

To the Editor:
Multiple myeloma (MM) is one of the most common hematological malignancies, accounting for 20% of all newly diagnosed hematological cancers [1]. The most recent data from Cancer Today show that in 2020 the number of new MM cases was 176,404 worldwide (https://gco.iarc.fr/today/home).
Established risk factors for MM include age, male sex, African ancestry, obesity, chronic inflammation, exposure to pesticides, organic solvents, and radiation [2]. Familial aggregation of MM and its precursor monoclonal gammopathy of undetermined significance (MGUS) suggests that genetic factors play a role in risk of MM as well [3]. Genetic variability has been identified as a risk factor for MM, including 25 common genetic loci identified in genome-wide association studies (GWAS). However, estimates of heritability show that many more loci remain to be found [4].
A key question is therefore how to find new causative variants. The stringent significance threshold usually used in GWAS (p < 5 × 10−8) accounts for the many statistical tests being performed but may result in false negatives. Reducing the number of tests will relax the required significance threshold, thereby increasing statistical power to detect associations with MM risk for each SNP. One strategy for reducing the number of tests is to examine SNPs with higher prior probability of association according to meaningful biological criteria. We looked for novel MM risk loci using a two-phase large-scale association study, prioritizing polymorphisms with predicted functional impact, a strategy that has been used for other cancers and led to the discovery of new loci [5–7] It is well known that functional variants are indeed more likely to be associated with disease development [8].
We used data from the International Lymphoma Epidemiology Consortium (InterLymph) for discovery and from the German-speaking Myeloma Multicenter Group (GMMG), the International Multiple Myeloma rESEarch (IMMEnSE) consortium, as well as summary statistics from the FinnGen study for replication, for a total of 5982 MM cases and 266,173 controls. Detailed characteristics of the study populations are shown in the supplementary methods and supplementary table 1.
Candidate SNPs to be replicated were selected based on their association with MM risk and their functional role. First, we obtained summary results including odds ratios (OR), 95% confidence intervals (95%-CI), and p-values of the top SNPs of the InterLymph GWAS. Subsequently, all SNPs in the MM data set from InterLymph with p < 5 × 10−4 (N = 4396) were looked up in the first replication dataset, the GMMG GWAS. We did not consider SNPs from 15 loci that were reported to be associated at genome-wide significance level in previous GWASs. All SNPs with significant p-values (p < 0.05) in the GMMG GWAS and ORs going in the same direction in both datasets were selected. The next step was annotating the selected SNPs (N = 136) for their predicted function, using several suitable bioinformatic tools and databases. Specifically, we looked at expression and splicing quantitative trait loci (eQTLs and sQTLs), SNPs located in transcription factor binding sites (TFBS), long non-coding RNA (lncSNPs), SNPs that are located within enhancers, and polymorphisms located in gene coding regions (missense, stop-gain, stop-loss, synonymous SNPs). Supplementary Table 2 shows the details of the 136 SNPs and their predicted functional characterization. The resulting list from all annotations was pruned for linkage disequilibrium (LD) using the LDlink portal (https://ldlink.nci.nih.gov/). Only SNPs with r2 < 0.6 among them were kept, resulting in a total of 12 independent loci on 9 chromosomes. Replication in IMMEnSE and FinnGen was attempted for SNPs showing association with risk in the meta-analysis between InterLymph and GMMG GWAS and at least one in silico functional annotation. After exclusion of SNPs that had already been analysed in IMMEnSE in the context of previous projects and already shown not to be significantly associated with MM risk (on chromosomes 6, 8, 12 and 21), 4 SNPs showed to have low p-value of association with MM risk and had at least one functional prediction annotation (rs12038685, rs2664188, rs12652920, rs28199), which were therefore chosen for replication in IMMEnSE (Supplementary Table 3). An in-depth description of the SNP functional annotation and selection, as well as the technical details of the genotyping and quality control, can be found in the Supplementary Methods.
Analysis of association between each SNP and MM risk was carried out with logistic regression models, by estimating ORs, their 95%-CI, and associated p-value. The analyses were adjusted for age (at diagnosis for MM cases and recruitment for controls), sex, and the 10 first principal components for GWAS data, or country of origin in IMMEnSE, which lacks GWAS data. All meta-analyses were conducted with R, using a fixed-effect model between summary statistics of the different studies. The I² statistic was computed to quantify heterogeneity across studies.
rs28199, on chromosome 5, was associated with MM risk in IMMEnSE (OR = 1.19, 95% C.I. = 0.72–0.97, p = 0.018) and FinnGen (1.17, 95% C.I. = 1.05–1.31, p = 0.014). The G allele of this SNP resulted to be significantly associated with increased MM risk at a genome-wide level in the meta-analysis of the four datasets (OR = 1.18, 95% C.I. = 1.11–1.23, p = 3.18 × 10−10) with no heterogeneity among the studies (I2 = 0) (Table 1, Fig. 1).
Table 1.
Association results of the SNPs selected for replication in IMMENSE.
| SNP | Study | ORa | 95% CIb | p-value | Functional annotationc |
|---|---|---|---|---|---|
| rs12038685 | InterLymph | 1.22 | 1.11–1.34 | 5.04 × 10-5 | TBFS |
| GMMG | 1.11 | 1.00–1.23 | 0.046 | ||
| FinnGen | 1.2 | 1.07–1.32 | 0.004 | ||
| IMMEnSE | 0.94 | 0.81–1.09 | 0.409 | ||
| Meta-analysis | 1.18 | 1.10–1.25 | 2.07 × 107 | ||
| rs2664188 | InterLymph | 1.18 | 1.09–1.28 | 8.14 × 10−5 | eQTL |
| GMMG | 1.09 | 1.00–1.09 | 0.048 | ||
| FinnGen | 1.01 | 0.89–1.12 | 0.808 | ||
| IMMEnSE | 0.96 | 0.84–1.10 | 0.58 | ||
| Meta-analysis | 0.91 | 0.85–0.97 | 0.002 | ||
| rs12652920 | InterLymph | 0.83 | 0.75–0.91 | 1.62 × 10−4 | TBFS |
| GMMG | 0.90 | 0.81–0.99 | 0.031 | ||
| FinnGen | 1.09 | 0.96–1.22 | 0.174 | ||
| IMMEnSE | 0.99 | 0.84–1.15 | 0.875 | ||
| Meta-analysis | 0.91 | 0.86–0.97 | 0.0001 | ||
| rs28199 | InterLymph | 1.18 | 1.10–1.28 | 7.31 × 10−5 | TBFS |
| GMMG | 1.14 | 1.04–1.25 | 0.004 | ||
| FinnGen | 1.18 | 1.05–1.31 | 0.014 | ||
| IMMEnSE | 1.20 | 1.03–1.39 | 0.018 | ||
| Meta-analysis | 1.18 | 1.11–1.23 | 3.18 × 10−10 |
aOR odds ratio.
b95%C.I. 95% confidence interval.
cTFBS: the SNP is predicted to alter one or more transcription factor binding site(s); eQTL: the SNP is predicted to be a quantitative trait locus in whole blood or EBV-transformed B-lymphocyte cell lines.
Fig. 1. Meta-analysis result of rs28199.
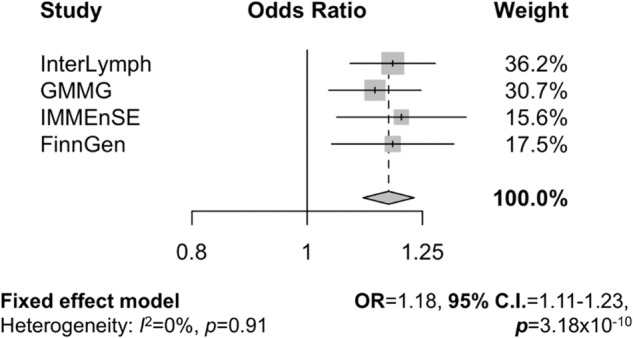
Forest plot of the meta-analysis using a fixed effects model across all four datasets. Heterogeneity was assessed using the I2 statistic. OR = odds ratio, 95% CI = 95% confidence interval.
This polymorphism was selected for being predicted to affect the binding site of three transcription factors: IRF1, STAT2_STAT1 and FOXP1. The strongest effect of the SNP was calculated for IRF1 (interferon regulatory factor 1), a protein member of the IRF family which was first recognized for its role as activator of genes involved in both innate and acquired immune responses. IRF-1 activates a set of target genes associated with regulation of cell cycle, apoptosis and the immune response [9, 10]. According to the SNP2TFBS database, rs28199 is predicted to modify a binding site of IRF1 leading to a stronger bond, which could in turn result in oncogenesis considering the set of genes that IRF1 regulates. The minor allele of rs28199 is located within a regulatory region which according to the variant effect predictor tool (https://www.ensembl.org/info/docs/tools/vep/index.html) and HaploReg (https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php) binds the CTCF protein, a highly conserved zinc finger with various cellular regulatory role. CTCF binding perturbations cause different types of 3D genome reorganization and may cause the activation of the neighboring oncogenes [11]. Among the genes that CTCF regulates there is STK10, which encodes for a serine/threonine-protein kinase, highly expressed in hematopoietic tissue [12]. In various lymphoid cells rs28199-G is associated with an increased expression of STK10. Overexpression of STK10 has been reported in several cancer types, including acute myeloid leukemia (AML), another blood malignancy [13, 14].
We used data from the 500 Functional Genomics cohort from the Human Functional Genomics Project (HFGP; http://www.humanfunctionalgenomics.org/site/) to explore the possible role in modulating immune response of the four SNPs selected for the final replication steps. Namely, we tested if any of the SNPs of interest were cytokine expression quantitative trait loci (cQTL) using data from in vitro stimulation experiments, as well as absolute numbers of 91 blood-derived cell populations and 103 serum or plasmatic inflammatory proteins. The cQTL analyses showed that rs28199-G is also associated with an increased blood level of Interleukin-6 (IL-6) (beta=0.075, p = 0.002). IL-6 is a cytokine with a well established role as a growth and survival factor in MM [15]. Specifically, in line with our results, an increased level of IL-6 contributes to the pleiotropic effects of IL-6 regarding proliferation, survival, drug resistance, and migration of MM cells, thereby facilitating disease progression [16]. The counts of cell populations and the levels of serum or plasmatic inflammatory proteins were not significantly associated with the SNPs of interest.
In conclusion, we identified a new genetic association for MM, supported by functional biological explanations, thus highlighting the importance of secondary analysis using functional approaches for GWAS.
Supplementary information
Acknowledgements
The authors wish to thank the International Myeloma Society (IMS) for its travel grant support to present this study during the 18th International Myeloma Workshop (IMW).
Author contributions
FC, DC and AM conceived and designed the study. KB and AM performed lab work. FC, AM, DC and AC performed data quality control and statistical analyses. FC, AM and DC drafted the manuscript. All other authors provided samples and data. All authors critically read, commented, and approved the manuscript.
Funding
This work was partially supported by: intramural funding of DKFZ; Horizon 2020 of the European Union, grant 856620; the Intramural Research Program of the National Cancer Institute, National Institutes of Health, grant number U01CA271014 and U01 CA249955. Open Access funding enabled and organized by Projekt DEAL.
Data availability
The primary data for this work will be made available to researchers who submit a reasonable request to the corresponding author, conditional to approval by the Steering Committees of the respective studies (InterLymph, GMMG, IMMEnSE) and Ethics Commission of the Medical Faculty of the University of Heidelberg, Germany. Data will be stripped from all information allowing identification of study participants. The summary statistics of the FinnGen GWAS are publicly available at https://www.finngen.fi. Functional data of the Human Functional Genomics Project used in this project have been catalogued and archived in the BBMRI-NL data infrastructure (https://hfgp.bbmri.nl/) using the MOLGENIS open-source platform for scientific data, which allows flexible data querying and download, including sufficiently rich metadata and interfaces for machine processing (R statistics, REST API) and using FAIR principles to optimize Findability, Accessibility, Interoperability and Reusability.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Celine Vachon, Daniele Campa, Federico Canzian.
Supplementary information
The online version contains supplementary material available at 10.1038/s41375-023-02022-8.
References
- 1.Kumar SK, Rajkumar V, Kyle RA, van Duin M, Sonneveld P, Mateos MV, et al. Multiple myeloma. Nat Rev Dis Prim. 2017;3:17046. doi: 10.1038/nrdp.2017.46. [DOI] [PubMed] [Google Scholar]
- 2.Georgakopoulou R, Fiste O, Sergentanis TN, Andrikopoulou A, Zagouri F, Gavriatopoulou M, et al. Occupational Exposure and Multiple Myeloma Risk: An Updated Review of Meta-Analyses. J Clin Med. 2021;10:4179. doi: 10.3390/jcm10184179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kristinsson SY, Bjorkholm M, Goldin LR, Blimark C, Mellqvist UH, Wahlin A, et al. Familial Aggregation of Multiple Myeloma and Its Precursor Monoclonal Gammopathy of Undetermined Significance (MGUS): A Population-Based Study in Sweden. Blood. 2008;112:1678. doi: 10.1182/blood.V112.11.1678.1678. [DOI] [Google Scholar]
- 4.Pertesi M, Went M, Hansson M, Hemminki K, Houlston RS, Nilsson B. Genetic predisposition for multiple myeloma. Leukemia. 2020;34:697–708. doi: 10.1038/s41375-019-0703-6. [DOI] [PubMed] [Google Scholar]
- 5.Campa D, Gentiluomo M, Stein A, Aoki MN, Oliverius M, Vodičková L, et al. The PANcreatic Disease ReseArch (PANDoRA) consortium: Ten years’ experience of association studies to understand the genetic architecture of pancreatic cancer. Crit Rev Oncol Hematol. 2023;186:104020. doi: 10.1016/j.critrevonc.2023.104020. [DOI] [PubMed] [Google Scholar]
- 6.Yu Y, Mao L, Cheng Z, Zhu X, Cui J, Fu X, et al. A novel regQTL-SNP and the risk of lung cancer: a multi-dimensional study. Arch Toxicol. 2021;95:3815–27. doi: 10.1007/s00204-021-03170-5. [DOI] [PubMed] [Google Scholar]
- 7.Iversen ES, Lipton G, Clyde MA, Monteiro ANA. Functional annotation signatures of disease susceptibility loci improve SNP association analysis. BMC Genom. 2014;15:398. doi: 10.1186/1471-2164-15-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu W, Li M, Zhang W, Zhou G, Wu X, Wang J, et al. Leveraging functional annotation to identify genes associated with complex diseases. PLOS Comput Biol. 2020;16:e1008315. doi: 10.1371/journal.pcbi.1008315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim PKM, Armstrong M, Liu Y, Yan P, Bucher B, Zuckerbraun BS, et al. IRF-1 expression induces apoptosis and inhibits tumor growth in mouse mammary cancer cells in vitro and in vivo. Oncogene. 2004;23:1125–35. doi: 10.1038/sj.onc.1207023. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong MJ, Stang MT, Liu Y, Gao J, Ren B, Zuckerbraun BS, et al. Interferon Regulatory Factor 1 (IRF-1) induces p21WAF1/CIP1 dependent cell cycle arrest and p21WAF1/CIP1 independent modulation of survivin in cancer cells. Cancer Lett. 2012;319:56–65. doi: 10.1016/j.canlet.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Segueni J, Noordermeer D. CTCF: A misguided jack-of-all-trades in cancer cells. Comput Struct Biotechnol J. 2022;20:2685–98. doi: 10.1016/j.csbj.2022.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walter SA, Cutler RE, Martinez R, Gishizky M, Hill RJ. Stk10, a new member of the polo-like kinase kinase family highly expressed in hematopoietic tissue. J Biol Chem. 2003;278:18221–8. doi: 10.1074/jbc.M212556200. [DOI] [PubMed] [Google Scholar]
- 13.Bi L, Jia S, Hu W, Su X, Chen X, Tang H. Systematic analysis of prognostic significance, functional enrichment and immune implication of STK10 in acute myeloid leukemia. BMC Med Genom. 2022;15:101. doi: 10.1186/s12920-022-01251-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Lu SY, Guo R, Ma JX, Tang LY, Wang JJ, et al. STK10 knockout inhibits cell migration and promotes cell proliferation via modulating the activity of ERM and p38 MAPK in prostate cancer cells. Exp Ther Med. 2021;22:851. doi: 10.3892/etm.2021.10283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burger R. Impact of Interleukin-6 in Hematological Malignancies. TMH. 2013;40:336–43. doi: 10.1159/000354194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harmer D, Falank C, Reagan MR. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma. Front Endocrinol. 2019;9:788. doi: 10.3389/fendo.2018.00788. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The primary data for this work will be made available to researchers who submit a reasonable request to the corresponding author, conditional to approval by the Steering Committees of the respective studies (InterLymph, GMMG, IMMEnSE) and Ethics Commission of the Medical Faculty of the University of Heidelberg, Germany. Data will be stripped from all information allowing identification of study participants. The summary statistics of the FinnGen GWAS are publicly available at https://www.finngen.fi. Functional data of the Human Functional Genomics Project used in this project have been catalogued and archived in the BBMRI-NL data infrastructure (https://hfgp.bbmri.nl/) using the MOLGENIS open-source platform for scientific data, which allows flexible data querying and download, including sufficiently rich metadata and interfaces for machine processing (R statistics, REST API) and using FAIR principles to optimize Findability, Accessibility, Interoperability and Reusability.