

Abstract
Breast cancer is a heterogeneous disease consisting of a diverse set of genomic mutations and clinical characteristics. The molecular subtypes of breast cancer are closely tied to prognosis and therapeutic treatment options. We investigate using deep graph learning on a collection of patient factors from multiple diagnostic disciplines to better represent breast cancer patient information and predict molecular subtype. Our method models breast cancer patient data into a multi-relational directed graph with extracted feature embeddings to directly represent patient information and diagnostic test results. We develop a radiographic image feature extraction pipeline to produce vector representation of breast cancer tumors in DCE-MRI and an autoencoder-based genomic variant embedding method to map variant assay results to a low-dimensional latent space. We leverage related-domain transfer learning to train and evaluate a Relational Graph Convolutional Network to predict the probabilities of molecular subtypes for individual breast cancer patient graphs. Our work found that utilizing information from multiple multimodal diagnostic disciplines improved the model’s prediction results and produced more distinct learned feature representations for breast cancer patients. This research demonstrates the capabilities of graph neural networks and deep learning feature representation to perform multimodal data fusion and representation in the breast cancer domain.
Keywords: graph neural network, multimodal machine learning, radiogenomics
1. INTRODUCTION
Current estimates predict in 2022 there will be 287,850 new cases of breast cancer in women and 43,250 deaths caused by breast cancer in the United States [1]. This continues the trend of breast cancer being the second most common cancer in women, behind skin cancer, and the second leading cause of cancer death for women. Breast cancer is heterogeneous in its molecular composition and clinical characteristics, representing a suitable application for the use of precision medicine to develop patient-specific treatment plans [2], [3]. Hierarchical clustering utilizing gene expression profiles of genes displaying larger variations in expression led to the current molecular subset classification of breast cancer: luminal A, luminal B, HER2+, basal-like, claudin-low, and normal-like [4], [5], [6], [7], [8]. The molecular subtypes serve as robust prognostic indicators in both guiding long-term outcomes and treatments. Identification of the molecular subtypes in patients is frequently done utilizing transcriptome analysis or immunohistochemical (IHC) staining to determine the presence of biomarker expression such as estrogen receptor (ER), progesterone receptor (PR), human epidermal growth receptor (HER) 1 and 2, and cytokeratin 5/6 [4], [9]. The expression levels of these biomarkers are closely tied to treatment options. However, a single biopsy may not capture the genetic heterogeneity of the entire tumor [10]. Our work investigates leveraging a wide breadth of patient information from multiple diagnostic disciplines and data modalities to predict molecular subtypes using deep graph learning. Graph-structured patient data representation integrates multiple data sources into a single, coherent patient representation.
Precision medicine guides long-term treatment strategies specific to individual patient needs utilizing personal genomic, phenotypic, and clinical factors [10], [11]. Breast cancer tumors exhibit large amounts of heterogeneity both within single lesions and across different lesions [12], [13], [14]. Intertumor heterogeneity in breast cancer induced the development of many different therapies targeting specific aspects of each patient’s breast cancer [15]. Intratumor heterogeneity due to variability in different tumor areas (spatial) and tumor progression over time (temporal) complicates the treatment of breast cancer; heterogenous tumors are more resistant to therapies, may evolve under selection pressure of therapeutic intervention, and are more difficult to treat [12], [16]. Tumor heterogeneity manifests itself in tumor morphology, biomarker expression, genetic mutations, and histopathologic characteristics [17]. We hypothesize that using a combination of data capturing these characteristics will produce a holistic representation of the individual patient’s breast cancer and improve predictions of molecular subtypes.
Radiographic imaging in breast cancer has been at the forefront of reducing mortality, with early mammographic screening and routine MRI screening for high-risk individuals becoming more common [1]. Computer-aided detection and diagnosis (CAD) systems are used at 91% of US digital mammography facilities, but several studies have found at large scale that CAD systems have shown mixed results in improving diagnostic accuracy [18], [19], [20], [21], [22]. Radiogenomics searches for relationships between the quantitative imaging features representing phenotypical tumor characteristics and the genomic profile of the cancer biopsy. Tumor imaging provides non-invasive measures for capturing the phenotypical traits that may be linked to underlying genomic characteristics [21]. Knowledge of genomic variants is crucial for the optimal care of cancer patients, enabling oncologists to refine prognosis and inform their treatment decisions. The detection of ‘actionable’ mutations allows the selection of targeted agents tailored to the patient’s particular molecular profile, including the possible use of off-label drugs that target specific molecules and pathways crucial for cancer survival [23]. Previous works have utilized multimodal data from singular diagnostic disciplines or from multiple diagnostic disciplines, but they have not utilized all the available patient information including clinical, radiological, pathological, and genomic sources. Our motivation is to learn an improved representation of the patient by fusing information from different diagnostic sources through the use of a graph convolutional neural network (GCNN). GCNNs capture the relationships between patient feature nodes by applying local filters, thus increasing the interpretability of the model by tracing back learning to the graph structure. This can provide an improved patient representation that can lead to better predictions for breast cancer subtype. Our work, as shown in Fig. 1, investigates the use of patient graph deep learning to predict molecular subtypes and the ability of deep embeddings representing individual diagnostic disciplines to separate patients by molecular subtype. Our contributions include the following:
Fig. 1.
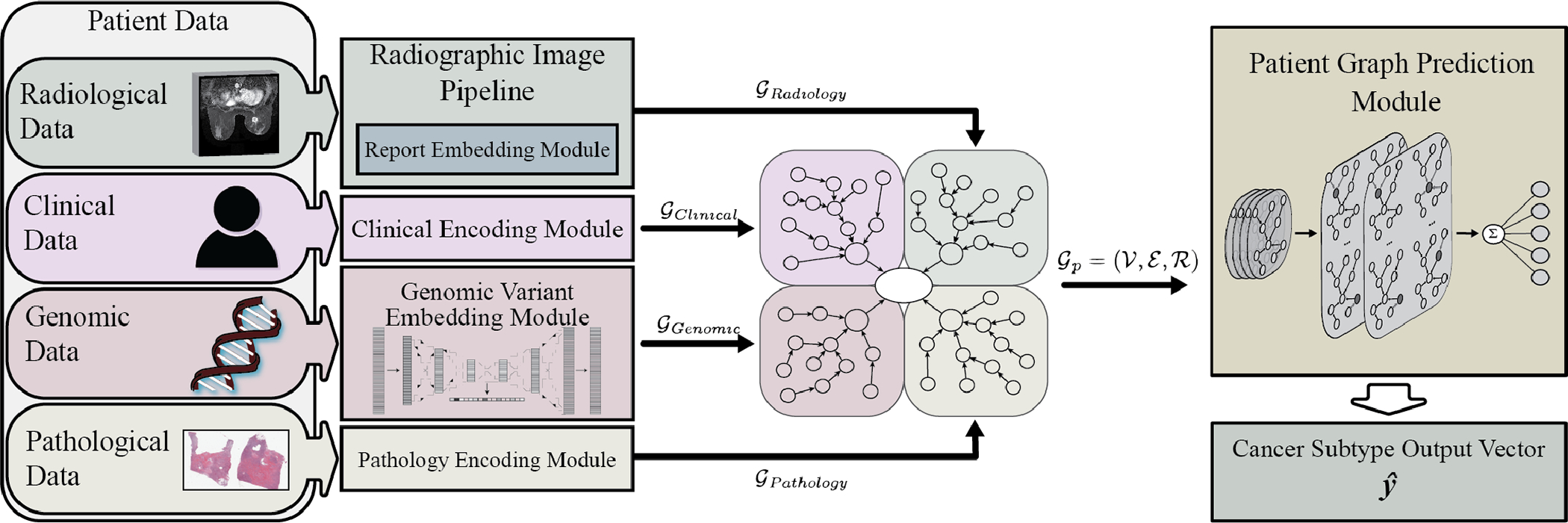
System architecture overview. The data from each patient diagnostic discipline is processed and embedded into feature vectors within graph nodes. Edges connect the nodes within diagnostic subgraphs to latent nodes and the central patient node prior to prediction by the relational graph convolution network module.
We construct an end-to-end trainable graph deep learning solution for utilizing supervised and semi-supervised data to classify breast cancer patient molecular subtypes with routinely collected patient data.
We provide a radiographic feature extraction pipeline to leverage information from radiologists’ annotations that produces high quality learned image embeddings from dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI).
Our research demonstrates the value of transfer learning from pan-cancer genomic assays in training autoencoders to extract feature vectors.
2. RELATED STUDIES
Advances in whole genome sequencing, exome sequencing, and panel specific targeted genes has pushed precision medicine research forward by providing large quantities of diverse data that require computationally efficient analysis for scalable clinical applicability [24]. Machine learning and deep learning-based approaches have already proven successful in providing clinical decision support for breast cancer molecular subtyping and predicting other clinically relevant features for patients.
Radiogenomic research in breast cancer has used breast MRI to predict breast cancer subtype with classic computer vision algorithms [25], [26], [27]. These studies found correlations between extracted MRI features and breast cancer heterogenous subtypes, specifically Luminal A and B. Wu et al. utilized various subsets of extracted imaging features to perform multivariate logistic regression for binary classification of Luminal A, Luminal B, and basal-like cancer with a respective area under the receiver operator curve () of 0.73, 0.69, and 0.79 on validation cohorts within the TCGA-BRCA dataset [28]. Other works focus on classifying breast cancer subtype biomarkers such as ER status, PR status, HER2 status, and triple-negative receptor status. Li et al. performed binary classification of one-vs-rest on these biomarkers with linear discriminant analysis, selecting radiomic features extracted from a subset of their results [29]. This work utilized a common subset of 91 TCGA-BRCA patients. Castaldo et al. improved upon this work by introducing normalization methods on the radiomic features and experimenting with simple machine learning classifiers including support vector machines, random forests, and Naïve Bayesian classifier [30]. Fan et al. performed radiomic analysis using a combination of features extracted from DCE-MRI and diffusion-weighted imaging (DWI) to predict the prognostic indicators, Ki-67 expression level and tumor grade. This showed the benefits of combining analysis from multiple imaging source domains and the ability to provide other useful prognostic information beyond molecular subtyping [31].
Early works in using deep learning for computer-aided diagnosis were able to identify clustered microcalcifications in mammograms using a convolutional neural network (CNN) over two decades ago [32]. CNNs have made significant progress in other fields to achieve top results in image classification challenges, image segmentation, and object localization, all of which are relevant applications in radiographic image analysis [33], [34], [35]. CNNs assist in clinical decision support by detecting and classifying breast lesions, predicting breast cancer biomarkers, and performing mass classification. Antropova et al. combined traditional radiomic features with features produced using a pretrained CNN to predict breast mass malignancy [36]. This study’s generalizable method performed well on full-field digital mammography, ultrasound, and DCE-MRI with values of 0.86, 0.90, and 0.89, respectively. Large-scale deep learning model analysis for dismissing breast MRI scans without lesions reduced the number of scans by 40% with 100% sensitivity for malignant scans [37]. Many of these radiographic analysis methods focus on using only the radiographic imaging to make predictions rather than integrating with other patient information in the decision-making process.
Other areas for breast cancer clinical decision support rely on using multi-omics data and integrating analysis with other patient information for predicting molecular subtypes and their biomarkers. Guo et al. used a subset of the TCGA-BRCA dataset and combined radiomic features with genomic features to predict stage, lymph node metastasis, ER status, PR status, and HER2 status [38]. This study performed feature selection using LASSO regularization within a two-tier cross-validation to obtain high values of 0.877, 0.695, 0.916, 0.775, and 0.641, respectively. Their results showed that frequently only a few features were selected to make the prediction and that genomic features provided better results than radiomic features when predicting molecular biomarkers. This study integrated the use of multiple genomic features: copy number, gene expression, and DNA methylation. Cristovao et al. compared the use of simpler models versus deep learning approaches to integrate the analysis of multi-omic data and predict breast cancer subtypes [39]. Their work fused multiple types of data by integrating RNA-seq gene expression, microRNA expression, and somatic copy number alteration data. They found that simpler models performed equal if not better than deep learning techniques but did suggest that variational autoencoders may have the potential to provide deep embeddings for extracting features from genomic data given further investigation. However, Byra et al. found the CNN models outperformed simpler models in predicting response to neoadjuvant chemotherapy using ultrasound images [40]. Other works have found using fully-connected neural networks and CNN models were able to integrate gene expression, copy number, and clinical data to predict breast cancer survival prognosis [41], [42].
Several studies attempted to tackle the problem of integrating more modalities of data into clinical decision support. Deepr provides an end-to-end system for utilizing a CNN to predict risk stratification from medical records [43]. MGNN is a framework for cancer survival prediction that combines genomic information with clinical information in a graph neural network (GNN) [44]. Parisot et al. utilized graph convolutional networks on population graphs to combine imaging and phenotype features to classify subjects with Autism Spectrum Disorders [45]. These methods combining multimodal data using deep learning showed more robust prediction abilities when compared to other works.
3. DATASETS
Our work utilizes The Cancer Genome Atlas Breast Invasive Carcinoma (TCGA-BRCA) dataset, which contains clinical, genomic, and pathological data in the Genomic Data Commons and radiological data from The Cancer Imaging Archive [46], [47], [48]. The TCGA-BRCA dataset contains genomic DNA copy number arrays, DNA methylation, exome sequencing, mRNA arrays, microRNA sequencing, and proteomic data from reverse-phase protein arrays [49]. We use the genomic variant information, radiographic information from DCE-MRI, clinical attributes, radiologist reports, and pathological testing results. Our combined subset of patients with molecular subtype information results in 1,040 patients, of which 108 patients have DCE-MRI and radiologist tumor measurements. Table 1 shows the breakdown of each cohort and the distribution of molecular subtypes. It shows the significant data imbalance between the different molecular subtypes. The cohort of patients with DCE-MRI has a much higher proportion of patients with Luminal A versus the other cohorts, 65% compared to approximately 50%. This difference in proportion is evenly divided amongst the other subtypes.
TABLE 1.
TCGA-BRCA Data Cohorts
| Patients with MRI | Patients w/o MRI | All Patients | |
|---|---|---|---|
|
| |||
| Luminal A | 70 | 466 | 536 |
| Luminal B | 17 | 184 | 201 |
| Basal-like | 14 | 170 | 184 |
| HER2 | 5 | 77 | 82 |
| Normal-like | 2 | 35 | 37 |
| Total | 108 | 932 | 1040 |
For supplementary validation of our framework, we apply it to predict the breast cancer molecular subtype of patients participating in the I-SPY2 trial. This ongoing, multi-center study aims to establish predictive models for neoadjuvant therapy response through the use of imaging, molecular, and clinical biomarkers [48], [50], [51], [52]. The I-SPY2 trial dataset encompasses molecular subtypes, DCE-MRI scans at various time points during the trial, clinical information, and micro array expression counts. We select a subset of 987 patients with available PAM50 molecular subtype results. Within this group, 562 patients have DCE-MRI scans before treatment and 974 patients have micro array expression data.
The distribution of molecular subtypes in the I-SPY2 dataset differs significantly from that of the TCGA-BRCA dataset, with 195 cases of Luminal A, 205 cases of Luminal B, 415 cases of Basal-like, 145 cases of HER2, and 27 cases of Normal-like. This disparity is due to the fact that neoadjuvant therapy is frequently used for patients with triple-negative receptor status. Furthermore, we utilize genomic data from the American Association for Cancer Research Project GENIE. The AACR Project GENIE is a pan-cancer registry of real-world oncology data assembled through data sharing between 19 leading international cancer centers (111,222 cases; 12,654 breast cancer cases, 5,381 metastasis) [53]. The dataset used for pretraining our genomic feature extraction method consists of genomic variant information from 100,099 patients, of which 13,599 have breast cancer.
4. METHODOLOGY
Patient healthcare data formalized into a graphical model represents the implicit relationships between patient diagnostic information and resulting outcomes. The graph model generalizes to differing modalities by embedding information in feature vectors that can vary by data node type. This modeling provides an inductive bias into the data to prioritize learning overall patient representation from the different diagnostic subgraphs as shown in Fig. 2. We formally define each patient graph where vertices of the graph represent nodes each containing a representation of a specific single aspect of patient information, such as age or estrogen receptor status. The set of directed edges connects these nodes where edges are of different relation types depending on the type of data represented in their respective node endpoints, typed from the set . We define the center of each patient graph as a patient node with learnable embeddings connected to latent nodes containing learnable embeddings representing each diagnostic discipline. Each latent node connects to a varying number of nodes depending on the data available for that patient. The typical node in a patient graph includes a feature vector representing a single diagnostic test or feature from that patient. Nodes are connected with a directed edge to either the central node for their diagnostic discipline or a node responsible for the feature it contains, such as linking multiple pathology test results to a single histology sample node. Our approach divides patient information into subgraphs based on the different diagnostic disciplines involved to represent the relationships these contribute to understanding the overall patient representation. Each subgraph feeds into a central node to aggregate the information from the different diagnostic fields. Table 2 indicates the average sizes of the patient graphs for the two datasets.
Fig. 2.
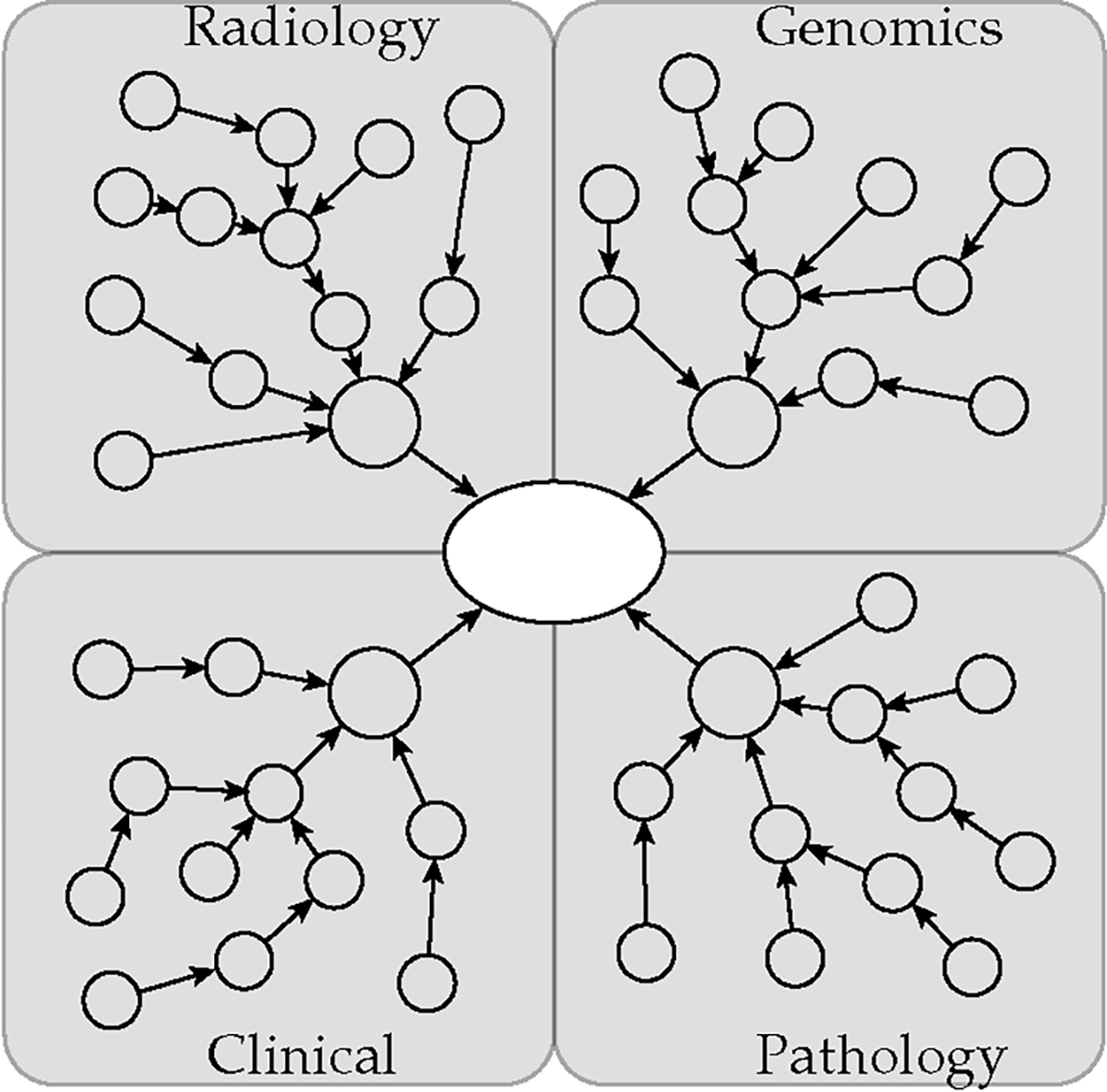
Patient graph representation.
TABLE 2.
Patient Graph Metrics
| TCGA | I-SPY2 | |
|---|---|---|
|
| ||
| Mean # Nodes | 19.15 | 11.03 |
| Mean # Edges | 37.30 | 21.06 |
4.1. Graph Neural Network
The graph neural network is based on the relational graph convolutional network (RGCN), which utilizes the benefits of convolutional graph filters on multi-relational, complex graph networks. The RGCN model extends the previous work on graph convolutional networks, which implements an approximation of spectral graph convolutions on local neighbors [54], [55]. RGCN adapts to multiple types of relations by aggregating the messages passed across all relations in the set of neighbors as shown in
| (1) |
The node embedding for layer is updated by aggregating the results across all relations in for each neighboring node connected to node . Our method utilizes two layers, allowing information from neighbors up to two connections away from node to impact the embedding values. The ReLU function, , provides the nonlinear activation by taking the element-wise maximum and achieves higher accuracy than other activation functions in our experiments. Our method trains the RGCN model for graph classification to predict the probability of five molecular subtypes by mean pooling the hidden node embeddings and applying the softmax function
| (2) |
where is the number of molecular subtypes. The output from the final mean pooling hidden layer feeds into the softmax layer and the linear combination with weights . Equation (3) shows the computation of the graph embedding from the final layer of node embeddings . This calculates the average of the node embeddings for each node in and pools the embeddings across all the node types in .
| (3) |
The patient graphs are grouped into batches for training. We calculate the loss for updating the graph embeddings in the GNN using the cross-entropy loss function. Equation (4) computes the loss difference between the predicted values and actual molecular subtype values .
| (4) |
The final output of the network both identifies the most probable molecular subtype and shows potential heterogeneity in a patient when the difference in probability of the most probable molecular subtypes is insignificant. Fig. 3 shows a batch of patient graphs being fed into the network for predicting molecular subtypes. The graph convolutional layers update the hidden embeddings by passing information from neighboring nodes. The GNN pools the last layer embeddings from the entire patient graph to make its prediction.
Fig. 3.
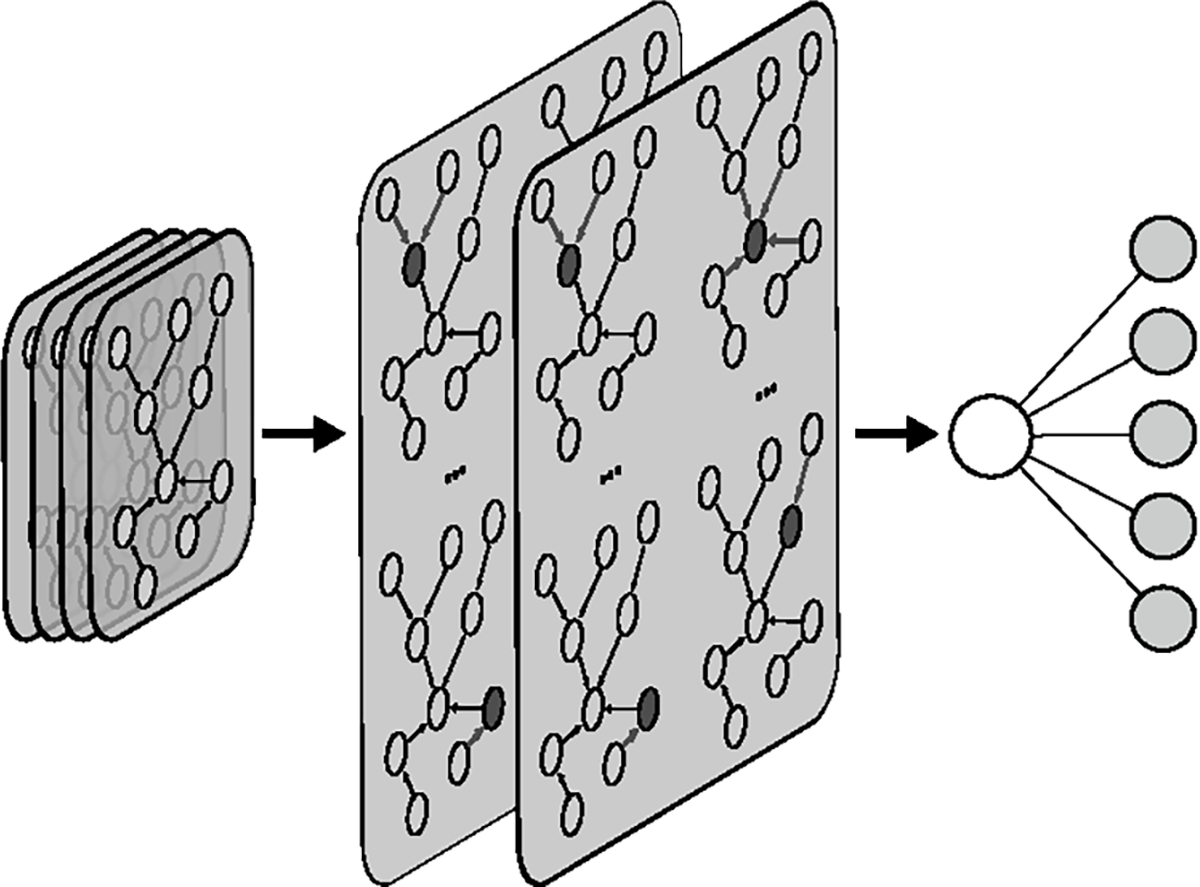
Patient graph neural network predicting subtype on a batch of patient graphs.
4.2. Node Features
Each breast cancer patient is linked to several categories of data that further consist of sub-attributes. We create a data graph for each patient consisting of a central patient node connected to nodes representing information in radiology, pathology, clinical attributes, and genomic features. These latent nodes have directed connections from their respective diagnostic subgraph nodes, which directly represent patient information. The patient node and latent nodes are fed through a trainable embedding layer, which utilizes the individual node identification numbers to produce an embedding vector fed through a fully-connected layer with ReLU activation. The patient node and latent node embeddings are 8×1 and 16×1 respectively. These both connect into a 16×1 dense ReLU layer.
Large sparse vectors represent mutations found in gene panels. We combined several gene panels from the GENIE Project using the HUGO symbols, which are standardized unique symbols for human genes [56]. We used symbols from the DFCI OncoPanels [57], MSK-IMPACT [58], and VICC-01-T7 [59] and filtered out silent and intron variants. These symbols represent genes commonly linked with cancer. This results in 158 potential gene symbols with variants. Fig. 4 shows the six-layer fully-connected autoencoder trained to recreate normalized mutation frequency vectors. The embedded low-dimensional vectors extracted from the latent space in the autoencoder provide inputs into the graph neural network. An autoencoder is an unsupervised neural network that learns to produce a low-dimensional representation of data while reconstructing the original input data. It consists of an encoder section and decoder section. The encoder layers consist of 64, 32, and 16 neurons, and the decoder section is the same design with the layer sizes in reverse order and the last layer being 158 neurons. The autoencoder uses the ReLU activation function and softmax for the final prediction layer. We pretrained the autoencoder on the GENIE Project cancer patients prior to use with the TCGA-BRCA patient data. The output from the encoder section creates a 16×1 embedding representation of the gene variants for use in the patient graphs.
Fig. 4.
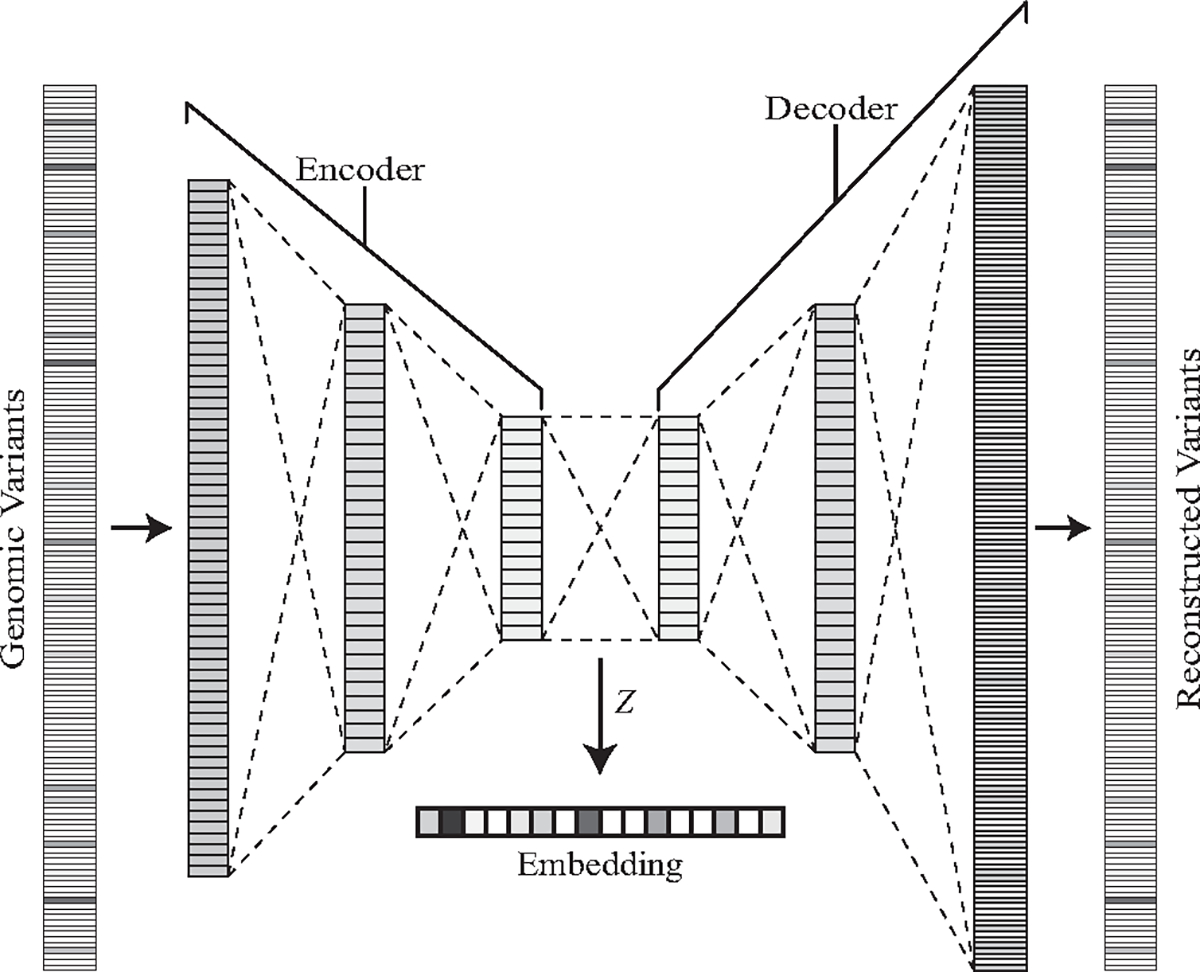
Genomic variant embeddings extracted from the latent space of the pretrained autoencoder.
The clinical and pathological data for each patient is encoded into one-hot vectors, which are binary vectors using values of 1 to represent the true value for a specific feature. Categorical information is binned based on the category, and numeric features such as age are grouped into ranges of values. These vectors cover relevant patient attributes, immunohistochemistry results, and findings from histopathological slides.
4.3. Image Feature Extraction
The TCGA-BRCA dataset consists of many types of MR and mammogram images. We utilize the DCE-MRI to provide image phenotype information to the graph neural network. Dynamic contrast-enhanced (DCE) imaging uses T1-weighted images at multiple time points as contrast agent passes from blood to tissues. Tumor tissues with more permeable blood vessels have higher signal intensity in the DCE-MRI. The TCGA Breast Phenotype Research Group provides radiologist annotations for 108 patients, with three independent radiologists identifying the largest length of the tumor in a single DCE-MRI DICOM slice and other Breast Imaging Reporting and Data System (BI-RADS) related characteristics, which are commonly included in radiologist reports [48], [46]. Our radiographic image pipeline presented in Fig. 5 converts the image into a feature vector to represent the key features in the graph. An EfficientNet-B0 CNN pretrained on the ImageNet dataset reduces regions-of-interest in patient MRI slices to compact feature vectors [60]. The EfficientNet architecture family balances model depth, width, and resolution to provide increased performance over similarly sized deep learning models. The smallest model, EfficientNet-B0 uses input images with 224×224 resolution, has only 5.3 million parameters, which is one fifth that of ResNet-50, and provides higher accuracy on ImageNet. The EfficientNet models reached state-of-the-art results on several transfer learning problems while utilizing significantly less parameters than competing models. The EfficentNet main blocks are inverted bottleneck MBConv blocks, which were originally invented for the compact MobileNetV2 [61]. These blocks have residual connections between the bottleneck layers with two expansion blocks containing the high number of channels in the middle. This reduces the memory requirements of the block. The model also uses Squeeze-and-Excitation blocks to perform dynamic channel-wise feature recalibration which allows it to use global information to selectively emphasize informative features [62]. These blocks aggregate the spatial information features to create a channel descriptor. This channel descriptor feeds into a gating mechanism to control the excitation of the channels. We replace the original classification head with a 16-neuron dense sigmoid layer to encode the extracted feature at the end of the base network and convert the values to the same range of the feature vectors within the graph. The CNN is trained to predict molecular subtypes to fine-tune these dense layer weights, with the classification layer removed when generating the features for the graph. This training requires two-dimensional regions of interest around the tumor in a single DICOM slice.
Fig. 5.
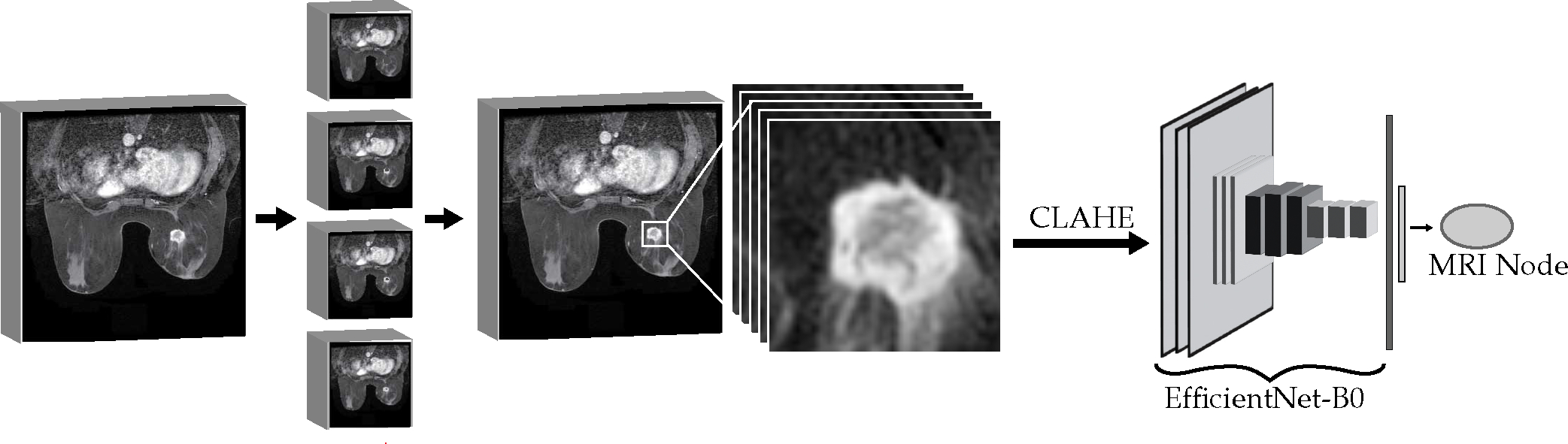
Radiographic image feature extraction.
We implement a region-growing algorithm to isolate the three-dimensional volume of interest (VOI) around the tumor. The algorithm utilizes a seed point closest to the midpoint along the radiologist measurement line with an intensity in the upper quartile of the line’s pixel values. This ensures the seed point is in the center of a bright spot of the tumor in the image slice. The region grows in all directions from its boundary each iteration, with a threshold to expand the region equal to half the pixel intensity standard deviation for the radiologist-identified bounding box. This algorithm automatically ends when there are no more pixels on the boundary of the region within the range of the intensity threshold or when the diagonal length of the center of the region reaches twice that of the radiologist annotated length. The second case captures potential failure cases arising from the tumor intensity being close in value to the radiologist identified region intensity, which can be an issue in images with especially low contrast. This increases the robustness of the segmentation algorithm. The region is converted to a bounding cube with a fifteen-pixel border on each slice to include local tumor environment and reduced in the z-axis to require at least six tumor pixels in a slice to be included in the volume. This method prioritizes quick segmentation by simultaneously expanding in all neighboring directions rather than a single pixel at a time and ensures regions include tumor pixels with surrounding environment. The DICOM VOIs for each patient are separated into the individual image slices for training the CNN. These slices are scaled and stacked to three channel gray scale, undergo contrast-limited adaptive histogram equalization (CLAHE) to increase contrast, and resized to 224×224 with padding for use with EfficientNetB0. CLAHE improves the local contrast and enhances edges within the regions of an image by transforming pixels in proportion to the cumulative distribution function of pixel intensities in the region surrounding the pixel [63]. The contrast-limited adjustment lessens the enhancement in homogenous areas by clipping the pixel intensity histogram. We train the CNN for a fixed number of epochs with the same cross-validation folds for the graph neural network to prevent leakage between train and test groups. The training time, feature vector size, batch size, and other hyperparameters for the CNN were determined through tuning with a Bayesian Optimization tuner that averaged cross-validation metrics as the tuning goal. During training the CNN, the images pass through multiple augmentation layers to add random intensity shifting, cropping, rotation, translation, and flipping to the images. However, the patient graphs only contain image nodes using the image slices identified by radiologists to ensure that image features used in the graph prediction contain the most accurate representation of the breast cancer tumor.
4.4. Method Validation
Stratified cross-validation splits the dataset into different folds that maintain the same proportion of patient breast cancer molecular subtypes as the original dataset. We perform stratified five-fold cross-validation to better demonstrate the generalizability of our classification framework and to reduce the variance of the model performance estimates. This provides an improved evaluation over splitting the datasets into fixed training, validation, and testing groups since we perform held-out testing on all patients in the dataset with the primary cost being more time spent training and testing the models. We compare the performance of the RGCN model with random forest, logistic regression with LASSO regularization, fully-connected multilayer perceptron, and convolutional neural network. These methods are not easily adaptable for taking inputs from multiple data modalities, so we feed the extracted patient node features from our framework as a flattened one-dimensional vector input. We keep the order of the features grouped by diagnostic discipline in such a manner to keep related features adjacent in the vector. This results in an input size of 149×1 and 71×1 for TCGA and I-SPY2, respectively. Missing patient features are set to default values of zero. For the I-SPY2 dataset, we retrain the genomic autoencoder model on the normalized micro array expression data to extend our method to the new data modality.
4.5. Hyperparameter Settings
The hyperparameters for the feature embedding modules were optimized using cross-validation to find the optimal design. The image feature extraction involved predicting patient characteristics using only the radiographic imaging to optimize the final layer embedding size. The optimal final layer embedding size was determined to be 16×1 prior to the classification head after testing sizes of 8, 16, 32, or zero trained embedding nodes. The layer size and depth of the genomic autoencoder model were selected from 2 to 3 layers of sizes 16, 32, and 64, and the model with the best performance in predicting molecular subtype using only the genomic embeddings was selected. The optimal results were obtained when the autoencoder was trained at a low learning rate of 0.00001 for 4,000 epochs, as experiments with different learning rates between 0.00001 and 0.01 and epochs between 16 and 4,000 showed. We searched for the best RGCN model depth between 2 and 3 layers of sizes 16 to 128 at step size intervals of 16. We trained the TCGA RGCN model for 35 epochs with a learning rate of 0.01 and a batch size of 50 and trained the I-SPY2 RGCN model for 20 epochs with the same learning rate and batch size. Our selection was based on evaluating a range of epoch values from 16 to 100 and learning rates from 0.00001 to 0.01.
The random forest and logistic regression are evaluated using a two-tier cross-validation to optimize the internal hyperparameters, replicating previous state-of-the-art methods using logistic regression to predict breast cancer features [38]. A Bayesian optimization tuner found the optimal layer sizes and depth and training hyperparameters of the MLP and CNN for each dataset. We utilized dropout layers and batch normalization in both the MLP and CNN. Dropout layers increase the robustness of the weight learning by dropping 50% of the neurons in the layer so the model does not rely too heavily on specific neurons [64]. Batch normalization layers normalize the intermediate layer inputs to reduce the internal covariate shift, which has shown benefits in reducing training time and increasing accuracy [65]. We found that two-layer MLP outperformed one-layer MLP, with the optimal models consisting of 64 neurons in the first layer and 256 neurons in the hidden layer for both I-SPY2 and TCGA data. The CNN model search consisted of a first layer one dimensional convolution followed by a fully-connected hidden layer before the classification output layer. The TCGA CNN has a convolutional layer with 4 filters, kernel size of 16, and stride of 2 followed by 16 fully-connected neurons. The I-SPY2 CNN has a convolutional layer with 4 filters, kernel size of 64, and stride of 16 followed by 512 fully-connected neurons. All baseline neural networks have a dropout and batch normalization layer prior to the final softmax classification output layer.
5. RESULTS
We performed stratified five-fold cross-validation using the TCGA-BRCA dataset which consists of 1,040 patients with clinical, pathological, and genomic information. A subset of 108 patients with DCE-MRI and radiologist annotations makes up the MRI Patients cohort. We stratify these subsets to provide equal proportions of patients with each molecular subtype in each fold. These proportions are approximately equal to the dataset proportions in Table 1. The results in Table 3 show the mean values from the cross-validation averaged over five repeated cross-validation experiments, meaning we retrained the graph neural network twenty-five times when collecting our results. The standard deviations represent the average standard deviation within a cross-validation experiment. We report both the micro-averaged and macro-averaged utilizing the equations below. The micro-averaged weights the of each class based on the number of samples in the class. This allows each patient to contribute equally to the calculations by summing the class variable across all the patients in the dataset and dividing by the total number of patients. The macro-averaged equally weights each class , which causes minority classes to have larger impacts on the metric. We compute the AUC of the precision recall curve () since it is an especially useful metric when the proportion of positive and negative cases is imbalanced during classification [66]. The precision or positive predictive value (PPV) is the proportion of true positive predictions among all the positive predictions; the recall is equivalent to the true positive rate. Table 4 includes the for each method.
TABLE 3.
Molecular Subtype Classification Results
| Validation Cohort | Micro Avg AUC-ROC | Macro Avg AUC-ROC | ||
|---|---|---|---|---|
| μ | σ | μ | σ | |
|
| ||||
| Patients with MRI | 0.912 | 0.028 | 0.710 | 0.046 |
| Patients w/o MRI | 0.867 | 0.019 | 0.778 | 0.028 |
| All Patients | 0.871 | 0.018 | 0.772 | 0.026 |
TABLE 4.
Model Performance Comparison
| Dataset | Model | Micro AUC-ROC | Macro AUC-ROC | Micro AUCPR | Macro AUCPR |
|---|---|---|---|---|---|
|
| |||||
| TCGA | RGCN | 0.871 | 0.772 | 0.646 | 0.500 |
| CNN | 0.871 | 0.751 | 0.665 | 0.498 | |
| MLP | 0.892 | 0.766 | 0.693 | 0.493 | |
| LR | 0.790 | 0.725 | 0.444 | 0.386 | |
| RF | 0.773 | 0.631 | 0.571 | 0.359 | |
|
| |||||
| I-SPY2 | RGCN | 0.895 | 0.838 | 0.724 | 0.531 |
| CNN | 0.841 | 0.740 | 0.611 | 0.427 | |
| MLP | 0.848 | 0.739 | 0.631 | 0.424 | |
| LR | 0.713 | 0.669 | 0.355 | 0.328 | |
| RF | 0.643 | 0.570 | 0.461 | 0.290 | |
| (5) |
| (6) |
| (7) |
| (8) |
Our results found our framework achieves the highest value of 0.912 when patient graphs included radiographic features extracted from DCE-MRI scans. This cohort of patients results in the highest standard deviations and lowest macro averaged . The cohort has a very small number of patients with more rare molecular subtypes, which significantly affects the macro averaged . When comparing the cohorts of patients with missing radiographic features versus the entire dataset of TCGA-BRCA, the classification results and standard deviations are approximately equivalent between the two groups. It is difficult to directly compare these results with other related studies, since many studies used a reduced subset of 91 TCGA-BRCA patients to report metrics, did not perform cross validation, or utilized different multi-omic data such as RNA data.
The comparison of the results of various machine learning methods, namely multilayer perceptron (MLP), convolutional neural network (CNN), logistic regression (LR) and random forest (RF), is presented on two datasets, TCGA-BRCA and I-SPY2, using features extracted from our embedding modules. Our findings indicate that the MLP and CNN methods perform similarly to the RGCN model on the TCGA-BRCA dataset with the extracted features, however, the RGCN outperforms the other methods significantly in all metrics on the I-SPY2 patients. The results of the ablation study, presented in Table 5, demonstrate the impact of removing different components from patient graphs. The removal of these components is equivalent to removing framework components, and the results indicate that performance decreases in general when diagnostic patient data is removed, except for a slight improvement in two cases in the TCGA results and I-SPY2 macro-averaged results. The degree of performance impact varies depending on the metric used and the dataset. On the TCGA-BRCA dataset, removal of the Pathology subgraph leads to a substantial decrease in performance, whereas the patients in the I-SPY2 dataset show much less impact.
TABLE 5.
Model Graph Ablation Results
| Dataset | Missing Component | Micro AUC-ROC | Macro AUC-ROC | Micro AUCPR | Macro AUCPR |
|---|---|---|---|---|---|
|
| |||||
| TCGA | None | 0.871 | 0.772 | 0.646 | 0.500 |
| Pathology | 0.787 | 0.613 | 0.507 | 0.287 | |
| Genomic | 0.859 | 0.761 | 0.599 | 0.434 | |
| Radiology | 0.873 | 0.766 | 0.623 | 0.464 | |
| Clinical | 0.880 | 0.779 | 0.631 | 0.492 | |
|
| |||||
| I-SPY2 | None | 0.895 | 0.838 | 0.724 | 0.531 |
| Pathology | 0.886 | 0.813 | 0.709 | 0.491 | |
| Genomic | 0.829 | 0.772 | 0.567 | 0.418 | |
| Radiology | 0.886 | 0.844 | 0.679 | 0.542 | |
| Clinical | 0.879 | 0.830 | 0.664 | 0.523 | |
The outputs from the different diagnostic subgraphs demonstrated varying contributions in separating the molecular subtype information. Fig. 6 shows the visualization of the patient node outputs after the second RGCN layer in the graph neural network. The plot is the two-dimensional t-distributed stochastic neighbor embedding (t-SNE) representation for each patient. The t-SNE algorithm is a nonlinear dimensionality reduction method developed to visualize high dimensional data [67]. The t-SNE figures lack axis markings because the nonlinear mapping results in the real values having no attached meanings. This figure demonstrates separation of clusters with notable Luminal A, Basal, HER2, and Luminal B groupings. The t-SNE visualization for the outputs from each diagnostic latent node is shown in Fig. 7. Each subgraph node representation provides less defined groupings. The pathology latent node exhibits the most defined groups correlated to subtypes and most closely resembles the overall patient representation. The radiology latent node did provide separation between Luminal A and Luminal B patient groups.
Fig. 6.
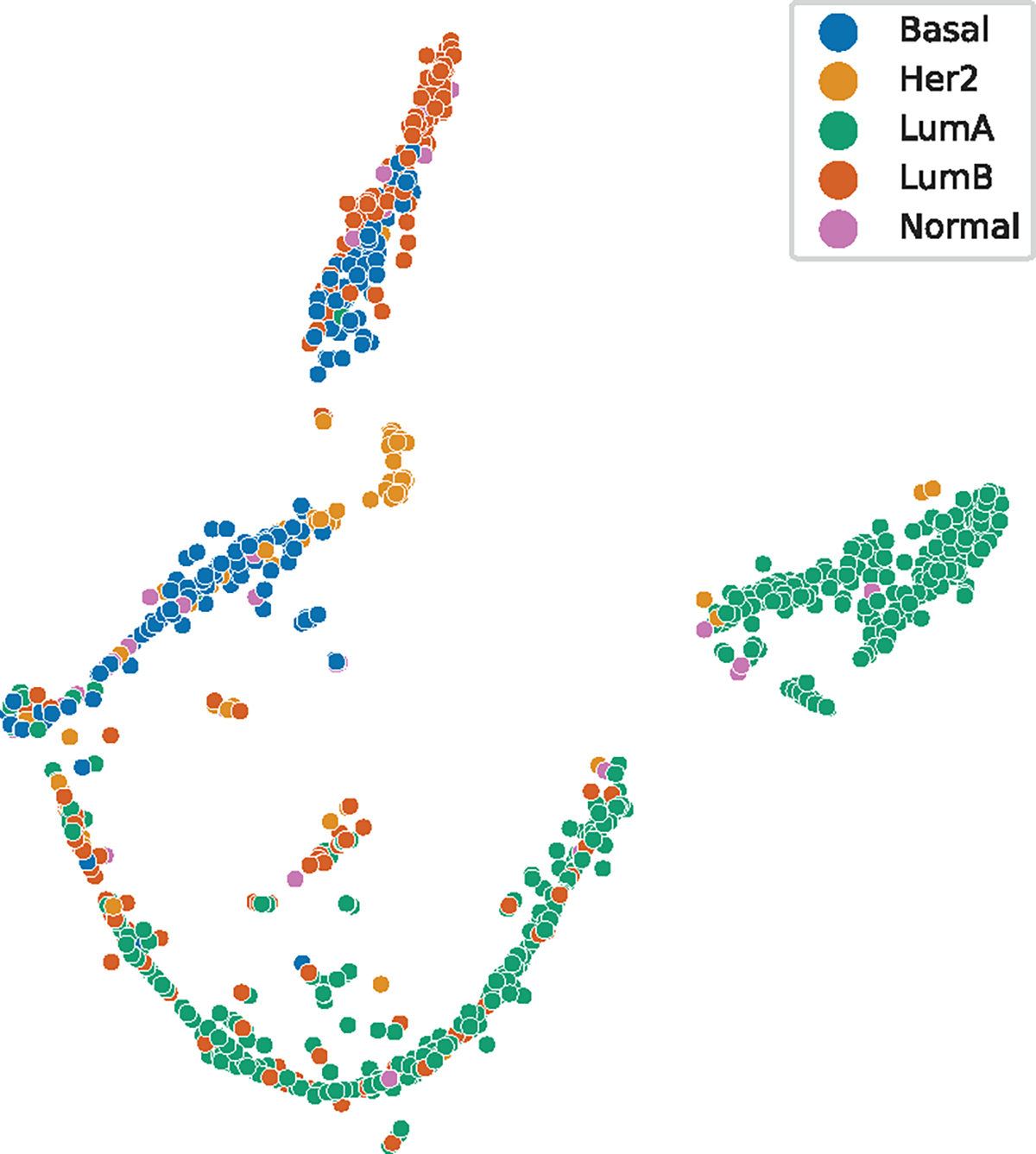
Plot of the patient RGCN output vector using t-SNE.
Fig. 7.
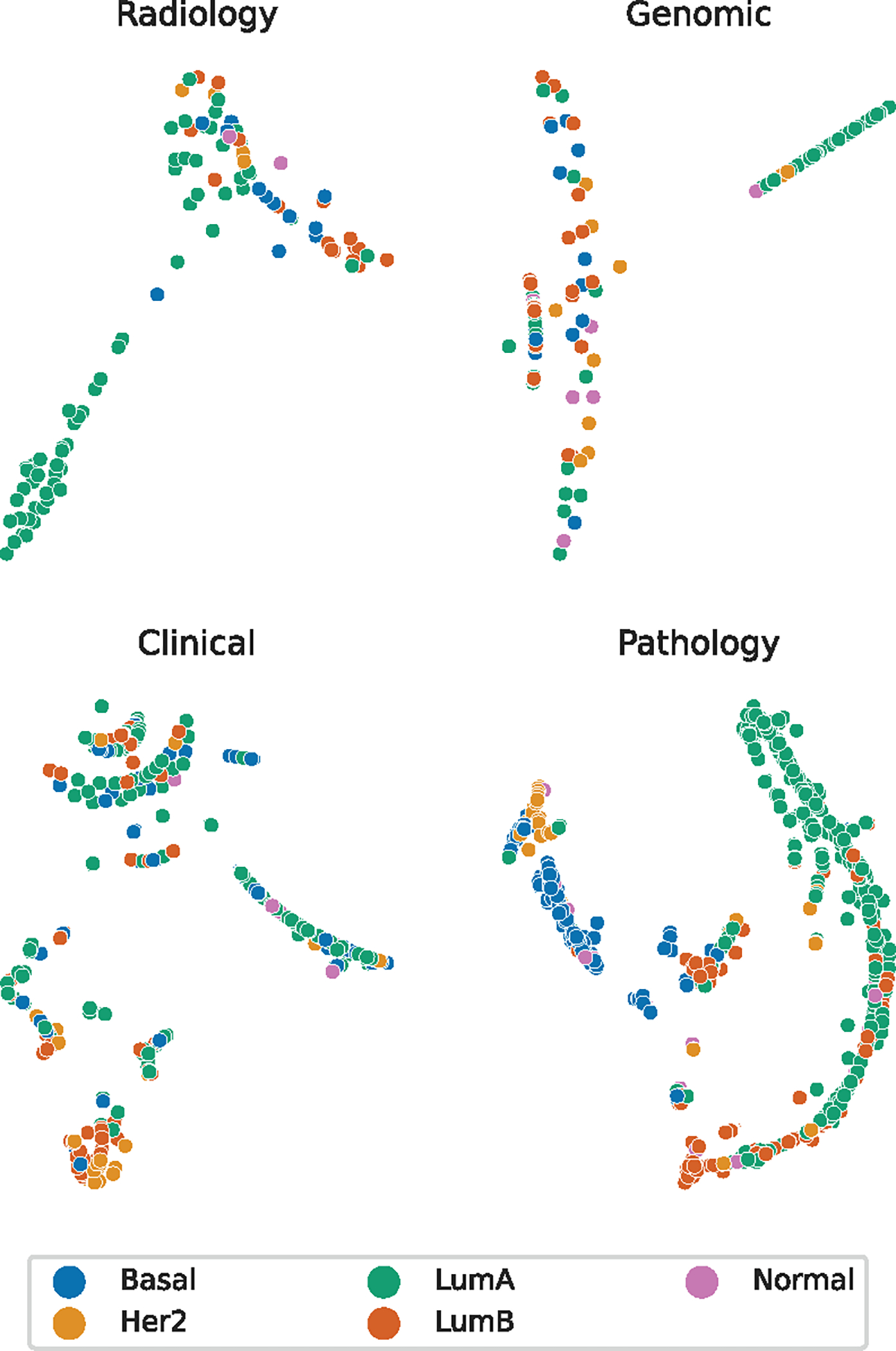
Plots of the latent node RGCN output vectors using t-SNE.
6. DISCUSSION
Our work explores the use of modeling patient information into directed, fully connected graphs to perform molecular subtype classification for breast cancer patients. Our framework achieves the best results when including image features extracted from DCE-MRI compared to subsets lacking radiographic imaging. The RGCN model also performs best on the I-SPY2 dataset, which contains a significantly larger proportion of patients with DCE-MRI. We found during hyperparameter searches that the baseline deep learning models had plateaued performance across several configurations for the TCGA dataset, which may indicate performance is limited by patient features and data proportions rather than model capacity. We note that our ablation results indicate the framework still performs well when missing entire subgraphs of diagnostic data. The most impactful diagnostic subgraph differs between datasets, implicating that future patient data may rely on different diagnostic subgraphs and none can be eliminated from the framework. The performance measured by the metric shows the most potential for improvement, indicating the models predict with lower positive predict value. The small number of patients with Normal-like breast cancer resulted in a low number of patients with positive predictions for this category, reducing the precision. The macro-averaged results are consistently inferior to the micro-averaged results; this is likely related to the large imbalance in molecular subtype patients contained in both datasets. We trained the graph model both using individual patient graphs and using batches of patient graphs. The batch training method achieves equivalent results with smoother loss curves but no significant increase in .
The TCGA-BRCA dataset presents an extremely imbalanced proportion of molecular subtypes, with a majority being Luminal A. The normal-like patients make up less than 5% of all the patients, and we did not have enough with DCE-MRI to provide adequate training on the final weights of the CNN during image feature extraction. We attempted to train the CNN on only the radiologist annotated DCE-MRI slices to ensure that all pretraining images had high quality views of the tumor but found reduced from the graph neural network predictions. Increasing the number of available DCE-MRI slices to train the CNN despite having slices with smaller amounts of tumor present improved the overall performance of the framework. As shown in Fig. 6 and 7, the RGCN layers extracted embeddings that produced separatable groupings in the patient data, but not all of the groupings separated the molecular subtype information. The Luminal A patients tend to form the most consistent groups among the radiology and genomic subgraphs. The radiology subgraph does appear to separate Luminal A and B subtypes, which reflects the success of the related radiomic research in also separating these subtypes in TCGA-BRCA. The clinical subgraph did lead to specific groupings for most HER2 patients. The pathology subgraph outputs clearly perform the best in separating the molecular subtypes and are supported by their impact on the ablation results, likely due to the biomarker information included in these graphs and its known correlation to molecular subtypes. The patient node output exhibits the most separation between molecular subtypes, supporting our hypothesis that utilizing the information from multiple diagnostic disciplines would outperform using a single discipline. The HER2 patients and Luminal B patients have notably more distinct clustering in the patient-wise representation than the pathology subgraph representations. The Normal-like patients do not exhibit clear clustering in any of the t-SNE plots. Although this may indicate it is simply more difficult to distinguish from other molecular subtypes, the group having the smallest number of patients would contribute to the difficulty in learning to represent the patient information for this category.
We utilized a modified version of the update-aggregate formula in the RGCN without degree normalization as shown in (1) rather than the degree normalized versions. The normalized aggregation provided worse performance on the small patient graphs, but normalization may be necessary if patient graphs have more variation in the magnitude of the node degrees.
7. CONCLUSION
We investigated using graph neural networks to predict molecular subtypes of breast cancer patients by embedding their information in feature vectors within graph models. Our approach used a CNN to extract features from DCE-MRI scans and an autoencoder to represent genomic variant results or micro array expression features in a condensed latent space. We found that the combination of radiographic data and genomic data improved the graph neural networks prediction abilities. Further ablation testing demonstrated that different diagnostic disciplines had varying impacts across datasets in predicting breast cancer molecular subtype, with all disciplines increasing the value of at least one performance metric, if not all metrics.
Pretraining the feature extraction models with related domain data improved the of the graph neural network predictions. We found that prediction abilities significantly decreased when TCGA patient graphs did not include information regarding IHC biomarkers. Therefore, the graph neural network is significantly relying on the information previously known to correlate with molecular subtypes. The genomic variants are utilized in the final graph predictions but not as the primary contributors. The micro array expression data in I-SPY2 significantly boosted prediction results. Our work may prove more beneficial in predicting metrics less correlated with specific patient features to take full advantage of the heterogeneous, multimodal data source.
This graph-based method can be further extended with more patients from other datasets because it utilizes commonly collected breast cancer patient information. We used MRI scans from multiple manufacturers and clinical providers as well as genomic variant assays with differing protocols to demonstrate the generalization potential of the method. We note that the method’s performance suffered most on less common molecular subtypes due to the small number of patients provided in the dataset, especially in the patient cohort with MRI. This issue is most simply resolved by including more patients from the less common molecular groups to create a balanced dataset for training and evaluation. Our framework demonstrates the potential benefits of modeling patient information into graphs using extracted deep learning features and performing multimodal fusion of breast cancer patient data with graph convolutional neural networks.
ACKNOWLEDGMENTS
This work was supported in part by the National Institute of Health NCI under contract 75N91021C00025. The results shown here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. The authors would also like to acknowledge the American Association for Cancer Research and its financial and material support in the development of the AACR Project GENIE registry, as well as members of the consortium for their commitment to data sharing.
Biographies
Isaac Furtney recieved the BS degree in computer engineering and MS degree in electrical and computer engineering at the University of Miami. He is currently working toward the PhD degree in electrical and computer engineering at the University of Miami. His research interests include deep learning and its applications in bioinformatics and radiographic image analysis.
Ray Bradley received the BS degree in computer engineering from the University of Miami in 2005. He is currently with INFOTECH Soft, Inc. His current research and development interests include bioinformatics, knowledge management and machine learning.
Mansur R. Kabuka received the PhD degree from the University of Virginia, Charlottesville, VA. He is a professor at the University of Miami, FL and the founder and director of INFOTECH Soft, Inc. His research interests include information integration, big data, and medical informatics.
Contributor Information
Isaac Furtney, Department of Electrical and Computer Engineering, University of Miami, Coral Gables, FL, 33146.
Ray Bradley, INFOTECH Soft, Inc., Miami, FL, 33131.
Mansur R. Kabuka, University of Miami, Coral Gables, FL, 33146
REFERENCES
- [1].American Cancer Society, “Cancer Facts & Figures 2022,” https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2022/2022-cancer-facts-and-figures.pdf.
- [2].Polyak K, “Breast cancer: origins and evolution,” J Clin Invest, vol. 117, no. 11, pp. 3155–3163, Nov. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rivenbark AG, O’Connor SM, and Coleman WB, “Molecular and Cellular Heterogeneity in Breast Cancer,” Am J Pathol, vol. 183, no. 4, pp. 1113–1124, Oct. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Perou CM et al. , “Molecular portraits of human breast tumours,” Nature, vol. 406, no. 6797, pp. 747–752, Aug. 2000. [DOI] [PubMed] [Google Scholar]
- [5].Hu Z et al. , “The molecular portraits of breast tumors are conserved across microarray platforms,” BMC Genomics, vol. 7, p. 96, Apr. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sørlie T et al. , “Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: gene expression analyses across three different platforms,” BMC Genomics, vol. 7, p. 127, May 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Herschkowitz JI et al. , “Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors,” Genome Biol, vol. 8, no. 5, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Prat A et al. , “Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer,” Breast Cancer Res, vol. 12, no. 5, p. R68, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Picornell AC et al. , “Breast cancer PAM50 signature: correlation and concordance between RNA-Seq and digital multiplexed gene expression technologies in a triple negative breast cancer series,” BMC Genomics, vol. 20, 2019, publisher: BioMed Central. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Arimura H, Soufi M, Kamezawa H, Ninomiya K, and Yamada M, “Radiomics with artificial intelligence for precision medicine in radiation therapy,” Journal of Radiation Research, vol. 60, no. 1, pp. 150–157, Jan. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pinker K, Chin J, Melsaether AN, Morris EA, and Moy L, “Precision Medicine and Radiogenomics in Breast Cancer: New Approaches toward Diagnosis and Treatment,” Radiology, vol. 287, no. 3, pp. 732–747, Jun. 2018. [DOI] [PubMed] [Google Scholar]
- [12].Bi WL et al. , “Artificial intelligence in cancer imaging: Clinical challenges and applications,” CA A Cancer J Clin, p. caac.21552, Feb. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Napel S, Mu W, Jardim-Perassi BV, Aerts HJWL, and Gillies RJ, “Quantitative imaging of cancer in the postgenomic era: Radio(geno)mics, deep learning, and habitats,” Cancer, vol. 124, no. 24, pp. 4633–4649, Dec. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nishino M, “Tumor Response Assessment for Precision Cancer Therapy: Response Evaluation Criteria in Solid Tumors and Beyond,” American Society of Clinical Oncology Educational Book, no. 38, pp. 1019–1029, May 2018. [DOI] [PubMed] [Google Scholar]
- [15].Polyak K, “Heterogeneity in breast cancer,” The Journal of Clinical Investigation, vol. 121, no. 10, p. 3786, Oct. 2011, publisher: American Society for Clinical Investigation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bettaieb A, Paul C, Plenchette S, Shan J, Chouchane L, and Ghiringhelli F, “Precision medicine in breast cancer: reality or utopia?” Journal of Translational Medicine, vol. 15, no. 1, p. 139, Jun. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Turashvili G and Brogi E, “Tumor Heterogeneity in Breast Cancer,” Frontiers in Medicine, vol. 4, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Keen JD, Keen JM, and Keen JE, “Utilization of Computer-Aided Detection for Digital Screening Mammography in the United States, 2008 to 2016,” Journal of the American College of Radiology, vol. 15, no. 1, Part A, pp. 44–48, Jan. 2018. [DOI] [PubMed] [Google Scholar]
- [19].Lehman CD et al. , “Diagnostic Accuracy of Digital Screening Mammography With and Without Computer-Aided Detection,” JAMA Internal Medicine, vol. 175, no. 11, pp. 1828–1837, Nov. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cole EB, Zhang Z, Marques HS, Edward Hendrick R, Yaffe MJ, and Pisano ED, “Impact of Computer-Aided Detection Systems on Radiologist Accuracy With Digital Mammography,” American Journal of Roentgenology, vol. 203, no. 4, pp. 909–916, Oct. 2014, publisher: American Roentgen Ray Society. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fenton JJ et al. , “Short-Term Outcomes of Screening Mammography Using Computer-Aided Detection,” Ann Intern Med, vol. 158, no. 8, pp. 580–587, Apr. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Freer TW and Ulissey MJ, “Screening Mammography with Computer-aided Detection: Prospective Study of 12,860 Patients in a Community Breast Center,” Radiology, vol. 220, no. 3, pp. 781–786, Sep. 2001, publisher: Radiological Society of North America. [DOI] [PubMed] [Google Scholar]
- [23].Schilsky RL, “Implementing personalized cancer care,” Nat Rev Clin Oncol, vol. 11, no. 7, pp. 432–438, Jul. 2014, publisher: Nature Publishing Group. [Online]. Available: https://www.nature.com/articles/nrclinonc.2014.54 [DOI] [PubMed] [Google Scholar]
- [24].Stover DG and Wagle N, “Precision Medicine in Breast Cancer: Genes, Genomes, and the Future of Genomically-Driven Treatments,” Curr Oncol Rep, vol. 17, no. 4, p. 15, Apr. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yamamoto S, Maki DD, Korn RL, and Kuo MD, “Radiogenomic analysis of breast cancer using MRI: a preliminary study to define the landscape,” AJR Am J Roentgenol, vol. 199, no. 3, pp. 654–663, Sep. 2012. [DOI] [PubMed] [Google Scholar]
- [26].Mazurowski MA, Zhang J, Grimm LJ, Yoon SC, and Silber JI, “Radiogenomic Analysis of Breast Cancer: Luminal B Molecular Subtype Is Associated with Enhancement Dynamics at MR Imaging,” Radiology, vol. 273, no. 2, pp. 365–372, Nov. 2014, publisher: Radiological Society of North America. [DOI] [PubMed] [Google Scholar]
- [27].Grimm LJ, Zhang J, and Mazurowski MA, “Computational approach to radiogenomics of breast cancer: Luminal A and luminal B molecular subtypes are associated with imaging features on routine breast MRI extracted using computer vision algorithms,” Journal of Magnetic Resonance Imaging, vol. 42, no. 4, pp. 902–907, 2015. [DOI] [PubMed] [Google Scholar]
- [28].Wu J et al. , “Identifying relations between imaging phenotypes and molecular subtypes of breast cancer: model discovery and external validation,” Journal of magnetic resonance imaging : JMRI, vol. 46, no. 4, p. 1017, Oct. 2017, publisher: NIH Public Access. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li H et al. , “Quantitative MRI radiomics in the prediction of molecular classifications of breast cancer subtypes in the TCGA/TCIA data set,” npj Breast Cancer, vol. 2, no. 1, pp. 1–10, May 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Castaldo R, Pane K, Nicolai E, Salvatore M, and Franzese M, “The Impact of Normalization Approaches to Automatically Detect Radiogenomic Phenotypes Characterizing Breast Cancer Receptors Status,” Cancers, vol. 12, no. 2, p. 518, Feb. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fan M et al. , “Joint Prediction of Breast Cancer Histological Grade and Ki-67 Expression Level Based on DCE-MRI and DWI Radiomics,” IEEE Journal of Biomedical and Health Informatics, vol. 24, no. 6, pp. 1632–1642, Jun. 2020. [DOI] [PubMed] [Google Scholar]
- [32].Chan H-P, Lo S-CB, Sahiner B, Lam KL, and Helvie MA, “Computer-aided detection of mammographic microcalcifications: Pattern recognition with an artificial neural network,” Medical Physics, vol. 22, no. 10, pp. 1555–1567, 1995. [DOI] [PubMed] [Google Scholar]
- [33].Krizhevsky A, Sutskever I, and Hinton GE, “ImageNet Classification with Deep Convolutional Neural Networks,” in Advances in Neural Information Processing Systems, vol. 25. Curran Associates, Inc., 2012. [Google Scholar]
- [34].Ronneberger O, Fischer P, and Brox T, “U-Net: Convolutional Networks for Biomedical Image Segmentation,” arXiv, Tech. Rep. arXiv:1505.04597, May 2015, arXiv:1505.04597 [cs] type: article. [Google Scholar]
- [35].Redmon J, Divvala S, Girshick R, and Farhadi A, “You Only Look Once: Unified, Real-Time Object Detection,” arXiv, Tech. Rep. arXiv:1506.02640, May 2016, arXiv:1506.02640 [cs] type: article. [Google Scholar]
- [36].Antropova N, Huynh BQ, and Giger ML, “A Deep Feature Fusion Methodology for Breast Cancer Diagnosis Demonstrated on Three Imaging Modality Datasets,” Med Phys, vol. 44, no. 10, pp. 5162–5171, Oct. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Verburg E et al. , “Deep Learning for Automated Triaging of 4581 Breast MRI Examinations from the DENSE Trial,” Radiology, vol. 302, no. 1, pp. 29–36, Jan. 2022, publisher: Radiological Society of North America. [DOI] [PubMed] [Google Scholar]
- [38].Guo W et al. , “Prediction of clinical phenotypes in invasive breast carcinomas from the integration of radiomics and genomics data,” Journal of Medical Imaging, vol. 2, no. 4, Oct. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cristovao F et al. , “Investigating Deep Learning based Breast Cancer Subtyping using Pan-cancer and Multi-omic Data,” IEEE/ACM Trans Comput Biol Bioinform, vol. PP, Dec. 2020. [DOI] [PubMed] [Google Scholar]
- [40].Byra M, Dobruch-Sobczak K, Klimonda Z, Piotrzkowska-Wroblewska H, and Litniewski J, “Early Prediction of Response to Neoadjuvant Chemotherapy in Breast Cancer Sonography Using Siamese Convolutional Neural Networks,” IEEE Journal of Biomedical and Health Informatics, vol. 25, no. 3, pp. 797–805, Mar. 2021. [DOI] [PubMed] [Google Scholar]
- [41].Sun D, Wang M, and Li A, “A multimodal deep neural network for human breast cancer prognosis prediction by integrating multi-dimensional data,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 16, no. 3, pp. 841–850, 2019. [DOI] [PubMed] [Google Scholar]
- [42].Arya N and Saha S, “Multi-modal classification for human breast cancer prognosis prediction: Proposal of deep-learning based stacked ensemble model,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 19, no. 2, pp. 1032–1041, 2022. [DOI] [PubMed] [Google Scholar]
- [43].Nguyen P, Tran T, Wickramasinghe N, and Venkatesh S, “Deepr: A convolutional net for medical records,” IEEE Journal of Biomedical and Health Informatics, vol. 21, no. 1, pp. 22–30, 2017. [DOI] [PubMed] [Google Scholar]
- [44].Gao J, Lyu T, Xiong F, Wang J, Ke W, and Li Z, “MGNN: A Multimodal Graph Neural Network for Predicting the Survival of Cancer Patients,” in Proceedings of the 43rd International ACM SIGIR Conference on Research and Development in Information Retrieval. New York, NY, USA: Association for Computing Machinery, Jul. 2020, pp. 1697–1700. [Online]. Available: 10.1145/3397271.3401214 [DOI] [Google Scholar]
- [45].Parisot S et al. , “Spectral Graph Convolutions for Population-Based Disease Prediction,” in Medical Image Computing and Computer Assisted Intervention MICCAI 2017, ser. Lecture Notes in Computer Science, Descoteaux M, Maier-Hein L, Franz A, Jannin P, Collins DL, and Duchesne S, Eds. Cham: Springer International Publishing, 2017, pp. 177–185. [Google Scholar]
- [46].Lingle W et al. , “Radiology Data from The Cancer Genome Atlas Breast Invasive Carcinoma [TCGA-BRCA] collection,” 2016, version Number: 3 Type: dataset. [Online]. Available: https://wiki.cancerimagingarchive.net/x/GQE2 [Google Scholar]
- [47].Grossman RL et al. , “Toward a shared vision for cancer genomic data,” New England Journal of Medicine, vol. 375, no. 12, pp. 1109–1112, 2016, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Clark K et al. , “The Cancer Imaging Archive (TCIA): Maintaining and Operating a Public Information Repository,” J Digit Imaging, vol. 26, no. 6, pp. 1045–1057, Dec. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].The Cancer Genome Atlas Network, “Comprehensive molecular portraits of human breast tumours,” Nature, vol. 490, no. 7418, pp. 61–70, Oct. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Newitt DC et al. , “Acrin 6698/i-spy2 breast dwi,” 2021.
- [51].Hu Z et al. , “Predicting breast cancer response to neoadjuvant treatment using multi-feature mri: results from the i-spy 2 trial,” npj Breast Cancer, vol. 6, no. 1, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li W et al. , “I-spy 2 breast dynamic contrast enhanced mri (i-spy2 trial) (version 1),” 2022.
- [53].The AACR Project GENIE Consortium, “AACR Project GENIE: Powering Precision Medicine through an International Consortium,” Cancer Discovery, vol. 7, no. 8, pp. 818–831, Aug. 2017. [Online]. Available: 10.1158/2159-8290.CD-17-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kipf TN and Welling M, “Semi-Supervised Classification with Graph Convolutional Networks,” arXiv:1609.02907 [cs, stat], Feb. 2017, arXiv: 1609.02907. [Google Scholar]
- [55].Schlichtkrull M, Kipf TN, Bloem P, Berg R. v. d., Titov I, and Welling M, “Modeling Relational Data with Graph Convolutional Networks,” arXiv:1703.06103 [cs, stat], Oct. 2017, arXiv: 1703.06103. [Google Scholar]
- [56].International H, “HUGO Nomenclature Standards.” [Online]. Available: https://www.hugo-international.org/standards/
- [57].Garcia EP et al. , “Validation of OncoPanel: A Targeted Next-Generation Sequencing Assay for the Detection of Somatic Variants in Cancer,” Archives of Pathology & Laboratory Medicine, vol. 141, no. 6, pp. 751–758, Mar. 2017. [Online]. Available: 10.5858/arpa.2016-0527-OA [DOI] [PubMed] [Google Scholar]
- [58].Cheng DT et al. , “Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT),” J Mol Diagn, vol. 17, no. 3, pp. 251–264, May 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wagner AH et al. , “A harmonized meta-knowledgebase of clinical interpretations of somatic genomic variants in cancer,” Nat Genet, vol. 52, no. 4, pp. 448–457, Apr. 2020, publisher: Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tan M and Le QV, “EfficientNet: Rethinking Model Scaling for Convolutional Neural Networks,” arXiv:1905.11946 [cs, stat], Nov. 2019, arXiv: 1905.11946. [Google Scholar]
- [61].Sandler M, Howard A, Zhu M, Zhmoginov A, and Chen L-C, “Mobilenetv2: Inverted residuals and linear bottlenecks,” in 2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition, 2018, pp. 4510–4520. [Google Scholar]
- [62].Hu J, Shen L, and Sun G, “Squeeze-and-excitation networks,” in 2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition, 2018, pp. 7132–7141. [Google Scholar]
- [63].Pizer S, Johnston R, Ericksen J, Yankaskas B, and Muller K, “Contrast-limited adaptive histogram equalization: speed and effectiveness,” in [1990] Proceedings of the First Conference on Visualization in Biomedical Computing, 1990, pp. 337–345. [Google Scholar]
- [64].Srivastava N, Hinton GE, Krizhevsky A, Sutskever I, and Salakhutdinov R, “Dropout: a simple way to prevent neural networks from overfitting,” The journal of machine learning research, vol. 15, no. 1, pp. 1929–1958, 2014. [Google Scholar]
- [65].LeCun YA, Bottou L, Orr GB, and Müller K-R, “Efficient backprop,” Neural networks: Tricks of the trade: Second edition, pp. 9–48, 2012. [Google Scholar]
- [66].Saito T and Rehmsmeier M, “The precision-recall plot is more informative than the roc plot when evaluating binary classifiers on imbalanced datasets,” PLoS one, vol. 10, no. 3, p. e0118432, 2015. [Online]. Available: 10.1371/journal.pone.0118432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Maaten L. v. d. and Hinton G, “Visualizing Data using t-SNE,” Journal of Machine Learning Research, vol. 9, no. 86, pp. 2579–2605, 2008. [Online]. Available: http://jmlr.org/papers/v9/vandermaaten08a.html [Google Scholar]