

Abstract
Stroke induces a plastic state in the brain. This period of enhanced plasticity leads to the sprouting of new axons, the formation of new synapses and the remapping of sensory-motor functions, and it is associated with motor recovery. This is a remarkable process in the adult brain, which is normally constrained in its levels of neuronal plasticity and connectional change. Recent evidence indicates that these changes are driven by molecular systems that underlie learning and memory, such as changes in cellular excitability during memory formation. This Review examines circuit changes after stroke, the shared mechanisms between memory formation and brain repair, the changes in neuronal excitability that underlie stroke recovery, and the molecular and pharmacological interventions that follow from these findings to promote motor recovery in animal models. These concepts build a framework for recovery after stroke in the concept of neuronal allocation in damaged circuits. The translation of the concepts discussed here to recovery in humans is under way in clinical trials for stroke recovery drugs.
Introduction
Stroke [G] causes death of brain tissue. The prevalence and limited recovery after acute brain injury make stroke one of the leading causes of adult disability worldwide1,2. Current therapies for stroke include clot removal within the first day after stroke in carefully selected patients, but these individuals represent a minority of people with stroke3–6. Furthermore, in cases of successful endovascular clot retrieval, 50% of patients are still left with neurological deficits7,8. Outside of this limited window for a therapy that promotes reperfusion of brain blood vessels in stroke, neurorehabilitative practices are the most widely applied therapies.
Initially, stroke produces cell injury and death with clearly evident neurological deficits. However, weeks and months after the acute events of injury and death, there exists profound plasticity [G] in brain circuits during the limited processes of recovery. These plasticity processes include turnover of local synaptic contacts adjacent to the lesion, altered excitability of neuronal circuits [G] that are adjacent to and connected with the area of damage, and the formation of new functional neuronal connections, seen in remapping of motor, sensory and language functions9,10. Studies on post-stroke plasticity events have identified important principles governing their timing, their role in the recovery of injured brain circuits and, most importantly, that these biological processes of recovery after acute brain injury are manipulable — they can be controlled by drugs and brain stimulation protocols to enhance recovery in animal models9,10. This Review focuses on the mechanisms of circuit plasticity after stroke, with an emphasis on changes in neuronal excitability [G] systems that lead to the reformation of brain circuits after injury and can be enhanced to promote recovery. We develop the concept of neuronal allocation [G] for recovery, in which we draw on parallels between the mechanisms of memory formation in the normal brain and of recovery after stroke.
Cellular events after stroke
There are four distinct time epochs after stroke: the hyperacute, acute, subacute and chronic phases. The hyperacute phase of stroke is characterized by cellular processes that mediate excitotoxity and cause cell death in the hours of the initial loss of blood flow. Excitotoxicity in injured neurons establishes a feedforward mechanism of depolarization, glutamate release, further depolarization and progressive neuronal cell death11–14. There is also an initial intrinsic inflammatory response involving brain microglia that results in inflammatory cytokine release15. This hyperacute phase of extensive cell death in the day after stroke is followed by an acute phase in the week after stroke of delayed neuronal cell death (apoptosis), an influx of peripheral immune cells and the further activation of local immune responses in microglia and astrocytes. Reactive immune cells migrate into the area of the stroke and secrete pro-inflammatory factors16. In this acute phase, the brain is sensitive to further stress caused by systemic infection, elevated temperature, alterations in physical activity levels and/or enhanced neuronal excitability, any of which may potentiate further injury17–20.
The hyperacute and acute phases of stroke are followed by a subacute phase of reduced inflammatory responses and maximal plasticity, which lasts in the rodent for approximately one month and in humans for up to three months after stroke21,22. This window of plasticity diminishes as the stroke progresses from the subacute phase to the chronic phase, in which there is a limited potential to induce recovery. The chronic phase of stroke is present from 3 months after stroke onset in humans and is characterized by an absence of spontaneous recovery21,22. Recovery is still possible in chronic stroke requires intensive neurorehabilitation therapy and substantial focus by the patient. Even with these practices, the amount of recovery in the chronic phase, as measured with scales of neurological impairment, appears to be roughly 10% of the recovery seen in the sub-acute phase24–27. This difference in the level of recovery in chronic versus subacute stroke is seen in motor outcome measures, such as the Fugl-Meyer scale28. A substantial recovery in the chronic period has recently been shown with an intensively focused 90 hours of training, exercise and behavioural shaping over a 6-week period23 or 300 hours of intensive training over a 12-week period29. What these and other studies in the chronic phase of stroke have in common is the point that spontaneous recovery is not seen to any substantial degree three months after stroke onset, but that rehabilitation-induced recovery can occur in this chronic period with great effort.
Endogenous plasticity responses in the subacute phase.
The events after a stroke are influenced by time-varying changes in gene expression and cellular responses (FIG. 1). Stroke induces a transcriptional growth program that promotes plasticity in the subacute phase30. This transcriptional program is robust, with more than 500 different neuronal genes differentially regulated. Neurons in the major area of functional motor recovery [G] after stroke, the peri-infarct [G] tissue, and its connected areas upregulate signalling pathways that promote axon guidance, growth factor responses, intracellular growth (such as upregulated expression of adhesion molecules) and cytoskeletal re-arrangement30–33. These induced gene systems in neurons adjacent to the infarct [G] lead to axonal growth30–33 and synapse formation34. Neurons in peri-infarct tissue have demonstrable new axonal projections that project by several millimetres into nearby cortical areas, including premotor, motor, sensory31–33 and retrosplenial cortices35, where presumably new functional synaptic connections are formed. Blockage of these new connections prevents stroke recovery in animal models36. Cortico-cortical connectivity is mostly mediated through excitatory synaptic contacts on dendritic spines [G]. In the subacute phase of stroke, there is enhanced dendritic spine turnover35,37–39, providing a substrate for the synaptic termination of new connections.
Fig. 1 |. Endogenous plasticity in sub-acute stroke.
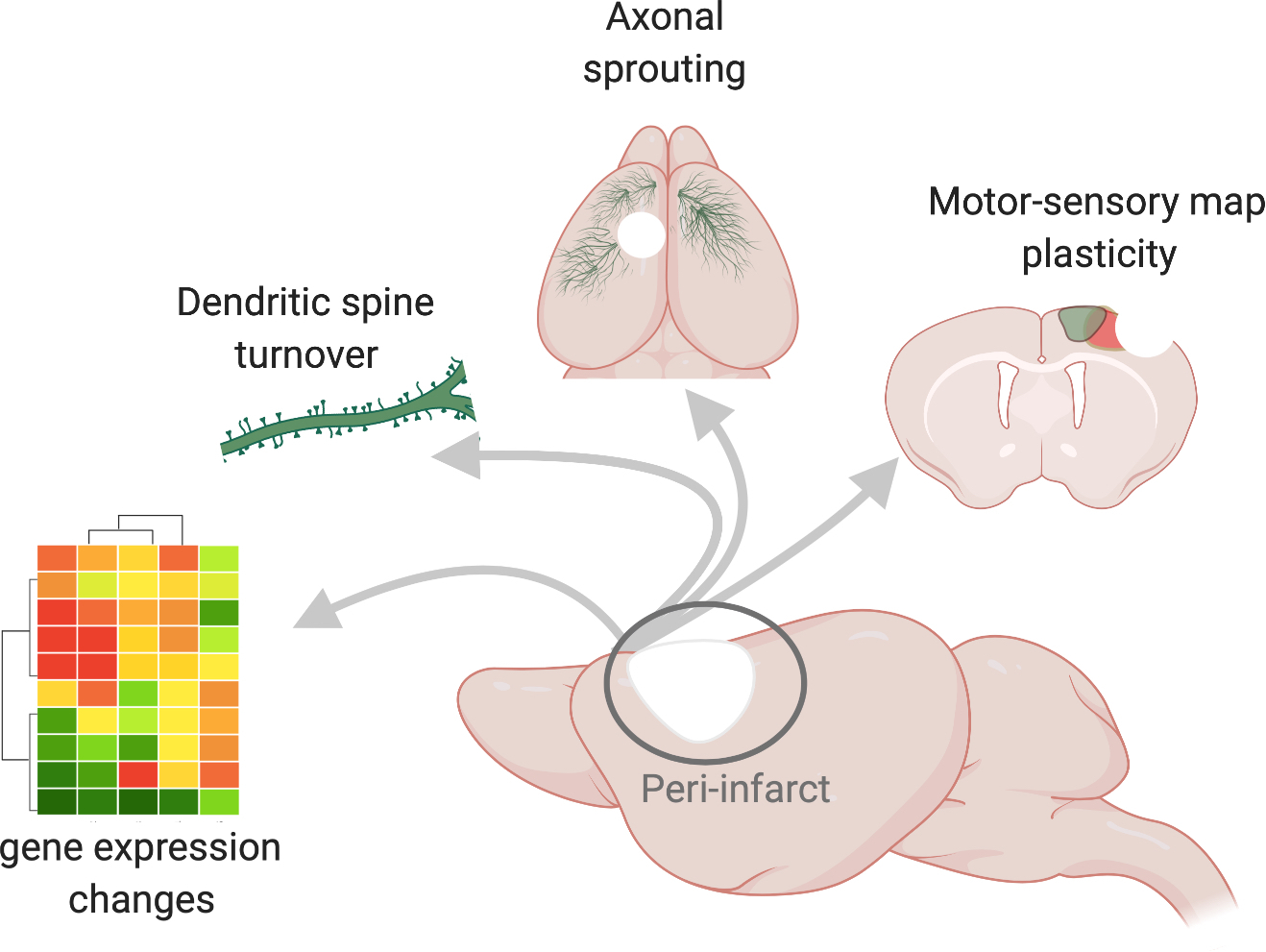
Sagittal view of a mouse brain (centre) in which the site of a stroke is depicted by the white area and the peri-infarct region is demarcated by a circle. The arrows point to molecular and cellular hallmarks of the peri-infarct region, including changes in gene expression. Here, these changes are denoted as a heatmap schematic in which the magnitudes of the changes in gene expression are visualized as differently coloured modules. New molecular programs support structural connectivity such as strengthening of connections through the growth and turnover of new spines, which are post-synaptic protrusions on dendrites, and the growth of new axons, depicted as blue fibres that sprout from the peri-infarct cortex and travel to intact motor, somatosensory and contralateral cortices. Changes in structural connectivity may underlie re-organization of functional motor (blue) and somatosensory (green) limb representations that re-appear at new locations several weeks after a stroke.
The period of synaptic plasticity in axonal connections and their post-synaptic partners occur during a period of substantial change in the sensory and motor maps of the body. A stroke that occurs in the motor cortex causes remapping of movement representations in rodent models, non-human primates and humans40–44. Newly reorganized maps show substantial changes in their spatial location and are dependent on the cortical structure that was damaged. This spatial change is mostly restricted to reorganization within the primary motor regions, which re-map over 8 weeks after stroke in rodent models35,42. By contrast, somatosensory limb maps lost after stroke that re-map over time do not show this substantial change in map location, as shown in rodents42 and in humans44. Concepts of the limb map in the primary motor and sensory areas, derived from decades of results of brain stimulation studies, imply a one to one functional structure that is sort of a ‘ground truth’ for movement and touch: the homunculus (human) or rodunculus (rodent) directly represent and control the mapped body part, and so a change in that brain representation means a change in that function. Changes in motor and sensory limb maps were thought to directly indicate and faithfully track the changes of recovery after stroke. However, changes in the brain limb map may actually follow functional recovery45, indicating that the underlying synaptic plasticity of axonal sprouting [G] and dendritic spine morphogenesis are the primary events of the subacute phase of stroke that lead to recovery, and motor and sensory maps may follow.
Priming plasticity: the concept of a critical window.
The opening up of a plasticity window in the subacute phase of stroke, termed the sensitive period in stroke recovery46–49, shares similarities with the concept of heightened plasticity during development, termed the critical period [G] 50–52. In both periods, the brain shows substantial and parallel changes in cellular and molecular systems, but only within a time-restricted interval. In the critical period in development and sensitive period after stroke, changes in sensory or motor activity patterns induce widespread changes in synaptic plasticity and cortical maps. In the critical period, changes in activity resulting from forced eye closure53,54 or whisker trimming55 in rodents cause changes in cortical responsiveness and mapping of the periphery. In stroke, high levels of motor activity of the impaired limb induce similar widespread changes in sensory and motor maps during the sensitive period, but not after it49,56. Characteristic patterns of low frequency and synchronized neuronal discharges are present during both the critical period in development and the sensitive period after stroke and are responsible for substantial cortical remapping and changes in axonal connections57,58.
Similar cellular events define these two epochs (FIG. 2). For example, during development, the critical period occurs during a time of reduced accumulation of extracellular matrix structures such as peri-neuronal nets59,60. Digestion of perineuronal nets reopen the critical period60. In stroke, the sensitive period corresponds to a time in which peri-neuronal nets are degraded locally in areas that experience robust spontaneous plasticity61,62. Heightened plasticity in the developmental critical period and in the adult brain after lesions are both promoted by cholinergic signalling63,64. The transcriptional profile of neurons that mediate recovery after stroke and that of the cortex during the critical period overlap in molecular systems, but with important differences in excitatory signalling in stroke that may reflect a direct injury signal30.
Fig. 2 |. Parallels between windows of plasticity in development and stroke.
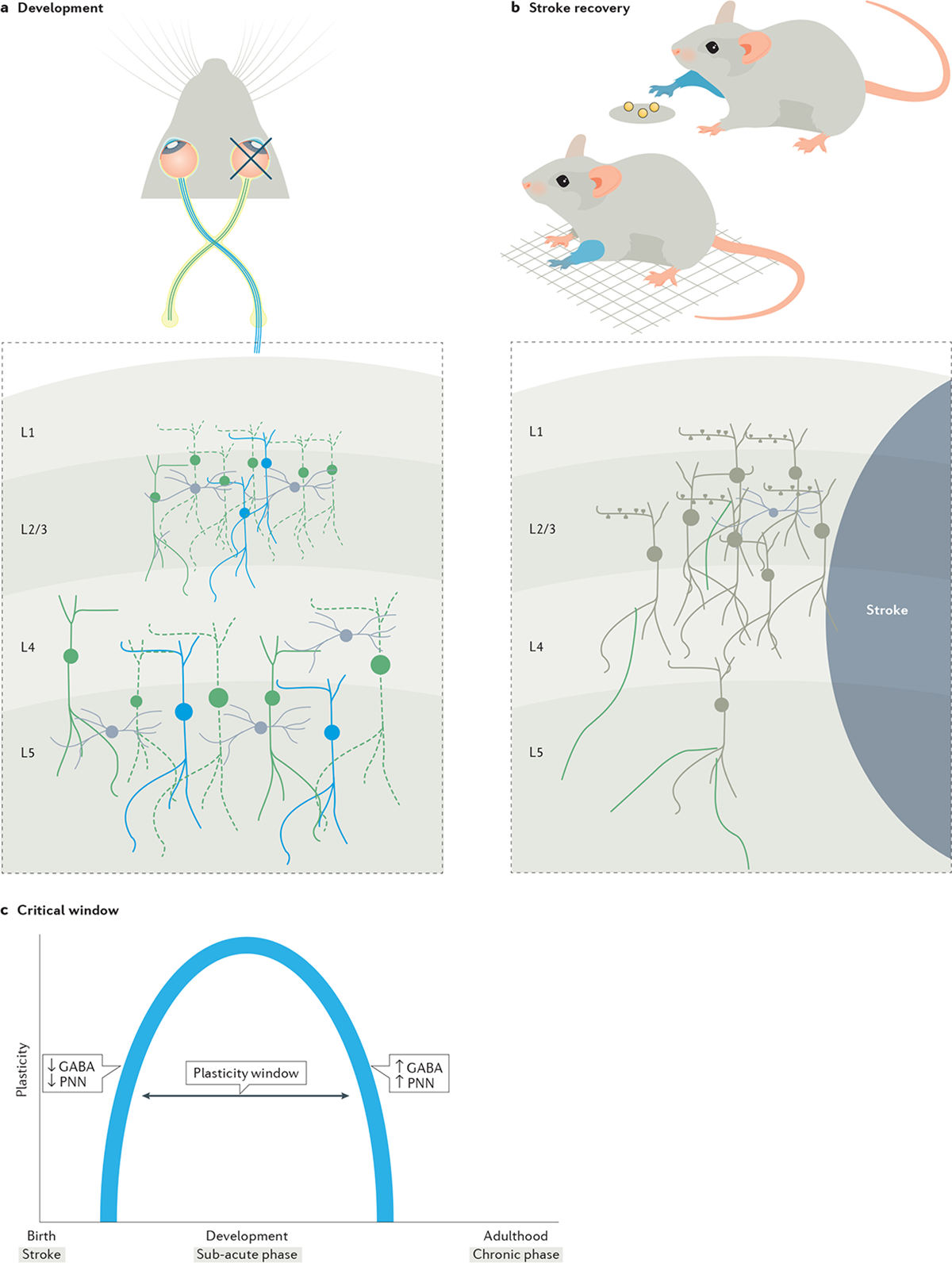
The critical period in development and the sensitive period in stroke share molecular and connectional principles. a | Visual experience during the critical period shapes neuronal connectivity. Temporary deprivation of visual experience, only during the critical period, by unilateral eye closure (depicted by the cross on one eye), can cause a decrease in the responsiveness of neurons (dotted neurons in green) to a visual stimulus across different layers of the visual cortex pertaining to the closed eye. Here, during this window, neural connectivity is amenable to plastic changes and shaped by visual experience. b | After a stroke, a similar window of plasticity exists. Stroke impairs motor function such as in the ability of a stroke-induced mouse to locomote on a grid or in skilled reaching of a food pellet with the impaired limb (depicted as blue forelimbs). The sub-acute phase of stroke is marked by a sensitive period similar to the critical period in development in which turnover of dendritic spines in the peri-infarct cortex and the sprouting of new axons (green axons on neurons) provide a neural substrate for functional reorganization of limb representations (not shown). These endogenous mechanisms are associated with spontaneous incomplete recovery of motor function, in which the impaired limb gains limited ability or compensates to perform a motor task. c | The opening and closing of the critical window during development and the sensitive period in stroke as it progresses from acute to chronic are regulated by similar molecular mechanisms, such as changes in excitatory to inhibitory GABA signalling and the maturation of peri-neuronal nets (PNNs).
The similarities between the developmental critical period and sensitive period after stroke also extend to the molecular systems that close these windows. The critical period in development is in part closed by maturation of the GABA signalling system, such that tighter inhibitory tone is present in local cortical circuits51. In the sensitive period after stroke, cortical recovery is inhibited by increased GABAergic tone through extrasynaptic GABA signalling65–68. Notably, intracellular chloride gradients that predicate an inhibitory tone during GABA signalling remain unaltered during the sensitive period in stroke65,68, unlike during early development in which increased chloride gradients can lead to GABA-induced excitatory currents that depreciate over time. The critical period during development is also terminated by increased expression of myelin and neuronal signalling through Nogo-A69. Nogo-A limits brain plasticity after stroke and blocking Nogo-A signalling enhances functional plasticity and stroke recovery70–73.
The similarities between the developmental critical period and the sensitive period after stroke indicate that stroke opens a window of plasticity in the adult brain. This heightened and short-lived brain plasticity in the adult offers a platform in which neuromodulation or pharmacological therapies might further stimulate recovery in injured circuits.
Manipulating neuronal excitability
The subacute period after stroke is associated with heightened plasticity overall, but locally depressed neuronal signalling35,74. In humans and animal models, there is decreased neuronal excitability in the peri-infarct cortex and connected cortical areas on ipsilesional and contralesional sides, as measured by cortical activity evoked by sensory stimulation35,74–77. This depressed signalling across regions is a direct measure of lost connectivity, as remote sites such as the contralesional visual cortex that share fewer connections show little or no depression in sensory-motor signalling74. At 8 weeks in the rodent, there is partial return of normal excitatory cortical signalling patterns to afferent stimulation. This is also reflective at the single-neuron level, at which neuronal activity underlying limb selectivity (that is, neurons that respond to either the forelimb or hindlimb) lose responsiveness initially after the stroke, then show mixed responsiveness (that is, neurons expand their functional repertoire to respond to both limbs78). This loss of neuronal selectivity at 1 month after stroke transitions to responsiveness to a preferred limb at 2 months post-stroke in rodent models78, and at 3 months in humans75. In parallel with the concept of a critical period during neurodevelopment, the stroke itself induces a response in the subacute phase, in which neurons with initially reduced excitability undergo functional reorganization. At first, there is expansion of the neurons’ functional repertoires, which are then refined to meet the functional output of the cortical area, such as somatosensation or motor output, as the window closes with time.
Manipulating neurotransmitter signalling for motor recovery.
A major mechanism for reduced neuronal excitability after stroke arises from changes in inhibitory neurotransmitter signalling, such as tonic GABAergic signalling in tissue adjacent to the infarct17. Tonic GABAergic signalling occurs through GABA receptors at extrasynaptic sites that have high affinity for GABA and produce slow (tonic) and persistent inhibition79. This inhibition, also known as the shunt current, sets the action potential threshold for pyramidal neurons79. In the peri-infarct cortex after stroke, tonic GABAergic signalling is elevated in the first weeks, during the period of reduced cortical excitability noted above65. Stroke downregulates the expression of the astrocytic GABA transporter GAT3, causing GABA to accumulate at extrasynaptic sites that bind to high-affinity GABAA receptors (GABAARs), producing slow and persistent inhibition65. The distinct molecular composition of GABAARs, which comprise α and δ subunits, allows for specific molecular and pharmacological targeting of these tonic receptor subunits, such that undesirable adverse effects such as seizures can be avoided80. Genetic or pharmacological blockade of the GABAAR α5 subunit after stroke restores pyramidal neuron excitability and enhances functional recovery65,66,68,81,82. Reducing tonic inhibition promotes recovery, whereas targeting fast synaptic inhibition (phasic) and tonic signalling deteriorates motor performance over time65. Moreover, suspending treatment that blocks the α5 subunit reverses the beneficial effects on recovery, suggesting that in the absence of treatment, the effects of tonic GABA inhibition on motor function persist for weeks after stroke65.
In addition to approaches that block increased inhibitory signaling after stroke, enhancing excitatory signalling in the tissue adjacent to the infarct promotes recovery. Treatment with positive allosteric modulators of AMPA-type glutamate receptors (AMPARs), known as AMPAkines, facilitate AMPAR signalling. Binding of glutamate to AMPARs causes cation influx, depolarization and downstream gene expression, such as the expression of brain-derived neurotrophic factor (BDNF)83. AMPAkines promote recovery of motor function in stroke by selectively boosting BDNF signalling in the peri-infarct motor cortex20,84. An alternative approach to boosting AMPAR signalling, leading to enhanced AMPAR levels at post-synaptic sites, also improves motor recovery after stroke. This effect is mediated through collapsin response mediator protein 2 (CRMP2), which drives greater numbers of AMPARs to the cell surface and enhances recovery after cortical lesions85.
The findings described above in GABAergic and glutamatergic signalling indicate that promoting neuronal excitability after stroke in highly selective ways enhances recovery in rodent models of disease. However, this recovery is only promoted in the subacute period after stroke. The acute period of stroke is characterized by ongoing cell death, and the brain is still sensitive to manipulations that promote neuronal excitability, which can result in an enlargement of infarct size17.
Excitability principles from learning to motor control.
The principle of co-active, or mutually excited, synaptic connections on ensembles of cells is a common cellular feature underlying stroke recovery and memory formation. Connectivity between neurons and neuronal excitability work mutually through mechanisms of Hebbian plasticity, long identified with the phrase “cells that fire together wire together”86. In Hebbian plasticity, neurons that are excited during a narrow time window will strengthen their interconnections. These strengthened connections underlie memory formation and store task memories87. Co-active or strengthened connections are then consolidated and stabilized through structural changes at the synapse, such as with changes in dendritic spines88,89. In the event of a stroke, connectivity between neurons is lost, dendritic spines are unstable and the overall excitability of neurons in areas adjacent to the stroke is reduced30,35,78. Performance of the motor function that is lost in stroke requires re-establishing this lost connectivity. Restoration of lost connectivity requires the availability of a new neural substrate and the re-establishment of temporal activation patterns within these newly formed circuits.
The growth of new connections can be induced by increasing the excitability threshold of specific circuits that are local and distant to the stroke site. Optogenetic manipulations that increase neuronal firing synchronously across a population of neurons after stroke improves motor function90–92. This improvement in motor function is associated with the formation of new dendritic spines91 and axons92 in local and distant sites and the release of neurotrophic factors that support a state of enhanced plasticity90. A notable factor is that circuit-specific remodelling of long-range projections accounts for gains in motor function whereas increasing the excitability of non-relevant circuits can lead to undesirable manifestations on motor control93 and mood disorders94. Hence, excitability increases directed towards specific circuits allow for the innervation of appropriate local and distant targets and formation of synapses that result in restoring motor function.
Neuromodulation strategies aim to elevate neuronal excitability, in which timing an excitable signal with motor behaviours or cellular states enhances connectivity after stroke. After a stroke, delivering a timed electrical stimulus at the onset of skilled reaching for a target enables successful completion of the task95. A key aspect is timing: the electrical stimulus is timed to events, here the onset of reach, that as a result of the stroke have disordered timing in their activity. At a behavioural level, timing a neuromodulatory stimulus in stroke patients or animal models to the period of behavioural activity, which pairs excitability from neuromodulation with a motor action, leads to a greater recovery than delivering a stimulus without this timing element95,96. This is evident in studies of vagal nerve stimulation for stroke recovery, an approach with a still indeterminate mechanism of action but that likely increases excitability in motor circuits97. Pairing vagal nerve stimulation with neurorehabilitative activity of the impaired limb, but not after that behavioural activity, enhances recovery98. In individuals with stroke and rodent models, synchronous activity within brain oscillations, measured as a composite neural signal with local field potentials, can be used to track recovery95,99. This is a measure of excitability in specific time epochs of cohorts of neurons. In the peri-infarct cortex, the local field potentials within such oscillations are diminished after a stroke95,99. With recovery99, local field potentials re-appear, suggesting a return of synchronous neuronal activity underlying a motor task.
Outside of local connectivity, applying this principle of inducing excitability within pre- and post-synaptic neurons in long-range intercortical circuits can restore motor function through Hebbian principles. In a model of traumatic brain injury100, which shares several pathophysiological characteristics with stroke, recovery of skilled motor function can be induced by delivering a timed electrical signal to the somatosensory cortex based on firing patterns from the premotor cortex. These observations in recovery from traumatic brain injury parallel observations with restoration of local field potentials in stroke recovery.
In summary, co-activation requires simultaneous firing of neurons, predicated on an excitability threshold — neurons have to be excitable enough to fire together. Stroke diminishes neuronal excitability whereas spontaneous recovery is seen with the return of indicators of normal neuronal excitability, such as brain oscillations in recovering brain areas. Enhanced recovery can be produced by timed stimulation of brain areas or to entrain neurons to fire together in injured areas. Both systems of learning and recovery after stroke share common ground in that both depend on cellular excitability for formation and consolidation of a motor skill or recovery of motor function, suggesting that this common ground extends from excitability principles to shared molecular pathways.
Molecular memory systems that enhance neuronal excitability.
Synchronous neuronal firing, enhanced excitability and long-term potentiation are cellular features that are shared between memory formation and motor recovery. The overlap at the cellular level extends to the overlap in the molecular mechanisms that are shared between the two systems of memory formation and motor recovery. Changes in neurotransmitter signalling that positively modulate AMPAR-mediated BDNF signalling20,84 or dampen tonic GABAergic signalling65 promote memory formation and recovery of motor function. At the transcriptional level, cAMP-binding response element (CREB) is a key signalling molecule that enhances cellular excitability and is critical for memory formation101,102. Increasing CREB signalling after stroke enhances cellular excitability and improves the recovery of motor function103. CREB expression can be induced by promoting cAMP or cGMP signalling in a cell101. In addition to direct induction of CREB expression, inhibiting cAMP and cGMP degradation through a phosphodiesterase inhibitor promotes recovery in animal models of stroke104. The activation of a specific chemokine receptor, C-C chemokine receptor 5 (CCR5), downregulates CREB33. In normal learning, CCR5 downregulation leads to enhanced long-term potentiation, increased spike-timing dependent plasticity and enhanced spatial, social and associative learning105. Inhibiting the same CCR5 signalling system after stroke induces early and robust recovery of motor function33. The overlap in the many molecular systems that underlie memory formation and recovery of motor function suggest that targets that enhance learning also enhance motor recovery. Intersectional principles between learning, memory and motor recovery and converging roles of CREB–CCR5 signalling in both systems for selecting circuits are discussed in subsequent sections.
Effective circuit selection
The assembly of an effective motor program requires the recruitment of the appropriate circuits106. This recruitment means that a selection process is initiated and based on a defined set of properties such as the intrinsic state or the activity profile of a neuron in response to an external cue107–109. The mechanisms that partake in selection are active in normal processes of learning. Such processes have been well-described in motor learning of a skill, defined by a process of active searching, selection and refinement110–113. At the cellular level, early exposure to an external cue, such as a new context or environment, triggers activity in a large population of neurons. This is the active searching phase in which neurons within a motor network exhibit spatiotemporal activity profiles that are loosely correlated with a motor action. Here, individual neurons initially explore many activation profiles that vary in their temporal relationship to the cue and to each other. Over the course of learning, the variance in patterns from individual neurons decreases with an increase in successful attempts. This decrease marks the next phase, which requires effective selection of neurons that have low variability in patterns; that is, neurons that exhibit temporal activation profiles that encode for correct movements. From a population perspective, neurons with similar profiles, such as co-active patterns or co-varying patterns, are selected114. The selected neurons then undergo a period of refinement in which spatial and temporal activation patterns compress over time, facilitating sparse coding of a motor skill in a set of highly efficient neurons. Experimental and computational models support the occurrence of sparse coding in visual and motor systems underlying task performance115–119. Moreover, the surveying of different activation patterns of a neuron during searching followed by constrainment of the neuron to a task-relevant network during refinement has been described in models of flexible learning networks114. This initial flexibility and then refinement comprise circuit selection.
The process of circuit selection is not limited to local intracortical circuits but involves connected intercortical120–122 and sub-cortical targets123–125. Presumably, active searching in one cortical region (for example, the primary motor cortex (M1)) in turn activates larger intercortical and subcortical targets that refine as the skill is acquired. This is evident in normal motor learning in humans in which the initial phase of skill acquisition involves activation of large cortical and subcortical regions that refine over time to include a set of discrete regions that show tight spatial–temporal activation as the skill is acquired120,126,127. The process of circuit selection is active after a stroke. In individuals with stroke, functional MRI (fMRI) studies show that with time, complex skills are associated with large activations across brain regions (active searching) that refine to a discrete set of regions that show compressed and higher activity (selection and refinement)128–132.
For circuit refinement to be meaningful in terms of building an effective motor program, selection needs to be effective. The appropriate circuits need to be selected, as sparse coding requires ‘efficient’ neurons117. This feature is compromised after a stroke and in cortical lesions. In the event of a large cortical lesion or after a stroke, learning new motor sequences is severely impaired133. This means that the process of effective circuit selection cannot be executed with the absence of the appropriate neural substrate. After a stroke, even in the presence of endogenous plasticity with the growth of new axons and new connections, recovery of motor function is impaired or is incomplete92,134. This limitation in recovery despite a structural platform for the formation of new circuits (new axonal connections) means that even with the availability of a neural substrate, the selection of circuits that do not efficiently encode behaviour results in compromised motor function.
Finally, the selectivity or specificity of circuit selection is further supported by structural remodelling, in which motor learning or task-specific neurorehabilitation drives changes in only task-specific circuits, resulting in overall strengthened connectivity of the network36,135–137. With task-specific training, corticospinal neurons undergo remodelling with increased axonal outgrowth36, spine density and/or dendritic branching in only a subset of corticospinal neurons137. New dendritic spines are formed on specific output neurons, and these newly formed spines are then selectively stabilized by rehabilitative or task-specific activity138,139. In further parsing of the anatomical specificity of structural remodeling of brain circuits by activity in some experimental systems, dendritic spine changes are localized to corticospinal neurons projecting to the C8 cervical segment that control the distal forelimb involved in fine motor control such as grasping; and are absent in C4-projecting neurons that control proximal forelimb musculature. Within these corticospinal populations, a connectional bias from subcortical targets is present, in which motor learning increases synaptic connectivity from thalamocortical inputs to C8-projecting and not C4-projecting neurons136. Here, skill acquisition is accompanied by structural and synaptic changes in only those circuits that are relevant (selected). A stroke or a cortical lesion that impairs thalamocortical connectivity impairs motor function91, whereas task-specific neurorehabilitation that drives structural and synaptic changes in C8-projecting neurons leads to an improvement in motor function136.
In studies of normal motor learning and stroke recovery, effective circuit selection is key to producing a functional motor outcome. What process selects a neuron into a specific motor circuit? A main determinant, as in the studies discussed below, is the intrinsic excitability of a neuron, such that neurons with higher excitability are preferentially allocated.
The engram in stroke recovery
A key element in the formation of new memories involves the expression of excitatory signalling in the brain in co-active and functionally connected sets of neurons that store a memory trace, the memory engram [G] 140,141. During the formation of new memories, linked inputs are stored in the co-activation patterns of neurons in regions such as the hippocampus, lateral amygdala and cortex. There is substantial experimental literature on the cellular mechanics of memory engrams, especially as they have been defined with recent intersectional genetic labelling and neuronal stimulation tools140–142. Networks of neurons store a memory when they are linked as, or allocated into, a circuit of neurons that are co-activated by the sensory inputs of that memory (such as a novel environment paired with electrical shock)143,144. This allocation of neurons into an engram is competitive — neurons can outcompete their neighbours and link up into this network145. This competition is based on neuronal excitability, such that the more excitable a cell is, the more likely it is to fire in close relation to other neurons during an arriving stimulus and form the engram that will store that stimulus146. In addition to competition, one engram can compensate for the loss of a memory that is stored in a different engram of cells if that engram is inhibited147,148. Engrams that store a memory trace are formed by strengthening the connections among cells, seen by enhanced dendritic spines on engram cells149–151. These points define four principles of the engram that relate to stroke recovery: neuronal allocation, competition influenced by excitability state, compensation for disrupted engrams and dendritic spine morphogenesis in engram formation.
Neuronal allocation.
In an engram, a memory is stored in coactive neuronal networks and their associated connections. During the formation of a conditioned fear memory, the storage of the environmental context (the cage) paired with an electrical shock (the stimulus) is made within neurons that were activated by the experience of these paired stimuli. These co-active cells are then activated by the context (the cage) to produce the memory of the shock and activate a behavioural freezing in the mouse to the cage. Ablation or inactivation of the cells in the engram, such as with diphtheria toxin152, an inhibitory ion channel153 or an inhibitory DREADD (designer receptors exclusively activated by designer drugs)154,155, destroys the memory. By contrast, artificial activation, with optogenetics, of the cells of the engram expresses the memory156,157. The memory engram has been thus experimentally shown to be necessary and sufficient to store a memory because its cells can be mapped, inactivating them eliminates the memory, and artificially activating the cells in the engram artificially expresses the memory.
With respect to skilled motor movements, unlike episodic memory, evidence for a ‘motor engram’ is less developed. Learning and execution of skilled movement is a result of reproducible spatiotemporal activations encoded in coordinated cortical110 and subcortical neurons158. The motor cortex is a driver of these actions and is dependent on the inputs received from the motor thalamus159. In the motor cortex, collections of neurons store the patterns of limb movement. This stored limb movement in the mouse motor cortex relates specifically to learned, skilled behaviours, such as reaching for a food pellet, and not routine behaviours, such as licking or grooming160. When neurons in motor cortex are artificially inhibited at different epochs of a skilled reaching behaviour, performance on the task is severely impaired or completely inhibited. The inhibition of neurons in a motor circuit reveals that the motor program not only initiates skilled reach but also is important for the distinct elements of the task, such as lifting, grabbing and retrieval, suggesting the continued involvement of the motor engram from initiation to completion160. Similarly, inactivation of these neurons does not block routine, non-skilled motor actions160. During motor learning, as neuronal activity in the motor cortex is followed over time, acquisition of a skilled task is developed through co-active networks of neurons that have a tight correlation to that movement pattern 110,111,113,161,162. These features of co-active networks of neurons that emerge with motor learning and organize a specific motor behaviour suggest similar principles between the motor system and episodic memory systems.
Competition and compensation in the engram.
The well-known concept that “neurons that fire together wire together” applies to the competition among neighbours to link into a memory engram. When CREB is expressed in a neuron, there is an elevation in the excitability of that cell102,143,145. Viral-mediated overexpression of Creb in random neurons in the lateral amygdala biases those cells to allocate into a memory engram. The Creb-overexpressing neurons are more likely than their neighbours to be activated by, and then out-compete, their neighbours to be part of a functional network to store associations of incoming sensory stimuli152. This competition is specific, as these neurons do not form associations outside of the incoming input163. Ablation or inactivation of the Creb-expressing neurons that have been allocated to a memory knocks out the memory152.
Similar to associative learning, the preference for allocating function to neurons with higher intrinsic excitability resulting from Creb induction is present in skilled motor control103. Creb overexpression in a small number of cortical neurons in the motor cortex biases a task function to be allocated to these neurons, such that silencing the activity of these neurons impairs task performance103. Here, neurons with higher excitability from Creb overexpression outcompete neighbouring neurons to form a circuit that underlies motor control. Interestingly, this feature of preferential allocation based on CREB excitability is not limited to a learned skilled behaviour such as handling a piece of pasta, but also non-learned fine motor behaviour such as navigation on a wired grid103. Together, this principle of allocation based on the excitability status of competing neighbouring neurons is generalizable across engrams for associative learning and fine motor control.
In addition to neurons competing to be allocated into a memory engram, memory engrams themselves can compensate to store incoming stimuli if there is dysfunction in the primary circuit that might store that memory147,148,164. Prolonged inhibition of cells normally allocated into a memory engram when paired associations occur to an animal, can be compensated by other groups of cells, to form an engram and store that memory. These studies reveal a degree of flexibility in brain regions to allocate sets of neurons to a brain function, to overcome local dysfunction in the primary area for memory storage.
In motor control, compensation [G] has been observed in stroke recovery165,166. With loss of the inherent motor engram from a stroke, a return of motor function can be a result of true recovery of the previous motor activity and the pattern of integrated movements92, or a process of compensation, whereby alternative movement patterns are integrated into a motor activity that is different from that of the normal, pre-stroke baseline167–171. Compensation might be seen in a patient167–169,172 or experimental animal92,134 adopting more trunk and shoulder use, for example, in an extended upper extremity reach task, rather than the distal limb extension and selective wrist movement that would be normally used to execute the task; or in the use of more proximal joints in the hand in a movement in grasp that is produced after stroke than before stroke. A distinctive feature of this compensation process is that during spontaneous motor recovery, compensation evolves over time and develops new strategies for movement. In other words, compensation is the development of new motor control programs, even if these are not optimal for the particular motor task. As with normal motor learning or movement control, compensation in motor control after stroke probably involves ensembles of cells being allocated into a motor engram or motor neuronal representation. This form of compensation is reflected at the behavioural level in the form of suboptimal performance and is possibly a consequence of ineffective circuit selection. These ineffective circuits that drive a compensatory instead of a recovered behaviour might be in motor circuits in cortex ipsilateral to the affected limb (contralateral to the stroke)173, or in different parts of sensorimotor areas of the surviving cortex on the side of the stroke, as compared to movement-related neuronal networks in the normal pre-stroke condition174. These compensatory motor patterns result in more errors and greater energy consumption, with poorer performance for skilled tasks. The development of compensation in motor control after stroke is a common occurrence and suggests that motor engrams or motor ensembles may compete for movement control, and that a partially damaged motor engram can be compensated by an intact motor engram, just as occurs in memory. Put another way, compensation involves neuronal allocation, and likely selection of co-active neurons into a new circuit. The problem is that, in compensation, the new compensatory motor engram often leads to maladaptive motor patterns that compromise movement quality.
Dendritic spine morphogenesis in engram formation.
Excitatory connections among engram cells are mediated through synaptic contacts on dendritic spines149,151,175,176. Synapses present on engram cells during learning are preferentially stabilized. This same process is not limited to episodic memories. Motor learning promotes stabilization of specific sets of dendritic spines in motor cortex176. Selective silencing of newly formed spines created during learning erases task memory151, providing further evidence of how engrams can be stabilized by means of their strengthened synaptic contacts.
In disease, spine stabilization on memory engram cells is disturbed, and this process can be reversed with artificial activation of the engram cells, such as with optical simulation150,177. These data derive from mouse models of Alzheimer disease but have relevance to recovery from stroke. Dendritic spines are lost in motor cortex after stroke37–39, even in cells distant from the infarct38, and rehabilitation training alters activity patterns in motor cortex to promote spine formation178. Dendritic spine plasticity after stroke may be a potent target to promote behavioural recovery and, as noted below, is likely to follow the rules of the engram in neuronal allocation, competition and compensation.
Stroke and the recovery engram
Emerging evidence suggests that motor recovery from stroke may involve the allocation of neurons into motor engrams with features of these four principles of the memory engram field: allocation, competition, compensation and dendritic spine morphogenesis. Motor recovery after stroke is enhanced when Creb is induced in a subset of neurons in the motor cortex adjacent to the stroke site103. This recovery occurs even though the number of Creb-induced neurons is small (<16% of all motor cortical neurons) and spatially restricted to a local area near the infarct103. Interestingly, this percentage of motor neurons with Creb-induction that enhance motor recovery is similar to that of neuronal allocation into memory engrams143–145. The recovery after stroke mediated by a small number of cells suggests that, as in memory formation, functional recovery after stroke may be controlled through sparse coding of recovered movement control patterns (FIG. 3).
Fig. 3 |. Storage of memory in co-active neuronal networks, or engrams.
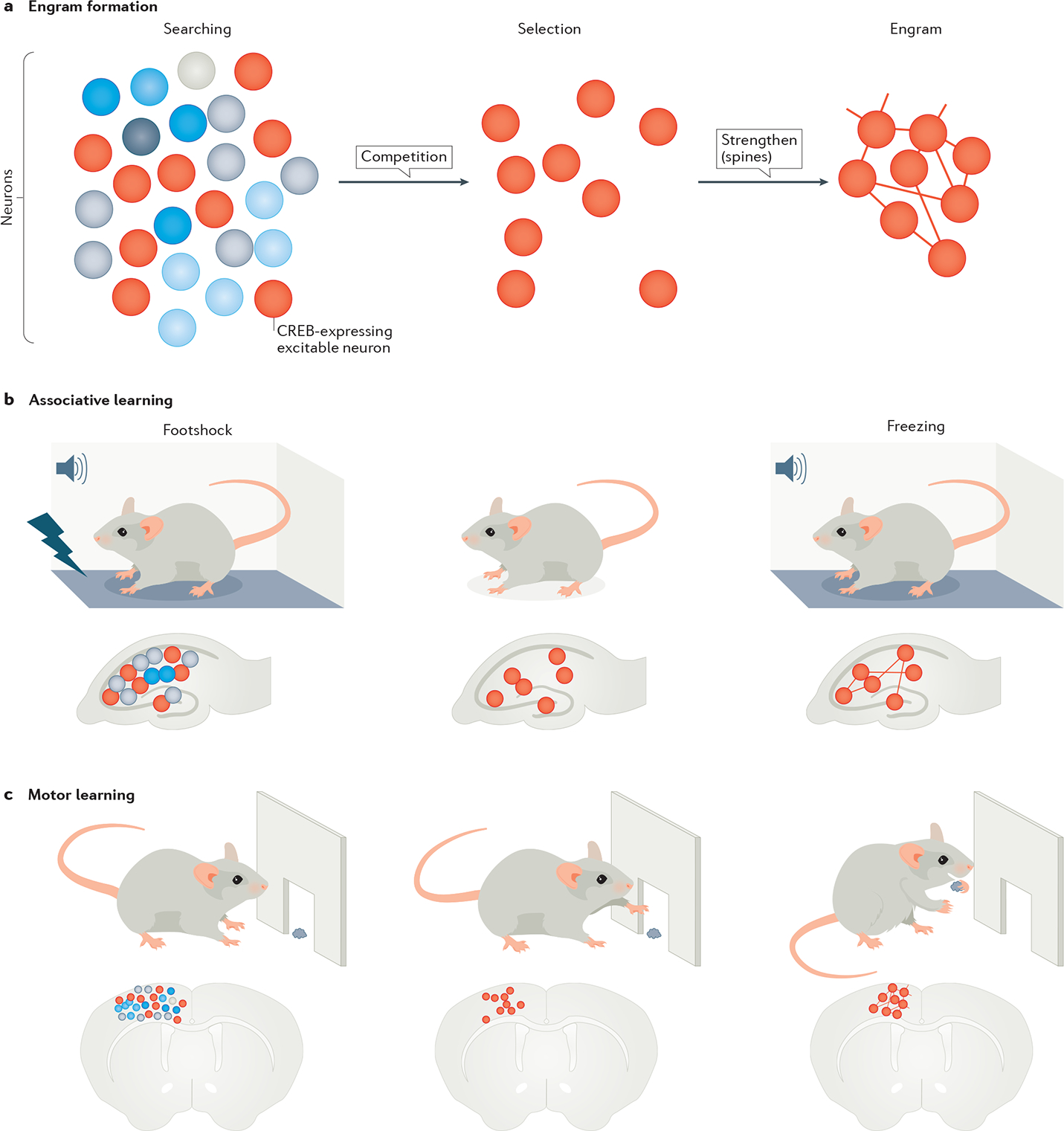
The figure shows the stages of engram formation that store a fear memory or motor memory. a | Engram formation begins with active searching for neurons that are co-active during a behavioural event. From this pool of neurons with similar spatiotemporal activation patterns, neurons with higher cAMP-binding response element (CREB) expression (red circles) are selected to form an engram. The connections within an engram are further strengthened through their dendritic spines that carry post-synaptic information. b | This part shows a collection of neurons in the hippocampus, with those showing CREB expression (red circles) storing a fear memory by associating foot shock with a tone. The fear memory can be recalled by reproducing the tone in the absence of shock and leads to freezing behaviour. The process of recall involves reactivating the memory engram associated with the foot shock. c | This part shows a similar engram in the motor cortex, which stores a motor skill as the motor task is being learnt. The learned function is allocated to a sparse and specific set of neurons initially selected from a large network.
A further element of support in the concept of neuronal allocation in recovery is seen when Creb-induced neurons are inactivated in the motor cortex103. Direct viral gene delivery into the motor cortex near a stroke site, using a virus that delivers Creb and an inhibitory DREADD, allows overexpression of Creb in transduced neurons that can then be selectively silenced when activating a DREADD through administration of its ligand. Inducing Creb induces motor recovery. However, if neurons induced with Creb are then inhibited, the motor deficit of the stroke reappears, and in fact is much worse. Once the Creb-induced neurons escape from this inhibition, enhanced recovery returns. This indicates that Creb-induced neurons influence motor circuits to enhance recovery and that recovery after stroke itself can be turned on and off by inhibiting and releasing Creb-induced neurons. This effect is not seen when this same DREADD inhibitory receptor is activated in neurons that have not been first induced with Creb. This means that Creb induction in a neuron boosts its excitability and allows it to outcompete its neighbours, enhancing this circuity to more fully control the impaired forelimb after stroke. Neuronal allocation and competition in recovering motor circuits are revealed as important principles through this study of the effects of Creb on motor recovery103.
Memory engrams can compensate for dysfunction or inactivation of a brain area. Creb induction in stroke also has an adaptive or compensatory effect if neighbouring brain regions are damaged103. Stroke occurring in the somatosensory cortex alters the cortical mapping of forelimbs and hindlimbs, specifically disrupting limb representation within the stroke area. Spontaneous recovery occurs with a new representation in a more distant location over a delayed period of time31,33. When Creb is induced in the cortex adjacent to stroke, this recovery of the lost limb representation occurs more quickly and in the cortex adjacent to the original representation103. Thus, motor circuits that are enhanced in their excitability and function by Creb induction induce compensation in brain areas adjacent to the site of an infarction, such that body map reorganization [G] occurs more quickly and in a spatially more appropriate manner, possibly enhancing the integrated sensorimotor structure of movement control after stroke179. This aspect of compensation in a motor recovery circuit parallels the compensation of memory engrams during memory formation and retrieval.
Stroke produces an increase in spine turnover in the adjacent cortex that undergoes recovery. As measured by in vivo two-photon imaging, stroke causes either a loss of dendritic spines and then a recovery in the weeks after stroke or an increase in the overall rate of spine turnover37. Motor training promotes spine stabilization in the motor cortex176 and rehabilitative training after stroke promotes stabilization of newly formed dendritic spines178. Hence, similar to dendritic spine formation in engram cells during memory storage, dendritic spine turnover and stabilization in specific motor cortical circuits plays a role in recovery. This concept is strengthened in observations of the effect of molecular systems that play a role in both motor recovery and memory formation. Activation of CCR5 blocks CREB signalling and antagonizes memory formation105. Blocking CCR5 after stroke dramatically promotes recovery of motor function33. One of the mechanisms of action of CCR5 blockade is to reduce dendritic spine loss in the early stages after stroke33. A retention in spines suggests a retention in synaptic connectivity in the region, an overlap with the mechanism of neuronal allocation and an underlying mechanism that leads to improved motor performance. At the molecular level, retention would require a molecular pathway that supports signalling of effector molecules at synaptic sites as a means to stabilize synapses post-stroke. Such mechanisms have been described in the roles of CREB in normal processes of learning as well as dual-leucine zipper kinase (DLK) signalling at synapses180 in other injury systems. CCR5 knockdown induces upregulation of both CREB and DLK proteins in the premotor cortex. These observations point to a phenomenon whereby early recovery can be achieved by stabilizing molecular components for synaptic signalling in the ipsilesional premotor cortex. A conceptual working model could be that neurons with CCR5 knockdown are selectively integrated into a motor circuit by virtue of increased CREB function.
The parallels between the memory engram and recovering circuits after stroke suggest the principle of the motor recovery engram. This similarity highlights the principles of neuronal allocation, competition, compensation and dendritic spine morphogenesis in recovering motor circuits after stroke (FIG. 4). Further studies are required to define specific engrams or a reproducibly identifiable set of motor neurons necessary and sufficient for recovery. Moreover, memory engrams involve changes in interneuron function, such as parvalbumin and somatostatin control of local inhibition181,182. Reductions in parvalbumin interneuron function occur after stroke183 and are apparent with recovery-enhancing drugs in animal models of stroke (such as fluoxetine184), and an interaction of local inhibitory signalling with the allocation of sets of neurons into a recovering motor circuit is likely, but not yet studied. Finally, motor recovery after stroke involves more than local circuits; it also includes the recruitment of adjacent brain areas, and brainstem and spinal circuit reorganization. It is possible that principles of a recovery motor engram mean co-active neuronal networks that competitively associate and recover lost motor function across brain regions.
Fig. 4 |. Recovery engram in stroke.
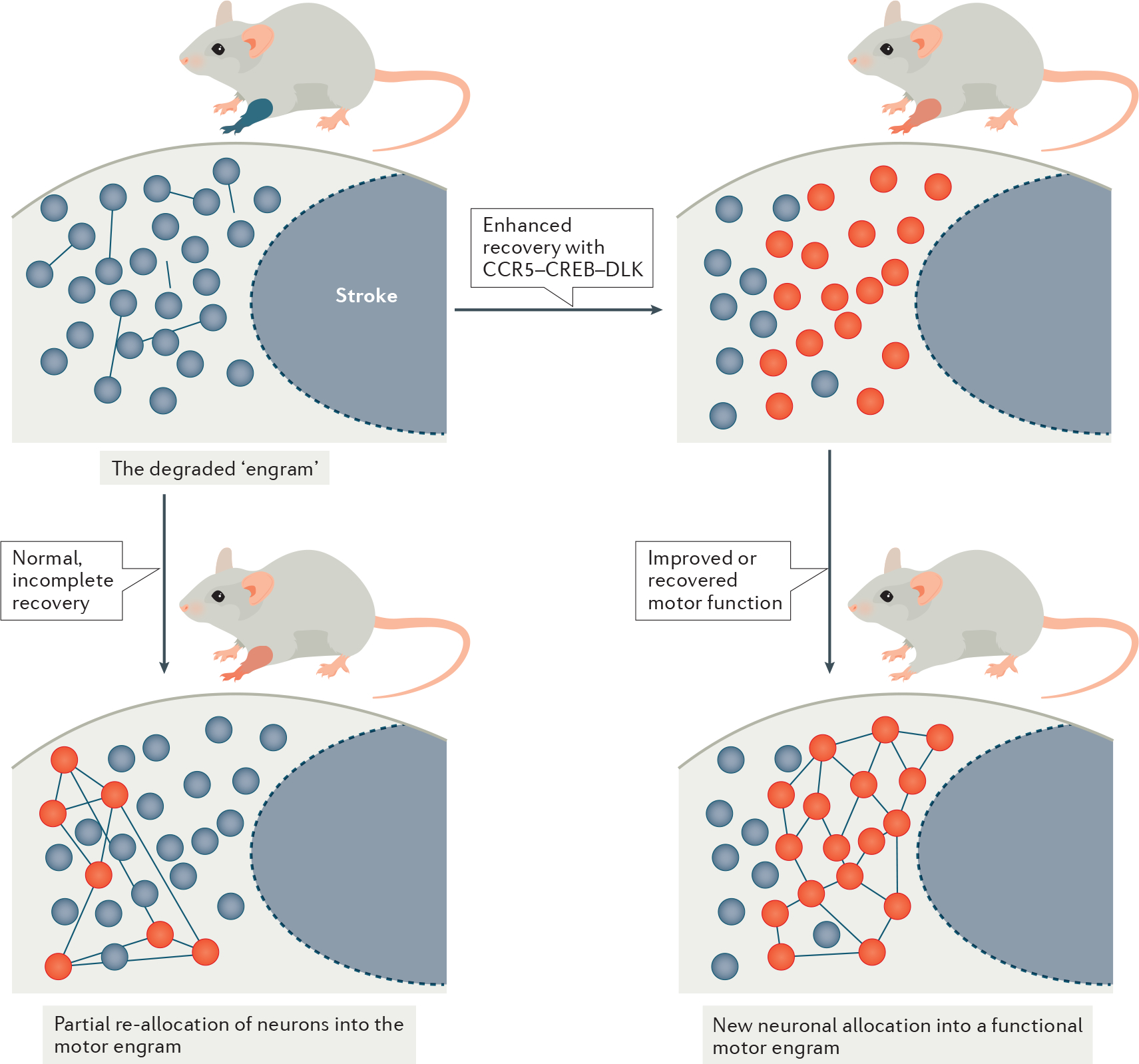
A conceptual model of the engram in stroke recovery. Top left panel: stroke induces a loss of neural connectivity, disrupting the engram that encodes a motor behaviour and, hence, causing a motor impairment. Note that the level of functional impairment is depicted by the colour of the mouse forelimb, ranging from blue (no function) to shades of red (partial recovery) and grey (normal movement). Bottom left panel: endogenous plasticity during the critical period provides a new neural substrate where lost function is partially allocated to a sub-set of neurons (red circles) in a motor circuit. Inefficient allocation leads to spontaneous partial recovery or compensation of motor function. Right panels: increasing the excitability threshold of neurons in peri-infarct by increasing cAMP-binding response element (CREB) expression captures excitable and efficient neurons to allocate into a functional motor circuit for complete motor recovery. CREB–C-C chemokine receptor 5 (CCR5)–dual-leucine zipper kinase (DLK) signalling, which can mediate allocation, may be a target in developing drugs for stroke recovery.
Clinical translation
In line with the concepts of and evidence for learning, recovery and neuronal allocation regulated by tonic GABA signalling, CREB and CCR5 in motor control and motor recovery, pharmacological targeting of these pathways may be potential drug treatment strategies for human stroke recovery. CCR5 is an attractive, druggable target, given that FDA-approved CCR5 antagonists are in clinical use for HIV treatment185. Moreover, humans who carry a 32-base pair deletion in CCR5, rendering the receptor inactive186, have better cognitive outcomes after stroke than individuals without such a deletion33. Additionally, in rodent models of stroke, treatment with the FDA-approved CCR5 antagonist maraviroc (Selzentry®, Pfizer)187 produces functional motor recovery and promotes neural plasticity, such as on dendritic spine preservation and axonal sprouting, as discussed above33. Finally, maraviroc is effective in enhancing cognitive function in rodent models of traumatic brain injury, which shares a similar health burden as stroke33, has limited treatments and shares similar pathological and neurophysiological principles during stroke and stroke recovery. A clinical trial of maraviroc in stroke recovery is under way (NCT03172026)188. Trials of tonic GABAAR inhibitors after stroke have also been planned or undertaken (NCT02928393 (REF.189), NCT02877615 (REF.190)). Studies that target CREB induction with selective phosphodiesterase (PDE)191 inhibition (NCT02013310)192 may provide a pathway forward in stroke, as these enzymes are selectively expressed in distinct regions of the brain, such as the striatum, cortex or hippocampus. Recently, inhibition of PDE10a, which is selectively expressed in the striatum, was shown to improve functional recovery in a mouse model of stroke in the striatum104, with a new drug that has already passed a Phase I clinical trial193. As noted above, fluoxetine may have a mechanism of action that increases the excitatory/inhibitory ratio in the peri-infarct cortex,194 and has gone to clinical trials for stroke recovery, with mixed results184,195(BOX 1).
Box 1 |. Key issues in the translation of stroke repair therapies.
The molecular and cellular concepts of stroke neural repair are poised to translate to the clinic. However, the stroke field has been here before. The last time that a major intellectual understanding of the brain mechanisms of stroke pathophysiology progressed to the clinic, the ship really hit the reef. The failures in the field of stroke neuroprotection occurred at both pre-clinical and clinical ends and have been reviewed extensively197,198. These failures have poisoned the waters for pharmaceutical development in stroke for many years. Unfortunately, the field of stroke neural repair is starting to make the same mistakes. There are several areas of concern, and possible solutions, two of which are highlighted here. There is time to right the ship.
Careful understanding of genetic models in stroke
Mouse genetics has advanced considerably and now allows us to model co-morbid conditions (for example, hypertension and diabetes) in stroke, and to selectively study specific molecular systems in stroke. If used improperly, they will lead to errors. Constitutive knockout mouse models have a selected gene inactivated for the life of the animal in all cell types. This leads to compensation from related gene systems, and also produces its effects in all stages of stroke, early cell stages of cell death and later stages of recovery. Both effects — compensation and the lack of temporal control — prohibit inferences on that gene’s or it’s encoded protein’s effect on stroke recovery. One example of this problem is provided by ephrin A5 signalling. When ephrin A5 expression is selectively reduced only after stroke, there is a potent improvement in axonal sprouting and recovery32. However, when the gene encoding ephrin A5 is constitutively knocked out, there is no effect199 on stroke recovery — probably owing to compensation from other ephrins, as occurs with other axonal growth inhibitors200. Knockout of other neural repair gene systems show profound differences on stroke recovery, depending on whether it is done prior to, during or after stroke201,202. Knockout of a specific chemokine receptor gene produces compensation from related chemokine receptor genes during development and in adulthood203. This means that a constitutive knockout of, for example, the gene encoding C-C chemokine receptor 5 (CCR5) will lead to a lifetime of opportunity for compensation by other signalling systems in mice. This may be why studies in mice in which Ccr5 is constitutively knocked out have produced a confusing picture of enlarged204 or reduced205 stroke size. When Ccr5 is knocked down after the initial stroke, stroke size is unaffected and recovery is enhanced33—indicating the profound difference between a genetic manipulation that occurs indiscriminately through all phases of stroke, versus a targeted genetic manipulation to only the phases of stroke after the initial cell death. A misunderstanding of the role of genetic systems in stroke recovery can influence clinical translation in drug design and development (for example, in ephrin receptor antagonists206), in the rapid development of clinical trials from a discovery study33 or even in the fundamental understanding of whether a specific molecular system has a positive or negative effect on stroke initial damage versus stroke recovery202.
Clinical trial outcome measures
In pre-clinical stroke models, recovery is measured in specific domains of function, such as sensory, motor, spatial and contextual memory areas. However, human stroke recovery trials often default to large-scale behavioural overviews, often in the form of disability scales, that are not sensitive to recovery in specific domains. These disability scales in human recovery outcomes are not aligned with what was demonstrated in stroke recovery trials in the pre-clinical models. Furthermore, these general overview scales lack sensitivity to specific impairments in stroke and their change in stroke recovery. The most potent example of this is the use of two disability scales in human stroke recovery trials, the Modified Rankin Scale (mRS)207 and the Barthel Index208. There is a lack of correlation of these two disability scales with more fine-grained measures of impairment after stroke in humans209,210, and studies have shown that there may be poor recovery by patient self-report yet seeming normalization of function according to the Barthel211 or mRS212. Disability scales measure global function, such as whether a person needs an assistive device to walk or to use their arm, rather than the actual function of the leg or the arm. Promising pre-clinical therapies, such as tonic GABA antagonists65,82 or fluxetine194, the results for which have been replicated in independent labs, have been associated with behavioural recovery according to more fine-grained impairment scales in humans184 but they have shown no effects on disability in human trials with the more global and less sensitive disability scales195,213.
Although therapeutic feasibility is supported through pre-clinical findings, effective clinical translation [G] requires factoring in important considerations for efficacy. The first consideration is the timing of delivery. Targeting excitatory signalling in the acute phase after a stroke exacerbates the stroke response owing to enhancement of excitotoxic signalling pathways17,20. In other words, targeting of a neural repair drug during the cell death phase of stroke may lead to more cell death. The second consideration is the appropriate measurement of outcomes in stroke recovery. Ideally, recovery in a clinical trial of a neural repair therapeutic would track a biomarker of neural repair. Despite intensive research, there is no such biomarker in brain imaging or serum. Instead, recovery in a clinical trial of a neural repair therapeutic relies on behavioural measures. If the behavioural measures are not sensitive to motor control or are influenced by compensation, they will miss the effects on recovery (BOX 1). Nevertheless, insensitive behavioural outcome measures have been applied to neural repair therapy [G] trials. This issue has been extensively discussed196.
Conclusions
As clinical trials progress with drugs that enhance learning and memory, determining the mechanisms through which recovery is manifested and appropriate considerations on the timing, dosage and delivery of these agents will allow for better therapies. These mechanisms include the excitability level of a neuron that leads to selection of that neuron into a circuit of motor recovery—a motor recovery engram. In addition, recovery after stroke is strongly influenced by neurorehabilitation and the behavioral activity of the recovering circuits. More studies that pair motor behaviour with the underlying neural and molecular changes in systems will allow us to define the role of neurorehabilitation in circuit plasticity and recovery after stroke, and delineate adaptive from maladaptive circuit organizations. Moreover, further studies on how circuit function is modulated with learning after injury in a closed-loop system that employs feedback will give us better working models on how the brain rewires in functionally meaningful ways.
Acknowledgements
We thank the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the American Heart Association and the NIH (grant number NS085019) for financial support.
Glossary
- Stroke
Disease caused by the blockade or rupture of a blood vessel to the brain. Loss of blood flow causes death of brain tissue, resulting in long-term neurological impairments.
- Plasticity
Structural or functional changes (or both) within neurons that affect the connectivity of neurons with each other in a network serving a function. Thses changes are usually in response to a change in neuronal input or firing patterns induced by this input, such as during learning or after injury.
- Neuronal circuits
Neurons from either the same brain region or different regions that are connected to each other via synapses to form networks that subserve different brain functions.
- Neuronal excitability
Changes in the electrical properties of a neuron in response to a stimulus.
- Neuronal allocation
The property by which neurons are selected to integrate into a circuit that stores information on a particular stimulus.
- Motor recovery
The return of a motor function where the initial motor patterns prior to injury or a stroke are regained.
- Peri-infarct
Brain tissue that borders the infarct.
- Infarct
The site of tissue loss in the brain from a stroke..
- Dendritic spines
Sub-micron protrusions on dendrites of a neuron that receive synaptic input as post-synaptic elements in the synaptic connection.
- Axonal sprouting
The growth and extension of a new axon and its connections. This may occur from an injured or damaged axon, or through collateralization or extension from an existing axon. The ability to grow new axons is nearly absent in the adult brain except after injury.
- Critical period
A time window during development or after a stroke marked by significant changes in neuronal growth and connectivity shaped by sensory, motor and cognitive experiences.
- Engram
A collection of neurons that fire together in response to a stimulus with higher connectivity with each other and store the memory for that stimulus.
- Map reorganization
Maps are gross anatomical and functional readouts from the activity of a population of neurons that underlie or respond to movement or sensation. Maps can reorganize to take a different spatial location or expand in original territory during learning or in response to injury.
- Molecular memory systems
A network of signalling molecules that underlie memory formation and learning normally regulated by a key set of genes.
- Clinical translation
The transfer of an experimental paradigm identified in an animal or lab-based model of disease to a clinical setting in which patients are treated based on these pre-clinical experimental results.
- Neural repair therapy
A therapeutic strategy that targets genes or signalling molecules underlying plasticity such that stroke recovery can be induced through increased connectivity between relevant brain regions to execute lost function. Neural repair therapies do not minimize cell death that occurs in the initial phases of stroke, but rather utilize and enhance inherent plasticity present in the later stages of stroke by manipulating specific genes, allowing for more effective therapies and greater treatment accessibility to patients who do not qualify for clot removal therapies.
- Compensation (motor)
The return of a motor function using new movement patterns that allow complete or partial execution of a task, often at the expense of higher energy demands and lower movement quality.
- Genetic models in stroke
The use of animal models where a specific gene is inactivated, or its functioning is reduced or increased prior to stroke (usually at birth) or after a stroke.
Footnotes
Competing interests
S.T.C. has received research grant funding from Takeda Phamaceuticals. M.T.J. declares no competing interests.
References
- 1.Benjamin EJ et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 139, e56–e528 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Faul M, Xu L, Wald MM & Coronado VG Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. Atlanta (GA): Centers for Disease Control and Prevention. Natl. Cent. Inj. Prev. Control 2, (2010). [Google Scholar]
- 3.Song SS Advanced imaging in acute ischemic stroke. Semin. Neurol. 33, 436–440 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Fisher M & Albers GW Advanced imaging to extend the therapeutic time window of acute ischemic stroke. Ann. Neurol. 73: 4–9 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Sandhu GS & Sunshine JL Advanced neuroimaging to guide acute stroke therapy. Curr. Cardiol. Rep. 14, 741–753 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Yoo AJ, Pulli B & Gonzalez RG Imaging-based treatment selection for intravenous and intra-arterial stroke therapies: A comprehensive review. Expert Review of Cardiovascular Therapy vol. 9 857–876 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaechter JD Motor rehabilitation and brain plasticity after hemiparetic stroke. Progress in Neurobiology vol. 73 61–72 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Leng T & Xiong Z-G Treatment for ischemic stroke: From thrombolysis to thrombectomy and remaining challenges. Brain Circ. 5, 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cirillo C et al. Post-stroke remodeling processes in animal models and humans. J Cereb Blood Flow Metab 40, 3–22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carmichael ST The 3 Rs of Stroke Biology: Radial, Relayed, and Regenerative. Neurotherapeutics 3, 348–59 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharbrough FW, Messick JM & Sundt TM Correlation of continuous electroencephalograms with cerebral blood flow measurements during carotid endarterectomy. Stroke;4(4):674–683 (1973). [DOI] [PubMed] [Google Scholar]
- 12.Heiss WD, Hayakawa T & Waltz AG Cortical Neuronal Function During Ischemia: Effects of Occlusion of One Middle Cerebral Artery on Single-Unit Activity in Cats. Arch. Neurol;33(12):813–820 (1976). [DOI] [PubMed] [Google Scholar]
- 13.Branston NM, Symon L, Crockard HA & Pasztor E Relationship between the cortical evoked potential and local cortical blood flow following acute middle cerebral artery occlusion in the baboon. Exp. Neurol; 45(2):195–208 (1974). [DOI] [PubMed] [Google Scholar]
- 14.Mies G, Ishimaru S, Xie Y, Seo K & Hossmann KA Ischemic thresholds of cerebral protein synthesis and energy state following middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab;11(5):753–761 (1991) [DOI] [PubMed] [Google Scholar]
- 15.Taylor RA & Sansing LH Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin. Dev. Immunol. 2013, 746068 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anrather J & Iadecola C Inflammation and Stroke: An Overview. Neurotherapeutics 13, 661–670 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carmichael ST Brain excitability in stroke: The yin and yang of stroke progression. Archives of Neurology vol. 69 161–167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernhardt J et al. Efficacy and safety of very early mobilisation within 24 h of stroke onset (AVERT): A randomised controlled trial. Lancet 386, 46–55 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Dromerick AW et al. Very Early Constraint-Induced Movement during Stroke Rehabilitation (VECTORS): A single-center RCT. Neurology 73, 195–201 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarkson AN et al. AMPA receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J. Neurosci. 31, 3766–3775 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernhardt J et al. Agreed Definitions and a Shared Vision for New Standards in Stroke Recovery Research: The Stroke Recovery and Rehabilitation Roundtable Taskforce. Neurorehabil. Neural Repair 31, 793–799 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Corbett D et al. Enhancing the Alignment of the Preclinical and Clinical Stroke Recovery Research Pipeline: Consensus-Based Core Recommendations from the Stroke Recovery and Rehabilitation Roundtable Translational Working Group ∗. Neurorehabil. Neural Repair 31, 699–707 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Ward NS, Brander F & Kelly K Intensive upper limb neurorehabilitation in chronic stroke: outcomes from the Queen Square programme. J. Neurol. Neurosurg. Psychiatry 90, 498–506 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Lo AC et al. Robot-assisted therapy for long-term upper-limb impairment after stroke. N Engl J Med. 362, 1772–83 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duncan PW, Goldstein LB, Matchar D, Divine GW, Feussner J Measurement of motor recovery after stroke. Outcome assessment and sample size requirements. Stroke 23, 1084–1089 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Duncan PW, Lai SM, Keighley J Defining post-stroke recovery: implications for design and interpretation of drug trials. Neuropharmacology 39, 835–841 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Wolf SL et al. The EXCITE stroke trial: comparing early and delayed constraint-induced movement therapy. Stroke. 41, 2309–2315 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gladstone DJ, Danells CJ, Black SE. The fugl-meyer assessment of motor recovery after stroke: a critical review of its measurement properties. Neurorehabil Neural Repair. 16(3):232–240 (2002). [DOI] [PubMed] [Google Scholar]
- 29.McCabe J, Monkiewicz M, Holcomb J, et al. Comparison of robotics, functional electrical stimulation, and motor learning methods for treatment of persistent upper extremity dysfunction after stroke: a randomized controlled trial. Arch Phys Med Rehabil;96:981–90 (2015). [DOI] [PubMed] [Google Scholar]
- 30. Li S et al. An age-related sprouting transcriptome provides molecular control of axonal sprouting after stroke. Nat. Neurosci. 13, 1496–1506 (2010). This study provides evidence for a transcriptional program unique to post-stroke periods in the peri-infarct cortex that supports axonal outgrowth in the adult and aged brain.
- 31.Li S et al. GDF10 is a signal for axonal sprouting and functional recovery after stroke. Nat. Neurosci. 18, 1737–1745 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Overman JJ et al. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci U S A;109(33):E2230–E2239 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Joy MT et al. CCR5 Is a Therapeutic Target for Recovery after Stroke and Traumatic Brain Injury. Cell 176, 1143–1157.e13 (2019). Description of a gene system that is involved in learning and stroke recovery in mouse models and patients with stroke.
- 34.Luke LM, Allred RP & Jones TA Unilateral ischemic sensorimotor cortical damage induces contralesional synaptogenesis and enhances skilled reaching with the ipsilateral forelimb in adult male rats. Synapse 54, 187–199 (2004). [DOI] [PubMed] [Google Scholar]
- 35. Brown CE, Aminoltejari K, Erb H, Winship IR & Murphy TH In vivo voltage-sensitive dye imaging in adult mice reveals that somatosensory maps lost to stroke are replaced over weeks by new structural and functional circuits with prolonged modes of activation within both the peri-infarct zone and distant sites. J. Neurosci. 29, 1719–1734 (2009). This study reports re-organization of limb representations in the motor cortex following a stroke.
- 36.Wahl AS, Omlor W, Rubio JC, et al. Neuronal repair. Asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science. 344(6189):1250–1255 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Brown CE, Wong C & Murphy TH Rapid morphologic plasticity of peri-infarct dendritic spines after focal ischemic stroke. Stroke 39, 1286–1291 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Mostany R, Chowdhury TG, Johnston DG, Portonovo SA, Carmichael ST, Portera-Cailliau C. Local hemodynamics dictate long-term dendritic plasticity in peri-infarct cortex. J Neurosci;30(42):14116–14126 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown CE, Li P, Boyd JD, Delaney KR, Murphy TH. Extensive turnover of dendritic spines and vascular remodeling in cortical tissues recovering from stroke. J Neurosci;27(15):4101–4109 (2007). This study provides evidence for dendritic spine turn over after a stroke.
- 40.Bundy DT & Nudo RJ Preclinical Studies of Neuroplasticity Following Experimental Brain Injury. Stroke;50(9):2626–2633 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harrison TC, Silasi G, Boyd JD & Murphy TH Displacement of sensory maps and disorganization of motor cortex after targeted stroke in mice. Stroke 44, 2300–2306 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Nudo RJ & Milliken GW Reorganization of movement representations in primary motor cortex following focal ischemic infarcts in adult squirrel monkeys. J. Neurophysiol. 75, 2144–2149 (1996). [DOI] [PubMed] [Google Scholar]
- 43.Jaillard A, Martin CD, Garambois K, Lebas JF, Hommel M. Vicarious function within the human primary motor cortex? A longitudinal fMRI stroke study. Brain;128(Pt 5):1122–38 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Cramer SC & Crafton KR Somatotopy and movement representation sites following cortical stroke. Exp. Brain Res. 168(1–2):25–32. (2006) [DOI] [PubMed] [Google Scholar]
- 45.Nishibe M, Urban ETR, Barbay S & Nudo RJ Rehabilitative training promotes rapid motor recovery but delayed motor map reorganization in a rat cortical ischemic infarct model. Neurorehabil. Neural Repair 29, 472–482 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zeiler SR et al. Paradoxical motor recovery from a first stroke after induction of a second stroke: Reopening a postischemic sensitive period. Neurorehabil. Neural Repair 30, 794–800 (2016). This study provides evidence for the existence of a sensitive period after a stroke.
- 47.Dromerick A Critical periods after stroke study (CPASS): does a critical period exist during stroke recovery? Int. J. stroke (2016). [Google Scholar]
- 48.Dromerick AW et al. Critical periods after stroke study: Translating animal stroke recovery experiments into a clinical trial. Front. Hum. Neurosci;9:231 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kraft AW, Bauer AQ, Culver JP & Lee JM Sensory deprivation after focal ischemia in mice accelerates brain remapping and improves functional recovery through Arc-dependent synaptic plasticity. Sci. Transl. Med. (2018). [DOI] [PubMed] [Google Scholar]
- 50.Maravall M, Stern EA & Svoboda K Development of intrinsic properties and excitability of layer 2/3 pyramidal neurons during a critical period for sensory maps in rat barrel cortex. J. Neurophysiol.;92(1):144–156. (2004). [DOI] [PubMed] [Google Scholar]
- 51.Takesian AE, Bogart LJ, Lichtman JW & Hensch TK Inhibitory circuit gating of auditory critical-period plasticity. Nat. Neurosci. 21, 218–227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hensch TK Critical period plasticity in local cortical circuits. Nature Reviews Neuroscience;6(11):877–888. (2005). [DOI] [PubMed] [Google Scholar]
- 53.Wiesel TN & Hubel DH Single-cell responses in striate cortex of kittens deprived of vision in one eye. J. Neurophysiol.;26:1003–1017 (1963). [DOI] [PubMed] [Google Scholar]
- 54.Wang BS, Sarnaik R & Cang J Critical Period Plasticity Matches Binocular Orientation Preference in the Visual Cortex. Neuron;65(2):246–256 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Che A et al. Layer I Interneurons Sharpen Sensory Maps during Neonatal Development. Neuron 2018;99(1):98–116.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Biernaskie J, Chernenko G & Corbett D Efficacy of Rehabilitative Experience Declines with Time after Focal Ischemic Brain Injury. J. Neurosci;24(5):1245–1254 (2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carmichael ST & Chesselet MF Synchronous neuronal activity is a signal for axonal sprouting after cortical lesions in the adult. J. Neurosci. 22, 6062–6070 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McLaughlin T, Torborg CL, Feller MB & O’Leary DDM Retinotopic map refinement requires spontaneous retinal waves during a brief critical period of development. Neuron; 40(6):1147–1160 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Miyata S, Komatsu Y, Yoshimura Y, Taya C & Kitagawa H Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nat. Neurosci.;15(3):414–S2 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Pizzorusso T et al. Reactivation of ocular dominance plasticity in the adult visual cortex. Science;298(5596):1248–1251 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Gherardini L, Gennaro M & Pizzorusso T Perilesional treatment with chondroitinase ABC and motor training promote functional recovery after stroke in rats. Cereb. Cortex; 25(1):202–212 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Hill JJ, Jin K, Mao XO, Xie L & Greenberg DA Intracerebral chondroitinase ABC and heparan sulfate proteoglycan glypican improve outcome from chronic stroke in rats. Proc. Natl. Acad. Sci. U. S. A; 109(23):9155–9160 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yaeger CE, Ringach DL & Trachtenberg JT Neuromodulatory control of localized dendritic spiking in critical period cortex. Nature; 567(7746):100–104 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Conner JM, Chiba AA & Tuszynski MH The basal forebrain cholinergic system is essential for cortical plasticity and functional recovery following brain injury. Neuron; 46(2):173–179 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Clarkson AN, Huang BS, MacIsaac SE, Mody I & Carmichael ST Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 468, 305–309 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Orfila JE et al. Delayed inhibition of tonic inhibition enhances functional recovery following experimental ischemic stroke. J. Cereb. Blood Flow Metab. 39(6):1005–1014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lake EMR et al. The effects of delayed reduction of tonic inhibition on ischemic lesion and sensorimotor function. J. Cereb. Blood Flow Metab. 35(10):1601–1609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takeshi H et al. Enhanced phasic GABA inhibition during the repair phase of stroke: a novel therapeutic target. BRAIN: 139; 468–480 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science;309(5744):2222–2226 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sozmen EG et al. Nogo receptor blockade overcomes remyelination failure after white matter stroke and stimulates functional recovery in aged mice. Proc. Natl. Acad. Sci. U. S. A. 113(52):E8453–E8462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Papadopoulos CM, Tsai S-Y, Alsbiei T, O’Brien TE, Schwab ME and Kartje GL Functional recovery and neuroanatomical plasticity following middle cerebral artery occlusion and IN-1 antibody treatment in the adult rat. Ann Neurol, 51: 433–441 (2002). [DOI] [PubMed] [Google Scholar]
- 72.Markus TM, Tsai SY, Bollnow MR, et al. Recovery and brain reorganization after stroke in adult and aged rats. Ann Neurol;58(6):950–953 (2005). [DOI] [PubMed] [Google Scholar]
- 73.Lindau NT, Bänninger BJ, Gullo M, et al. Rewiring of the corticospinal tract in the adult rat after unilateral stroke and anti-Nogo-A therapy. Brain;137(Pt 3):739–756 (2014). [DOI] [PubMed] [Google Scholar]
- 74.Lim DH, Ledue JM, Mohajerani MH & Murphy TH Optogenetic mapping after stroke reveals network-wide scaling of functional connections and heterogeneous recovery of the peri-infarct. J. Neurosci. 34, 16455–16466 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McDonnell MN, Stinear CM. McDonnell MN, Stinear CM. TMS measures of motor cortex function after stroke: A meta-analysis. Brain Stimul. 2017;10(4):721–734. [DOI] [PubMed] [Google Scholar]
- 76.Shohami E & Biegon A Shohami E, Biegon A. Novel approach to the role of NMDA receptors in traumatic brain injury. CNS Neurol Disord Drug Targets. 2014;13(4):567–573. [DOI] [PubMed] [Google Scholar]
- 77.Di Lazzaro V et al. Motor cortex plasticity predicts recovery in acute stroke. Cereb. Cortex;20(7):1523–1528 (2010). [DOI] [PubMed] [Google Scholar]
- 78. Winship IR & Murphy TH In vivo calcium imaging reveals functional rewiring of single somatosensory neurons after stroke. J. Neurosci. 28(26):6592–6606 (2008). This report provides evidence for neural activity underlying limb selectivity and re-organization in the periods after a stroke.
- 79.Glykys J & Mody I Activation of GABAA Receptors: Views from Outside the Synaptic Cleft.;56(5):763–770 Neuron (2007). [DOI] [PubMed] [Google Scholar]
- 80.Atack JR. Preclinical and clinical pharmacology of the GABAA receptor alpha5 subtype-selective inverse agonist alpha5IA. Pharmacol Ther. 2010;125(1):11–26 (2010). [DOI] [PubMed] [Google Scholar]
- 81.Kim YK, Yang EJ, Cho K, Lim JY & Paik NJ Functional recovery after ischemic stroke is associated with reduced gabaergic inhibition in the cerebral cortex: A GABA PET study. Neurorehabil Neural Repair;28(6):576–583 (2014). [DOI] [PubMed] [Google Scholar]
- 82.Wang YC, Dzyubenko E, Sanchez-Mendoza EH, Sardari M, Silva de Carvalho T, Doeppner TR, Kaltwasser B, Machado P, Kleinschnitz C, Bassetti CL, Hermann DM. Postacute Delivery of GABAA α5 Antagonist Promotes Postischemic Neurological Recovery and Peri-infarct Brain Remodeling. Stroke;49(10):2495–2503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jourdi H, Hsu YT, Zhou M, Qin Q, Bi X, Baudry M. Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J Neurosci;29(27):8688–97 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Clarkson AN, Parker K, Nilsson M, Walker FR & Gowing EK Combined ampakine and BDNF treatments enhance poststroke functional recovery in aged mice via AKT-CREB signaling. J. Cereb. Blood Flow Metab. 35(8):1272–1279 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abe H et al. CRMP2-binding compound, edonerpic maleate, accelerates motor function recovery from brain damage. Science 360(6384):50–57 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Attneave F The organization of behavior: a neuropsychological theory. Amer J Psych.;63:633–5 (1950). [Google Scholar]
- 87.Rioult-Pedotti MS, Friedman D, Hess G & Donoghue JP Strengthening of horizontal cortical connections following skill learning. Nat. Neurosci. 1(3):230–234 (1998). [DOI] [PubMed] [Google Scholar]
- 88.Xu T et al. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature 462, 915–919 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bailey CH, Kandel ER & Harris KM Structural components of synaptic plasticity and memory consolidation. Cold Spring Harb. Perspect. Biol;7(7):a021758 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cheng MY, Woodson WJ, Wang EH, Wang S, Sun G, Lee AG, Arac A, Fenno L, Deisseroth K, Steinberg GK. Optogenetic neuronal stimulation promotes functional recovery after stroke. Proceedings of the National Academy of Sciences 2;111(35):12913–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tennant KA, Taylor SL, White ER, Brown CE. Optogenetic rewiring of thalamocortical circuits to restore function in the stroke injured brain. Nat Commun. 2017;8:15879 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wahl AS, Büchler U, Brändli A et al. Optogenetically stimulating intact rat corticospinal tract post-stroke restores motor control through regionalized functional circuit formation. Nat Commun 8, 1187 (2017). This study shows that optogenetic silencing of corticospinal neurons that have sprouted new connections after a stroke impairs motor function that was regained.
- 93.Blomstedt P., Hariz MI. Are complications less common in deep brain stimulation than in ablative procedures for movement disorders? Stereotact Funct Neurosurg 84(2–3):72–81 (2006). [DOI] [PubMed] [Google Scholar]
- 94.Mandat TS., Hurwitz T., Honey CR. Hypomania as an adverse effect of subthalamic nucleus stimulation: Report of two cases. Acta Neurochir (Wien) 148(8):895–897, discussion 898 (2006). [DOI] [PubMed] [Google Scholar]
- 95. Ramanathan DS et al. Low-frequency cortical activity is a neuromodulatory target that tracks recovery after stroke. Nat. Med. 24, 1257–1267 (2018). This study reports that timed electrical stimulation in rodent models and in a stroke patient can restore lost motor function.
- 96.Biasiucci A et al. Brain-actuated functional electrical stimulation elicits lasting arm motor recovery after stroke.Nat Commun;9(1):2421, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Engineer ND et al. Targeted vagus nerve stimulation for rehabilitation after stroke. Frontiers in Neuroscience; 13:280 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hays SA et al. The timing and amount of vagus nerve stimulation during rehabilitative training affect poststroke recovery of forelimb strength. Neuroreport, 25(9):676–682 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bönstrup M et al. Low-Frequency Brain Oscillations Track Motor Recovery in Human Stroke. Ann. Neurol. 86, 853–865 (2019). [DOI] [PubMed] [Google Scholar]
- 100.Guggenmos DJ et al. Restoration of function after brain damage using a neural prosthesis. Proc. Natl. Acad. Sci. U. S. A. 110(52):21177–21182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kandel ER The molecular biology of memory: CAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Molecular Brain vol. 5; 5:14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sano Y et al. CREB regulates memory allocation in the insular cortex. Curr. Biol. 24, 2833–2837 (2014). This study details neuronal allocation as a result of CREB function.
- 103. Caracciolo L et al. CREB controls cortical circuit plasticity and functional recovery after stroke. Nat. Commun 9(1):2250 (2018). This study shows that overexpression of CREB improves motor function presumably through neuronal allocation.
- 104.Birjandi SZ, Abduljawad N, Nair S, Dehghani M, Suzuki K, Kimura H Carmichael ST Phosphodiesterase 10A Inhibition Leads to Brain Region-Specific Recovery Based on Stroke Type. Transl Stroke Res. 10.1007/s12975-020-00819-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou M et al. CCR5 is a suppressor for cortical plasticity and hippocampal learning and memory. Elife 5:e20985, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Arber S Motor Circuits in Action: Specification, Connectivity, and Function. Neuron 74(6):975–989 (2012). [DOI] [PubMed] [Google Scholar]
- 107.Ohashi H, Gribble PL & Ostry DJ Somatosensory cortical excitability changes precede those in motor cortex during human motor learning. J. Neurophysiol. 122(4):1397–1405 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Biane JS, Scanziani M, Tuszynski MH & Conner JM Motor cortex maturation is associated with reductions in recurrent connectivity among functional subpopulations and increases in intrinsic excitability. J. Neurosci. 2015;35(11):4719–4728 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Costa RM, Cohen D & Nicolelis MAL Differential corticostriatal plasticity during fast and slow motor skill learning in mice. Curr. Biol. 14, 1124–1134 (2004). [DOI] [PubMed] [Google Scholar]
- 110. Peters AJ, Chen SX & Komiyama T Emergence of reproducible spatiotemporal activity during motor learning. Nature 510, 263–267 (2014). This study details searching, selection and refinement of the motor engram during stages of motor learning.
- 111.Li Q et al. Refinement of learned skilled movement representation in motor cortex deep output layer. Nat. Commun. 8:15834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Masamizu Y et al. Two distinct layer-specific dynamics of cortical ensembles during learning of a motor task. Nat. Neurosci. 17(7):987–994 (2014). [DOI] [PubMed] [Google Scholar]
- 113.Makino H, Hwang EJ, Hedrick NG & Komiyama T Circuit Mechanisms of Sensorimotor Learning. Neuron vol. 92 705–721 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Costa RM, Ganguly K, Costa RM & Carmena JM Emergence of Coordinated Neural Dynamics Underlies Neuroprosthetic Learning and Skillful Control. Neuron 93(4):955–970.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 115.Kern M, Bert S, Glanz O, Schulze-Bonhage A & Ball T Human motor cortex relies on sparse and action-specific activation during laughing, smiling and speech production. Commun. Biol; 2:118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chalk M, Marre O & Tkačik G Toward a unified theory of efficient, predictive, and sparse coding. Proc. Natl. Acad. Sci. U. S. A. 115(1):186–191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tang E et al. Effective learning is accompanied by high-dimensional and efficient representations of neural activity. Nat. Neurosci. 22(6):1000–1009 (2019). [DOI] [PubMed] [Google Scholar]
- 118.Simmons PJ & De Ruyter Van Steveninck, R. R. Sparse but specific temporal coding by spikes in an insect sensory-motor ocellar pathway. J. Exp. Biol. 213(Pt 15):2629–2639 (2010). [DOI] [PubMed] [Google Scholar]
- 119.Clemens J, Kutzki O, Ronacher B, Schreiber S & Wohlgemuth S Efficient transformation of an auditory population code in a small sensory system. Proc. Natl. Acad. Sci. U. S. A. 108(33):13812–13817 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wiestler T & Diedrichsen J Skill learning strengthens cortical representations of motor sequences. Elife 2:e00801 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vahdat S, Darainy M, Milner TE & Ostry DJ Functionally specific changes in resting-state sensorimotor networks after motor learning. J. Neurosci. 31(47):16907–16915 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Makino H et al. Transformation of Cortex-wide Emergent Properties during Motor Learning. Neuron 94(4):880–890.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lewis JE & Maler L Synaptic Dynamics on Different Time Scales in a Parallel Fiber Feedback Pathway of the Weakly Electric Fish. J. Neurophysiol 91(2):1064–1070 (2004). [DOI] [PubMed] [Google Scholar]
- 124.Mooney R Neurobiology of song learning. Current Opinion in Neurobiology 19(6):654–660 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pidoux L, Leblanc P, Levenes C & Leblois A A subcortical circuit linking the cerebellum to the basal ganglia engaged in vocal learning. Elife 7:e32167 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ghilardi MF et al. Patterns of regional brain activation associated with different forms of motor learning. Brain Res. 871(1):127–145 (2000). [DOI] [PubMed] [Google Scholar]
- 127.Seidler RD et al. Neural correlates of encoding and expression in implicit sequence learning. Exp. Brain Res. 165(1):114–124 (2005). [DOI] [PubMed] [Google Scholar]
- 128.Ward NS, Brown MM, Thompson AJ & Frackowiak RSJ Neural correlates of outcome after stroke: A cross-sectional fMRI study. Brain 126, 1430–1448 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ward NS, Brown MM, Thompson AJ & Frackowiak RSJ The influence of time after stroke on brain activations during a motor task. Ann. Neurol. 55(6):829–834 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carey LM et al. Evolution of brain activation with good and poor motor recovery after stroke. Neurorehabil. Neural Repair 20(1):24–41 (2006). [DOI] [PubMed] [Google Scholar]
- 131.Askim T, Indredavik B, Vangberg T & Håberg A Motor network changes associated with successful motor skill relearning after acute ischemic stroke: A longitudinal functional magnetic resonance imaging study. Neurorehabil. Neural Repair 23(3):295–304 (2009). [DOI] [PubMed] [Google Scholar]
- 132.Rehme AK, Eickhoff SB, Wang LE, Fink GR & Grefkes C Dynamic causal modeling of cortical activity from the acute to the chronic stage after stroke. Neuroimage 55(3):1147–1158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kawai R et al. Motor Cortex Is Required for Learning but Not for Executing a Motor Skill. Neuron 86(3):800–812 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Whishaw IQ Loss of the innate cortical engram for action patterns used in skilled reaching and the development of behavioral compensation following motor cortex lesions in the rat. Neuropharmacology 39(5):788–805 (2000). This report provides a description of compensated skilled behaviour following a stroke to the rodent cortex.
- 135.Biane JS, Takashima Y, Scanziani M, Conner JM & Tuszynski MH Reorganization of recurrent layer 5 corticospinal networks following adult motor training. J. Neurosci. 39(24):4684–4693 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Biane JS, Takashima Y, Scanziani M, Conner JM & Tuszynski MH Thalamocortical Projections onto Behaviorally Relevant Neurons Exhibit Plasticity during Adult Motor Learning. Neuron 89(6):1173–1179 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang L, Conner JM, Rickert J & Tuszynski MH Structural plasticity within highly specific neuronal populations identifies a unique parcellation of motor learning in the adult brain. Proc. Natl. Acad. Sci. U. S. A. 108(6):2545–2550 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xu T, Yu X, Perlik AJ, Tobin WF, Zweig JA, Tennant K, Jones T, Zuo Y. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 17;462(7275):915–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang L, Conner JM, Nagahara AH & Tuszynski MH Rehabilitation drives enhancement of neuronal structure in functionally relevant neuronal subsets. Proc. Natl. Acad. Sci. U. S. A. 113(10):2750–2755 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tonegawa S, Liu X, Ramirez S & Redondo R Memory Engram Cells Have Come of Age. Neuron 87(5):918–931 (2015). [DOI] [PubMed] [Google Scholar]
- 141.Josselyn SA, Köhler S & Frankland PW Finding the engram. Nature Reviews Neuroscience 16(9):521–534 (2015). [DOI] [PubMed] [Google Scholar]
- 142. Josselyn SA & Tonegawa S Memory engrams: Recalling the past and imagining the future. Science 367(6473):eaaw4325 (2020). A thorough review on memory engrams during normal learning.
- 143.Zhou Y et al. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat. Neurosci. 12, 1438–1443 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gouty-Colomer L, Hosseini B, Marcelo I et al. Arc expression identifies the lateral amygdala fear memory trace. Mol Psychiatry 21, 364–375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Han JH et al. Neuronal competition and selection during memory formation. Science (80-. ). 316, 457–460 (2007). [DOI] [PubMed] [Google Scholar]
- 146.Yiu AP et al. Neurons Are Recruited to a Memory Trace Based on Relative Neuronal Excitability Immediately before Training. Neuron 83(3):722–735 (2014). [DOI] [PubMed] [Google Scholar]
- 147.Cowansage KK et al. Direct Reactivation of a Coherent Neocortical Memory of Context. Neuron 84(2):432–441 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Goshen I et al. Dynamics of retrieval strategies for remote memories. Cell 2011;147(3):678–689 (2011). [DOI] [PubMed] [Google Scholar]
- 149.Frank AC et al. Hotspots of dendritic spine turnover facilitate clustered spine addition and learning and memory. Nat. Commun. 9(1):422 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Roy DS, Muralidhar S, Smith LM & Tonegawa S Silent memory engrams as the basis for retrograde amnesia. Proc. Natl. Acad. Sci. U. S. A. 114(46):E9972–E9979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Hayashi-Takagi A et al. Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525, 333–338 (2015). This study shows that dendritic spine plasticity is an anatomical manifestation of a motor memory, the inhibition of which impairs motor memory and performance.
- 152.Han JH et al. Selective erasure of a fear memory. Science (80-. ). 323, 1492–1496 (2009). [DOI] [PubMed] [Google Scholar]
- 153.Kheirbek MA et al. Differential control of learning and anxiety along the dorsoventral axis of the dentate gyrus. 77(5):955–968 Neuron (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Matos MR et al. Memory strength gates the involvement of a CREB-dependent cortical fear engram in remote memory. Nat. Commun. 10(1):2315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Park S et al. Neuronal Allocation to a Hippocampal Engram. Neuropsychopharmacology 41(13):2987–2993 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu X et al. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature 484(7394):381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ramirez S et al. Activating positive memory engrams suppresses depression-like behaviour. Nature 522(7556):335–339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tanaka YH et al. Thalamocortical Axonal Activity in Motor Cortex Exhibits Layer-Specific Dynamics during Motor Learning. Neuron 100, 244–258.e12 (2018). [DOI] [PubMed] [Google Scholar]
- 159.Sauerbrei BA et al. Cortical pattern generation during dexterous movement is input-driven. Nature 577(7790):386–391 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Guo JZ et al. Cortex commands the performance of skilled movement. Elife 4:e10774 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Peters AJ, Lee J, Hedrick NG, O’neil K & Komiyama T Reorganization of corticospinal output during motor learning. Nat. Neurosci. 20, 1133–1141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Omlor W et al. Context-dependent limb movement encoding in neuronal populations of motor cortex. Nat. Commun. 10(1):4812 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Park A et al. A time-dependent role for the transcription factor CREB in neuronal allocation to an engram underlying a fear memory revealed using a novel in vivo optogenetic tool to modulate CREB function. Neuropsychopharmacology 45(6):916–924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tanaka KZ et al. Cortical Representations Are Reinstated by the Hippocampus during Memory Retrieval. Neuron;84(2):347–354 (2014). [DOI] [PubMed] [Google Scholar]
- 165.Jones TA Motor compensation and its effects on neural reorganization after stroke. Nature Reviews Neuroscience 18(5):267–280 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kwakkel G, Kollen B & Lindeman E Understanding the pattern of functional recovery after stroke: Facts and theories. Restor. Neurol. Neurosci. 22(3–5):281–299 (2004). [PubMed] [Google Scholar]
- 167.Levin MF, Michaelsen SM, Cirstea CM & Roby-Brami A Use of the trunk for reaching targets placed within and beyond the reach in adult hemiparesis. Exp. Brain Res;143(2):171–180. (2002). [DOI] [PubMed] [Google Scholar]
- 168.Shaikh T, Goussev V, Feldman AG & Levin MF Arm-trunk coordination for beyond-the-reach movements in adults with stroke. Neurorehabil. Neural Repair;28(4):355–366 (2014). [DOI] [PubMed] [Google Scholar]
- 169.Michaelsen SM, Jacobs S, Roby-Brami A & Levin MF Compensation for distal impairments of grasping in adults with hemiparesis. Exp. Brain Res. 157(2):162–173 (2004). [DOI] [PubMed] [Google Scholar]
- 170.Nowak DA et al. Dexterity is impaired at both hands following unilateral subcortical middle cerebral artery stroke. Eur. J. Neurosci. 25(10):3173–3184 (2007). [DOI] [PubMed] [Google Scholar]
- 171.Van Kordelaar J, Van Wegen EEH, Nijland RHM, Daffertshofer A & Kwakkel G Understanding adaptive motor control of the paretic upper limb early poststroke: The EXPLICIT-stroke program. Neurorehabil. Neural Repair 27(9):854–863 (2013). [DOI] [PubMed] [Google Scholar]
- 172.Subramanian SK, Yamanaka J, Chilingaryan G & Levin MF Validity of movement pattern kinematics as measures of arm motor impairment poststroke. Stroke 41(10):2303–2308 (2010). [DOI] [PubMed] [Google Scholar]
- 173.Harris-Love ML, Morton SM, Perez MA & Cohen LG Mechanisms of short-term training-induced reaching improvement in severely hemiparetic stroke patients: A TMS study. Neurorehabil. Neural Repair 25(5):398–411 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Grefkes C & Ward NS Cortical reorganization after stroke: How much and how functional? Neuroscientist 20(1):56–70. (2014). [DOI] [PubMed] [Google Scholar]
- 175.Sargin D, Mercaldo V, Yiu AP, et al. CREB regulates spine density of lateral amygdala neurons: implications for memory allocation. Front Behav Neurosci;7:209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fu M, Yu X, Lu J & Zuo Y Repetitive motor learning induces coordinated formation of clustered dendritic spines in vivo. Nature 483, 92–96 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Roy DS et al. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature 531(7595):508–512 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Clark TA et al. Rehabilitative Training Interacts with Ischemia-Instigated Spine Dynamics to Promote a Lasting Population of New Synapses in Peri-Infarct Motor Cortex. J. Neurosci. 39(43):8471–8483. (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cramer SC Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Annals of Neurology vol. 63 272–287 (2008). [DOI] [PubMed] [Google Scholar]
- 180.Goel P & Dickman D Distinct homeostatic modulations stabilize reduced postsynaptic receptivity in response to presynaptic DLK signaling. Nat. Commun. 9(1):1856 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rashid AJ et al. Competition between engrams influences fear memory formation and recall. Science 353(6297):383–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sun X, Bernstein MJ, Meng M, et al. Functionally Distinct Neuronal Ensembles within the Memory Engram. Cell.; 181(2):410–423.e17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zeiler SR et al. Medial premotor cortex shows a reduction in inhibitory markers and mediates recovery in a mouse model of focal stroke. Stroke 44(2):483–489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chollet F, Tardy J, Albucher JF, Thalamas C, Berard E, Lamy C, Bejot Y, Deltour S, Jaillard A, Niclot P, Guillon B, Moulin T, Marque P, Pariente J, Arnaud C, Loubinoux I. Fluoxetine for motor recovery after acute ischaemic stroke (FLAME): a randomised placebo-controlled trial. Lancet Neurol.10(2):123–30 (2011). [DOI] [PubMed] [Google Scholar]
- 185.MacArthur RD & Novak RM Maraviroc: The first of a new class of antiretroviral agents. Clin. Infect. Dis. 47(2):236–241 (2008). [DOI] [PubMed] [Google Scholar]
- 186.Samson M et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382(6593):722–725 (1996) [DOI] [PubMed] [Google Scholar]
- 187.Kuritzkes D, Kar S & Kirkpatrick P Maraviroc. Nat. Rev. Drug Discov. 7, 15–16 (2008). [Google Scholar]
- 188.US National Library of Medicine. CinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03172026 [DOI] [PubMed] [Google Scholar]
- 189.US National Library of Medicine. CinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02928393 (2019). [DOI] [PubMed] [Google Scholar]
- 190.US National Library of Medicine. CinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02877615 (2020). [DOI] [PubMed] [Google Scholar]
- 191.Prickaerts J, Heckman PRA & Blokland A Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opinion on Investigational Drugs 26(9):1033–1048 (2017). [DOI] [PubMed] [Google Scholar]
- 192.US National Library of Medicine. CinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02013310 (2015). [DOI] [PubMed] [Google Scholar]
- 193.Yurgelun-Todd DA, Renshaw PF, Goldsmith P, Uz T, Macek TA. A randomized, placebo-controlled, phase 1 study to evaluate the effects of TAK-063 on ketamine-induced changes in fMRI BOLD signal in healthy subjects. Psychopharmacology (Berl). 237(2):317–328 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ng KL, Gibson EM, Hubbard R, Yang J, Caffo B, O’Brien RJ, Krakauer JW, Zeiler SR. Fluoxetine Maintains a State of Heightened Responsiveness to Motor Training Early After Stroke in a Mouse Model. Stroke;46(10):2951–60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.FOCUS Trial Collaboration. Effects of fluoxetine on functional outcomes after acute stroke (FOCUS): a pragmatic, double-blind, randomised, controlled trial. Lancet. 19;393(10168):265–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Dobkin BH & Carmichael ST The Specific Requirements of Neural Repair Trials for Stroke. Neurorehabil. Neural Repair 30(5):470–478 (2016). This report provides discussions on the design of neural repair trials in stroke research.
- 197.Savitz SI, Fisher M. Future of neuroprotection for acute stroke: in the aftermath of the SAINT trials. Ann Neurol. 2007;61(5):396–402 (2007). [DOI] [PubMed] [Google Scholar]
- 198.O’Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol;59(3):467–77 (2006). [DOI] [PubMed] [Google Scholar]
- 199.de Boer A, Storm A, Rué L, Poppe L, Robberecht W, Lemmens R. Heterozygous Deletion of EphrinA5 Does Not Improve Functional Recovery After Experimental Stroke. Stroke;50(4):e101 (2019). [DOI] [PubMed] [Google Scholar]
- 200.Kempf A, Montani L, Petrinovic MM, Schroeter A, Weinmann O, Patrignani A, Schwab ME. Upregulation of axon guidance molecules in the adult central nervous system of Nogo-A knockout mice restricts neuronal growth and regeneration. Eur J Neurosci. Dec;38(11):3567–79 (2013). [DOI] [PubMed] [Google Scholar]
- 201.Lee JK, Kim JE, Sivula M, Strittmatter SM. Nogo receptor antagonism promotes stroke recovery by enhancing axonal plasticity. J Neurosci.7;24(27):6209–17 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kilic E, ElAli A, Kilic U, Guo Z, Ugur M, Uslu U, Bassetti CL, Schwab ME, Hermann DM. Role of Nogo-A in neuronal survival in the reperfused ischemic brain. J Cereb Blood Flow Metab;30(5):969–84 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Dyer DP, Medina-Ruiz L, Bartolini R, Schuette F, Hughes CE, Pallas K, Vidler F, Macleod MKL, Kelly CJ, Lee KM, Hansell CAH, Graham GJ. Chemokine Receptor Redundancy and Specificity Are Context Dependent. Immunity. 19;50(2):378–389.e5 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sorce S, Bonnefont J, Julien S, Marq-Lin N, Rodriguez I, Dubois-Dauphin M, Krause KH. Increased brain damage after ischaemic stroke in mice lacking the chemokine receptor CCR5. Br J Pharmacol.;160(2):311–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Victoria ECG, de Brito Toscano EC, de Sousa Cardoso AC, da Silva DG, de Miranda AS, da Silva Barcelos L, Sugimoto MA, Sousa LP, de Assis Lima IV, de Oliveira ACP, Brant F, Machado FS, Teixeira MM, Teixeira AL, Rachid MA. Knockdown of C-C Chemokine Receptor 5 (CCR5) is Protective Against Cerebral Ischemia and Reperfusion Injury. Curr Neurovasc Res.;14(2):125–131 (2017). [DOI] [PubMed] [Google Scholar]
- 206.Giorgio C, Incerti M, Pala D, Russo S, Chiodelli P, Rusnati M, Cantoni AM, Di Lecce R, Barocelli E, Bertoni S, Ravassard P, Manenti F, Piemonti L, Ferlenghi F, Lodola A, Tognolini M. Inhibition of Eph/ephrin interaction with the small molecule UniPR500 improves glucose tolerance in healthy and insulin-resistant mice. Pharmacol Res; 141:319–330 (2019). [DOI] [PubMed] [Google Scholar]
- 207.Broderick JP, Adeoye O, Elm J. Evolution of the Modified Rankin Scale and Its Use in Future Stroke Trials. Stroke.; 48(7):2007–2012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Quinn TJ, Langhorne P, Stott DJ. Barthel index for stroke trials: development, properties, and application. Stroke;42(4):1146–1151. doi: 10.1161/STROKEAHA.110.598540 (2011). [DOI] [PubMed] [Google Scholar]
- 209.Steinberg GK, Kondziolka D, Wechsler LR, Lunsford LD, Coburn ML, Billigen JB, Kim AS, Johnson JN, Bates D, King B, Case C, McGrogan M, Yankee EW, Schwartz NE. Clinical Outcomes of Transplanted Modified Bone Marrow-Derived Mesenchymal Stem Cells in Stroke: A Phase 1/2a Study. Stroke. Jul;47(7):1817–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wu WX, Zhou CY, Wang ZW, Chen GQ, Chen XL, Jin HM, He DR. Effect of Early and Intensive Rehabilitation after Ischemic Stroke on Functional Recovery of the Lower Limbs: A Pilot, Randomized Trial. J Stroke Cerebrovasc Dis.;29(5):104649 (2020). [DOI] [PubMed] [Google Scholar]
- 211.Mayo NE, Wood-Dauphinee S, Côté R, Durcan L, Carlton J. Activity, participation, and quality of life 6 months poststroke. Arch Phys Med Rehabil;83(8):1035–42 (2002). [DOI] [PubMed] [Google Scholar]
- 212.Stewart JC, Cramer SC. Patient-reported measures provide unique insights into motor function after stroke. Stroke;44(4):1111–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Chabriat H, Bassetti CL, Marx U, Audoli-Inthavong ML, Sors A, Lambert E, Wattez M, Hermann DM; RESTORE BRAIN study investigators. Safety and efficacy of GABAA α5 antagonist S44819 in patients with ischaemic stroke: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet Neurol.;19(3):226–233 (2020). [DOI] [PubMed] [Google Scholar]