

Abstract
Ruthenium(II) polypyridyl complexes form a vast family of molecules characterized by their finely tuned photochemical and photophysical properties. Their ability to undergo excited-state deactivation via photosubstitution reactions makes them quite unique in inorganic photochemistry. As a consequence, they have been used, in general, for building dynamic molecular systems responsive to light but, more particularly, in the field of oncology, as prodrugs for a new cancer treatment modality called photoactivated chemotherapy (PACT). Indeed, the ability of a coordination bond to be selectively broken under visible light irradiation offers fascinating perspectives in oncology: it is possible to make poorly toxic agents in the dark that become activated toward cancer cell killing by simple visible light irradiation of the compound inside a tumor. In this Perspective, we review the most important concepts behind the PACT idea, the relationship between ruthenium compounds used for PACT and those used for a related phototherapeutic approach called photodynamic therapy (PDT), and we discuss important questions about real-life applications of PACT in the clinic. We conclude this Perspective with important challenges in the field and an outlook.
1. Phototherapies: An Introduction
1.1. Phototherapies in Medicine
Our eyes are not the only light-sensitive organs in humans. Our moods, our sleep, and our skin are also sensitive to sunlight. Artificial light sources entered clinical practice a long time ago, for example, to treat smallpox.1 Newborn jaundice treatment is one of the best-known clinical application of phototherapy,2 while skin tumors were treated with phototherapy in ancient Egypt.3 Inspired in part by naturally photoactive compounds,4 and driven by the development of antibiotic-resistant bacteria, new applications of phototherapy have developed rapidly, such as antibacterial photodynamic therapy (aPDT).5 However, the most developed application of medicinal phototherapy targets tumors.
1.2. Anticancer Phototherapies
Techniques to treat cancer patients using light-sensitive compounds have emerged to circumvent the toxicity of conventional treatments. In photodynamic therapy (PDT), a clinically approved treatment of pre-cancerous diseases of the skin or esophagus or of more advanced cancers of the brain or lungs, the photosensitive compound is called a “photosensitizer” (PS). Upon light excitation followed by spin flip, the PS is promoted into a triplet excited state (3PS*) that transfers an electron (PDT type I) or energy (PDT type II) to the O2 molecules present in the irradiated tissues. Such a transfer produces high local doses of reactive oxygen species (ROS) that generate three effects. First, they kill cancer cells by oxidative damage to nucleic acids, proteins, and lipid membranes. Second, they consume oxygen and damage blood vessels, thus generating hypoxia. Third, they trigger the immune system.6 Altogether, PDT often generates a strong antitumor effect with minimal side-effects for the patients. These factors explain, in part, the fast growth of clinical PDT and the number and quality of reviews dedicated to it.7,8
A second form of anticancer phototherapy involves organic protein inhibitors covalently functionalized with reversible photoswitches such as azobenzene or diarylethene.9,10 In azobenzene conjugates, the dark form of the prodrug has a trans azo bond, while its light-activated form is cis. Both forms show different interactions of the inhibitor with the target protein, which modulates protein activity and sometimes kills cancer cells upon light irradiation. With azo compounds, however, the cis form is thermally unstable and reverts to the more thermodynamically stable trans form, thereby leading to reversible prodrug light activation.
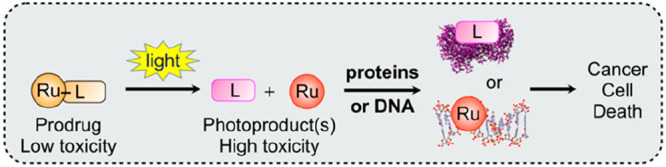
The third main form of anticancer phototherapy, which is the focus of this Perspective, is called PhotoActivated ChemoTherapy (PACT). For clinicians, PACT may look like PDT (Figure 1): the patient receives a non-active prodrug, which distributes in the body and inside the tumor without causing harm. After some time, called the drug-to-light interval (DLI), light is shone onto the tumor, where it activates the prodrug. Finally, the activated compound and tumor debris are excreted outside the body. Chemically speaking, however, PACT addresses both the oxygen dependence of PDT and the reversibility of photoswitches in photopharmacology. It relies on the irreversible and oxygen-independent photochemical bond cleavage of either a metal–ligand coordination bond11 or a carbon–oxygen bond.12 In fact, PACT is similar to photocaging, a technique that uses a non-toxic “caging” group to “hide” the biological activity of a molecule, for example, ATP13 or morphin.14 When “caged”, the bioactive compound cannot interact with its target because the photocage ruins the precise key–lock fit built by medicinal chemists. Upon photochemical “uncaging”, the bioactive compound recovers its ability to interact with its biological target. Importantly, for PACT treatment of cancer, the prodrug in its photocaged form should be poorly toxic, while after light activation, at least one of the two photoreleased fragments should be very toxic to cancer cells. Inorganic photochemists have used different metal centers to prepare photocaged compounds that are activated with visible or near-infrared (NIR) light.15,16 Ruthenium-based photocages for the PACT treatment of cancer are the focus of this Perspective.
Figure 1.

Principle of ruthenium-based photoactivated chemotherapy (PACT). Top: light-induced bond cleavage reaction in the prodrug. Either the photoreleased ligand (L) or the metal fragment (Ru), or both, interact(s) with biomolecules, leading to cell death. Bottom: PACT treatment of a patient with a lung tumor (in purple). The prodrug (orange) is injected intravenously, distributes in the body, and reaches the tumor in its non-toxic form. After the drug-to-light interval (DLI), light is shone onto the tumor, activating the prodrug and destroying the tumor. Finally, the body excretes the excess drug. Image courtesy Bianka Siewert.
1.3. Early Developments of Metal-Based PACT Compounds
Historically, PACT is based on the concomitant development of PDT and platinum chemotherapy drugs. While Figge (1955) first detected tumor fluorescence upon hematoporphyrin injection,18 Dougherty shone light on hematoporphyrin-injected mice and patients (1975–1979) and demonstrated that singlet oxygen (1O2) was the cytotoxic agent.19 In parallel, cisplatin was discovered as a potent chemotherapeutic agent20 and was approved for clinical use in 1978. While Malik and Kennedy developed PDT using 5-aminolevulinic acid (1987–1990), Photofrin was approved by the FDA in 1995. The first article mentioning a “photo cisplatinum reagent”, from Morrison,21 dealt with the rhodium(III) polypyridyl complex [Rh(phen)2Cl2] (phen = 1,10-phenanthroline). This complex was able to photosubstitute one of its chloride ligand by a DNA base pair upon UV light irradiation.22 Though no biological experiments were initially performed, UV light irradiation was suggested to trigger metal coordination to DNA with light, which opened the door to photoinorganic therapeutic approaches.
The first platinum-based PACT prodrugs originated with Bednarski and Sadler.23,24 The activation mechanism for these thermally inert Pt(IV) compounds is different from that of rhodium(III) and ruthenium(II) polypyridyl complexes: upon light irradiation in cells, Pt(IV) compounds are photoreduced into a more labile platinum(II) photoproduct capable of exchanging ligands with biomolecules and finally binding to DNA. Finally, the first ruthenium(II) polypyridyl PACT compounds working by photosubstitution were proposed by Etchenique and Turro in 2003 and 2004, respectively.25,69 For Turro’s compound, the cytotoxic species was the metal-containing photoproduct, while in Etchenique’s case, the bioactive compound was the photosubstituted ligand. In parallel, thorough understanding of the photochemistry of ruthenium(II) polypyridyl complexes initiated by Sauvage,26,27 Balzani,11,28 McMillin,29 and Meyer30 led to fast developments of ruthenium-based PACT, leading to the first in vivo experiment by the Wu and Bonnet groups in 2016 and 2019, respectively. The term “photoactivated chemotherapy” (PACT) was proposed by Salder in 2009.16
2. Ruthenium-Based PACT Compounds
2.1. Photochemical Activation Mechanisms
In ruthenium-based PACT compounds, a coordination bond between the ruthenium center and an organic ligand is broken via a photosubstitution reaction (Figure 2). In order to show this type of reactivity, the ruthenium center should be in the oxidation state +II and bound to a so-called “polypyridyl” chelate comprising at least two pyridyl rings connected to each other via a C–C bond. Both the 2,2′;6′,2″-terpyridine (tpy) and 2,2′-bipyridine (bpy) chelates in Figure 2 are typical examples of such polypyridyl ligands. Initially seen as a detrimental decomposition pathway for ruthenium-based photosensitizers in photocatalysis,30 photosubstitution reactions have since then proven to be useful tools for the controlled activation of molecular machines31,32 or anticancer drugs.33 In fact, photosubstitution reactions in ruthenium(II) polypyridyl complexes are rather unique, because they occur with good quantum yields upon irradiation with visible light, while the complexes are usually thermally inert. However, photosubstitution is not strictly reserved to Ru(+II) complexes: it has also been reported for low-spin polypyridyl d6 transition metal centers based on Ir(+III), Rh(+III), or Re(I) for example. Fe(+II) complexes are difficult to use for PACT because they are thermally labile, though photosubstitution with strong ligands (CO, CN–, or NO) has been described.34 Finally, photosubstitution on Os(+II) complexes is very rare and very slow.35
Figure 2.

Example of a photosubstitution reaction used in PACT with ruthenium photocage [Ru(tpy)(bpy)(Hmte)]2+. The blue peak in the UV–vis spectrum shows the emission of the light source used to trigger photosubstitution, centered at 450 nm. The bottom graph shows the time evolution of the absorption spectrum of the solution during light irradiation. The low α angle (∼160°) in the terpyridine ligand distorts the first coordination sphere of the metal center compared to a perfect octahedron (180°), which facilitates photosubstitution. Data adapted from ref (17).
In polypyridyl ligands, conjugation leads to low-lying π* orbitals ending up as the lowest unoccupied molecular orbital (LUMO) of their ruthenium(II) complexes. Upon photon absorption, the octahedral complex promotes an electron from a metal-centered t2g (HOMO) orbital into the ligand-centered LUMO, thereby generating a metal-to-ligand charge-transfer singlet excited state (1MLCT) that efficiently spin-flips to a triplet (3MLCT, Figure 3a,b). The classical mechanism of photosubstitution in polypyridyl ruthenium(II) compounds starts from these 3MLCT states. While they are typically responsible for the phosphorescence, electron-transfer, or energy-transfer processes observed with photoinert compounds such as [Ru(bpy)3]2+, they can also be thermally promoted to a metal-centered (3MC) triplet excited states that lies close enough in energy (Figure 3a). Usually, while 1MLCT-to-3MLCT transitions are very fast (<100 fs) and thermally non-activated, 3MLCT-to-3MC conversions take time and occur via an activation barrier.36 The corresponding triplet transition state (3TS) is represented in Figure 3a. This thermal barrier is due to the different geometries of the 3MLCT and 3MC states: in the 3MC state, the electron promoted in an antibonding (eg*) metal–ligand orbital elongates the Ru–ligand bond distance, compared to 3MLCT states (Figure 3b,c). Such a longer distance facilitates substitution of the ligand by a solvent molecule before decaying to the ground state of the photosubstituted product. This mechanism derives from ancient37,38 temperature-dependent phosphorescence lifetime and photosubstitution measurements39 and has been confirmed by multiple reports.40−42,36 In short, enhanced quenching of the phosphorescence of [Ru(bpy)3]2+ at high temperatures suggested that nearby 3MC states may be thermally populated from the photochemically generated 3MLCT states. From the temperature dependence of the phosphorescence lifetime and quantum yield, Watts and Houten derived an excess energy of ΔG0 ≈ +43 kJ/mol.37 This value was later confirmed by measuring the increase of photosubstitution quantum yield with temperature.38 Sauvage’s observation that more sterically hindered compounds showed more pronounced photosubstitution at the cost of phosphorescence43 confirmed the role played by ligand-field 3MC states in photosubstitution. It also demonstrated that ligand design can fine-tune the relative energies of the 3MC and 3MLCT states to favor photosubstitution.
Figure 3.

Classical model for photosubstitution reactions in ruthenium(II) polypyridyl complexes. (a) Molecular energy of the different states involved in the photochemistry of ruthenium(II) polypyridyl complexes. Gray pathways generate 1O2 in the presence of dioxygen; black pathways remain in the absence of O2. kISC, knr, k′nr, kP, and kTTET are rate constants for intersystem crossing, non-radiative decay, phosphorescence, and triplet–triplet energy transfer, respectively. ΔG⧧a is the activation barrier for the conversion of the 3MLCT to the 3MC state, and ΔG0 = G(3MC) – G(3MCLT). 3SA represents a Solvent Adduct of the complex in the triplet state. (b) Orbital energy scheme of the excited states involved in photosubstitution. Numerical values for bond lengths are indicated for [Ru(tpy)(bpy)(Hmte)]2+ (Figure 2), as reported in ref (50).
As a note, different 3MC states may exist, characterized by different geometries and energies, in particular when different ligands may be photosubstituted.44,36 Recently, Elliott and Dixon suggested that, for tris-diimine complexes such as [Ru(bpy)3]2+, 3MC states reminiscent of trans bond activation may be responsible for non-radiative decay of the 3MLCT states (knr in Figure 3a), while photosubstitution may preferentially occur from cis3MC states.45,46Cis and trans3MC states are unrelated to cis- and trans-[Ru(bpy)2(OH2)2]2+ or [Ru(bpy)2(MeCN)2]2+ photoproducts, which may also interconvert upon prolonged light irradiation in water or acetonitrile, respectively, after initial photosubstitution of a bidentate ligand.47−49 Overall, the triplet hypersurface of ruthenium polypyridyl complexes is topologically complicated, and several 3MLCT, 3MC, intraligand charge transfer (3ILCT), or more localized (3π–π*) excited states may coexist and interchange upon light irradiation of a complex, thus leading to a wide range of photochemistries.
Recently, the Turro group found that the photosubstitution of monodentate nitriles in [Ru(tpy)(acac)(RCN)]+ complexes (1+ for R = Me, acac– = acetylacetonate, see Figure 5) was possible using far-red light (655 nm), while in [Ru(tpy)(bpy)(RCN)]+ complexes one should irradiate in the blue region (450 nm) to obtain photosubstitution.51 Further mechanistic studies demonstrated that photosubstitution in [Ru(tpy)(acac)(RCN)]+ did not follow the energy gap law.52 In other words, the 3MLCT lifetimes increased as its energy went down, thus making these states less prone to deactivate via 3MC states. In parallel, the complexes with the lowest 3MLCT had surprisingly the highest photosubstitution quantum yields. Thus, the acetylacetonate chelate, which is known to generate low-lying, poorly distorted MLCT states, led to longer 3MLCT lifetimes and higher photosubstitution quantum yields, while in the classical mechanism (Figure 3) lower photosubstitution quantum yields (φPS) would be expected for low-lying 3MLCT states. This observation suggested that photosubstitution may also occur directly from the 3MLCT state, without thermal promotion to the 3MC. This striking observation was qualitatively confirmed by the Bonnet group in a series of [Ru(tpy)(N-N)(Hmte)]2+ (Hmte = 2-methylthioethanol) complexes53 and recently more directly demonstrated by Turro et al.54 As shown in Figure 4, the φPS values in Turro’s complexes increased when the activation barrier Ea (∼ΔG⧧a in Figure 3) increased. There is currently no solid theoretical model that explains this recent observation. However, additional discussion on this topic can be found in a recent review.33
Figure 5.

Selection of ruthenium-based PACT compounds. The first photosubstituted ligand is highlighted in blue.
Figure 4.

Relationship between the photosubstitution quantum yield in water (φPS) and the activation energy (Ea) to promote the 3MCLT state to the 3MC state, in [Ru(tpy)(L)(MeCN)]n+ (n = 1 or 2), where L is a bidentate ligand. Each dot represents a metal complex. Adapted from ref (54). Copyright 2022 American Chemical Society.
2.2. Molecular Design of Ruthenium-Based PACT Compounds
Despite these recent results, the current understanding of the photochemistry of ruthenium polypyridyl complexes is good, and several molecular design principles for PACT compounds have been established (Figure 5). The molecular structure influences both the photosubstitution mechanism and they quantum efficiency, but also the light absorption properties of the complex. One of the most studied families of ruthenium complexes investigated for PACT is based on complexes bound to three diimine chelates.55−57 The reference compound, [Ru(bpy)3]2+, is weakly phosphorescent (φP ≈ 0.02) and a good generator of 1O2 (φΔ ≈ 0.73), but it is not a PACT compound: The cis3MC states are high in energy, which prevents photosubstitution at body temperatures. Notably, bpy photosubstitution does occur in near-boiling (90 °C) HCl aqueous solutions because ΔG⧧a is not infinite.38 The Sauvage group first reported that introducing steric hindrance in such complexes triggered photosubstitution at room temperature.43 Inspired by this approach, the Glazer group has developed, since 2012,55 a series of sterically hindered tris-diimine complexes for PACT.58,59 In those compounds (22+, Figure 5), steric hindrance comes from the methyl groups ortho to the nitrogen bpy (or phen) atoms. For example, the sterically hindering 6,6′-dimethyl-2,2′-bipyridine chelate (dmbpy) introduces distortion of the first coordination sphere of the complex, which lowers the 3MC energy and triggers efficient photosubstitution. The Papish group developed analogous complexes (32+, Figure 5) based on 6,6′-dihydroxy-2,2′-bipyridine (dOHbpy). 32+ photosubstitutes dOHbpy in acidic conditions (pH 5.0) where phenols are protonated. At pH 7.5, however, the phenol groups become deprotonated, leading to a major shift of the photochemistry of the complex that becomes photostable and a good PDT sensitizer (Table 1).56,60
Table 1. Photochemical Properties of Selected Ruthenium(II) Polypyridyl Complexes Used in PACTa.
| λmaxMLCT | φPS (λexc in nm)b | φPc | φΔd | ref | |
|---|---|---|---|---|---|
| [Ru(bpy)3]2+ | 450 | 0 (450)f | 0.015 | 0.73i | (66) |
| 0.00053 (436)e | (38) | ||||
| [Ru(bpy)2(dmbpy)]2+ (22+) | 450 | 0.05 (413)f | 0.00003 | 0.023i | (67) |
| [Ru(dpp)(bpy)(mtmp)]2+ (52+) | 430 | 0.111 (521)f | n.d. | 0.03i | (64) |
| [Ru(phpy)(bpy)(mtep)+ (23+) | 526 | 0.00035 (521)f | n.d. | n.d. | (68) |
| [Ru(bpy)2(6,6′-dOHbpy)]2+ (32+) | 462 | 0.0058 (450)g | n.d. | 0.041i | (60) |
| 493 | 0.0012 (450)h | n.d. | 0.18i | (60) | |
| [Ru(bpy)2(NH3)2]2+ (82+) | 490 | 0.024 (350)j | 0.002 | n.d. | (69) |
| [Ru(bpy)2(MeCN)2]2+ | 427 | 0.21 (40098)j | n.d. | n.d. | (70) |
| 0.22 (450)j | |||||
| [Ru(bpy)(dppn)(MeCN)2]2+ (172+) | 430 | 0.002 (400)j | n.d. | 0.72i | (71) |
| cis-[Ru(bpy)2(PMe3)(ImH)]2+ (cis-92+) | 432 | 0.10j | n.d. | n.d. | (72) |
| trans-[Ru(bpy)2(PMe3)(ImH)]2+ (trans-92+) | 464 | 0.23j | n.d. | n.d. | (72) |
| [Ru(bapbpy)(dmso)(OH2)]+ (102+) | 308 | 0.003 (450)j | n.d. | 0.013i | (73) |
| [Ru(tpy)(bpy)(dmso)]2+ | 411 | 0.016 (450)f | (74) | ||
| [Ru(phbpy)(bpy)(dmso)]+ (22+) | 476 | 0.000041 (450)f | 0.00016 | 0.032i | (74) |
| [Ru(tpy)(bpy)(Hmte)]2+ | 450 | 0.022 (452)j | <10–4 | <0.005i | (75) |
| [Ru(tpy)(bpy)(R-SCH3)]2+ (132+) | 454 | 0.0038 (530)f | n.d. | n.d. | (76) |
| 0.0055 (450) | |||||
| [Ru(tpy)(dppn)(R-SCH3)]2+ | 458 | 0.00095 (450)fj | 0.000037 | 0.71i | (77) |
| [Ru(tpy)(biq)(R-py)]2+ (122+) | 531 | 0.013 (625)j | n.d. | 0.0036i | (78) |
| [Ru(tpy)(acac)(MeCN)]+ (1+) | 505 | 0.014 (450)j | n.d. | n.d. | (51, 52) |
| [Ru(tpy)(bca)(R-py)]0 (15) | 550 | 0.0081 (470)k | n.d. | n.d. | (79) |
| [Ru(tpy)(dmbpy)(R-py)]2+ (112+) | 474 | 0.15 (500)j | n.d. | n.d. | (80) |
| 0.31 (500)f | (80) |
In the chemical formulas, R represents different substituents (see original publications); the ligand abbreviations are indicated in the main text.
Quantum yield for photosubstitution measured at the indicated excitation wavelength λexc and, unless otherwise noted, at room temperature.
Phosphorescence quantum yield.
Singlet oxygen (1O2) generation quantum yield. n.d. = not determined.
Measured at 363 K in 0.1 M aqueous HCl.
In acetonitrile.
In aqueous solution at pH 5.0.
In aqueous solution at pH 7.5.
In CD3OD solution.
In water.
In H2O containing 5% DMSO.
Next to steric hindrance, electronic effects in tris-diimine ruthenium complexes may also trigger photosubstitution. The Elliott group developed a series of analogues of 22+ where the dmbpy chelate was replaced by a bis-triazole derivative (42+, Figure 5).61 These ligands destabilize 3MLCT states rather than lowering 3MC states, which accelerates photosubstitution. Following studies from Jouvenot et al.,62 the Bonnet group replaced dmbpy by thioether-containing bidentate chelates such as 2-(methylthiomethyl)pyridine (mtmp)63,64 (52+, Figure 5) or 1,3-bis(methylthio)-2-propane, which also led to efficient photosubstitution.47 Turro suggested65 that greater photosubstitution quantum yields were obtained with bis-thioethers due to the longer Ru–S bond distance elongation in the lowest triplet-state geometry. On the other hand, there was no indication about the nature (3MLCT vs 3MC) of these lowest triplet states. As pyridyl–thioether chelates have not yet been included in detailed theoretical studies, it is unclear at that stage why they lead to such good photosubstitution quantum yields. However, it is clear that they form excellent caging groups for ruthenium-based PACT compounds.64
A second family of ruthenium-based PACT compounds consists of complexes containing two cis monodentate ligands. Initially introduced by Etchenique,25 [Ru(bpy)2(L)(L′)]2+ compounds may release either one or two monodentate ligand(s), L and L′, depending on their chemical nature and on irradiation times. For example, monodentate phosphines (L = PPh3 or PMe3, see 62+ in Figure 5) are usually photostable, but they allow efficient photorelease of monodentate amines, pyridines, or nitriles (L′). Alternatively, two identical pyridines, primary amines or imidazoles (L = L′, 72+ and 82+Figure 5), may be photosubstituted successively. The second photosubstitution is much slower than the first one due to excited-state deactivation in the monoaqua intermediate [Ru(bpy)2(OH2)(L′)]2+. These compounds have been initially introduced for the photocaging of neurotransmitters (4-aminopyridine in 72+, γ-aminobutyric acid in 62+)25,81−83 but later on served as phototoxic warheads,84 as suggested by Turro.69 These compounds also exist in a trans form. Though less information is available on trans isomers, recently trans-[Ru(bpy)2(PMe3)(ImH)]2+ (trans-92+, ImH = imidazole, Figure 5) was shown to have red-shifted absorption, compared to its cis analogue, and also higher photosubstitution quantum yields (Table 1).72 This observation opens new design opportunities toward ruthenium-based PACT compounds with red-shifted activation.
Recently, the Bonnet group introduced a new family of tetrapyridyl complexes that, upon coordinating the basal plane of ruthenium, leave two trans coordination sites (102+, Figure 5).73 In these compounds, light irradiation led to photosubstitution of the axial dmso ligand, which, when performed in cancer cells, led to cell death. The Glazer group recently demonstrated that trans ruthenium polypyridyl complexes may have improved toxicity compared to cis analogues, unlike for platinum compounds, for which transplatin is less active than cisplatin.85 Clearly, for ruthenium polypyridyl complexes, the relationship between the poorly toxic cis photocages and their more toxic trans analogues needs to be further investigated.
Another important family of ruthenium polypyridyl compounds used in PACT contains molecules based on tpy ligands (Figure 2).86,87 This scaffold generates N–Ru–N angles between N atoms of the terminal pyridyl rings of tpy that are much lower (α ≈ 150–160° in Figure 2) than the 180° angle expected in a perfect coordination octahedron. This low angle represents a significant distortion of the first coordination sphere of the metal, which significantly lowers the 3MC energy, thus shifting photoreactivity toward photosubstitution.88 A prototypic example is the [Ru(tpy)(bpy)(OH2)]2+ complex, which is poorly toxic by itself89 but binds to many monodentate ligands L that can thereafter be photosubstituted with good quantum yields (Figures 2 and 3b).50,90 This scaffold forms an excellent photocaging group, and a wide range of photocaged complexes of the type [Ru(tpy)(N-N)(L)]2+ have been published with different cytotoxic organic inhibitors L, and different bidentate spectator ligands N-N, such as dmbpy, 2,2′-biquinoline (biq), di(isoquinolin-3-yl)amine (i-Hdiqa), or bicinchoninic acid (H2bca), some of which (112+–142+, 15) are shown in Figure 5 and Table 1. Four types of monodentate ligands L were considered for these photocages: thioether, nitriles, pyridines, and pyrazines.15 Importantly, the steric hindrance of the chelate N-N must be adjusted to the steric requirements of the monodentate ligand L. With L = thioethers, for example, the CH2 or CH3 substituents on sulfur come close to ruthenium upon coordination, which requires limited steric hindrance on the bidentate chelate: N-N should be an unsubstituted bipyridine (132+) to keep good thermal stability.17 i-Hdiqa (142+) provides higher steric hindrance and photosubstitution quantum yields without jeopardizing thermal stability (Table 1),53 but biq is too sterically hindered, resulting in a Ru–S bond that is thermally unstable in water.17 By contrast, monodentate L = pyridine ligands make thermally and photochemically non-labile complexes when N-N = bpy. To obtain efficient pyridine photolabilization, steric hindrance on the bidentate chelate N-N is required. The N-N = dmbpy, biq, and bca2– chelates have allowed the caging of a wide series of pyridine-based inhibitors (112+, 122+, or 15, Figure 5).78,80,79 The advantage of the [Ru(tpy)(N-N)(L)]2+ scaffold is its great versatility and tunability. On the other hand, most of these complexes are activated by blue or green light, and only a small subset is sensitive to red light (usually 630 nm, but 15 is sensitive to 660 nm).78 Replacing the N-N chelate by an oxygen-based, monoanionic acetylacetonate ligand (1+) recently made it possible to shift the activation wavelength to the NIR region of the spectrum.51 The biology of these compounds has not been extensively evaluated, but apparently 1+ is quite toxic in the dark.91 The Sun group recently made use of a similar ruthenium cage for activating tumor-targeted nanoparticles at 760 nm via a combination of PDT and PACT.92 This work demonstrates the high potential of [Ru(tpy)(O-O)(L)]2+ for anticancer phototherapy.
Finally, next to changing the first coordination sphere of ruthenium by fine-tuning the denticity, steric hindrance, and electronic effects of the ligands, PACT compounds can be functionalized on one of the “spectator” ligands by π-extended functional groups (162+–192+, Figure 5). Extended π substituents can have a profound influence on the photobiology of ruthenium polypyridyl complexes, and notably on their behavior in hypoxic cancer cells. In many cases, the extended π substituents introduce localized (3π–π*, 162+) or intraligand charge-transfer (3ILCT, 172+–192+) excited states (Figure 3a) that generate new pathways for 1O2 or radical generation via energy or electron transfer, respectively. These effects were recently discussed in papers from Glazer and McFarland describing sterically hindered ruthenium complexes functionalized with naphthalene (172+)93,59 or oligothiophene (182+, 192+)94 groups, respectively. As first highlighted by Turro with 162+,71 extended π ligands often result in mixed photoreactivity combining PACT and PDT mechanisms. The photoindex values in so-called “dual action” phototherapeutic ruthenium compounds can be extremely high (up to 103–106 in hypoxic SKMEL28 cells94), which represents one of the great advances in the field of ruthenium-based anticancer phototherapy in the past few years.
2.3. About Photosubstitution Quantum Yields and Irradiation Times
One central question in PACT is what the photosubstitution quantum yield of a good compound should be. Photochemists are often used to compounds with 1O2 quantum yields or fluorescence quantum yields that are close to 1. However, in PACT, a photosubstitution quantum yield of 1 would be detrimental to preclinical developments, as photosubstitution is a decomposition reaction that changes the chemical structure of the molecule. A near-unity photosubstitution quantum yield means that each absorbed photon activates the molecule. In practice, chemists, biologists, or doctors studying such compounds would need to work in absolute darkness. In addition, tuning complexes toward higher photosubstitution quantum yields often lowers their dark stability, which lowers their photoindexes. As an example, compounds based on the [Ru(tpy)(dmbpy)(R-py)]2+ scaffold have been proposed that have photosubstitution quantum yields above 0.10; in our hands, the thermal stability of such compounds is insufficient.78 Overall, the most useful PACT compounds have photosubstitution quantum efficiencies of a few percent (0.01 to 0.10, see Table 1), while a few well-studied compounds have even lower φPS values (down to 0.001, see Table 1). Such quantum yields are excellent for activation in vitro or in vivo because LEDs and lasers are cheap and powerful: it is always possible to increase the number of photons shone onto a tissue and hence to activate a compound with a low φPS value. On the other hand, a photosubstitution quantum yield of a few percent is low enough to allow chemists to isolate compounds and study their biology in low-light conditions by protecting flasks, samples, or 96-well plates with opaque foil and brown glassware or Eppendorf.
In fact, the real question is how high the irradiation time and power density can go when performing in vitro and in vivo PACT experiments. In vitro irradiation of living cancer cells is typically performed using LED arrays placed above or below 96-well plates.95 It is difficult to irradiate such a plate for more than 60–90 min, as in most setups cells are deprived of CO2 and controlled humidity during light irradiation. As typical light intensities of LED devices are 10–50 mW/cm2, maximum light doses in vitro lie around 300 J/cm2. With such light doses, compounds with a quantum yield of 0.01 or more are perfectly activated in vitro,73 while quantum yields of 0.001 may start posing a problem. It should be noted that divergent beams from extremely powerful lasers (e.g., 5–15 W) can also be used to irradiate 96-well plates. Such setups allow for reaching much higher light intensities in vitro (e.g., 700 mW/cm2), which may be used to activate compounds with low photosubstitution quantum yields.96
In vivo, the irradiation time depends on the animal model. In mice or rats, the animal should not move during irradiation, so in most PDT or PACT studies it is anesthetized during light irradiation. The maximum irradiation time is hence determined by the maximum time allowed by ethical committees to keep an animal anesthetized, which is typically 15–20 min. We use typical values of fluence rate (or light intensity) of 50–150 mW/cm2in vivo; the maximum laser intensity allowed for medicine depends on the wavelength and organ irradiated, but for the skin intensities typical values of 0.7 W/cm2 are acceptable for visible light. The corresponding light doses (or fluence values) would be typically 50–100 J/cm2, and the maximal fluence values would be 500–800 J/cm2. In our experience in mice tumor models, we see activation of compound 132+ (Figure 5) at doses around 38 J/cm2,76 but to our knowledge there is no published paper yet quantifying the necessary light dose for a PACT compound to be activated in vivo. In the zebrafish embryo, the whole animal (including eyes) is irradiated, which may generate light toxicity. In a recent PACT study using green light activation (520 nm), our group determined that the maximum tolerated irradiation time under a light intensity of 21 mW/cm2 was 6 h, which corresponded to a light dose of 450 J/cm2.64 After 12 or 24 h irradiation at the same intensity, 50% or 100% dead embryos were observed, respectively. There is hence a limit to the amount of light that a zebrafish embryo can handle. Here as well, at much lower light doses we saw clear activation for 52+ (Figure 5) in an orthotopic model of an eye tumor following four consecutive PACT treatments of 90 min irradiation time (114 J/cm2) each. For this compound, the photosubstitution quantum yield in deaerated acetonitrile was 0.11 (Table 1). Overall, our current experience in vivo is that light is usually not a problem for activating PACT compounds with photosubstitution quantum yields of a few percent. However, more in vivo data are needed to determine the link between the photosubstitution quantum yield of Ru-based PACT compounds, measured with chemical methods in aqueous or acetonitrile conditions, and in vivo activation of their antitumor properties.
2.4. Toward Red or Near-Infrared Light Activation
In phototherapy, the wavelength necessary for triggering light activation is very important. It should be part of the so-called first PDT window (600–1000 nm), a region of the spectrum where light penetrates optimally (up to 1 cm) in biological tissues.97 Most ruthenium polypyridyl complexes have their lowest-energy absorption maximum in the blue or green region of the spectrum, which is perfect in vitro but suboptimal in vivo. Only a few PACT complexes were demonstrated to be photoactivated using red or near-infrared light, though the library of available compound is increasing steadily.92 It should be noted that it has remained impossible to shift the lowest energy 1MLCT absorption maximum itself, λmax (Figure 6), to the red of the NIR region of the spectrum. A preferred strategy is to bathochromically shift this absorption maximum as much as possible, which increases εexc, the molar absorption coefficient of the compound at the excitation wavelength λexc (Figure 6). One can then activate the complex with excitation wavelengths λexc that are red-shifted compared to the absorption maximum λmax of the compound, and located in the red or NIR region of the spectrum. φPS poorly depends indeed on the excitation wavelength. This observation is in line with Vavilov’s rule, which states that the fluorescence spectrum and quantum yield of a fluorophore are independent of the excitation wavelength. For photoreactivities based on triplet states, the topology of the excited-state hypersurface may be more complex than for a fluorophore, but the few photosubstitutionally active compounds for which φPS has been measured at different wavelengths usually confirmed this principle. For example, for 132+ (Figure 5), φPS was 0.0055 with blue light and 0.0038 with green light,76 and [Ru(tpy)(MeCN)3]2+ photosubstitutes one of the axial MeCN by chloride at 298 K, with a quantum yield φPS = 0.040 at 436 nm and 0.041 at 480 nm in CH2Cl2.88 Overall, provided that the molar absorption coefficient at the excitation wavelength is not zero, it is possible to excite a PACT compound on the right edge of its main absorption band without paying too much penalty on φPS; one only needs to provide enough photons.
Figure 6.

For ruthenium-based PACT compounds, the activation wavelength λexc does not have to coincide with the absorption maximum λmax. Top: 132+ (see Figure 5) has a maximum in the blue (λmax = 452 nm) but was activated with green light (λexc = 520 nm, εexc = 1510 M–1·cm–1) in vitro and in vivo. Red light (630 nm) hardly activated the compound. Image developed using data from ref (76). Copyright 2019 American Chemical Society. Bottom: 122+ (see Figure 5) has λmax shifted to the green (531 nm), which allowed red light activation (λexc = 625 nm, εexc = 379 M–1·cm–1). Image developed using data from ref (78). Copyright 2017 Wiley-VCH.
One classical strategy to implement a red shift in the absorption maximum of a polypyridyl compound is to prepare a “cyclometalated” analogue, in which one of the metal–pyridyl bonds is replaced by a metal–phenylene bond (Figure 7). As phenylene ligands are π-donors while polypyridyl ligands are π-acceptors, cyclometalated compounds have higher t2g orbitals, which shifts their MLCT states toward lower energies, and hence their absorption bands to higher wavelengths. Red or NIR light activation was obtained, for example, at 690 nm for the aquation of [Ru(phpy)(phen)(MeCN)2]+ (20+, phen = 1,10-phenanthroline, Hphpy = 2-phenylpyridine).98 Cyclometalated complexes have also a lower charge, which helps them penetrate cell membranes. They can hence be excellent anticancer drugs, PDT sensitizers, or protein inhibitors. On the other hand, cyclometalation is often detrimental for the photosubstitution efficacy of bidentate chelates;99 for example, [Ru(phpy)(biq)2]+ (21+, biq = 2,2′-biquinoline) is completely photoinert. The negative charge borne by phenylene increases the ligand field splitting energy of the complex, which increases the 3MC energy and ΔG0 (Figure 3a) and lowers φPS. To keep the dissociative 3MC states low enough for photosubstitution to occur, the coordination octahedron should be distorted. Our group demonstrated this principle for the cyclometalated complex [Ru(phbpy)(bpy)(dmso)]+ (22+, Hphbpy = 6-phenyl-2,2′-bipyridine), in which the terpyridine effect generated enough distortion in the coordination octahedron to allow photosubstitution of dmso by acetonitrile. The quantum yield of this reaction was low, however (0.00041 at 450 nm, compared to 0.016 for its terpyridine analogue).74 Steric hindrance could also be introduced in the form of a six-membered metallacycle obtained by coordinating the N,S chelate 2-(methylthio)ethyl-2-pyridine (mtep) to obtain the heteroleptic complexes [Ru(bpy)(phpy)(mtep)]+ (23+).68 Mtep photosubstitution worked in acetonitrile (φPS = 0.00035 at 521 nm) but was 1 order of magnitude slower than for the bipyridyl analogue [Ru(bpy)2(mtep)]2+ (φPS = 0.0030). These compounds, together with 20+, belong to the few reported cyclometalated complexes capable of photosubstitution. Their biological properties remain unknown, however, and in general the cyclometalation strategy has not been shown (yet) to lead to efficient PACT compounds.
Figure 7.
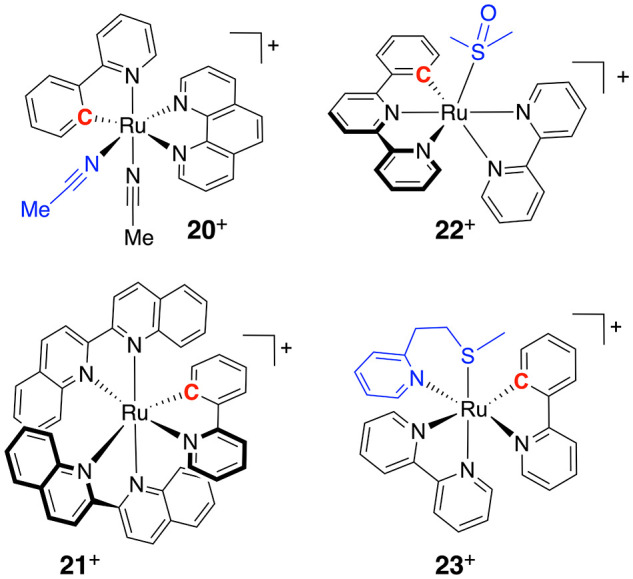
Examples of cyclometalated complexes investigated in Ru-based PACT. The ligand that is photosubstituted first is colored in blue and the carbon atom bound to ruthenium in red.
As discussed above, a more successful strategy to bathochromically shift the absorbance maximum of ruthenium polypyridyl compounds and obtain light activation with red or NIR light was introduced by Turro.51 It consisted of changing bpy in [Ru(tpy)(bpy)(L)]2+ into an oxygen-based acac– chelate to obtain [Ru(tpy)(acac)(RCN)]+ complexes (1+, Figure 5). By doing so, the π-accepting bipyridyl ligands are replaced by a σ-donor chelate with weak π-donor properties, which comparatively increases the energy of the t2g orbitals. The resulting 3MLCT states are dramatically lowered in energy, compared to bipyridine analogues, which shifts absorption toward the NIR region of the spectrum. This strategy was recently extended by the Glazer and Sun groups, who provided phototoxic compounds biologically activated by NIR light.100,92 In contrast to [Ru(tpy)(acac)(MeCN)]+, with Glazer’s [Ru(bpy)2(acac)]+ and analogues, there was no reported sign of photosubstitution, and the compound worked via a PDT mechanism. This observation fits with the classical mechanism of photosubstitution, where low 3MLCT states and lack of distortion of the coordination sphere are detrimental for the thermal promotion of 3MLCT states to 3MC states. It is unclear at this stage why photosubstitution from 3MLCT states would work with terpyridine-based complexes [Ru(tpy)(O-O)(L)]+ and not with bis-bipyridine complexes [Ru(bpy)2(O-O)]+. It should be noted, however, that similar deviations from the classical model of photosubstitution have also been observed by Etchenique’s groups. Although trans-92+ has a red-shifted absorption maximum (464 nm) compared to cis-92+ (432 nm), and hence a lower 1MLCT state, it also has a higher photosubstitution quantum yield (Table 1).
2.5. Photocaging: A Working Strategy ... with a Twist
In principle, in PACT the photocaged compound cannot inhibit its targeted protein at all, nor bind to DNA. This is a simple idea, but a great majority of the work discussed in this Perspective has effectively shown experimentally that the photocaged inhibitor was (much) less active in the dark than after light activation. For example, compounds 132+ and 15, shown in Figure 5, really cannot inhibit tubulin polymerization and CYP1B1, respectively. Other works, for example, from Etchenique on neurotransmitters,101 Turro on cathepsin inhibitors,102 Glazer on P450 inhibitors,103 or more recently Zhang on the kinase inhibitor sorafenib,104 have demonstrated similarly that the ruthenium caging groups do their caging job properly.
In some cases, however, the dark toxicity of non-activated ruthenium-based PACT prodrugs, or the protein inhibition properties of the caged compound in the dark, were reported to be significant.78 Recently, the Turro and Kodanko groups found out an explanation for this observation: their caged molecule 112+ (Figure 5) was found to be a better inhibitor of the major human drug-metabolizing enzyme CYP3A4 than the uncaged ligand 24 (Figure 8a).80 CYP3A4 belongs to the large P450 family of heme proteins capable of oxidizing hydrophobic drugs to increase their water solubility. Classical CYP3A4 inhibitors such as 24 contain a coordinating pyridine ligand which binds to the iron heme center, thereby blocking the catalytic center (Figure 8b). The ruthenium caged inhibitor 112+ was shown via an enzyme activity assay (Figure 8c) and an X-ray structure of the protein (Figure 8d) to better fill the protein binding pocket than the uncaged inhibitor itself. Of course, in the caged compound 112+, the pyridine ligand of the inhibitor 24 is engaged in coordination to ruthenium, so it cannot bind to heme, but the shape of the ruthenium prodrug turned out to be ideal for filling the catalytic pocket and preventing substrates from reaching the catalytic center. At this stage, this unexpected effect was clearly demonstrated in only one case. However, it is probable that it may play a role in other ruthenium-based PACT compounds as well. As an example, when the NAMPT inhibitor called STF31 (IC50 = 0.25 μM) was caged into compound [Ru(tpy)(biq)(STF31)]2+ (122+ in Figure 5), it became 18 times less potent (IC50,dark = 4.8 μM).78 Such a caging effect is significant and corresponds to expectations. On the other hand, the IC50 value for NAMPT inhibition by 122+ in the dark was not negligible, which suggested some form of interaction between the Ru-caged prodrug and the protein. Like for 112+, the enzyme inhibition properties of 122+ in the dark may also explain, at least in part, the significant dark toxicity of this compound toward cancer cells (e.g., EC50,dark = 23.6 μM in A431 skin cancer cells in normoxia). Although similar unwanted inhibitory properties are observed every now and then with metal complexes,105 it should be noted that they are more an exception than the rule. In addition, it should be possible to correct them by changing the ruthenium cage or the place where it is installed on the inhibitor, to lower interaction with the targeted protein. Overall, the principle of photocaging, where installing the ruthenium photocage suppresses the biological properties of an inhibitor, is a simple and working idea.
Figure 8.

(a) Formula of the CYP3A4-inhibiting ritonavir analogue 24 photocaged by Turro et al. with the [Ru(tpy)(dmbpy)]2+ moiety. (b) X-ray structure of the uncaged CYP3A4 inhibitor shown in (a) bound via pyridine coordination to the heme iron center (PDB: 4D78). (c) Protein activity dose–response curves for the uncaged inhibitor 24, the ruthenium-caged inhibitor 112+ (Figure 5) in the dark and after light activation, and the control ruthenium caging group [Ru(tpy)(dmbpy)Cl]Cl. (d) X-ray structure of the caged CYP3A4 inhibitor 112+ interacting with CYP3A4 without pyridine coordination to the heme iron center (PDB: 7KS8). Color code: pyridine nitrogen atoms are in green, heme is in red, the inhibitor 24 is in blue, and the ruthenium caging group of 112+ is in orange. Adapted from ref (80). Copyright 2021 American Chemical Society.
2.6. What Do We Need to Optimize?
In most PACT studies, researchers tend to maximize the photoindex (PI) value of their compounds in vitro. Although this idea seems reasonable, the relationship between the PI value in vitro and the light-activated antitumor activity in vivo is, however, not straightforward to establish. In fact, there are many molecular parameters other than the PI value that need to be optimized to obtain good ruthenium-based compounds for in vivo PACT tumor treatment. In a recent collaboration with the Snaar-Jagalska group,64 our group studied in zebrafish embryo tumor models the antitumor activity of 52+ under green light activation (Figure 9). In vitro, the PI value of this compound in PC3Pro4 prostate cancer cell lines was >31, which is much better than the PI value found in conjunctival melanoma cancer cell lines CRMM1 and CRMM2 (8.5 and 8.8, respectively). However, in vivo, we observed no antitumor activity in the prostate tumor model used, while good antitumor activity was observed in the eye tumor model. An important difference in the type of tumor models should be highlighted here. In the prostate cancer model used, which is called “ectopic”, the human prostate cells were injected intravenously and settled as a tumor in the tail fin of the embryo, which does not correspond to the (prostate) tissue from which the cells originate. The PACT compound was administered either in the water in which the embryo swum, which would require compound uptake through the skin and/or the bronchia, or by intravenous injection. In both cases, no antitumor effect was observed, despite the excellent in vitro properties of this compound. Probably, 52+ never reached the tumor or reached it in too small quantities. By contrast, in the eye cancer model, which is called “orthotopic”, the retro-orbital tumor was installed by injection of the cancer cells behind the eye, which corresponds to the origin of the tumor cells. The compound was also injected retro-orbitally, which led to a clear antitumor effect (Figure 9). Clearly, access of the prodrug to the tumor worked better in the second case, despite the lower PI value in vitro. These results highlight that drug delivery is critical in vivo. Though they cannot be modeled easily in vitro, drug delivery aspects must be considered as well when designing new ruthenium-based PACT compounds. It should also be highlighted that it will be impossible to progress in the field of ruthenium-based PACT without more in vivo data.
Figure 9.

Non-trivial relationship between in vitro and in vivo performances of the PACT compound [5](PF6)2. (a) Photosubstitution reaction for 52+ irradiated with green light in acetonitrile. (b) In vitro dose–response curves for 52+ in CRMM1 eye cancer cell (PI ≈ 8.5) and PC3Pro4 prostate cancer cells (PI > 31). (c) In vivo performance of 52+ under green light activation (520 nm, 114 J/cm2) in an orthotopic CRMM1 eye tumor model in zebrafish embryo (left) and in a PC3Pro4 ectopic prostate zebrafish tumor model (right). Green fluorescence shows blood vessels, and red fluorescence shows the tumor cells. ****p < 0.0001. Reproduced with permission from ref (64). Copyright 2022 Royal Society of Chemistry.
2.7. General Considerations on the Design of Ru-Based PACT Compounds
Overall, the design of ruthenium-based PACT compounds must address several issues altogether (Figure 10). Some of the questions regard the ground-state chemical properties of the molecule, such as its synthetic availability, solubility in water (log P), aggregation properties, and dark stability. The next set of questions concern the photochemical properties of the molecules: its absorbance spectrum, whether it absorbs red or NIR light, the photosubstitution quantum efficiency at different wavelengths, as well as the quantum efficiency of 1O2 generation. Last but not least, the biological properties are critical: whether the metal- or ligand-based photoproduct generates phototoxicity,63 whether or not the molecule targets the tumor, whether and how well the compound is taken up by cancer cells in the non-activated form or in the activated form, whether the molecule is photoactivated under normoxia, whether it also works under hypoxia, whether it works in vivo, and how toxic it is to the animal, in particular to the kidneys and liver. PACT represents an advanced form of traditional chemotherapy, and the number of properties to be optimized altogether makes the clinical development of PACT a challenging process.
Figure 10.

General design aspects for the ruthenium-based PACT compound.
3. Biological and Clinical Considerations of Ru-Based PACT
3.1. Which Clinical Application for PACT?
In principle, PACT is a very general technology, as a large variety of protein inhibitors can be photocaged with a large variety of ruthenium complexes. On the other hand, Ru-based PACT is still in its infancy as it is not yet applied in clinics. In fact, its potential for real-life applications relies on the immense experience of PDT oncologists throughout the world who have performed phototherapeutic cancer treatment using clinically approved sensitizers such as Photofrin (skin, esophagus), 5-ALA (glioblastoma), mTHPC (head-and-neck), Visudyne (eye), or Padeliporfin (prostate).8 According to clinicians using PDT, for many tumors, surgical removal is preferred to phototherapeutic treatment. However, specific types of tumors are more relevant for phototherapeutic treatment than for surgery. First, in patients with multiple small tumors, such as basal cell carcinomas (BCCs) on the back, surgical removal can be problematic, and PDT is often preferred.106 In general, early tumors represent an interesting field of application for phototherapy, as a cream application or intravenous injection, followed by light irradiation, is usually simpler than surgery. For example, retinoblastoma in the eyes of infants can be treated by PDT with fewer side-effects than surgery,107 and Barrett’s esophagus has been one of the main clinical applications of PDT since 1994. Second, PDT is also used for tumors where surgery is too debilitating, such as for patients with non-resectable brain tumors,108 Paget’s disease of the vulva,109 sinus tumors,110 or tongue tumors.111 Last but not least, PDT is currently being developed in clinics as an adjuvant treatment to surgery, notably to prevent recurrences. For example, clinical trials are currently undergoing for brain tumors (NCT05363826), lung cancer (NCT02662504), or Paget’s disease.112 The idea in this strategy is to insert the phototherapeutic treatment in the standard-of-care procedure with minimal discomfort and minimal safety issues for the patient, while disease control is improved. Altogether, the currently used or new applications of PDT should be taken as inspiration for the future development of Ru-based PACT in the clinics.
As noted, there have been intense clinical efforts toward the industrial development of medical lasers and light-irradiation devices to bring light into the human body. Therefore, it is nowadays possible to shine light on most parts of the body. A few representative examples of light-irradiation devices are shown in Figure 11. Because of these developments, PACT researchers do not need to develop new light delivery techniques for clinical applications. They could use existing devices and techniques for the development of PACT compounds. On the other hand, the development of new light-irradiation devices represents an interesting avenue for the industrial development of new, integrated phototherapeutic solutions for cancer treatment. Such products may contain both the PACT compound itself, appropriately formulated, and a device to shine light in an appropriate manner (shape, wavelength, light intensity, protocol) on the organ targeted by the compound. These matters should be taken seriously by PACT researchers, considering the time and money needed for developing any new molecule toward the market.
Figure 11.

Lasers and devices used to shine light in patients for anticancer phototherapy. (a) Fabric-based biophotonic device used for Phosistos photodynamic therapy of actinic keratosis.113 (b) Frontal light delivery using an optical fiber, for example, for irradiation of skin tumors. (c) Radial light distribution for interstitial photodynamic therapy using an optical fiber terminated by a light diffuser. (d) The TLC-3200 system for simultaneous green light irradiation of bladder tumors (532 nm) and light dosimetry during PDT treatment with TLD-1433.114 (e) Clinical setup of PDT bladder cancer treatment using TLD-1433 and green light. Image courtesy Lothar Lilge. (f) PAGETEX device for controlled vulvar illumination with 635 nm light in the PDT treatment of primary extramammary Paget’s disease of the vulva. Reprinted with permission under a Creative Commons CC BY 4.0 from ref (109). Copyright 2020 The Authors, published by Wiley-VCH. (g) Cevira device for cervix illumination with light. Image courtesy Serge Mordon. (h) Homogeneous light diffusion with a light-scattering balloon inflated in the excised primary tumor cavity for intraoperative PDT treatment of glioblastoma (INDYGO trial). Reprinted with permission under a Creative Commons CC BY 4.0 from ref (115). Copyright 2020 The Authors, published by Springer.
3.2. Hypoxia in Oncology
PDT is extremely efficient in a range of diseases when surgery is either impossible (some forms of liver metastases, pancreas tumors, etc.) or strongly debilitating (brain, genitals, face, etc.). PDT is also ideally suited for treating cancer in developing countries, where access to last-generation chemotherapy, antibiotics, or radiation therapy equipment is insufficient.116 Ideally, Ru-based PACT should not compete with PDT but bring new solutions where PDT is not working. An essential question in the field of PACT is which application is the most promising for this new technology, and for which disease PACT represents a solution that the clinically more advanced techniques (i.e., PDT) cannot address efficiently. If this question can be answered, PACT has a chance to reach the clinics.
Following this approach, our group introduced the idea that the non-dependence of photosubstitution reactions on molecular oxygen may make PACT ideal for the treatment of hypoxic tumors,78 which are difficult to treat with approved PDT sensitizers.117 Hypoxia is qualitatively defined as an abnormally low concentration of molecular oxygen in biological tissue. It can be a state of disease, but it also occurs as part of the natural evolution of embryo development or tissue regeneration in healthy individuals. Hypoxia occurs whenever the number of cells around a blood vessel becomes too large for available O2 delivery (Figure 12a). Cells might develop too quickly for the available blood supply, such as in an embryo or in a tumor, but in other cases, the blood supply may become suddenly or chronically impaired because of a wound, of PDT treatment, of ischemia, of high altitudes, or of apnea.118 As hypoxia is a natural phenomenon, cells have evolved different mechanisms to cope with it, which are primarily controlled by the transcription factors HIF1α and HIF2α.119 These mechanisms are highjacked in cancer cells,120 which must always face an abnormally low state of oxygenation during early tumor development. Following HIF1α activation, hypoxia leads to the overexpression of different factors, such as the vascular endothelial growth factor (VEGF) to grow new blood vessels, carbonic anhydrase,121 the glucose receptor (GLUT1), and different genes related to the reprogramming of cell metabolism toward glycolysis, a phenomenon called the Warburg effect. Hypoxia also pushes cancer cells to seek for better oxygenated tissues, which drives the metastasis cascade: the cells undergo the epithelial–mesenchymal transition (EMT), escape the primary tumor into the bloodstream, and re-settle in a distant tissue where they start to grow again and generate more blood vessels.
Figure 12.

Hypoxia in oncology. (a) Three regions around a blood vessel in a tumor. Image courtesy Iris Kort. (b) Association between tumor hypoxia and overall survival in advanced cancer of the uterine cervix. Reprinted with permission from Vaupel et al., Association between Tumor Hypoxia and Malignant Progression in Advanced Cancer of the Uterine Cervix. Cancer Res., 1996, 56, 4509–4515. Copyright 1996 American Association for Cancer Research. (c) Clinical response to 5-ALA PDT treatment in tongue/floor of mouth tumor patients. Reproduced with permission from Busch et al., Lesion oxygenation associates with clinical outcomes in premalignant and early stage head and neck tumors treated on a phase 1 trial of photodynamic therapy. Photodiagnosis Photodynamic Therapy2018, 21, 28–35. Copyright 2018 Elsevier.
Clinically speaking, it is possible to measure, upon cancer diagnosis, the percentage of the tumor volume that shows low O2 concentration.122 This percentage has been shown to be positively correlated to the efficiency of anticancer therapies: the less O2 present in the tumor tissue at diagnosis, the lower the patient’s chance of survival, all treatment considered (see an example for cervical cancer in Figure 12b).45,46 Importantly, this correlation is not only observed for PDT, where a lack of dioxygen obviously leads to less ROS and hence less antitumor effect (Figure 12c), but it also holds for radiation therapy,47 immunotherapy,48 or chemotherapy.49Solving the hypoxia problem is hence an important issue in oncology,50 and we are convinced that developing Ru-based PACT specifically to address this problem can be a clinically relevant approach.
3.3. Ruthenium-Based PACT for the Treatment of Hypoxic Tumors
In fact, hypoxia is understood differently by chemists and biologists. Chemically speaking, hypoxia represents a lower O2 concentration (typically 1% O2) compared to that present in “normoxic” incubators (21% O2). In principle, such low O2 concentrations lower, at a given excited-state photosensitizer concentration, the rate of 1O2 formation123 and hence the efficacy of PDT. This effect has been reported multiple times for PDT type II photosensitizers,124,125 but things get more complicated for PDT type I, which is notoriously less sensitive to hypoxia.126−128 Recently, studies by the McFarland and Glazer groups have confirmed these trends for ruthenium-based PDT sensitizers, which in spite of an unknown but 1O2-independent mechanism behave extremely well under hypoxia.94,129,130 Usually, it is argued that PDT type I involves photoredox chemistry: electron transfer from the excited state of the photosensitizer occurs to biomolecules other than O2 because the excited states of ruthenium-based sensitizers are particularly good oxidizing and reducing agents. A photoreduced sensitizer may further transfer its strongly reductive electron to H2O2 to produce hydroxy radicals OH• without the involvement of molecular oxygen, which explains the comparatively low sensitivity of PDT type I to hypoxia. In PACT, the photosubstitution reaction quenches the triplet states responsible for 1O2 formation (usually the 3MLCT, 3π–π*, or 3ILCT states), which makes most PACT compounds too short-lived to be good sensitizers for PDT type II.78,53 As a consequence, photosubstitution reactions are typically not efficiently quenched by O2, so that they remain more or less effective under hypoxia as under normoxia.69
Biologically speaking, however, hypoxia represents something else: essentially a tougher environment for cancer cells. Surviving hypoxia during tumor development selects the most resistant cells, which become biologically different compared to cells that always lived in a normoxic area. Hypoxic cells import and burn glucose via different pathways,131 they express many proteins differently, and their sensitivity to apoptosis132 or other forms of cell death133 is very different. Therefore, even if the photosubstitution reaction in a PACT compound is as efficient under hypoxia as under normoxia, the killing of a cancer cell by the chemical action of a photoreleased cytotoxic molecule may be very dependent on local O2 concentrations.
Experimentally speaking, two observations are typically made for most ruthenium-based PACT compounds. On one hand, the PI value remains identical or very similar between normoxia (21% O2) and hypoxia (1% O2). For example, 122+ (Figure 5) showed a PI value of 3.6 in hypoxic A431 skin cancer cells (1% O2) vs 3.3 in normoxia (21% O2).78 Other compounds, such as 132+ (Figure 5), showed a PI of 4.0 vs 4.1 in normoxic vs hypoxic A549 cells, respectively.76 On the other hand, the cell growth inhibition effective concentration (EC50) values in the dark and under light activation of these compounds became higher under hypoxia, which means that PACT molecules become less toxic in hypoxic cells. For 132+ the identical PI values obtained at 21% and 1% O2 were in fact obtained from different EC50,dark and EC50,light values: 35 and 9.2 μM in normoxia vs 55 and 14 μM in hypoxia, respectively. Thus, though it is possible to affirm that the activation of a ruthenium-based PACT compound is independent of the O2 concentration, it would be incorrect to say that the phototoxicity of a ruthenium-based PACT compound does not depend on the O2 concentration.
In fact, chemically speaking, the distinction of a PDT vs PACT compound is easy to establish by measuring the quantum yields of photosubstitution and of 1O2 generation (Table 1): PDT compounds have high ΦΔ (>0.20) and low ΦPS (<0.001) values, whereas PACT molecules have a low ΦΔ (<0.10) and a high ΦPS (>0.001). However, making a clear-cut experimental distinction between PDT and PACT in biological conditions is much more difficult, in particular for compounds such as 162+ (Figure 5) that can do both.77,134,94In vitro, for PDT compounds the lower efficacy of light-induced cell killing at low O2 concentrations, compared to normoxia, leads to a dramatically lower photoindex under hypoxic conditions. This observation is usually interpreted as a chemical consequence of the low O2 concentration—at least for PDT type II compounds. For PACT compounds, light-induced cell killing under hypoxic conditions is also often less efficient as EC50,light becomes higher, but the interpretation of this observation is usually different: the cells have, biologically speaking, become more resistant to chemotherapy due to the activation of the hypoxia response of the cell—not due to the chemical unavailability of 3O2. Experimentally speaking, a difference between the two effects can be made, as the PI values of typical PACT compounds remain similar at low vs high O2 concentration, while the PI values of PDT compounds usually become much lower in hypoxia. It is also possible to experimentally observe another difference between PDT and PACT: intracellular ROS generation is negligible for PACT compounds, while it is strong for PDT compounds. In fact, a similar PI value in normoxic and hypoxic conditions in vitro may be considered a hallmark of PACT compounds, while PI values that vary with the O2 concentration may be signs of a cell-killing mechanism involving a photodynamic effect. However, these effects were recently shown to be dependent on the activation wavelength,94 while little data exists for PDT type I compounds. At this stage, this statement should be only taken as a proposition, and the distinction between PDT and PACT mechanisms in biological conditions remains an open question.
In vivo, the situation is more complicated. First, each tumor contains both regions of normoxia and regions of different levels of hypoxia (Figure 13). Therefore, for compounds combining PDT and PACT, different modes of cell killing might take place within the same tumor, depending on the local O2 concentration. In addition, the difficulty of killing intrinsically more resistant hypoxic cancer cells should not hide the problem of drug penetration into hypoxic regions of a tumor, which is a serious issue both for PDT and PACT compounds. Hypoxic regions in tumors are hypoxic precisely because they are badly vascularized, which prevents not only O2 delivery but also drug delivery via blood circulation. Overall, solving the hypoxia problem in phototherapy will require not only developing photocaged compounds that can kill more resistant cells but also looking for innovative drug delivery strategies that can bring such drugs into hypoxic areas. Molecular compounds that diffuse efficiently into hypoxic areas are, in fact, needed here as well. Solving this difficulty will require more in vivo studies.
Figure 13.
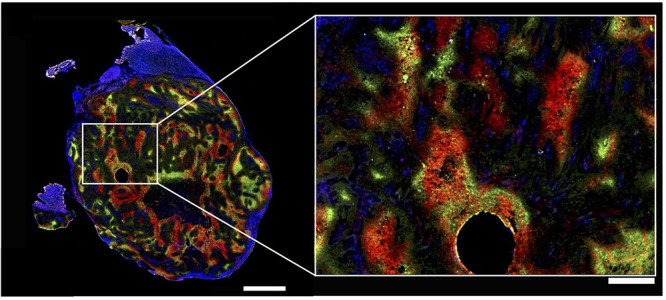
Heterogeneity of hypoxia in hind limb SQ20b human squamous cell carcinoma subcutaneous xenograft in mice shown by three-color hypoxia imaging. Blue is Hoechst 33342 (nuclei), green is pimonidazole (hypoxia marker 1), and red is carbonic anhydrase (hypoxia marker 2). Left bar is 2 mm, right bar is 500 μm. The right image shows high magnification of the region of interest shown on the left by a white rectangle. The circular hole was caused by angiocatheter placement before tumor sectioning. This research was originally published in ref (135). Copyright 2014 Society of Nuclear Medicine and Molecular Imaging, Inc.
3.4. Tissue Necrosis or Apoptosis?
As highlighted above, phototherapy modalities are more promising for two types of clinical issues. The first are non-resectable tumors, for which surgery cannot be considered because the danger for the patient is too high—for example, non-resectable brain tumors or tumors located near essential blood vessels such as the aorta, the portal vein, or the liver. Second, phototherapy may also be useful in cases where the tumor is small or badly located for surgery, for example, on the tongue, the face, the bladder, the colon, or the genitals. Next to the lower quality of life, surgery at a hospital can be costly and lead to serious risk for resistant infection, while a simple injection or local application of a light-activated prodrug followed by visible light irradiation of the tumor might represent attractive therapeutic approaches due to their low side effects, low costs (light sources are comparatively cheap), and low risk for contracting resistant infection. Typical examples can be superficial mouth tumors or suspect nevi in the retina, which are precursors for more life-threatening uveal melanoma tumors.136
For both types of applications, tissue conservation may be seen as an essential feature of phototherapy. One of the potential issues in clinical PDT is that efficient treatment typically leads to tissue necrosis. On the one hand, tissue necrosis is good, because it triggers the immune system and generates serious antitumor immunity.137 On the other hand, necrosis leads to inflammation and pain, which has been reported by many PDT patients. Pain can be managed by lowering the light intensity (in mW/cm2) while increasing irradiation time to keep the light dose (in J/cm2) constant.138−140 However, if necrosis reaches essential tissues, clinical success of a PDT treatment may be an issue for the patient, notably if PDT is performed on tissues that should be conserved. Such effects have been observed in red-light PDT treatment of the bladder using Photofrin in the 1990s, which destroyed part of the healthy muscle tissues underlying the tumor, which stopped clinical trials.141 Photoactivated technologies that trigger other forms of cell death, such as apoptosis, ferroptosis, or immunologic cell death, may lead to tumor eradication without pain and without tissue necrosis, which could benefit the development of Ru-based PACT. Apoptosis may not necessarily generate as much immune response as that generated by PDT, which some see as detrimental to the development of PACT. On the other hand, many metal-based drugs have been shown to trigger significant immune response even in the absence of necrotic cell death.142 Overall, the immune aspects of Ru-based PACT have not yet been studied and may require more attention in the future.
4. Conclusions and Outlook
Ru-based PACT represents a fantastic opportunity for the development of bioinorganic photochemistry. It is a very general approach for cancer therapy, but it may also be relevant for diseases different from cancer. For example, the Etchenique group focuses on the light-induced delivery of neurotransmitters. Recent photopharmacological studies on, for example, light-activated morphin derivatives that alleviate pain by local remote activation without opioid-related adverse effects14 may inspire new opportunities for ruthenium-based photocages. Other fields of applications, such as antibiotics, may benefit from light activation as well whenever side effects of antibacterial treatment are problematic.
From the fundamental point of view, the development of a new ruthenium-based scaffold with improved photosubstitution properties at wavelengths closer to the NIR region of the spectrum is still needed. The very recent development of PACT compounds performing photosubstitution from their 3MLCT state, rather than via the classical mechanism involving 3MC excited states, is still poorly understood and will need additional (theoretical) studies. New developments in the theoretical description of photosubstitution reactions, and notably on the involvement of solvent molecules, are also needed.50 In terms of efficacy, very few PACT compounds show photoindexes higher than 50 in vitro, in particular, under hypoxia, while PI values of thousands have been reported for PDT compounds in normoxia. Improved PI values may be obtained by combining a toxic ligand and a toxic ruthenium cage that cancel each other when bound in the dark and kill cells synergistically after uncaging, but the validity of this approach has not yet been addressed convincingly. In general, we lack a thorough understanding of the efficacy of PDT vs PACT compounds in the context of hypoxia. As noted, the question of the link between in vitro efficacy (i.e., the PI value) and in vivo antitumor properties remains open, mostly due to the lack of animal data. Though most researchers in the field try to maximize the photoindexes of new PACT compounds in 2D and 3D cancer cell cultures, a single study from our group compared the efficacy of a PACT compound in two different tumor models and found no correlation between the PI value in vitro and the antitumor efficacy in vivo.64 More studies are clearly needed on this essential topic, in particular, in mice.
On the impact side, the PACT community may also need to look for more clinical relevance, not only to avoid sterile competition with PDT, a technique that is often very efficient and already in the clinics, but also to avoid the “me too” approach, which is detrimental to realistic technological development. In general, too few in vivo preclinical studies have been published for Ru-based PACT compounds, which does not allow for answering essential questions such as the biodistribution and systemic toxicity of ruthenium polypyridyl compounds (pharmacokinetic/pharmacodynamic effect), the link between Ru-based PACT and the immune system, or the necessity of making actively tumor-targeted PACT compounds. We should also name the question of the combination of PACT compounds with approved chemotherapy drugs, which has been addressed, to our knowledge, only once, while it may be the only way to reach the clinics.80 In order to move toward randomized clinical trials, it would indeed be necessary to compare a group treated with the new technique (Ru-based PACT) plus the best-available treatment (e.g., chemotherapy) to a control group receiving the best-available treatment only. If one wants to ever see Ru-based PACT compounds in the clinics, more pre-clinical data combining ruthenium compounds and approved chemotherapy will be needed.
Acknowledgments
NWO is kindly acknowledged for a VICI grant to S.B.
The author declares no competing financial interest.
References
- Finsen N. R. Remarks on the Red-Light Treatment of Small-Pox: Is the Treatment of Small-Pox Patients in Broad Daylight Warrantable?. BMJ 1903, 1 (2214), 1297–1298. 10.1136/bmj.1.2214.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip S.; Chung M.; Kulig J.; O’Brien R.; Sege R.; Glicken S.; Maisels M. J.; Lau J. Subcommittee on Hyperbilirubinemia. An Evidence-Based Review of Important Issues Concerning Neonatal Hyperbilirubinemia. Pediatrics 2004, 114 (1), e130–e153. 10.1542/peds.114.1.e130. [DOI] [PubMed] [Google Scholar]
- Abdel-kader M. H.The Journey of PDT Throughout History: PDT from Pharos to Present. In Comprehensive Series in Photochemical & Photobiological Sciences; Kostron H., Hasan T., Eds.; Royal Society of Chemistry: Cambridge, 2016; Chap. 1, pp 1–21. 10.1039/9781782626824-00001. [DOI] [Google Scholar]
- Hammerle F.; Bingger I.; Pannwitz A.; Magnutzki A.; Gstir R.; Rutz A.; Wolfender J.-L.; Peintner U.; Siewert B. Targeted Isolation of Photoactive Pigments from Mushrooms Yielded a Highly Potent New Photosensitizer: 7,7′-Biphyscion. Sci. Rep. 2022, 12 (1), 1108. 10.1038/s41598-022-04975-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youf R.; Müller M.; Balasini A.; Thétiot F.; Müller M.; Hascoët A.; Jonas U.; Schönherr H.; Lemercier G.; Montier T.; Le Gall T. Antimicrobial Photodynamic Therapy: Latest Developments with a Focus on Combinatory Strategies. Pharmaceutics 2021, 13 (12), 1995. 10.3390/pharmaceutics13121995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weijer R.; Broekgaarden M.; Kos M.; van Vught R.; Rauws E. A. J.; Breukink E.; van Gulik T. M.; Storm G.; Heger M. Enhancing Photodynamic Therapy of Refractory Solid Cancers: Combining Second-Generation Photosensitizers with Multi-Targeted Liposomal Delivery. J. Photochem. Photobiol. C Photochem. Rev. 2015, 23, 103–131. 10.1016/j.jphotochemrev.2015.05.002. [DOI] [Google Scholar]
- Mari C.; Pierroz V.; Ferrari S.; Gasser G. Combination of Ru(Ii) Complexes and Light: New Frontiers in Cancer Therapy. Chem. Sci. 2015, 6 (5), 2660–2686. 10.1039/C4SC03759F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frochot C.; Mordon S. Update of the Situation of Clinical Photodynamic Therapy in Europe in the 2003–2018 Period. J. Porphyr. Phthalocyanines 2019, 23 (4–5), 347–357. 10.1142/S1088424619300027. [DOI] [Google Scholar]
- Broichhagen J.; Frank J. A.; Trauner D. A Roadmap to Success in Photopharmacology. Acc. Chem. Res. 2015, 48 (7), 1947–1960. 10.1021/acs.accounts.5b00129. [DOI] [PubMed] [Google Scholar]
- Velema W. A.; Szymanski W.; Feringa B. L. Photopharmacology: Beyond Proof of Principle. J. Am. Chem. Soc. 2014, 136 (6), 2178–2191. 10.1021/ja413063e. [DOI] [PubMed] [Google Scholar]
- Balzani V.; Bergamini G.; Campagna S.; Puntoriero F.. Photochemistry and Photophysics of Coordination Compounds: Overview and General Concepts. In Photochemistry and Photophysics of Coordination Compounds I; Balzani V., Campagna S., Eds.; Topics in Current Chemistry 280; Springer: Berlin, Heidelberg, 2007; , pp 1–36. 10.1007/128_2007_132. [DOI] [Google Scholar]
- Joshi T.; Pierroz V.; Mari C.; Gemperle L.; Ferrari S.; Gasser G. A Bis(Dipyridophenazine)(2-(2-Pyridyl)Pyrimidine-4-Carboxylic Acid)Ruthenium(II) Complex with Anticancer Action upon Photodeprotection. Angew. Chem., Int. Ed. 2014, 53 (11), 2960–2963. 10.1002/anie.201309576. [DOI] [PubMed] [Google Scholar]
- Kaplan J. H.; Forbush B. I.; Hoffman J. F. Rapid Photolytic Release of Adenosine 5′-Triphosphate from a Protected Analog: Utilization by the Sodium:Potassium Pump of Human Red Blood Cell Ghosts. Biochemistry 1978, 17 (10), 1929–1935. 10.1021/bi00603a020. [DOI] [PubMed] [Google Scholar]
- López-Cano M.; Font J.; Aso E.; Sahlholm K.; Cabré G.; Giraldo J.; De Koninck Y.; Hernando J.; Llebaria A.; Fernández-Dueñas V.; Ciruela F. Remote Local Photoactivation of Morphine Produces Analgesia without Opioid-related Adverse Effects. Br. J. Pharmacol. 2021, bph.15645. 10.1111/bph.15645. [DOI] [PubMed] [Google Scholar]
- Havrylyuk D.; Stevens K.; Parkin S.; Glazer E. C. Toward Optimal Ru(II) Photocages: Balancing Photochemistry, Stability, and Biocompatibility Through Fine Tuning of Steric, Electronic, and Physiochemical Features. Inorg. Chem. 2020, 59 (2), 1006–1013. 10.1021/acs.inorgchem.9b02065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer N. J.; Salassa L.; Sadler P. J. Photoactivated Chemotherapy (PACT): The Potential of Excited-State d-Block Metals in Medicine. Dalton Trans. 2009, (48), 10690–10701. 10.1039/b917753a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahreman A.; Limburg B.; Siegler M.; Bouwman E.; Bonnet S. Spontaneous Formation in the Dark, and Visible Light-Induced Cleavage, of a Ru-S Bond in Water: A Thermodynamic and Kinetic Study. Inorg. Chem. 2013, 52 (16), 9456–9469. 10.1021/ic401105v. [DOI] [PubMed] [Google Scholar]
- Rassmussen-Taxdal D. S.; Ward G. E.; Figge F. H. J. Fluorescence of Human Lymphatic and Cancer Tissues Following High Doses of Intravenous Hematoporphyrin. Cancer 1955, 8 (1), 78–81. . [DOI] [PubMed] [Google Scholar]
- Weishaupt K. R.; Gomer C. J.; Dougherty T. J. Identification of Singlet Oxygen as the Cytotoxic Agent in Photo-Inactivation of a Murine Tumor1. Cancer Res. 1976, 36 (7_Part_1), 2326–2329. [PubMed] [Google Scholar]
- Rosenberg B.; Vancamp L.; Trosko J. E.; Mansour V. H. Platinum Compounds: A New Class of Potent Antitumour Agents. Nature 1969, 222 (5191), 385–386. 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- Mahnken R.; Billadeau M.; Nikonowicz E.; Morrison H. Organic-Photochemistry.98. toward the Development of Photo Cisplatinum Reagents - Reaction of Cis-Dichlorobis(1,10-phenanthroline)rhodium(III) with Calf Thymus DNA, Nucleotides, and Nucleosides. J. Am. Chem. Soc. 1992, 114 (24), 9253–9265. 10.1021/ja00050a002. [DOI] [Google Scholar]
- Muir M. M.; Huang W.-L. Photoaquation of Some Complexes of Rhodium(III). Inorg. Chem. 1973, 12 (8), 1831–1835. 10.1021/ic50126a025. [DOI] [Google Scholar]
- Kratochwil N.; Guo Z.; Murdoch P.; Parkinson J.; Bednarski P.; Sadler P. Electron-Transfer-Driven Trans-Ligand Labilization: A Novel Activation Mechanism for Pt(IV) Anticancer Complexes. J. Am. Chem. Soc. 1998, 120 (32), 8253–8254. 10.1021/ja980393q. [DOI] [Google Scholar]
- Kratochwil N.; Parkinson J.; Bednarski P.; Sadler P. Nucleotide Platination Induced by Visible Light. Angew. Chem., Int. Ed. 1999, 38 (10), 1460–1463. . [DOI] [PubMed] [Google Scholar]
- Zayat L.; Calero C.; Alborés P.; Baraldo L.; Etchenique R. A New Strategy for Neurochemical Photodelivery: Metal-Ligand Heterolytic Cleavage. J. Am. Chem. Soc. 2003, 125 (4), 882–883. 10.1021/ja0278943. [DOI] [PubMed] [Google Scholar]
- Mobian P.; Kern J.; Sauvage J. Light-Driven Machine Prototypes Based on Dissociative Excited States: Photoinduced Decoordination and Thermal Recoordination of a Ring in a Ruthenium(II)-Containing [2]Catenane. Angew. Chem., Int. Ed. 2004, 43 (18), 2392–2395. 10.1002/anie.200352522. [DOI] [PubMed] [Google Scholar]
- Laemmel A.; Collin J.; Sauvage J. Photosubstitution of Ancillary Ligands in Octahedral Mono-Terpyridine Ruthenium (II) Complexes. Comptes Rendus Acad. Sci. Ser. II Fasc. C-Chim. 2000, 3 (1), 43–49. 10.1016/S1387-1609(00)00102-X. [DOI] [Google Scholar]
- Juris A.; Balzani V.; Barigelletti F.; Campagna S.; Belser P.; von Zelewsky A. Ru(II) Polypyridine Complexes: Photophysics, Photochemistry, Eletrochemistry, and Chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. 10.1016/0010-8545(88)80032-8. [DOI] [Google Scholar]
- Hecker C. R.; Fanwick P. E.; McMillin D. R. Evidence for Dissociative Photosubstitution Reactions of (Acetonitrile)(Bipyridine)(Terpyridine)Ruthenium(2+). Crystal and Molecular Structure of [Ru(Trpy)(Bpy)(Py)](PF6)2·(CH3)2CO. Inorg. Chem. 1991, 30 (4), 659–666. 10.1021/ic00004a013. [DOI] [Google Scholar]
- Meyer T. J. Photochemistry of Metal Coordination Complexes: Metal to Ligand Charge Transfer Excited States. Pure Appl. Chem. 1986, 58 (9), 1193–1206. 10.1351/pac198658091193. [DOI] [Google Scholar]
- Bonnet S.; Collin J. Ruthenium-Based Light-Driven Molecular Machine Prototypes: Synthesis and Properties. Chem. Soc. Rev. 2008, 37 (6), 1207–1217. 10.1039/b713678c. [DOI] [PubMed] [Google Scholar]
- Collin J.; Sauvage J. Transition Metal-Complexed Catenanes and Rotaxanes as Light-Driven Molecular Machines Prototypes. Chem. Lett. 2005, 34 (6), 742–747. 10.1246/cl.2005.742. [DOI] [Google Scholar]
- White J. K.; Schmehl R. H.; Turro C. An Overview of Photosubstitution Reactions of Ru(II) Imine Complexes and Their Application in Photobiology and Photodynamic Therapy. Inorg. Chim. Acta 2017, 454, 7–20. 10.1016/j.ica.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford P. C.; Wink D.; Dibenedetto J.. Mechanistic Aspects of the Photosubstitution and Photoisomerization Reactions of d6 Metal Complexes. In Progress in Inorganic Chemistry; Lippard S. J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp 213–271. 10.1002/9780470166314.ch5. [DOI] [Google Scholar]
- Wang R.; Eberspacher T. A.; Hasegawa T.; Day V.; Ware D. C.; Taube H. Tmtacn, Tacn, and Triammine Complexes of (η 6 -Arene)Os II : Syntheses, Characterizations, and Photosubstitution Reactions (Tmtacn = 1,4,7-Trimethyl-1,4,7-Triazacyclononane; Tacn = 1,4,7-Triazacyclononane). Inorg. Chem. 2001, 40 (4), 593–600. 10.1021/ic000028b. [DOI] [PubMed] [Google Scholar]
- Hakkennes M. L. A.; Meijer M. S.; Menzel J. P.; Goetz A.-C.; Van Duijn R.; Siegler M. A.; Buda F.; Bonnet S. Ligand Rigidity Steers the Selectivity and Efficiency of the Photosubstitution Reaction of Strained Ruthenium Polypyridyl Complexes. J. Am. Chem. Soc. 2023, 145, 13420. 10.1021/jacs.3c03543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houten J.; Watts R. J. Temperature Dependence of the Photophysical and Photochemical Properties of the Tris(2,2’-Bipyridyl)Ruthenium(II) Ion in Aqueous Solution. J. Am. Chem. Soc. 1976, 98 (16), 4853–4858. 10.1021/ja00432a028. [DOI] [Google Scholar]
- Van Houten J.; Watts R. J. Photochemistry of Tris(2,2’-Bipyridyl)Ruthenium(II) in Aqueous Solutions. Inorg. Chem. 1978, 17 (12), 3381–3385. 10.1021/ic50190a016. [DOI] [Google Scholar]
- Durham B.; Caspar J. V.; Nagle J. K.; Meyer T. J. Photochemistry of Tris(2,2’-Bipyridine)Ruthenium(2+) Ion. J. Am. Chem. Soc. 1982, 104 (18), 4803–4810. 10.1021/ja00382a012. [DOI] [Google Scholar]
- Zayat L.; Filevich O.; Baraldo L.; Etchenique R. Ruthenium Polypyridyl Phototriggers: From Beginnings to Perspectives. Philos. Trans. R. Soc. -Math. Phys. Eng. Sci. 2013, 371 (1995), 330. 10.1098/rsta.2012.0330. [DOI] [PubMed] [Google Scholar]
- Pinnick D. V.; Durham B. Temperature Dependence of the Quantum Yields for the Photoanation of Ru(Bpy)2L22+ Complexes. Inorg. Chem. 1984, 23 (24), 3841–3842. 10.1021/ic00192a002. [DOI] [Google Scholar]
- Wacholtz W. M.; Auerbach R. A.; Schmehl R. H.; Ollino M.; Cherry W. R. Correlation of Ligand Field Excited-State Energies with Ligand Field Strength in (Polypyridine)Ruthenium(II) Complexes. Inorg. Chem. 1985, 24 (12), 1758–1760. 10.1021/ic00206a009. [DOI] [Google Scholar]
- Laemmel A.; Collin J.; Sauvage J. Efficient and Selective Photochemical Labilization of a given Bidentate Ligand in Mixed Ruthenium(II) Complexes of the Ru(Phen)(2)L2+ and Ru(Bipy)(2)L2+ Family (L = Sterically Hindering Chelate). Eur. J. Inorg. Chem. 1999, 1999 (3), 383–386. . [DOI] [Google Scholar]
- Tu Y.; Mazumder S.; Endicott J.; Turro C.; Kodanko J.; Schlegel H. Selective Photodissociation of Acetonitrile Ligands in Ruthenium Polypyridyl Complexes Studied by Density Functional Theory. Inorg. Chem. 2015, 54 (16), 8003–8011. 10.1021/acs.inorgchem.5b01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soupart A.; Alary F.; Heully J.; Elliott P.; Dixon I. Exploration of Uncharted (PES)-P-3 Territory for [Ru(Bpy)(3)](2+): A New (MC)-M-3 minimum Prone to Ligand Loss Photochemistry. Inorg. Chem. 2018, 57 (6), 3192–3196. 10.1021/acs.inorgchem.7b03229. [DOI] [PubMed] [Google Scholar]
- Soupart A.; Alary F.; Heully J.; Elliott P.; Dixon I. Theoretical Study of the Full Photosolvolysis Mechanism of [Ru(Bpy)(3)] (2+): Providing a General Mechanistic Roadmap for the Photochemistry of [Ru(N Boolean AND N)(3)](2+)-Type Complexes toward Both Cis and Trans Photoproducts. Inorg. Chem. 2020, 59 (20), 14679–14695. 10.1021/acs.inorgchem.0c01843. [DOI] [PubMed] [Google Scholar]
- Meijer M. S.; Bonnet S. Diastereoselective Synthesis and Two-Step Photocleavage of Ruthenium Polypyridyl Complexes Bearing a Bis(Thioether) Ligand. Inorg. Chem. 2019, 58 (17), 11689–11698. 10.1021/acs.inorgchem.9b01669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham B.; Wilson S. R.; Hodgson D. J.; Meyer T. J. Cis-Trans Photoisomerization in Ru(Bpy)2(OH2)22+. Crystal Structure of Trans-[Ru(Bpy)2(OH2)(OH)](ClO4)2. J. Am. Chem. Soc. 1980, 102 (2), 600–607. 10.1021/ja00522a026. [DOI] [Google Scholar]
- Gama Sauaia M.; Tfouni E.; Helena de Almeida Santos R.; Teresa do Prado Gambardella M.; Del Lama M. P. F. M.; Fernando Guimarães L.; Santana da Silva R. Use of HPLC in the Identification of Cis and Trans-Diaquabis(2,2′-Bipyridine)Ruthenium(II) Complexes: Crystal Structure of Cis-[Ru(H2O)2(Bpy)2](PF6)2. Inorg. Chem. Commun. 2003, 6 (7), 864–868. 10.1016/S1387-7003(03)00131-X. [DOI] [Google Scholar]
- Gottle A.; Alary F.; Boggio-Pasqua M.; Dixon I.; Heully J.; Bahreman A.; Askes S.; Bonnet S. Pivotal Role of a Pentacoordinate (MC)-M-3 State on the Photocleavage Efficiency of a Thioether Ligand in Ruthenium(II) Complexes: A Theoretical Mechanistic Study. Inorg. Chem. 2016, 55 (9), 4448–4456. 10.1021/acs.inorgchem.6b00268. [DOI] [PubMed] [Google Scholar]
- Loftus L. M.; Al-Afyouni K. F.; Turro C. New RuII Scaffold for Photoinduced Ligand Release with Red Light in the Photodynamic Therapy (PDT) Window. Chem. - Eur. J. 2018, 24 (45), 11550–11553. 10.1002/chem.201802405. [DOI] [PubMed] [Google Scholar]
- Loftus L. M.; Rack J. J.; Turro C. Photoinduced Ligand Dissociation Follows Reverse Energy Gap Law: Nitrile Photodissociation from Low Energy 3MLCT Excited States. Chem. Commun. 2020, 56 (29), 4070–4073. 10.1039/C9CC10095D. [DOI] [PubMed] [Google Scholar]
- Busemann A.; Flaspohler I.; Zhou X.; Schmidt C.; Goetzfried S.; van Rixel V.; Ott I.; Siegler M.; Bonnet S. Ruthenium-Based PACT Agents Based on Bisquinoline Chelates: Synthesis, Photochemistry, and Cytotoxicity. J. Biol. Inorg. Chem. 2021, 26 (6), 667–674. 10.1007/s00775-021-01882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinke S. J.; Piechota E. J.; Loftus L. M.; Turro C. Acetonitrile Ligand Photosubstitution in Ru(II) Complexes Directly from the 3 MLCT State. J. Am. Chem. Soc. 2022, 144 (44), 20177–20182. 10.1021/jacs.2c07209. [DOI] [PubMed] [Google Scholar]
- Howerton B. S.; Heidary D. K.; Glazer E. C. Strained Ruthenium Complexes Are Potent Light-Activated Anticancer Agents. J. Am. Chem. Soc. 2012, 134 (20), 8324–8327. 10.1021/ja3009677. [DOI] [PubMed] [Google Scholar]
- Hufziger K. T.; Thowfeik F. S.; Charboneau D. J.; Nieto I.; Dougherty W. G.; Kassel W. S.; Dudley T. J.; Merino E. J.; Papish E. T.; Paul J. J. Ruthenium Dihydroxybipyridine Complexes Are Tumor Activated Prodrugs Due to Low pH and Blue Light Induced Ligand Release. J. Inorg. Biochem. 2014, 130, 103–111. 10.1016/j.jinorgbio.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welby C.; Rice C.; Elliott P. Unambiguous Characterization of a Photoreactive Ligand-Loss Intermediate. Angew. Chem., Int. Ed. 2013, 52 (41), 10826–10829. 10.1002/anie.201304219. [DOI] [PubMed] [Google Scholar]
- Wachter E.; Heidary D. K.; Howerton B. S.; Parkin S.; Glazer E. C. Light-Activated Ruthenium Complexes Photobind DNA and Are Cytotoxic in the Photodynamic Therapy Window. Chem. Commun. Camb 2012, 48 (77), 9649–9651. 10.1039/c2cc33359g. [DOI] [PubMed] [Google Scholar]
- Roque J. III; Havrylyuk D.; Barrett P. C.; Sainuddin T.; McCain J.; Colón K.; Sparks W. T.; Bradner E.; Monro S.; Heidary D.; Cameron C. G.; Glazer E. C.; McFarland S. A. Strained, Photoejecting Ru(II) Complexes That Are Cytotoxic Under Hypoxic Conditions. Photochem. Photobiol. 2020, 96 (2), 327–339. 10.1111/php.13174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu F.; Lamb R.; Cameron C.; Park S.; Oladipupo O.; Gray J.; Xu Y.; Cole H.; Bonizzoni M.; Kim Y.; McFarland S.; Webster C.; Papish E. Singlet Oxygen Formation vs Photodissociation for Light- Responsive Protic Ruthenium Anticancer Compounds: The Oxygenated Substituent Determines Which Pathway Dominates. Inorg. Chem. 2021, 60 (4), 2138–2148. 10.1021/acs.inorgchem.0c02027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welby C.; Armitage G.; Bartley H.; Sinopoli A.; Uppal B.; Elliott P. Photochemical Ligand Ejection from Non- Sterically Promoted Ru(II)Bis(Diimine) 4,4′-Bi-1,2,3-Triazolyl Complexes. Photochem. Photobiol. Sci. 2014, 13 (5), 735–738. 10.1039/c3pp50437a. [DOI] [PubMed] [Google Scholar]
- Collin J.; Jouvenot D.; Koizumi M.; Sauvage J. Ru(Phen)(2)(Bis-Thioether)(2+) Complexes: Synthesis and Photosubstitution Reactions. Inorg. Chim. Acta 2007, 360 (3), 923–930. 10.1016/j.ica.2006.06.028. [DOI] [Google Scholar]
- Cuello-Garibo J.-A.; Meijer M. S.; Bonnet S. To Cage or to Be Caged? The Cytotoxic Species in Ruthenium-Based Photoactivated Chemotherapy Is Not Always the Metal. Chem. Commun. 2017, 53 (50), 6768–6771. 10.1039/C7CC03469E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; Cuello-Garibo J.; Bretin L.; Zhang L.; Ramu V.; Aydar Y.; Batsiun Y.; Bronkhorst S.; Husiev Y.; Beztsinna N.; Chen L.; Zhou X.; Schmidt C.; Ott I.; Jager M.; Brouwer A.; Snaar-Jagalska B.; Bonnet S. Photosubstitution in a Trisheteroleptic Ruthenium Complex Inhibits Conjunctival Melanoma Growth in a Zebrafish Orthotopic Xenograft Model. Chem. Sci. 2022, 13 (23), 6899–6919. 10.1039/D2SC01646J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner R.; Joyce L.; Turro C. Effect of Electronic Structure on the Photoinduced Ligand Exchange of Ru(II) Polypyridine Complexes. Inorg. Chem. 2011, 50 (10), 4384–4391. 10.1021/ic102482c. [DOI] [PubMed] [Google Scholar]
- Garcìa-Fresnadillo D.; Georgiadou Y.; Orellana G.; Braun A. M.; Oliveros E. Singlet-Oxygen (1Δg) Production by Ruthenium(II) Complexes Containing Polyazaheterocyclic Ligands in Methanol and in Water. Helv. Chim. Acta 1996, 79 (4), 1222–1238. 10.1002/hlca.19960790428. [DOI] [Google Scholar]
- Meijer M. S.Photo-Activation of Ruthenium-Decorated Upconverting Nanoparticles. Ph.D. Thesis, Universiteit Leiden, The Netherlands, 2018. [Google Scholar]
- Cuello-Garibo J.; James C.; Siegler M.; Hopkins S.; Bonnet S. Selective Preparation of a Heteroleptic Cyclometallated Ruthenium Complex Capable of Undergoing Photosubstitution of a Bidentate Ligand. Chem. - Eur. J. 2019, 25 (5), 1260–1268. 10.1002/chem.201803720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T. N.; Turro C. Photoinitiated DNA Binding by Cis-[Ru(Bpy)2(NH3)2]2+. Inorg. Chem. 2004, 43 (23), 7260–7262. 10.1021/ic049075k. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Turner D. B.; Singh T. N.; Angeles-Boza A. M.; Chouai A.; Dunbar K. R.; Turro C. Ultrafast Ligand Exchange: Detection of a Pentacoordinate Ru(II) Intermediate and Product Formation. J. Am. Chem. Soc. 2009, 131 (1), 26–27. 10.1021/ja806860w. [DOI] [PubMed] [Google Scholar]
- Albani B.; Pena B.; Leed N.; de Paula N.; Pavani C.; Baptista M.; Dunbar K.; Turro C. Marked Improvement in Photoinduced Cell Death by a New Tris-Heteroleptic Complex with Dual Action: Singlet Oxygen Sensitization and Ligand Dissociation. J. Am. Chem. Soc. 2014, 136 (49), 17095–17101. 10.1021/ja508272h. [DOI] [PubMed] [Google Scholar]
- Perez Y.; Slep L.; Etchenique R. Cis-Trans Interconversion in Ruthenium(II) Bipyridine Complexes. Inorg. Chem. 2019, 58 (17), 11606–11613. 10.1021/acs.inorgchem.9b01485. [DOI] [PubMed] [Google Scholar]
- van Rixel V. H. S.; Siewert B.; Hopkins S. L.; Askes S. H. C.; Busemann A.; Siegler M. A.; Bonnet S. Green Light-Induced Apoptosis in Cancer Cells by a Tetrapyridyl Ruthenium Prodrug Offering Two Trans Coordination Sites. Chem. Sci. 2016, 7 (8), 4922–4929. 10.1039/C6SC00167J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lameijer L.; van de Griend C.; Hopkins S.; Volbeda A.; Askes S.; Siegler M.; Bonnet S. Photochemical Resolution of a Thermally Inert Cyclometalated Ru(Phbpy)(N-N)(Sulfoxide)(+) Complex. J. Am. Chem. Soc. 2019, 141 (1), 352–362. 10.1021/jacs.8b10264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahreman A.; Cuello-Garibo J.; Bonnet S. Yellow-Light Sensitization of a Ligand Photosubstitution Reaction in a Ruthenium Polypyridyl Complex Covalently Bound to a Rhodamine Dye (Vol 43, Pg 4494, 2014). Dalton Trans. 2014, 43 (11), 4494–4505. 10.1039/C3DT52643G. [DOI] [PubMed] [Google Scholar]
- van Rixel V. H. S.; Ramu V.; Auyeung A. B.; Beztsinna N.; Leger D. Y.; Lameijer L. N.; Hilt S. T.; Le Dévédec S. E.; Yildiz T.; Betancourt T.; Gildner M. B.; Hudnall T. W.; Sol V.; Liagre B.; Kornienko A.; Bonnet S. Photo-Uncaging of a Microtubule-Targeted Rigidin Analogue in Hypoxic Cancer Cells and in a Xenograft Mouse Model. J. Am. Chem. Soc. 2019, 141 (46), 18444–18454. 10.1021/jacs.9b07225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lameijer L.; Hopkins S.; Breve T.; Askes S.; Bonnet S. D- Versus L-Glucose Conjugation: Mitochondrial Targeting of a Light-Activated Dual-Mode-of-Action Ruthenium-Based Anticancer Prodrug. Chem.- Eur. J. 2016, 22 (51), 18484–18491. 10.1002/chem.201603066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lameijer L. N.; Ernst D.; Hopkins S. L.; Meijer M. S.; Askes S. H. C.; Le Dévédec S. E.; Bonnet S. A Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells. Angew. Chem., Int. Ed. 2017, 56 (38), 11549–11553. 10.1002/anie.201703890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havrylyuk D.; Hachey A. C.; Fenton A.; Heidary D. K.; Glazer E. C. Ru(II) Photocages Enable Precise Control over Enzyme Activity with Red Light. Nat. Commun. 2022, 13 (1), 3636. 10.1038/s41467-022-31269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toupin N.; Steinke S.; Nadella S.; Li A.; Rohrabaugh T.; Samuels E.; Turro C.; Sevrioukova I.; Kodanko J. Photosensitive Ru(II) Complexes as Inhibitors of the Major Human Drug Metabolizing Enzyme CYP3A4. J. Am. Chem. Soc. 2021, 143 (24), 9191–9205. 10.1021/jacs.1c04155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera R.; Filevich O.; Garcia-Acosta B.; Athilingam J.; Bender K.; Poskanzer K.; Etchenique R. A Visible-Light-Sensitive Caged Serotonin. ACS Chem. Neurosci. 2017, 8 (5), 1036–1042. 10.1021/acschemneuro.7b00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filevich O.; Etchenique R. RuBiGABA-2: A Hydrophilic Caged GABA with Long Wavelength Sensitivity. Photochem. Photobiol. Sci. 2013, 12 (9), 1565–1570. 10.1039/c3pp25248e. [DOI] [PubMed] [Google Scholar]
- Salierno M.; Marceca E.; Peterka D.; Yuste R.; Etchenique R. A Fast Ruthenium Polypyridine Cage Complex Photoreleases Glutamate with Visible or IR Light in One and Two Photon Regimes. J. Inorg. Biochem. 2010, 104 (4), 418–422. 10.1016/j.jinorgbio.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanquist A.; Gupta S.; Al-Afyouni K.; Al-Afyouni M.; Kodanko J.; Turro C. Trifluoromethyl Substitution Enhances Photoinduced Activity against Breast Cancer Cells but Reduces Ligand Exchange in Ru(II) Complex. Chem. Sci. 2021, 12 (36), 12056–12067. 10.1039/D1SC03213E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachter E.; Zamora A.; Heidary D.; Ruiz J.; Glazer E. Geometry Matters: Inverse Cytotoxic Relationship for Cis/Trans-Ru(II) Polypyridyl Complexes from Cis/Trans-[PtCl2(NH3)(2)]. Chem. Commun. 2016, 52 (66), 10121–10124. 10.1039/C6CC04813G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgambellone M. A.; David A.; Garner R. N.; Dunbar K. R.; Turro C. Cellular Toxicity Induced by the Photorelease of a Caged Bioactive Molecule: Design of a Potential Dual-Action Ru(II) Complex. J. Am. Chem. Soc. 2013, 135 (30), 11274–11282. 10.1021/ja4045604. [DOI] [PubMed] [Google Scholar]
- Goldbach R.; Rodriguez-Garcia I.; van Lenthe J.; Siegler M.; Bonnet S. N-Acetylmethionine and Biotin as Photocleavable Protective Groups for Ruthenium Polypyridyl Complexes. Chem. - Eur. J. 2011, 17 (36), 9924–9929. 10.1002/chem.201101541. [DOI] [PubMed] [Google Scholar]
- Suen H. F.; Wilson S. W.; Pomerantz M.; Walsh J. L. Photosubstitution Reactions of Terpyridine Complexes of Ruthenium(II). Inorg. Chem. 1989, 28 (4), 786–791. 10.1021/ic00303a034. [DOI] [Google Scholar]
- Novakova O.; Kasparkova J.; Vrana O.; van Vliet P. M.; Reedijk J.; Brabec V. Correlation between Cytotoxicity and DNA Binding of Polypyridyl Ruthenium Complexes. Biochemistry 1995, 34 (38), 12369–12378. 10.1021/bi00038a034. [DOI] [PubMed] [Google Scholar]
- Bonnet S.; Collin J.; Sauvage J.; Schofield E. Photochemical Expulsion of the Neutral Monodentate Ligand L in Ru(Terpy*)(Diimine)(L)(2+): A Dramatic Effect of the Steric Properties of the Spectator Diimine Ligand. Inorg. Chem. 2004, 43 (26), 8346–8354. 10.1021/ic0491736. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Vandevord J.; Loftus L.; Toupin N.; Al-Afyouni M.; Rohrabaugh T.; Turro C.; Kodanko J. Ru(II)-Based Acetylacetonate Complexes Induce Apoptosis Selectively in Cancer Cells. Inorg. Chem. 2021, 60 (24), 18964–18974. 10.1021/acs.inorgchem.1c02796. [DOI] [PubMed] [Google Scholar]
- He G.; He M.; Wang R.; Li X.; Hu H.; Wang D.; Wang Z.; Lu Y.; Xu N.; Du J.; Fan J.; Peng X.; Sun W. A Near-Infrared Light-Activated Photocage Based on a Ruthenium Complex for Cancer Phototherapy. Angew. Chem., Int. Ed. 2023, 62, e202218768 10.1002/anie.202218768. [DOI] [PubMed] [Google Scholar]
- Cole H.; Roque J.; Lifshits L.; Hodges R.; Barrett P.; Havrylyuk D.; Heidary D.; Ramasamy E.; Cameron C.; Glazer E.; McFarland S. Fine-Feature Modifications to Strained Ruthenium Complexes Radically Alter Their Hypoxic Anticancer Activity(Dagger). Photochem. Photobiol. 2022, 98 (1), 73–84. 10.1111/php.13395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque J.; Cole H.; Barrett P.; Lifshits L.; Hodges R.; Kim S.; Deep G.; Frances-Monerris A.; Alberto M.; Cameron C.; McFarland S. Intraligand Excited States Turn a Ruthenium Oligothiophene Complex into a Light-Triggered Ubertoxin with Anticancer Effects in Extreme Hypoxia. J. Am. Chem. Soc. 2022, 144 (18), 8317–8336. 10.1021/jacs.2c02475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins S. L.; Siewert B.; Askes S. H.; Veldhuizen P.; Zwier R.; Heger M.; Bonnet S. An in Vitro Cell Irradiation Protocol for Testing Photopharmaceuticals and the Effect of Blue, Green, and Red Light on Human Cancer Cell Lines. Photochem. Photobiol. Sci. 2016, 15 (5), 644–653. 10.1039/c5pp00424a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J.; Ding Y.; Zhu H.; Li Z.; Dai R.; Zhao H.; Hong X.; Zhang H. Emitter-Active Shell in NaYF 4 :Yb,Er/NaYF 4 :Er Upconversion Nanoparticles for Enhanced Energy Transfer in Photodynamic Therapy. ACS Appl. Nano Mater. 2022, 5 (1), 559–568. 10.1021/acsanm.1c03377. [DOI] [Google Scholar]
- Fan W.; Huang P.; Chen X. Overcoming the Achilles’ Heel of Photodynamic Therapy. Chem. Soc. Rev. 2016, 45 (23), 6488–6519. 10.1039/C6CS00616G. [DOI] [PubMed] [Google Scholar]
- Palmer A.; Pena B.; Sears R.; Chen O.; El Ojaimi M.; Thummel R.; Dunbar K.; Turro C. Cytotoxicity of Cyclometallated Ruthenium Complexes: The Role of Ligand Exchange on the Activity. Philos. Trans. R. Soc. -Math. Phys. Eng. Sci. 2013, 371 (1995), 135. 10.1098/rsta.2012.0135. [DOI] [PubMed] [Google Scholar]
- Albani B. A.; Peña B.; Dunbar K. R.; Turro C. New Cyclometallated Ru(II) Complex for Potential Application in Photochemotherapy?. Photochem. Photobiol. Sci. 2014, 13 (2), 272–280. 10.1039/c3pp50327e. [DOI] [PubMed] [Google Scholar]
- Ryan R.; Havrylyuk D.; Stevens K.; Moore L.; Parkin S.; Blackburn J.; Heidary D.; Selegue J.; Glazer E. Biological Investigations of Ru(II) Complexes with Diverse Beta-Diketone Ligands. Eur. J. Inorg. Chem. 2021, 2021 (35), 3611–3621. 10.1002/ejic.202100468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E.; Araya R.; Peterka D.; Salierno M.; Etchenique R.; Yuste R. RuBi-Glutamate: Two-Photon and Visible-Light Photoactivation of Neurons and Dendritic Spines. Front. Neural Circuits 2009, 3, 2. 10.3389/neuro.04.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Respondek T.; Sharma R.; Herroon M.; Garner R.; Knoll J.; Cueny E.; Turro C.; Podgorski I.; Kodanko J. Inhibition of Cathepsin Activity in a Cell-Based Assay by a Light-Activated Ruthenium Compound. ChemMedChem 2014, 9 (6), 1306–1315. 10.1002/cmdc.201400081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora A.; Denning C.; Heidary D.; Wachter E.; Nease L.; Ruiz J.; Glazer E. Ruthenium-Containing P450 Inhibitors for Dual Enzyme Inhibition and DNA Damage. Dalton Trans. 2017, 46 (7), 2165–2173. 10.1039/C6DT04405K. [DOI] [PubMed] [Google Scholar]
- Lai Y.; Lu N.; Luo S.; Wang H.; Zhang P. A Photoactivated Sorafenib-Ruthenium(II) Prodrug for Resistant Hepatocellular Carcinoma Therapy through Ferroptosis and Purine Metabolism Disruption. J. Med. Chem. 2022, 65 (19), 13041–13051. 10.1021/acs.jmedchem.2c00880. [DOI] [PubMed] [Google Scholar]
- Denison M.; Ahrens J. J.; Dunbar M. N.; Warmahaye H.; Majeed A.; Turro C.; Kocarek T. A.; Sevrioukova I. F.; Kodanko J. J. Dynamic Ir(III) Photosensors for the Major Human Drug-Metabolizing Enzyme Cytochrome P450 3A4. Inorg. Chem. 2023, 62 (7), 3305–3320. 10.1021/acs.inorgchem.3c00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmur E. S.; Schmults C. D.; Goldberg D. J. A Review of Laser and Photodynamic Therapy for the Treatment of Nonmelanoma Skin Cancer. Dermatol. Surg. 2004, 30 (s2), 264–271. 10.1111/j.1524-4725.2004.30083.x. [DOI] [PubMed] [Google Scholar]
- Cerman E.; Çekiç O. Clinical Use of Photodynamic Therapy in Ocular Tumors. Surv. Ophthalmol. 2015, 60 (6), 557–574. 10.1016/j.survophthal.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Beck T. J.; Kreth F. W.; Beyer W.; Mehrkens J. H.; Obermeier A.; Stepp H.; Stummer W.; Baumgartner R. Interstitial Photodynamic Therapy of Nonresectable Malignant Glioma Recurrences Using 5-Aminolevulinic Acid Induced Protoporphyrin IX. Lasers Surg. Med. 2007, 39 (5), 386–393. 10.1002/lsm.20507. [DOI] [PubMed] [Google Scholar]
- Mordon S.; Thécua E.; Ziane L.; Lecomte F.; Deleporte P.; Baert G.; Vignion-Dewalle A. Light Emitting Fabrics for Photodynamic Therapy: Technology, Experimental and Clinical Applications. Transl. Biophotonics 2020, 2 (3), e202000005. 10.1002/tbio.202000005. [DOI] [Google Scholar]
- Indrasari S. R.; Timmermans A. J.; Wildeman M. A.; Karakullukcu M. B.; Herdini C.; Hariwiyanto B.; Tan I. B. Remarkable Response to Photodynamic Therapy in Residual T4N0M0 Nasopharyngeal Carcinoma: A Case Report. Photodiagnosis Photodyn. Ther. 2012, 9 (4), 319–320. 10.1016/j.pdpdt.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Karakullukcu B.; Nyst H. J.; van Veen R. L.; Hoebers F. J. P.; Hamming-Vrieze O.; Witjes M. J. H.; de Visscher S. A. H. J.; Burlage F. R.; Levendag P. C.; Sterenborg H. J. C. M.; Tan I. B. mTHPC Mediated Interstitial Photodynamic Therapy of Recurrent Nonmetastatic Base of Tongue Cancers: Development of a New Method. Head Neck 2012, 34 (11), 1597–1606. 10.1002/hed.21969. [DOI] [PubMed] [Google Scholar]
- Wang D.; Wang P.; Li C.; Zhou Z.; Zhang L.; Zhang G.; Wang X. Efficacy and Safety of HpD-PDT for Extramammary Paget’s Disease Refractory to Conventional Therapy: A Prospective, Open-Label and Single Arm Pilot Study. Photodiagnosis Photodyn. Ther. 2022, 37, 102670. 10.1016/j.pdpdt.2021.102670. [DOI] [PubMed] [Google Scholar]
- Mordon S.; Vignion-Dewalle A. s.; Abi-Rached H.; Thecua E.; Lecomte F.; Vicentini C.; Deleporte P.; Béhal H.; Kerob D.; Hommel T.; Duhamel A.; Szeimies R. m.; Mortier L. The Conventional Protocol vs. a Protocol Including Illumination with a Fabric-Based Biophotonic Device (the Phosistos Protocol) in Photodynamic Therapy for Actinic Keratosis: A Randomized, Controlled, Noninferiority Clinical Study. Br. J. Dermatol. 2019, 182 (1), 76–84. 10.1111/bjd.18048. [DOI] [PubMed] [Google Scholar]
- Kulkarni G. S.; Lilge L.; Nesbitt M.; Dumoulin-White R. J.; Mandel A.; Jewett M. A.S. A Phase 1b Clinical Study of Intravesical Photodynamic Therapy in Patients with Bacillus Calmette-Guérin-Unresponsive Non-Muscle-Invasive Bladder Cancer. Eur. Urol. Open Sci. 2022, 41, 105–111. 10.1016/j.euros.2022.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermandel M.; Dupont C.; Lecomte F.; Leroy H.; Tuleasca C.; Mordon S.; Hadjipanayis C.; Reyns N. Standardized Intraoperative 5-ALA Photodynamic Therapy for Newly Diagnosed Glioblastoma Patients: A Preliminary Analysis of the INDYGO Clinical Trial. J. Neurooncol. 2021, 152 (3), 501–514. 10.1007/s11060-021-03718-6. [DOI] [PubMed] [Google Scholar]
- Hopper C.; Tan B.; Hasan T.; Kurachi C.; Bagnato V.; Milanesi E.; Siddiqui S.. Developing World Applications of PDT in Head and Neck Cancer. Presented at the 17th International Photodynamic Association World Congress, Cambridge, Massachusetts, 2019; p 110703U. 10.1117/12.2526613. [DOI]
- Larue; Myrzakhmetov; Ben-Mihoub; Moussaron; Thomas; Arnoux; Baros; Vanderesse; Acherar; Frochot Fighting Hypoxia to Improve PDT. Pharmaceuticals 2019, 12 (4), 163. 10.3390/ph12040163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Palmgren V. A. C.; Danen E. H.; Le Dévédec S. E. Acute vs. Chronic vs. Intermittent Hypoxia in Breast Cancer: A Review on Its Application in in Vitro Research. Mol. Biol. Rep. 2022, 49 (11), 10961–10973. 10.1007/s11033-022-07802-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B.; Johnson R. S.; Simon M. C. HIF1α and HIF2α: Sibling Rivalry in Hypoxic Tumour Growth and Progression. Nat. Rev. Cancer 2012, 12 (1), 9–22. 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaupel P.; Harrison L. Tumor Hypoxia: Causative Factors, Compensatory Mechanisms, and Cellular Response. Oncologist 2004, 9 (S5), 4–9. 10.1634/theoncologist.9-90005-4. [DOI] [PubMed] [Google Scholar]
- Wykoff C. C.; Beasley N. J. P.; Watson P. H.; Turner K. J.; Pastorek J.; Sibtain A.; Wilson G. D.; Turley H.; Talks K. L.; Maxwell P. H.; Pugh C. W.; Ratcliffe P. J.; Harris A. L. Hypoxia-Inducible Expression of Tumor-Associated Carbonic Anhydrases1. Cancer Res. 2000, 60 (24), 7075–7083. [PubMed] [Google Scholar]
- Flood A. B.; Satinsky V. A.; Swartz H. M.. Comparing the Effectiveness of Methods to Measure Oxygen in Tissues for Prognosis and Treatment of Cancer. In Oxygen Transport to Tissue; Luo Q., Li L. Z., Harrison D. K., Shi H., Bruley D. F., Eds.; Advances in Experimental Medicine and Biology XXXVIII; Springer International Publishing: Cham, 2016; Vol. 923, pp 113–120. 10.1007/978-3-319-38810-6_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Lin L.; Li Y.; Lin H.; Qiu Z.; Gu Y.; Li B. Effect of Oxygen Concentration on Singlet Oxygen Luminescence Detection. J. Lumin. 2014, 152, 98–102. 10.1016/j.jlumin.2013.10.034. [DOI] [Google Scholar]
- Ahn P. H.; Finlay J. C.; Gallagher-Colombo S. M.; Quon H.; O’Malley B. W.; Weinstein G. S.; Chalian A.; Malloy K.; Sollecito T.; Greenberg M.; Simone C. B.; McNulty S.; Lin A.; Zhu T. C.; Livolsi V.; Feldman M.; Mick R.; Cengel K. A.; Busch T. M. Lesion Oxygenation Associates with Clinical Outcomes in Premalignant and Early Stage Head and Neck Tumors Treated on a Phase 1 Trial of Photodynamic Therapy. Photodiagnosis Photodyn. Ther. 2018, 21, 28–35. 10.1016/j.pdpdt.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas I. Facing Hypoxia: A Must for Photodynamic Therapy. J. Photochem. Photobiol., B 1988, 2 (2), 281–282. 10.1016/1011-1344(88)80011-3. [DOI] [PubMed] [Google Scholar]
- Li M.; Xia J.; Tian R.; Wang J.; Fan J.; Du J.; Long S.; Song X.; Foley J. W.; Peng X. Near-Infrared Light-Initiated Molecular Superoxide Radical Generator: Rejuvenating Photodynamic Therapy against Hypoxic Tumors. J. Am. Chem. Soc. 2018, 140 (44), 14851–14859. 10.1021/jacs.8b08658. [DOI] [PubMed] [Google Scholar]
- Lv W.; Zhang Z.; Zhang K. Y.; Yang H.; Liu S.; Xu A.; Guo S.; Zhao Q.; Huang W. A Mitochondria-Targeted Photosensitizer Showing Improved Photodynamic Therapy Effects Under Hypoxia. Angew. Chem., Int. Ed. 2016, 55 (34), 9947–9951. 10.1002/anie.201604130. [DOI] [PubMed] [Google Scholar]
- Teng K.-X.; Niu L.-Y.; Yang Q.-Z. A Host-Guest Strategy for Converting the Photodynamic Agents from a Singlet Oxygen Generator to a Superoxide Radical Generator. Chem. Sci. 2022, 13 (20), 5951–5956. 10.1039/D2SC01469F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas Y.; Monro S.; Shi G.; Mandel A.; McFarland S.; Lilge L. Photodynamic Inactivation of Staphylococcus Aureus and Methicillin-Resistant Staphylococcus Aureus with Ru(II)-Based Type I/Type II Photosensitizers. Photodiagnosis Photodyn. Ther. 2013, 10 (4), 615–625. 10.1016/j.pdpdt.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Cole H.; Roque J.; Shi G.; Lifshits L.; Ramasamy E.; Barrett P.; Hodges R.; Cameron C.; McFarland S. Anticancer Agent with Inexplicable Potency in Extreme Hypoxia: Characterizing a Light-Triggered Ruthenium Ubertoxin. J. Am. Chem. Soc. 2022, 144 (22), 9543–9547. 10.1021/jacs.1c09010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denko N. C. Hypoxia, HIF1 and Glucose Metabolism in the Solid Tumour. Nat. Rev. Cancer 2008, 8 (9), 705–713. 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- Dong Z.; Wang J. Z.; Yu F.; Venkatachalam M. A. Apoptosis-Resistance of Hypoxic Cells. Am. J. Pathol. 2003, 163 (2), 663–671. 10.1016/S0002-9440(10)63693-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q.; Wang M.; Yu M.; Zhang J.; Bristow R. G.; Hill R. P.; Tannock I. F. Role of Autophagy as a Survival Mechanism for Hypoxic Cells in Tumors. Neoplasia 2016, 18 (6), 347–355. 10.1016/j.neo.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Turro C.; Kodanko J.; Dunbar K. Dual Action Complexes That Target Cancer: Drug Photorelease and Singlet Oxygen Production. J. Biol. Inorg. Chem. 2017, 22, S156–S156. [Google Scholar]
- Carlin S.; Zhang H.; Reese M.; Ramos N. N.; Chen Q.; Ricketts S.-A. A Comparison of the Imaging Characteristics and Microregional Distribution of 4 Hypoxia PET Tracers. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2014, 55 (3), 515–521. 10.2967/jnumed.113.126615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields C. L. Choroidal Nevus Transformation Into Melanoma: Analysis of 2514 Consecutive Cases. Arch. Ophthalmol. 2009, 127 (8), 981. 10.1001/archophthalmol.2009.151. [DOI] [PubMed] [Google Scholar]
- Hernandez I.; Yu Y.; Ossendorp F.; Korbelik M.; Oliveira S. Preclinical and Clinical Evidence of Immune Responses Triggered in Oncologic Photodynamic Therapy: Clinical Recommendations. J. Clin. Med. 2020, 9 (2), 333. 10.3390/jcm9020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecomte F.; Thecua E.; Ziane L.; Deleporte P.; Duhamel A.; Maire C.; Staumont-Salle D.; Mordon S.; Mortier L. Photodynamic Therapy Using a New Painless Light-Emitting Fabrics Device in the Treatment of Extramammary Paget Disease of the Vulva (the PAGETEX Study): Protocol for an Interventional Efficacy and Safety Trial. JMIR Res. Protoc. 2019, 8 (12), 15026. 10.2196/15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordon S. Painless and Efficient ALA-PDT and MAL-PDT of Actinic Keratosis Can Be Achieved by Drastically Reducing the Drug-Light Interval. Dermatol. Ther. 2020, 33 (3), 13423. 10.1111/dth.13423. [DOI] [PubMed] [Google Scholar]
- Ang J. M.; Riaz I. B.; Kamal M. U.; Paragh G.; Zeitouni N. C. Photodynamic Therapy and Pain: A Systematic Review. Photodiagnosis Photodyn. Ther. 2017, 19, 308–344. 10.1016/j.pdpdt.2017.07.002. [DOI] [PubMed] [Google Scholar]
- Rahman K. M. M.; Giram P.; Foster B. A.; You Y. Photodynamic Therapy for Bladder Cancers, A Focused Review. Photochem. Photobiol. 2022, 13726. 10.1111/php.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englinger B.; Pirker C.; Heffeter P.; Terenzi A.; Kowol C. R.; Keppler B. K.; Berger W. Metal Drugs and the Anticancer Immune Response. Chem. Rev. 2019, 119 (2), 1519–1624. 10.1021/acs.chemrev.8b00396. [DOI] [PubMed] [Google Scholar]