

Abstract
Alzheimer’s disease (AD) research has entered a new era with the recent positive phase 3 clinical trials of the anti-Aβ antibodies lecanemab and donanemab. Why did it take 30 years to achieve these successes? Developing potent therapies for reducing fibrillar amyloid was key, as was selection of patients at relatively early stages of disease. Biomarkers of the target pathologies, including amyloid and tau PET, and insights from past trials were also critical to the recent successes. Moving forward, the challenge will be to develop more efficacious therapies with greater efficiency. Novel trial designs, including combination therapies, umbrella and basket protocols, will accelerate clinical development. Better diversity and inclusivity of trial participants are needed, and blood-based biomarkers may help to improve access for medically underserved groups. Incentivizing innovation in both academia and industry through public-private partnerships, collaborative mechanisms and creating new career paths will be critical to build momentum in these exciting times.
Introduction
After decades of disappointing attempts, the field of Alzheimer’s Disease (AD) research achieved an important milestone in 2022, the positive result of a Phase 3 clinical trial of a potentially disease modifying treatment for AD. The anti-Aβ monoclonal antibody (mAb) lecanemab demonstrated significant beneficial effects on multiple clinical and biomarker endpoints.1 In a large, randomized, placebo-controlled clinical trial involving 1,795 patients with prodromal to mild AD, lecanemab was able to slow the rate of decline of a global measure of clinical status by 27% as compared to placebo (−0.45 [95%CI: −0.67 to −0.23] Clinical Dementia Rating Scale sum of boxes [CDR-SB]) after 18 months of treatment. Similar benefits were observed on all other clinical outcome measures and were supported by substantial reductions or attenuations of multiple AD pathobiology biomarkers including plasma measures of phosphorylated tau (P-tau181) and glial fibrillary acid protein (GFAP), as well as reductions in Aβ plaques and attenuation of insoluble tau deposition in the brain measured by PET. In 2023, a positive Phase 3 trial of another amyloid plaque-clearing mAb, donanemab, was also reported, with a similar 29% (combined population: −0.7 [95%CI, −0.95 to −0.45] CDR-SB) reduction in disease progression, supporting the validity of the lecanemab results. 2 In the primary analysis focused on individuals with low/intermediate levels of insoluble tau by PET, a larger effect of donanemab was observed (40%; low/medium tau: −0.67 [95%CI, −0.95 to −0.40]). Lecanemab received an accelerated FDA approval in January 2023 based on the substantial reduction in amyloid plaques, followed by a full approval based on slowing of clinical progression in June 2023, but whether the modest degree of slowing in clinical progression is sufficiently clinically meaningful (producing a noticeable benefit to patients and caregivers) 3 to outweigh the risks of side effects and costs of treatment is still a topic of active debate among clinicians, public health officials and insurance companies. Nonetheless, the two Phase 3 results were important because these interventional human experiments proved that the amyloid hypothesis of AD must be at least partially correct and demonstrated that creating disease modifying drugs for AD and related disorders is feasible. This proof of concept provides a much-needed shot of encouragement to drug development researchers in academia and at pharmaceutical companies, some of whom have abandoned CNS work due to previous failed or negative AD clinical trials. The consistency of the lecanemab and donanemab results also help put to rest some of the criticisms about anti-Aβ therapies that arose after the highly controversial accelerated approval of the related anti-Aβ mAb, aducanumab, which showed inconsistent findings across two interrupted Phase 3 trials in 2021. 4
While drugs such as acetylcholinesterase inhibitors and memantine that transiently stabilize the cognitive and functional consequences of AD have been approved and widely used for decades, they don’t reduce the accumulation of underlying AD neuropathology and would not have much of impact on the predicted steep increase in the number of AD patients in the US (and elsewhere) due to the aging of the Baby Boomer population. 5 Approximately three decades ago, soon after the formulation of the amyloid cascade hypothesis 6 and construction of the first human transgenic mouse models of AD that overexpressed the human amyloid precursor protein (APP), 7 Schenk and co-workers demonstrated that immunization of the mice with a full length human Aβ1–42 peptide could substantially reduce the accumulation of insoluble deposits of human Aβ in the mouse brain. 8 This seminal experiment spurred the development of the first human immunotherapeutic approaches to AD including the active vaccine AN1792 that was abandoned shortly after the initiation of human clinical trials due to the emergence of a severe adverse event (side effect), meningoencephalitis, that led to death in handful of patients, 9. Nevertheless, this motivated the development of a series of anti-amyloid mAbs, designed to be safer and able to deliver higher levels of antibody titers than the vaccine, culminating in the more recent amyloid plaque-clearing mAbs, aducanumab, gantenerumab, donanemab and lecanemab, that have been the first to produce large amyloid PET changes and positive clinical findings in early AD.
Why did it take almost 30 years to achieve this milestone? While developing the right drugs was clearly important, much of the recent success is also attributable to advances in AD biomarkers and clinical trial methods that occurred in parallel with or often as a result of learning from failed trials that didn’t adequately test the therapeutic hypothesis (for example, in the case of tarenflurbil that did not achieve sufficient CNS drug concentrations), 10 or negative trials that demonstrated CNS target engagement and pharmacodynamic effects, but still did not lead to a clinical benefit. Therefore, past AD trials cannot be written off purely as failures, and when analyzed in comparison to the more recent plaque targeting mAb trials, the reasons for their lack of success are now relatively well-understood. 11
Even if lecanemab and donanemab become widely used, the public health burden of AD and related dementias is rapidly growing, and it is unclear how quickly these and related anti-Aβ therapies can reduce that burden. How do we achieve the remaining ~70% slowing of disease progression to completely arrest the disease? Will it take another 30 years to find other treatments that have substantially larger clinical benefits and fewer side effects? The average Phase 3 AD clinical trial costs approximately $370M (adjusted for inflation), involves hundreds to thousands of participants and lasts 3–4 years or longer. 12 13 What therapeutic approaches should be prioritized? Can we conduct more efficient clinical trials that are less expensive, faster to complete and less burdensome to participants? In this perspective, we highlight some of the barriers and new technologies that have played a role in the development of anti-Aβ therapies from academic clinical trialists’ perspectives and suggest strategies to accelerate development of more effective treatments. One of this perspective’s goals is to highlight advances in biomarkers and clinical trial insights that enabled the recent amyloid immunotherapy milestones (Figure 1). We refer the reader to recent reviews for information on other unsuccessful anti-Aβ therapeutic approaches such as gamma and beta secretase inhibitors, clinical trials of which have produced important biological and therapeutic insights based on unexpected treatment-related worsening of cognition in some individuals. 14
Figure 1. Insights from past clinical trials and new approaches.
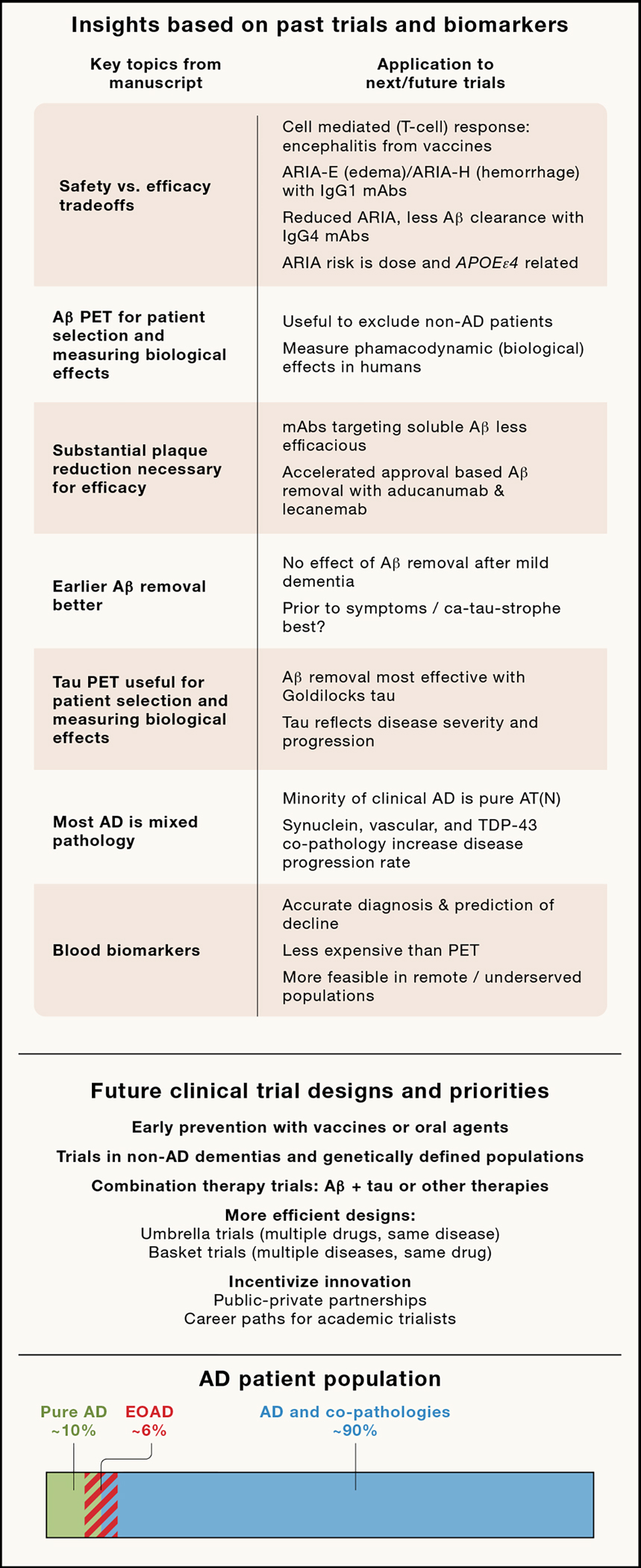
(Top panel) Insights from past biomarker development and clinical trials. (Middle panel) New trial designs and priorities to accelerate clinical development. (Bottom panel) Bar represents percent estimates based on autopsy cohorts of clinical AD cases with pure AD (ATN pathology without co-pathology), AD with co-pathology (e.g., ATN plus vascular, α-synuclein and/or TDP-43) 75 and relative percentage of Early Onset AD (EOAD; first symptoms before age 65).
Key insights from previous AD clinical trials
Safety versus therapeutic efficacy tradeoffs.
AN1792, the first attempt at active immunization in AD, was abandoned due to concerns regarding meningoencephalitis that likely emerged due to reformulation of the vaccine with a denaturing preservative that exposed epitopes and generated cytotoxic t-cell responses in some individuals (Figure 2A) 15. Still, post hoc analyses of clinical measures in patients who mounted high titer vaccine responses showed that there were clearly observable trends toward clinical benefits along with paradoxically increased brain atrophy measured on longitudinal volumetric MRI scans. 16,17 This potential clinical benefit of AN1792 suggested that amyloid removal could be a viable treatment approach, but that safer therapies were needed. One subsequent approach was to design mAbs that bound to Aβ peptide epitopes that would not produce a cell-mediated response, but could still clear amyloid, with bapineuzumab, a humanized IgG1 backbone mAb that neutralized a variety of soluble Aβ species being the first to reach late-stage trials (Figure 3). 18,19 In the Phase 1 single ascending dose study, the drug was safe and well tolerated, but three out of ten participants who received the highest dose developed vasogenic edema (mild brain swelling visible on MRI that looked similar to other diseases with leaky blood vessels), with two having no symptoms, but one having mild, transient confusion. 19 This adverse event (AE) was later termed Amyloid Related Imaging Abnormalities-Edema (ARIA-E) to differentiate it from rare hemorrhagic events observed on blood sensitive MRI sequences termed ARIA-H 20.
Figure 2. History of Aβ immunotherapy.
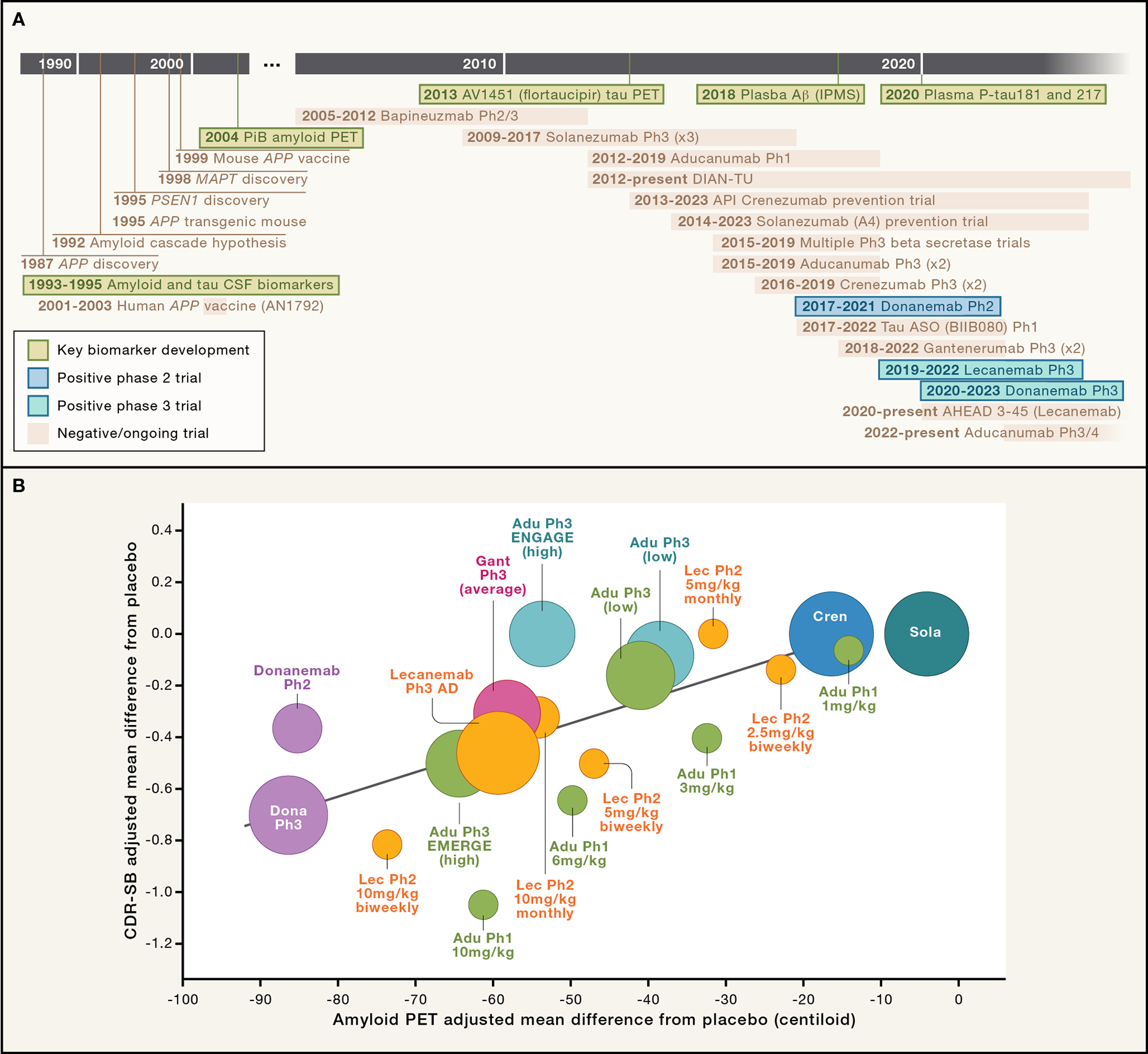
(A) Timeline from discovery of amyloid precursor protein (APP) mutations as a cause of early onset AD, nonclinical proof of concept in PDAPP mouse model, showing selected clinical trial programs and biomarker discoveries (green boxes) discussed in the text through the present day lecanemab and donanemab programs. Timeline is not meant to be comprehensive and does not include many important biomarker and clinical trial programs. PiB = Pittsburgh Compound B [first widely used amyloid PET ligand]; P-tau181 and 217 = phosphorylated tau at residues 181 or 217; ASO= antisense oligonucleotide; IPMS = immunoprecipitation mass spectrometry. Clinical trial dates from clinical trials.gov: NCT00112073, NCT00574132, NCT02760602, NCT00905372, NCT01900665, NCT01677572, NCT01760005, NCT01998841, NCT02008357, NCT02484547, NCT02477800, NCT03114657, NCT02670083, NCT03367, NCT03186989, NCT03887455, NCT04437511, NCT04468659, NCT05310071. (B) Amyloid plaque reduction is correlated with clinical benefit. Treatment versus placebo difference in change in CDR-SB correlated with amyloid PET centiloid difference in change over time with various anti-Aβ mAbs including lecanemab (Lec), aducanumab (Adu), donanemab (Dona) and solanezumab (Sola). Note that solanezumab Phase 3 estimates are from previous Phase 3 studies in sporadic AD, as well as the A4 Study and subset of DIAN-TU study that was conducted in a preclinical (asymptomatic) AD population. Crenezumab (Cren) estimates includes the API Study in preclinical AD population. Adapted with recent updates from similar depictions in. 11,36
Figure 3: Stages of clinical development for typical AD new molecular entity drugs.
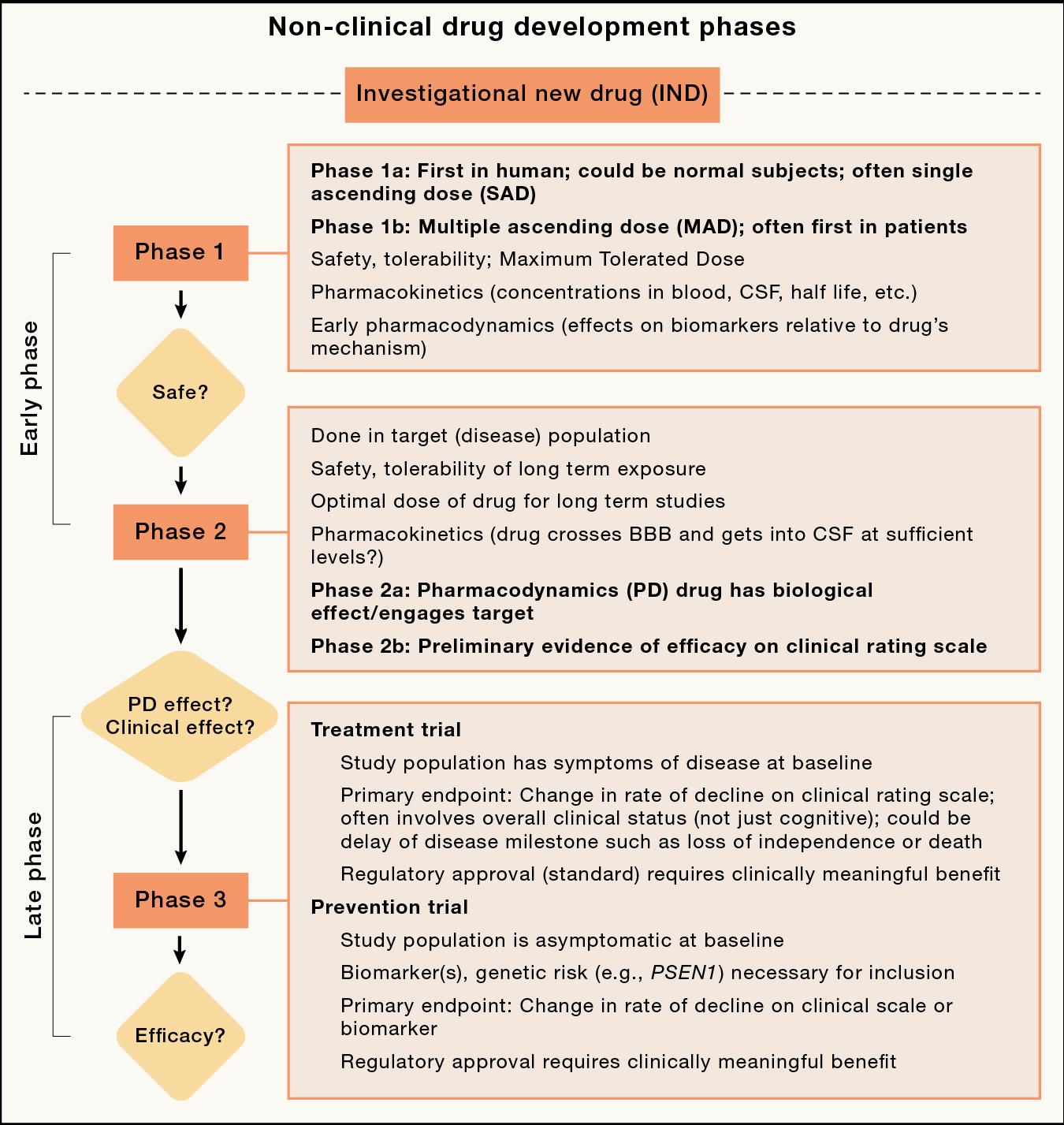
Nonclinical refers to in vitro, cell culture and animal model studies prior to moving forward with first in human clinical trials through an Investigational New Drug (IND) or equivalent process.
ARIA was recognized as an AE of special interest and was closely monitored in subsequent studies of anti-Aβ therapies as a potential dose-limiting toxicity. In early clinical trials such as the bapineuzumab Phase 2 and 3 studies, individuals who developed even mild ARIA were not allowed to receive further treatment. 18 Further trials demonstrated that ARIA risk increased both with the number of APOEe4 alleles present in each individual as well as the dose of drug administered, and that it was associated with greater amyloid reduction on PET. 21 ARIA’s similar neuroradiological appearance and the presence of auto immune anti-Aβ mAbs in some cases of inflammatory cerebral amyloid angiopathy further suggested that ARIA may be mediated by an inflammatory response 22. From these findings, a number of strategies were adopted (often at the request of regulators such as the EMA and FDA) to lower the risk of ARIA in subsequent clinical trials including: 1) limiting the dose of mAb administered, with lower maximal doses in APOEe4 carriers; 2) developing new mAbs that were genetically engineered to reduce the risk of ARIA by reducing immune effector function, for example by engineering IgG4 backbones that do not activate microglia; 23 and 3) limiting participation in subsequent trials of participants who already had evidence of significant vascular amyloid (cerebral amyloid angiopathy) detectable as previous micro- or macro-hemorrhages on iron-sensitive MRI sequences. The first two strategies were generally successful at reducing ARIA incidence, however despite the ability to use much higher doses of the newer IgG4 antibodies, such as crenezumab23 and solanezumab, 24 these trials produced little evidence of amyloid reduction or clinical efficacy. This lack of efficacy was likely multifactorial, related to both targeting the wrong Aβ species as well as the lack of an effector domain to help engage the immune system in Aβ clearance.
ARIA-related dosing challenges.
More problematic for recent clinical trials was the requirement to use lower doses of anti-Aβ mAbs in APOEe4 carriers, for example after the emergence of higher rates of ARIA in the Phase 2 lecanemab trial in a small number of participants who initially received a dose of 10 mg/kg biweekly, to mitigate further ARIA risk. 25 Although this strategy successfully reduced ARIA rates, there was also less clinical effect at the lower dose. Fortunately, in the Phase 3 lecanemab trials, both carriers and non-carriers received the higher dose of lecanemab with a treatment related Aβ reduction of 59 centiloids (CL, a standardized PET measure of amyloid levels).1 Similarly, in the Phase 3 aducanumab trials, the initial protocols required lower doses of aducanumab in APOEe4 carriers. After the trials were underway, a greater understanding of the relatively rare clinical sequelae of most ARIA-E led to protocols to continue treatment with mild ARIA. Subsequently, the protocols were revised further to allow higher dose treatment in APOEe4 carriers. 26 The aducanumab Phase 3 trials were stopped early after interim analyses suggested that the drug was not effective. These futility analyses evaluated pooled results across the two Phase 3 trials, but in one of the trials (EMERGE) APOEe4 carrier individuals only received limited higher doses of drug. When these additional data were included in the analyses at the time of study close-out, the other Phase 3 study (ENGAGE) demonstrated substantial slowing of disease progression and was thought to be a positive trial. 26 These analyses and the subsequent FDA accelerated approval were highly controversial because the approval was based on a surrogate endpoint, amyloid PET-measured plaque reduction, that many experts felt did not meet the requirement that a surrogate endpoint be “sufficiently predictive of clinical benefit” (many people have high levels of amyloid PET-measured plaques, but no clinical symptoms of AD). 27 There were also a variety of other deviations from normal FDA practices that were subsequently described in a detailed report after a Congressional investigation of aducanumab’s accelerated approval. 28 Some experts felt that the analyses departed from standard clinical trial practice (and regulatory policies) that key analyses must be pre-specified at the beginning of a trial (usually as the primary endpoint), and not done post hoc after the data have been collected. Finally, many felt that Biogen’s subsequent decision to initially set the price of aducanumab at $56,000/year was unjustified since clinical benefits of treatment were not convincingly demonstrated. 4
More work is still needed to fully understand the pathogenesis of ARIA-E and in particular, to develop accurate predictors of risk for the relatively rare symptomatic cases. It remains unknown whether ARIA is primarily an inflammatory response, related to movement of Aβ from plaque to perivascular space, or direct removal of amyloid from leptomeningeal and cortical arterioles resulting in transient “leaky vessels”, or some combination of pathophysiological processes. 22 It is clear that the peak blood concentration (C-max) of antibody dose and APOE genotype influence risk of ARIA-E, but there are differential rates of ARIA among plaque clearing antibodies. The finding that lecanemab, based on the APP Artic mutation that is associated with less CAA, 29 has somewhat lower rates of ARIA-E with similar levels of amyloid reduction on PET than what has been reported with donanemab and aducanumab may be informative.
Importance of amyloid PET for selecting patients and measuring biological effects.
Another key insight from the Phase 2 bapineuzumab trial, which was the first to use amyloid PET in a substantial number of participants, was that approximately 30% of the APOEe4 non-carriers did not have substantial amyloid plaques in their brains, and therefore likely didn’t have AD. 30 Some may have had an AD-related dementia, such as frontotemporal dementia (FTD), or even non-neurodegenerative causes of impairment. Subsequent AD clinical trials have used either amyloid PET or CSF measures of Aβ and tau as key inclusion criteria (diagnostic biomarkers) to ensure that AD is the cause of patients’ clinical syndrome or more recently, in prevention clinical trials, such as the Anti-Aβ Treatment in Asymptomatic Alzheimer’s study (A4)31 and AHEAD 32, to identify asymptomatic elderly participants who are at risk for developing AD due to the presence of substantial amounts of amyloid plaques in their brains.
Substantial plaque reduction required for efficacy.
Amyloid PET has also been highly informative as a treatment response (pharmacodynamic) biomarker to identify clinically relevant effects of anti-Aβ therapies. The phase 1b multiple ascending dose trial of aducanumab demonstrated substantial slowing of clinical disease progression in patients treated with higher doses of drug, and this closely corresponded to the amount of amyloid plaque removed as measured by longitudinal amyloid PET scans over the course of a year 33. Only individuals with substantial reductions in brain amyloid experienced slowing of clinical progression. Because there is greater power to measure changes on Aβ PET than clinical measures (due to a much better signal/noise ratio with imaging versus clinical assessments), this key demonstration enabled others to use amyloid PET changes as a key proof of concept biomarker for the development of other anti-Aβ mAbs. Similarly, donanemab, which demonstrated clinical efficacy in a Phase 2b clinical trial 34 and the recent Phase 3 trial, 2 showed dramatic reductions in amyloid PET signal in early-stage trials, providing confidence that it should be tested for clinical benefits 35.
Recent retrospective analyses of amyloid PET data comparing the results of the four anti-Aβ mAb trials with the newer “plaque-busting” mAbs that demonstrated substantial treatment-related amyloid PET signal reductions, including aducanumab, donanemab, gantenerumab and lecanemab, appear to have clarified a key relationship between brain amyloid reduction and clinical efficacy (Figure 2B). 11 Significant clinical efficacy was strongly related to the amount of amyloid (measured in centiloids) removed by the brain. The clinical trials, including the donanemab phase 2 and 3, the positive EMERGE Phase 3 aducanumab and the Phase 3 lecanemab trials, that reduced amyloid PET signal from high levels seen in symptomatic AD, often 70–120 CL, to normal or low levels seen very early in preclinical AD, below 24 CL (on average a >71% reduction over 18 months of treatment), demonstrated clinical efficacy. The negative phase 3 aducanumab ENGAGE and Phase 3 gantenerumab trials reduced brain amyloid, as measured by PET, but not sufficiently to lower levels below the 24 CL threshold in most patients. Applying a similar amyloid PET signal reduction metric to previous negative phase 3 clinical trials, such as with crenezumab and solanezumab, shows that these two drugs produced marginal 18-month amyloid reductions in Phase 3 trials, providing additional support for the hypothesis that substantial reduction of amyloid plaque levels is likely necessary for clinical efficacy. 11,36 The supposition that amyloid removal would be reasonably likely to predict clinical benefit was the basis for the FDA’s controversial accelerated approval of aducanumab and less controversial accelerated approval of lecanemab for early AD, and fortunately the recent Phase 3 trials of lecanemab and donanemab clearly demonstrated this relationship.
Who and when to treat? Selecting the right population at the right stage of disease.
While differences in amyloid binding, immune activation and other characteristics of the various anti-Aβ mAbs are clearly important for determining clinical efficacy, particularly for the more recently developed “plaque-busting” mAbs that all have the potential to produce substantial amyloid reduction, recent clinical trial results suggest that differences in clinical trial design and conduct also impact whether a drug is ultimately found to be clinically efficacious. Many of these differences relate to timing of brain Aβ removal during the clinical trial. If patients are too advanced at the initiation of Aβ removal, or if their underlying disease progressed to a more advanced state during the course of treatment because amyloid was not removed quickly enough, treatments are less effective.
The current NIA-Alzheimer’s Association (“AT(N)”) diagnostic criteria for AD require evidence of both brain amyloid (A) as well as tau (T) deposition with evidence of neurodegeneration (N) frequently present, but not necessary 37. The ATN criteria operationalize the amyloid cascade hypothesis that insoluble amyloid plaque accumulation leads to insoluble tau neurofibrillary tangle accumulation that causes neurodegeneration leading to clinical symptoms and death (A→T→N). More recent data suggest that amyloid and tau, at least in the medial temporal lobe, may accumulate simultaneously, reaching a threshold that enables rapid neocortical tau spreading, a so called “ca-tau-strophe” 38. Data from both clinical neuropathologic comparisons, such as the Braak staging system, 39 as well as more recent studies using tau PET ligands 40, show that both the amount and anatomical location of insoluble tau in the brain is strongly correlated with an individual’s type and severity of clinical symptoms during life 41. In the typical, amnestic clinical AD phenotype experienced by many older individuals, when tau is confined to the medial temporal lobe, people experience predominantly memory problems, but when it accumulates in neocortical regions, other symptoms of AD emerge, such as language, navigation or executive functioning (planning, multitasking, etc.) problems 42. When the disease has progressed too far, past the ca-tau-strophe phase, the recent Phase 3 donanemab and lecanemab trial results suggest that anti-Aβ therapies in isolation may be less clinically efficacious 2.
Data from previous trials of both successful and unsuccessful anti-Aβ therapies suggested that there was little clinical effect of amyloid removal if patients progressed past the mild dementia phase. 31,43 From there, the idea originated that early removal of Aβ, before the onset of symptoms, might produce a larger benefit than waiting for symptom development. This led to prevention trials in both sporadic, late onset AD — including A431 and AHEAD44 — where asymptomatic individuals with PET evidence of brain amyloid were randomized to receive an anti-Aβ therapy or in rare, early-onset, autosomal dominant AD mutation carriers (e.g., PSEN1 or APP), anti-Aβ therapy just prior to predicted age of onset based on the mean familial age of onset (API45 and DIAN-TU46). Of the trials that have reported results to date, including API, DIAN-TU and A4, none have produced substantial clinical benefit, although each used anti-Aβ mAbs that were not efficacious in phase 3 trials in symptomatic late onset AD (crenezumab, solanezumab/ganterenumab or solanuzemab respectively). Prevention studies have been challenging because measurable changes using clinical endpoints are often subtle prior to the onset of clinical impairment. For sporadic AD prevention trials (A4 and AHEAD), measuring a clinical effect often requires large sample sizes and >4 years of follow up because there is heterogeneity in the rates of cognitive decline during the preclinical stage. In the ADAD trials (API and DIAN-TU), the rarity of the population limits the numbers of participants available to enroll in the trial.
Comparisons of results from the lecanemab and gantenerumab Phase 3 trials also suggest that the lecanemab population was on average slightly earlier in the disease course due to the enrollment criteria requiring less severe short-term memory impairments for lecanemab. 47 The higher incidence of ARIA with gantenerumab, which required gradual titration of the drug to mitigate risk compounded this limitation. The unintended result was that participants randomized to drug in the gantenerumab trials did not receive the potentially efficacious dose until months later than they did in the lecanemab and donanemab trials. Thus, the lecanemab and donanemab trial participants received higher relative doses of drug at earlier stages of disease, which may have explained the better clinical response to a similar degree of Aβ removal.
Goldilocks tau and the timing of therapeutic interventions.
If the amount of insoluble tau in the brain is a measure of disease status, it could potentially be used to identify people at an early enough stage of disease (early ca-tau-strophe phase) to respond to anti-Aβ therapy, but late enough to have confidence that they will progress clinically over the course of an 18-month trial, giving greater power to detect treatment effects. In the phase 2 and 3 donanemab trials, the key biomarker inclusion criterion was the pattern of tau PET (flortaucipir [FTP]) uptake, requiring participants to have a “Goldilocks” amount of tau in the brain: enough medial temporal lobe uptake to give confidence that they had AD and would progress clinically during the trial, but not too much to be past the stage of responsiveness to amyloid removal. 2,34 The phase 3 trial that prespecified low/intermediate and high baseline tau groups showed substantially larger clinical effects in the low/intermediate as compared to high tau groups thereby supporting this hypothesis.
Insoluble tau reflects more than just disease stage.
Whether insoluble tau deposition is a measure of latent disease age 48 or other individual differences in disease biology remains an open question. In the donanemab Phase 3 trial, the clinical disease severity of the low/intermediate tau and combined high-low/intermediate tau burden groups were nearly identical 2, and previous studies have identified modest correlations between regional tau PET uptake and clinical disease severity or neuropsychological impairment in the range of rho=0.4–0.6. 49 There are also other important drivers of tau pathology that are not fully understood. Women are known to have higher AD risk than men, which may be at least partially explained by genetic differences in tau metabolism. 50,51 Individuals with sporadic early-onset AD (EOAD) — where disease onset occurs before age 65 — are also known to have more severe tau pathology and faster rates of accumulation particularly in cortical regions. 52 With this in mind, it is notable that individuals who were less than 65 years old in the lecanemab Phase 3 trial did not experience a significant benefit of Aβ removal possibly because this younger group (also enriched in APOEƐ4 homozygotes) had already had too much tau accumulation 1.
Tau PET as a response biomarker.
Although tau PET has been used less extensively in clinical trials, in both the Phase 2 donanemab 34 and Phase 3 lecanemab trials 1 that showed clinically meaningful treatment effects, there were significant attenuations of tau PET signal accumulation, suggesting that PET may reflect clinically meaningful treatment effects. However, donanemab had no effect on tau PET uptake in the Phase 3 trial. 2 There are at least three potential explanations for the lack of a reported effect. First, it is possible that there were medial temporal lobe (MTL) tau reductions from donanemb that could not be measured because the tau PET tracer used, FTP, has limited ability to measure MTL tau, whereas the tracer used for the lecanemab trial, MK6240, can measure MTL changes. Second, tau PET signal differences in cortical regions measurable using FTP might have taken longer than 18 months to accumulate and would have become more prominent as the populations were followed for longer durations in the open label extension. Finally, less likely but more troubling for the current model of AD pathophysiology, it remains possible that insoluble tau accumulation is a marker of but not critical for neurodegeneration in AD, and therefore clinical benefits of amyloid plaque removal can occur even in the absence of an effect on neurofibrillary tangle accumulation. If the results of an ongoing phase 2 trial of a tau antisense oligonucleotide for early AD (NCT05399888) demonstrate no clinical effects despite reduced tau PET signal, there may be added support for this last hypothesis.
Insights from new AD blood tests.
New sensitive blood tests that detect phosphorylated fragments of tau (P-tau) at the typical epitope measured for CSF diagnosis of AD (P-tau181), as well as other epitopes including P-tau217, have recently been shown to be useful tools in clinical trials.53,54 Initially, these tests were developed as an attempt to reduce the reliance on expensive and logistically difficult to collect tau (and amyloid) PET scans for clinical trials of anti-Aβ therapies. In addition to being substantially less expensive than PET, they also spare participants from radiation exposure, and can be used with medically underserved populations including remote locations that may not have ready access to PET scanners. The blood tests have performed better than expected in many contexts, particularly in the differential diagnosis of AD vs. other forms of dementia, but also in pre-screening individuals for disease modifying clinical trials such as the AHEAD prevention trial with lecanemab.32 Remarkably, plasma P-tau181 and −217 are also exquisitely sensitive to treatment effects of anti-Aβ therapies, such as aducanumab, donanemab and lecanemab, with large reductions in plasma concentrations reaching steady state approximately 6–12 months after the initiation of treatment. 1,2,55 However, similar to anti-Aβ therapy induced reductions in amyloid plaque load measured by PET, these reductions have occurred even in clinical trials where clinical efficacy was not demonstrated, such as the aducanumab ENGAGE Phase 3 study 26 and gantenerumab Phase 3 studies. This suggests that plasma P-tau may be sensitive to even small (not clinically meaningful) effects of amyloid reduction. Plasma P-tau likely reflects (and is strongly correlated with) CSF N-terminal P-tau fragment concentrations, which rise earlier in the course of AD 56 prior to the ca-tau-strophe. After the ca-tau-strophe, concentrations of CSF microtubule binding region (MTBR) tau fragments are strongly correlated with tau PET uptake and clinical status. 57
The relative ease of addition of plasma biomarkers to AD therapeutic trials has also revealed potential mechanistic insights into the link between amyloid and tau pathology. Strong treatment-related reductions in plasma GFAP levels that parallel reductions in plasma P-tau concentrations have been observed with donanemab and lecanemab treatment 1,55 and could potentially support a mediating role of astrocytes in the generation of toxic tau species, or alternatively, astrocytic changes could be a response to amyloid or tau pathologic changes. In contrast, the nonspecific biomarker, neurofilament light chain (NfL), has shown variable responses to different anti-Aβ therapies. Likely reflecting the aggressiveness of neurodegeneration, plasma NfL is less prominently elevated in AD than other neurodegenerative diseases, 58 which may explain the lower sensitivity of this biomarker to treatment effects as opposed to with SOD1-related ALS, where reductions in plasma NfL were found to be compelling evidence of disease modification by the antisense oligonucleotide, tofersen and supported an accelerated FDA approval of the drug 59.
Non-amyloid therapeutic targets
There are many ongoing clinical development programs with novel, promising disease-modifying approaches, and with the lecanemab and donanemab results, there is a renewed sense of optimism that one or more of these programs may be successful. 60 With the new empirical evidence supporting the amyloid hypothesis, maximizing the efficacy of anti-Aβ treatments using new, more efficacious mAbs and prevention approaches are already underway. Alternate approaches that focus on upstream causes and downstream consequences of amyloid pathology, most importantly tau protein accumulation, are also increasingly being pursued. Given the high costs and long duration of current AD development programs, there are active discussions about which treatment approaches to prioritize next and how to more efficiently determine which will produce clinically meaningful benefits.
Targeting tau protein: the tau hypothesis.
Given the central role of tau in the ATN research framework for AD, 37 it is arguably the most promising next target on which to focus clinical trial efforts. Tau is theoretically a better AD therapeutic target than amyloid because, although many years of normal cognition are possible in the setting of high brain amyloid plaque burden, the emergence of insoluble tau pathology coincides with the onset of clinical symptoms of AD and strongly predicts patterns of clinical decline 61. The emergence of insoluble tau deposition measured by tau PET strongly correlates with the onset of clinical symptoms in autosomal dominant AD (ADAD), 62 and temporal lobe tau PET uptake also predicts cortical spread of tau pathology, 38 brain atrophy, and subsequent clinical decline in sporadic AD. 63 Autosomal dominant mutations in MAPT gene lead to severe, early onset forms of frontotemporal lobar degeneration (FTLD), and the severity and types of symptoms from AD and other tauopathies, such as progressive supranuclear palsy (PSP) and chronic traumatic encephalopathy, closely reflect the distribution of insoluble tau in the brain, suggesting that interventions that reduce tau have the potential to produce large clinical effects 61. A corollary of the amyloid hypothesis, the tau hypothesis, states that tau protein abnormalities are necessary for initiating clinical symptoms of AD and other non-AD tauopathies. Whether additional factors are also necessary to initiate neurodegeneration, such as amyloid accumulation in AD or other undetermined factors in MAPT mutation carriers, is a topic of active investigation.
Consistent with the tau hypothesis, nonclinical tau models that express mutant human tau have identified multiple therapies that reduce tau or levels of toxic tau species 61,64. Clinical trials of early tau therapies, such as anti-aggregation and microtubule stabilizing agents, as well as the ‘first generation’ N-terminal anti-tau monoclonal antibodies, including tilavonemab, gosuranemab, 65 zagotenemab and semorinemab,66 failed to demonstrate clinical benefits in AD and or other tauopathies 67,68. Although all four mAbs had evidence of target engagement of N-terminal tau fragments in CSF, none had any reported pharmacodynamic effects on tau PET. In prodromal-mild AD, semorinemab modestly reduced CSF P-tau181 levels by ~15% (average of all doses), which is less than the ~25% reductions reported for patients who responded to high-dose aducanumab and less than the substantial reductions seen with lecanemab, possibly explaining the lack of clinical effect of semorinemab in this population. 26,69
First generation tau mAbs may be ineffective because they target presumably non-pathogenic, N-terminal tau fragments. Second generation anti-tau mAbs and an active vaccine 70 that target MTBR and C-terminal regions have entered clinical trials 71. In addition, multiple small molecule approaches that alter post-translational modification of tau (e.g., kinase or o-GlcNACase inhibitors) are also in clinical trials, with other approaches such as agents that reduce aggregation or target tau clearance in late pre-clinical or Phase 1 development 61. Recently, a tau targeted antisense oligonucleotide (ASO), MAPTRx, was able to reduce CSF tau concentrations by over 50% after 24 weeks with sustained reductions in CSF tau after up to 16 months at the highest doses in a long-term extension study, suggesting that substantial long term tau reductions may be safe and well tolerated 72. In a small number of participants who had longitudinal tau PET scans, substantial, dose-related reductions in insoluble tau measured by PET (recall that anti-Aβ therapies have only attenuated tau accumulation) were observed after 100 weeks of therapy. 73
Demonstration of clinically meaningful benefits of tau reduction in AD would be another significant milestone for the field and potentially of great benefit for patients. However, if no clinical benefits are observed with tau reduction, it will be important to understand the reasons why. One interpretation might be that the tau hypothesis is incorrect. For example, the distribution of insoluble tau pathology could be a correlate or predictor, but not a cause of neurodegeneration. However, given what has been learned from early anti-Aβ therapy trials, where failures prematurely signaled the demise of the amyloid hypothesis, 14 there could be other explanations, such as intervening too late or too early during disease, picking the wrong population (too much or too little tau at baseline), not reducing insoluble tau levels sufficiently, or even because the presence of amyloid might block the effects of tau reduction. The planned NIH funded Alzheimer’s Tau Platform (ATP) trial will test some of these hypotheses by combining anti-Aβ with multiple tau therapies in a very early AD population (Figure 4).
Figure 4. Amyloid and tau biomarker changes in relation to symptom onset with predicted effects of amyloid and tau targeted therapies in current and planned trials.
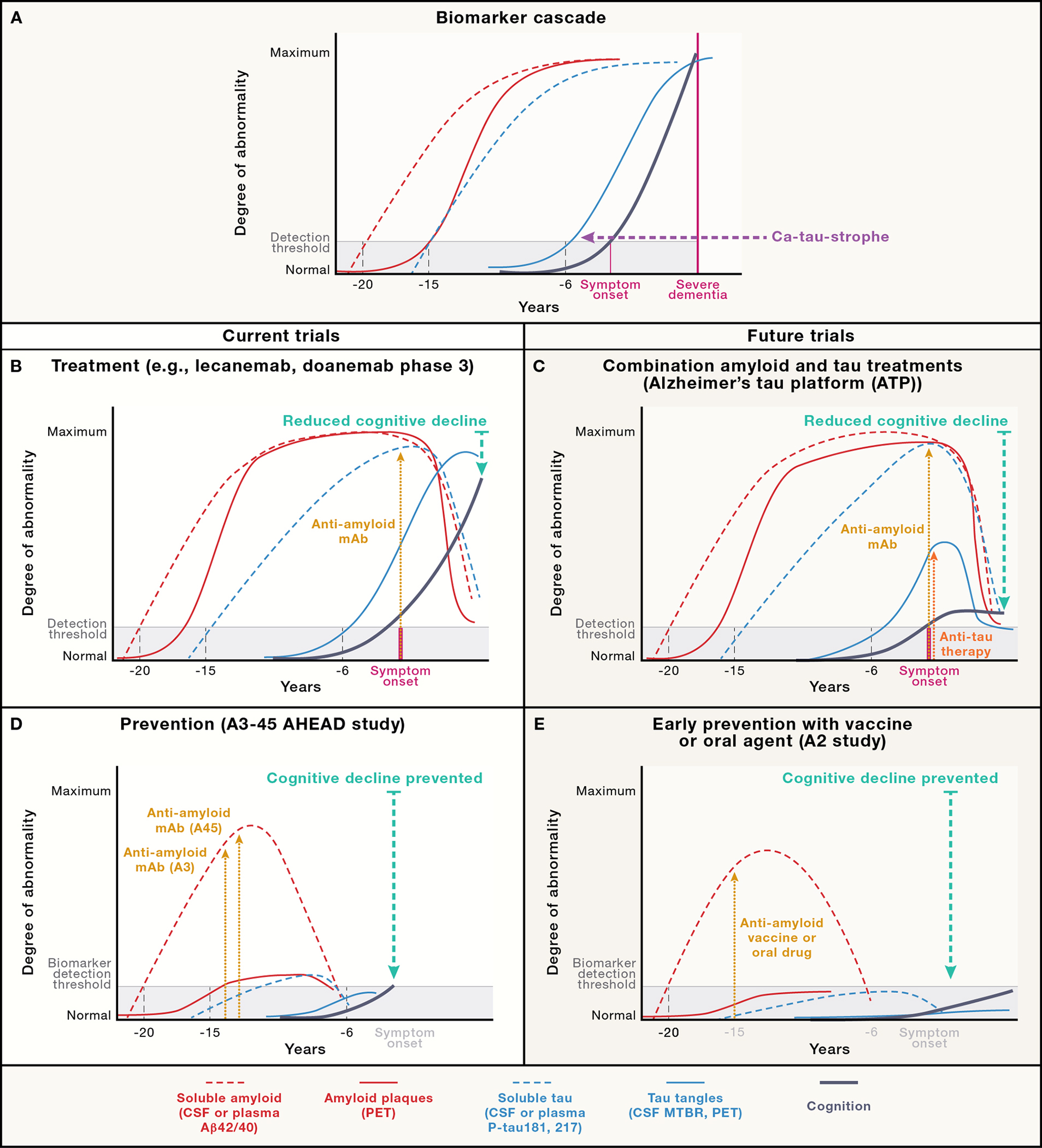
(A) Hypothetical pattern of soluble and insoluble amyloid and tau biomarker changes in relationship to symptom onset based on work from the Dominantly Inherited Alzheimer’s Network (DIAN; Barthelemy et al, 2019) modified from a slide presented at AD/PD2022 by Dr. Randall Bateman. (B) Representation of biomarker and cognitive changes after treatment with lecanemab or donanemab in recent Phase 3 studies. (C) Predicted biomarker changes if current A3–45 (AHEAD) study is positive. (D) Predicted biomarker changes if planned Alzheimer’s Tau Platform (ATP) study is positive. (E) Predicted biomarker changes if a hypothetical “A2” even earlier intervention study to prevent “amyloid positivity” succeeds.
Non-amyloid, non-tau co-pathologies as therapeutic targets.
The majority of clinically probable AD cases do not have pure ATN pathology, with various forms of vascular disease (macro and microinfarcts, arteriosclerosis and cerebral amyloid angiopathy), TDP-43 (Limbic-Predominant Age-Related TDP-43 Encephalopathy [LATE] 74 and α-synuclein (Lewy Bodies) commonly present 75. It is likely that such co-pathologies contribute to disease severity or progression rate and may explain why some participants in the lecanemab and donanemab Phase 3 trials experienced less disease slowing despite substantial plaque reduction. The presence of TDP-43 may have independent effects on clinical symptoms (and progression rate) that add to Aβ- and tau-associated effects to produce the overall clinical picture 76. Recent work with a new CSF α-synuclein RT-QUIC biomarker in the cognitively unimpaired 77 and individuals with mild dementia 78 demonstrates that the presence of α-synuclein aggregates influences both the symptoms (e.g., more hallucinations and motor symptoms) and rate of disease progression. This means that even if Aβ (and possibly tau) are reduced, other proteinopaties such as TDP-43 or Lewy Bodies could continue to drive some patients’ clinical decline.
Potential roles of proteostasis.
Individuals with high levels of insoluble tau pathology at baseline who don’t respond as well to Aβ removal may also have greater levels of non-AD co-pathology due to cellular effects of tau leading to impaired proteostasis that in turns promotes the accumulation of non-AD co-pathologies. 79 In support of this hypothesis, recent GWAS studies in PSP implicate both ubiquitin ligases (TRIM11/17) and LRRK2 80 in mediating disease phenotype and survival, and female AD patients may have higher levels of tau due to X chromosome inactivation of a ubiquitin ligase gene important for clearing tau 51. TRIM11 is downregulated in AD brains and reduces tau in animal models 81. Finally, approaches that target more ubiquitous protein degradation or clearance mechanisms, perhaps through reversal of neuroimmunosenence, may serve to impact multiple proteinopathies simultaneously 79,82–84.
Rare, genetic causes and protective factors for dementia may help to inform AD therapeutics.
Although TDP-43 co-pathology is common in AD, studying other diseases might be a more effective means for developing TDP-43 specific biomarkers and therapeutic targets. ALS and frontotemporal dementia (FTD) are associated with relatively pure TDP-43 pathology and are often caused by autosomal dominant mutations in chromosome 9 open reading frame 72 (C9orf72), progranulin (GRN), the TDP-43 gene itself, TARBP. Studies in these disorders are rapidly producing new biomarkers and therapeutic targets that might also be useful in AD with TDP-43 co-pathology. RNA processing and transport abnormalities, 85 as well as lysosomal dysfunction, are strongly implicated in TDP-43 proteinopathies 86. As lysosomal dysfunction is one potential mechanism that could explain the AD ca-tau-strophe, it may be of interest to investigate lysosomally-targeted therapies such as the PIKFYVE kinase inhibitor, apilimod, which recently demonstrated biological proof of concept in C9orf72 ALS, 87 and also mitigates tau toxicity in an organotypic iPSC MAPT V337M mutation model 88.
In addition to therapies targeting known protein co-pathologies in AD, a variety of other disease modifying approaches targeting AD-related immune and lysosomal dysfunction have been identified through the study of human mutation carriers, some of which are now in clinical trials. One target is the lysosomal/anti-inflammatory protein, progranulin, which in addition to being an important cause of autosomal dominant FTD, allowed the identification of a FTD-protective allele in TMEM106B 89 that has since been implicated in LATE as well. 90 More recently, study of the large Colombian PSEN1-E280A mutation cohort identified two protective mutations in individuals who carried the mutation but had attenuated symptoms. Both the APOE-Christchurch mutation 91 and the Reelin-COLBOS mutation 92 were shown to partially block the effects of PSEN1-induced tau accumulation and cognitive decline possibly through biological effects on the same ApoE-related signaling pathway 93.
New clinical trial design approaches
Currently, most AD clinical trials compare new or repurposed drugs with inactive placebo, but if lecanemab, donanemab and similar agents become widely used as standard of care, it may become increasingly difficult to recruit willing trial participants and eventually be considered unethical to compare novel treatments with placebo alone in symptomatic patients. Combination therapy approaches are common in epilepsy and have been critical for development of effective cancer and cardiovascular treatment strategies. True combination therapy trials may not be necessary in some individuals who respond well to amyloid removal, as has been suggested by the recent donanemab phase 3 study, where treatment was stopped in 52% of participants when their amyloid PET scans showed plaque clearance. If such individuals remain stable for sufficient time, it may be that a second therapy could be tested after the initial amyloid reduction. Even if such an approach is feasible, it might be limited to a subset of individuals previously treated with an anti-Aβ therapy, and by analogy to the negative gantenerumab phase 3 trial results where efficacy may have been limited due to delays in reaching a therapeutic dose, the potential delays introduced by first removing amyloid in combination trials could limit the efficacy of add-on therapies if such therapies are also more effective at earlier stages of disease. In addition, management of ARIA and other potential adverse events that arise from combinations of amyloid and non-amyloid treatment approaches, as well as the expense associated with paying for background anti-Aβ therapy for all trial participants, introduce added complexity and cost to the clinical trial procedures.
In a standard, AD phase 3, placebo controlled clinical trial where individuals are randomized 1:1 to receive a treatment or matched placebo, the main hypothesis to be tested is usually whether or not the treatment is clinically efficacious. This is done by comparing the difference in longitudinal change in the primary endpoint (usually a clinical measure) over the duration of each individual’s participation. A 2022 analysis found that such trials require an average of 4.4 years (recruitment plus treatment), 674 participants (both arms combined) and ~$368M (2023 dollars) to complete. 13 Planning and fundraising add additional time and cost to the process. To successively test 3 new therapies (or one new therapy, alone and in combination with two different anti-Aβ drugs) would therefore take 3 × 4.4 = 13.2 years, 2,022 participants, and $1.1B. In the worst (or best) case scenarios, if 50% of the 31 drugs in Phase 2 trials in 202260 were to move on to Phase 3 where a substantial subset of the agents would need to be tested alone and in combination with an anti-Aβ therapy, this would necessitate decades, involve thousands of participants and cost billions of dollars to adequately evaluate these drugs!
Master clinical trial protocols to increase efficiency: baskets and umbrellas
With large numbers of novel drugs to test, oncology has faced similar clinical drug development challenges as those that now loom for AD and has developed master protocols to increase efficiency. 94 Master protocols define the overall clinical parameters and participant flow through a trial and are most commonly focused on testing different treatments for the same clinical population (“umbrella trials”) or the same treatment in multiple populations (“basket” trials). Establishing the master trial creates multiple efficiencies: it builds trial infrastructure, improves regulatory agreement negotiation and standardizes data to allow more direct comparisons between therapeutic outcomes.
For umbrella trials, testing multiple or combination therapies simultaneously saves time, reduces costs and decreases the number of participants necessary to test a therapeutic hypothesis, since placebo group data can often be pooled across multiple therapeutic regimens. Therefore, as compared to the example of 3 successive Phase 3 clinical trials in the section above, a single umbrella trial protocol could theoretically be completed in approximately half the amount of time (5–6 years to allow for more enrollment time), with two thirds the number of participants (448 participants per evaluated therapy [336 drug : 112 placebo] for a total of 1,344 participants to test three drugs) with a cost savings of at least $400M (not accounting for shared infrastructure savings, which could further reduce costs). Because there is a much higher chance of receiving active drug — 75% in a three-arm umbrella design as compared to 50% in a standard parallel design trial — such designs are also preferred by research participants. One such umbrella platform trial in ADAD is DIAN-TU that previously simultaneously evaluated solanezumab and ganterenumab 46 and has now moved on to test a combination of lecanemab and the MTBR targeted tau mAb, E2814. Platform trials can also incorporate adaptive components whereby specific trial parameters may be altered during the course of the study if planned in advance. For example, if one of the therapies is found to be ineffective, that arm can be selectively stopped and participants assigned to the remaining arms, or alternatively, new drugs can be added. Additionally, such a “rolling platform” can move on to test new drugs without stopping the trial or seamlessly include multiple stages of development 95. The Healey ALS Platform trial began with four treatments, some of which were abandoned, and has moved on to test three new drugs 96.
Basket trials.
Basket trials have been successful in oncology for example by identifying multiple different types of cancer with the same mutated oncoprotein (e.g., HER2) that is targeted by a particular drug and testing all patients, regardless of cancer type, in the same trial 97. Genetic testing or use of biomarkers or biopsies that allow selection of patients based on a common molecular target have enabled such trials. This is more difficult to do in neurodegenerative disease due to the lack of sufficient biomarkers for most molecular pathologies other than Aβ and tau. However, because both AD and certain non-AD neurodegenerative syndromes are strongly linked to underlying tau pathology, it has been possible to combine populations such as AD, PSP, and corticobasal syndrome (CBS) in a single clinical trial of a tau-targeted intervention. In one such study, although the intervention was not found to be effective, the safety and tolerability were vastly different in the primary and secondary (AD) tauopathy populations, potentially indicating key biological differences in the underlying etiologies of the different tauopathies 67. Such designs may be particularly important for tau-targeted therapies because current cell culture (e.g., iPSC) and transgenic tau models generally incorporate autosomal dominant MAPT mutations that produce predominantly 4 microtubule binding domain (4R) tau and cause human frontotemporal dementia, such as P301L, and yet drugs developed in these models are often intended for use in AD, a tauopathy with mixed 3R/4R tau that is not caused by MAPT mutations. Tau aggregate structures also differ in different tauopathies 98, and it may be difficult to know whether a tau drug, such as a mAb that targets a particular epitope, fails to demonstrate effects in AD due to targeting the wrong tau species or an inherent lack of efficacy. Basket trial approaches often focus on early phases of development (Phase 1b/2a) where the primary endpoint may be a pharmacodynamic signal on a biomarker in addition to safety.
Incentivizing innovation in clinical AD therapeutic development
Industry incentives.
Most late-stage AD clinical trials of new molecular entities or repurposed drugs are funded by industry because the costs of developing such interventions generally exceed those available to the NIH and other government agencies or private foundations. PET scans, if used as treatment response biomarkers requiring multiple repeated scans in each individual, can further increase the costs of trials past the financial means of all but the largest pharmaceutical companies (the recent aducanumab, lecanemab, donanemab and gantenerumab Phase 3 studies were all sponsored by large pharmaceutical companies). Many industry researchers are highly motivated to develop effective treatments for altruistic reasons. Still, companies are responsible for generating value for their investors and reducing the risk of investment loss. The ultimate financial upside of drug development for a pharmaceutical company is an approved drug that goes to market and continues to generate revenue for the duration of its patent life. This creates incentive, particularly with late-stage clinical trials, to collect sufficiently compelling efficacy data to reach approval as quickly and inexpensively as possible.
In the US, all clinical trials involving drugs or devices are monitored by the US Food and Drug Administration which ultimately decides whether a drug is sufficiently safe and effective to be marketed for a particular indication. A drug that meets criteria for efficacy and safety can be approved for marketing. This generally requires demonstrating a clinically meaningful effect (a phrase which can be hard to define, but generally involves an outcome that is noticeable or appreciable by a patient and/or family as impactful in her/his/their lives)3 and sufficient safety data (both an acceptable side effect profile and enough patients treated for a long enough period of time). Regardless of a clinical trial’s outcome, the data and human biospecimens from the trial are valuable commodities that can be used in multiple ways including refining the treatment approach or developing new treatments. In the case of industry-sponsored clinical trials, the clinical trial data are most often proprietary to the company and give it a competitive advantage over other researchers. This can create disincentive to share data. There may also be fewer incentives to publish expeditiously if an approval is unlikely, and conversely, there are incentives to limit the data and analyses to protect a company’s intellectual property and competitive advantage. These factors may limit the ability of other researchers to learn from negative trials.
Academic incentives.
Many physician-scientists dream of discovering a potentially effective treatment approach in their laboratory, translating it to a human therapy and leading the pivotal clinical trial(s) that will establish clinical efficacy. However, since the number of publications and their impact can be key criteria for career advancement, there are disincentives to focus research efforts on clinical trials since they can be slow to produce results and given that negative clinical trials can be harder to publish in high impact journals. Trial manuscripts often include many authors, creating potential challenges to receiving credit for intellectually valuable contributions. As late-stage clinical trial manuscripts are often led by industry investigators who strictly adhere to the International Committee of Medical Journal Editors (ICMJE) authorship criteria, academic investigators responsible for clinical trial conduct (including patient management, which may be intellectually challenging and time consuming) as local site principal investigators are often not considered to have provided sufficient intellectual contribution to warrant authorship. As a result, they are not credited and are less able to rely on their clinical trial activities for promotion. There are many important studies to be done with generic or repurposed drugs or non-pharmacologic treatment approaches. However, working with novel, potentially disease modifying drugs can be a challenge because many are owned by industry, thereby requiring collaborations. Because of historical concerns regarding financial conflict of interest and corporate profit motives, working with industry can be reputationally risky creating further disincentive for some academic researchers to work on clinical trials.
Value of public-private partnerships for incentivizing trial innovation.
Industry often does not prioritize innovative trial designs such as prevention, basket and umbrella trials, because they are viewed as financially or strategically risky since they require relying on a master protocol generated by others that could contain potentially unfavorable elements, such as requiring direct comparisons to competitors’ drugs. In the case of basket trials involving rare populations, even if one treatment group arm identifies a responsive population leading to approval, the number of patients who would pay for the drug may be too small to produce sufficient return on investment. Similarly, the long duration and relatively large sample sizes required for prevention trials are generally viewed as financially risky. Most of the large, innovative clinical trial programs mentioned above have been successful by raising funds from the NIH and/or private foundations in the case of the prevention trials, including A4 31 AHEAD, 32 the Amyloid Prevention Initiative (API) trial, 45 and DIAN-TU, 99 or philanthropic foundations in the case of the Healey ALS Platform. The public and philanthropic funds can decrease financial risk and incentivize industry to provide compounds for testing often with additional funding necessary to complete the trial. The funds lower the costs for companies to test drugs in these settings, provide unique populations to investigate treatment effects (in the case of API and DIAN-TU), and provide valuable scientific expertise from academic investigators. By providing seed funding, public-private partnerships reduce the perceived risk to companies of investing in innovative trial design programs and can bring together multiple competing companies with different compounds to promote collaboration under the setting of umbrella trials. For small companies with limited resources or academic investigators wishing to develop their own therapies, public or philanthropic funds can facilitate clinical trials without requiring substantial fundraising or successfully competing for grants. Public-private partnerships can ensure that clinical trial data and biospecimens are widely shared with other researchers and trial results are published expeditiously, thereby benefiting the scientific community at large. For example, the A4 study has shared screening data with over 1,500 researchers 31, and AHEAD will share longitudinal trial data within 1 year of completion 32. Public-private partnerships also provide more opportunities for patient and public involvement in trial design and conduct, as well as novel strategies to increase the ethnoracial diversity of trial participants. The cost (lecanemab will cost $26.5K per patient per year in the US) and complexity of new AD therapies creates barriers to their use in medically underserved populations and low-and-middle-income countries. Partnerships with public health and other international organizations will likely be helpful in lowering these barriers.
Training and career paths for academic clinical trial researchers.
Another mechanism for increasing the speed of clinical development is to conduct earlier, biomarker-focused “proof of concept” trials to identify compounds that may potentially have large clinical effects that might be predicted by robust pharmacodynamic effects or early signs of clinical benefit. Facilitating the conduct of such trials by academic researchers is important but faces two important barriers. First, few academic researchers have clinical trial experience or formal training in clinical trial methods. This is beginning to change with new programs such as the NIA-Alzheimer’s Association sponsored Institute on Methods and Protocols for Advancement of Clinical Trials in ADRD (IMPACT-AD). 100 There are also concerns regarding adequate numbers of clinical trial expert researchers in industry; training more such individuals would facilitate industry-sponsored clinical trials as well. Finally, academic promotion committees need to give more consideration for participation as a productive site investigator in clinical trials. For laboratory-based or observational clinical researchers, academic productivity may be assessed during the promotion process by the number of first or last-authored manuscripts. Since opportunities to generate such manuscripts are more limited for clinical trialists, other means to assess productivity should be developed to incentivize academics to focus their careers on clinical trials. For example, productivity could be based on the number of participants enrolled in various clinical trials, even if the trials are organized by other investigators, including industry-sponsored trials.
Conclusion
The recent positive Phase 3 clinical trials with the anti-Aβ mAbs lecanemab and donanemab are important milestones for AD research because they demonstrate that rational approaches to drug development for complex, sporadic neurodegenerative diseases are possible and potentially critical to success. The two and a half decades of human clinical trials that took place between when the first anti-Aβ immunotherapies were shown to reduce amyloid pathology in mice and the recent, successful Phase 3 trials generated much insight. The emergence of worrisome side effects, including meningoencephalitis from the original active amyloid vaccine, and more importantly, ARIA from mAb therapies, introduced long delays that took years to understand how to manage. The recent successes also relied on the development of new biomarkers, including amyloid and tau PET, and have been further accelerated by the development of plasma biomarkers of different tau species, Aβ and other neurodegenerative proteins. While there is cause for celebration, there are also concerns that the clinical benefits associated with anti-Aβ therapy are modest, particularly given their high costs, and may be limited to certain highly selected populations. Potential paths to development of more efficacious AD therapies include combination therapy trials testing anti-Aβ plus other therapies that could have additive or synergistic effects including tau, ApoE, reelin and proteostasis enhancing approaches. Since most clinically diagnosed AD is associated with both AD and non-AD co-pathologies, combination therapy targeting α-synuclein, vascular disease and TDP-43 will likely also be necessary for many patients. Initiating Aβ and other pathology reduction earlier in prevention trials might also lead to larger clinical benefits. These exciting new treatment approaches will require new, more efficient clinical trial designs, such as umbrella and basket trials, that will in turn require increasing support from the NIH and philanthropic funders for public-private partnerships that can bring together the necessary partners from academia and industry to ensure their success. The next era of AD therapeutic development has begun and seems likely to produce clinically meaningful advances at a much faster rate than in the past.
Acknowledgments
We thank the reviewers for providing a suggested format for Figure 1 and Eden Barragan, PhD for help with generating Figure 2A.
Footnotes
Declaration of Interests
Adam Boxer has served as a consultant to AGTC, Alector, Alzprotect, Amylyx, Arkuda, Arrowhead, Arvinas, Aviado, Boehringer Ingelheim, Denali, Eli Lilly, GSK, Humana, Life Edit, Merck, Modalis, Oligomerix, Oscotec, Roche, Transposon and Wave. He has received research support from Biogen and Eisai for serving as a site investigator for clinical trials, as well as from Regeneron. He has received research support from the National Institutes on Aging: NIH U19AG063911, R01AG073482, R56AG075744, R01AG038791, RF1AG077557, R01AG071756, U24AG057437; Rainwater Charitable Foundation, Bluefield Project to Cure FTD, GHR Foundation, Alzheimer’s Association, Association for Frontotemporal Degeneration, Gates Ventures, Alzheimer’s Drug Discovery Foundation, UCSF Parkinson’s Spectrum Disorders Center and the University of California Cures AD Program.
Reisa Sperling has served as a consultant to AC Immune, Acumen, Genentech, Ionis, Janssen, Oligomerix, Prothena, Roche, Shionogi and Vaxxinity. Her spouse has been a consultant to Merck and Novartis. She has received research funding from the National Institute on Aging, NIH: P01AG036694, U24AG057437; R01AG063689; R01AG054029; R01AG061848; Alzheimer’s Association, GHR Foundation, Gates Ventures, an Anonymous Foundation, Accelerating Medicines Partnership FNIH, and public partnership trial funding from Eli Lilly, and Eisai.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Adam L. Boxer, Memory and Aging Center, Department of Neurology, Weill Institute of Neuroscience, University of California, San Francisco, CA.
Reisa Sperling, Center for Alzheimer Research and Treatment, Department of Neurology, MassGeneral Brigham, Harvard Medical School, Boston, MA.
References
- 1.van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, et al. (2022). Lecanemab in Early Alzheimer’s Disease. N Engl J Med. 10.1056/NEJMoa2212948. [DOI] [PubMed] [Google Scholar]
- 2.Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, Wessels AM, Shcherbinin S, Wang H, Monkul Nery ES, et al. (2023). Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA. 10.1001/jama.2023.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rentz DM, Wessels AM, Annapragada AV, Berger AK, Edgar CJ, Gold M, Miller DS, Randolph C, Ryan JM, Wunderlich G, et al. (2021). Building clinically relevant outcomes across the Alzheimer’s disease spectrum. Alzheimers Dement (N Y) 7, e12181. 10.1002/trc2.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabinovici GD (2021). Controversy and Progress in Alzheimer’s Disease - FDA Approval of Aducanumab. N Engl J Med. 10.1056/NEJMp2111320. [DOI] [PubMed] [Google Scholar]
- 5.Association A.s. (2023). 2023 Alzheimer’s disease facts and figures. Alzheimers Dement 19, 1598–1695. 10.1002/alz.13016. [DOI] [PubMed] [Google Scholar]
- 6.Hardy JA, and Higgins GA (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 7.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, and et al. (1995). Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373, 523–527. 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 8.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, et al. (1999). Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 9.Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, and Donoghue S (2005). Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology 64, 94–101. [DOI] [PubMed] [Google Scholar]
- 10.Cummings J, Feldman HH, and Scheltens P (2019). The “rights” of precision drug development for Alzheimer’s disease. Alzheimers Res Ther 11, 76. 10.1186/s13195-019-0529-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haass C, and Selkoe D (2022). If amyloid drives Alzheimer disease, why have anti-amyloid therapies not yet slowed cognitive decline? PLoS Biol 20, e3001694. 10.1371/journal.pbio.3001694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott TJ, O’Connor AC, Link AN, and Beaulieu TJ (2014). Economic analysis of opportunities to accelerate Alzheimer’s disease research and development. Ann N Y Acad Sci 1313, 17–34. 10.1111/nyas.12417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cummings J, Bauzon J, and Lee G (2021). Who funds Alzheimer’s disease drug development? Alzheimers Dement (N Y) 7, e12185. 10.1002/trc2.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karran E, and De Strooper B (2022). The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov 21, 306–318. 10.1038/s41573-022-00391-w. [DOI] [PubMed] [Google Scholar]
- 15.Pride M, Seubert P, Grundman M, Hagen M, Eldridge J, and Black RS (2008). Progress in the active immunotherapeutic approach to Alzheimer’s disease: clinical investigations into AN1792-associated meningoencephalitis. Neurodegener Dis 5, 194–196. 10.1159/000113700. [DOI] [PubMed] [Google Scholar]
- 16.Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, and Koller M (2005). Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 64, 1563–1572. [DOI] [PubMed] [Google Scholar]
- 17.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, and Orgogozo JM (2005). Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64, 1553–1562. [DOI] [PubMed] [Google Scholar]
- 18.Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, et al. (2014). Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370, 322–333. 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Dyck CH (2018). Anti-Amyloid-beta Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol Psychiatry 83, 311–319. 10.1016/j.biopsych.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sperling RA, Jack CR Jr., Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, et al. (2011). Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement 7, 367–385. 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Filippi M, Cecchetti G, Spinelli EG, Vezzulli P, Falini A, and Agosta F (2022). Amyloid-Related Imaging Abnormalities and beta-Amyloid-Targeting Antibodies: A Systematic Review. JAMA Neurol 79, 291–304. 10.1001/jamaneurol.2021.5205. [DOI] [PubMed] [Google Scholar]
- 22.Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, and van Veluw SJ (2020). Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol 16, 30–42. 10.1038/s41582-019-0281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cummings JL, Cohen S, van Dyck CH, Brody M, Curtis C, Cho W, Ward M, Friesenhahn M, Rabe C, Brunstein F, et al. (2018). ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 90, e1889–e1897. 10.1212/WNL.0000000000005550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, et al. (2014). Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370, 311–321. 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- 25.Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, Lannfelt L, Bradley H, Rabe M, Koyama A, et al. (2021). A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimers Res Ther 13, 80. 10.1186/s13195-021-00813-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Budd Haeberlein S, Aisen PS, Barkhof F, Chalkias S, Chen T, Cohen S, Dent G, Hansson O, Harrison K, von Hehn C, et al. (2022). Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J Prev Alzheimers Dis 9, 197–210. 10.14283/jpad.2022.30. [DOI] [PubMed] [Google Scholar]
- 27.Administration, F.a.D. (2018). Surrogate Endpoint Resources for Drug and Biologic Development. https://www.fda.gov/drugs/development-resources/surrogate-endpoint-resources-drug-and-biologic-development.
- 28.Belluck P (2022). Congressional Inquiry into Alzheimer’s Drug Faults Its Maker and F.D.A. The New York Times, December 30, 2022. [Google Scholar]
- 29.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, et al. (2001). The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat Neurosci 4, 887–893. 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 30.Liu E, Schmidt ME, Margolin R, Sperling R, Koeppe R, Mason NS, Klunk WE, Mathis CA, Salloway S, Fox NC, et al. (2015). Amyloid-beta 11C-PiB-PET imaging results from 2 randomized bapineuzumab phase 3 AD trials. Neurology 85, 692–700. 10.1212/WNL.0000000000001877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sperling RA, Donohue MC, Raman R, Rafii MS, Johnson K, Masters CL, van Dyck CH, Iwatsubo T, Marshall GA, Yaari R, et al. (2023). Trial of Solanezumab in Preclinical Alzheimer’s Disease. N Engl J Med. 10.1056/NEJMoa2305032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rafii MS, Sperling RA, Donohue MC, Zhou J, Roberts C, Irizarry MC, Dhadda S, Sethuraman G, Kramer LD, Swanson CJ, et al. (2023). The AHEAD 3–45 Study: Design of a prevention trial for Alzheimer’s disease. Alzheimers Dement 19, 1227–1233. 10.1002/alz.12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, et al. (2017). Addendum: The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 546, 564. 10.1038/nature22809. [DOI] [PubMed] [Google Scholar]
- 34.Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, Shcherbinin S, Sparks J, Sims JR, Brys M, et al. (2021). Donanemab in Early Alzheimer’s Disease. N Engl J Med 384, 1691–1704. 10.1056/NEJMoa2100708. [DOI] [PubMed] [Google Scholar]
- 35.Lowe SL, Duggan Evans C, Shcherbinin S, Cheng YJ, Willis BA, Gueorguieva I, Lo AC, Fleisher AS, Dage JL, Ardayfio P, et al. (2021). Donanemab (LY3002813) Phase 1b Study in Alzheimer’s Disease: Rapid and Sustained Reduction of Brain Amyloid Measured by Florbetapir F18 Imaging. J Prev Alzheimers Dis 8, 414–424. 10.14283/jpad.2021.56. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y (2023). An insider’s perspective on FDA approval of aducanumab. Alzheimers Dement (N Y) 9, e12382. 10.1002/trc2.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, et al. (2018). NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14, 535–562. 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanchez JS, Becker JA, Jacobs HIL, Hanseeuw BJ, Jiang S, Schultz AP, Properzi MJ, Katz SR, Beiser A, Satizabal CL, et al. (2021). The cortical origin and initial spread of medial temporal tauopathy in Alzheimer’s disease assessed with positron emission tomography. Sci Transl Med 13. 10.1126/scitranslmed.abc0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braak H, and Braak E (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- 40.Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, Shankle WR, Elizarov A, and Kolb HC (2013). Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis 34, 457–468. 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- 41.Pascoal TA, Benedet AL, Tudorascu DL, Therriault J, Mathotaarachchi S, Savard M, Lussier FZ, Tissot C, Chamoun M, Kang MS, et al. (2021). Longitudinal 18F-MK-6240 tau tangles accumulation follows Braak stages. Brain. 10.1093/brain/awab248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Therriault J, Pascoal TA, Lussier F, Tissot C, Chamoun M, Bezgin G, Sevaes S, Benedet AL, Ashton NJ, Karikari TK, et al. (2022). Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nature Aging. 10.1038/s43587-022-00204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH, et al. (2009). A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 73, 2061–2070. 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rafii MS, Sperling RA, Donohue MC, Zhou J, Roberts C, Irizarry MC, Dhadda S, Sethuraman G, Kramer LD, Swanson CJ, et al. (2022). The AHEAD 3–45 Study: Design of a prevention trial for Alzheimer’s disease. Alzheimers Dement. 10.1002/alz.12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tariot PN, Lopera F, Langbaum JB, Thomas RG, Hendrix S, Schneider LS, Rios-Romenets S, Giraldo M, Acosta N, Tobon C, et al. (2018). The Alzheimer’s Prevention Initiative Autosomal-Dominant Alzheimer’s Disease Trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement (N Y) 4, 150–160. 10.1016/j.trci.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salloway S, Farlow M, McDade E, Clifford DB, Wang G, Llibre-Guerra JJ, Hitchcock JM, Mills SL, Santacruz AM, Aschenbrenner AJ, et al. (2021). A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat Med 27, 1187–1196. 10.1038/s41591-021-01369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bateman R, Smith J, Donohue M, Delmar P, Gruendl E, Boada M, Rutten-Jacobs L, Lane C, Cummings J, Fontoura P, et al. (2023). GRADUATE I AND II: EFFICACY RESULTS FROM TWO PHASE III TRIALS OF SUBCUTANEOUS GANTENERUMAB IN EARLY ALZHEIMER’S DISEASE. [Google Scholar]
- 48.Staffaroni AM, Quintana M, Wendelberger B, Heuer HW, Russell LL, Cobigo Y, Wolf A, Goh SM, Petrucelli L, Gendron TF, et al. (2022). Temporal order of clinical and biomarker changes in familial frontotemporal dementia. Nat Med 28, 2194–2206. 10.1038/s41591-022-01942-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanner JA, Iaccarino L, Edwards L, Asken BM, Gorno-Tempini ML, Kramer JH, Pham J, Perry DC, Possin K, Malpetti M, et al. (2022). Amyloid, tau and metabolic PET correlates of cognition in early and late-onset Alzheimer’s disease. Brain 145, 4489–4505. 10.1093/brain/awac229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coughlan GT, Betthauser TJ, Boyle R, Koscik RL, Klinger HM, Chibnik LB, Jonaitis EM, Yau WW, Wenzel A, Christian BT, et al. (2023). Association of Age at Menopause and Hormone Therapy Use With Tau and beta-Amyloid Positron Emission Tomography. JAMA Neurol 80, 462–473. 10.1001/jamaneurol.2023.0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yan Y, Wang X, Chaput D, Shin MK, Koh Y, Gan L, Pieper AA, Woo JA, and Kang DE (2022). X-linked ubiquitin-specific peptidase 11 increases tauopathy vulnerability in women. Cell 185, 3913–3930 e3919. 10.1016/j.cell.2022.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pontecorvo MJ, Devous MD, Kennedy I, Navitsky M, Lu M, Galante N, Salloway S, Doraiswamy PM, Southekal S, Arora AK, et al. (2019). A multicentre longitudinal study of flortaucipir (18F) in normal ageing, mild cognitive impairment and Alzheimer’s disease dementia. Brain 142, 1723–1735. 10.1093/brain/awz090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, Su Y, Chen Y, Serrano GE, Leuzy A, et al. (2020). Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA 324, 772–781. 10.1001/jama.2020.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thijssen EH, La Joie R, Strom A, Fonseca C, Iaccarino L, Wolf A, Spina S, Allen IE, Cobigo Y, Heuer H, et al. (2021). Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol 20, 739–752. 10.1016/S1474-4422(21)00214-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pontecorvo MJ, Lu M, Burnham SC, Schade AE, Dage JL, Shcherbinin S, Collins EC, Sims JR, and Mintun MA (2022). Association of Donanemab Treatment With Exploratory Plasma Biomarkers in Early Symptomatic Alzheimer Disease: A Secondary Analysis of the TRAILBLAZER-ALZ Randomized Clinical Trial. JAMA Neurol 79, 1250–1259. 10.1001/jamaneurol.2022.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barthelemy NR, Li Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS, Buckles V, Fagan AM, Perrin RJ, Goate AM, et al. (2020). A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med 26, 398–407. 10.1038/s41591-020-0781-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Horie K, Salvado G, Barthelemy NR, Janelidze S, Li Y, He Y, Saef B, Chen CD, Jiang H, Strandberg O, et al. (2023). CSF MTBR-tau243 is a specific biomarker of tau tangle pathology in Alzheimer’s disease. Nat Med. 10.1038/s41591-023-02443-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansson O (2021). Biomarkers for neurodegenerative diseases. Nat Med 27, 954–963. 10.1038/s41591-021-01382-x. [DOI] [PubMed] [Google Scholar]
- 59.Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, Chio A, Van Damme P, Ludolph AC, Glass JD, et al. (2022). Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med 387, 1099–1110. 10.1056/NEJMoa2204705. [DOI] [PubMed] [Google Scholar]
- 60.Cummings J, Lee G, Nahed P, Kambar M, Zhong K, Fonseca J, and Taghva K (2022). Alzheimer’s disease drug development pipeline: 2022. Alzheimers Dement (N Y) 8, e12295. 10.1002/trc2.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samudra N, Lane-Donovan C, VandeVrede L, and Boxer AL (2023). Tau pathology in neurodegenerative disease: disease mechanisms and therapeutic avenues. J Clin Invest 133. 10.1172/JCI168553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gordon BA, Blazey TM, Christensen J, Dincer A, Flores S, Keefe S, Chen C, Su Y, McDade EM, Wang G, et al. (2019). Tau PET in autosomal dominant Alzheimer’s disease: relationship with cognition, dementia and other biomarkers. Brain 142, 1063–1076. 10.1093/brain/awz019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.La Joie R, Visani AV, Baker SL, Brown JA, Bourakova V, Cha J, Chaudhary K, Edwards L, Iaccarino L, Janabi M, et al. (2020). Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med 12. 10.1126/scitranslmed.aau5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang CW, Shao E, and Mucke L (2021). Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 371. 10.1126/science.abb8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bright J, Hussain S, Dang V, Wright S, Cooper B, Byun T, Ramos C, Singh A, Parry G, Stagliano N, and Griswold-Prenner I (2015). Human secreted tau increases amyloid-beta production. Neurobiology of aging 36, 693–709. 10.1016/j.neurobiolaging.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 66.Ayalon G, Lee SH, Adolfsson O, Foo-Atkins C, Atwal JK, Blendstrup M, Booler H, Bravo J, Brendza R, Brunstein F, et al. (2021). Antibody semorinemab reduces tau pathology in a transgenic mouse model and engages tau in patients with Alzheimer’s disease. Sci Transl Med 13. 10.1126/scitranslmed.abb2639. [DOI] [PubMed] [Google Scholar]
- 67.Tsai RM, Miller Z, Koestler M, Rojas JC, Ljubenkov PA, Rosen HJ, Rabinovici GD, Fagan AM, Cobigo Y, Brown JA, et al. (2020). Reactions to Multiple Ascending Doses of the Microtubule Stabilizer TPI-287 in Patients With Alzheimer Disease, Progressive Supranuclear Palsy, and Corticobasal Syndrome: A Randomized Clinical Trial. JAMA Neurol 77, 215–224. 10.1001/jamaneurol.2019.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dam T, Boxer AL, Golbe LI, Hoglinger GU, Morris HR, Litvan I, Lang AE, Corvol JC, Aiba I, Grundman M, et al. (2021). Safety and efficacy of anti-tau monoclonal antibody gosuranemab in progressive supranuclear palsy: a phase 2, randomized, placebo-controlled trial. Nat Med 27, 1451–1457. 10.1038/s41591-021-01455-x. [DOI] [PubMed] [Google Scholar]
- 69.Teng E, Manser PT, Pickthorn K, Brunstein F, Blendstrup M, Sanabria Bohorquez S, Wildsmith KR, Toth B, Dolton M, Ramakrishnan V, et al. (2022). Safety and Efficacy of Semorinemab in Individuals With Prodromal to Mild Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 10.1001/jamaneurol.2022.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Novak P, Kovacech B, Katina S, Schmid R, Scheltens P, Kontsekova E, Ropele S, Fialova L, Kramberger M, Paulenka-Ivanovova N, et al. (2021). ADAMANT: a placebo-controlled randomized phase 2 study of AADvac1, an active immunotherapy against pathological tau in Alzheimer’s disease. Nat Aging 1, 521–534. [DOI] [PubMed] [Google Scholar]
- 71.Ji C, and Sigurdsson EM (2021). Current Status of Clinical Trials on Tau Immunotherapies. Drugs 81, 1135–1152. 10.1007/s40265-021-01546-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mummery CJ, Borjesson-Hanson A, Blackburn DJ, Vijverberg EGB, De Deyn PP, Ducharme S, Jonsson M, Schneider A, Rinne JO, Ludolph AC, et al. (2023). Tau-targeting antisense oligonucleotide MAPT(Rx) in mild Alzheimer’s disease: a phase 1b, randomized, placebo-controlled trial. Nat Med. 10.1038/s41591-023-02326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alzforum (2023). First Hit on Aggregated Tau: Antisense Oligonucleotide Lowers Tangles. [Google Scholar]
- 74.Nelson PT, Brayne C, Flanagan ME, Abner EL, Agrawal S, Attems J, Castellani RJ, Corrada MM, Cykowski MD, Di J, et al. (2022). Frequency of LATE neuropathologic change across the spectrum of Alzheimer’s disease neuropathology: combined data from 13 community-based or population-based autopsy cohorts. Acta Neuropathol 144, 27–44. 10.1007/s00401-022-02444-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mehta RI, and Schneider JA (2021). What is ‘Alzheimer’s disease’? The neuropathological heterogeneity of clinically defined Alzheimer’s dementia. Curr Opin Neurol 34, 237–245. 10.1097/WCO.0000000000000912. [DOI] [PubMed] [Google Scholar]
- 76.Josephs KA, Martin PR, Weigand SD, Tosakulwong N, Buciuc M, Murray ME, Petrucelli L, Senjem ML, Spychalla AJ, Knopman DS, et al. (2020). Protein contributions to brain atrophy acceleration in Alzheimer’s disease and primary age-related tauopathy. Brain 143, 3463–3476. 10.1093/brain/awaa299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Palmqvist S, Rossi M, Hall S, Quadalti C, Mattsson-Carlgren N, Dellavalle S, Tideman P, Pereira JB, Nilsson MH, Mammana A, et al. (2023). Cognitive effects of Lewy body pathology in clinically unimpaired individuals. Nat Med. 10.1038/s41591-023-02450-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Quadalti C, Palmqvist S, Hall S, Rossi M, Mammana A, Janelidze S, Dellavalle S, Mattsson-Carlgren N, Baiardi S, Stomrud E, et al. (2023). Clinical effects of Lewy body pathology in cognitively impaired individuals. Nat Med. 10.1038/s41591-023-02449-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wilson DM 3rd, Cookson MR, Van Den Bosch L, Zetterberg H, Holtzman DM, and Dewachter I (2023). Hallmarks of neurodegenerative diseases. Cell 186, 693–714. 10.1016/j.cell.2022.12.032. [DOI] [PubMed] [Google Scholar]
- 80.Jabbari E, Koga S, Valentino RR, Reynolds RH, Ferrari R, Tan MMX, Rowe JB, Dalgard CL, Scholz SW, Dickson DW, et al. (2021). Genetic determinants of survival in progressive supranuclear palsy: a genome-wide association study. Lancet Neurol 20, 107–116. 10.1016/S1474-4422(20)30394-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang ZY, Harischandra DS, Wang R, Ghaisas S, Zhao JY, McMonagle TP, Zhu G, Lacuarta KD, Song J, Trojanowski JQ, et al. (2023). TRIM11 protects against tauopathies and is down-regulated in Alzheimer’s disease. Science 381, eadd6696. 10.1126/science.add6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bourdenx M, Martin-Segura A, Scrivo A, Rodriguez-Navarro JA, Kaushik S, Tasset I, Diaz A, Storm NJ, Xin Q, Juste YR, et al. (2021). Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 184, 2696–2714 e2625. 10.1016/j.cell.2021.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim J, de Haro M, Al-Ramahi I, Garaicoechea LL, Jeong HH, Sonn JY, Tadros B, Liu Z, Botas J, and Zoghbi HY (2023). Evolutionarily conserved regulators of tau identify targets for new therapies. Neuron 111, 824–838 e827. 10.1016/j.neuron.2022.12.012. [DOI] [PubMed] [Google Scholar]
- 84.Guerrero A, De Strooper B, and Arancibia-Carcamo IL (2021). Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci 44, 714–727. 10.1016/j.tins.2021.06.007. [DOI] [PubMed] [Google Scholar]
- 85.Mehta PR, Brown AL, Ward ME, and Fratta P (2023). The era of cryptic exons: implications for ALS-FTD. Mol Neurodegener 18, 16. 10.1186/s13024-023-00608-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Keating SS, San Gil R, Swanson MEV, Scotter EL, and Walker AK (2022). TDP-43 pathology: From noxious assembly to therapeutic removal. Prog Neurobiol 211, 102229. 10.1016/j.pneurobio.2022.102229. [DOI] [PubMed] [Google Scholar]
- 87.Hung ST, Linares GR, Chang WH, Eoh Y, Krishnan G, Mendonca S, Hong S, Shi Y, Santana M, Kueth C, et al. (2023). PIKFYVE inhibition mitigates disease in models of diverse forms of ALS. Cell 186, 786–802 e728. 10.1016/j.cell.2023.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bowles KR, Silva MC, Whitney K, Bertucci T, Berlind JE, Lai JD, Garza JC, Boles NC, Mahali S, Strang KH, et al. (2021). ELAVL4, splicing, and glutamatergic dysfunction precede neuron loss in MAPT mutation cerebral organoids. Cell 184, 4547–4563 e4517. 10.1016/j.cell.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, et al. (2010). Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42, 234–239. ng.536 [pii] 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Neumann M, Perneel J, Cheung S, Van den Broeck M, Nygaard H, Hsiung GR, Wynants S, Heeman B, Rademakers R, and Mackenzie IRA (2023). Limbic-predominant age-related TDP-43 proteinopathy (LATE-NC) is associated with abundant TMEM106B pathology. Acta Neuropathol. 10.1007/s00401-023-02580-2. [DOI] [PubMed] [Google Scholar]
- 91.Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, et al. (2019). Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nature Medicine 25, 1680–1683. 10.1038/s41591-019-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lopera F, Marino C, Chandrahas AS, O’Hare M, Villalba-Moreno ND, Aguillon D, Baena A, Sanchez JS, Vila-Castelar C, Ramirez Gomez L, et al. (2023). Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nature Medicine. 10.1038/s41591-023-02318-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wasser CR, and Herz J (2017). Reelin: Neurodevelopmental Architect and Homeostatic Regulator of Excitatory Synapses. J Biol Chem 292, 1330–1338. 10.1074/jbc.R116.766782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Woodcock J, and LaVange LM (2017). Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. N Engl J Med 377, 62–70. 10.1056/NEJMra1510062. [DOI] [PubMed] [Google Scholar]
- 95.Noor NM, Love SB, Isaacs T, Kaplan R, Parmar MKB, and Sydes MR (2022). Uptake of the multi-arm multi-stage (MAMS) adaptive platform approach: a trial-registry review of late-phase randomised clinical trials. BMJ Open 12, e055615. 10.1136/bmjopen-2021-055615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Paganoni S, Berry JD, Quintana M, Macklin E, Saville BR, Detry MA, Chase M, Sherman AV, Yu H, Drake K, et al. (2022). Adaptive Platform Trials to Transform Amyotrophic Lateral Sclerosis Therapy Development. Ann Neurol 91, 165–175. 10.1002/ana.26285. [DOI] [PubMed] [Google Scholar]
- 97.Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, Juric D, Quinn DI, Moreno V, Doger B, et al. (2018). HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 554, 189–194. 10.1038/nature25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shi Y, Zhang W, Yang Y, Murzin AG, Falcon B, Kotecha A, van Beers M, Tarutani A, Kametani F, Garringer HJ, et al. (2021). Structure-based classification of tauopathies. Nature 598, 359–363. 10.1038/s41586-021-03911-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bateman RJ, Benzinger TL, Berry S, Clifford DB, Duggan C, Fagan AM, Fanning K, Farlow MR, Hassenstab J, McDade EM, et al. (2017). The DIAN-TU Next Generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimers Dement 13, 8–19. 10.1016/j.jalz.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berkness T, Carrillo MC, Sperling R, Petersen R, Aisen P, Flournoy C, Snyder H, Raman R, and Grill JD (2021). The Institute on Methods and Protocols for Advancement of Clinical Trials in ADRD (IMPACT-AD): A Novel Clinical Trials Training Program. The Journal of Prevention of Alzheimer’s Disease 8, 286–291. 10.14283/jpad.2021.12. [DOI] [PMC free article] [PubMed] [Google Scholar]