide (DMSO) (Veh) or 2H3 for 24 h. The viable and dead cells were determined by trypan blue exclusion assay. Data shown are the mean ± SD of triplicates. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle control (two-tailed t test). (F) Cpd 2H3 inhibits the cell viability of MDA-MB-468 cells. Cells were treated with DMSO or 2H3 for 48 h. Cell viability was determined by MTT assay. Data shown are the mean ± SD from three biological replicates. (G) Cpd 2H3 induces apoptosis in MDAH-2774, TOV-112D, and MDA-MB-468 cancer cells but not nontransformed AML12 mouse hepatocytes or MCF-10A cells. Cells were treated with DMSO or 2H3 for 20 h. Active caspase-3/7 was determined by Caspase-Glo® 3/7 Assay. Data shown are the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle control (two-tailed t test). (H) Cpd 2H3 inhibits clonogenic survival of TOV-112D cells. Cells were treated with DMSO or 2H3 for 24 h. After PBS washing, cells were cultured in fresh growth medium for another 5 d. Viable cells were fixed with 3% formaldehyde followed by staining with 0.5% crystal violet.
Hit Expansion Identifies Cpd 5D4 As a More Potent TopBP1 Inhibitor.
To determine whether there are common features among the lead compounds capable of targeting TopBP1-BRCT7/8, we compared the docking of these lead compounds. Indeed, 2H3 and calcein possess multibenzene rings to dock into a pocket within TopBP1-BRCT7/8, which normally binds to the +4 Tryptophan residue of pS1159-TopBP1 phosphopeptide (21) or the +4 Tyrosine of pT1133-BACH1 phosphopeptide (SI Appendix, Fig. S3). Comparing the sequences from different species, this TopBP1-BRCT7/8 pocket binds a conserved S/TxxxΩ motif with an aromatic a.a. (Ω, such as W/Y/F) in the +4 position. The structure-based compound docking suggests that the polycyclic aromatic hydrocarbons of the lead compounds mimic +4 aromatic a.a. when they bind TopBP1-BRCT7/8. Calcein and Cpd 2H3 also compete with pS1159-TopBP1 phosphopeptides (1) for hydrogen bonding with the critical a.a. such as S1273, S1274, and K1317 and therefore block the phophoserine-binding pocket of TopBP1-BRCT7/8 (Fig. 2A). With the guide of docking, we performed hit expansion through Pubchem and ZINC docking servers and identified analogs/derivatives of Cpd 2H3 with modified side chains on the desired positions. These compounds were tested by docking with TopBP1-BRCT7/8 in mcule 1-Click Docking online server. Through this exercise, we identified 28 derivatives of Cpd 2H3 which show improved docking scores and favorable drug-like properties as analyzed in FAF-Drugs3 server and Chembioserver (Dataset S2). We then performed experiments to characterize these derivatives. Indeed, most derivatives exhibited more potent anticancer activities than the parental Cpd 2H3 (SI Appendix, Fig. S4), particularly 5H3 and 5D4 (Fig. 2 A and B). 5D4 bound TopBP1-BRCT7/8 and blocked its binding to pS1159-TopBP1 phosphopeptide, mutp53-R273H(DBD) and WT p53(DBD) with IC50 around 10 nM (Fig. 2 C and D and SI Appendix, Fig. S5). Structure–Activity Relationship (SAR) analysis from totally 64 analogs of Cpd 2H3 (Fig. 2E and Dataset S3) revealed that a naphthalene on the R1 position is essential for their cytotoxic activities. Different side chains on the R2 position also affect their activities. The naphthalene on the R1 position corresponds to the +4 aromatic a.a. of the pT1133-BACH or pS1159-TopBP1 phosphopeptide (Fig. 2F, yellow solid circles). On the other hand, the R2 side chain of the compounds corresponds to the phosphorylated threonine or serine residue in the docking structure (Fig. 2F, yellow dotted circles).
Fig. 2.
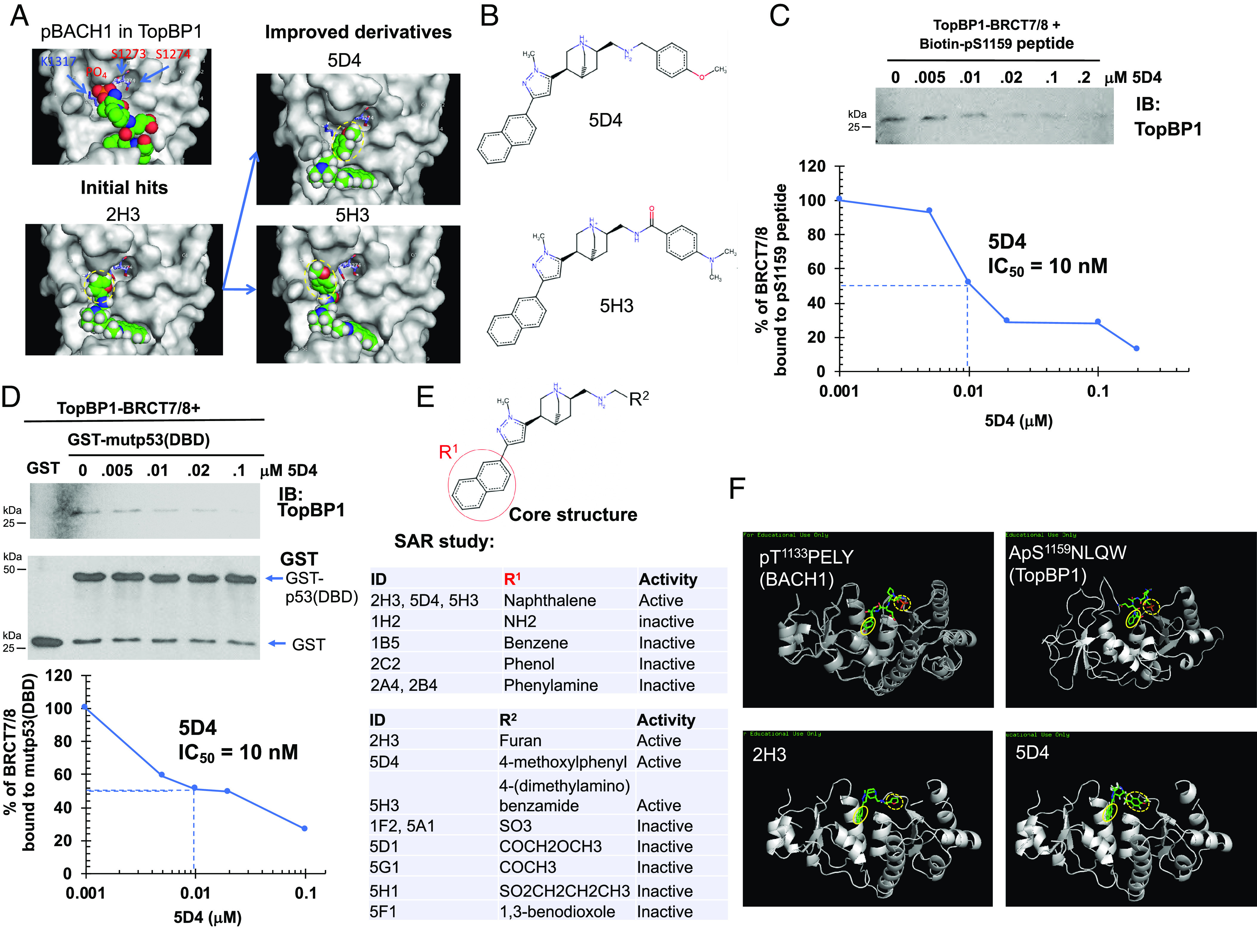
Hit expansion and SAR studies identify Cpd 5D4 as a TopBP1-BRCT7/8 inhibitor. (A and B) Structures of initial hit 2H3 and its derivatives 5D4 and 5H3. (A) Ball-and-stick structure of each compound in TopBP1-BRCT7/8 pocket. Yellow dotted circles indicate the modified side chains from parental compounds. (B) Chemical structures of 5D4 and 5H3. (C and D) Cpd 5D4 inhibits TopBP1-BRCT7/8 binding to the pS1159 peptide or mutp53(DBD). (C) The effect of 5D4 on the in vitro binding of biotinylated pS1159 peptide to purified TopBP1-BRCT7/8 was determined by Streptavidin Sepharose pulldown as described in Fig. 1B. (D) Purified TopBP1-BRCT7/8 was incubated with GST-mutp53-R273H(DBD) in the presence of 5D4, and GST pulldown assay was performed. The pulldown of TopBP1-BRCT7/8 was detected by immunoblotting using an antibody against the C terminus of TopBP1. (E) Summary of SAR study. The details of these derivatives are listed in Datasets S2 and S3. The cytotoxic activity of each compound was evaluated in MDA-MB-468, BT549, MDAH-2774, and TOV-112D. (F) Structure of TopBP1-BRCT7/8 complexed with BACH1 phosphopeptide (PDB: 3AL3), TopBP1 phosphopeptide (21), Cpd 2H3 or 5D4. Yellow solid circles indicate the position of tyrosine (in BACH1), tryptophan (in TopBP1), or a multibenzene ring in 2H3 and 5D4. Yellow dotted circles indicate the position of pT1133 (BACH1), pS1159 (TopBP1), or a side chain of 2H3 and 5D4.
It appears that Cpd 5H3 and 5D4 showed stronger cytotoxic activities than Cpd 2H3 in MDA-MB-468 cells but barely had any effect in nontransformed MCF10A cells or AML12 mouse hepatocytes (Fig. 3A). Likewise, 5D4 also showed strong cytotoxic activities against OVCAR8 ovarian carcinoma cells, MDAH-2774 and TOV-112D endometrioid ovarian cancer cells, and cisplatin-resistant A2780cis ovarian cancer cells (Fig. 3B). The clonogenic cell survival assay showed that the IC50 of 5D4 and 5H3 in blocking MDA-MB-468 cell growth was within the nanomolar range (Fig. 3C and SI Appendix, Fig. S6). The 3D culture showed that 5D4 effectively inhibited tumor sphere formation of MDA-MB-468 (Fig. 3D), BT549, and T47D breast cancer cells as well as TOV-112D ovarian cancer cells (SI Appendix, Fig. S7). Consistently, 5D4 also decreased the viability of tumor spheres, which were established from TNBC patient-derived xenografts (PDX), BCM2665 (22) (Fig. 3E).
Fig. 3.
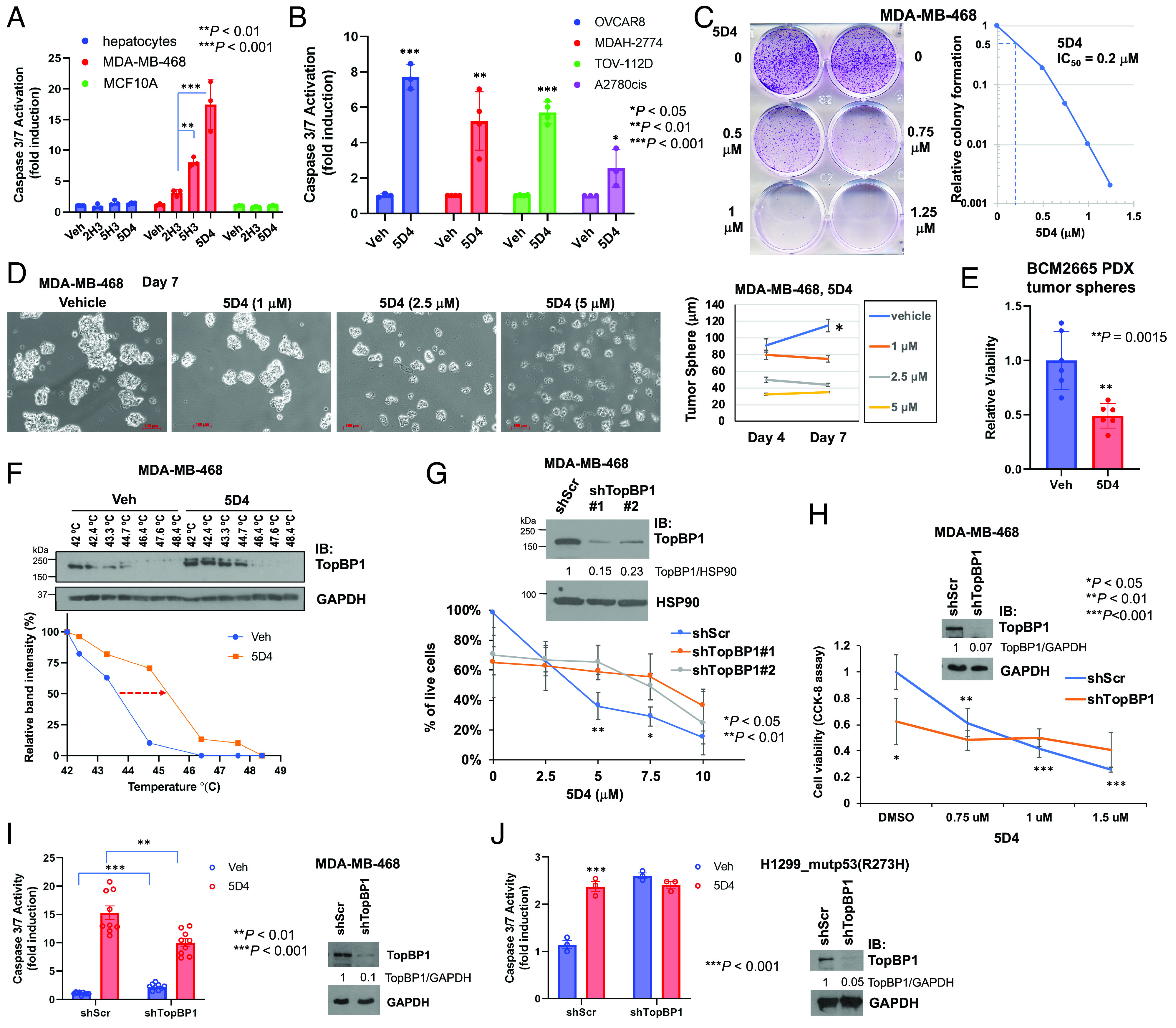
Cpd 5D4 induces apoptosis and inhibits cell viability in 2D and 3D cultured breast and ovarian cancer cells. (A and B) Cpd 5D4 induces apoptosis in breast and ovarian cancer cells but only has minimal effect on nontransformed AML12 mouse hepatocytes and MCF10A. Different cell lines as indicated were treated with DMSO or 5 µM 2H3, 5H3, or 5D4 for 18 to 22 h. Apoptosis was determined by Caspase-Glo® 3/7 Assay. Data shown are the mean ± SD of three or four independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle control (two-tailed t test). (C) Cpd 5D4 inhibits clonogenic survival of MDA-MB-468 cells. Cells were treated with DMSO or 5D4 for 24 h. After PBS washing, cells were cultured in fresh growth medium for another 4 d and then fixed and stained with 0.5% crystal violet. The right panel shows the relative colony formation compared to the vehicle control. The relative colony formation was analyzed using ImageJ to determine the IC50 of 5D4 (=0.2 µM). (D and E) Cpd 5D4 inhibits the growth of breast cancer tumor spheres. MDA-MB-468 cells were seeded on 6-well plates with cell-repellent surface. When the diameter of tumor spheres reached 30 to 50 µm, spheres were treated with DMSO or 5D4 on days 0, 3, and 6 (D). The images were captured 24 h after each treatment, and the diameter of tumor spheres was measured (scale bar, 100 μm). Data shown are the mean ± SEM of 40 to 60 spheres (*P < 0.05 vs. 5D4-treated cells). (E) BCM2665 PDX tissues were trypsinized and cells were cultured in Complete DMEM/F12 Media for Patient-Derived In Vitro and Organoid Cultures as described in NCI Patient-Derived Models Repository (SOP30101). After 10 d of culturing, tumor spheres were treated with DMSO or 5 µM 5D4 every other day for 6 d, followed by CCK-8 assay to determine the viability of tumor spheres. Data shown are the mean ± SD of two experiments done in triplicates. (F) Cellular thermal shift assay. MDA-MB-468 cells were treated with DMSO or 5D4 (10 µM) at 37 °C for 2 h. Cell lysates were heated for 3 min at temperature ranging from 42 to 48.4 °C, followed by immunoblotting. Quantification is shown in the Bottom panel. (G and H) TopBP1 depletion decreases cell viability and blunts the response to 5D4 in MDA-MB-468 cells. (G) The early-passage MDA-MB-468 cells stably harboring a scrambled shRNA (shScr) or one of the TopBP1 shRNAs (shTopBP1 #1 or #2) were treated with 5D4 for 19 h. The percentage of live cells was measured by trypan blue exclusion assay. Data shown are the mean ± SD of four biological replicates. *P < 0.05 and **P < 0.01 compared with shTopBP1 (two-tailed t test). (Upper) immunoblots confirming the depletion of TopBP1. The relative intensities of TopBP1 were quantified by ImageJ and normalized to loading control HSP90. (H) The early-passage MDA-MB-468 cells harboring shScr or shTopBP1 (#1) were treated with 5D4 for 24 h. Cell viability was determined by CCK-8 assay. Data shown are the mean ± SD of four biological replicates. **P < 0.01 and ***P < 0.001 vs. vehicle control (two-tailed t test). The intensities of TopBP1 on the immunoblot were quantified by ImageJ and normalized to GAPDH. (I) Depletion of TopBP1 increases the basal caspase-3/7 activity but attenuates 5D4-induced caspase-3/7 activation. MDA-MB-468 cells stably expressing shScr or shTopBP1 were treated with 3 µM 5D4 for 20 h, followed by caspase-3/7 activity assay. Data shown are the mean ± SD of nine biological replicates from four independent experiments. **P < 0.01 and ***P < 0.001 vs. treated and vehicle control cells, respectively (two-tailed t test). (J) Transient depletion of TopBP1 elevates the basal caspase-3/7 activity and blocks the further response to 5D4. H1299 cells stably expressing mutp53(R273H) were transiently transfected with a pSUPER vector harboring shScr or shTopBP1. After 48 h, cells were treated with 5 µM 5D4 for 21 h, followed by caspase-3/7 activity assay. Data shown are the mean ± SD of three biological replicates and are representative out of three independent experiments. ***P < 0.001 vs. vehicle control (two-tailed t test).
To further validate whether TopBP1 is a specific target of 5D4, we assessed the ability of 5D4 to bind endogenous TopBP1 in MDA-MB-468 cells by performing a cellular thermal shift assay (CETSA). CETSA showed that 5D4 shifted the melting temperature (Tm) of TopBP1 protein toward a higher temperature, providing evidence for the binding of 5D4 to TopBP1 within the cells (Fig. 3F). On the other hand, depletion of TopBP1 decreased the cell viability (Fig. 3 G and H) and blunted the effect of 5D4 on the growth inhibition in MDA-MB-468 cells as assayed by trypan blue exclusion (Fig. 3G) or CCK-8 cell viability assay (Fig. 3H). Moreover, depletion of TopBP1 mildly elevated the basal caspase-3/7 activity in MDA-MB-468 cells but attenuated 5D4-induced caspase-3/7 activation (Fig. 3I). Considering that these experiments were performed in cells stably harboring a TopBP1 shRNA, which might already undergo phenotypic changes due to the antiproliferative effect of TopBP1 knockdown, we also assessed the short-term impact of TopBP1 depletion on 5D4-induced caspase-3/7 activation by transiently transfecting a TopBP1 shRNA in H1299 cells stably expressing mutp53(R273H). Indeed, transient depletion of TopBP1 increased the basal cacspase-3/7 activity and blocked the further response to 5D4 (Fig. 3J), suggesting that the anticancer activity of 5D4 is dependent on TopBP1 expression. Cpd 5D4 and 5H3 have favorable predicted drug-like properties (SI Appendix, Table S1 and Dataset S2).
Cpd 5D4 Treatment Disrupts the Interaction of TopBP1 with E2F1, mutp53, MIZ1, PLK1, or CIP2A.
TopBP1 interacts with several proteins, such as E2F1 (15), mutp53 (13), MIZ1 (20), and PLK1 in a BRCT7/8-dependent manner (11), but binds to other proteins, such as RPA2, Treslin or Rad9 through BRCT1/2 domains (3, 23). Meanwhile, molecular docking showed that among all of the available tandem BRCT structures, TopBP1-BRCT7/8 domains show the best score for 5D4 docking (SI Appendix, Fig. S8A). We therefore investigated whether 5D4 treatment specifically disrupts the BRCT7/8-mediated protein–protein interactions. Indeed, 5D4 inhibited the TopBP1 binding to E2F1 (Fig. 4A). 5D4 also blocked the interaction of TopBP1 with mutp53, MIZ1 or PLK1 in MDA-MB-468 and MDAH-2774 cells (Fig. 4 B, C, E, and F). In contrast, 5D4 did not interfere with the TopBP1 binding to RPA2, Rad9 or Treslin (Fig. 4 D and E), and also did not affect the BRCT domain-mediated interaction of BRCA1 with BACH1 (SI Appendix, Fig. S8B). Together with the results shown in Fig. 3 F–J, these data strongly suggest that TopBP1-BRCT7/8 is a cellular target of 5D4.
Fig. 4.

Cpd 5D4 treatment blocks the interaction of TopBP1 with E2F1, mutp53, MIZ1, CIP2A, or PLK1 but has no impact on its binding to RPA2, Rad9, or Treslin. (A and B) Coimmunoprecipitation experiments show 5D4 treatment inhibits TopBP1 interaction with E2F1 (A) or mutp53 (B) in MDA-MB-468. MDA-MB-468 cells were treated with vehicle DMSO or 5D4 (5 µM) for 22 to 24 h. Cells were harvested and subjected to immunoprecipitation using an anti-TopBP1 mouse monoclonal antibody. Coimmunoprecipitated E2F1 or mutp53 was detected by immunoblotting using a rabbit antibody specific to E2F1 or mutp53. The relative intensities of immunoprecipitated E2F1 or mutp53 were quantified using ImageJ and normalized to the immunoprecipitated TopBP1. (C–F) MDA-MB-468 cells (C and D) and MDAH-2774 cells (F) were treated with vehicle DMSO or 5D4 (2 µM in C and D and 3 µM in F) for 20 h, and then harvested for coimmunoprecipitation. Endogenous TopBP1 in the whole cell lysates was immunoprecipitated with an anti-TopBP1 mouse monoclonal antibody or a control mouse IgG, followed by immunoblotting using a rabbit antibody specific to the indicated protein. Data shown in E are the relative fold changes of TopBP1 binding to its interacting partners in 5D4-treated MDA-MB-468 cells compared to that in vehicle-treated cells (n = 4 biological replicates for MIZ1, CIP2A, PLK1, or mutp53; n = 5 for RPA2; n = 2 for Rad9; n = 1 for Treslin). ***P < 0.001 vs. vehicle (two-tailed t test).
Recently, CIP2A was identified as another TopBP1 interacting protein (24) and their interaction was proposed to serve as a synthetic lethal target in BRCA-mutated cancer (25). It was reported that CIP2A binds to a TopBP1 peptide (a.a. 740 to 899) comprising the region between BRCT5 and 6 (9). However, we found that 5D4 could inhibit TopBP1/CIP2A interaction in both MDA-MB-468 and MDAH-2774 cells (Fig. 4 C, E, and F). Our result therefore suggests that either BRCT7/8 domains are also involved in the association with CIP2A, or the binding of 5D4 to TopBP1-BRCT7/8 results in a conformational change of TopBP1, thereby hindering its binding to CIP2A.
Transcriptome Profiling Reveals that TopBP1 Inhibitors Possess the Anti-MYC Activity.
To gain mechanistic insights into the anticancer actions of TopBP1 inhibitors, we performed RNA-seq transcriptome profiling of MDA-MB-468 cells treated with either dimethyl sulfoxide (DMSO) vehicle, CalAM or 5D4 for 6 h. Consistent with prior results (1), both CalAM and 5D4 activated E2F1 target genes in apoptosis and autophagy as well as p53/p63/p73 target genes (Fig. 5A), supporting the notion that disruption of the TopBP1/E2F1 and TopBP1/mutp53 complex formation by these inhibitors can reactivate E2F1-induced apoptosis and autophagy, and block mutp53 GOF. The transcriptome profile of CalAM or 5D4-treated MDA-MB-468 shares significant similarities with that of TopBP1-depleted MDA-MB-468 (SI Appendix, Fig. S9), supporting that the changes of the gene expression are on-target effects of these compounds. The data also showed that 5D4 significantly inhibited the MYC gene signature (Fig. 5B). The anti-MYC activities of these compounds were further verified by immunoblotting (IB), which showed the induction of p21Cip1 by 5D4 and decreased protein expression of several MYC targets, including Cyclin A2, Cyclin D1, ACTL6A (Actin-like protein 6A, also named BAF53A) (18, 26), PLK1 and Rad51 (27), by 5D4 or CalAM in MDA-MB-468, MDAH-2774 and A2780cis cells, respectively (Fig. 5 C–E). Consistent with the RNA-seq results, 5D4 also increased the protein levels of SQSTM1/p62 (Fig. 5 D and F). We next performed IB to determine the effect of TopBP1 depletion on the expression of these proteins in MDA-MB-468 cells. As expected, depletion of TopBP1 decreased the basal levels of PLK1, Cyclin D1 and Rad51 (Fig. 5F). Since the TopBP1-depleted cells still expressed low levels of TopBP1, they were able to respond, albeit at a lower degree, to 5D4. As a result, 5D4 could further attenuate the expression of these proteins in TopBP1-depleted cells. On the other hand, depletion of TopBP1 elevated the basal levels of p21Cip1 but did not further induce its expression after 5D4 treatment. Depletion of TopBP1 also slightly increased the basal levels of p62 but significantly attenuated 5D4-induced p62 expression. (Fig. 5F). Together, these results demonstrate that 5D4 effectively inhibits the TopBP1 target gene expression and exhibits anti-MYC activity.
Fig. 5.
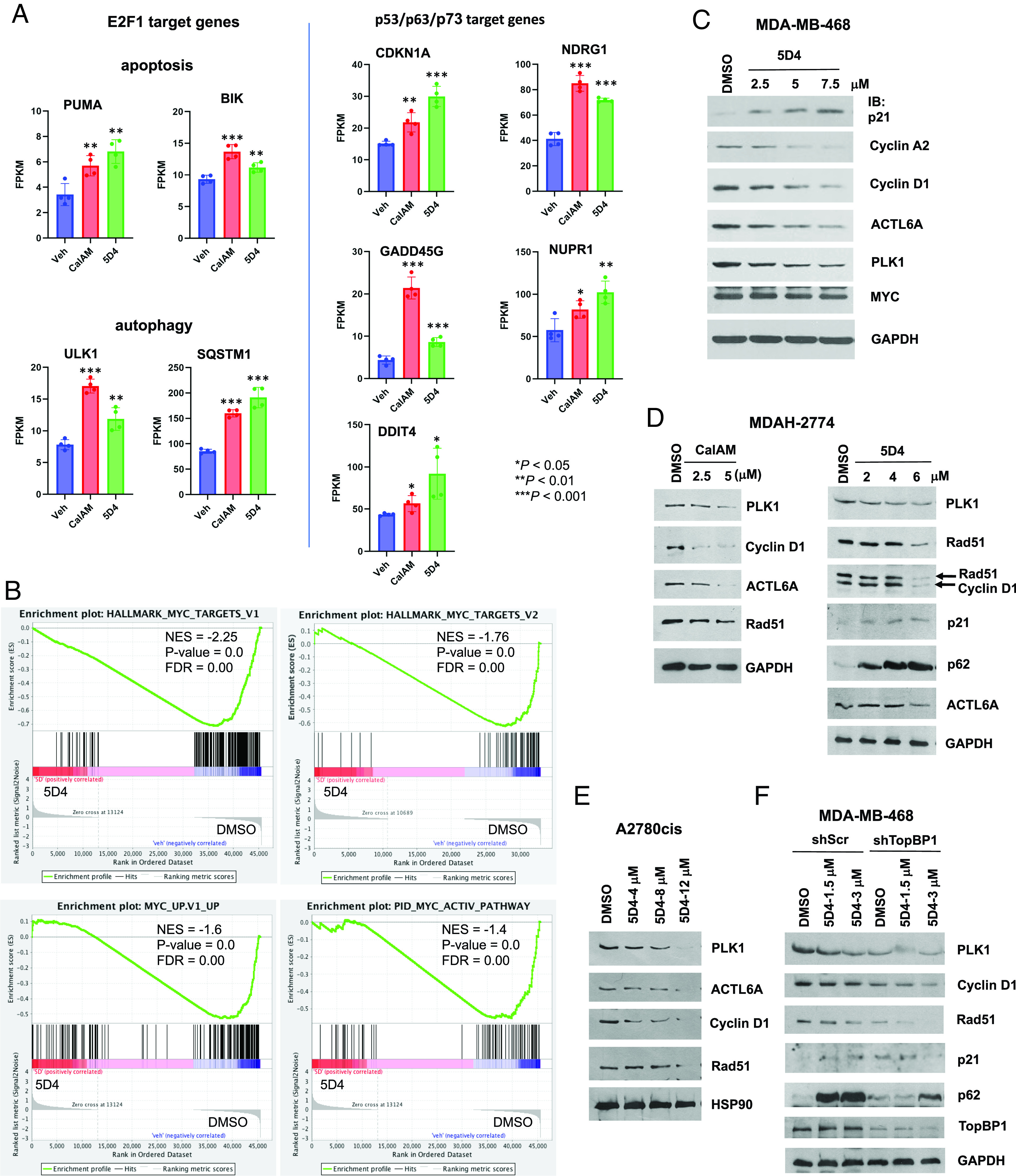
CalAM and 5D4 enhance the expression of E2F1 target genes in apoptosis and autophagy as well as p53/p63/p73 target genes but reduce the expression of MYC target genes. (A) MDA-MB-468 were treated with CalAM or 5D4 (5 µM) for 6 h. RNA was isolated and subjected to RNA-seq. FPKM: fragments per kilobase of exon per million mapped fragments. Shown are mean ± SD (n = 4 biological replicates). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle control (two-tailed t test). (B) Gene-set enrichment analysis shows that 5D4 treatment inhibits the mRNA expression of MYC target genes in MDA-MB-468 cells. NES: normalized enrichment score; FDR: false discovery rate. (C–E) MDA-MB-468 (C), MDAH-2774 (D), or A2780cis (E) cells were treated with CalAM or 5D4 for 18 h. The whole cell lysates were subjected to IB using antibodies specific to the indicated proteins. (F) Depletion of TopBP1 increases p21Cip1 expression but decreases the levels of PLK1, Rad51, and Cyclin D1 and attenuates 5D4-induced p62 expression. MDA-MB-468 cells stably expressing shScr or shTopBP1 were treated with 1.5 µM or 3 µM 5D4 for 20 h, followed by IB.
Treatment with 5D4 Promotes MIZ1 Binding to the p21Cip1 Promoter.
TopBP1 binds MIZ1 and inhibits its function in p21Cip1 transactivation (6, 20). Thus, TopBP1-BRCT7/8 inhibitors may release MIZ1 from TopBP1 (Fig. 4), thereby promoting its activity. To investigate this potential mechanism of action, we performed chromatin immunoprecipitation (ChIP) assay and found that indeed, 5D4 induced the MIZ1 chromatin binding to the p21Cip1 promoter in MDA-MB-468, MDAH-2774 and two lung cancer cell lines, HCC95 and NCI-H2170 (harboring MYC gene amplification) (Fig. 6 A–D).
Fig. 6.

Cpd 5D4 treatment enhances the MIZ1 binding to the p21Cip1 promoter. (A–D) ChIP assay was performed to determine the MIZ1 binding to the p21Cip1 promoter in vehicle (Veh)-treated control cells, or MDA-MB-468 cells treated with 2 μM 5D4 for 6 h (A), MDAH-2774 cells treated with 4 μM 5D4 for 6 h (B), HCC95 cells treated with 4 μM 5D4 for 4 h (C), or H2170 cells treated with 2 μM 5D4 for 4 h (D). MIZ1 binding to β-Actin promoter serves as a negative control (n = 4). ***P < 0.001 vs. vehicle control (two-tailed t test). (E and F) Depletion of MIZ1 attenuates the effect of 5D4 on the inhibition of cell viability. MDA-MB-468 (E) or MDAH-2774 (F) cells stably expressing shScr or a MIZ1 shRNA (shMIZ1#1 or #2) were treated with DMSO or 5D4 for 45 h. The relative cell viability was determined by CCK-8 assay. Data shown are the mean ± SD done in quadruplicates. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. shScr (two-tailed t test). (G and H) Depletion of MIZ1 dampens the effect of 5D4 on the induction of p21Cip1. MDA-MB-468 (G) or H2170 cells (H) stably expressing shScr or shMIZ1 (#1 or #2) were treated with DMSO or 5D4 for 20 h followed by IB.
MIZ1 accumulates on MYC target genes globally and contributes to the repression of MYC-activated target genes, particularly when HUWE1, an E3 ubiquitin ligase that targets MIZ1 for degradation, is inhibited (19). Thus, the release of MIZ1 from TopBP1 by BRCT7/8 inhibitors may contribute to the observed inhibition of MYC activity by 5D4. To determine a role for MIZ1 in the anticancer activity of 5D4, we depleted MIZ1 in MDA-MB-468 and MDAH-2774 cells. Indeed, MIZ1 depletion dampened the effect of 5D4 on the inhibition of cell viability in both cell lines (Fig. 6 E and F) and the induction of p21Cip1 in MDA-MB-468 and H2170 cells (Fig. 6 G and H).
Cpd 5D4 Inhibits the Growth of Ovarian Cancer and Breast Cancer Xenografts.
To investigate the in vivo activity of 5D4, we next established MDAH-2774 ovarian cancer and MDA-MB-468 breast cancer xenografts, respectively, in NSG mice, and injected intraperitoneally (i.p.) with 5D4 (40 mg/kg) or vehicle (DMSO) every 3 d for three doses. Indeed, 5D4 significantly reduced tumor growth in both models (Fig. 7 A and B). We next implanted BCM2665 breast cancer PDX into NSG mice. BCM2665 is a TNBC PDX harboring mutp53-R249S, MYC amplification (4 copies) and WT BRCA1/2 (BCM PDX Portal). Consistently, 5D4 also significantly reduced tumor growth in the PDX model (Fig. 7C). These mice tolerated 5D4 well without changes of body weight (SI Appendix, Fig. S10) or apparent side effects.
Fig. 7.

Cpd 5D4 significantly decreases tumor growth of ovarian and breast cancer xenografts. (A and B) NSG mice bearing MDAH-2774 (n = 5 in vehicle group; n = 6 in 5D4 group) or MDA-MB-468 (n = 5/group) xenografts were administered with 5D4 (40 mg/kg, i.p., q3d × 3 doses) or vehicle. (Left) the mean tumor volumes ± SD; (Right) the mean tumor weights ± SD. (C) NSG mice bearing BCM2665 PDX were administered with 5D4 (40 mg/kg, i.p., twice weekly × 3 wk) or vehicle; n = 7/group. ***P < 0.001 vs. vehicle (two-tailed t test). The x axis represents the day after cell injection or PDX implantation.
Synergism between TopBP1 Inhibitors and PARPi.
PARP inhibitors (PARPi) are the first clinically approved synthetic lethal therapy for the patients with HR deficiency. The interaction between TopBP1 and PLK1 is important for Rad51 chromatin recruitment and HR DNA repair (11). As such, CalAM, the TopBP1-BRCT7/8 inhibitor that we identified previously, has been shown to block TopBP1/PLK1 interaction and sensitize U2OS cells to PARPi olaparib (11). Likewise, 5D4 can also block TopBP1/PLK1 interaction (Fig. 4). Therefore, we next investigated whether 5D4 and PARPi have synergistic anticancer effect. The HR-proficient MDA-MB-468, MDAH-2774 and A2780cis cells, as expected, responded to 5D4 or CalAM but not PARPi rucaparib. Nonetheless, combined treatment with rucaparib and either 5D4 or CalAM rendered these cells even more responsive than individual drug treatment (Fig. 8 A–D and SI Appendix, Figs. S11A and S12 A–D). The combination index (CI) analysis showed that indeed, both 5D4 and CalAM synergized (CI < 1) with rucaparib in MDA-MB-468, MDAH-2774 and A2780cis cells. Likewise, this synergy was also observed in HCC95 and HCC2814 lung cancer cells (SI Appendix, Fig. S13). In addition to rucaparib, another PARPi, talazoparib, also showed similar synergy when combined with 5D4 (SI Appendix, Fig. S11B) or CalAM (SI Appendix, Fig. S12 E and F). Accordingly, CalAM and 5D4 also synergized with PARPi veliparib (SI Appendix, Fig. S14). On the contrary, treatment with 5D4, rucaparib or both did not elicit any effect in nontransformed AML12 mouse hepatocytes (SI Appendix, Fig. S15).
Fig. 8.
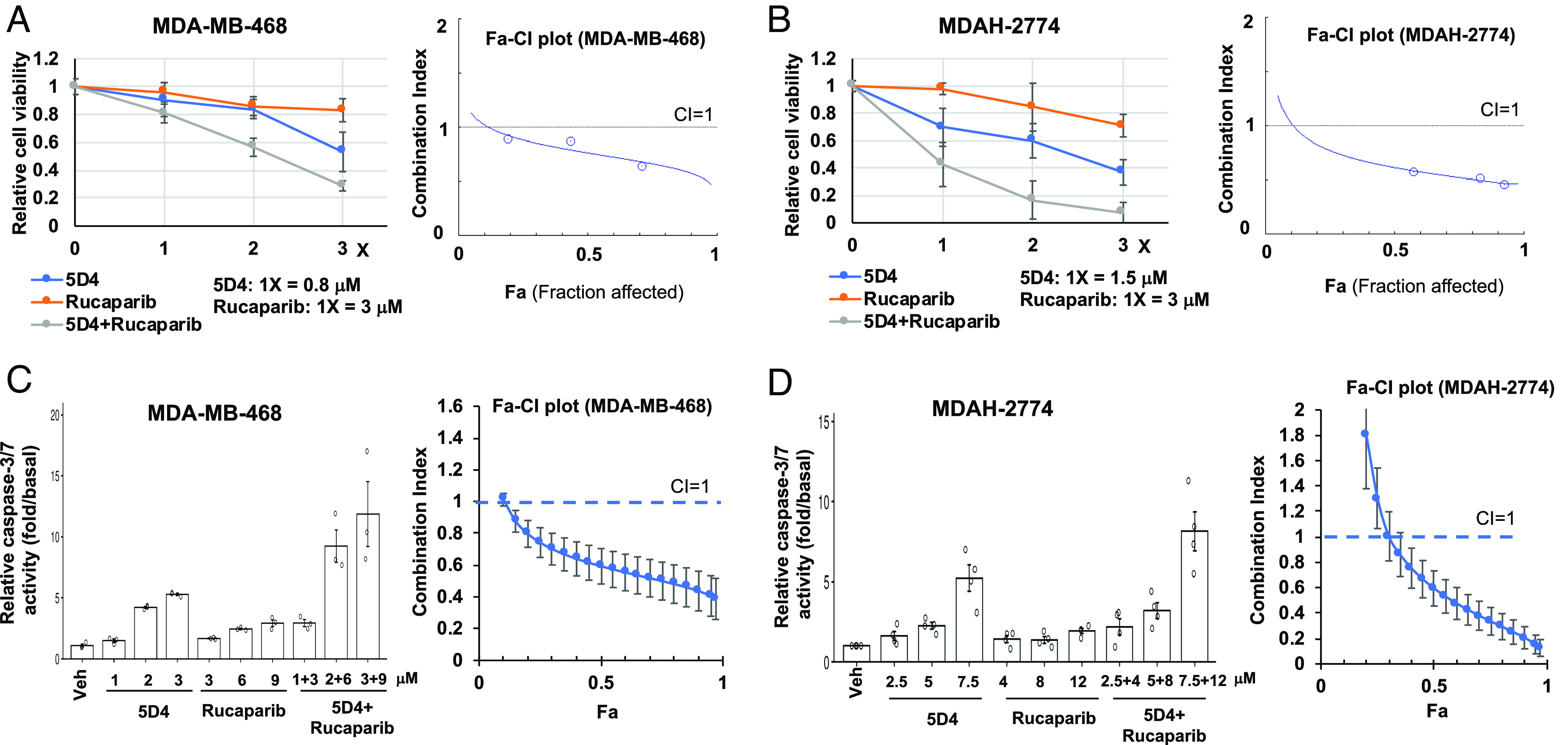
Cpd 5D4 and PARP1/2 inhibitors synergistically inhibit cell viability in breast and ovarian cancer cells. MDA-MB-468 or MDAH-2774 cells were treated with 5D4, rucaparib, or a combination of 5D4 with rucaparib for 45 h (A and B) or 20 h (C and D). Cell viability was determined by CCK-8 assay. Data shown in A and B are the mean ± SD done in quadruplicates. Active caspase-3/7 was determined by Caspase-Glo® 3/7 Assay. Data shown in C and D are the mean ± SEM from three or four independent experiments. Combination index (CI) values and Fa (Fraction affected)-CI plots were generated using CompuSyn software. CI < 1 indicates synergism.
The binding of TopBP1-BRCT7/8 to PLK1 is required for Rad51 recruitment and foci formation (11), a key step for HR. Our data showed that 5D4 or CalAM treatment decreased the expression of Rad51 and PLK1 (Fig. 5 C–F). Moreover, treatment with CalAM or 5D4 greatly prevented rucaparib-induced Rad51 foci formation in MDAH-2774 cells (SI Appendix, Fig. S16). Together, these data demonstrate that TopBP1 inhibitors dampen HR and show synergistic effect with PARPi in HR-proficient cancer cells.
Synergism between TopBP1 Inhibitors and a PARP14 Inhibitor.
Using a genome-wide CRISPR dropout screen to identify synthetic lethal targets in ADP-ribosyltransferase PARP14 knockout cells, TopBP1 was identified to have a synthetic lethal interaction with PARP14 (28). Therefore, we next tested the combinational effect between 5D4 and a PARP14 inhibitor GeA-69 (29). Indeed, 5D4 and GeA-69 synergistically inhibited cell viability (Fig. 9 A and B) and promoted apoptosis (Fig. 9 C and D) in both MDA-MB-468 and MDAH-2774 cells. The immmunoblotting also showed that 5D4 and GeA-69 synergistically repressed the expression of MYC and its targets, such as ACTL6A, Cyclin D1 and Rad51 (SI Appendix, Fig. S17).
Fig. 9.
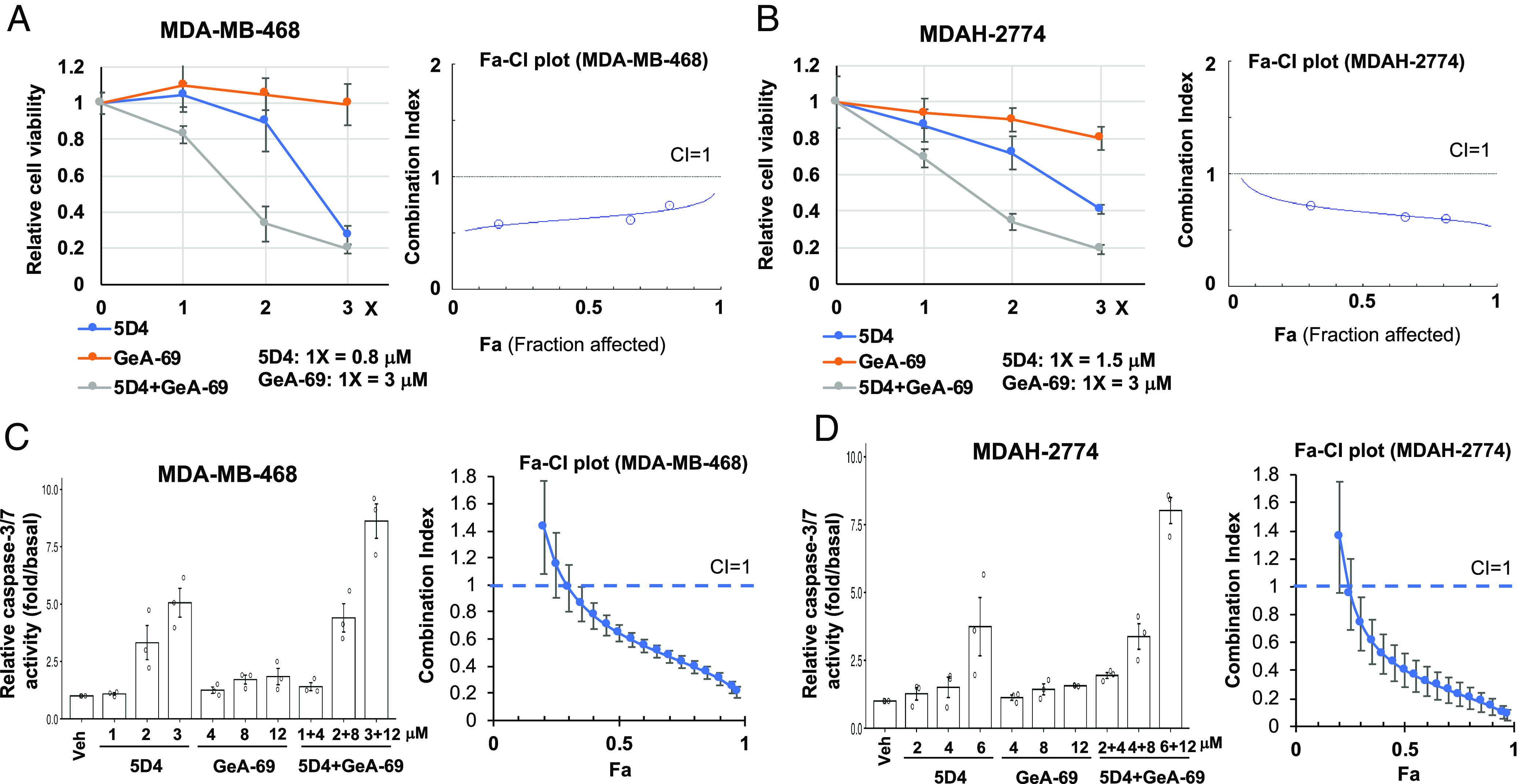
Cpd 5D4 and PARP-14 inhibitor GeA-69 synergistically inhibit cell viability in breast and ovarian cancer cells. MDA-MB-468 or MDAH-2774 cells were treated with 5D4, GeA-69, or both for 45 h (A and B) or 20 h (C and D). Cell viability and active caspase-3/7 were determined as described in Fig. 8. Data shown in A and B are the mean ± SD done in quadruplicates. Data shown in C and D are the mean ± SEM from three independent experiments. Right panels are corresponding Fa-CI plots.
Synergism between Cpd 5D4 and Talazoparib in Two Breast Cancer PDX Models.
To further determine the in vivo efficacy of the combination between TopBP1 inhibitors and PARPi, we implanted BCM2665 PDX into NSG mice. Indeed, combination of 5D4 and talazoparib synergistically inhibited the tumor growth of BCM2665 PDX without apparent toxicity or changes in mouse body weight (Fig. 10A and SI Appendix, Fig. S18A). We performed linear mixed model analysis using tumor size as the dependent variable and adding fixed effects of time of measurement, treatment, and the interaction between time and treatment. The biological replicates were treated as random effects. Indeed, there was a significant effect of treatment (P < 0.001) and an interaction between time and treatment (P < 0.001). The comparison between two treatments also showed statistically significant difference (P < 0.001) (Fig. 10A). We further performed similar experiments in another TNBC PDX BCM3107 (22), which harbors mutp53-T155N, MYC amplification (5 copies) and BRCA1-Q1395* mutation (BCM PDX Portal). BCM3107 PDX also shows a loss of one allele of 53BP1 and DYNLL1, which may confer resistance to PARPi in BRCA1-mutated cancers (30). Our data showed that BCM3107 was resistant to talazoparib but responded well to 5D4 (Fig. 10B). Moreover, combination of 5D4 and talzoparibe further inhibited tumor growth without apparent toxicity or changes in mouse body weight (Fig. 10B and SI Appendix, Fig. S18B).
Fig. 10.
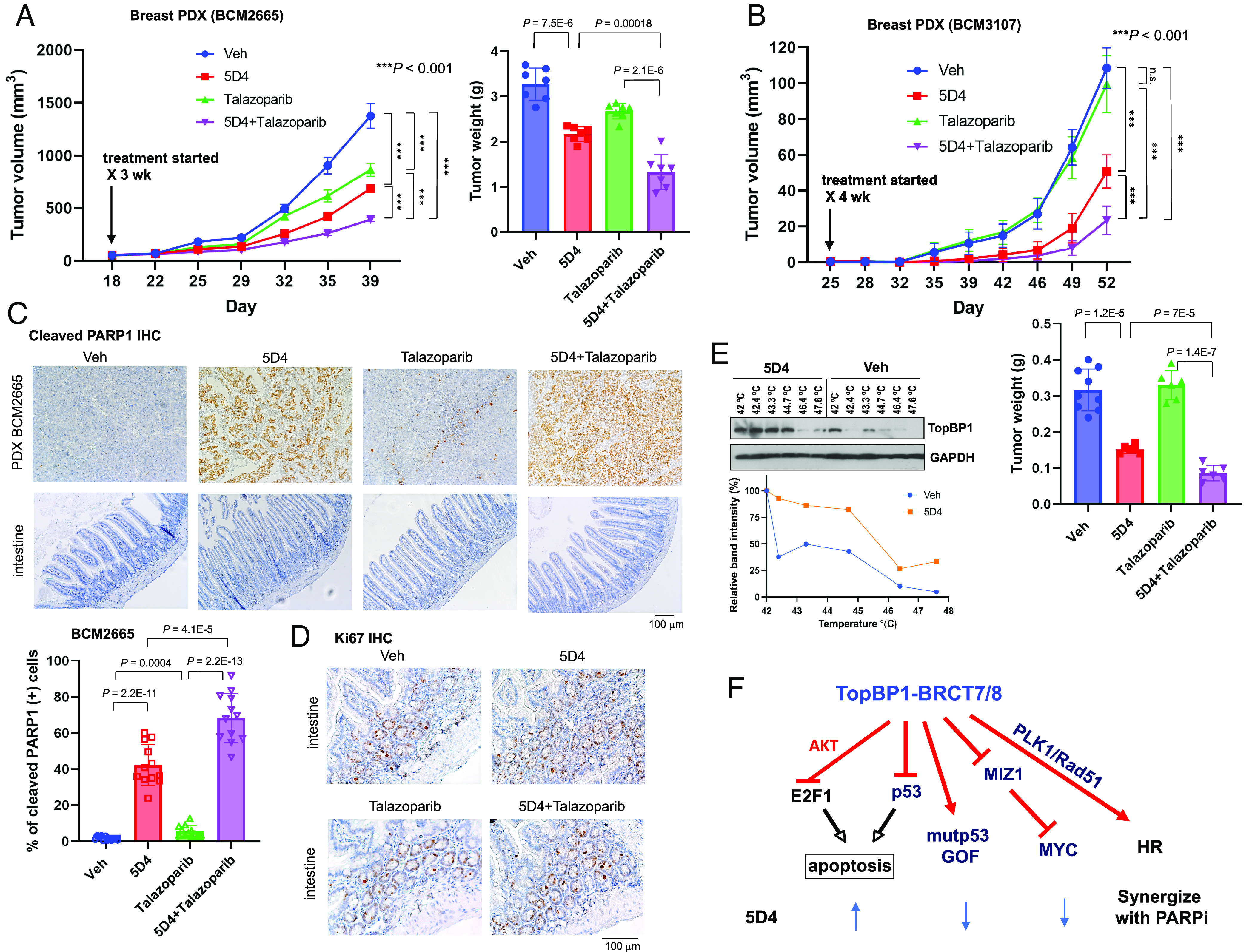
Cpd 5D4 and talazoparib synergistically inhibit tumor growth in two breast cancer PDX models. (A and B) NSG mice bearing BCM2665 or BCM3107 PDX were administered with vehicle, 5D4 (40 mg/kg, i.p., twice weekly) and/or talazoparib (0.5 mg/kg, daily × 5 per week) for 3 or 4 wk. Shown are mean tumor volumes ± SD (P values calculated for linear mixed models) and mean tumor weights ± SD (two-tailed t test). In the BCM2665 PDX experiment, n = 7 in each group; in the BCM3107 PDX experiment, n = 9 in the Veh group and 6 in the other groups; n.s, not significant. The x axis represents the day after PDX implantation. (C–E) After administration with the last dose of vehicle, 5D4 and/or talazoparib in A, mice were euthanized complying with AVMA guidelines for the euthanasia of animals on the following day. Mouse intestine and PDX tumor samples were subjected to IHC staining of cleaved PARP1 (C). Shown are representative images using 10× objective. (Lower) The percentage of cleaved PARP1-positive cells was determined by ImageJ. Twelve images from three xenografts per group were assessed. (D) Ki67 IHC staining of mouse intestinal epithelium using 20× objective. (E) CETSA was performed using the lysates of BCM2665 PDX from mice treated with 5D4 or vehicle. (F) A schematic diagram showing the anticancer activity of 5D4 and its synergy with PARPi.
We next performed immunohistochemistry in BCM2665 PDXs (Fig. 10 C and D). As expected, talazoparib only had mild proapoptotic effect. 5D4 induced apoptosis in breast cancer PDXs, as indicated by elevated PARP-1 cleavage; and when combined with talazoparib, this further enhanced apoptosis (Fig. 10C). In contrast, treatment with 5D4, talazoparib or both neither induced apoptosis nor affected Ki67 staining in mouse intestinal epithelium (Fig. 10 C and D). We then performed CETSA in the BCM2665 xenograft tumor tissues. Consistent with the results shown in Fig. 3F, 5D4 shifted the Tm of TopBP1 protein, providing evidence for the binding of 5D4 to TopBP1 in vivo (Fig. 10E).
Taken together, 5D4 targets TopBP1-BRCT7/8 and impedes its interactions with E2F1, mutp53, MIZ1, CIP2A, and PLK1. As a result, the E2F1-mediated proapoptotic function is enhanced, and the mutp53 GOF, Rad51 recruitment, and MYC activities are decreased by 5D4 treatment. Through these mechanisms of action, 5D4 exerts antitumor activities and shows synergy with PARPi (Fig. 10F).
Discussion
Through multiple rounds of compound screening, we identify a previously undescribed compound 5D4 that targets TopBP1-BRCT7/8 and exhibits anticancer activity in vivo. We further demonstrate that both TopBP1-BRCT7/8 inhibitors, CalAM and 5D4, exert anti-MYC activity and can synergize with inhibitors of PARP1/2 or PARP14 in a number of cancer types.
To verify the on-target effects of 5D4, we provide multiple independent evidences. First, 5D4 can block the TopBP1-BRCT7/8 binding to pS1159-TopBP1 phosphopeptide or mutp53(DBD) in vitro. Second, the CETSA results confirm the 5D4 binding to TopBP1 in breast cancer cells and PDX tumors. Third, 5D4 treatment inhibits the interactions of TopBP1with several BRCT7/8-interacting partners, such as E2F1, mutp53, PLK1, MIZ1, and CIP2A. This leads to the induction of E2F1 proapoptotic targets and p53/p63/p73 targets, as well as the inhibition of MYC targets. Fourth, depletion of TopBP1 or MIZ1 blunts the anticancer effect of 5D4. Fifth, there is a correlation between the response to 5D4 and the levels of TopBP1 mRNA and protein in the cancer cell lines that we examined (SI Appendix, Fig. S19). Sixth, 5D4 can synergize with inhibitors of PARP1/2 or PARP14, which is likely attributed to the disruption of TopBP1-BRCT7/8 interaction with PLK1 by 5D4. It is worth noting that both CalAM (1) and 5D4 do not affect the epithelial cell proliferation of mouse intestines, indicating a specific anticancer effect of TopBP1-BRCT7/8 inhibitors.
MYC is the most frequently amplified gene in human cancers. However, direct inhibition of MYC is widely recognized as a challenge due to its disordered structure and lack of targetable pockets. Our study uncovers an alternative approach to target MYC with small-molecule inhibitors against TopBP1-BRCT7/8. Our SAR analysis and molecular docking identify a pS/TxxxΩ motif-binding pocket in TopBP1-BRCT7/8 as a targetable site. Both MYC and TopBP1 compete for MIZ1 binding (6, 20, 31); thus, overexpressed TopBP1 might sequester MIZ1 and tip the balance toward MYC activation. In fact, TopBP1 expression is highly correlated with MYC target gene signature in many types of cancer (TCGA datasets) (SI Appendix, Fig. S20). Thus, small-molecule inhibitors that bind to TopBP1-BRCT7/8 might release MIZ1 from the TopBP1 complex, allowing it to bind and repress MYC activity in cancer. Together with the effects of TopBP1-BRCT7/8 inhibitors on the activation of E2F1-mediated apoptosis and inhibition of mutp53 GOF (1), these events contribute to the anticancer activities of TopBP1-BRCT7/8 inhibitors.
The synergy between TopBP1-BRCT7/8 inhibitors and PARPi may be attributed to several mechanisms. First, given a role of TopBP1-BRCT7/8 in Rad51 recruitment (11), TopBP1-BRCT7/8 inhibitors are able to prevent Rad51 foci formation and thus synergize with PARPi through the inhibition of HR. Second, many proteins involved in DNA repair are MYC targets, such as Rad51, PLK1 (27), and ACTL6A (18, 26, 32). Indeed, TopBP1-BRCT7/8 inhibitors can down-regulate the expression of these proteins, thereby enhancing the drug synergism with PARPi. Moreover, 5D4 can overcome acquired PARPi resistance in BRCA1-mutated breast cancer harboring DYNLL1 loss. DYNLL1 suppresses the activity of several components of the end-resection machinery involved in HR, including the MRN complex. Thus, loss of DYNLL1 in BRCA1-mutated cells enhances end resection, and restores Rad51 foci formation and HR-mediated DSB repair (30). Since 5D4 inhibits Rad51 expression and recruitment, it can prevent DYNLL1 loss-mediated HR restoration and overcome PARPi resistance in BRCA1-mutated cancer.
Our study further extends the synergy of TopBP1-BRCT7/8 inhibitors with PARP14 inhibitors. It has been shown that defective DNA replication is responsible for the synthetic lethal interaction between PARP14 and either ATR or TopBP1 (28). PARP14 is involved in the processing of the stalled replication forks (33). Unlike PARP1/2, PARP14 is a mono-ADP-ribosyltransferase. While PARP1, 2, and 14 are all involved in the repair of DNA damage especially on the replication forks, PARP14 also plays a role in cell structure, adhesion, motility, and innate immunity (34). In addition, PARP14 is required for Cyclin D1 3′UTR mRNA stability and Cyclin D1 expression (35). Indeed, combined treatment with 5D4 and PARP14 inhibitor GeA-69 synergistically decreased the levels of c-MYC, Cyclin D1, ACTL6A, and Rad51. Thus, it is possible that the synergy between TopBP1-BRCT7/8 inhibitors and PARP14 inhibitors also involves multiple mechanisms.
A limitation of the NSG mouse model is the lack of the immune system. MYC has been known to suppress the immune response (36), which may potentially be restored by inhibiting MYC activity with 5D4. On the other hand, PARPi can induce double-strand breaks and trigger immune response (37). As such, the combination of 5D4 and PARPi may be even more effective in immune-competent mice than in NSG mice. This possibility deserves future investigation. In summary, our study identifies an inhibitor of TopBP1-BRCT7/8 and demonstrates its anticancer activity and synergy with PARPi in HR-proficient cancer cells. These data support the potential use of TopBP1-BRCT7/8 inhibitors for targeted cancer therapy.
Materials and Methods
Virtual screening was performed in Schrödinger suite (version 2010, Schrödinger, LLC, New York, NY, 2010) as described previously (1). TopBP1-BRCT7/8 protein (PDB code: 3AL3) was prepared using the protein preparation wizard in Maestro 9.1 with default protein parameters: water molecules were removed, hydrogen atoms added and the BACH1 peptide ligand extracted for docking. Over 200,000 compounds from three selected libraries (drug-like, nature product derivatives and compounds tested in men) were used for the initial virtual screening. The subsequent molecular docking study for positive hits and hit expansion was performed using 1-Click Docking of Mcule software. Compound 5D4 (purity 99%) was resynthesized by AnalytiCon Discovery. The purity and structure characterization of 5D4 were analyzed by LCMS (SI Appendix, Fig. S21), 1D- and 2D NMR (SI Appendix, Fig. S22), and HRESIMS (High-resolution electrospray ionization mass spectrometry) (SI Appendix, Fig. S23). Other methods are described in SI Appendix, Materials and Methods.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Acknowledgments
This work was supported by funding from NIH R01CA203824, R01CA269971, Department of Defense Grants W81XWH-18-1-0329, W81XWH-19-1-0369, W81XWH-22-1-0226, W81XWH-22-1-0534 (to F.-T.L. and W.-C.L.), and Rivkin Center for Ovarian Cancer Pilot Award (to W.-C.L.). H.F.-K. was supported by T32CA174647, and L.A.W.G. was supported by T32GM136560. We also acknowledge the support from the Cytometry and Cell Sorting Core and Pathology & Histology Core at BCM.
Author contributions
F.-T.L. and W.-C.L. designed research; F.-T.L., K.L., L.A.W.G., H.F.-K., Y.S., S.-J.L., and W.-C.L. performed research; W.-C.L. contributed new reagents/analytic tools; F.-T.L. and W.-C.L. analyzed data; and F.-T.L. and W.-C.L. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Fang-Tsyr Lin, Email: flin@bcm.edu.
Weei-Chin Lin, Email: weeichil@bcm.edu.
Data, Materials, and Software Availability
The RNA-seq data have been submitted to the NCBI Gene Expression Omnibus (accession number GSE227406) (38). All other data are included in the article and supporting information.
Supporting Information
References
- 1.Chowdhury P., et al. , Targeting TopBP1 at a convergent point of multiple oncogenic pathways for cancer therapy. Nat. Commun. 5, 5476 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin W. C., et al. , “Method of treating cancer that overexpresses TopBP1.” US Patent No: US 9,636,323 (2017). US Patent & Trademark Office, Patent Full Text and Image Database United States.
- 3.Wardlaw C. P., Carr A. M., Oliver A. W., TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair (Amst) 22, 165–174 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Liu K., Luo Y., Lin F. T., Lin W. C., TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev. 18, 673–686 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumagai A., Shevchenko A., Dunphy W. G., Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 140, 349–359 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu K., Paik J. C., Wang B., Lin F. T., Lin W. C., Regulation of TopBP1 oligomerization by Akt/PKB for cell survival. Embo J. 25, 4795–4807 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu K., Graves J. D., Lee Y. J., Lin F. T., Lin W. C., Cell cycle-dependent switch of TopBP1 functions by Cdk2 and Akt. Mol. Cell Biol. 40, e00599-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bagge J., Oestergaard V. H., Lisby M., Functions of TopBP1 in preserving genome integrity during mitosis. Semin. Cell Dev. Biol. 113, 57–64 (2021). [DOI] [PubMed] [Google Scholar]
- 9.De Marco Zompit M., et al. , The CIP2A-TOPBP1 complex safeguards chromosomal stability during mitosis. Nat. Commun. 13, 4143 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumagai A., Lee J., Yoo H. Y., Dunphy W. G., TopBP1 activates the ATR-ATRIP complex. Cell 124, 943–955 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Moudry P., et al. , TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J. Cell Biol. 212, 281–288 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu K., et al. , Regulation of p53 by TopBP1: A potential mechanism for p53 inactivation in cancer. Mol. Cell Biol. 29, 2673–2693 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu K., Ling S., Lin W. C., TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell Biol. 31, 4464–4481 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan T. Z., et al. , CSIOVDB: A microarray gene expression database of epithelial ovarian cancer subtype. Oncotarget 6, 43843–43852 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu K., Lin F. T., Ruppert J. M., Lin W. C., Regulation of E2F1 by BRCT-domain containing protein TopBP1. Mol. Cell Biol. 23, 3287–3304 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu K., Graves J. D., Lin F. T., Lin W. C., Overexpression of TopBP1, a canonical ATR/Chk1 activator, paradoxically hinders ATR/Chk1 activation in cancer. J. Biol. Chem. 296, 100382 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu K., Lin F. T., Graves J. D., Lee Y. J., Lin W. C., Mutant p53 perturbs DNA replication checkpoint control through TopBP1 and Treslin. Proc. Natl. Acad. Sci. U.S.A. 114, E3766–E3775 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walz S., et al. , Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511, 483–487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peter S., et al. , Tumor cell-specific inhibition of MYC function using small molecule inhibitors of the HUWE1 ubiquitin ligase. EMBO Mol. Med. 6, 1525–1541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herold S., et al. , Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol. Cell 10, 509–521 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Liu K., Graves J. D., Scott J. D., Li R., Lin W. C., Akt switches TopBP1 function from checkpoint activation to transcriptional regulation through phosphoserine binding-mediated oligomerization. Mol. Cell Biol. 33, 4685–4700 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X., et al. , A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 73, 4885–4897 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acevedo J., Yan S., Michael W. M., Direct binding to replication protein A (RPA)-coated single-stranded DNA allows recruitment of the ATR activator TopBP1 to sites of DNA damage. J. Biol. Chem. 291, 13124–13131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laine A., et al. , CIP2A interacts with TopBP1 and drives basal-like breast cancer tumorigenesis. Cancer Res. 81, 4319–4331 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adam S., et al. , The CIP2A-TOPBP1 axis safeguards chromosome stability and is a synthetic lethal target for BRCA-mutated cancer. Nat. Cancer 2, 1357–1371 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Schlosser I., et al. , Dissection of transcriptional programmes in response to serum and c-Myc in a human B-cell line. Oncogene 24, 520–524 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Kim J., Lee J. H., Iyer V. R., Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One 3, e1798 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dhoonmoon A., Schleicher E. M., Clements K. E., Nicolae C. M., Moldovan G. L., Genome-wide CRISPR synthetic lethality screen identifies a role for the ADP-ribosyltransferase PARP14 in DNA replication dynamics controlled by ATR. Nucleic Acids Res. 48, 7252–7264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuller M., et al. , Discovery of a selective allosteric inhibitor targeting macrodomain 2 of polyadenosine-diphosphate-ribose polymerase 14. ACS Chem. Biol. 12, 2866–2874 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee E. K., Matulonis U. A., PARP inhibitor resistance mechanisms and implications for post-progression combination therapies. Cancers (Basel) 12, 2054 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herold S., et al. , Miz1 and HectH9 regulate the stability of the checkpoint protein, TopBP1. EMBO J. 27, 2851–2861 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao Y., Lin F. T., Lin W. C., ACTL6A promotes repair of cisplatin-induced DNA damage, a new mechanism of platinum resistance in cancer. Proc. Natl. Acad. Sci. U.S.A. 118, e2015808118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhoonmoon A., Nicolae C. M., Moldovan G. L., The KU-PARP14 axis differentially regulates DNA resection at stalled replication forks by MRE11 and EXO1. Nat. Commun. 13, 5063 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richard I. A., Burgess J. T., O’Byrne K. J., Bolderson E., Beyond PARP1: The potential of other members of the poly (ADP-Ribose) polymerase family in DNA repair and cancer therapeutics. Front. Cell Dev. Biol. 9, 801200 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Connor M. J., Thakar T., Nicolae C. M., Moldovan G. L., PARP14 regulates cyclin D1 expression to promote cell-cycle progression. Oncogene 40, 4872–4883 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casey S. C., Baylot V., Felsher D. W., The MYC oncogene is a global regulator of the immune response. Blood 131, 2007–2015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim C., Wang X. D., Yu Y., PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. Elife 9, e60637 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin W. C., Lin F. T., RNA-seq data of 5D4- or Calcein AM-treated MDA-MB-468. NCBI Gene Expression Omnibus (GEO). https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227406. Deposited 15 March 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Data Availability Statement
The RNA-seq data have been submitted to the NCBI Gene Expression Omnibus (accession number GSE227406) (38). All other data are included in the article and supporting information.