

Abstract
Objectives
Neurodegeneration is considered a relevant pathophysiologic feature in neurologic disorders associated with antibodies against glutamic acid decarboxylase 65 (GAD65). In this study, we investigate surrogates of neuroaxonal damage in relation to disease duration and clinical presentation.
Methods
In a multicentric cohort of 50 patients, we measured serum neurofilament light chain (sNfL) in relation to disease duration and disease phenotypes, applied automated MRI volumetry, and analyzed clinical characteristics.
Results
In patients with neurologic disorders associated with GAD65 antibodies, we detected elevated sNfL levels early in the disease course. By contrast, this elevation of sNfL levels was less pronounced in patients with long-standing disease. Increased sNfL levels were observed in patients presenting with cerebellar ataxia and limbic encephalitis, but not in those with stiff person syndrome. Using MRI volumetry, we identified atrophy predominantly of the cerebellar cortex, cerebellar superior posterior lobe, and cerebral cortex with similar atrophy patterns throughout all clinical phenotypes.
Discussion
Together, our data provide evidence for early neuroaxonal damage and support the need for timely therapeutic interventions in GAD65 antibody-associated neurologic disorders.
Introduction
Antibodies (Abs) against glutamic acid decarboxylase 65 (GAD65) are associated with different neurologic disorders, including stiff person syndrome (SPS), cerebellar ataxia (CA), (limbic) encephalitis (LE), and overlap syndromes,1 together referred to as GAD65-Ab spectrum disorders (GAD65-Ab-SD). Neuroinflammation is suggested as relevant pathophysiologic feature at early disease stages,2-4 whereas later disease stages clinically seem reminiscent to classical neurodegenerative disorders. Currently, there are only insufficient data regarding the timing of most pronounced neuroaxonal damage throughout the disease course.
Neurofilament light chain (NfL) is a marker of neuroaxonal damage5 with elevated serum levels being observed in several neurologic conditions including other forms of autoimmune encephalitis6,7 and multiple sclerosis (MS), where they seem to reflect the rate of tissue degeneration.8
Here, we investigated surrogates of neurodegeneration in a multicentric cohort of 50 GAD65-Ab-SD patients by analyzing serum (s)NfL levels, GAD65-Ab-levels, MRI volumetric data, and relevant clinical information.
Methods
Study Population
Data were collected from the GENERATE registry (generate-net.de). Eleven GENERATE centers participated in the study. Inclusion criteria were (1) presentation with a typical GAD65-Ab–associated neurologic syndrome without evidence of coexisting antineuronal Abs; (2) evidence for intrathecal GAD65-Ab production; and (3) availability of a serum sample and relevant clinical data. Intrathecal GAD65-Ab production was defined by a specific antibody index of >4 (cell-based assay [CBA], indirect immunofluorescence testing [IIFT]) or >1.4 (radioimmunoassay [RIA], ELISA) for patients with CA and LE; patients with SPS were included if GAD65-Ab–positive in serum and CSF in CBA, IIFT, RIA, or ELISA. In addition, patients with available MRI data ±6 months from the date of serum sampling were included as a subcohort. Figure, A summarizes the flow chart of inclusions and exclusions. As control cohorts, we recruited age-matched and sex-matched healthy participants at the Institute of Clinical Neuroimmunology (LMU Munich, Germany) for the analysis of serum parameters (GAD65-Abs, sNfL) and at the German Center for Neurodegenerative Diseases (DZNE, Bonn, Germany) for the MRI study.
Figure. Analysis of Serum Biomarkers Reveals Elevated Levels of sNfL Early in the Disease Course.
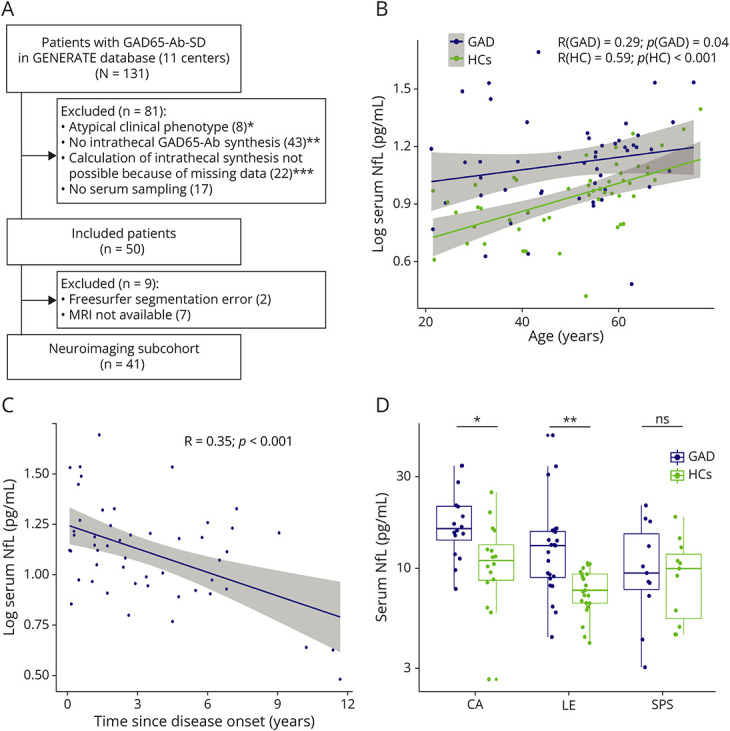
(A) Flowchart of the study cohort; * 2 patients additionally showed no intrathecal synthesis of GAD65-Abs, and 4 other patients had missing parameters to calculate the latter; ** criteria for intrathecal GAD65-Ab synthesis—CA, LE: GAD65 antibody index >4 (CBA, IIFT), >1.4 (RIA, ELISA); SPS: GAD65-Ab positive in serum and CSF in CBA, IIFT, RIA, or ELISA; in 2 patients additionally, no serum was sampled; *** in one patient additionally no serum was sampled. (B) Association of sNfL levels and age in patients with GAD65-Ab-SD and age-matched and sex-matched HCs. (C) Association of sNfL levels with disease duration in GAD65-Ab-SD patients. Both (B) and (C) show a linear regression model adjusted for age and sex with 95% confidence intervals depicted in gray. Spearman's R is given. The Shapiro-Wilk test was applied to test for normality and was significant; thus, log transformation (base 10) was used to achieve normal distribution in (B) and (C). (D) sNfL levels were compared in patients with different clinical phenotypes (CA, LE, and SPS) to age-matched and sex-matched controls by the Kruskal-Wallis test followed by Dunn's multiple comparisons test. CA = cerebellar ataxia; HC = healthy controls; LE = limbic encephalitis; NfL = neurofilament light chain; SPS = stiff person syndrome.
Standard Protocol Approvals, Registrations, and Patient Consents
GENERATE was approved by the institutional review boards of all participating centers and all patients or their legal representatives as well as healthy controls gave written informed consent.
Serum Analysis
sNfL was measured in duplicates using the Simoa NF‐light Advantage kit (Quanterix, #103186). Serum GAD65-Abs were remeasured centrally and quantified by human anti-GAD65 ELISA (IgG) (Euroimmun). All measurements were performed according to the manufacturer's instructions.
MRI Volumetry and Analysis
T1w MR images of each participant were processed with a fully automated image-processing pipeline using FreeSurfer (version 6.0) and CerebNet,9-11 which is further delineated in the eMethods (links.lww.com/NXI/A927).
Statistics
Statistical analyses were performed with R software (version 4.2.3) or GraphPad Prism (version 9.0.2) and are described in the eMethods (links.lww.com/NXI/A927).
Data Availability
Data are available to qualified researchers based on reasonable request.
Results
Fifty GAD65-Ab-SD patients were included—CA: n = 16, LE: n = 23, and SPS: n = 11 (Figure, A). 14% of patients showed an overlap phenotype and were categorized according to the predominant clinical phenotype. Clinical characteristics of the study cohort are specified in Table 1. Patients with CA were oldest at the time of sampling (median (IQR))—CA: 62 (55–67), SPS: 53 (41–57), LE: 41 (31–55) years). Serum GAD65-Ab levels were high in our patient cohort and very low in HCs (median (IQR))—GAD: 15 × 104 (5 × 104–18 × 104), HC: 1 (0–2) IU/mL). sNfL levels were significantly higher in patients with GAD65-Ab-SD than in HCs (median (IQR))—GAD: 13 (9–17), HC: 9 (7–11) pg/mL) (Table 1). The increase of sNfL with age was less pronounced in patients with GAD65-Ab-SD (R = 0.29, p = 0.04; in comparison, R = 0.59, p < 0.001 in HC; Figure, B). sNfL levels in GAD65-Ab-SD were highest at disease onset and lower with increasing disease duration (R = 0.35, p < 0.001; Figure, C). When looking at the different GAD65-Ab disease phenotypes, sNfL levels were elevated in patients with CA and LE, but not in patients with SPS (Figure, D). Notably, patients with SPS exhibited the longest disease duration at the time of sampling (median (IQR))—SPS: 6 (2–7) vs CA: 3 (1–6); LE: 2 (1–4) years) (Table 1).
Table 1.
Characterization of the Study Cohort

| n = 100 | Patients (n = 50) | CA (n = 16)a | LE (n = 23)b | SPS (n = 11) | Controls (n = 50) | p-valuec |
| Female, n (%) | 38 (76) | 13 (81) | 17 (74) | 8 (73) | 38 (76) | — |
| Age at onset, median (IQR), y | 51 (35–56) | 60 (53–63) | 39 (28–53) | 46 (39–51) | NA | NA |
| Age at assessment, median (IQR), y | 54 (37–61) | 62 (55–67) | 41 (31–55) | 53 (41–57) | 54 (39–61) | 0.85 |
| Assessment after onset, median (IQR), y | 3 (1–6) | 3 (1–6) | 2 (1–4) | 6 (2–7) | NA | NA |
| Overlap syndrome, n (%) | 7 (14) | 5 (31) | 2 (9) | 0 (0) | NA | NA |
| Tumord, n (%) | 5 (10) | 3 (19) | 0 (0) | 2 (18) | NA | NA |
| Other autoimmune disease diagnosed, n (%) | NA | NA | ||||
| All | 32 (64) | 13 (81) | 13 (57) | 6 (55) | NA | NA |
| Type 1 diabetes | 15 (30) | 7 (44) | 6 (26) | 2 (18) | ||
| GAD65 AI at diagnosis, median (IQR)e | 39 (9–130) | 116 (38–143) | 32 (12–190) | 9 (4–47) | NA | NA |
| CSF-specific OCB at diagnosise, n (%) | 29 (58) | 6 (38) | 16 (70) | 7 (64) | NA | NA |
| CSF cell count at diagnosis, median (IQR)e, cells/μL | 2 (1–4) | 2 (0.3–4) | 3 (1–5) | 2 (2–3) | NA | NA |
| mRS at assessment, median (IQR) | 2 (2–3) | 2 (2–3) | 2 (1–3) | 2 (2–3) | NA | NA |
| mRS at assessment >2, n (%) | 15 (30) | 5 (31) | 6 (26) | 4 (36) | NA | NA |
| Ever received IT before assessment, n (%) | 48 (96) | 16 (100) | 22 (96) | 10 (91) | NA | NA |
| Time between onset and first IT, median (IQR), mo | 11 (4–30) | 10 (5–29) | 10 (2–23) | 26 (7–92) | NA | NA |
| Received IT before assessment, n (%) | ||||||
| Azathioprine | 9 (18) | 4 (25) | 4 (17) | 1 (9) | NA | NA |
| Corticosteroids | 38 (76) | 14 (88) | 18 (78) | 6 (55) | NA | NA |
| Cyclophosphamide | 4 (8) | 1 (6) | 2 (9) | 1 (9) | NA | NA |
| IVIG | 16 (32) | 6 [38) | 4 (17) | 6 (55) | NA | NA |
| MMF | 4 (8) | 3 (19) | 1 (4) | 0 | NA | NA |
| MTX | 2 (4) | 1 (6) | 1 (4) | 0 | NA | NA |
| PLEX/IA | 19 (38) | 7 (44) | 9 (39) | 3 (27) | NA | NA |
| Rituximab | 16 (32) | 5 (31) | 8 (35) | 3 (27) | NA | NA |
| Serum GAD65 Ab at assessment, median (IQR), IU/mL | 15 × 104 (5 × 104–18 × 104) | 13 × 104 (4 × 104–35 × 104) | 11 × 104 (1 × 104–17 × 104) | 17 × 104 (15 × 104–20 × 104) | 1 (0–2) | <0.0001 |
| Serum NfL at assessment, median (IQR), pg/mL | 13 (9–17) | 16 (13–21) | 13 (9–16) | 9 (7–18) | 9 (7–11) | <0.0001 |
Abbreviations: AI = antibody index; CA = cerebellar ataxia; IA = immunoadsorption; IQR = interquartile range; IT = immunotherapy; IVIG = IV immunoglobulines; mRS = modified Rankin Scale; MMF = mycophenolate mofetil; MTX = methotrexate; NfL = neurofilament light chain; OCB = oligoclonal bands; PLEX = plasma exchange; SPS = stiff person syndrome.
One patient with CA and LE overlap syndrome developed hippocampal sclerosis.
Twelve patients presented with temporal-lobe epilepsy; no LE patient exhibited hippocampal sclerosis at the time of assessment.
Comparisons of all GAD patients and all controls. Statistics: Continuous variables were compared by the Wilcoxon test. Ordinal variables were compared by Fisher's exact test.
2x breast cancer (10 y before and 12 y after onset of disease), 1x AML (9 y after onset of disease), 1x CLL (1 y before onset of disease), 1x ovarian teratoma (10 y before onset of disease).
If not available at diagnosis, data from first spinal tap at study site.
To identify brain regions with most pronounced neuronal loss, we applied automated MRI volumetry, including a cerebellar subsegmentation, in a subcohort of 41 patients. Here, we observed cerebellar atrophy with predominance of the cerebellar gray matter (median (IQR)) [relative to estimated total intracranial volume]—GAD: 6.1 (4.5–6.8)% and HC: 6.9 (6.1–7.2)% as well as superior posterior lobe—GAD: 3.8 (2.6–4.4)% and HC: 4.4 (0.39–0.46)%. In addition, we found a reduction of cerebral cortical volume in GAD65-Ab-SD patients—GAD: 26.7 (19.6–30.6)% and HC: 29.3 (28.6–30.1)%. Throughout different clinical phenotypes, similar reductions of cerebellar volumes and cerebral cortex volume were observed with an elevation of hippocampal volume only in patients with LE (Table 2).
Table 2.
Brain Segmentation Including Cerebellar Subsegmentation

| Metric | All GAD (n = 41) mediana (IQR), % | All HC (n = 41) mediana (IQR), % | p b | CA GAD (n = 13) mediana (IQR), % | CA HC (n = 13) mediana (IQR), % | p b | LE GAD (n = 20) mediana (IQR), % | LE HC (n = 20) mediana (IQR), % | p b | SPS GAD (n = 8) mediana (IQR), % | SPS HC (n = 8) mediana (IQR), % | p b |
| Brain segmentation | ||||||||||||
| Cortex | 26.7 (19.6–30.6) | 29.3 (28.6–30.1) | 0.011 | 27.8 (21.2–31.0) | 29.2 (27.0–30.1) | 3.436 | 27.4 (19.5–31.3) | 29.4 (28.7–30.2) | 1.366 | 21.8 (16.3–28.8) | 29.4 (28.7–29.9) | 0.704 |
| Amygdala | 0.2 (0.2–0.2) | 0.2 (0.2–0.2) | 0.238 | 0.2 (0.2–0.2) | 0.2 (0.2–0.2) | 13.237 | 0.2 (0.2–0.2) | 0.2 (0.2–0.2) | 0.149 | 0.2 (0.2–0.2) | 0.2 (0.2–0.2) | 6.890 |
| Hippocampus | 0.6 (0.5–0.6) | 0.5 (0.4–0.5) | 0.016 | 0.5 (0.5–0.6) | 0.5 (0.4–0.5) | 2.290 | 0.6 (0.5–0.6) | 0.5 (0.5–0.5) | 0.022 | 0.6 (0.5–0.6) | 0.5 (0.5–0.6) | 3.515 |
| Cerebellar subsegmentation | ||||||||||||
| Cerebellar gray matter | 6.1 (4.5–6.8) | 6.9 (6.1–7.2) | 0.005 | 6.0 (5.0–6.4) | 6.2 (5.8–6.2) | 5.497 | 6.4 (4.6–7.0) | 7.0 (6.9–7.3) | 0.275 | 4.9 (1.8–7.0) | 7.2 (6.0–7.5) | 0.421 |
| Anterior lobec | 0.8 (0.7–0.9) | 0.9 (0.8–1.0) | 0.059 | 0.8 (0.8–1.0) | 0.9 (0.8–0.9) | 8.951 | 0.8 (0.8–0.9) | 0.9 (0.9–1.0) | 0.688 | 0.7 (0.6–0.9) | 0.9 (0.8–1.0) | 0.140 |
| Superior posterior lobed | 3.8 (2.6–4.4) | 4.4 (0.4–0.5) | 0.007 | 3.6 (2.9–4.1) | 3.9 (3.7–4.0) | 6.113 | 4.1 (2.6–4.7) | 4.5 (4.3–4.6) | 0.590 | 3.2 (1.0–4.4) | 4.5 (3.9–4.8) | 0.421 |
| Inferior posterior lobee | 1.3 (1.0–1.4) | 1.4 (1.3–1.5) | 0.081 | 1.4 (1.2–1.5) | 1.3 (1.2–1.4) | 6.763 | 1.3 (1.0–1.4) | 1.5 (1.4–1.6) | 5.328 | 0.9 (0.3–1.6) | 1.4 (1.3–1.5) | 1.969 |
| Lobule I-IV | 0.4 (0.4–0.5) | 0.5 (0.4–0.5) | 0.029 | 0.4 (0.4–0.5) | 0.4 (0.4–0.5) | 9.751 | 0.4 (0.4–0.5) | 0.5 (0.4–0.5) | 0.245 | 0.3 (0.3–0.4) | 0.5 (0.4–0.5) | 0.281 |
| Lobule V | 0.4 (0.4–0.5) | 0.4 (0.4–0.5) | 0.238 | 0.4 (0.4–0.5) | 0.4 (0.4–0.5) | 10.582 | 0.4 (0.4–0.5) | 0.5 (0.4–0.5) | 1.157 | 0.4 (0.2–0.5) | 0.5 (0.4–0.5) | 0.281 |
| Lobule VI | 0.9 (0.7–1.1) | 1.0 (0.9–1.1) | 0.493 | 0.9 (0.8–1.0) | 0.9 (0.8–1.0) | 17.998 | 0.9 (0.8–1.1) | 1.1 (1.0–1.1) | 1.615 | 0.9 (0.3–1.1) | 1.0 (0.9–1.1) | 1.969 |
| Crus I | 1.5 (1.2–1.7) | 1.5 (1.4–1.7) | 0.400 | 1.4 (1.3–1.6) | 1.5 (1.3–1.5) | 15.109 | 1.6 (1.0–1.8) | 1.6 (1.5–1.7) | 8.548 | 1.3 (0.4–1.6) | 1.6 (1.4–1.7) | 0.985 |
| Crus II | 0.9 (0.5–1.1) | 1.1 (1.0–1.3) | 0.002 | 0.8 (0.5–0.9) | 1.0 (0.9–1.1) | 0.146 | 1.0 (0.6–1.2) | 1.2 (1.1–1.3) | 0.193 | 0.7 (0.2–1.0) | 1.2 (1.0–1.4) | 0.140 |
| Lobule VIIb | 0.5 (0.2–0.6) | 0.6 (0.5–0.7) | 0.002 | 0.5 (0.3–0.6) | 0.5 (0.5–0.6) | 4.921 | 0.5 (0.3–0.6) | 0.7 (0.6–0.7) | 0.067 | 0.3 (0.1–0.6) | 0.6 (0.6–0.7) | 0.704 |
| Lobule VIIIa | 0.5 (0.3–0.6) | 0.6 (0.6–0.6) | 0.011 | 0.6 (0.5–0.6) | 0.6 (0.5–0.6) | 13.237 | 0.5 (0.3–0.6) | 0.6 (0.6–0.7) | 0.218 | 0.3 (0.1–0.7) | 0.6 (0.5–0.6) | 1.969 |
| Lobule VIIIb | 0.4 (0.3–0.5) | 0.4 (0.4–0.5) | 0.567 | 0.5 (0.4–0.5) | 0.4 (0.4–0.5) | 13.237 | 0.4 (0.3–0.5) | 0.5 (0.4–0.5) | 6.278 | 0.3 (0.1–0.5) | 0.5 (0.4–0.5) | 0.985 |
| Lobule IX | 0.4 (0.3–0.4) | 0.4 (0.3–0.4) | 1.485 | 0.4 (0.3–0.4) | 0.3 (0.3–0.4) | 8.951 | 0.4 (0.3–0.4) | 0.4 (0.4–0.4) | 1.751 | 0.3 (0.2–0.4) | 0.4 (0.4–0.4) | 1.969 |
| Lobule X | 0.07 (0.06–0.08) | 0.08 (0.07–0.08) | 0.839 | 0.07 (0.05–0.09) | 0.07 (0.07–0.08) | 15.109 | 0.08 (0.07–0.08) | 0.08 (0.07–0.09) | 4.149 | 0.06 (0.03–0.08) | 0.08 (0.08–0.09) | 0.140 |
| Vermis | 0.3 (0.3–0.3) | 0.3 (0.3–0.4) | 0.045 | 0.3 (0.3–0.3) | 0.3 (0.3–0.4) | 3.019 | 0.3 (0.3–0.3) | 0.3 (0.3–0.4) | 1.253 | 0.3 (0.2–0.4) | 0.4 (0.3–0.4) | 0.985 |
Abbreviations: CA = cerebellar ataxia; HC = healthy controls; IQR = interquartile range; LE = limbic encephalitis; SPS = stiff person syndrome.
Values of each participant were normalized to the individual estimated total intracranial volume (eTIV) and shown as percentage of eTIV.
Bonferroni-corrected p-values are depicted.
Compound volume: cerebellar lobules I–V.
Compound volume: cerebellar lobules VI, VIIA [crus I, crus II], and VIIB.
Compound volume cerebellar lobules VIIIA, VIIIB, and IX. HCs are age-matched and sex-matched. Statistics: Variables were compared by the Wilcoxon test.
Correlations of clinical, serologic, and imaging findings revealed strong correlation between cerebellar gray matter and cerebral cortex volume (eFigure 1, links.lww.com/NXI/A927). sNfL levels and GAD65-Ab levels showed a negative, however nonsignificant, correlation with cerebellar and cerebral volumes. Disease duration until an immunotherapy was initiated negatively correlated with volumetric parameters (nonsignificant) (eFigure 1).
Discussion
We performed a multimodal analysis of surrogates for neuroaxonal damage in a multicentric cohort of 50 GAD65-Ab-SD patients and report several important aspects: (1) sNfL levels are increased in patients with GAD65-Ab-SD particularly in patients presenting with CA and LE; (2) elevation of sNfL levels is pronounced in the early phase of GAD65-Ab-SD; and (3) volume loss in GAD65-Ab-SD predominantly affects the cerebellar and cerebral cortex. Similar atrophy patterns are found throughout all disease phenotypes.
The observed association of sNfL levels with disease duration argues for at least a relevant proportion of neuroaxonal damage occurring already early in the disease course. In line with this, recent neuropathologic evidence suggests that the disease is initially dominated by acute neuroinflammation, characterized by lymphocyte infiltration with a predominance of CD8+ T cells and evidence of T-cell–mediated neuronal destruction.2 Despite their still unsolved relevance, the presence of plasma cells in the brain and GAD65-Ab producing B cells in the CSF is also more pronounced early in the disease.2,3 In contrast to this initial phase, where immunotherapy shows at least some effect,4,12 later disease stages remain largely refractory to therapeutic interventions.13
The observed influence of timing of sampling on sNfL levels limits the value of sNfL as a biomarker to predict clinical outcome. Similarly, in patients with anti–N-methyl-d-aspartate receptor encephalitis, an impact of sampling time on sNfL levels was proposed.6 However, early measurement of sNfL levels could potentially assist in patient stratification to identify those who would particularly benefit from prompt and intensive immunosuppressive treatment. The undetectable increase of sNfL levels in patients with SPS can be explained by the long disease duration at the time of sampling in this patient group. Alternatively, a rather reversible deficit in patients with SPS vs irreversible neuronal injury in other phenotypes may be discussed.
As described previously, we found similar atrophy patterns throughout all disease phenotypes with atrophy of cerebellar and cortical volumes14 and elevated hippocampal volume in patients with LE.15 Strikingly, cerebellar degeneration is even observed in patients without clinically evident cerebellar ataxia. These findings support the concept of GAD65-Ab–associated neurologic syndromes as a continuous disease spectrum, also reflected by the high rate of overlap phenotypes.
Limitations of our study include the limited patient number because of the rarity of the disease and the strict inclusion criteria, the cross-sectional and retrospective study design, the multicentric recruitment, the lack of a standardized protocol for MRI acquisition, and multiple comparisons in the MRI study.
To conclude, our study provides insights into sNfL dynamics throughout the disease course of GAD65-Ab-SD patients. Imaging data reveal a comparable atrophy profile throughout all clinical phenotypes. We provide evidence for early neuroaxonal damage in GAD65-Ab-SD. This finding has important clinical implications and underlines the need for timely initiation of effective therapy.
Acknowledgment
The authors thank Prof. Christian Haass for providing us the infrastructure for the NfL measurements in his department.
Appendix 1. Authors

| Name | Location | Contribution |
| Katharina Eisenhut, MD | Institute of Clinical Neuroimmunology, University Hospital, Ludwig-Maximilians-Universität Munich; Biomedical Center (BMC), Medical Faculty, Ludwig-Maximilians-Universität Munich, Martinsried, Germany; Graduate School of Sy | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Jennifer Faber, MD | German Center for Neurodegenerative Diseases (DZNE); Department of Neurology, University Hospital Bonn, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data |
| Daniel Engels, MD, PhD | Institute of Clinical Neuroimmunology, University Hospital, Ludwig-Maximilians-Universität Munich; Biomedical Center (BMC), Medical Faculty, Ludwig-Maximilians-Universität Munich, Martinsried, Germany | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Ramona Gerhards, MD, PhD | Institute of Clinical Neuroimmunology, University Hospital, Ludwig-Maximilians-Universität Munich; Biomedical Center (BMC), Medical Faculty, Ludwig-Maximilians-Universität Munich, Martinsried, Germany | Major role in the acquisition of data |
| Jan Lewerenz, MD | Department of Neurology, Ulm University, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Kathrin Doppler, MD | Department of Neurology, University Hospital Würzburg, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Claudia Sommer, MD | Department of Neurology, University Hospital Würzburg, Germany | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Robert Markewitz, MD | Institute of Clinical Chemistry, University Hospital Schleswig-Holstein, Lübeck, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Kim K. Falk, MD | Institute of Clinical Chemistry, University Hospital Schleswig-Holstein, Kiel, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Rosa Rössling, MD | Department of Neurology and Experimental Neurology, Charité - Universitätsmedizin Berlin; German Center for Neurodegenerative Diseases (DZNE) Berlin, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Harald Pruess, MD | Department of Neurology and Experimental Neurology, Charité - Universitätsmedizin Berlin; German Center for Neurodegenerative Diseases (DZNE) Berlin, Germany | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Carsten Finke, MD | Department of Neurology and Experimental Neurology, Charité - Universitätsmedizin Berlin, Germany | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Jonathan Wickel, MD | Section of Translational Neuroimmunology, Department of Neurology, Jena University Hospital, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Christian Geis, MD | Section of Translational Neuroimmunology, Department of Neurology, Jena University Hospital, Germany | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Dominica Ratuszny, MD | Department of Neurology, Hannover Medical School, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Lena K. Pfeffer, MD | Institute of Neuroimmunology and Multiple Sclerosis, Center for Molecular Neurobiology Hamburg, University Medical Center Hamburg-Eppendorf, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Stefan Bittner, MD | Department of Neurology, University Medical Center of the Johannes Gutenberg University Mainz, Germany | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Johannes Piepgras, MD | Department of Neurology, University Medical Center of the Johannes Gutenberg University Mainz, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Andrea Kraft, MD | Department of Neurology, Martha-Maria Hospital Halle, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Jaqueline Klausewitz, MD | Department of Neurology, University Hospital Bochum, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Brigitte Nuscher | German Center for Neurodegenerative Diseases (DZNE), Munich; Metabolic Biochemistry, Faculty of Medicine, Biomedical Center (BMC), Ludwig-Maximilians-Universität München, Munich, Germany | Major role in the acquisition of data |
| Tania Kümpfel, MD | Institute of Clinical Neuroimmunology, University Hospital, Ludwig-Maximilians-Universität Munich; Biomedical Center (BMC), Medical Faculty, Ludwig-Maximilians-Universität Munich, Martinsried, Germany | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design; analysis or interpretation of data |
| Franziska S. Thaler, MD | Institute of Clinical Neuroimmunology, University Hospital, Ludwig-Maximilians-Universität Munich; Biomedical Center (BMC), Medical Faculty, Ludwig-Maximilians-Universität Munich, Martinsried, Germany; Munich Cluster for Sys | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
Appendix 2. Coinvestigators

| Coinvestigators are listed at links.lww.com/NXI/A926. |
Study Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy ID 390857198) and Project ID 408885537 - TRR 274, the Gemeinnützige Hertie Stiftung, LMUexcellent, and the German Federal Ministry of Education and Research (BMBF) through a grant Forschungsverbund CONNECT-GENERATE (01GM1908 and 01GM2208).
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NN for full disclosures.
References
- 1.Budhram A, Sechi E, Flanagan EP, et al. . Clinical spectrum of high-titre GAD65 antibodies. J Neurol Neurosurg Psychiatry. 2021;92(6):645-654. doi: 10.1136/jnnp-2020-325275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Troscher AR, Mair KM, Verdú de Juan L, et al. . Temporal lobe epilepsy with GAD antibodies: neurons killed by T cells not by complement membrane attack complex. Brain. 2022;146(4):1436-1452. doi: 10.1093/brain/awac404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biljecki M, Eisenhut K, Beltran E, et al. . Antibodies against glutamic acid decarboxylase 65 are locally produced in the csf and arise during affinity maturation. Neurol Neuroimmunol Neuroinflamm. 2023;10(3):e200090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arino H, Gresa-Arribas N, Blanco Y, et al. . Cerebellar ataxia and glutamic acid decarboxylase antibodies: immunologic profile and long-term effect of immunotherapy. JAMA Neurol. 2014;71(8):1009-1016. doi: 10.1001/jamaneurol.2014.1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zetterberg H, Blennow K. Fluid biomarkers for mild traumatic brain injury and related conditions. Nat Rev Neurol. 2016;12(10):563-574. doi: 10.1038/nrneurol.2016.127 [DOI] [PubMed] [Google Scholar]
- 6.Brenner J, Mariotto S, Bastiaansen AEM, et al. . Predictive value of serum neurofilament light chain levels in anti-NMDA receptor encephalitis. Neurology. 2023;100(21):e2204-e2213. doi: 10.1212/WNL.0000000000207221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Day GS, Yarbrough MY, Kortvelyessy P, et al. . Prospective quantification of CSF biomarkers in antibody-mediated encephalitis. Neurology. 2021;96(20):e2546-e2557. doi: 10.1212/WNL.0000000000011937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuhle J, Kropshofer H, Haering DA, et al. . Blood neurofilament light chain as a biomarker of MS disease activity and treatment response. Neurology. 2019;92(10):e1007-e1015. doi: 10.1212/WNL.0000000000007032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faber J, Schaprian T, Berkan K, et al. . Regional brain and spinal cord volume loss in spinocerebellar ataxia type 3. Mov Disord. 2021;36(10):2273-2281. doi: 10.1002/mds.28610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buckner RL, Head D, Parker J, et al. . A unified approach for morphometric and functional data analysis in young, old, and demented adults using automated atlas-based head size normalization: reliability and validation against manual measurement of total intracranial volume. Neuroimage. 2004;23(2):724-738. doi: 10.1016/j.neuroimage.2004.06.018 [DOI] [PubMed] [Google Scholar]
- 11.Faber J, Kugler D, Bahrami E, et al. . CerebNet: a fast and reliable deep-learning pipeline for detailed cerebellum sub-segmentation. Neuroimage. 2022;264:119703. doi: 10.1016/j.neuroimage.2022.119703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalakas MC, Fujii M, Li M, Lutfi B, Kyhos J, McElroy B. High-dose intravenous immune globulin for stiff-person syndrome. N Engl J Med. 2001;345(26):1870-1876. doi: 10.1056/NEJMoa01167 [DOI] [PubMed] [Google Scholar]
- 13.Thaler FS, Zimmermann L, Kammermeier S, et al. . Rituximab treatment and long-term outcome of patients with autoimmune encephalitis: real-world evidence from the GENERATE registry. Neurol Neuroimmunol Neuroinflamm. 2021;8(6):e1088. doi: 10.1212/NXI.0000000000001088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dade M, Giry M, Berzero G, et al. . Quantitative brain imaging analysis of neurological syndromes associated with anti-GAD antibodies. Neuroimage Clin. 2021;32:102826. doi: 10.1016/j.nicl.2021.102826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harms A, Bauer T, Witt JA, et al. . Mesiotemporal volumetry, cortical thickness, and neuropsychological deficits in the long-term course of limbic encephalitis. Neurol Neuroimmunol Neuroinflamm. 2023;10(4):e200125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available to qualified researchers based on reasonable request.